

Abstract
Epidemiological studies have strongly linked caffeine consumption with a reduced risk of developing Parkinson's disease (PD) in men. Interestingly, in women, this inverse association is present only in those who have not taken postmenopausal estrogens, suggesting an interaction between the influences of estrogen and caffeine use on the risk of PD. To explore a possible biological basis for this interaction, we systematically investigated how the neuroprotective effect of caffeine is influenced by gender, ovariectomy (OVX), and then exogenous estrogen in the mouse 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) model of PD. (1) Caffeine treatment produced a dose-dependent attenuation of MPTP-induced striatal dopamine loss in both young and retired breeder (RB) male, but not female, mice. (2) In female mice (both young and RB), caffeine was less potent or altogether ineffective as a neuroprotectant after sham surgery compared to OVX or after OVX plus estrogen replacement compared to OVX plus placebo treatment. (3) Estrogen treatment also prevented the protection of caffeine against dopamine loss in young male mice. (4) Consistent with the putative protective effect of estrogen, female and OVX plus estrogen mice were relatively resistant to MPTP toxicity compared to male and OVX plus placebo mice, respectively. (5) There was no overall difference in brain levels of caffeine and its metabolites between OVX plus placebo and OVX plus estrogen mice. Together, these results suggest that estrogen can occlude and thereby prevent the neuroprotective effect of caffeine in a model of PD neurodegeneration, supporting a biological basis for the interaction between estrogen and caffeine in modifying the risk of PD.
Keywords: adenosine A2A receptor, dopamine, gender, methylxanthine, ovariectomy, striatum
Introduction
Parkinson's disease (PD) is a progressive neurodegenerative disorder that is pathologically well characterized. However, the etiology of PD remains unclear. Twin studies (Tanner et al., 1999; Vieregge et al., 1999; Wirdefeldt et al., 2004) have suggested that nongenetic factors, such as environmental exposures or random cellular events that occur during aging, play a prominent role in promoting the development of typical PD.
A major negative risk factor for PD has been identified recently as the consumption of caffeine. Multiple retrospective as well as several large prospective epidemiological studies have demonstrated that among dietary factors, previous coffee or tea drinking are consistently associated with a reduced risk of developing PD even after accounting for smoking and other potential confounding factors (Benedetti et al., 2000; Ross et al., 2000; Ascherio et al., 2001, 2004). The incidence of PD declines steadily with increasing intake of caffeine or coffee (but not decaffeinated coffee).
The mechanisms that underlie this epidemiological correlation remain unclear. One hypothesis that caffeine might represent a protective environmental factor in PD is supported by our findings that caffeine can protect against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced nigrostriatal neurodegeneration in mice (Chen et al., 2001; Oztas et al., 2002). Moreover, the caffeine metabolites paraxanthine and theophylline provide similar attenuations of MPTP-induced dopaminergic toxicity (Xu et al., 2002b). In contrast to its locomotor stimulant effect, the neuroprotectant effect of caffeine does not show tolerance after chronic caffeine exposure (Xu et al., 2002a). Recently, the protection of caffeine against dopaminergic neuron loss and associated behavior changes was confirmed in the 6-OHDA rat model of PD (Joghataie et al., 2004). Together, the protective effects of caffeine and its metabolites in rodent models of PD support a causal basis for the inverse relationship between human caffeine consumption and the risk of subsequently developing PD.
Interestingly, the negative link between caffeine intake and risk of developing PD has been observed consistently in men but not in women (Benedetti et al., 2000; Ascherio et al., 2001, 2004). Overall, there was no clear relationship between PD and caffeine intake in two large prospectively followed populations of women (Ascherio et al., 2001, 2004). When these women were further divided by their estrogen use status after menopause, a negative association between caffeine intake and risk of PD similar to men was observed in those women who had never used estrogen replacement therapy but not in those who had ever used it (Ascherio et al., 2003, 2004). These results suggest that estrogen replacement therapy may prevent the beneficial effect of caffeine in reducing the risk of developing PD.
To address the possibility that estrogen, which can serve as a neuroprotectant in its own right, may interfere with neuroprotection by caffeine against dopaminergic neurotoxicity, we investigated their interaction in the MPTP mouse model of PD. We first assessed the difference in neuroprotection by caffeine in male and female mice. Then we systematically investigated the effect of exogenous estrogen on the neuroprotection of caffeine. Finally, we explored the effect of estrogen on the metabolism of caffeine as a possible mechanism of interaction between estrogen and caffeine.
Materials and Methods
Animals, ovariectomy, and estrogen replacement. Young (∼10 weeks old) or retired breeder (6–9 months old) male and female C57BL/6 mice (Charles River Laboratories, Wilmington, MA) were used in these experiments. All experiments were performed in accordance with Massachusetts General Hospital and National Institutes of Health guidelines on the ethical use of animals. The mice were housed five per cage with ad libitum access to food and water and were maintained at a constant temperature and humidity with a 12 h light/dark cycle. To remove the main source of female hormones, bilateral ovariectomies were performed under anesthesia using Avertin (2% 2,2,2-tribromoethanol and 1% amyl alcohol; 20 ml/kg, i.p.) at either Charles River Laboratories or Massachusetts General Hospital. Sham operations were also included, in which all of the other procedures were the same, except for removal of the ovaries. In the experiments in which estrogen replacement was used, placebo or estrogen pellets (17β-estradiol, 0.1 mg per pellet, 21 d release; Innovative Research of America, Sarasota, FL) were implanted at the neck of mice under anesthesia 7–10 d after ovariectomy. This estrogen regimen was used because it produces an approximate replacement of physiological levels within the serum in ovariectomized (OVX) mice (Gao and Dluzen, 2001).
Caffeine and MPTP treatment. Different doses of caffeine (5, 10, 20, or 40 mg/kg) or saline were injected intraperitoneally 10 min before MPTP (40 mg/kg, i.p.) or saline injection (n = 3–7 for saline treatments and n = 4–15 for MPTP treatments). The extent of the caffeine dose range used across experiments varied depending on the number of mice available. In experiments in which ovariectomy and/or estrogen pellets were implanted, caffeine and MPTP treatments were performed 10 d after ovariectomy or estrogen implantation, whichever came later, to assure that estrogen levels were depleted or maintained at a constant level, respectively.
Dopamine and 3,4-dihydroxyphenylacetic acid measurement. One week after MPTP treatment, mice were killed by rapid cervical dislocation. The striatum was dissected out from the right cerebral hemisphere, frozen on dry ice, and stored at –80°C until use. Each striatum was weighed and extracted with 150 mm phosphoric acid and 0.2 mm EDTA. The striatum was homogenized and centrifuged at 12,000 × g for 15 min at 4°C. Supernatants were analyzed for dopamine and 3,4-dihydroxyphenylacetic acid (DOPAC) content using standard reverse-phase HPLC with electrochemical detection (ESA, Chelmsford, MA). Biogenic amines were separated on a C-18 5 μm sphere column (Varian, Palo Alto, CA). The mobile phase consisted of 0.1 m sodium phosphate monobasic, 0.1 mm EDTA, 0.18 mm sodium octyl sulfate, and 8% methanol in filtered distilled water. The final pH of 3.3 was obtained with the addition of concentrated phosphoric acid, and the mobile phase was filtered and degassed before use. The dopamine and DOPAC contents were calculated as picomoles per milligram of tissue, and these values are presented within the figures as percentage of change from respective saline–saline-treated controls.
Measurement of caffeine and its metabolites. Ten days after estrogen or placebo pellet implantation, OVX retired breeder female mice were treated with saline or caffeine (5 or 40 mg/kg, i.p.; n = 1 for saline and n = 5 for caffeine). The mice were killed at 10, 30, 60, 120, 180, 240, or 360 min after injection by rapid cervical dislocation. The right cerebral hemisphere was dissected out, frozen on dry ice, and stored at –80°C until use. Each brain tissue was homogenized in 0.1 m monobasic sodium phosphate with a volume 10 times of tissue weight and centrifuged at 12,000 × g for 15 min at 4°C. Supernatants were analyzed using liquid chromatography/mass spectrometry for determination of caffeine and its three metabolites, paraxanthine, theophylline, and theobromine. The lower limit of quantitation was 30 ng/ml. The analysis of caffeine and its metabolites was gratefully performed by Drs. R. L. Foltz and D. Andrenyak (Center for Human Toxicology, University of Utah, Salt Lake City, UT).
Statistical analyses. The data from striatal dopamine and DOPAC content as well as caffeine/metabolite measurements were analyzed by two-way ANOVA. Post hoc comparisons were performed using Fisher's least significant difference test. Data values in the figures represent the group mean ± SEM.
Results
Gender differences in the attenuation of MPTP toxicity by caffeine
The dose dependence of the neuroprotective effect of caffeine in the MPTP model of PD was first compared between intact male and female mice.
Young mice
MPTP treatment depleted the striatal dopamine level measured 1 week later down to 38% of control (saline treated) in ∼10-week-old male mice (Fig. 1A). Caffeine pretreatment attenuated MPTP-induced dopamine loss in a dose-dependent manner in these young males, with a maximal effect (of doubling residual dopamine levels) achieved at 10 mg/kg. In female mice of the same age, MPTP depleted striatal dopamine levels to 62% of control levels (in saline-treated females). This reduction is significantly less than that observed in their male counterparts (p < 0.05), agreeing with previous reports (Brooks et al., 1989; Freyaldenhoven et al., 1996; Miller et al., 1998) that MPTP induced less dopaminergic toxicity in female than male mice. However, in contrast to male mice, female mice showed no attenuation of MPTP toxicity after lower doses (5, 10, or 20 mg/kg) of caffeine pretreatment. Only the highest dose (40 mg/kg) of caffeine protected against MPTP. Simultaneous measurement of DOPAC (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material), the major metabolite of dopamine in mouse, revealed that lower doses of caffeine pretreatment (10 or 20 mg/kg) significantly attenuated MPTP-induced depletion in male mice, whereas only the highest dose of caffeine pretreatment (40 mg/kg) provided similar protection in female mice.
Figure 1.

Caffeine dose-dependently attenuates MPTP-induced dopamine depletion in male but not female C57BL/6 mice, either young (∼10 weeks old; A) or retired breeder (6–9 months old; B). Caffeine (5–40 mg/kg, i.p.) or saline was administered 10 min before a single dose of MPTP (40 mg/kg, i.p.; n = 4–12) or saline (n = 3–6). One week later, striatal dopamine content was determined. The bars represent striatal dopamine levels (mean ± SEM) calculated as a percentage of their respective controls (i.e., S+S group). The dopamine concentrations (in picomoles per milligram of tissue) of these controls are 71.8 ± 2.4 and 70.6 ± 2.6 (A) and 80 ± 9 and 92 ± 9.8 (B) for males and females, respectively. Data were analyzed by two-way ANOVA, followed by Fisher's LSD test. A, *p < 0.05, **p < 0.01, and ***p < 0.001 compared with the respective S+M group; #p < 0.05 compared with the respective C5+M group; +p < 0.05 compared with S+M in male mice; @p < 0.01 compared with C40+Min male mice.B, *p<0.05 and **p<0.01 compared with the respective S+M group; #p<0.05 compared with the respective C10+M group; @p<0.05 compared with C40+M in male mice. S, Saline; M, MPTP; C5–40, caffeine at 5–40 mg/kg.
Retired breeders
We also examined the neuroprotection of caffeine against MPTP toxicity in older (6–9 months old) retired breeder mice (Fig. 1B). Because these mice have passed their peak reproductive age, their comparison may be more relevant to gender differences in epidemiological studies of PD, which is usually diagnosed in people >50 years old. Not surprisingly, MPTP treatment induced a greater biochemical lesion in older mice for either gender (Fig. 1, compare A, B), as reported previously (Irwin et al., 1992). In male retired breeders, MPTP treatment depleted striatal dopamine levels to 8% of unlesioned controls (Fig. 1B). As in the younger male mice, caffeine pretreatment ≥10 mg/kg again significantly attenuated MPTP-induced dopamine depletion. In female retired breeders, MPTP treatment depleted striatal dopamine levels to 23% of control levels. Again, female retired breeder mice also showed less MPTP toxicity compared with that of male mice. However, just as in young mice, pretreatment with only the highest dose of caffeine (40 mg/kg) significantly attenuated the dopamine loss. Caffeine pretreatment, at all doses tested (10, 20, or 40 mg/kg), also attenuated loss of DOPAC in male mice, whereas only higher doses provided similar protection in female mice (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material).
Ovariectomy increases the potency of the protective effect of caffeine
To investigate whether the gender difference in the neuroprotective effects of caffeine may relate to differences in female hormone status, we examined the effect of caffeine on MPTP toxicity in young female mice whose ovaries, the main site of female hormone production, were removed (OVX) or in sham-operated female littermates (Fig. 2). MPTP treatment depleted striatal dopamine levels in both OVX and sham-operated mice. However, the lower dose of caffeine tested significantly increased residual striatal dopamine levels only in OVX females (Fig. 1, males). In contrast, sham-operated females (like intact females) required pretreatment with a higher dose of caffeine for attenuation of MPTP-induced dopamine loss.
Figure 2.

A low dose of caffeine attenuates MPTP-induced dopamine depletion in OVX but not sham-operated young female mice. Ten days after ovariectomy or sham operation, mice received caffeine (5 or 20 mg/kg, i.p.) or saline administration 10 min before a single dose of MPTP (40 mg/kg, i.p.; n = 8) or saline (n = 5–7). One week later, striatal dopamine content was determined. The bars represent striatal dopamine levels (mean ± SEM) calculated as a percentage of their respective controls (i.e., S+S group). The dopamine concentrations of these controls are 61 ± 3.7 and 67 ± 2.7 pmol/mg of tissue for OVX and sham mice, respectively. Data were analyzed by two-way ANOVA, followed by Fisher's LSD test. **p < 0.01 and ***p < 0.001 compared with the respective S+M group; #p < 0.001 compared with the respective C5+M group; @p < 0.01 compared with C5+M in OVX mice. S, Saline; M, MPTP; C5 or C20, caffeine at 5 or 20 mg/kg.
Estrogen attenuates the neuroprotection of caffeine in ovariectomized female mice
Young mice
To determine whether estrogen, the main female sex hormone, can account for the above-identified ovarian influence on neuroprotection by caffeine, we examined the effect of prolonged estrogen replacement on neuroprotection by caffeine in female mice depleted of endogenous estrogen (Fig. 3A). These OVX mice were implanted with subcutaneous pellets containing placebo or estrogen (17β-estradiol), which is continuously released to maintain steady-state concentrations for 21 d. MPTP treatment reduced striatal dopamine content to 27% of control in placebo-treated mice (OVX+P). The same MPTP exposure in OVX mice treated with estrogen pellets (OVX+E) depleted striatal dopamine levels, but to a significantly lesser extent (down to 39% of control), consistent with previous reports (Dluzen et al., 1996; Miller et al., 1998). In these OVX+P mice, pretreatment with caffeine, both at 10 and 20 mg/kg, significantly attenuated MPTP-induced striatal dopamine loss. In mice that were receiving estrogen replacement (OVX+E), however, only the higher dose of caffeine (20 mg/kg) provided significant protection. The striatal levels of DOPAC (supplemental Fig. 2A, available at www.jneurosci.org as supplemental material) demonstrated a similar phenomenon.
Figure 3.

Caffeine attenuates MPTP-induced dopamine depletion in OVX female mice receiving placebo (OVX+P) but not estrogen (OVX+E) replacement (A, young mice; B, retired breeders). Ten days after ovariectomy, mice were implanted with placebo or estrogen pellets (17β-estradiol, 0.1 mg per pellet, 21 d release) subcutaneously. Ten days later, caffeine (5–40 mg/kg, i.p.) or saline was administered 10 min before a single dose of MPTP (40 mg/kg, i.p.; n = 6–15) or saline (n = 3–6). Striatal dopamine content was determined 1 week after MPTP. The bars represent striatal dopamine levels (mean ± SEM) calculated as a percentage of their respective controls (i.e., S+S group). The dopamine concentrations (in picomoles per milligram of tissue) of these controls are 72 ± 4 and 78 ± 4.7 (A) and 78.9 ± 7.3 and 73.9 ± 7.7 (B) for males and females, respectively. Data were analyzed by two-way ANOVA, followed by Fisher's LSD test. A, **p < 0.01 and ***p < 0.001 compared with the respective S+M group; #p < 0.05 compared with the respective C5+M and C10+M groups; +p < 0.05 compared with the respective S+M or C5+M in OVX+P mice. B, *p < 0.05, **p < 0.01, and ***p < 0.001 compared with the respective S+M group; #p < 0.001 compared with the respective C5+M, C10+M, or C20+M group; @p < 0.05 compared with C40+M in OVX+P mice. S, Saline; M, MPTP; C5–40, caffeine at 5–40 mg/kg.
Retired breeders
To better model estrogen replacement in the menopausal state, a parallel experiment was conducted in retired breeder (rather than young) mice (Fig. 3B). Again, estrogen by itself significantly attenuated MPTP-induced striatal dopamine loss. MPTP depleted dopamine levels to 23% of control in OVX+P mice, whereas it reduced them to 36% of control in OVX+E mice. In keeping with observations in young mice, caffeine pretreatments (5, 20, or 40 mg/kg) significantly attenuated MPTP-induced dopamine loss in the OVX+P retired breeder mice, with complete reversal achieved at the highest dose of caffeine. However, in the setting of estrogen replacement in these OVX retired breeders, caffeine pretreatment did not confer protection at any dose tested. The measurement of DOPAC (supplemental Fig. 2B, available at www.jneurosci.org as supplemental material) yielded a similar set of findings, with caffeine pretreatment (20 or 40 mg/kg) significantly reducing DOPAC loss in OVX+P but not OVX+E mice. Together, these results demonstrate that estrogen replacement can reduce or abolish the neuroprotective effect of caffeine on MPTP toxicity in young and older OVX females.
Estrogen can prevent neuroprotection by caffeine in male mice
The above effects of manipulating endogenous and exogenous estrogen in female mice suggest that estrogen status may be the key factor accounting for the gender difference in the neuroprotective action of caffeine in the MPTP model of PD. To determine whether the neuroprotective effect of caffeine in males indeed relies on the absence or relatively low levels of estrogen, we assessed the effects of caffeine on MPTP toxicity in male mice implanted with placebo or estrogen pellets (Fig. 4). MPTP treatment depleted striatal dopamine levels to 44% and 55% of their respective control level in male mice pretreated for 21 d with placebo or estrogen, suggestive of a mild protective estrogen effect as repeatedly observed above. In the placebo-implanted males, caffeine pretreatments significantly attenuated striatal dopamine loss. However, no dose of caffeine pretreatment protected the male mice treated with estrogen pellets. These results demonstrate that estrogen can abolish the neuroprotection of caffeine not only in female mice but also in male mice.
Figure 4.

Caffeine attenuates MPTP-induced dopamine depletion in young male mice receiving placebo but not estrogen treatment. Mice were implanted with placebo or estrogen pellets. Ten days later, caffeine (10–40 mg/kg, i.p.) or saline was administered 10 min before a single dose of MPTP (40 mg/kg, i.p.; n = 8–9) or saline (n = 3). Striatal dopamine content was determined 1 week after MPTP. The bars represent striatal dopamine levels (mean ± SEM) calculated as a percentage of their respective controls (i.e., S+S group). The dopamine concentrations of these controls are 79 ± 10.6 and 80.6 ± 4.2 pmol/mg of tissue for placebo- and estrogen-treated mice, respectively. Data were analyzed by two-way ANOVA, followed by Fisher's LSD test. *p < 0.05 and **p < 0.01 compared with the respective S+M group. S, Saline; M, MPTP; C10–40, caffeine at 10–40 mg/kg.
The effect of estrogen on caffeine metabolism in ovariectomized female mice
As a first step to understanding the mechanism of the influence of estrogen on neuroprotection by caffeine, we measured the brain levels of caffeine (trimethylxanthine) and its major demethylation metabolites (dimethylxanthines; i.e., paraxanthine, theophylline, and theobromine) at multiple time points after treatment with caffeine in OVX retired breeder female mice treated with placebo or estrogen pellets. Because estrogen status most consistently modulated the protective effect of lower doses of caffeine, we assessed the effect of estrogen on the CNS pharmacokinetics of caffeine administered intraperitoneally at 5 mg/kg (Fig. 5A). Brain concentrations of caffeine, which were maximal within 10 min of injection and declined with a half-life of ∼50 min, were indistinguishable between the estrogen and placebo groups at all time points measured. When caffeine was administered at this low dose, the concentrations of its metabolites were below the limits of reliable detection. There was no measurable caffeine after saline injection.
Figure 5.
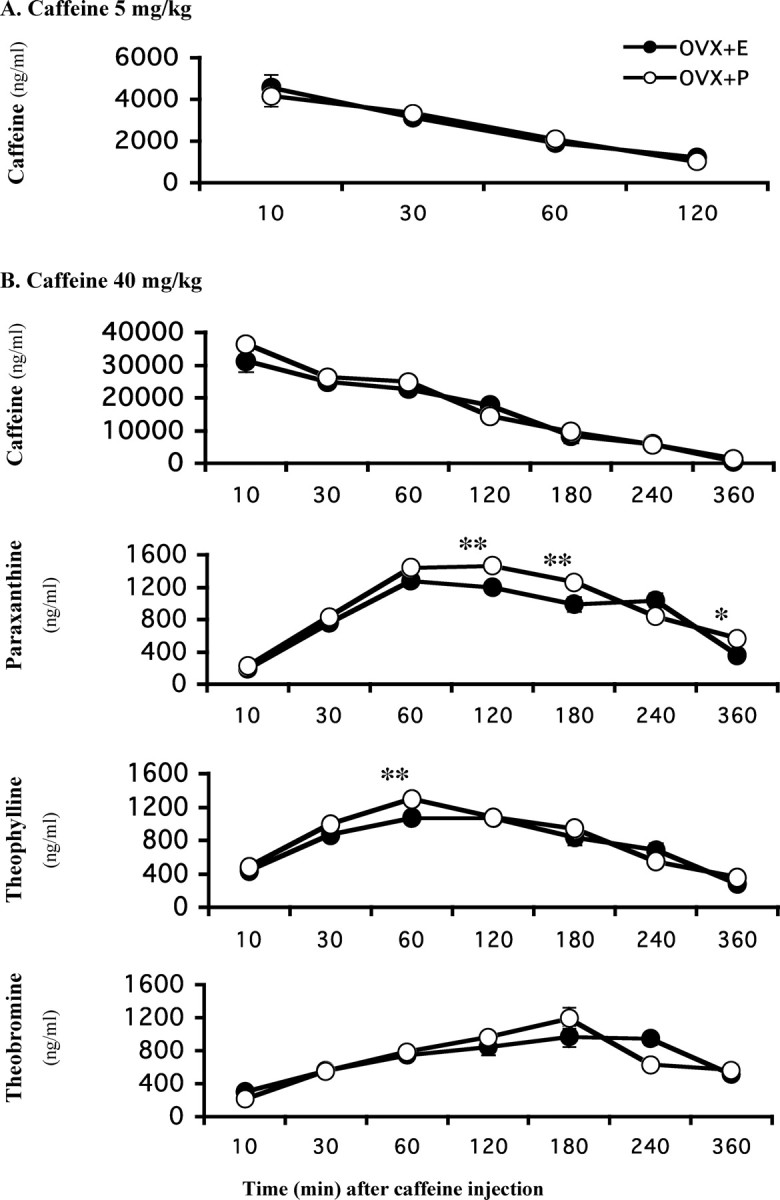
Caffeine metabolism is not changed by estrogen replacement in OVX retired breeder female mice. Ten days after ovariectomy, mice were implanted with placebo or estrogen pellets. Caffeine (5 or 40 mg/kg, i.p.) was administered 10 d after pellet implantation. Brain concentrations of caffeine and its metabolites were measured at 10, 30, 60, 120, 180, 240, or 360 min after injection (n = 5 for each time point). A, Brain caffeine concentration after intraperitoneal 5 mg/kg caffeine. The levels of metabolites are below the detection limit. B, Brain concentrations of caffeine, paraxanthine, theophylline, and theobromine, respectively, after intraperitoneal 40 mg/kg caffeine. Data were analyzed by two-way ANOVA, followed by Fisher's LSD test. *p < 0.05 and **p < 0.01 compared with the respective estrogen-treated group.
As demonstrated above (Fig. 3B), in these OVX retired breeder mice, caffeine pretreatments attenuated dopamine depletion in placebo-treated but not in estrogen-treated mice, with the biggest difference found in the 40 mg/kg caffeine group. Therefore, we also determined brain concentrations of caffeine and its metabolites after intraperitoneal injection of caffeine at 40 mg/kg (Fig. 5B). There was no difference in brain caffeine concentrations between OVX+E and OVX+P mice. There was a small effect of estrogen on brain paraxanthine concentrations, which were slightly but significantly higher in OVX+P than in OVX+E mice at 120, 180, and 360 min. There was also a minor effect on brain theophylline concentrations, which were slightly higher in OVX+P than OVX+E mice 60 min after caffeine injection. There was no difference in brain theobromine concentrations after caffeine injection. Thus, exposure to estrogen, under conditions that attenuated the neuroprotective effect of caffeine, had no effect on brain concentrations of caffeine. Moreover, the overall concentrations of the three caffeine dimethyl metabolites even when combined are much (an order of magnitude) lower than that of caffeine such that the slight differences in metabolite concentrations found at some time points are not sufficient to explain the difference in caffeine neuroprotection between OVX+E and OVX+P mice.
Discussion
The present data reveal a novel interaction between estrogen and caffeine in a mouse model of PD, an interaction that recapitulates the relationship between estrogen and caffeine exposures in the human epidemiology of PD. In male but not female mice, low doses of caffeine attenuated MPTP-induced depletion of striatal dopamine and DOPAC content, functional markers of the nigrostriatal neurons that degenerate in PD. Remarkably, neuroprotection by low doses of caffeine was abolished in the presence of estrogen (either endogenous or exogenous) in young and retired breeder OVX female mice. Moreover, estrogen placement in male mice effectively replicated the female phenotype of attenuated neuroprotection by caffeine.
Estrogen treatment by itself protects against MPTP toxicity
Our data, in agreement with previous reports (Brooks et al., 1989; Freyaldenhoven et al., 1996; Miller et al., 1998), demonstrated that MPTP induced greater striatal dopamine loss in male than female mice. These experimental findings are consistent with epidemiological studies, which have generally demonstrated a higher prevalence of PD in men than women (Dluzen et al., 1998; Baldereschi et al., 2000; Ascherio et al., 2001, 2004). The gender differences observed in PD patients and animal models support the possibility that estrogen may play a protective role in dopaminergic neurodegeneration. Indeed, our current data confirm the neuroprotective effect of estrogen replacement therapy in this mouse model of PD (Dluzen et al., 1996; Callier et al., 2001; Ramirez et al., 2003).
Attenuation of MPTP toxicity by caffeine is diminished in the presence of estrogen
The current data confirm our previous findings (Chen et al., 2001; Xu et al., 2002a,b) that caffeine dose-dependently attenuates MPTP-induced striatal dopamine loss in male mice. Moreover, our data demonstrate that this phenomenon persists as animals age. These findings support a biological basis for a causal epidemiological correlation between caffeine consumption and reduced risk of developing PD.
Strikingly, the protection by lower doses of caffeine against MPTP-induced dopamine depletion was lost in female mice. The gender difference in the dose–response relationship of caffeine with neuroprotection can be described pharmacologically as a reduction in the potency of caffeine in female compared with male mice. In both young and retired breeder mice (Fig. 1), an ED50 value of ≤5 mg/kg is apparent in males compared with ≥20 mg/kg in females. As we discussed above, female mice are less vulnerable than males to MPTP toxicity, raising the possibility that attenuated protection by caffeine in females may also reflect a “ceiling effect” rather than reduced potency. However, the loss of protection in both moderate and severe lesions together with repeatedly significant protection achieved by the highest dose of caffeine in female mice of different ages argue against an impenetrable ceiling and support a lower potency in females. The current data correlate well with the epidemiological studies that showed no inverse association between caffeine or coffee intake and risk of PD in women (Benedetti et al., 2000; Ascherio et al., 2001). The neuroprotection observed in female mice was restricted to the highest caffeine dose of 40 mg/kg and may not be relevant to PD epidemiology. This dose in rodents may be comparable to a human exposure to the caffeine in some 10 cups of coffee (Fredholm et al., 1999), which exceeds the daily consumption of almost any human subject in PD epidemiology studies.
The most notable finding of the current study is that the presence of the major female steroid hormone estrogen prevents neuroprotection by caffeine (or reduces its potency) in female and male mice. The estrogen effects observed in young mice were more robust in mice past their reproductive prime, highlighting the relevance of these findings to epidemiological studies linking PD risk to caffeine and estrogen exposures of middle-aged adults. These data systematically demonstrate that estrogen treatment attenuates the protection of caffeine against MPTP toxicity in this mouse model of PD, providing a possible biological basis for the epidemiological findings that an interaction between estrogen and caffeine exposure modulates the risk of PD (Ascherio et al., 2003, 2004).
Mechanism of interaction between caffeine and estrogen
In pursuing the mechanism, a pharmacokinetic or metabolic interaction is an important consideration. Human data (Patwardhan et al., 1980; Abernethy and Todd, 1985; Pollock et al., 1999) have demonstrated that caffeine metabolism is inhibited in women taking estrogen-containing oral contraceptives or estrogen replacement therapy after menopause, suggesting that the actions of caffeine would be enhanced rather than attenuated by estrogen. However, our data show no difference in brain caffeine concentrations over the time course (extending beyond three half-lives) of measurement in OVX retired breeder female mice treated with either estrogen or placebo.
Interestingly, two metabolites of caffeine (paraxanthine and theophylline) have been shown to provide protection against MPTP-induced dopaminergic toxicity with potencies comparable to caffeine (Xu et al., 2002b). So paradoxically, delaying or inhibiting the metabolism of caffeine could result in a decreased level of these metabolites, which might cause reduced protection against neurotoxicity after estrogen therapy in human or animals. In the present study, however, although there were slight differences in the brain concentrations of paraxanthine and theophylline between estrogen- and placebo-treated mice, the levels of these metabolites were much lower (>25 times) than that of caffeine, therefore arguing against their involvement.
It is interesting that although both estrogen and caffeine individually protect nigrostriatal dopaminergic neurons, when combined, estrogen diminished the potency (rather than enhancing the efficacy) of the neuroprotective action of caffeine. This finding suggests that these two agents may work through a common mechanism to prevent MPTP toxicity. Estrogen may compete with caffeine for its activation of a protective pathway, effectively “occluding” the pathway and shifting the dose–response curve of caffeine for neuroprotection to the right. It is worth noting that estrogen and caffeine as well as MPTP share a cytochrome P450, CYP1A2, for their metabolism or detoxification (Gu et al., 1992; Tassaneeyakul et al., 1994; Yamazaki et al., 1998; Forsyth et al., 2000). Therefore, metabolism of caffeine and/or estrogen could alter MPTP metabolism. However, because CYP1A2 contributes to hepatic detoxification of MPTP (Forsyth et al., 2000), competition for this enzyme by caffeine or estrogen would be expected to exacerbate rather than ameliorate MPTP toxicity. Moreover, because the brain MPTP levels were unaffected by acute caffeine treatment (Chen et al., 2001), it is unlikely that estrogen modulates the protection of caffeine through its action on MPTP metabolism. Alternatively, estrogen may noncompetitively inhibit the neuroprotective effect of caffeine while it provides its own independent protection effect. Our findings that higher doses of caffeine generally produce additional protection, however, argue against a simple noncompetitive inhibition.
Caffeine at the doses used in the present study, which produced peak brain concentrations of ∼4–40 μg/ml, most likely functions as an antagonist of adenosine A1 and A2A receptors (Fredholm et al., 1999). Our previous study (Chen et al., 2001) indicates that the attenuated neurotoxicity of caffeine likely involves its antagonism at the A2A receptor (A2AR). Therefore, estrogen might directly modulate the neuroprotection by caffeine through an action on the A2AR, although there is no information currently on such an interaction. It is reported (Rose'Meyer et al., 2003) that A2AR expression is significantly decreased after OVX. However, we found no difference in striatal A2AR binding densities between OVX mice treated with estrogen and placebo (data not shown). Among the potential mechanisms for neuroprotection by caffeine (and more specific A2A antagonists (for review, see Xu et al., 2005), the attenuation of glutamate release and of resultant excitotoxicity may be a protective mechanism shared with estrogen (O'Neill et al., 2004; Ritz et al., 2004; Saleh et al., 2004). Interestingly, it has been reported that estrogen can alter dopamine D2 receptor (D2R)/G-protein coupling (Levesque and Di Paolo, 1993; Thompson and Certain, 2005) and D2R affinity (Di Paolo et al., 1988). Because it is well known that A2ARs colocalize with D2Rs in the basal ganglia and interact with each other closely (for review, see Xu et al., 2005), it is possible that estrogen could modulate A2AR through its action on D2R. Whether these or other shared mechanisms of neuroprotection by caffeine and estrogen can account for their interaction in the MPTP model and in the epidemiology of PD remains to be clarified.
Relevance of an estrogen–caffeine interaction for PD
The attenuation of the neuroprotective effect of caffeine by estrogen in the MPTP model of PD establishes a potential neural basis for the epidemiological association between estrogen replacement therapy, caffeine consumption, and risk of developing PD in women. This convergence of laboratory and human epidemiological findings strengthens the contention that widely used exogenous estrogen, which may provide neuroprotection by itself, interacts with the nearly ubiquitously consumed A2A antagonist caffeine to modify the risk of developing PD. The present study also establishes an animal model of this interaction in humans, along with the opportunity to pursue its molecular mechanism(s). In addition, the cumulative evidence for this interaction is sufficiently compelling to influence the design and interpretation of neuroprotection trials of estrogen or caffeine currently underway or under consideration (Ravina et al., 2003). Ultimately, a better understanding of the interplay between environmental factors like caffeine and estrogen may suggest effective preventative as well as therapeutic strategies for this neurodegenerative disorder.
Footnotes
This work was supported by National Institutes of Health Grant ES10804, Department of Defense Grant W81XWH-04-1-0881, and the Paul Beeson Faculty Scholars Program. We thank Angela Scibelli for excellent technical assistance. We thank Drs. Hari Singh, Rodger Foltz, and David Andrenyak for analysis of caffeine and its metabolites under the National Institute on Drug Abuse Drug Supply and Analytical Services program/DA013508.
Correspondence should be addressed to Dr. Kui Xu, Molecular Neurobiology Laboratory, Department of Neurology, MassGeneral Institute for Neurodegenerative Disease, Massachusetts General Hospital and Harvard Medical School, 114 16th Street, Room 2925, Charlestown, MA 02129. E-mail: xu@helix.mgh.harvard.edu.
DOI:10.1523/JNEUROSCI.3008-05.2006
Copyright © 2006 Society for Neuroscience 0270-6474/06/260535-07$15.00/0
References
- Abernethy DR, Todd EL (1985) Impairment of caffeine clearance by chronic use of low-dose oestrogen-containing oral contraceptives. Eur J Clin Pharmacol 28: 425–428. [DOI] [PubMed] [Google Scholar]
- Ascherio A, Zhang SM, Hernan MA, Kawachi I, Colditz GA, Speizer FE, Willett WC (2001) Prospective study of caffeine consumption and risk of Parkinson's disease in men and women. Ann Neurol 50: 56–63. [DOI] [PubMed] [Google Scholar]
- Ascherio A, Chen H, Schwarzschild MA, Zhang SM, Colditz GA, Speizer FE (2003) Caffeine, postmenopausal estrogen, and risk of Parkinson's disease. Neurology 60: 790–795. [DOI] [PubMed] [Google Scholar]
- Ascherio A, Weisskopf MG, O'Reilly EJ, McCullough ML, Calle EE, Rodriguez C, Thun MJ (2004) Coffee consumption, gender, and Parkinson's disease mortality in the cancer prevention study II cohort: the modifying effects of estrogen. Am J Epidemiol 160: 977–984. [DOI] [PubMed] [Google Scholar]
- Baldereschi M, Di Carlo A, Rocca WA, Vanni P, Maggi S, Perissinotto E, Grigoletto F, Amaducci L, Inzitari D (2000) Parkinson's disease and parkinsonism in a longitudinal study: two-fold higher incidence in men. ILSA Working Group. Italian Longitudinal Study on Aging. Neurology 55: 1358–1363. [DOI] [PubMed] [Google Scholar]
- Benedetti MD, Bower JH, Maraganore DM, McDonnell SK, Peterson BJ, Ahlskog JE, Schaid DJ, Rocca WA (2000) Smoking, alcohol, and coffee consumption preceding Parkinson's disease: a case-control study. Neurology 55: 1350–1358. [DOI] [PubMed] [Google Scholar]
- Brooks WJ, Jarvis MF, Wagner GC (1989) Influence of sex, age and strain on MPTP-induced neurotoxicity. Res Commun Subst Abuse 10: 181–184. [Google Scholar]
- Callier S, Morissette M, Grandbois M, Pelaprat D, Di Paolo T (2001) Neuroprotective properties of 17beta-estradiol, progesterone, and raloxifene in MPTP C57Bl/6 mice. Synapse 41: 131–138. [DOI] [PubMed] [Google Scholar]
- Chen JF, Xu K, Petzer JP, Staal R, Xu YH, Beilstein M, Sonsalla PK, Castagnoli K, Castagnoli Jr N, Schwarzschild MA (2001) Neuroprotection by caffeine and A2A adenosine receptor inactivation in a model of Parkinson's disease. J Neurosci 21: RC143(1–6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Paolo T, Falardeau P, Morissette M (1988) Striatal D-2 dopamine agonist binding sites fluctuate during the rat estrous cycle. Life Sci 43: 665–672. [DOI] [PubMed] [Google Scholar]
- Dluzen DE, McDermott JL, Liu B (1996) Estrogen alters MPTP-induced neurotoxicity in female mice: effects on striatal dopamine concentrations and release. J Neurochem 66: 658–666. [DOI] [PubMed] [Google Scholar]
- Dluzen DE, Disshon KA, McDermott JL (1998) Estrogen as a modulator of striatal dopaminergic neurotoxicity. In: Recent advances in neurodegenerative disorders (Marwah J, Tietelbaum H, eds), pp 149–192. Scottsdale, AZ: Prominent.
- Forsyth JT, Grunewald RA, Rostami-Hodjegan A, Lennard MS, Sagar HJ, Tucker GT (2000) Parkinson's disease and CYP1A2 activity. Br J Clin Pharmacol 50: 303–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredholm BB, Battig K, Holmen J, Nehlig A, Zvartau EE (1999) Actions of caffeine in the brain with special reference to factors that contribute to its widespread use. Pharmacol Rev 51: 83–133. [PubMed] [Google Scholar]
- Freyaldenhoven TE, Cadet JL, Ali SF (1996) The dopamine-depleting effects of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine in CD-1 mice are gender-dependent. Brain Res 735: 232–238. [DOI] [PubMed] [Google Scholar]
- Gao X, Dluzen DE (2001) Tamoxifen abolishes estrogen's neuroprotective effect upon methamphetamine neurotoxicity of the nigrostriatal dopaminergic system. Neuroscience 103: 385–394. [DOI] [PubMed] [Google Scholar]
- Gu L, Gonzalez FJ, Kalow W, Tang BK (1992) Biotransformation of caffeine, paraxanthine, theobromine and theophylline by cDNA-expressed human CYP1A2 and CYP2E1. Pharmacogenetics 2: 73–77. [DOI] [PubMed] [Google Scholar]
- Irwin I, Finnegan KT, Delanney LE, Di Monte D, Langston JW (1992) The relationships between aging, monoamine oxidase, striatal dopamine and the effects of MPTP in C57BL/6 mice: a critical reassessment. Brain Res 572: 224–231. [DOI] [PubMed] [Google Scholar]
- Joghataie MT, Roghani M, Negahdar F, Hashemi L (2004) Protective effect of caffeine against neurodegeneration in a model of Parkinson's disease in rat: behavioral and histochemical evidence. Parkinsonism Relat Disord 10: 465–468. [DOI] [PubMed] [Google Scholar]
- Levesque D, Di Paolo T (1993) Modulation by estradiol and progesterone of the GTP effect on striatal D-2 dopamine receptors. Biochem Pharmacol 45: 723–733. [DOI] [PubMed] [Google Scholar]
- Miller DB, Ali SF, O'Callaghan JP, Laws SC (1998) The impact of gender and estrogen on striatal dopaminergic neurotoxicity. Ann NY Acad Sci 844: 153–165. [PubMed] [Google Scholar]
- O'Neill K, Chen S, Brinton RD (2004) Impact of the selective estrogen receptor modulator, raloxifene, on neuronal survival and outgrowth following toxic insults associated with aging and Alzheimer's disease. Exp Neurol 185: 63–80. [DOI] [PubMed] [Google Scholar]
- Oztas E, Kalda A, Xu K, Irrizary MC, Schwarzschild MA, Chen JF (2002) Caffeine attenuates MPTP-induced loss of dopaminergic neurons in substantia nigra in mice. Soc Neurosci Abstr 28: 487.6. [Google Scholar]
- Patwardhan RV, Desmond PV, Johnson RF, Schenker S (1980) Impaired elimination of caffeine by oral contraceptive steroids. J Lab Clin Med 95: 603–608. [PubMed] [Google Scholar]
- Pollock BG, Wylie M, Stack JA, Sorisio DA, Thompson DS, Kirshner MA, Folan MM, Condifer KA (1999) Inhibition of caffeine metabolism by estrogen replacement therapy in postmenopausal women. J Clin Pharmacol 39: 936–940. [DOI] [PubMed] [Google Scholar]
- Ramirez AD, Liu X, Menniti FS (2003) Repeated estradiol treatment prevents MPTP-induced dopamine depletion in male mice. Neuroendocrinology 77: 223–231. [DOI] [PubMed] [Google Scholar]
- Ravina BM, Fagan SC, Hart RG, Hovinga CA, Murphy DD, Dawson TM, Marler JR (2003) Neuroprotective agents for clinical trials in Parkinson's disease: a systematic assessment. Neurology 60: 1234–1240. [DOI] [PubMed] [Google Scholar]
- Ritz MF, Schmidt P, Mendelowitsch A (2004) Acute effects of 17betaestradiol on the extracellular concentration of excitatory amino acids and energy metabolites during transient cerebral ischemia in male rats. Brain Res 1022: 157–163. [DOI] [PubMed] [Google Scholar]
- Rose'Meyer RB, Mellick AS, Garnham BG, Harrison GJ, Massa HM, Griffiths LR (2003) The measurement of adenosine and estrogen receptor expression in rat brains following ovariectomy using quantitative PCR analysis. Brain Res Brain Res Protoc 11: 9–18. [DOI] [PubMed] [Google Scholar]
- Ross GW, Abbott RD, Petrovitch H, Morens DM, Grandinetti A, Tung KH, Tanner CM, Masaki KH, Blanchette PL, Curb JD, Popper JS, White LR (2000) Association of coffee and caffeine intake with the risk of Parkinson disease. JAMA 283: 2674–2679. [DOI] [PubMed] [Google Scholar]
- Saleh TM, Connell BJ, Legge C, Cribb AE (2004) Estrogen attenuates neuronal excitability in the insular cortex following middle cerebral artery occlusion. Brain Res 1018: 119–129. [DOI] [PubMed] [Google Scholar]
- Tanner CM, Ottman R, Goldman SM, Ellenberg J, Chan P, Mayeux R, Langston JW (1999) Parkinson disease in twins: an etiologic study. JAMA 281: 341–346. [DOI] [PubMed] [Google Scholar]
- Tassaneeyakul W, Birkett DJ, McManus ME, Veronese ME, Andersson T, Tukey RH, Miners JO (1994) Caffeine metabolism by human hepatic cytochromes P450: contributions of 1A2, 2E1 and 3A isoforms. Biochem Pharmacol 47: 1767–1776. [DOI] [PubMed] [Google Scholar]
- Thompson TL, Certain ME (2005) Estrogen mediated inhibition of dopamine transport in the striatum: regulation by G alpha i/o. Eur J Pharmacol 511: 121–126. [DOI] [PubMed] [Google Scholar]
- Vieregge P, Hagenah J, Heberlein I, Klein C, Ludin HP (1999) Parkinson's disease in twins: a follow-up study. Neurology 53: 566–572. [DOI] [PubMed] [Google Scholar]
- Wirdefeldt K, Gatz M, Schalling M, Pedersen NL (2004) No evidence for heritability of Parkinson disease in Swedish twins. Neurology 63: 305–311. [DOI] [PubMed] [Google Scholar]
- Xu K, Xu YH, Chen JF, Schwarzschild MA (2002a) Caffeine's neuroprotection against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine toxicity shows no tolerance to chronic caffeine administration in mice. Neurosci Lett 322: 13–16. [DOI] [PubMed] [Google Scholar]
- Xu K, Xu YH, Chen JF, Schwarzschild MA (2002b) Neuroprotection by metabolites of caffeine may prolong its neuroprotection in the MPTP model of Parkinson's disease. Soc Neurosci Abstr 28: 487.5. [Google Scholar]
- Xu K, Bastia E, Schwarzschild M (2005) Therapeutic potential of adenosine A2Areceptor antagonists in Parkinson's disease. Pharmacol Ther 105: 267–310. [DOI] [PubMed] [Google Scholar]
- Yamazaki H, Shaw PM, Guengerich FP, Shimada T (1998) Roles of cytochromes P450 1A2 and 3A4 in the oxidation of estradiol and estrone in human liver microsomes. Chem Res Toxicol 11: 659–665. [DOI] [PubMed] [Google Scholar]