

Abstract
A corollary of the neuroinflammation hypothesis is that selective suppression of neurotoxic products produced by excessive glial activation will result in neuroprotection. We report here that daily oral administration to mice of the brain-penetrant compound 4,6-diphenyl-3-(4-(pyrimidin-2-yl)piperazin-1-yl)pyridazine (MW01-5-188WH), a selective inhibitor of proinflammatory cytokine production by activated glia, suppressed the human amyloid-β (Aβ) 1-42-induced upregulation of interleukin-1β, tumor necrosis factor-α, and S100B in the hippocampus. Suppression of neuroinflammation was accompanied by restoration of hippocampal synaptic dysfunction markers synaptophysin and postsynaptic density-95 back toward control levels. Consistent with the neuropathophysiological improvements, MW01-5-188WH therapy attenuated deficits in Y maze behavior, a hippocampal-linked task. Oral MW01-5-188WH therapy begun 3 weeks after initiation of intracerebroventricular infusion of human Aβ decreased the numbers of activated astrocytes and microglia and the cytokine levels in the hippocampus without modifying amyloid plaque burden or altering peripheral tissue cytokine upregulation in response to an in vivo inflammatory challenge. The results provide a novel integrative chemical biology proof in support of the neuroinflammation hypothesis of disease progression, demonstrate that neurodegeneration can be attenuated independently of plaque modulation by targeting innate brain proinflammatory cytokine responses, and indicate the feasibility of developing efficacious, safe, and selective therapies for neurodegenerative disorders by targeting key glial activation pathways.
Keywords: neuroinflammation, neurodegeneration, glia, cytokines, behavior, Alzheimer's disease
Introduction
Alzheimer's disease (AD) and related neurodegenerative disorders represent a major cause of disability and death, with an unmet need for therapies that alter disease progression. Clinical and preclinical studies revealed a previously underappreciated role of excessive glial activation, or neuroinflammation, in the pathophysiology of these progressive neurodegenerative disorders (Griffin et al., 1989, 1998; Wright, 2002; Craft et al., 2005a,b,c; Miller, 2005; Tuppo and Arias, 2005; Weydt and Moller, 2005) and raised the hypothesis that targeting of neuroinflammation might be a therapeutic approach to altering disease progression (Frautschy et al., 2001; Jantzen et al., 2002; Yan et al., 2003; Craft et al., 2004a,b, 2005c,d). However, most investigations testing this hypothesis used drugs approved for other clinical indications or developed using approaches that did not have suppression of glia activation as the primary focus, yielding mixed results (Aisen et al., 2003; Breitner, 2003; Cole et al., 2004; Reines et al., 2004). It is becoming more generally appreciated that a drug targeting peripheral inflammation may not be the most appropriate therapeutic for neuroinflammation and that a new paradigm starting with a focus on selective modulation of activated glia mechanisms relevant to disease is needed. However, it is not clear if selective targeting of proinflammatory glia responses is a possibility.
The lack of a precedent in neurosciences reflects the difficulty in meeting a perhaps insurmountable set of demands. For example, analysis of previous failures in CNS drug development identifies several key criteria that should be addressed in preclinical investigations. First, rapid blood-brain barrier penetration of compounds is necessary to attain a pharmacologically relevant drug concentration within a useful time window. Lack of brain penetration is a major point of failure for compounds identified by activity in ex vivo studies (Pardridge, 2005). Second, oral administration of chronically administered drugs is desirable for improved patient compliance, so oral bioavailability should be high (Wilkinson, 2001). Third, efficacy with compound administered at a dose of ≤5 mg/kg body weight is ideal (Galli and Faller, 2003). Appropriate pharmacokinetics and dosing portend well for efficacy without idiopathic toxic effects (e.g., liver injury). Fourth, minimal suppression of peripheral inflammatory responses at a drug dose that produces CNS therapeutic effects is desirable but has not been achieved by current anti-inflammatory drugs. A de novo focus on suppression of selective, disease-linked glia responses and the availability of animal models that exhibit disease-relevant pathophysiology allow for the possible discovery and use of such novel bioavailable compounds.
To address this unmet scientific and medical need, we developed a unique, orally bioavailable and brain-penetrant compound [4,6-diphenyl-3-(4-(pyrimidin-2-yl)piperazin-1-yl)pyridazine (MW01-5-188WH)] that is an inhibitor of glial activation. We show here that chronic treatment of mice with MW01-5-188WH suppresses proinflammatory cytokine responses linked to AD-related pathophysiology, with resultant protection against hippocampus synaptic dysfunction and hippocampal-dependent behavioral deficits. These integrative chemical biology results add a causal link to the increasing evidence that couples increased proinflammatory cytokine production with neurodegenerative pathophysiology, and demonstrate the feasibility of developing new therapies for AD by targeting glia activation pathways.
Materials and Methods
MW01-5-188WH. The compound MW01-5-188WH was synthesized in the production of a proprietary small molecule library designed by chemical diversification of an inactive pyridazine fragment (Hu et al., 2005; Wing et al., 2006). MW01-5-188WH was initially selected based on endpoints in a hierarchal, cell-based screen (Mirzoeva et al., 1999, 2002; Hu et al., 2005) that identifies concentration-dependent, selective inhibitors of activated glia. Specifically, MW01-5-188WH was made by first synthesizing 3-chloro-4,6-diphenylpyridazine, starting with commercially available 4-oxo-2,4-diphenylbutanoic acid and then using a previously described multistep synthesis (Sircar, 1983; Coudert et al., 1988). The resultant, purified product of the multistep synthesis was reacted with commercially available 2-(piperazin-1-yl)pyrimidine in 1-BuOH at 130°C to give the novel 4,6-diphenyl-3-(4-(pyrimidin-2-yl)piperazin-1-yl)pyridazine. The solvent was removed by evaporation under vacuum, the residue was suspended in water, collected by filtration, washed with water, and dried over a filter funnel to give a light pink solid. The isolated yield was 81.1%. Homogeneity was >95%, as judged by HPLC, and the experimentally determined mass of 395.1973, detected by high-resolution mass spectrometry, was in good agreement with the computed expected mass of 395.1979 (M+H+). Structure was confirmed by 1H NMR (CDCl3): 8.356 (d, J = 4.5, 2H), 8.011 (d, J = 7.5, 2H), 7.692 (d, J = 9.5, 1H), 7.468 (t, J = 6.0, 2H), 7.417 (d, J = 7.5, 1H), 7.047 (d, J = 9.5, 1H), 6.546 (t, J = 4.5, 1H), 4.013 (t, J = 5.0, 4H), 3.826 (t, J = 5.0, 4H).
Cell culture assays. Cell-based assays of the concentration-dependent activity of MW01-5-188WH were done as described previously (Mirzoeva et al., 2002; Hu et al., 2005). BV-2 mouse microglial cells (1.25 × 104 cells per well in a 48-well plate) were cultured for 1 d in αMEM media containing 10% fetal bovine serum and then treated in serum-free media for 16 h with either control buffer or the standard glial-activating stimulus lipopolysaccharide (LPS; from Salmonella typhimurium; 100 ng/ml final concentration) in the presence of diluent or MW01-5-188WH. Stock solutions (20 mm) of MW01-5-188WH are prepared in DMSO. Solutions for cell treatments are prepared by dilution of stock solutions into serum-free media immediately before adding to the cells. Control wells contain the same final concentration of DMSO as the compound-containing wells, and we have determined previously that this concentration of DMSO is not toxic to the cells (Mirzoeva et al., 1999). The accumulation of nitrite, the stable metabolite of nitric oxide (NO), was measured in BV-2-conditioned media by the Griess assay as described previously (Mirzoeva et al., 1999, 2002; Hu et al., 2005). Levels of interleukin-1β (IL-1β) in cell lysates and tumor necrosis factor-α (TNF-α) in conditioned media were measured by ELISA (Biosource International, Camarillo, CA) as per the manufacturer's instructions. Cell lysates were analyzed by Western blots as described previously (Mirzoeva et al., 2002) to determine the levels of inducible nitric oxide synthase (iNOS), cyclooxygenase-2 (COX-2), and apolipoprotein E (apoE). For apoE measurements, rat primary mixed glia were prepared and stimulated with human oligomeric amyloid-β 1-42 (Aβ1-42; 10 μm) as described previously (Mirzoeva et al., 2002). Antibodies and dilutions used for Western blots were anti-COX-2 (1:1000; Santa Cruz Biotechnology, Santa Cruz, CA), anti-iNOS (1:1000; Transduction Laboratories, Lexington, KY), and anti-apoE (1:1000). Antibody against β-actin (1:500,000 dilution; Sigma, St. Louis, MO) was used to confirm equal protein loading among the samples.
In vitro stability, oral bioavailability, and brain uptake. The stability of MW01-5-188WH (1 μm) in a standard incubation with rat liver microsomes (BD Biosciences, Bedford, MA) and an NADPH-regenerating system was done at 37°C for 30 and 120 min. Reactions were stopped by acetonitrile, and the reaction mixture was centrifuged at 16,000 × g for 10 min. Ten microliters of the supernatant were analyzed by calibrated HPLC to quantify the percentage of the initial amount of MW01-5-188WH remaining after the incubation. The HPLC system (Dionex, Sunnyvale, CA) includes a Dionex P680 pump, a Phenomenex (Torrance, CA) Luna C18 column (250 × 2.0 mm; 5 μm) with a guard column, and a Dionex UVD340U ultraviolet detector. The mobile phase consisted of 0.1% formic acid as reagent A and 0.08% formic acid/water in 80% acetonitrile as reagent B at a flow rate of 0.2 ml per minute. The gradient consisted of the following linear and isocratic gradient elution changes in reagent B: isocratic at 60% from 0 to 5 min, 60-90% from 5 to 39 min, isocratic at 90% until 44 min. Peak quantification was done based on absorption measured at 260 nm relative to a standard curve obtained by using serial dilutions of MW01-5-188WH.
To estimate oral bioavailability (concentration of compound in the blood as a function of time after oral administration) and to gain insight into potential brain uptake, MW01-5-188WH (2.5 mg/kg) was administered to mice by oral gavage in a 0.5% (w/v) carboxymethylcellulose suspension. At 5, 15, 60, and 120 min after compound administration, the animals were anesthetized with pentobarbital (50 mg/kg). Blood was harvested by intracardiac puncture, collected in heparinized tubes, and plasma obtained by centrifugation. Mice were perfused with PBS. Brain homogenates were centrifuged at 12,000 × g for 10 min and the supernatant acidified by diluting 1:3 with 0.1% formic acid (Fluka, Sigma-Aldrich, St. Louis, MO). Solid phase extraction followed by HPLC analysis was used to quantify the amount of compound in brain supernatants. Briefly, cartridges (Sep-Pak C18; Waters Associates, Milford, MA) were conditioned with 1 ml of acetonitrile (HPLC grade; EMD Biosciences, San Diego, CA) and equilibrated with 1 ml of water. A structural analog of MW01-5-188WH was used as an internal standard. The acidified brain supernatant was added to the cartridge followed by a 1 ml wash with 30% acetonitrile. MW01-5-188WH was eluted from the cartridge using 80% acetonitrile. The eluate was evaporated to dryness, reconstituted in 0.08% formic acid/water in 80% acetonitrile, and analyzed by HPLC using the following gradient in reagent B: 0-60% from 2 to 5 min, isocratic at 65% until 7 min, 65-80% from 7 to 12 min, isocratic at 80% until 15 min, 89-100% from 15 to 18 min, and isocratic at 100% until 23 min. Plasma samples were deproteinized in 0.1 m perchloric acid and centrifuged at 12,000 × g for 10 min. The supernatant was neutralized with 1 m NaOH, then extracted with dichloromethane, and the layers separated at 3000 × g for 5 min. The organic phases from three successive extractions were pooled and then evaporated to dryness under reduced pressure. The dried residue was reconstituted in 50 μl of reagent B, and 10 μl of the reconstituted material was analyzed by HPLC using the gradient described above for brain supernatants.
Suppression of CNS versus peripheral inflammation. Mice were administered by oral gavage MW01-5-188WH (2.5 mg/kg) or diluent (10% DMSO) in a 0.5% (w/v) carboxymethylcellulose suspension once daily for 2 weeks. After the last administration, mice were injected intraperitoneally with 10 mg/kg LPS. Control mice were injected with saline. Six hours after the LPS challenge, mice were anesthetized with pentobarbital (50 mg/kg), and blood was drawn by intracardiac puncture, allowed to clot, and centrifuged for serum preparation. Mice were perfused with a HEPES buffer (10 mm, pH 7.2) containing a protease inhibitor mixture (1 μg/ml leupeptin, 1 μm dithithreitol, 2 mm sodium vanadate, 1 μm phenylmethylsulphonylfluoride), and brains were removed and weighed. Brain homogenates were prepared by dounce and sonication in the HEPES buffer containing a protease inhibitor mixture. Levels of IL-1β and TNF-α in brain supernatants and serum were measured using a multiplex assay per the manufacturer's instructions (Meso-Scale Discovery, Gaithersburg, MD).
Liver toxicity after chronic in vivo administration of MW01-5-188WH. Mice were administered by oral gavage either MW01-5-188WH (2.5 mg/kg) or diluent (10% DMSO) in a 0.5% (w/v) carboxymethylcellulose suspension once daily for 2 weeks. Mice were anesthetized and killed as described above. Livers were removed, fixed in 4% (v/v) paraformaldehyde, and paraffin-embedded for histology. To assess histological toxicity, 4 μm liver sections were stained with hematoxylin and eosin. Two independent observers blinded to the treatment groups performed microscopic assessment of the tissue for injury.
In vivo efficacy studies in mice. The study design and treatment paradigm for intracerebroventricular (ICV) infusion of human oligomeric Aβ1-42 into the mouse were as described previously (Craft et al., 2004b), except that compound administration was by mouth. Female C57BL/6 mice (Harlan Sprague Dawley, Indianapolis, IN) weighing 20-25 g (3-4 months of age) were housed in a pathogen-free facility under an approximate 12 h light/dark cycle and had access ad libitum to food and water. All animal procedures were approved by the Northwestern Animal Care and Use Committee.
Mice were administered by oral gavage either MW01-5-188WH (2.5 mg/kg) or solvent control (10% DMSO) in a 0.5% (w/v) carboxymethylcellulose suspension. Once daily treatment began at day 21 after start of Aβ ICV infusion and continued for 14 d. Beginning at day 50 after start of Aβ ICV infusion, the Y maze test of spontaneous alternation was used to evaluate hippocampus-dependent spatial learning as described previously (Craft et al., 2004a). Briefly, each mouse was placed in the start arm and then released to choose one of the two other arms. The mouse was blocked from exiting the chosen arm for 30 s, then placed back in the start arm and released again to choose one of the two other arms. If the second choice was different from the first one, the mouse is scored as alternating. Mice were tested for 10 d with one trial per day, and a mean percentage of alternation was calculated for each mouse. At day 60 after start of Aβ ICV infusion, mice were anesthetized with pentobarbital (50 mg/kg) and perfused with a HEPES buffer (10 mm, pH 7.2) containing a protease inhibitor mixture (1 μg/ml leupeptin, 1 μm dithiothreitol, 2 mm sodium orthovanadate, 1 μm phenylmethylsulphonylfluoride). The brain was removed and longitudinally bisected as described previously (Craft et al., 2004b). The right half of the brain was fixed in 4% (v/v) paraformaldehyde and paraffin-embedded for histology. The hippocampus was dissected from the left half of the brain and snap-frozen for subsequent biochemical evaluation. Hippocampal extract supernatants were prepared by dounce and sonication in the HEPES buffer containing a protease inhibitor mixture, followed by centrifugation as described (Craft et al., 2004b).
Levels of IL-1β and TNF-α in hippocampal supernatants were measured by ELISA (Biosource International) per the manufacturer's instructions. S100B levels in hippocampal supernatants were measured by a europium-based ELISA essentially as described previously (Van Eldik and Griffin, 1994). Synaptophysin levels in hippocampal supernatants were quantified by ELISA following the procedure described previously (Craft et al., 2004b). Postsynaptic density-95 (PSD-95) levels were determined by Western blots using anti-PSD-95 antibodies (1:100,000 dilution; Upstate Biotechnology, Charlottesville, VA) as described previously (Craft et al., 2004b).
Immunohistochemical detection of activated astrocytes and microglia was performed on 10 μm sections as described previously (Craft et al., 2004b, 2005a,b), with anti-GFAP (1:1500; Sigma) and anti-F4/80 (1:100; Serotek, Raleigh, NC) antibodies, respectively, using the mouse on mouse or Vectastain Universal Elite ABC immunodetection kits (Vector Laboratories, Burlingame, CA) and development with diaminobenzidine (DAB) substrate. Cell bodies were counted manually in the hippocampus of three GFAP- and F4/80-labeled sections positioned at -1.8, -2.1, and -2.3 mm from bregma. Aβ immunohistochemistry was done with a rabbit anti-human Aβ antibody as described previously (Craft et al., 2004b). Cell counts and amyloid plaque counts were determined by two blinded observers, and amyloid plaque area was determined as described previously (Craft et al., 2004b). Peroxynitrite-mediated neuronal damage was measured with an anti-nitrotyrosine antibody (1:125; Chemicon, Temecula, CA), using the Vectastain Rabbit Elite ABC kit. For nitrotyrosine cell counts, all DAB-stained cell bodies in the neuronal layers of the hippocampus and subiculum were counted on three sections approximately adjacent to those used for F4/80 and GFAP analysis, as described previously (Craft et al., 2004b).
Statistical analyses. Experimental and control groups were compared using one-way ANOVA with Newman-Keuls post hoc analysis using a statistical software package (GraphPad Prism, version 4.00; GraphPad Software, San Diego CA). Statistical significance was assumed when p < 0.05.
Results
Selective, concentration-dependent suppression of increased cytokine production by activated glial cell cultures
Compound MW01-5-188WH (Fig. 1A) shows concentration-dependent suppression of glia activation endpoints relevant to AD pathology. MW01-5-188WH inhibited, in a concentration-dependent manner, the increased production of the proinflammatory cytokines IL-1β (Fig. 1B) and TNF-α (Fig. 1C) induced by LPS treatment of the microglia-like BV-2 cell line. In contrast, MW01-5-188WH did not block the increased NO production, as measured by the accumulation of its stable metabolite nitrite (Fig. 1D), over the same concentration range that inhibited the increased proinflammatory cytokine production or at higher concentrations (up to 33 μm). Consistent with this lack of effect on NO production, MW01-5-188WH did not inhibit the upregulation of iNOS in LPS-activated glia (Fig. 1E). These results revealed that MW01-5-188WH might be a valuable and novel integrative chemical biology tool for the in vivo dissection of proinflammatory cytokine contribution to in vivo pathophysiology in the absence of perturbing oxidative stress-related pathways involving upregulation of iNOS.
Figure 1.

Ex vivo inhibition of proinflammatory cytokine production by MW01-5-188WH and stability after incubation with rat liver microsomes. A, Structure of MW01-5-188WH. Concentration-dependent inhibition by MW01-5-188WH of LPS-induced increases of IL-1β (B) and TNF-α (C) levels in the BV-2 microglial cell line is shown. D, LPS-stimulated accumulation of the NO metabolite nitrite was not inhibited by MW01-5-188WH at concentrations up to 33 μm. MW01-5-188WH (188) does not suppress LPS-induced production of iNOS (E) or COX-2 (F) in activated BV-2 cells. The compound also does not block Aβ-induced apoE upregulation (G) in activated rat primary glia cultures. Cultures were treated with control buffer (C) or activated in the absence (A) or presence (A+188) of 7.5 μm MW01-5-188WH, then analyzed by Western blots. H, Amount of remaining MW01-5-188WH (1 μm) after 30 and 120 min incubation with rat liver microsomes. MW01-5-188WH is stable at 30 min but is able to be metabolized by 120 min. Error bars represent SEM.
As secondary screens for selectivity, we investigated the effects of MW01-5-188WH on COX-2 and apoE production by activated glia. We wanted to avoid compounds that blocked COX-2 production, because the induction of COX-2 is linked to peripheral tissue inflammatory response pathways, which we sought to avoid in our search for compounds with a bias toward suppression of glia activation pathways over pathways that predominate in peripheral tissue activation responses. In addition, some compounds that inhibit COX-2 production or activity have exhibited side effects in human trials. Similarly, apoE has divergent effects on the susceptibility to Aβ-induced pathophysiology and can modulate Aβ aggregation and deposition into amyloid plaques (Poirier et al., 1995; Manelli et al., 2004). As shown in Figure 1F, MW01-5-188WH did not inhibit the LPS-induced upregulation of COX-2 levels in BV-2 cells. Similarly, MW01-5-188WH did not block the Aβ-induced increase in apoE levels in rat primary glial cultures (Fig. 1G). Overall, the ex vivo results show that MW01-5-188WH is a novel, selective suppressor of activated glia responses, especially key proinflammatory cytokines that have been linked to AD pathology.
Stability of MW01-5-188WH in liver microsome incubations
A critical issue that prevents the transition of many neuroscience studies from ex vivo to in vivo is the need for bioavailable reagents. Although small molecules are preferred to macromolecules for in vivo studies, many small molecule compounds are, like macromolecules, too labile for in vivo use. Small molecules are especially subject to modification or degradation by liver enzymes, often referred to as susceptibility to first pass metabolism. Therefore, we analyzed the stability of MW01-5-188WH in a standard liver microsome incubation assay as a prelude to determination of the utility of the compound for in vivo chemical biology investigations. As shown in Figure 1H, the majority of MW01-5-188WH remains after 30 min of incubation, a time course over which a useful compound would have reached a peak concentration in blood and brain. The lower concentration of MW01-5-188WH after 120 min is indicative of its ability to be metabolized, an important feature that facilitates the use of daily administration regimens over the extended time courses required for in vivo investigations, especially in AD-relevant chronic injury and treatment models.
Oral bioavailability and brain uptake of MW01-5-188WH
Integrative chemical biology tools for neurosciences and CNS-targeted drugs must exhibit appropriate bioavailability and brain uptake or penetration of the blood-brain barrier. Daily oral administration is the preferred method of administration for longer-term and time-delimited in vivo studies using animal models and is the preferred mode in drug development for a variety of reasons, including better patient compliance. In this regard, it is critical to demonstrate bioavailability and appropriate rates of initial brain uptake for an inhibitor, to fully interpret the outcomes from in vivo studies. Therefore, we determined the rate of MW01-5-188WH concentration change in the blood after oral administration (oral bioavailability) and its rate of change in the brain. Using the protocols described in Materials and Methods for the quantitative analysis of MW01-5-188WH extracted from biological samples, we examined the rates of appearance in blood and brain after a low-dose oral administration (2.5 mg/kg) to mice. The appearance of MW01-5-188WH in the blood (Fig. 2A) is readily detected within the earliest possible time point, 5 min, with a peak concentration being reached within 15 min and bulk clearance happening within 120 min after oral administration. This demonstrates that MW01-5-188WH has good oral bio-availability properties. A similar pattern of time-dependent change in concentration is seen for the brain (Fig. 2B), indicative of MW01-5-188WH initial brain uptake reflecting that of the blood. However, the MW01-5-188WH peak brain/blood concentration ratio is >3.3, comparable with those of CNS drugs in clinical use. For example, the brain/blood ratio for minaprine, a 6-phenylaminopyridazine CNS drug, is ∼2 (Caccia et al., 1985). These results demonstrate that MW01-5-188WH fulfills criteria that typically exclude many compounds that are active in cell culture from being used for in vivo investigations and indicate its potential to work in vivo after oral administration and within the experimental constraints imposed by the human Aβ ICV infusion model. Additional pharmacokinetic analyses of MW01-5-188WH were not required for the subsequent experiments reported below and are not reported here.
Figure 2.
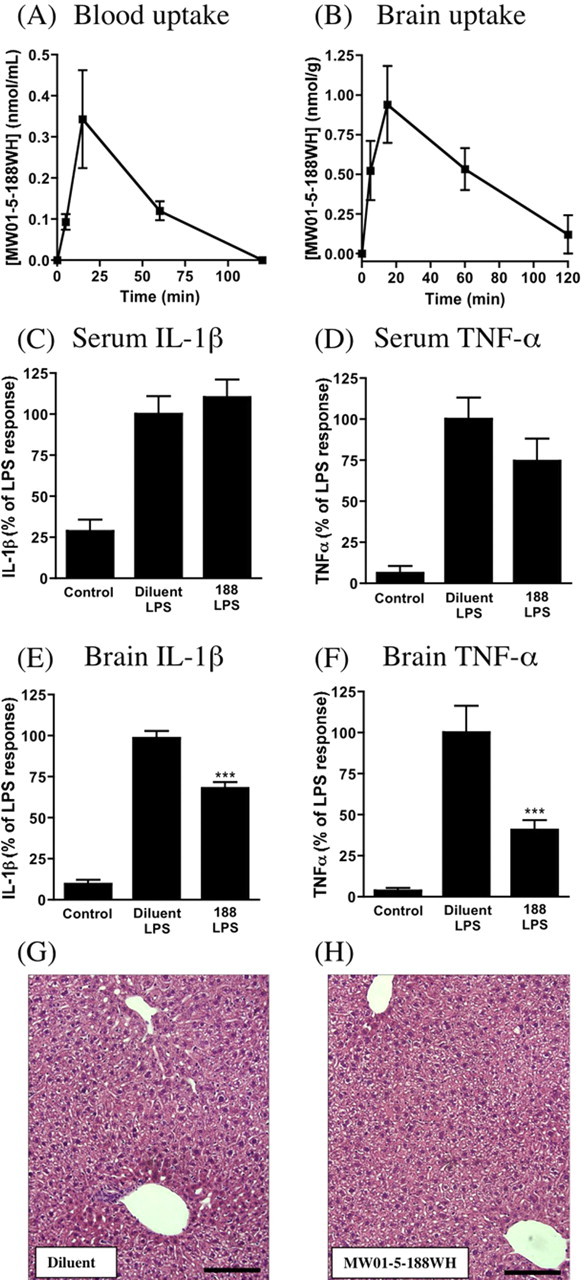
MW01-5-188WH is readily detected in the plasma and the brain after a single oral dose administration and does not suppress peripheral tissue inflammatory responses or cause liver injury after chronic oral administration. C57BL/6 mice were administered MW01-5-188WH (2.5 mg/kg) by oral gavage, blood and brain processed at different times after administration, and compound levels in plasma and brain determined as described in Materials and Methods. MW01-5-188WH rapidly appears in plasma (A) and brain (B), reaches a peak at 15 min, and then slowly declines to basal levels by 120 min. Data are the mean± SEM from three to six mice at each time point. MW01-5-188WH does not inhibit increased production of IL-1β (C) and TNF-α (D) in the serum but does suppress the cytokine response in the brains from the same mice (E, F). Mice (n = 3-6 per group) were administered by oral gavage either diluent or MW01-5-188WH (2.5 mg/kg) once daily for 2 weeks and then challenged with LPS (10 mg/kg, i.p.) for 6 h. Control mice were injected with saline. IL-1β and TNF-α levels in the serum and in brain supernatants were determined. Data represent mean ± SEM. ***p < 0.001, significantly different from diluent. Daily oral administration of diluent (G) or MW01-5-188WH (H) (2.5 mg/kg) does not result in any histological liver toxicity. Representative liver sections from mice treated as in C-F were stained with hematoxylin and eosin. Scale bar, 125 μm. 188, MW01-5-188WH.
MW01-5-188WH dosing is selective for CNS inflammation
Our de novo focus on suppression of selected glia activation pathways and the excellent brain uptake properties of orally administered MW01-5-188WH raised the possibility that the compound might exhibit selectivity for CNS proinflammatory cytokine suppression versus suppression of proinflammatory cytokine production by peripheral tissues. To examine this possibility, MW01-5-188WH was administered daily at a standard therapeutic dose (2.5 mg/kg) by oral gavage for 2 weeks, and then mice were challenged with an intraperitoneal injection of bacterial LPS. Six hours after the LPS challenge, the serum and brain levels of IL-1β and TNF-α were measured. As anticipated, the LPS challenge induced an increase in the levels of IL-1β and TNF-α in the serum (Fig. 2C,D) and brain (Fig. 2E, F), compared with control mice injected with saline. The interesting finding was that treatment with MW01-5-188WH for 2 weeks suppressed the LPS-induced upregulation of IL-1β and TNF-α production in the brain (Fig. 2E, F) but did not suppress the serum response (Fig. 2C,D). The suppression of brain cytokine responses by MW01-5-188WH is consistent with its ability to suppress proinflammatory cytokine production by activated glia and its oral bioavailability and brain uptake properties shown above.
MW01-5-188WH does not exhibit tissue toxicity
Liver toxicity is an especially important initial consideration for orally administered compounds, because the liver is critical to in vivo drug metabolism, and to overall metabolism and homeostasis of an animal. Liver injury is also a component of idiopathic tissue injury seen in certain chronically administered drugs. Therefore, it was important to know whether chronic administration of MW01-5-188WH at potential therapeutic doses might alter animal metabolism and response to stress unexpectedly via liver injury. However, histological assessment of liver tissue showed that oral administration of MW01-5-188WH at 2.5 mg/kg daily for 2 weeks did not induce any indices of hepatotoxic tissue injury compared with mice treated with the diluent (Fig. 2G,H).
Suppression of Aβ-induced proinflammatory cytokine production in vivo
The preceding ex vivo and in vivo analyses of MW01-5-188WH demonstrate its novel nature and potential for in vivo testing of hypotheses, including causal linkage of increased proinflammatory cytokine levels to neuropathology outcomes and the potential for targeting glia in the development of new therapies. An essential aspect of testing these hypotheses is showing that a selective suppressor of increased proinflammatory cytokine production by activated glia can selectively attenuate proinflammatory cytokine production and neurophysiological injury endpoints induced by a disease-relevant stimulus in vivo. Therefore, MW01-5-188WH was tested for efficacy in a mouse assay of human Aβ-induced neuroinflammation with neurodegeneration outcomes, including synaptic dysfunction, neuronal death, and hippocampus-dependent behavioral deficits (Craft et al., 2004a,b, 2005a,b,d). This model has high phenotypic penetrance of pathophysiology endpoints and uses infusion of human Aβ, not amyloid precursor protein (APP). Therefore, the contribution of APP processing to biological outcomes is minimized.
The infusion of Aβ induced an increase in the levels of the proinflammatory cytokines IL-1β, TNF-α, and S100B (Fig. 3A-C) in the hippocampus and activation of glia, as seen by an increase in the number of GFAP-positive astrocytes and F4/80-positive microglia (Fig. 3D, E). Daily oral administration of MW01-5-188WH at a low dose (2.5 mg/kg) once daily for 2 weeks, beginning 21 d after initiation of the ICV infusion of human Aβ, effectively suppressed Aβ-induced glial activation and the increased production of proinflammatory cytokines in the hippocampus. This is evidenced by inhibition of the increased levels of IL-1β (Fig. 3A), TNF-α (Fig. 3B), and S100B (Fig. 3C) in the hippocampus, as well as suppression of the number of activated GFAP-positive astrocytes (Fig. 3D) and F4/80-positive microglia (Fig. 3E). The inhibition of increased IL-1β and TNF-α production in the hippocampus is consistent with the inhibition of these same endpoints in activated glia cultures in the initial cell-based screens that identified MW01-5-188WH as a potentially useful reagent (Fig. 1A, B). Under the same in vivo experimental conditions, MW01-5-188WH did not produce a detectable effect on the Aβ-induced increase in nitrotyrosine-positive neurons (Fig. 3F). These in vivo findings are also consistent with the cell-based assays (Fig. 1C,D) that showed no inhibition of iNOS induction or NO accumulation over the same concentrations of MW01-5-188WH that inhibited cytokine upregulation. MW01-5-188WH treatment had no effect on the amyloid plaque number (Fig. 3G) or amyloid load (Fig. 3H), consistent with no effect on apoE production by activated glia in the cell-based screen (Fig. 1F).
Figure 3.
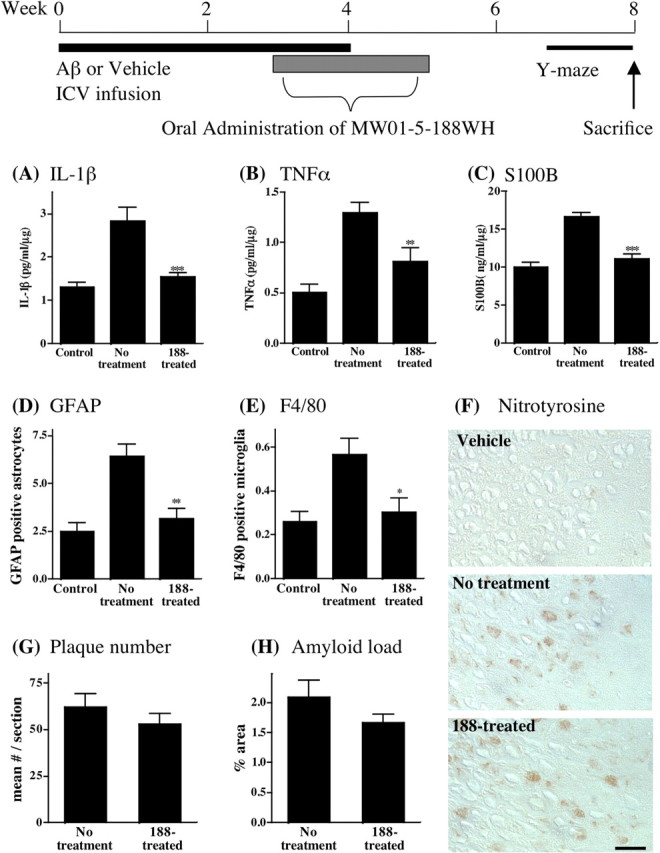
Oral administration of MW01-5-188WH suppresses human Aβ-induced upregulation of proinflammatory cytokine production in mouse hippocampus without detectable effect on the profile of nitrotyrosine-labeled neurons or on amyloid plaque deposition. A schematic of the experimental paradigm is shown. Mice were subjected to vehicle infusion (Control) or human oligomeric Aβ1-42 infusion (No treatment) for 4 weeks. At 3 weeks after the start of infusion and continuing once daily for 2 weeks thereafter, mice were administered by oral gavage either 2.5 mg/kg MW01-5-188WH (188-treated) or diluent (No treatment). MW01-5-188WH treatment resulted in significant suppression of the Aβ-induced increase in IL-1β (A), TNF-α (B), and S100B (C) levels in hippocampal supernatants (n = 10 mice per group). MW01-5-188WH treatment also decreased the number of GFAP-positive activated astrocytes (D) and F4/80-positive microglia (E) in hippocampus. F, MW01-5-188WH treatment did not alter the profile of nitrotyrosine-stained neurons, an indicator of oxidative stress-linked injury. Representative micrographs are shown for hippocampus sections stained for nitrotyrosine-positive neurons from vehicle-infused mice, Aβ-infused mice orally administered diluent (No treatment), and Aβ-infused mice orally administered MW01-5-188WH (188-treated) for 2 weeks. Scale bar, 25 μm. The number of amyloid plaques (G) or the area occupied by plaques (H) is not altered by MW01-5-188WH therapy. Data are mean ± SEM of n = 5 mice per group. Significantly different from Aβ infused: *p < 0.05, **p < 0.01, ***p < 0.001.
Attenuation of synaptic dysfunction and behavioral deficits
The symptomatic manifestation of human Aβ-induced brain injury in AD is usually related to effects on hippocampal-dependent functions. Therefore, it was of paramount importance to test the hypothesis that MW01-5-188WH suppression of glial activation and increased cytokine production could be accompanied by improvement in hippocampal-associated functioning. As shown in Figure 4, the in vivo inhibition of cytokine production by MW01-5-188WH was accompanied by significant suppression of synaptic dysfunction endpoints. Specifically, oral administration of MW01-5-188WH prevented the human Aβ-induced decrease in the presynaptic protein synaptophysin (Fig. 4A) and the postsynaptic protein PSD-95 (Fig. 4B). These proteins are biochemical markers of synaptic integrity, so restoration toward normal quantifies the ability of therapeutic intervention with a glial inhibitor to alleviate hippocampal injury. Consistent with the biochemical endpoint results from hippocampus homogenates, MW01-5-188WH also attenuated animal deficits in the Y maze (Fig. 4C), a hippocampal-dependent behavioral task. Because cognitive function test results are an established measure of clinical AD onset and progression, the linkage of MW01-5-188WH therapeutic intervention to improvement of a hippocampus-dependent behavioral task is especially interesting.
Figure 4.

Oral administration of MW01-5-188WH attenuates hippocampal synaptic dysfunction and hippocampus-linked behavioral deficits. A schematic of the experimental paradigm is shown. Mice were treated as in Figure 3. MW01-5-188WH administration significantly attenuated Aβ-induced loss of synaptophysin and PSD-95 and the behavioral deficit in the Y maze. Levels of the presynaptic protein synaptophysin (A) and the postsynaptic protein PSD-95 (B) were measured in hippocampal supernatants from vehicle-infused mice (Control), Aβ-infused mice administered diluent (No treatment), and Aβ-infused mice administered MW01-5-188WH (188-treated) at 2.5 mg/kg by oral gavage once daily for 2 weeks. C, Spontaneous alternation of mice in the Y maze, a hippocampus-dependent spatial learning task, was measured for 10 d during the seventh and eighth week after the start of Aβ infusion. Data are mean ± SEM of n = 5-10 mice per group. Significantly different from Aβ infused: *p < 0.05, **p < 0.01, ***p < 0.001.
Discussion
We report here several precedents and key observations. First, the in vivo results provide support for the neuroinflammation hypothesis of pathophysiology progression by using a novel integrative chemical biology approach that can be readily extended to diverse species, including primates. Much of the previous experimental evidence in support of the neuroinflammatory hypothesis has come either from cell culture studies using intervention approaches that are not readily transferable to in vivo studies across multiple species, from correlative clinical pathology outcomes analyses, or from in vivo studies using extant anti-inflammatory drugs that alter amyloid precursor protein processing or suppress peripheral tissue inflammation. Second, one outcome of this study is the first orally bioavailable lead compound amenable to medicinal chemistry refinement that has a selective effect on hippocampal proinflammatory cytokine production and related neuropathophysiologies without suppressing peripheral inflammatory responses over the same dose and time window. Third, the results demonstrate that one can alter disease-relevant pathophysiology endpoints with selective inhibition of proinflammatory cytokine production independent of direct modulation of oxidative stress pathways or effects on amyloid plaques. Overall, the results provide a precedent for neurophysiology, inflammation biology, and drug discovery, demonstrating effective alteration of clinically relevant endpoints using anti-proinflammatory cytokine therapy that fulfills key criteria for future clinical investigations.
An advantage of an integrative chemical biology approach with small molecules to test hypotheses about brain function and pathophysiology is the power for using these bioavailable tools to probe in vivo function across animal species. In addition, they offer a starting point for future proteomic analyses that can identify molecular differences between brain and peripheral tissues in regulating the same biological endpoint, proinflammatory cytokine upregulation. Integrative chemical biology cannot be done in the absence of the required bioavailable compound. MW01-5-188WH is a pyridazine derivative, a heteroaromatic chemical ring with multiple positions amenable to diversification using standard chemistries. Chemical diversification of pyridazines can result in divergent pharmacological effects (Wermuth, 1998), the definition of a privileged molecule in medicinal chemistry (Evans et al., 1988). Therefore, the combination of a neuropharmacology-focused synthetic design strategy based on diversification of a privileged structure for library construction (Wing et al., 2005), in conjunction with a glia-focused, hierarchal biology screen for function, allowed the successful discovery of this promising and novel compound that meets critical criteria for in vivo investigational use in systems biology and for future drug development.
The late stage in vivo results obtained with MW01-5-188WH are internally consistent with the early stage cell culture results and are congruent with relevant clinical findings. For example, treatment with MW01-5-188WH suppressed upregulation of proinflammatory cytokine production in the hippocampus and did not significantly decrease the number of nitrotyrosine-labeled neurons, consistent with the glial cell culture data. MW01-5-188WH also did not modify the amyloid plaque burden, documenting that the protective effects are not directly related to a decrease in plaques and consistent with the lack of effects in glial cell culture on apoE production, a molecule known to alter plaque dynamics and composition. It should be noted that the in vivo model minimizes the influence of APP processing because Aβ1-42 is infused, not APP. However, similar reductions in proinflammatory cytokine production are seen in a transgenic mouse expressing mutant human APP (data not shown). Whether MW01-5-188WH has an effect on soluble Aβ levels was not the goal of this study and remains to be determined. Our data are consistent with clinical findings that cognitively intact individuals can have considerable amyloid deposits and that relevant clinicopathological endpoints in the human disease do not correlate directly or solely with the pattern of amyloid plaque deposition (Crystal et al., 1988; Davis et al., 1999; Guillozet et al., 2003). Age-onset AD has many contributing and susceptibility factors, which will probably require the use of pharmaceutical combinations for therapy, dependent on patient history. The development of glia activation suppressors selective for different neurotoxic products is needed as part of this armamentarium. Our results show that inhibitors of proinflammatory cytokine upregulation in activated glia should be one of the multiple therapies considered in attempts to alter disease progression.
A key result is that MW01-5-188WH inhibits the proinflammatory response in the brain but not in the serum. This important finding using a common inflammatory stimulus (LPS) and the same molecular endpoints (IL-1β and TNF-α) in serum or brain from the same mouse demonstrates the selective nature of MW01-5-188WH for CNS inflammation compared with peripheral inflammation. MW01-5-188WH appears to inhibit cytokine response pathways that are quantitatively more important in CNS than in the periphery, thereby providing insight into tissue selectivity of responses. Based on precedents in biological regulatory pathways, these differences are probably not an all-or-none difference in the existence of a particular pathway but may be the outcome of a combinatorial array of pathways converging on the cytokine production, reflective of the cell context within a given tissue and the nature of the activating stimulus. These are points for consideration in future research now that the results with MW01-5-188WH have revealed the existence of these in vivo differences. Regardless of the details of pharmacological action, our results provide a precedent that CNS inflammation can be selectively targeted without necessarily resulting in a concomitant suppression of peripheral inflammation, an important consideration in chronic anti-inflammatory treatment of elderly individuals.
Our results showing that S100B levels are increased in the hippocampus of Aβ-infused mice and are restored back toward basal levels with MW01-5-188WH administration are especially interesting. There is compelling evidence that increases in S100B levels in human CSF and serum correlate with the severity of brain damage and may have predictive value for adverse clinical outcomes (Van Eldik and Wainwright, 2003). Furthermore, the levels of S100B are elevated in pathologically relevant brain regions from AD patients (Van Eldik and Griffin, 1994), S100B levels in CSF are increased in the earlier stages of AD (Peskind et al., 2001), and transgenic mice overproducing brain S100B are more susceptible to human Aβ-induced injury (Craft et al., 2005b). An increase in S100B levels in Aβ-injured hippocampus and a reduction back toward control in animals treated with MW01-5-188WH raise the logical possibility that S100B levels might be a translatable endpoint. Increased CSF concentration of S100B is a clinical endpoint for human brain injury, so its inclusion in the array of surrogate biochemical endpoints to complement cognitive function tests might allow the monitoring of response to therapy as well as the extent or progression of injury. Failure in preclinical studies to identify endpoints that have potential for transfer to the clinic is a major contributor to failure in translational studies, so our results also provide a starting point for translational research with a higher potential for success.
Rapid blood-brain barrier penetration is necessary to attain a pharmacologically relevant concentration of drug within a useful time window for in vivo investigations and is a critical pharmacokinetic issue in the development of new CNS therapeutics. Our results show that MW01-5-188WH rapidly appears in the mouse brain after oral administration, with a brain/blood ratio comparable with established CNS-targeted drugs (Caccia et al., 1985), and clears from the brain and blood within 2 h after administration. These preliminary insights into pharmacokinetics highlight the bioavailability and brain penetrance of MW01-5-188WH. This favorable bioavailability profile, along with the in vivo efficacy at a low therapeutic dose (2.5 mg/kg body weight) and the absence of liver histological toxicity after chronic administration, make MW01-5-188WH an attractive candidate for future medicinal chemistry refinement. Good brain uptake properties of a compound such as MW01-5-188WH will facilitate medicinal chemistry refinement in late stage drug development, because synthesis of analogs can be focused on improvement in therapeutic effect, formulation properties, or stability instead of improvement in blood-brain barrier penetration. Medicinal chemistry refinement during drug development often increases the molecular weight of a compound by ∼100 atomic mass units, and many compounds detected by activity screens have molecular weights already near the maximal desired molecular weight of 500. In this regard, MW01-5-188WH is also advantageous as a foundation for future refinement, because it has efficacy and good brain uptake in a compound with a molecular weight < 400.
In summary, our results provide an experimental precedent for neurophysiology, cytokine biology, and drug discovery. The most clinically relevant outcome is efficacy in attenuation of neurodegeneration-relevant pathophysiology after oral administration of a selective anti-neuroinflammatory agent. Therapeutic intervention with the orally bioavailable glia inhibitor resulted in the in vivo suppression of glia activation markers and inhibition of the overproduction of proinflammatory cytokines IL-1β, TNF-α, and S100B in the hippocampus. The effects on proinflammatory cytokine production provide a link to AD pathology and a prevailing hypothesis of AD progression. Suppression of proinflammatory cytokine production by MW01-5-188WH led to neuroprotection as evidenced by reduced synaptic damage and attenuation of behavioral deficits reflective of hippocampal injury, consistent with the clinical progression of AD symptoms. Overall, the results provide strong evidence that attempts to develop CNS-selective anti-neuroinflammatory compounds that attenuate neurodegeneration are feasible, and support the concept of glia as a therapeutic target for AD and related neurodegenerative disorders.
Footnotes
This work was supported in part by funds from the Institute for the Study of Aging, from the National Institutes of Health Grants (AG13939, NS47586, AG21184, NS46942, and AG00260), and from the PhRMA Foundation. H. R. R. and W. H. are postdoctoral scholars and J.M.C. and L.K.W. are predoctoral scholars in the Drug Discovery Training Program. We thank S. Medgyesi, J. Ford, J. Shavocky, and S. M. Roy for their assistance.
Correspondence should be addressed to D. Martin Watterson, Center for Drug Discovery and Chemical Biology, 303 East Chicago Avenue, Mail Code W896, Chicago, IL 60611. E-mail: m-watterson@northwestern.edu.
Copyright © 2006 Society for Neuroscience 0270-6474/06/260662-09$15.00/0
References
- Aisen PS, Schafer KA, Grundman M, Pfeiffer E, Sano M, Davis KL, Farlow MR, Jin S, Thomas RG, Thal LJ (2003) Effects of rofecoxib or naproxen vs placebo on Alzheimer disease progression: a randomized controlled trial. JAMA 289: 2819-2826. [DOI] [PubMed] [Google Scholar]
- Breitner JC (2003) NSAIDs and Alzheimer's disease: how far to generalise from trials? Lancet Neurol 2: 527. [DOI] [PubMed] [Google Scholar]
- Caccia S, Fossati T, Mancinelli A (1985) Disposition and metabolism of minaprine in the rat. Xenobiotica 15: 1111-1119. [DOI] [PubMed] [Google Scholar]
- Cole GM, Morihara T, Lim GP, Yang F, Begum A, Frautschy SA (2004) NSAID and antioxidant prevention of Alzheimer's disease: lessons from in vitro and animal models. Ann NY Acad Sci 1035: 68-84. [DOI] [PubMed] [Google Scholar]
- Coudert P, Couquelet J, Tronche P (1988) A new synthetic route to 4,6-diarylpyridazinones and some of their derivatives. J Heterocycl Chem 25: 799-802. [Google Scholar]
- Craft JM, Van Eldik LJ, Zasadzki M, Hu W, Watterson DM (2004a) Aminopyridazines attenuate hippocampus-dependent behavioral deficits induced by human β-amyloid in a murine model of neuroinflammation. J Mol Neurosci 24: 115-122. [DOI] [PubMed] [Google Scholar]
- Craft JM, Watterson DM, Frautschy SA, Van Eldik LJ (2004b) Aminopyridazines inhibit β-amyloid-induced glial activation and neuronal damage in vivo. Neurobiol Aging 25: 1283-1292. [DOI] [PubMed] [Google Scholar]
- Craft JM, Watterson DM, Hirsch E, Van Eldik LJ (2005a) Interleukin 1 receptor antagonist knockout mice show enhanced microglial activation and neuronal damage induced by intracerebroventricular infusion of human β-amyloid. J Neuroinflammation 2: 15-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craft JM, Watterson DM, Marks A, Van Eldik LJ (2005b) Enhanced susceptibility of S100B transgenic mice to neuroinflammation and neuronal dysfunction induced by intracerebroventricular infusion of human β-amyloid. Glia 51: 209-216. [DOI] [PubMed] [Google Scholar]
- Craft JM, Watterson DM, Van Eldik LJ (2005c) Neuroinflammation: a therapeutic target. Expert Opin Ther Targets 9: 887-900. [DOI] [PubMed] [Google Scholar]
- Craft JM, Watterson DM, Van Eldik LJ (2005d) Human β amyloid-induced neuroinflammation is an early event in neurodegeneration. Glia, in press. [DOI] [PubMed]
- Crystal H, Dickson D, Fuld P, Masur D, Scott R, Mehler M, Masdeu J, Kawas C, Aronson M, Wolfson L (1988) Clinico-pathologic studies in dementia: nondemented subjects with pathologically confirmed Alzheimer's disease. Neurology 38: 1682-1687. [DOI] [PubMed] [Google Scholar]
- Davis DG, Schmitt FA, Wekstein DR, Markesbery WR (1999) Alzheimer neuropathologic alterations in aged cognitively normal subjects. J Neuropathol Exp Neurol 58: 376-388. [DOI] [PubMed] [Google Scholar]
- Evans BE, Rittle KE, Bock MG, DiPardo RM, Freidinger RM, Whitter WL, Lundell GF, Veber DF, Anderson PS, Chang RS, Lotti VJ, Cerino DJ, Chen TB, Kling PJ, Kunkel KA, Springer JP, Hirshfield J (1988) Methods for drug discovery: development of potent, selective, orally effective cholecystokinin antagonists. J Med Chem 31: 2235-2246. [DOI] [PubMed] [Google Scholar]
- Frautschy SA, Hu W, Kim P, Miller SA, Chu T, Harris-White ME, Cole GM (2001) Phenolic anti-inflammatory antioxidant reversal of Aβ-induced cognitive deficits and neuropathology. Neurobiol Aging 22: 993-1005. [DOI] [PubMed] [Google Scholar]
- Galli B, Faller B (2003) Discover a drug substance, formulate and develop it to a product. In: The practice of medicinal chemistry, Ed 2 (Wermuth CG, ed), pp 687-695. London: Academic.
- Griffin WS, Stanley LC, Ling C, White L, MacLeod V, Perrot LJ, White III CL, Araoz C (1989) Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc Natl Acad Sci USA 86: 7611-7615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin WS, Sheng JG, Royston MC, Gentleman SM, McKenzie JE, Graham DI, Roberts GW, Mrak RE (1998) Glial-neuronal interactions in Alzheimer's disease: the potential role of a “cytokine cycle” in disease progression. Brain Pathol 8: 65-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillozet AL, Weintraub S, Mash DC, Mesulam MM (2003) Neurofibrillary tangles, amyloid, and memory in aging and mild cognitive impairment. Arch Neurol 60: 729-736. [DOI] [PubMed] [Google Scholar]
- Hu W, Ralay Ranaivo H, Craft JM, Van Eldik LJ, Watterson DM (2005) Validation of the neuroinflammation cycle as a drug discovery target using integrative chemical biology and lead compound development with an Alzheimer's disease-related mouse model. Curr Alzheimer Res 2: 197-205. [DOI] [PubMed] [Google Scholar]
- Jantzen PT, Connor KE, DiCarlo G, Wenk GL, Wallace JL, Rojiani AM, Coppola D, Morgan D, Gordon MN (2002) Microglial activation and β-amyloid deposit reduction caused by a nitric oxide-releasing nonsteroidal anti-inflammatory drug in amyloid precursor protein plus presenilin-1 transgenic mice. J Neurosci 22: 2246-2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manelli AM, Stine WB, Van Eldik LJ, LaDu MJ (2004) ApoE and Aβ1-42 interactions: effects of isoform and conformation on structure and function. J Mol Neurosci 23: 235-246. [DOI] [PubMed] [Google Scholar]
- Miller G (2005) Neuroscience: the dark side of glia. Science 308: 778-781. [DOI] [PubMed] [Google Scholar]
- Mirzoeva S, Koppal T, Petrova TV, Lukas TJ, Watterson DM, Van Eldik LJ (1999) Screening in a cell-based assay for inhibitors of microglial nitric oxide production reveals calmodulin-regulated protein kinases as potential drug discovery targets. Brain Res 844: 126-134. [DOI] [PubMed] [Google Scholar]
- Mirzoeva S, Sawkar A, Zasadzki M, Guo L, Velentza AV, Dunlap V, Bourguignon JJ, Ramstrom H, Haiech J, Van Eldik LJ, Watterson DM (2002) Discovery of a 3-amino-6-phenyl-pyridazine derivative as a new synthetic antineuroinflammatory compound. J Med Chem 45: 563-566. [DOI] [PubMed] [Google Scholar]
- Pardridge WM (2005) The blood-brain barrier: bottleneck in brain drug development. NeuroRx 2: 3-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peskind ER, Griffin WS, Akama KT, Raskind MA, Van Eldik LJ (2001) Cerebrospinal fluid S100B is elevated in the earlier stages of Alzheimer's disease. Neurochem Int 39: 409-413. [DOI] [PubMed] [Google Scholar]
- Poirier J, Delisle MC, Quirion R, Aubert I, Farlow M, Lahiri D, Hui S, Bertrand P, Nalbantoglu J, Gilfix BM, Gauthier S (1995) Apolipoprotein E4 allele as a predictor of cholinergic deficits and treatment outcome in Alzheimer disease. Proc Natl Acad Sci USA 92: 12260-12264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reines SA, Block GA, Morris JC, Liu G, Nessly ML, Lines CR, Norman BA, Baranak CC (2004) Rofecoxib: no effect on Alzheimer's disease in a 1-year, randomized, blinded, controlled study. Neurology 62: 66-71. [DOI] [PubMed] [Google Scholar]
- Sircar I (1983) Synthesis of 4-amino-6-phenyl-3 (2 H)-pyridazinones: a general procedure. J Heterocycl Chem 20: 1473-1476. [Google Scholar]
- Tuppo EE, Arias HR (2005) The role of inflammation in Alzheimer's disease. Int J Biochem Cell Biol 37: 289-305. [DOI] [PubMed] [Google Scholar]
- Van Eldik LJ, Griffin WS (1994) S100β expression in Alzheimer's disease: relation to neuropathology in brain regions. Biochim Biophys Acta 1223: 398-403. [DOI] [PubMed] [Google Scholar]
- Van Eldik LJ, Wainwright MS (2003) The Janus face of glial-derived S100B: beneficial and detrimental functions in the brain. Restor Neurol Neurosci 21: 97-108. [PubMed] [Google Scholar]
- Wermuth CG (1998) Search for new lead compounds: the example of the chemical and pharmacological dissection of aminopyridazines. J Heterocycl Chem 35: 1091-1100. [Google Scholar]
- Weydt P, Moller T (2005) Neuroinflammation in the pathogenesis of amyotrophic lateral sclerosis. NeuroReport 16: 527-531. [DOI] [PubMed] [Google Scholar]
- Wilkinson GR (2001) Pharmacokinetics: the dynamic of drug absorption, distribution and elimination. In: Goodman and Gilman's the pharmacological basis of therapeutics, Ed 10 (Hardman JG, Limbird LE, Gilman AG, eds), pp 3-29. New York: McGraw-Hill.
- Wing LK, Behanna HA, Van Eldik LJ, Watterson DM, Ralay Ranaivo H (2006) De novo and molecular target-independent discovery of orally bioavailable lead compounds for neurological disorders. Curr Alzheimer Res, in press. [DOI] [PubMed]
- Wright JM (2002) The double-edged sword of COX-2 selective NSAIDs. Can Med Assoc J 167: 1131-1137. [PMC free article] [PubMed] [Google Scholar]
- Yan Q, Zhang J, Liu H, Babu-Khan S, Vassar R, Biere AL, Citron M, Landreth G (2003) Anti-inflammatory drug therapy alters β-amyloid processing and deposition in an animal model of Alzheimer's disease. J Neurosci 23: 7504-7509. [DOI] [PMC free article] [PubMed] [Google Scholar]