

Abstract
Modulation of K+ channels is widely used to dynamically regulate neuronal membrane excitability. The voltage-gated K+ channel Kv2.1 is an abundant delayed rectifier K+ (IK) channel expressed at high levels in many types of mammalian central neurons where it regulates diverse aspects of membrane excitability. Neuronal Kv2.1 is constitutively phosphorylated, localized in high-density somatodendritic clusters, and has a relatively depolarized voltage dependence of activation. Here, we show that the clustering and voltage-dependent gating of endogenous Kv2.1 in cultured rat hippocampal neurons are modulated by cholinergic stimulation, a common form of neuromodulation. The properties of neuronal Kv2.1 are recapitulated in recombinant Kv2.1 expressed in human embryonic kidney 293 (HEK293) cells, but not COS-1 cells, because of cell background-specific differences in Kv2.1 phosphorylation. As in neurons, Kv2.1 in HEK293 cells is dynamically regulated by cholinergic stimulation, which leads to Ca2+/calcineurin-dependent dephosphorylation of Kv2.1, dispersion of channel clusters, and hyperpolarizing shifts in the voltage-dependent gating properties of the channel. Immunocytochemical, biochemical, and biophysical analyses of chimeric Kv channels show that the Kv2.1 cytoplasmic C-terminal domain can act as an autonomous domain sufficient to transfer Kv2.1-like clustering, voltage-dependent activation, and cholinergic modulation to diverse Kv channels. These findings provide novel mechanistic insights into cholinergic modulation of ion channels and regulation of the localization and voltage-dependent gating properties of the abundant neuronal Kv2.1 channel by cholinergic and other neuromodulatory stimuli.
Keywords: Kv2.1, calcineurin, neuromodulation, acetylcholine, hippocampus, intracellular signaling, calcium
Introduction
Modulation of neuronal ion channels allows for dynamic and reversible regulation of intrinsic membrane excitability. Altering channel phosphorylation state is a widely used mechanism to couple neuronal excitability to diverse cell-signaling events (Levitan, 1994, 1999). Potassium channels are the most diverse molecular and functional group of ion channels (Coetzee et al., 1999) and display the most varied responses to modulation by synaptic activity and neuromodulatory influences (Jonas and Kaczmarek, 1996). That phosphorylation modulates potassium-channel function, neuronal excitability, and animal behavior has been recognized for >20 years (Klein et al., 1982; Hochner and Kandel, 1992). The extent that phosphorylation impacts function of mammalian potassium channels in response to physiological and pathological conditions, and how this could be advantageously modified through pharmacological intervention, remains a major focus of investigation.
Voltage-gated potassium (Kv) channels play diverse and important roles in regulating neuronal membrane excitability (Pongs, 1999). Kv-channel modulation allows synaptic and neuromodulatory events to influence neuronal firing (Jonas and Kaczmarek, 1996). Kv2.1 constitutes the major delayed rectifier Kv channel expressed in mammalian central neurons (Murakoshi and Trimmer, 1999; Malin and Nerbonne, 2002). Knockdown of Kv2.1 in rat hippocampal neurons, where it contributes the majority of delayed rectifier K+ (IK) currents (Murakoshi and Trimmer, 1999), leads to enhanced excitability, especially during high-frequency synaptic transmission (Du et al., 2000). Kv2.1 in adult mammalian brain neurons is constitutively maintained in a highly phosphorylated state (Murakoshi et al., 1997; Misonou et al., 2004) and is localized in high-density somatodendritic clusters (Antonucci et al., 2001; Trimmer and Rhodes, 2004). Enhanced glutamatergic activity (Misonou et al., 2004) and ischemia (Misonou et al., 2005b) lead to rapid Ca2+/calcineurin-dependent dephosphorylation of Kv2.1 and a lateral dispersion of clustered Kv2.1 to a uniform distribution, in vivo and in vitro. Large, hyperpolarizing shifts in voltage-dependent activation of Kv2.1-based neuronal IK are observed under conditions leading to Kv2.1 dephosphorylation (Misonou et al., 2004, 2005b). Such changes in Kv2.1/IK significantly suppress neuronal firing (Surmeier and Foehring, 2004; Misonou et al., 2005b).
Acetylcholine, acting through muscarinic receptor-mediated modulation of ion channels, acts as a potent regulator of neuronal activity in many classes of mammalian neurons. Especially well known examples of muscarinic modulation of ion channels are for KCNQ/M-current potassium channels (Brown and Adams, 1980; Wang et al., 1998) and G-protein-coupled inwardly rectifying potassium channels (Dascal, 1997; Sadja et al., 2003). Here, we show that the localization and function of Kv2.1 in mammalian central neurons are also modulated by cholinergic signaling. Expression of recombinant Kv2.1 in different mammalian cell backgrounds leads to channels with distinct localization and functional properties that we can directly relate to the phosphorylation state. Analyses of chimeric Kv channels defined the Kv2.1 cytoplasmic C terminus as an autonomous domain that can transfer Kv2.1-like clustering, voltage-dependent gating, and cholinergic modulation to other Kv channels. That Kv2.1 is expressed at high levels in many mammalian central neurons suggests that Kv2.1 modulation by muscarinic signaling, and by other pathways that lead to Ca2+ mobilization and calcineurin activation, may provide a widespread mechanism for dynamically regulating neuronal excitability.
Materials and Methods
Cell culture, transient transfection, and drug treatments. Generation of chimeras between Kv2.1, Kv2.2, and Kv1.5 was described previously (Lim et al., 2000). COS-1 and human embryonic kidney 293 (HEK293) cells (American Type Culture Collection, Manassas, VA) were transiently transfected with recombinant cDNAs encoding wild-type (WT) or chimeric Kv2.1, Kv2.2, or Kv1.5 in pRBG4 or pCGN plasmids (cytomegalovirus promoter-controlled expression vectors) using Lipofectamine (Invitrogen, San Diego, CA) according to the manufacturer's instructions. All cells were used within 42-48 h after transfection. Embryonic rat hippocampal neurons (from embryonic day 18/19 embryos) were isolated and cultured on poly-l-lysine-coated glass coverslips or tissue culture-treated plastic Petri dishes as described previously (Misonou and Trimmer, 2005). Neurons were cultured in serum-free astrocyte-conditioned media and used within 13-20 d in vitro for all experiments.
Unless indicated otherwise, drug treatments were performed as follows. Cholinergic stimulation was performed using 100 μm carbachol for 15 min and Ca2+ ionophore treatment using 1 μm ionomycin for 10 min. Initial effects of these drug treatments on Kv2.1 electrophoretic mobility, clustering, and gating were observed within 5 min, and maximum effects were observed after 10-15 min (data not shown). Protein phosphatase (PP)/PP2B inhibition was performed using 5 μm FK520 (ascomycin) for 10 min.
Antibodies. All rabbit polyclonal and mouse monoclonal antibodies used in this study have been described previously. The rabbit polyclonal antibodies used were KC [generated against the distal C-terminal end of Kv2.1 (Trimmer, 1991)], anti-Kv2.1e [generated against the S1-S2 extracellular region of Kv2.1 (Lim et al., 2000)], and Kv1.5e (generated against the S1-S2 extracellular region of Kv1.5). The mouse monoclonal antibodies used were K89/41 [generated against the distal C-terminal domain of Kv2.1 (Antonucci et al., 2001)], K39/25 [generated against the S1-S2 extracellular loop of Kv2.1 (Lim et al., 2000)], and K37/89 [generated against the cytoplasmic N-terminal domain of Kv2.2 (Gelband et al., 1999)].
Biochemical analysis of Kv2.1 protein. SDS-PAGE and immunoblotting of detergent-soluble protein extracts from COS-1 or HEK293 cells transfected with recombinant rat Kv2.1 and from primary culture of rat hippocampal neurons were performed as described previously (Shi et al., 1994; Murakoshi et al., 1997; Misonou et al., 2004). Briefly, cells with or without various drug treatments were lysed with ice-cold 1% Triton X-100-containing buffer and centrifuged at 12,000 × g to collect the supernatants. Aliquots of these lysates were incubated with and without calf intestinal alkaline phosphatase (AP; 100 U/ml) in the presence of 0.1% SDS for 2 h at 37°C. Protein samples were separated on 7.5% SDS-PAGE gels, transferred to nitrocellulose membranes, and immunoblotted with anti-Kv2.1 mouse monoclonal antibody K89/41. Immunoblots were incubated with horseradish peroxidase-conjugated antimouse secondary antibodies (1:1000; ICN Biochemicals, Costa Mesa, CA), followed by enhanced chemiluminescence reagent (PerkinElmer, Wellesley, MA). The Kv2.1-immunoreactive bands on the membranes were visualized by exposing to x-ray film (Eastman Kodak, Rochester, NY). Immunoblots were repeated at least four times to ensure reproducibility of results.
Immunofluorescence staining of cultured hippocampal neurons or mammalian cell lines expressing Kv2.1. COS-1 and HEK293 cells transiently expressing the recombinant Kv channels or cultured rat hippocampal neurons expressing endogenous Kv2.1 were fixed with ice-cold, freshly prepared 4% paraformaldehyde. The cells were then permeabilized and stained with various anti-Kv2.1, anti-Kv1.5, and anti-Kv2.2 monoclonal and polyclonal antibodies and Alexa488- or Alexa594-conjugated antimouse or anti-rabbit secondary antibodies (1:2000; Molecular Probes, Eugene, OR). Immunofluorescence images were captured with a 24-bit color digital camera coupled to Axiovision software installed on an Axioskop2 microscope with a 63× objective (Zeiss, Thornwood, NY). Three-dimensional image analyses of the above-mentioned samples were performed on a Zeiss ApoTome confocal microscope with Axiovision software, as described previously (Yuste et al., 2000). X-Z sections for analyzing the lateral translocation of Kv2.1 clusters and detection of intracellular or cell-surface protein distributions after drug treatment(s) were generated using Axiovision software. All of the images were transferred to PhotoShop software (Adobe Systems, San Jose, CA) as JPEG files. For analyses of clustering, four independent blinded samples of at least 100 cells each were scored for having clustered or dispersed localization.
Electrophysiology and data analysis. Outward potassium currents were recorded from COS-1 or HEK293 cells transiently expressing the recombinant rat Kv2.1 subunit or from cultured rat hippocampal neurons using the whole-cell mode of the patch-clamp technique. Pipettes were pulled from borosilicate glass tubing to give a tip resistance of 1-3 MΩ when filled with the pipette solution. Currents were recorded with an EPC-10 patch-clamp amplifier (HEKA Elektronik, Lambrecht/Pfalz, Germany), sampled at 10 kHz and filtered at 2 kHz using a digital Bessel filter. All currents were capacitance and series resistance compensated, and leak was subtracted by the standard P/n procedure. The extracellular buffer contained (in mm) 140 NaCl, 5 KCl, 2 CaCl2, 2 MgCl2, 10 glucose, and 10 HEPES, pH 7.3. For Ca2+-free extracellular buffer, 2 mm CaCl2 was substituted with 10 mm EGTA. The pipette solution contained (in mm) 140 KCl, 2 MgCl2, 1 CaCl2, 5 EGTA, 10 glucose, and 10 HEPES, pH 7.3. Ionomycin, carbachol, and FK520 were diluted in the extracellular buffer to achieve the final desired concentrations. Calf intestinal AP (Roche, Basel, Switzerland) was dialyzed extensively with the pipette solution at 4°C overnight using 10 kDa pore size dialysis cassettes (Pierce, Rockford, IL) and diluted in the internal solution to give a final working concentration of 100 U/ml. Control experiments were performed using AP inactivated by boiling for 30 min (Murakoshi et al., 1997). Control and experimental extracellular buffers were applied using a polytetrafluorethylene glass multiple-barrel perfusion system. 1,2-Bis(2-aminophenoxy)ethane-N, N,N′, N′-tetraacetic acid (BAPTA) was diluted in the pipette solution to obtain the desired final concentration.
For voltage-dependent current activation experiments, the cells were held at -100 mV and step depolarized to +80 mV for 200 ms with depolarizing 10 mV increments. For neurons, a prepulse at -10 mV for 30 ms was given before each test pulse to inactivate the majority of transient outward K+ currents. For steady-state inactivation experiments, the cells were held at -100 mV and step depolarized to +40 mV for 10 s with 10 mV increments (conditioning steady pulse), followed by a test pulse at +10 mV (test pulse). The interpulse interval was 20 s. The Nernst K+ equilibrium potential EK was calculated as -84 mV. Voltage-dependent activation and steady-state inactivation curves were generated as described previously (VanDongen et al., 1990; Misonou et al., 2004), and the voltage-dependent parameters are detailed in the tables.
PULSE software (HEKA Elektronik) was used for acquisition and analysis of currents. IGOR Pro 4 (WaveMetrix, Lake Oswego, OR) and Origin 7 software (OriginLab, Northampton, MA) were used to perform least-squares fitting and to create the figures. Data are presented as mean ± SEM or fitted value ± SE of the fit. Paired or unpaired Student's t tests (Origin; OriginLab) were used to evaluate the significance of changes in mean values. p < 0.05 was considered statistically significant.
Results
Cholinergic stimulation of hippocampal neurons induces calcineurin-dependent dephosphorylation of Kv2.1, disruption of Kv2.1 clusters, and altered voltage-dependent gating via a mechanism distinct from glutamatergic stimulation
Recently, we found that endogenous Kv2.1 in cultured rat hippocampal neurons undergoes a Ca2+/calcineurin-dependent dephosphorylation after glutamate stimulation that leads to both dispersion of membrane Kv2.1 clusters and hyperpolarizing shifts in voltage-dependent activation (Misonou et al., 2004). Moreover, kainate-induced seizures induce similar changes in rat brain Kv2.1 in vivo (Misonou et al., 2004). Ischemic conditions induce similar changes in neurons in vivo and in vitro (Misonou et al., 2005b). To determine whether stimulation of cholinergic signaling pathways would induce similar modifications in the phosphorylation, localization, and voltage-dependent gating of Kv2.1, cultured rat hippocampal neurons were subjected to carbachol treatment with or without pretreatment with FK520, a specific inhibitor of the Ca2+/calmodulin-dependent PP2B (calcineurin), and analyzed by immunoblotting, immunofluorescence staining, and whole-cell voltage clamp. Under control conditions, Kv2.1 is found localized in the soma and proximal dendrites in high-density cell-surface clusters, which were present in ≈88.6 ± 1.3% (597 cells total in four independent samples of >100 cells each) of the neurons assayed. Cholinergic stimulation led to disruption of these large Kv2.1 clusters, resulting in partial fragmentation or complete disruption of Kv2.1 clusters such that only 42.0 ± 1.6% of the 599 neurons counted in four independent samples of >100 cells each (p < 0.01 vs control) exhibited clustering. Carbachol-induced dispersal of Kv2.1 clusters was antagonized by pretreatment with FK520, such that 82.9 ± 2.4% of neurons (610 counted in four independent samples of >100 neurons each) exhibited Kv2.1 clusters (Fig. 1A). These cultured neurons also have an additional pool of Kv2.1 on the axon initial segment, a culture artifact (Lim et al., 2000) not seen in brain neurons. Like glutamate treatment (Misonou et al., 2004), carbachol treatment did not affect Kv2.1 at these sites.
Figure 1.

Carbachol-treatment of cultured rat hippocampal neurons leads to calcineurin-dependent dephosphorylation and disruption of Kv2.1 clusters and hyperpolarizing shifts in voltage-dependent gating of IK currents. A, Calcineurin-dependent disruption of Kv2.1 clusters in cultured rat hippocampal neurons after carbachol treatment. Images of immunofluorescence staining of control and carbachol-treated (100 μm, 15 min) neurons, without or with pretreatment of FK520 (5 μm, 10 min) are shown. Neurons were immunostained with rabbit anti-Kv2.1 polyclonal antibody KC (green) and mouse anti-microtubule-associated protein 2 monoclonal antibody (red), as detailed in Materials and Methods. The arrows indicate high-density Kv2.1 clusters. Scale bar, 10 μm. B, Immunoblot analysis of native Kv2.1 from cultured rat hippocampal neurons (∼10 μg of crude lysate) without or with carbachol and FK520 treatments, as in A. Samples were size fractionated on 7.5% SDS-PAGE and analyzed by immunoblot/ECL using the anti-Kv2.1 mouse monoclonal antibody K89/41. Native Kv2.1 from rat brain [∼10 μg of crude rat brain membrane proteins (RBM)] without or with in vitro treatment with AP (100 U/ml) is presented to allow for a comparison of the extent of electrophoretic mobility shift obtained after in vivo carbachol treatment versus in vitro AP treatment. The numbers on left refer to the mobility of prestained molecular weight standards in kilodaltons. C, Voltage-dependent activation (filled symbols) and steady-state inactivation (open symbols) curves of outward currents from cultured rat hippocampal neurons without or with carbachol and FK520 treatments, as in A. See Table 1 for voltage-dependent activation and steady-state inactivation parameters. CCh, Carbachol. Error bars represent mean ± SEM.
We next investigated the effects of muscarinic stimulation on the overall phosphorylation state of Kv2.1 by analyzing relative electrophoretic mobility (Mr) on SDS-PAGE gels (Shi et al., 1994; Murakoshi et al., 1997). Cholinergic stimulation led to a pronounced shift in the Mr of Kv2.1 on SDS gels (from ≈125 to 110 kDa), consistent with a carbachol-induced dephosphorylation. The carbachol-induced dephosphorylation was inhibited by pretreatment of neurons with FK520 (Fig. 1B). We next determined whether cholinergic stimulation would affect the voltage-dependent properties of IK currents in these cultured neurons, the majority of which are contributed by Kv2.1 (Murakoshi and Trimmer, 1999). Large (≈22 mV) hyperpolarizing shifts in the voltage dependence of activation of steady-state inactivation were seen in neurons after carbachol treatment (Fig. 1C). The extent of the carbachol effects are revealed by significant shifts in the half-maximal conductance (G1/2) and steady-state inactivation (Vi1/2) membrane potentials (Table 1). The carbachol-induced shifts in voltage-dependent gating were inhibited by pretreating the neurons with FK520 (Fig. 1C, Table 1). Interestingly, cholinergic stimulation of neurons in Ca2+-free extracellular conditions still led to a similar magnitude of dephosphorylation and dispersal of Kv2.1 clusters (data not shown) as well as ≈19 mV hyperpolarizing shifts in the voltage-dependent activation and inactivation midpoint membrane potentials of neuronal IK currents (Table 1). In this regard, muscarinic modulation of Kv2.1 differs from that induced by glutamate stimulation, which depended on Ca2+ in the external medium (Misonou et al., 2004). These results together show that increased cholinergic activity in hippocampal neurons induces dephosphorylation of Kv2.1, dispersion of Kv2.1 clusters, and hyperpolarizing shifts in the voltage-dependent Kv2.1 gating. The muscarinic signaling pathway is distinct in its upstream events from that induced by glutamate but similar in its convergence on calcineurin-dependent Kv2.1 dephosphorylation.
Table 1.
Voltage-dependent gating properties of IK currents in cultured rat hippocampal pyramidal neurons and Kv2.1 currents in COS and HEK cells
|
|
Activation parameters |
Inactivation parameters |
||||||
|---|---|---|---|---|---|---|---|---|
| Drug treatments |
G½ (mV) |
k
|
Vi½ (mV) |
k
|
n
|
|||
| Hippocampal neuron IK | ||||||||
| Control | +16.7 ± 0.6 | 17.2 ± 0.5 | −25.7 ± 0.5 | 9.6 ± 0.4 | 6 | |||
| Plus CCh | −6.5 ± 0.4a | 13.2 ± 0.4 | −54.5 ± 0.6a | 9.3 ± 0.6 | 6 | |||
| Plus CCh (Ca2+-free external) | −2.7 ± 0.4a | 14.5 ± 0.4 | −54.1 ± 0.4a | 8.6 ± 0.6 | 3 | |||
| Plus FK520 plus CCh | +14.4 ± 0.6 | 16.4 ± 0.5 | −28.2 ± 0.4 | 9.4 ± 0.5 | 4 | |||
| COS-Kv2.1 | ||||||||
| Control | +1.4 ± 0.4 | 15.9 ± 0.3 | −29.5 ± 0.3 | 7.7 ± 0.3 | 6 | |||
| Plus AP | −21.3 ± 0.7a | 14.4 ± 0.6 | −55.0 ± 0.1a | 8.2 ± 0.4 | 6 | |||
| HEK-Kv2.1 | ||||||||
| Control | +16.4 ± 0.6 | 15.1 ± 0.6 | −26.2 ± 0.4 | 7.6 ± 0.3 | 8 | |||
| Plus AP | −19.2 ± 0.9a | 13.9 ± 0.8 | −61.0 ± 0.3a | 7.3 ± 0.1 | 7 | |||
| Plus Inm | −9.4 ± 0.7a | 11.8 ± 0.6 | −59.0 ± 0.3a | 7.3 ± 0.1 | 7 | |||
| Plus Inm (in Ca2+-free external) | +14.1 ± 0.6 | 15.2 ± 0.5 | −29.2 ± 0.7 | 7.4 ± 0.3 | 5 | |||
| Plus FK520 plus Inm | +14.6 ± 0.9 | 16.1 ± 0.5 | −28.4 ± 0.7 | 8.1 ± 0.5 | 6 | |||
| Plus CCh | −9.0 ± 0.5a | 12.7 ± 0.4 | −60.2 ± 0.5a | 9.9 ± 0.2 | 8 | |||
| Plus FK520 plus CCh | +15.1 ± 0.7 | 16.4 ± 0.6 | −27.2 ± 0.5 | 7.8 ± 0.3 | 5 | |||
| Plus BAPTA plus CCh | +17.6 ± 0.6 | 16.3 ± 0.4 | −25.8 ± 0.5 | 7.2 ± 0.5 | 6 | |||
| Plus 2-APB plus CCh | +13.1 ± 0.8 | 16.8 ± 0.7 | −30.2 ± 1.1 | 8.3 ± 0.6 | 4 | |||
| Plus FK520 |
+15.8 ± 0.5 |
15.5 ± 0.7 |
−26.4 ± 0.8 |
7.5 ± 0.4 |
5 |
|||
k denotes the slope of activation or inactivation curves. CCh, Carbachol; Inm, ionomycin; 2-APB, 2-aminoethoxydiphenyl borate.
Significantly different (p<0.05) from respective control values.
Phosphorylation-dependent differences in localization and function of Kv2.1 expressed in different mammalian cell lines
The detailed investigation of the mechanistic basis for the profound phosphorylation-dependent changes in Kv2.1 localization and function requires a reliable expression system that recapitulates the constitutive and regulated properties of endogenous Kv2.1 in neurons. Because the localization and function of Kv2.1 are so dramatically affected by posttranslational changes in the phosphorylation state, the cell repertoire of protein kinases and phosphatases should be critical to the properties of heterologously expressed Kv2.1. We therefore expressed recombinant WT rat Kv2.1 in two commonly used transformed primate kidney cell lines, green monkey COS-1 and human HEK293, to determine their suitability for such analyses. Immunofluorescence staining of exogenous Kv2.1 expressed in COS-1 cells revealed uniform plasma membrane staining of cell-surface Kv2.1 (Fig. 2A, left), similar to that shown previously (Shi et al., 1994). However, in HEK293 cells, exogenous Kv2.1 was localized in the form of distinct cell-surface clusters (Fig. 2A, right), similar to those seen for endogenous and exogenous Kv2.1 in mammalian neurons (Lim et al., 2000; Antonucci et al., 2001). This suggests that certain, but not all, mammalian cell lines contain the machinery for formation of plasma membrane Kv2.1 clusters.
Figure 2.
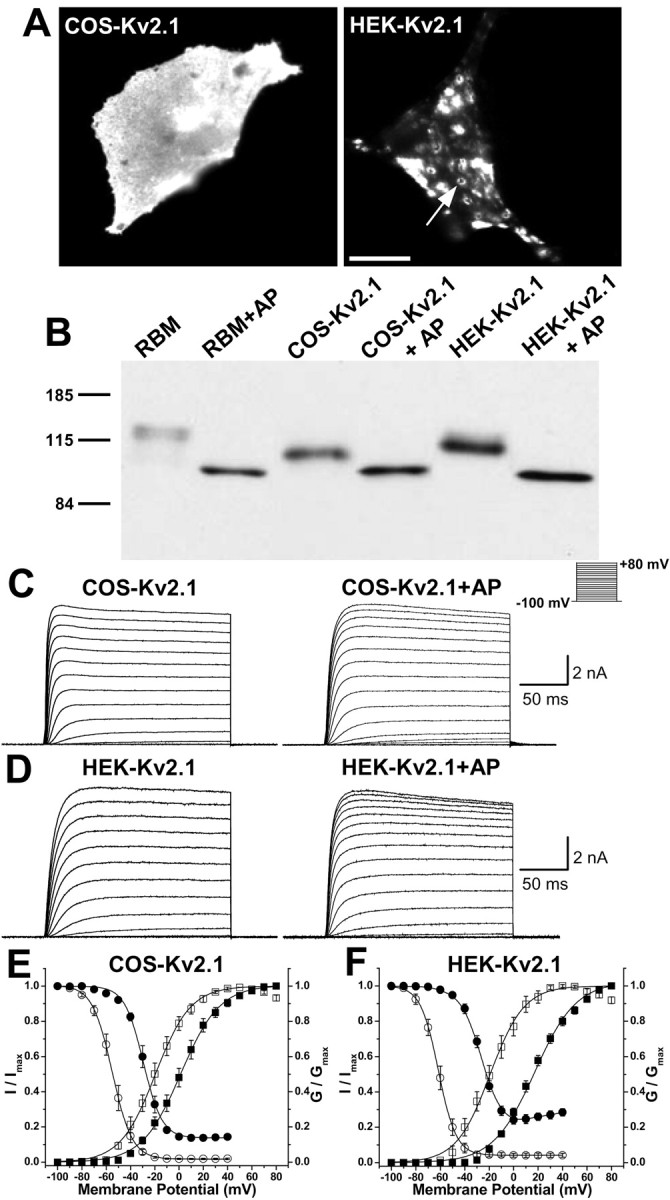
Phosphorylation-dependent differences in localization and function of recombinant Kv2.1 in different mammalian cell backgrounds. A, Immunofluorescence staining analysis of recombinant rat Kv2.1 expressed in COS-1 (left) and HEK293 (right) cells. The cells were immunostained with anti-Kv2.1 mouse monoclonal antibody K89/41, as detailed in Materials and Methods. The arrow indicates a high-density Kv2.1 cluster in an HEK293 cell; note the uniform localization in COS-1 cells. Scale bar, 10 μm. B, Immunoblot analysis of native Kv2.1 from rat brain [∼10 μg of crude rat brain membrane proteins (RBM)] and recombinant Kv2.1 in detergent extracts from COS-1 or HEK293 cells (∼5 μg of total protein each) without or with in vitro treatment with AP (100 U/ml), as detailed in Materials and Methods. Samples were size fractionated on 7.5% SDS-PAGE and analyzed by immunoblot using anti-Kv2.1 mouse monoclonal antibody K89/41, followed by ECL detection. The numbers on the left refer to the mobility of prestained molecular weight standards in kilodaltons. C, D, Representative whole-cell Kv2.1 current traces from COS-1 (C) and HEK293 (D) cells before and after 30 min intracellular dialysis of AP (100 U/ml) as per the shown pulse protocol. E, F, Voltage-dependent activation (squares) and steady-state inactivation (circles) relationships of Kv2.1 currents in COS-1 (E) and HEK293 (F) cells before (filled symbols) and after (open symbols) intracellular dialysis of AP for 30 min, as detailed in Materials and Methods. See Table 1 for voltage-dependent activation and steadystate inactivation parameters. Error bars represent mean ± SEM.
Analysis of the overall phosphorylation state of Kv2.1 using relative electrophoretic mobility (Mr) on SDS-PAGE gels (Shi et al., 1994; Murakoshi et al., 1997) revealed that recombinant Kv2.1 in HEK293 cells is more extensively phosphorylated (has a higher Mr ≈ 115 kDa) (Fig. 2B) than it is in COS-1 cells (Mr ≈ 105 kDa) (Fig. 2B). However, in both heterologous cell lines, Kv2.1 protein is overall less extensively phosphorylated than is native Kv2.1 protein in rat brain membrane preparations (Mr ≈ 125 kDa) (Fig. 2B). That these differences in Mr are, in fact, attributable to cell-specific differences in phosphorylation is shown by the fact that in vitro AP treatment of the different preparations removed all of the differences in Kv2.1 Mr (Fig. 2B). AP treatment yields in each case a Kv2.1 protein with an Mr ≈ 97 kDa, similar to that predicted (95.3 kDa) from the deduced amino acid sequence of the Kv2.1 cDNA. These data suggest that phosphorylation of Kv2.1 is the major posttranslational modification occurring in these different cell backgrounds, although the extent and/or nature of phosphorylation differs between cell types.
Whole-cell outward K+ currents recorded from COS-1 and HEK293 cells expressing recombinant Kv2.1 showed distinct voltage dependence of activation. Kv2.1 currents in HEK293 cells exhibited activation and steady-state inactivation parameters (Fig. 2D, F; Table 1) similar to that observed for endogenous Kv2.1 in cultured hippocampal neurons (Table 1) (Murakoshi and Trimmer, 1999; Misonou et al., 2004). In contrast, Kv2.1 currents in COS-1 cells showed a significant ≈15 mV hyperpolarizing shift in voltage-dependent activation relative to Kv2.1 in HEK293 cells and neurons but had similar steady-state inactivation parameters (Fig. 2C,E; Table 1). To experimentally address whether these cell type-specific differences in channel function were attributable to the distinct patterns of phosphorylation inferred from SDS-PAGE, we used intracellular dialysis of AP to induce channel dephosphorylation in vivo (Murakoshi et al., 1997). Dialysis of AP (100 U/ml for 30 min) through the patch pipette led to dramatic hyperpolarizing shifts in the voltage-dependent activation and inactivation properties of Kv2.1 currents in neurons, HEK293, and COS-1 cells (Table 1). Importantly, AP treatment removed all of the cell-specific differences in activation and steady-state inactivation properties of Kv2.1 currents, suggesting that the differences in Kv2.1 function in the different cell backgrounds were attributable to cell-specific differences in the phosphorylation state of the channel. As observed previously for COS-1 cells (Murakoshi et al., 1997), holding cells at 30 min without AP in the patch pipette, or with AP that had been inactivated by boiling, had no effect on the activation and steady-state inactivation of Kv2.1 in either COS-1 or HEK cells (supplemental Table 1, available at www.jneurosci.org as supplemental material).
Interestingly, Kv2.1 currents in HEK293 cells, but not in COS-1 cells, exhibit U-type voltage dependence of inactivation (Fig. 2F), as described previously for Kv2.1 (VanDongen et al., 1990; Klemic et al., 1998; Kurata et al., 2002). The U-type inactivation of Kv2.1 currents in HEK293 cells is abolished with intracellular AP treatment (Fig. 2F). Although the half-maximal inactivation potentials of Kv2.1 current are not significantly different in COS-1 and HEK293 cells, the percentage of total current that is resistant to steady-state inactivation differs in the different cell backgrounds. The fraction of Kv2.1 current resistant to inactivation in COS-1 and HEK293 cells was 24.2 ± 2.4 and 13.8 ± 0.3%, respectively, which shifted to similar values (4.1 ± 1.4 and 1.9 ± 0.4%, respectively) after AP treatment (Fig. 2E, F).
Muscarinic and ionophore induction of Ca2+/calcineurin-dependent dephosphorylation of Kv2.1 modulates channel clustering and gating in HEK293 cells
We next determined whether recombinant Kv2.1 expressed in HEK293 cells would be modulated by Ca2+-dependent dephosphorylation as is endogenous Kv2.1 in hippocampal neurons after treatment with glutamate (Misonou et al., 2004) and with carbachol (Fig. 1). Immunoblots of samples from Kv2.1-transfected HEK293 cells showed shifts in the Mr of Kv2.1 (from Mr ≈ 115 to ≈ 103 kDa) in samples from cells treated with the Ca2+ ionophore ionomycin, although the ionomycin-induced in vivo shift in the Mr of HEK cell Kv2.1 was not to the full extent observed after in vitro AP treatment (Fig. 3A). Higher concentrations of ionomycin (10 and 100 μm) did not yield further shifts in Kv2.1 Mr (data not shown), and in vitro AP treatment of samples from ionomycin-treated cells yielded a further shift in Mr to the same end point as observed for AP treatment of unstimulated cells (Fig. 3A). These data suggest that the ionomycin-induced changes in the Kv2.1 Mr are attributable to in vivo dephosphorylation at a subset of sites dephosphorylated by exhaustive AP treatment in vitro. Ionomycin treatment in Ca2+-free extracellular buffer did not lead to a shifted Mr of Kv2.1, indicating that ionophore-mediated Ca2+ influx underlies ionomycin-induced channel dephosphorylation (data not shown). Pretreatment of cells with the calcineurin inhibitor FK520 (5 μm, 10 min) blocked the ionomycin-induced shift in Kv2.1 Mr (Fig. 3B), suggesting that as in hippocampal neurons, the effects of increasing [Ca2+]i on Kv2.1 phosphorylation are mediated through calcineurin.
Figure 3.

Ionomycin and carbachol induce Ca2+/calcineurin-dependent dephosphorylation and disruption of Kv2.1 clusters in HEK293 cells. A, HEK293 cells expressing Kv2.1 were treated with 1 μm ionomycin or 100 μm carbachol. One aliquot each of the drug-treated cell lysates was treated with AP (100 U/ml). Approximately 5 μg of protein each from control and AP-treated samples were size fractionated on 7.5% SDS-PAGE and analyzed by immunoblot/ECL using anti-Kv2.1 mouse monoclonal antibody K89/41. B, HEK293 cells expressing Kv2.1 were treated with 1 μm ionomycin or 100 μm carbachol without or with pretreatment of 5 μm FK520, as detailed in Materials and Methods. Lysates from these drug-treated cells (∼5 μg of protein each) were size fractionated on 7.5% SDS-PAGE and analyzed by immunoblot/ECL using anti-Kv2.1 mouse monoclonal antibody K89/41. The numbers on the left refer to the mobility of prestained molecular weight standards in kilodaltons. C, Ca2+/calcineurin-dependent altered localization of Kv2.1 in HEK293 cells after ionomycin or carbachol treatment. Kv2.1-transfected cells were treated with ionomycin or carbachol, as in A and B, in either Ca2+-containing or Ca2+-free extracellular conditions and immunostained with anti-Kv2.1 antibody K89/41. The projected images (top) were reconstructed from ∼40 optical X-Z sections (0.35 μm) of the cells taken with the Zeiss ApoTome confocal microscope. The bottom panel in each cell is the cross-sectional view at the level of drawn line. The arrows indicate high-density Kv2.1 clusters. Scale bar, 10 μm. Quantitative analyses of clustering in these samples are presented in supplemental Table 2 (available at www.jneurosci.org as supplemental material). CCh, Carbachol; Inm, ionomycin.
We next investigated whether cholinergic stimulation acting through intracellular Ca2+ release could induce dephosphorylation of Kv2.1 in HEK293 cells as it does in hippocampal neurons. HEK293 cells endogenously express m1 and m3 isoforms of muscarinic acetylcholine receptors, which mediate carbachol-induced Ca2+ release from inositol 1,4,5-trisphosphate receptor (IP3R)-sensitive intracellular Ca2+ stores (Lee et al., 1998; Tong et al., 1999). Treatment of HEK293 cells expressing Kv2.1 with carbachol resulted in a shift in the electrophoretic mobility of Kv2.1 similar to that observed after ionomycin treatment (Fig. 3A). That the Mr of Kv2.1 in samples from carbachol-treated and control HEK cells reached the same end point after AP treatment shows that the altered electrophoretic mobility of Kv2.1 in carbachol-stimulated cells is attributable to altered phosphorylation. Unlike the results obtained with ionomycin treatment of HEK cells, extracellular Ca2+ was not required for carbachol-induced shifts in the Kv2.1 Mr (data not shown). Pretreatment of cells with FK520 completely blocked the carbachol-induced shift in the Mr of Kv2.1 (Fig. 3B), whereas 1 μm okadaic acid, a specific inhibitor of PP1/2A, did not (data not shown), indicating that muscarinic stimulation of HEK cells leads to Ca2+/calcineurin-dependent dephosphorylation of Kv2.1.
We next investigated whether treatments that modify the phosphorylation state of Kv2.1 in HEK293 cells (i.e., ionomycin, carbachol) would impact channel clustering. Kv2.1 clusters in HEK cells were disrupted (Fig. 3C) after treatment with either ionomycin, requiring extracellular Ca2+, or carbachol, not requiring extracellular Ca2+ (supplemental Table 2, available at www.jneurosci.org as supplemental material). Pretreatment of cells with FK520 inhibited the ionomycin- and carbachol-induced dispersal of Kv2.1 clusters (Fig. 3C), suggesting that calcineurin-mediated dephosphorylation of Kv2.1 leads to the induced dispersal of Kv2.1 clusters. As suggested by immunoblots (Fig. 3A, B), by confocal imaging (Fig. 3C) and by analyses of cell-surface versus total Kv2.1 staining (data not shown), these short (15 min) drug treatments did not lead to changes in either the overall or cell-surface expression of Kv2.1.
We next investigated whether Ca2+ ionophore- and cholinergic-mediated modulation of the voltage dependence of Kv2.1 gating occurred in HEK cells. Treatment of Kv2.1-transfected HEK293 cells with ionomycin or carbachol led to significant (≈25-30 mV) hyperpolarizing shifts in the voltage dependence of activation and steady-state inactivation of macroscopic Kv2.1 currents (Fig. 4, Table 1). Ionomycin and carbachol treatments also abolished the U-type inactivation of Kv2.1 currents observed in control cells (Fig. 4C). As such, the effects of Ca2+ ionophore and cholinergic stimulation were similar to those observed after dialysis of AP (Fig. 2E, F). These functional alterations in Kv2.1 channels were antagonized by pretreatment of cells with FK520 or by including 10 μm BAPTA in the pipette solution (to buffer intracellular Ca2+) before ionomycin or carbachol treatment (Fig. 4A, C; Table 1). It should be noted that all other recordings were performed with 5 mm EGTA in the patch pipette. Pretreatment of cells with pirenzipine (10 μm, 15 min), a selective blocker of m1 and m3 type muscarinic receptors (data not shown), or 2-aminoethoxy-diphenyl borate (10 μm, 15 min) (Table 1), a blocker of IP3Rs (Ma et al., 2000), also antagonized the carbachol-induced hyperpolarizing shifts in the voltage-dependent gating of HEK cell Kv2.1. Interestingly, treatment of COS-1 cells expressing Kv2.1 with ionomycin or carbachol did not yield any significant changes in the voltage-dependent activation and steady-state inactivation properties (data not shown), suggesting that they may lack sufficient calcineurin levels to mediate Ca2+-dependent Kv2.1 dephosphorylation. These data together suggest that muscarinic stimulation of HEK cells leads to m1/m3 muscarinic receptor-mediated, Gq-coupled phospholipase C (PLC) activation, resulting in an IP3R-mediated increase in [Ca2+]i and calcineurin activation and Kv2.1 dephosphorylation. That HEK cells resemble hippocampal neurons in the overall phosphorylation state, clustering, and voltage-dependent gating properties of Kv2.1, and in the modulation thereof by cholinergic and Ca2+ ionophore stimulation, allows for the use of this model system for analyses of the domains of Kv2.1 responsible for such modulation.
Figure 4.

Ionomycin and carbachol treatment of HEK293 cells expressing Kv2.1 lead to Ca2+/calcineurin-dependent hyperpolarizing shifts in voltage-dependent channel-gating properties. A, Representative Kv2.1 current traces from control cells and from cells treated with ionomycin (1 μm) or carbachol (100 μm) and with or without previous intracellular dialysis of FK520 (5 μm) or BAPTA (10 μm). Currents were recorded following the same pulse protocol as used in Figure 2C. B, C, Voltage-dependent activation (B) and steady-state inactivation (C) relationship of Kv2.1 currents obtained in experiments performed as in A. See Table 1 for voltage-dependent activation and steady-state inactivation parameters. Error bars represent mean ± SEM.
The cytoplasmic C terminus of Kv2.1 is a transferable, autonomous domain that is both necessary and sufficient for conferring Kv2.1-like constitutive and regulated properties to diverse Kv channels
To investigate the mechanism whereby the localization and function of Kv2.1 is regulated by muscarinic and Ca2+ ionophore stimulation, we analyzed a panel of chimeric Kv channels. Previous studies of Kv2.1 phosphorylation in COS-1 cells (Murakoshi et al., 1997) and immunoblot analyses of samples prepared from HEK cells (data not shown) suggest that the Ser/Thr-rich cytoplasmic C-terminal domain of Kv2.1 is the major substrate for phosphorylation. Moreover, this domain can act as an autonomous determinant of Kv2.1 channel clustering in hippocampal neurons (Lim et al., 2000). As such, we also investigated the role of the Kv2.1 C terminus as a mediator of cholinergic and Ca2+ ionophore-stimulated modulation of Kv channel localization and function.
The Kv2.1 and Kv2.2 channel proteins exhibit a high degree of sequence identity (87%) within the first 521 aa residues, after which there is a divergence in the amino acid sequence such that the remainder of the C terminus is only 24% identical. Chimeric Kv2.1-Kv2.2 channels were generated (Lim et al., 2000) by exchanging the divergent C termini of these otherwise highly related Kv2 channels (Fig. 5A). Immunostaining of the chimeric Kv2.2N-Kv2.1C channel showed clustered surface localization (Fig. 5B; supplemental Table 2, available at www.jneurosci.org as supplemental material) as opposed to dispersed surface localization of WT Kv2.2 (Fig. 5B; supplemental Table 2, available at www.jneurosci.org as supplemental material) and chimeric Kv2.1N-Kv2.2C channels (data not shown) in HEK293 cells. This pattern of clustering parallels that observed in hippocampal neurons expressing these same chimeras (Lim et al., 2000), providing additional support for the conserved mechanisms for Kv channel clustering between HEK cells and hippocampal neurons. Cholinergic stimulation led to a dispersal of surface clusters in the chimeric Kv2.2N-Kv2.1C channel (Fig. 5B; supplemental Table 2, available at www.jneurosci.org as supplemental material), similar to that observed for WT Kv2.1, but did not alter the dispersed plasma membrane localization pattern of WT Kv2.2 (Fig. 5B) and the Kv2.1N-Kv2.2C chimera (data not shown) in HEK293 cells. Ca2+ ionophore stimulation led to a similar pattern of modulation (data not shown). Thus, the Kv2.1 C terminus can transfer muscarinic modulation of localization to the highly related mammalian Shab/Kv2 channel.
Figure 5.

Immunocytochemical analyses of surface localization of the chimeric Kv2.2N-Kv2.1C and Kv1.5N-Kv2.1C channels in HEK293 cells without or with cholinergic stimulation. A, Schematic showing the construction of chimeric Kv2.2N-Kv2.1C and Kv1.5N-Kv1.5C channels. B, C, The chimeric Kv2.2N-Kv2.1C (B) and Kv1.5N-Kv2.1C (C) channels containing the cytoplasmic C-terminal domain of Kv2. 1 show clustered localizations that are disrupted after carbachol treatment. HEK293 cells expressing WT or chimeric channels were treated with 100 μm carbachol and immunostained with the Kv1.5e rabbit polyclonal antibody (A) and the anti-Kv2.2 mouse monoclonal antibody K37/89 (B). The projected images (top) were reconstructed from ∼40 optical X-Z sections (0.35 μm) of the cells taken with the Zeiss ApoTome confocal microscope. The bottom panel in each cell is the cross-sectional view at the level of drawn line. The arrows indicate high-density channel clusters. Scale bar, 10 μm. Quantitative analyses of clustering in these samples are presented in supplemental Table 2 (available at www.jneurosci.org as supplemental material). CCh, Carbachol.
To further investigate the autonomous nature of the Kv2.1 C terminus, we analyzed a second set of chimeric Kv channels (Fig. 5A), in this case between Kv2.1 and the much less related (27% overall amino acid identity) mammalian Shaker Kv1.5 channel. Immunostaining of the chimeric Kv1.5N-Kv2.1C channel showed distinct cell-surface clusters, unlike the dispersed surface localization of WT Kv1.5 (Fig. 5C; supplemental Table 2, available at www.jneurosci.org as supplemental material), again consistent with the pattern of localization observed in hippocampal neurons (Lim et al., 2000). Interestingly, carbachol treatment also led to the dispersal of surface clusters of the chimeric Kv1.5N-Kv2.1C channel (Fig. 5C; supplemental Table 2, available at www.jneurosci.org as supplemental material). Carbachol treatment did not lead to any detectable alteration in the dispersed surface localization pattern of WT Kv1.5 (Fig. 5C). We next analyzed a second Kv1.5N-Kv2.1C chimera, in which only a short (65 aa) stretch (amino acids 536-600) of the 443 aa Kv2.1 C terminus, and including the minimal 28 aa segment [the proxinal restriction and clustering (PRC) signal] previously shown to be necessary and sufficient for the proximal dendritic localization and clustering of Kv2.1 in hippocampal neurons (Lim et al., 2000), was transferred to Kv1.5 to generate the Kv1.5N-Kv2.1C-PRC chimera. Consistent with the previous results from hippocampal neurons (Lim et al., 2000), this 65 aa segment of Kv2.1 is sufficient to transfer Kv2.1-like clustering to Kv1.5 in HEK293 cells (Fig. 5C; supplemental Table 2, available at www.jneurosci.org as supplemental material). However, the Kv1.5N-Kv2.1C-PRC chimera is unresponsive to muscarinic stimulation, or to treatment with Ca2+ ionophore (Fig. 5C; supplemental Table 2, available at www.jneurosci.org as supplemental material). These results define the Kv2.1 cytoplasmic C terminus as an autonomous, transferable cholinergic response element and segregate the PRC signal itself from the domain conferring muscarinic (and Ca2+ ionophore) modulation of clustering.
Electrophysiological analyses of the voltage-dependent gating properties of these chimeric channels show that the voltage-dependent gating properties of WT Kv2.2 and Kv1.5 channels respond only minimally to cholinergic modulation, with 5-7 mV shifts in activation V1/2 (Fig. 6A, Table 2). However, the chimeras containing the Kv2.1 C terminus, Kv2.2N-Kv2.1C and Kv1.5-Kv2.1C, exhibit large and significantly different (≈37 and ≈18 mV, respectively) hyperpolarizing shifts in the G1/2 of activation after carbachol stimulation (Fig. 6A, Table 2). In contrast to the effects of muscarinic modulation on activation, no uniform pattern was observed for inactivation (Fig. 6B, Table 2). The other chimeric channels lacking the complete Kv2.1 cytoplasmic C terminus [Kv2.1N-Kv2.2C and Kv1.5-Kv2.1C(PRC)] did not exhibit carbachol-induced hyperpolarizing shifts in their voltage-dependent gating properties relative to WT Kv2.2 and Kv1.5 channels (Fig. 6, Table 2).
Figure 6.

Voltage-dependent biophysical properties of Kv2.2N-Kv2.1C and Kv1.5N-Kv2.1C chimeric channels without or with carbachol treatment. Voltage-dependent activation (A, C) and steady-state inactivation (B, D) curves of WT Kv2.2 (A, B) and Kv1.5 (C, D) and of chimeric Kv2.2N-Kv2.1C (A, B) and Kv1.5N-Kv2.1C (C, D) channels, without or with the extracellular application of 100 μm carbachol (CCh) for 15 min. The voltage-dependent parameters are detailed in Table 2. Error bars represent mean ± SEM.
Table 2.
Voltage-dependent activation and inactivation properties of WT and chimeric Kv2.1, Kv2.2, and Kv1.5 channels in HEK293 cells without or with carbachol treatment
|
|
|
Activation parameters |
Inactivation parameters |
|
||||
|---|---|---|---|---|---|---|---|---|
| WT/chimeric channel |
Drug treatment |
G½ (mV) |
k
|
Vi½ (mV) |
k
|
n
|
||
| Kv2.2 WT | Control | +16.0 ± 0.5 | 14.5 ± 0.4 | −26.4 ± 0.5 | 7.8 ± 0.4 | 7 | ||
| Plus CCh | +8.6 ± 1.0a | 15.9 ± 0.9 | −44.5 ± 0.3a | 4.8 ± 0.2 | 6 | |||
| Kv2.2N/2.1C | Control | +23.8 ± 1.1a | 18.6 ± 1.0 | −15.1 ± 0.8 | 6.6 ± 0.7 | 6 | ||
| Plus CCh | −13.5 ± 0.6a | 13.9 ± 0.5 | −37.5 ± 0.3a | 4.3 ± 0.2 | 6 | |||
| Kv2.1N/2.2C | Control | +17.5 ± 0.9 | 15.1 ± 0.7 | −29.8 ± 0.7 | 5.5 ± 0.6 | 6 | ||
| Plus CCh | +9.8 ± 0.4a | 11.6 ± 0.2 | −35.3 ± 0.6 | 9.8 ± 0.1 | 6 | |||
| Kv1.5 WT | Control | +10.2 ± 1.4 | 19.1 ± 1.2 | +3.7 ± 0.8 | 14.3 ± 0.5 | 7 | ||
| Plus CCh | +5.4 ± 0.9a | 15.3 ± 0.8 | −12.4 ± 0.4a | 9.8 ± 0.3 | 7 | |||
| Kv1.5N/2.1C | Control | −1.1 ± 1.1 | 11.6 ± 0.9 | −22.1 ± 0.9 | 6.1 ± 0.8 | 7 | ||
| Plus CCh | −19.8 ± 0.6a | 14.1 ± 0.6 | −37.2 ± 0.2a | 4.1 ± 0.2 | 6 | |||
| Kv1.5N/2.1C (PRC) | Control | +10.7 ± 0.7 | 18.8 ± 1.2 | +11.8 ± 1.8 | 15.4 ± 0.9 | 6 | ||
|
|
Plus CCh |
+4.8 ± 0.9a
|
17.0 ± 0.8 |
−7.8 ± 0.5a
|
12.7 ± 0.4 |
6 |
||
k denotes the slope of activation or inactivation curves. CCh, Carbachol.
Significantly different (p <0.05) from respective control values.
Discussion
Diverse modes of Kv2.1 modulation
The cholinergic modulation of Kv2.1 in cultured hippocampal neurons extends the spectrum of stimuli that can impinge on intrinsic neuronal excitability via calcineurin activation and Kv2.1 dephosphorylation. The finding that glutamate stimulation in vitro (and seizures in vivo) can induce Kv2.1 dephosphorylation via a mechanism requiring Ca2+ influx and calcineurin activation (Misonou et al., 2004) was followed by studies showing that hypoxia/ischemia in vivo and in vitro induce Ca2+ release from intracellular stores, apparently from mitochondria, leading to calcineurin activation and Kv2.1 modulation (Misonou et al., 2005b). We show here that cholinergic stimulation also leads to calcineurin-dependent Kv2.1 modulation in hippocampal neurons (as well as HEK293 cells), caused by Ca2+ release from intracellular stores. Hippocampal neurons express each of the muscarinic receptor subtypes (Levey et al., 1991, 1995), and activation of m1 or m3 muscarinic receptors in hippocampal neurons leads to IP3R-mediated intracellular Ca2+ release (Muller and Connor, 1991; Irving and Collingridge, 1998). That cholinergic stimulation induces Kv2.1 modulation through increased [Ca2+]i raises the question as to how widespread Kv2.1 modulation may be, given the high-level expression of Kv2.1 in many types of principal cells and interneurons throughout the brain (Hwang et al., 1993; Scannevin et al., 1996; Du et al., 1998) and the large number of G-protein-coupled receptor (GPCR) and receptor tyrosine kinase systems that activate PLC and lead to IP3R-dependent increases in [Ca2+]i. This includes, but is not limited to, the important neuromodulatory serotonergic (5-HT2), purinergic (P2Y), and metabotropic (mGluR1 and mGluR3) pathways (gpDB, a database of G-proteins and their interaction with GPCRs; http://bioinformatics2.biol.uoa.gr/gpDB/index.jsp). The overall effect of such modulation would be to allow for coupling of diverse neurotransmitter-induced increases in [Ca2+]i to enhanced activity of the abundant neuronal delayed rectifier Kv2.1 channel and suppression of neuronal excitability. However, the effects of cholinergic stimulation are complex and involve both positive and negative modulation of intrinsic excitability through the pleiotropic action of acetylcholine on a number of ion channels (Lucas-Meunier et al., 2003); as such, the specific role of Kv2.1 modulation in regulating overall neuronal excitability in response to cholinergic stimulation remains to be elucidated.
That Kv2.1 is regulated by Ca2+ release from intracellular stores fits well with the localization of Kv2.1 clusters over subsurface cisterns (modified smooth endoplasmic reticulum) in cortical neurons (Du et al., 1998). We also observed Kv2.1 clusters adjacent to ryanodine receptor- and calsequestrin-rich intracellular membranes in cultured hippocampal neurons (Antonucci et al., 2001). We show here that Kv2.1 modulation is sensitive to the presence of the fast Ca2+ chelator BAPTA in the patch pipette, but not to the slow chelator EGTA, which is consistent with a role for local Ca2+ in mediating Kv2.1 modulation (Marrion and Tavalin, 1998). Calcineurin-dependent dispersal of Kv2.1 from the clusters may facilitate eventual recovery to the constitutive state (i.e., after rephosphorylation) by removing Kv2.1 from the focal source of intracellular Ca2+ release, as well as the high levels of calcineurin that have been reported to be associated with the intracellular Ca2+ channels themselves (Cameron et al., 1995).
Mechanisms of muscarinic modulation
Muscarinic stimulation regulates a variety of other K+ channels through diverse mechanisms, including tyrosine phosphorylation (Huang et al., 1993), G-protein binding (Dascal, 1997), and phosphatidylinositol 4,5-bisphosphate binding (Mark and Herlitze, 2000). Muscarinic modulation of Kv2.1 occurs via a distinct mechanism, involving calcineurin-mediated dephosphorylation of the channel protein. We found that the Kv2.1 cytoplasmic C terminus can act as a transferable “muscarinic response element” that can confer muscarinic modulation of localization and function to otherwise insensitive K+ channels. This expands the known function of this domain that has already been identified as the main determinant of the constitutive polarized somatodendritic localization and clustering of Kv2.1 (Scannevin et al., 1996; Lim et al., 2000). The more distal portion of the Kv2.1 C terminus, from amino acids 521-853, is highly conserved among Kv2.1 orthologs (rat/mouse, 94%; rat/human, 86%) but poorly conserved between rat Kv2.1 and Kv2.2 (24%), or Kv2.1 and Kv1.5. The high degree of evolutionary conservation presumably reflects the important role of the Kv2.1 C terminus in determining subtype-specific characteristics of localization, gating, and neuromodulation.
The results presented here suggest that the muscarinic response elements within the Kv2.1 C terminus lie outside of the precise “PRC” signal for Kv2.1 clustering that was identified in previous studies (Lim et al., 2000), because transfer of a short segment containing the PRC domain transfers constitutive clustering to Kv1.5 but not muscarinic modulation. This finding may be consistent with the perplexing data of Lim et al. (2000), which revealed a surprising level of amino acid similarity between Kv2.1 and Kv2.2 within the PRC signal itself, but dramatic differences in the endogenous localization of these two channels in the same neurons (see also Hwang et al., 1993), and the ability of their respective C termini to mediate channel clustering (Lim et al., 2000). This suggests that although the primary determinant of clustering is the PRC domain, clustering is mediated by additional phosphorylation state-dependent determinants within the Kv2.1 C terminus, but outside of the PRC region. Analyses of the voltage-dependent gating and overall phosphorylation state of a large panel of Kv2.1 truncation and deletion mutants suggest a complex pattern of C-terminal phosphorylation (D. P. Mohapatra and J. S. Trimmer, unpublished data). The pattern of constitutive clustering (or lack thereof) of this large mutant pool (data not shown) was entirely consistent with that observed in hippocampal neurons (Lim et al., 2000), showing that the Kv2.1 PRC clustering signal operates reliably in HEK293 cells. However, analyses of carbachol responsiveness of these mutants, of their AP-induced shifts in Mr, and of their carbacholstimulated, voltage-dependent gating properties yielded a complex data set more consistent with phosphorylation at multiple, widely distributed sites in the cytoplasmic C terminus than with a single switch-like determinant of muscarinic responsiveness (data not shown). Phosphorylation site prediction programs (e.g., NetPhos; http://www.cbs.dtu.dk/services/NetPhos/) predict that of 160 Ser, Thr, and Tyr residues in Kv2.1, 68 score as strong consensus phosphorylation sites. The bulk of these consensus sites (45 of 68) are found dispersed throughout the 443 aa cytoplasmic C terminus. Given the large number of potential phosphorylation sites, it is not surprising that Kv2.1 localization and gating would be controlled by more than a single site. Direct chemical identification of sites phosphorylated in cells before and after muscarinic stimulation may allow for identification of the specific sites critical for mediating muscarinic modulation of Kv2.1
That HEK cells appear to constitutively phosphorylate exogenous Kv2.1 in a manner that allows for clustering and voltage-dependent gating similar to that observed for endogenous Kv2.1 in neurons, and also contain machinery for neuronal-like muscarinic modulation of these properties, is advantageous to studies such as those presented here to define the C terminus of Kv2.1 as a muscarinic response element. However, the dramatic cellspecific effects of posttranslational processing on Kv2.1 function, as revealed from comparison of exogenous Kv2.1 in COS-1 and HEK293 cells, reinforces that any data from heterologous expression systems should be analyzed carefully to ensure consistency with those from native cells. It is intriguing that such an array of diverse properties (localization, voltage-dependent gating, muscarinic modulation) are so dramatically affected by cell background-specific posttranslational processes in these two widely used mammalian cell lines, especially given that both the COS-1 and HEK293 cell lines were derived from viral transformation of primate kidney cells [African green monkey (Gluzman, 1981) and human (Graham et al., 1977), respectively]. A recent microarray analysis suggests that HEK293 cells may be neuronal in origin, because they express a number of neural-specific genes, including neuron-specific enolase, serotonin transporter, and a number of neurofilament isoforms (Shaw et al., 2002). As such, one can speculate that expression of specific neuronal kinases, or their requisite activators, in HEK293 but not COS-1 cells may underlie the dramatic phosphorylation-dependent phenotypic differences in recombinant Kv2.1 protein expressed in the two different cell backgrounds. Our attempts to pharmacologically stimulate COS-1 cells (e.g., with kinase activators and phosphatase inhibitors) to phosphorylate Kv2.1 to a more HEK293/neuronal-like phenotype have failed, suggesting that COS-1 cells are intrinsically deficient in the required kinase activities. That Kv2.1 is processed so differently in these heterologous cell backgrounds is intriguing given the widespread expression of Kv2.1 in the brain and in a variety of non-neuronal tissues, such as the heart, skeletal muscle, smooth muscle, and pancreas (Misonou et al., 2005a). Moreover, the Mr of Kv2.1 in rat brain changes during early postnatal development (Trimmer, 1993), suggesting that Kv2.1 phosphorylation state may be developmentally regulated in the brain. Different patterns of constitutive and regulated phosphorylation of Kv2.1 in different excitable cell backgrounds, or in different temporal, physiological, or pathological contexts may lead to diversity of Kv2.1 function that may profoundly affect membrane excitability but confound attempts to identify endogenous Kv2.1 currents by direct comparison with recombinant Kv2.1 expressed in heterologous expression systems.
Footnotes
This work was supported by National Institutes of Health Grant NS42225 to J.S.T. D.P.M. acknowledges postdoctoral fellowship support from the Epilepsy Foundation through the generous support of the American Epilepsy Society and the Milken Family Foundation. We thank Drs. R. W. Aldrich, J. Sack, K.-S. Park, H. Vacher, and M. Menegola for constructive suggestions, Dr. H. Misonou for assistance with confocal microscopy, and K. Brinckmann for technical assistance.
Correspondence should be addressed to Dr. James S. Trimmer, Department of Pharmacology, School of Medicine, 3502 GBSF, University of California, One Shields Avenue, Davis, CA 95616. E-mail: jtrimmer@ucdavis.edu.
Copyright © 2006 Society for Neuroscience 0270-6474/06/260685-11$15.00/0
References
- Antonucci DE, Lim ST, Vassanelli S, Trimmer JS (2001) Dynamic localization and clustering of dendritic Kv2.1 voltage-dependent potassium channels in developing hippocampal neurons. Neuroscience 108: 69-81. [DOI] [PubMed] [Google Scholar]
- Brown DA, Adams PR (1980) Muscarinic suppression of a novel voltagesensitive K+ current in a vertebrate neurone. Nature 283: 673-676. [DOI] [PubMed] [Google Scholar]
- Cameron AM, Steiner JP, Roskams AJ, Ali SM, Ronnett GV, Snyder SH (1995) Calcineurin associated with the inositol 1,4,5-triphosphate receptor-FKBP12 complex modulates Ca2+ flux. Cell 83: 463-472. [DOI] [PubMed] [Google Scholar]
- Coetzee WA, Amarillo Y, Chiu J, Chow A, Lau D, McCormack T, Moreno H, Nadal MS, Ozaita A, Pountney D, Saganich M, Vega-Saenz de Miera E, Rudy B (1999) Molecular diversity of K+ channels. Ann NY Acad Sci 868: 233-285. [DOI] [PubMed] [Google Scholar]
- Dascal N (1997) Signalling via the G protein-activated K+ channels. Cell Signal 9: 551-573. [DOI] [PubMed] [Google Scholar]
- Du J, Tao-Cheng JH, Zerfas P, McBain CJ (1998) The K+ channel, Kv2.1, is apposed to astrocytic processes and is associated with inhibitory postsynaptic membranes in hippocampal and cortical principal neurons and inhibitory interneurons. Neuroscience 84: 37-48. [DOI] [PubMed] [Google Scholar]
- Du J, Haak LL, Phillips-Tansey E, Russell JT, McBain CJ (2000) Frequency-dependent regulation of rat hippocampal somato-dendritic excitability by the K+ channel subunit Kv2.1. J Physiol (Lond) 522: 19-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelband CH, Warth JD, Mason HS, Zhu M, Moore JM, Kenyon JL, Horowitz B, Sumners C (1999) Angiotensin II type 1 receptor-mediated inhibition of K+ channel subunit kv2.2 in brain stem and hypothalamic neurons. Circ Res 84: 352-359. [DOI] [PubMed] [Google Scholar]
- Gluzman Y (1981) SV40-transformed simian cells support the replication of early SV40 mutants. Cell 23: 175-182. [DOI] [PubMed] [Google Scholar]
- Graham FL, Smiley J, Russell WC, Nairn R (1977) Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J Gen Virol 36: 59-74. [DOI] [PubMed] [Google Scholar]
- Hochner B, Kandel ER (1992) Modulation of a transient K+ current in the pleural sensory neurons of Aplysia by serotonin and cAMP: implications for spike broadening. Proc Natl Acad Sci USA 89: 11476-11480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang XY, Morielli AD, Peralta EG (1993) Tyrosine kinase-dependent suppression of a potassium channel by the G protein-coupled m1 muscarinic acetylcholine receptor. Cell 75: 1145-1156. [DOI] [PubMed] [Google Scholar]
- Hwang PM, Fotuhi M, Bredt DS, Cunningham AM, Snyder SH (1993) Contrasting immunohistochemical localizations in rat brain of two novel K+ channels of the Shab subfamily. J Neurosci 13: 1569-1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irving AJ, Collingridge GL (1998) A characterization of muscarinic receptor-mediated intracellular Ca2+ mobilization in cultured rat hippocampal neurones. J Physiol (Lond) 511: 747-759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas EA, Kaczmarek LK (1996) Regulation of potassium channels by protein kinases. Curr Opin Neurobiol 6: 318-323. [DOI] [PubMed] [Google Scholar]
- Klein M, Camardo J, Kandel ER (1982) Serotonin modulates a specific potassium current in the sensory neurons that show presynaptic facilitation in Aplysia Proc Natl Acad Sci USA 79: 5713-5717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemic KG, Shieh CC, Kirsch GE, Jones SW (1998) Inactivation of Kv2.1 potassium channels. Biophys J 74: 1779-1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurata HT, Soon GS, Eldstrom JR, Lu GW, Steele DF, Fedida D (2002) Amino-terminal determinants of U-type inactivation of voltage-gated K+ channels. J Biol Chem 277: 29045-29053. [DOI] [PubMed] [Google Scholar]
- Lee KB, Pals-Rylaarsdam R, Benovic JL, Hosey MM (1998) Arrestin-independent internalization of the m1, m3, and m4 subtypes of muscarinic cholinergic receptors. J Biol Chem 273: 12967-12972. [DOI] [PubMed] [Google Scholar]
- Levey AI, Kitt CA, Simonds WF, Price DL, Brann MR (1991) Identification and localization of muscarinic acetylcholine receptor proteins in brain with subtype-specific antibodies. J Neurosci 11: 3218-3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levey AI, Edmunds SM, Koliatsos V, Wiley RG, Heilman CJ (1995) Expression of m1-m4 muscarinic acetylcholine receptor proteins in rat hippocampus and regulation by cholinergic innervation. J Neurosci 15: 4077-4092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitan IB (1994) Modulation of ion channels by protein phosphorylation and dephosphorylation. Annu Rev Physiol 56: 193-212. [DOI] [PubMed] [Google Scholar]
- Levitan IB (1999) Modulation of ion channels by protein phosphorylation. How the brain works. Adv Second Messenger Phosphoprotein Res 33: 3-22. [DOI] [PubMed] [Google Scholar]
- Lim ST, Antonucci DE, Scannevin RH, Trimmer JS (2000) A novel targeting signal for proximal clustering of the Kv2.1 K+ channel in hippocampal neurons. Neuron 25: 385-397. [DOI] [PubMed] [Google Scholar]
- Lucas-Meunier E, Fossier P, Baux G, Amar M (2003) Cholinergic modulation of the cortical neuronal network. Pflügers Arch 446: 17-29. [DOI] [PubMed] [Google Scholar]
- Ma H-T, Patterson RL, van Rossum DB, Birnbaumer L, Mikoshiba K, Gill DL (2000) Requirement of the inositol trisphosphate receptor for activation of store-operated Ca2+ channels. Science 287: 1647-1651. [DOI] [PubMed] [Google Scholar]
- Malin SA, Nerbonne JM (2002) Delayed rectifier K+ currents, IK, are encoded by Kv2 α-subunits and regulate tonic firing in mammalian sympathetic neurons. J Neurosci 22: 10094-10105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mark MD, Herlitze S (2000) G-protein mediated gating of inward-rectifier K+ channels. Eur J Biochem 267: 5830-5836. [DOI] [PubMed] [Google Scholar]
- Marrion NV, Tavalin SJ (1998) Selective activation of Ca2+-activated K+ channels by co-localized Ca2+ channels in hippocampal neurons. Nature 395: 900-905. [DOI] [PubMed] [Google Scholar]
- Misonou H, Trimmer JS (2005) A primary culture system for biochemical analyses of neuronal proteins. J Neurosci Methods 144: 165-173. [DOI] [PubMed] [Google Scholar]
- Misonou H, Mohapatra DP, Park EW, Leung V, Zhen D, Misonou K, Anderson AE, Trimmer JS (2004) Regulation of ion channel localization and phosphorylation by neuronal activity. Nat Neurosci 7: 711-718. [DOI] [PubMed] [Google Scholar]
- Misonou H, Mohapatra DP, Trimmer JS (2005a) Kv2.1: a voltage-gated K+ channel critical to dynamic control of neuronal excitability. Neurotoxicology 26: 743-752. [DOI] [PubMed] [Google Scholar]
- Misonou H, Mohapatra DP, Menegola M, Trimmer JS (2005b) Calcium- and metabolic state-dependent modulation of the voltage-dependent Kv2.1 channel regulates neuronal excitability in response to ischemia. J Neurosci 25: 11184-11193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller W, Connor JA (1991) Cholinergic input uncouples Ca2+ changes from K+ conductance activation and amplifies intradendritic Ca2+ changes in hippocampal neurons. Neuron 6: 901-905. [DOI] [PubMed] [Google Scholar]
- Murakoshi H, Trimmer JS (1999) Identification of the Kv2.1 K+ channel as a major component of the delayed rectifier K+ current in rat hippocampal neurons. J Neurosci 19: 1728-1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakoshi H, Shi G, Scannevin RH, Trimmer JS (1997) Phosphorylation of the Kv2.1 K+ channel alters voltage-dependent activation. Mol Pharmacol 52: 821-828. [DOI] [PubMed] [Google Scholar]
- Pongs O (1999) Voltage-gated potassium channels: from hyperexcitability to excitement. FEBS Lett 452: 31-35. [DOI] [PubMed] [Google Scholar]
- Sadja R, Alagem N, Reuveny E (2003) Gating of GIRK channels: details of an intricate, membrane-delimited signaling complex. Neuron 39: 9-12. [DOI] [PubMed] [Google Scholar]
- Scannevin RH, Murakoshi H, Rhodes KJ, Trimmer JS (1996) Identification of a cytoplasmic domain important in the polarized expression and clustering of the Kv2.1 K+ channel. J Cell Biol 135: 1619-1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw G, Morse S, Ararat M, Graham FL (2002) Preferential transformation of human neuronal cells by human adenoviruses and the origin of HEK 293 cells. FASEB J 16: 869-871. [DOI] [PubMed] [Google Scholar]
- Shi G, Kleinklaus AK, Marrion NV, Trimmer JS (1994) Properties of Kv2.1 K+ channels expressed in transfected mammalian cells. J Biol Chem 269: 23204-23211. [PubMed] [Google Scholar]
- Surmeier DJ, Foehring R (2004) A mechanism for homeostatic plasticity. Nat Neurosci 7: 691-692. [DOI] [PubMed] [Google Scholar]
- Tong J, Du GG, Chen SR, MacLennan DH (1999) HEK-293 cells possess a carbachol- and thapsigargin-sensitive intracellular Ca2+ store that is responsive to stop-flow medium changes and insensitive to caffeine and ryanodine. Biochem J 343: 39-44. [PMC free article] [PubMed] [Google Scholar]
- Trimmer JS (1991) Immunological identification and characterization of a delayed rectifier K+ channel polypeptide in rat brain. Proc Natl Acad Sci USA 88: 10764-10768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trimmer JS (1993) Expression of Kv2.1 delayed rectifier K+ channel isoforms in the developing rat brain. FEBS Lett 324: 205-210. [DOI] [PubMed] [Google Scholar]
- Trimmer JS, Rhodes KJ (2004) Localization of voltage-gated ion channels in mammalian brain. Annu Rev Physiol 66: 477-519. [DOI] [PubMed] [Google Scholar]
- VanDongen AM, Frech GC, Drewe JA, Joho RH, Brown AM (1990) Alteration and restoration of K+ channel function by deletions at the N- and C-termini. Neuron 5: 433-443. [DOI] [PubMed] [Google Scholar]
- Wang HS, Pan Z, Shi W, Brown BS, Wymore RS, Cohen IS, Dixon JE, McKinnon D (1998) KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel. Science 282: 1890-1893. [DOI] [PubMed] [Google Scholar]
- Yuste R, Lanni F, Konnerth A (2000) Imaging neurons: a laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory.