

Abstract
Spinocerebellar ataxia type 8 (SCA8) patients typically have a slowly progressive, adult-onset ataxia. SCA8 is dominantly inherited and is caused by large CTG repeat expansions in the untranslated antisense RNA of the Kelch-like 1 gene (KLHL1), but the molecular mechanism through which this expansion leads to disease is still unknown. To more fully characterize the underlying molecular mechanisms involved in SCA8, we developed a mouse model in which Klhl1 is deleted in either all tissues or is deleted specifically in Purkinje cells only. We found that mice that are either homozygous or heterozygous for the Klhl1 deletion have significant gait abnormalities at an early age and develop a significant loss of motor coordination by 24 weeks of age. This loss progresses more rapidly in homozygous knock-outs. Mice with Klhl1 specifically deleted in only Purkinje cells had a loss of motor coordination that was almost identical to the total-tissue deletion mice. Finally, we found significant Purkinje cell dendritic deficits, as measured by the thickness of the molecular layer, in all mice in which Klhl1 was deleted (both total and Purkinje cell-specific deletions) and an intermediate reduction in molecular layer thickness in mice with reduced levels of Klhl1 expression (heterozygous deletions). The results from this mouse model show that even a partial loss of Klhl1 function leads to degeneration of Purkinje cell function and indicates that loss of KLHL1 activity is likely to play a significant part in the underlying pathophysiology of SCA8.
Keywords: ataxia, halpoinsufficiency, knock-out, neurodegeneration, Purkinje cell, SCA8
Introduction
Spinocerebellar ataxia type 8 (SCA8) is a dominantly inherited, adult-onset ataxia (Koob et al., 1999; Koob, 2003). SCA8 is caused by large CTG repeat expansions in the untranslated antisense RNA of the Kelch-like 1 gene (KLHL1) (Nemes et al., 2000), but the mechanism through which this expansion mutation causes disease is still unknown. The evolutionarily conserved KLHL1-antisense (KLHL1AS) RNA containing the expansion is transcribed from a promoter region in the first intron of KLHL1 and extends through the transcription and translation start sites as well as the first splice donor sequence of KLHL1 (Benzow and Koob, 2002). Although the SCA8 mutation is not within the coding region of the KLHL1 gene, analyses of the completed human genome sequence indicate that KLHL1 is the only protein-encoding gene within a megabase region containing the SCA8 mutation. These findings make KLHL1 a strong and obvious candidate for playing a central role the pathophysiology of the SCA8 form of ataxia.
The KLHL1 protein was named for its homology to the Drosophila protein kelch, which is an actin cross-linking component of the “ring canals” that are required for cytoplasm transport from nurse cells to the oocyte during oogenesis (Robinson and Cooley, 1997). Sequence analysis of the human genome has identified a superfamily of 51 proteins that contain the same general domain architecture as KLHL1 and kelch (Prag and Adams, 2003). Thirty-eight of these proteins have an almost identical domain structure consisting of a BTB/POZ multimerization domain and six kelch repeat units, which in KLHL1 apparently comprises an actin-binding domain but which in some proteins can have altered binding specificity. Other members of this family include gigaxonin, which is mutated in Giant Axonal Neuropathy (Bomont et al., 2000), actinfilin, which is an actin-binding protein present in dendritic spines (Chen and Li, 2005), and Mayven (KLHL2), which appears to be involved in process formation in oligodendrocytes (Jiang et al., 2005). The functions of most of the proteins in this superfamily, including KLHL1, are currently unknown.
We have shown previously that KLHL1 is primarily expressed in the brain, and here we report that the KLHL1 protein is located exclusively in the cell bodies and dendrites of Purkinje cells and other neurons. To understand the normal function of KLHL1 in these neurons and determine the role that it may play in SCA8 pathophysiology, we developed a mouse model in which the Klhl1 gene is deleted. We find that loss of expression from even a single allele causes abnormal gait, progressive loss of motor coordination, and Purkinje cell dendritic deficits. Mice with Klhl1 specifically deleted in only Purkinje cells have the same phenotype, indicating that Klhl1 is essential for normal motor coordination and for maintaining normal Purkinje cell functions.
Materials and Methods
Preparation of KLHL1-specific antibody 13D9 and immunohistochemistry
A 5′ fragment of human KLHL1 cDNA (Nemes et al., 2000), which expresses the N terminal to L308, was prepared by reverse transcription (RT)-PCR and KpnI and HindIII sites of the pET-37b (+) expression vector (Novagen, Madison, WI). The expressed KLHL1 fragment, including the N-terminal region and BTB/POZ domain (Bardwell and Treisman, 1994; Albagli et al., 1995) was purified and refolded according to the pET system manual. Female BALB/c mice were immunized by intraperitoneal injection of 100 μg of antigen in 50% complete Freund's adjuvant (Sigma, St. Louis, MO). Spleen cells were harvested and fused with mouse myeloma cells X63-Ag8.653 (American Type Culture Collection, Manassas, VA) as reported previously (Nelson et al., 2000). We performed ELISA to identify the KLHL1-specific monoclonal antibodies using Nunc-Immuno plates (Nunc, Naperville, IL). Positive wells were identified by the presence of colored reaction product and recloned individually at least three times by limiting dilution. The specificity for KLHL1 of the generated monoclonal antibodies (mAbs) was tested, and 13D9 was selected for the study.
FVB/NJ Mice (The Jackson Laboratory, Bar Harbor, ME) was used for Klhl1 distribution analysis by immunohistochemistry. Mice were perfused with 4% paraformaldehyde and then postfixed in 4% paraformaldehyde overnight at 4°C. The brains were processed by a Tissue-Tek VIP machine (Miles, Elkhart, IN), followed by embedding in paraffin. Sections were cut on a rotary microtome (American Optical, Leica, Deerfield, IL) at 6.0 μm in sagittal orientation and stained with monoclonal anti-calbindin-D-28K (Sigma) or monoclonal anti-KLHL1 13D9 antibodies. Avidin–biotin–peroxidase staining was performed using Vectastain Elite ABC kit (Vector Laboratories, Burlingame, CA). The products were visualized by staining with freshly prepared 3′,3′-diaminobenzidine tetrahydrochloride (Vector Laboratories), and finally the sections were counterstained with hematoxylin, dehydrated in graded alcohol, and mounted with Permount (Fisher Scientific, Houston, TX). Digital pictures were taken with a Leica Microsystems (Nussloch, Germany) light microscope with a resolution of 1300 × 1030 pixels.
Generation of Klhl1 knock-out mice and genotyping
Klhlflox/+ mice. An ∼10.2 kb fragment (4.1 and 2.6 kb for flanking arms and a 3.5 kb intervening fragment containing the promoter and exon 1 of Klhl1) was isolated by PCR from 129/SvJ mice (The Jackson Laboratory) genomic DNA and cloned into a derivative of pK11. The 5′ loxP site was inserted into the upstream of the polyadenylation signal of Klhl1as and the transcription start site of Klhl1. A neomycin expression cassette flanked with two flippase recombinase target (FRT) sites and the 3′ loxP site were inserted into the first intron of Klhl1, downstream of the transcription start site of Klhl1as (see Fig. 2A). The sequence of the targeting construct (Klhl1–2loxP) is available as supplemental data (available at www.jneurosci.org as supplemental material). The targeting vector was linearized with NheI and electroporated into CJ7 embryonic stem (ES) cells. Genomic DNA from 384 neomycin-resistant ES clones was analyzed by nested PCR using two primer pair sets: Neo1 (5′-AAGAACGAGATCAGCAGC CT-3′)/Gem1 (5′-CTGTCAGACCAAAAACCCTGT-3′); and Neo2 (5′-GCCTCTGTTCCACATACACT-3′)/Gem2–2 (5′-CTAACCCCTGGTTTTCACCAAACT-3′). We obtained seven correctly targeted ES clones, and clone 1H11 was selected for karyotype analysis and used for blastocyst injection. The resulting chimeric males were bred with C57BL/6NCr (National Cancer Institute, Bethesda, MD) females to identify germ-line transmission by coat color. Both the positive ES clones and agouti mice were confirmed by Southern blot. Therefore, Klhl1flox/+ mice were generated by mating the mice to FLP transgenic mice (ACTFLPe; The Jackson Laboratory) to delete the FRT flanking neo cassette.
Figure 2.
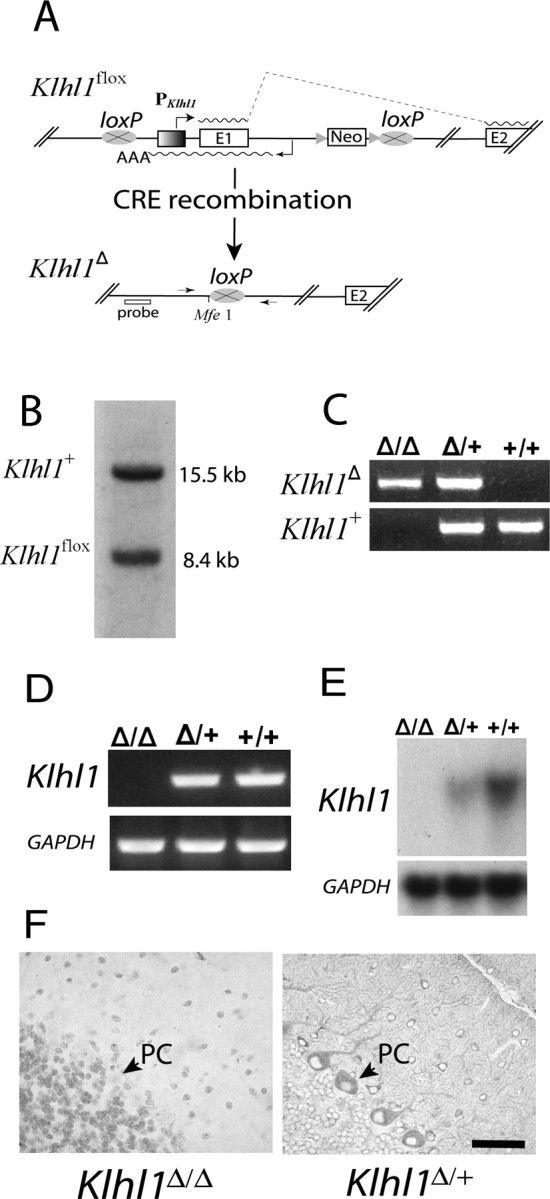
Expression of Klhl1 is eliminated in Klhl1Δ/Δ mice and is significantly reduced in Klhl1Δ/+ mice. A, Overview of the deletion strategy and of the modified loci Klhl1flox (floxed) and Klhl1Δ (deleted). Klhl1flox mice were generated by flanking the Klhl1 transcription start site and the first Klhl1 coding exon and Klhl1as gene with two loxP sites. The neomycin-resistant selection cassette (Neo) used in targeting was inserted into intron 1 of Klhl1 and flanked with two FRT sites, which were used to delete the cassette by mating the mice to FLP transgenic animals. The Klhl1flox allele was then modified by mating to cre transgenic mice to generate mouse lines carrying a Klhl1 null allele (Klhl1Δ) in either all tissues (ubiquitous CRE expression) or specific tissues only (tissue-specific CRE expression). E, Exon; PKlhl1, Klhl1 promoter; triangle, FRT site; oval with a cross, loxP recombination sites; waved line, mRNA, AAA, polyadenylation signal of Klhl1as. The arrows in the bottom indicate the two primers that cross the 5′ and 3′ loxP sites (MEX1–8R and MEX2–3, indicated by P1 and P2), and the approximate location of the probe by Southern blot analysis (below) is also shown. B, Southern blot analysis of a heterozygous mouse showing both the wild-type (Klhl1+) and the Klhl1flox allele. The Southern blot was performed on MfeI-digested tail DNA using a flanking probe generated the expected 8.4 and 15.5 kb fragments. C, The genotyping of Klhl1Δ/Δ, Klhl1Δ/+, and Klhl1+/+ mice was performed by PCR using mouse tail DNA. Primers MEX1–8R and MEX2–3 generate a 642 bp fragment, whereas primer set MEXKO5C and MEX1–8R generate a 557 bp product from the wild-type Klhl1+ allele. D, RT-PCR analysis. RNA isolated from cerebella of 3-week-old mice was reverse transcribed and assayed by PCR using primer pair Mus2–22 and Mus3–24R. The mRNA of Klhl1 is clearly absent in the homozygous Klhl1Δ/Δ mice (lane 1), whereas in the heterozygous Klhl1Δ/+ (lane 2) and wild-type Klhl1+/+ (lane 3) cerebella, the primers are able to detect a 682 bp amplification product representing the Klhl1 cDNA. E, Northern blot analysis of Klhl1 mRNA in adult knock-out mice and their wild-type controls. Messenger RNA of Klhl1 is clearly absent in Klhl1Δ/Δ mice, and the expression is dramatically decreased in the Klhl1Δ/+ mice compared with that of the wild-type Klhl1+/+ controls. A subsequent probe of GAPDH mRNA was used as a loading control. F, Paraffin cerebella sections from 3-week-old Klhl1Δ/Δ (left) and Klhl1Δ/+ (right) mice stained for Klhl1 using 13D9 mAb. Sections show loss of immunoreactivity in the Purkinje cells of homozygous Klhl1Δ/Δ mice (left), but Klhl1-positive staining is clearly visible in the cell soma and dendrites of Purkinje cells from the heterozygous Klhl1Δ/+ mice (right). Note that there is no hematoxylin counterstaining in the right. PC, Purkinje cell. Scale bars, 50 μm.
Klhl1Δ/Δ mice. Klhl1flox/+ mice were mated with ACTB-Cre mice (The Jackson Laboratory) to generate the deleted Klhl1Δ allele. Klhl1Δ/+ heterozygotes were inbred to generate Klhl1Δ/Δ, Klhl1Δ/+, and Klhl1+/+ mice. PCR assay was used to genotype those mice: a 557 bp band from primers MEX1–8R (5′-GAATGCTGATACCCAAACTACTC-3′) and KO5C (5′-CCACATATGATCCCTAAGTCTAA-3′) is generated from a wild-type Klhl1+ allele, and a 642 bp band from primers MEX1–8R and MEX2–3 (5′-GTAGCTCAGCCATCGGGTAAGA-3′) is generated from the Klhl1Δ allele. We confirmed the deletion by direct sequencing and Southern blot analyses. For Southern blot analysis, mouse tail DNA was digested with MfeI and transferred to Hybond-N+ membrane (Amersham Biosciences, Arlington Heights, IL), and a Klhl1 probe (323 bp), which is a PCR product from primer pair MEX1–3 (5′-GTGAGAGAAAATCCAAACACAAGGG-3′) and KO2 (5′-AGCTGCGAGTTCACTAC-3′) was used to detect the mutated (Klhl1Δ) and the wild-type (Klhl1+) alleles.
P-CRE-3 transgenic mice.
The transgene construct consisted of a 835 bp SalI–PvuI fragment of the Pcp2 (Purkinje cell protein 2) 5′ upstream regulatory region, 1.3 kb of Cre gene, and 240 bp of simian virus 40 polyadenylation signal. The intact transgene was separated from vector sequence and gel purified before microinjection, and three transgenic founders were identified after the embryo injection. Primers for transgenic mice genotyping are PC5–1023 (5′-CTATGGTGAGAGCAGAGATG-3′) and CRE3–78 (5′-GGTTCTTGCGAACCTCATCA-3′), and a 270 bp PCR product is generated from the Pcp2-Cre allele. The mouse line from founder P-CRE-3 were selected and crossed to R26R mice (Soriano, 1999) to determine the distribution and efficiency of the Cre/loxP recombination. Brains from Pcp2-Cre/R26R compound heterozygotes were examined for β-galactosidase (β-gal) activity as follows: the brains were perfused with PBS, pH 7.3, and then 4% paraformaldehyde in PBS. Brains were postfixed in the same fixative for an additional 30 min at room temperature, and brain sections (200 μm) were stained at 37°C for 2 h in 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-gal) solution [1 mg/ml X-gal, 4 mm K4Fe(CN)6, 4 mm K3Fe(CN)6, and 2 mm MgCl2 in PBS]. For whole-mount staining, brains were not postfixed and were stained for 2 d at 4°C in X-gal solution.
P-CRE-3/Klhl1flox/flox mice.
Klhl1flox mice were bred with P-CRE-3 transgenic heterozygotes to generate heterozygous P-CRE-3/Klhl1flox/flox compound mice, which were used for testing the Purkinje cell specificity.
Klhl1 expression analysis
Total RNA was isolated from the mice cerebellum using Trizol (Invitrogen, Carlsbad, CA), and Northern blot and RT-PCR analyses for Klhl1 expression were performed as described previously (Nemes et al., 2000). A 460 bp cDNA probe for the Northern blot was generated by PCR using primers musK1F5 (5′-CTCTATCCAGGCAACAGGTGAAG-3′) and musK1V (5′-GTTTCATGAGGAAGTGACAACAC-3′) and was used to detect the 3.5 kb of full-length Klhl1 mRNA. Primers for RT-PCR reactions are Mus2–22 (5′-AGTTGCCACGAGGCTGCAGCAAG-3′) and Mus3–24R (5′-TCCTTGAGCATCAGCAAATGCCCT-3′), which are located in exon 1 and exon 4 of Klhl1, respectively. The ubiquitously expressed glyceraldehyde-3-phosphate dehydrogenase (GAPDH) transcript was used as an expression control for both the Northern blots and RT-PCR reactions.
Rotarod test and treadmill gait analyses
ROTAMEX 418 (Columbus Instruments, Columbus, OH) was used to test mice motor ability as described previously (Clark et al., 1997; Zu et al., 2004). Mice were tested using four trials per day on 4 consecutive days. The rod accelerated from 4 to 40 rpm over 5 min and then remained at 40 rpm for an additional 5 min for each trial. During the trial, the time was recorded when the mice fell from the rod up to a maximum of 10 min. The mice rested 10 min between the trials. At the age of 12 weeks old, 9 homozygous and 11 heterozygous Klhl1 knock-out mice and 11 of their wild-type siblings were used for the rotarod test. For the groups of 18- and 24-week-old mice, 11, 12, 11 and 11, 16, 12 mice were used for each genotype, respectively. For 24-week-old conditional knock-out mice, 12 P-CRE-3/Klhl1flox/flox mice and 15 controls were used. The gait analyses of ACTB-Cre-mediated Klhl1 knock-out mice was performed using TreadScan (Clever Sys, Reston, VA) following the provided protocol and the recommendations of Wooley et al. (2005). A group of six mice (two mice for each genotype) were pretested for their performance on the treadmill at the speed of 10, 15, 20, and 25 m/s, and the speed of 20 m/s was selected after the test. Two groups of mice were pretrained two times a week for 2 consecutive weeks when they were 4 and 10 weeks old, respectively, with the speed of 20 m/s for 20 s each time. At the age of 6 or 12 weeks, the two group of mice were recorded a fixed period (20 s) at the same speed, and 2000 frames digital camera frames were captured. For the 12-week-old group, 10 homozygous (female/male, respectively, n = 5/5), 7 heterozygous (n = 2/5), and 7 wild-type siblings (n = 3/4) were tested. For the 6-week-old mice group, 5 homozygote (2/3), 12 heterozygote (6/6), and 9 wild-type controls (5/4) were used for the gait analysis. We analyzed at least five consecutive strides (minimum, 5; maximum, 20) by TreadScan 2.0 (Clever Sys). The right and left paws were averaged in each animal, using the average representative values of the four paws for stance and stride parameters. The body weight and size of the knock-out mice was not found to differ significantly from that of the wild-type sibling controls at any of the ages at which rotarod or TreadScan was performed. Weight data from a representative cohort is provided in supplemental Table 1 (available at www.jneurosci.org as supplemental material).
Molecular layer measurement
Molecular layer measurement of mouse cerebellum was performed as reported previously (Klement et al., 1998; Zu et al., 2004). Brains were perfused and fixed as above and then washed in PBS overnight at 4°C. Fifty micrometer midsagittal sections were cut by Vibratome (Vibratome, St. Louis, MO) and were stained with anti-calbindin antibody for 48 h at 4°C, followed by an anti-mouse-conjugated cyanine 3 secondary antibody (Jackson ImmunoResearch, West Grove, PA). After washing with PBS, the sections were mounted and pictures were taken of the cerebellum using an MRC 1024 confocal microscope (Bio-Rad, Hercules, CA) with a resolution of 512 × 768 pixels. Nine 6-week-old (homozygote/heterozygote/wild, respectively, n = 4/3/2), 8 12-week-old (3/3/2), and 13 24-week-old (6/4/3) mice were used for this study. For 24-week-old Purkinje cell knock-out mice, two mice were used for the measurement. All of the mice were measured three times individually. Thickness of the molecular layer was determined by measuring the distance between the Purkinje cell body layers and dividing in half as reported previously (Zu et al., 2004).
Statistical analysis
To analyze the data from each animal group at a specific age, one-way ANOVA was used in TreadScan studies, and body weight analyses and molecular layer measurements and two-way ANOVA was performed in rotarod tests, with genotype and day as factors. Tukey's honestly significant difference test was performed as post hoc analysis. p ≤ 0.05 was used as a limit for declaring statistical significance.
Results
Klhl1 protein is located in the stomata and dendrites of Purkinje cells
The cellular and subcellular localization of the Klhl1 protein in mouse adult cerebellum were studied by immunohistochemistry with KLHL1-specific monoclonal antibody mAb-13D9 (Fig. 1). Normal hematoxylin and eosin staining of the perfused and embedded tissues was performed before immunohistochemistry staining. The most prominent Klhl1 labeling in mouse cerebellar tissue was in the cell soma and dendrites of the Purkinje cells (Fig. 1A), but there is a very noticeable absence of staining of the nuclei and axons of these cells (Fig. 1B). For comparison, we stained identical cerebellar slices with mouse anti-calbindin monoclonal antibody (Fig. 1C), which labeled all parts of the Purkinje cells, including the nuclei, dendrites, and axons.
Figure 1.
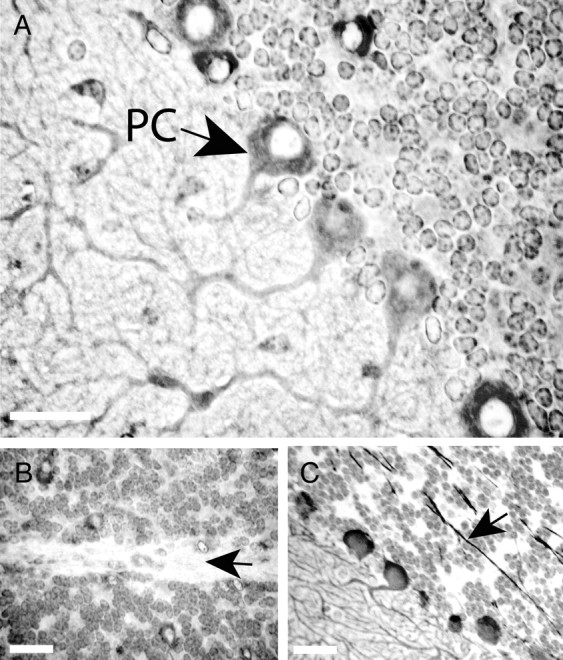
Cellular and subcellular localization of Klhl1 in mouse cerebellar tissue. Immunohistochemical staining of normal FVB mice at 4 weeks of age with KLHL1-specific monoclonal antibody 13D9 shows that Klhl1 is a prominent cytoplasmic protein in the soma and dendrites of cerebellar Purkinje cells but is not present in the nuclei (A) or the axons (B) of these cells. Both the axons and the nuclei of Purkinje cells stain positively with anti-calbindin antibody (C). PC, Purkinje cell. Black arrows show the axons of the Purkinje cells. Scale bars, 25 μm.
Establishing a conditional Klhl1 knock-out mouse model
To begin to determine the function of the Klhl1 protein in the mouse brain, we developed a mouse model in which a critical region of the Klhl1 gene is deleted (Fig. 2). We made a targeting construct in which the promoter region and first exon of Klhl1 are flanked with loxP recombination sites (Fig. 2A) and recombined this construct at the Klhl1 locus using standard mouse embryonic stem cell techniques (Fig. 2B). Mice generated from these cells were fully viable and did not have obvious phenotypic abnormalities. We then generated mice in which the region of Klhl1 flanked by loxP sites (flox) is deleted by mating the Klhl1flox mice to mice in which the CRE recombinase is constitutively expressed in all tissues (Fig. 2A,C). The resulting Klhl1 deletion (Klhl1Δ) allele no longer contains the CpG-rich promoter region, the transcription initiation site, the first splice donor site, or the first exon portion of the coding frame present in the wild-type Klhl1+ allele. In addition, the entire genomic sequence transcribed by the Klhl1as RNA and the predicted promoter region for this antisense transcript (Benzow and Koob, 2002) are no longer present at the Klhl1Δ allele (Fig. 2A).
We compared expression levels of the Klhl1 transcript in cerebellar tissue taken from mice that were either homozygous (Klhl1Δ/Δ) or heterozygous (Klhl1+/Δ) for the Klhl1Δ allele and compared these levels with those found in wild-type (Klhl1+/+) siblings. We found that the Klhl1 transcript was undetectable in the Klhl1Δ/Δ mice by either RT-PCR (Fig. 2D) or Northern analyses (Fig. 2E). The levels of Klhl1 transcript detected in the Klhl1+/Δ heterozygous mice by Northern blot analyses was also significantly lower than that found in the control samples (Fig. 2E).
The Klhl1 protein was undetectable in cerebellar slices from the Klhl1Δ/Δ mice stained with a monoclonal antibody specific for this protein (Fig. 2F). The Klhl1 protein was, however, clearly detected in slices from the Klhl1+/Δ heterozygous mice (Fig. 2F, right), and the distribution of the protein was similar to that found in the slices from wild-type controls (Fig. 1).
Mice homozygous or heterozygous for the Klhl1 deletion have significant gait abnormalities
For phenotype analyses, we bred Klhl1+/Δ heterozygous mice to generate age-matched groups of homozygous Klhl1Δ/Δ, heterozygous Klhl1+/Δ, and wild-type Klhl1+/+ siblings. We used the TreadScan system from Clever Sys to assay the gait of these mice. In this system, the mouse walks on a clear treadmill, and a digital camera is used to capture video of paw placements from a mirror beneath the treadmill (Wooley et al., 2005). The footprint locations are automatically determined by the system software, and gait measurements such as stance time (i.e., the amount of time that the paw is in contact with the treadmill), swing time (i.e., the amount of time that the paw is in the air), and stride length and width are calculated. This apparatus is particularly well suited for analyzing early signs of motor system disease in mice (Wooley et al., 2005).
We detected significant gait abnormalities in Klhl1+/Δ and Klhl1Δ/Δ mice as young as 4 weeks old with the TreadScan system. Both the front and rear paw of the homozygotes and heterozygotes show longer stance time and stride time compared with wild-type siblings, although these differences are in general more pronounced for the rear paw. We detected similar gait abnormalities through 12 weeks of age, at which time the mice grew too large for us to obtain meaningful measurements using the relatively small retention chamber in the TreadScan apparatus.
Figure 3 shows graphs comparing the rear paw stance time and stride time for the three groups of siblings at 6 weeks (Fig. 3A) and 12 weeks (Fig. 3B) of age. At both of these ages, the mice had been trained on the apparatus twice weekly during the preceding 2 weeks. All of these gait measurements for the Klhl1Δ/Δ mice are statistically longer than those of their Klhl1+/+ siblings at the same age. All of the gait measurements for the heterozygous Klhl1+/Δ mice are also statistically longer from the wild-type sibling controls except the stance time at 6 weeks of age (p = 0.06). Because the stride time is calculated as a combined time of the stance time and the swing time, we believe that a longer stance time is the most important and significant gait difference between the Klhl1 knock-out and wild-type mice at these relatively young ages.
Figure 3.

Gait analysis with a TreadScan device and performance on an accelerating rotarod detects progressive deficiencies in heterozygous Klhl1Δ/+ and homozygous Klhl1Δ/Δ mice. Two groups of mice were trained for 2 weeks, and the gait analysis was performed at a speed of 20 cm/s for 20 s. At 6 weeks of age (A), the stride time for heterozygous Klhl1Δ/+ and homozygous Klhl1Δ/Δ mice was significantly longer than wild-type siblings in both front paws and rear paws. At the age of 12 weeks (B), the abnormalities of gait is also significant for both front paws and rear paws. For stance time, 6-week-old homozygous mice have a significantly longer stance time than wild-type animals, whereas the heterozygous mice have an obvious but not yet quite significantly longer stance than the controls. For the 12-week-old group, gait abnormalities are more obvious; the homozygous and heterozygous mice have increased significant impairments of their stance time compared with wild-type controls. The gait results for the hindpaw only are shown in the graphs in this figure. Twelve-week-old (C), 18-week-old (D), and 24-week-old (E) mice were tested for four trials per day for 4 consecutive days on an accelerating rotarod. At 12 weeks of age, the performances of the homozygous Klhl1Δ/Δ and heterozygous Klhl1Δ/+ mice were indistinguishable with their wild-type controls, but, at 18 weeks of age, homozygous Klhl1Δ/Δ mice showed significantly impaired performance improvement (p = 0.002) whereas the heterozygous Klhl1Δ/+ mice showed obvious but not significant impairment (p = 0.052). At 24 weeks of age, both homozygous and heterozygous mice showed significant impaired performance improvements at day 4 (p < 0.001; p < 0.05 respectively). Error bars indicate SEM.
Klhl1 knock-out mice have a progressive loss of motor coordination
We used an accelerating rotarod apparatus to measure the motor coordination of the Klhl1 knock-out mice (Fig. 3C,D). At 12 weeks of age, siblings that were homozygous or heterozygous for the deletion performed as well as their wild-type littermates. By 18 weeks, however, the Klhl1Δ/Δ mice showed significant motor coordination deficits, and, by 24 weeks, these deficits were even more pronounced. Mice that were heterozygous for the deletion performed noticeably and consistently worse on the accelerating rotarod than their wild-type siblings by 18 weeks of age, but this difference did not quite reach a statistically significant value (p = 0.066). By 24 weeks of age, however, these heterozygous mice also showed motor coordination deficits that were statistically different than the control group of mice.
Cerebellar Purkinje cell-restricted Cre expression in transgenic mice
Previous studies have shown that the 5′ regulatory sequences of the Pcp2 gene are capable of directing transgene expression specifically to Purkinje cells (Vandaele et al., 1991). To direct the expression of Cre recombinase in Purkinje cells, we generated transgenic mice in which Cre was expressed from the Pcp2 promoter (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material). We identified three pups as positive for the transgene after implantation of microinjected embryos, two of which bred successfully (P-CRE-1 and P-CRE-3). To determine the distribution and efficiency of the Cre/loxP recombination, each Cre transgenic mouse line was crossed to R26R mice, in which the reporter lacZ gene is expressed when Cre excises a strong translation termination sequence flanked by two loxP sites (Soriano, 1999). We examined brain sections from P-CRE/R26R compound heterozygotes at 16 weeks of age for β-gal activity and found that X-gal staining was highly restricted to the Purkinje cells. The frequency of stained cells in 16-week-old P-CRE-3/R26R mice was much higher than P-CRE-1/R26R. We next examined P-CRE-3/R26R transgenic mice at 4, 8, and 12 weeks to determine the time course of Purkinje cell specific Cre excision. Supplemental Figure 1B (available at www.jneurosci.org as supplemental material) shows that the frequency and density of stained Purkinje cells increased from 4 to 12 weeks. β-gal-positive cells appear less frequent at 4 weeks. Most Purkinje cells eventually become positive for β-gal activity. Whole-brain staining from 14-week-old P-CRE-3/R26R mice shows a cerebellum-specific Cre activity (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material).
Loss of Klhl1 only in Purkinje cells leads to progressive loss of motor coordination
By mating Klhl1flox mice with Pcp2-CRE mice, we were able to generate a mouse model in which the Klhl1 gene is specifically deleted only in Purkinje cells. Northern blot analyses of RNA isolated from cerebellar tissue from these mice confirmed that the overall expression level of Klhl1 is dramatically reduced in the Pcp2-CRE/Klhl1flox/flox mice, whereas the Klhl1flox/flox mice expressed Klhl1 at the normal wild-type level (Fig. 4A). Immunohistochemical staining of cerebellar slices from these mice showed that Klhl1 is not detected in Purkinje cells (Fig. 4B) but is still present in other neurons that typically express Klhl1. Rotarod analyses of the Purkinje cell-specific Klhl1 knock-out mice showed that, by 24 weeks of age, the Purkinje cell-specific knock-out mice have a loss of motor coordination (Fig. 4C) that is almost identical to that of the complete knock-out mice (Fig. 3E).
Figure 4.

Analyses of mice in which Klhl1 is deleted only in cerebellar Purkinje cells. A, Northern blot analysis demonstrated that the mRNA Klhl1 is dramatically decreased in cerebellar tissue from 24-week-old homozygous P-CRE-3/Klhl1flox/flox mice (lane 1) compared with the controls (Klhl1flox/flox; lane 2). The RNA loading levels of each lane were measured by a subsequent hybridization with GAPDH probe. B, Paraffin-embedded sections of brain from homozygous P-CRE-3/Klhl1flox/flox mice stained for Klhl1 using 13D9 mAb show cerebellar Purkinje cell-specific Klhl1 deficits. There was an absence of staining of Klhl1 in the soma and dendrites of Purkinje cells (left), but Klhl1 is normally expressed in the other neurons in this brain (neocortex; right). PC, Purkinje cell. Scale bar, 25 μm. C, Motor coordination performance deficits of 24-week-old P-CRE-3/Klhl1flox/flox mice on accelerating rotarods. Their performance was significantly worse (p = 0.011 at day 4) than the performance of controls. Error bars indicate SEM.
Loss of Klhl1 leads to Purkinje cell dendritic atrophy but not Purkinje cell loss
Brain tissues from Klhl1 knock-out mice were examined by a neuropathologist with extensive experience with mouse models of ataxia (H.B.C.). No abnormal loss of Purkinje cells or other neurons was found, even in mice older than 1 year of age, and the overall Purkinje cell morphology was preserved. However, the Purkinje cells did show signs of dendritic atrophy as assessed by a thinning of the molecular layer. We measured the thickness of the molecular layer in groups of homozygous Klhl1Δ/Δ, heterozygous Klhl1+/Δ, and wild-type Klhl1 +/+ siblings at 6, 12, and 24 weeks of age and of a group of Pcp2-CRE/Klhl1 flox/flox mice at 24 weeks of age (Fig. 5). We found no significant difference in thickness between these groups at 6 weeks of age, but, by 24 weeks, the molecular layer thickness of both the Klhl1+/Δ and Klhl1Δ/Δ as well as that of the Purkinje cell-specific knock-out mice was significantly thinner than in the wild-type mice controls. The molecular layer was generally thinner in the knock-out mice at 12 weeks of age as well, but these size differences were not yet statistically different than the controls at this age. The dendritic atrophy in the heterozygous mice also generally appeared to be somewhat less severe than in the homozygous knock-outs, but the measured difference in molecular layer thickness was not statistically different between these groups at this younger age.
Figure 5.
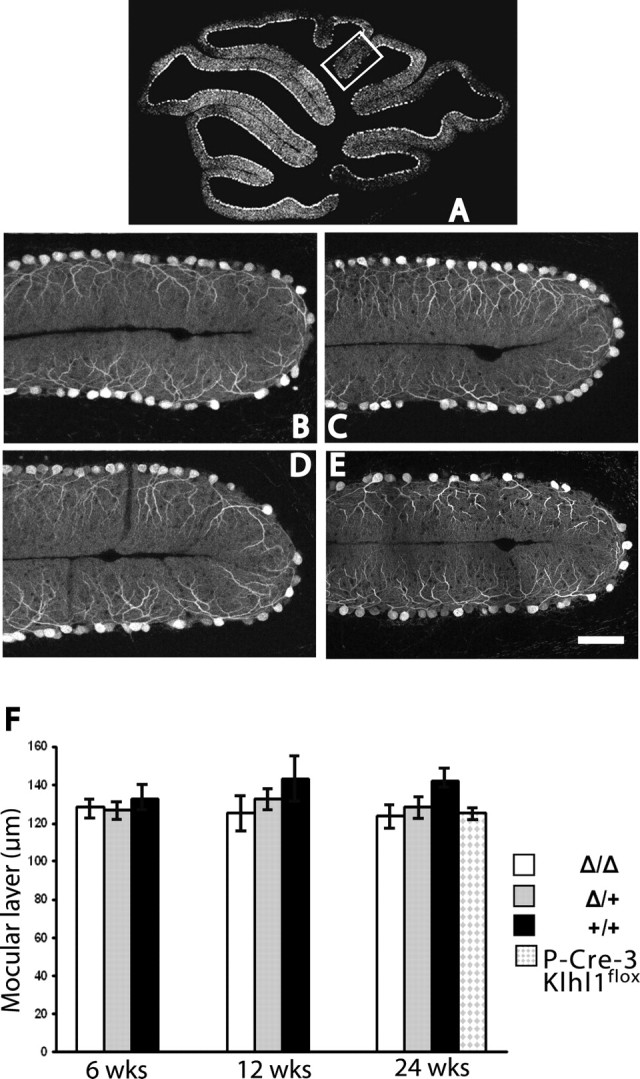
Klhl1 knock-out mice have significant thinning of the cerebellar molecular layer. A–E, Immunohistochemical analysis of cerebellar sections of Klhl1 knock-out mice. A, Diagram of a midsagittal cerebellar section indicating the posterior location of the folia presented in B–E. B–E, Calbindin immunoassays of 24-week-old mice were used measure the thickness of cerebellar molecular layers of Klhl1Δ/Δ (B), Klhl1Δ/+ (C), and P-CRE-3/Klhl1flox/flox (E) mice were significantly thinner than in the wild-type controls (D). Scale bar: B–E, 50 μm. F, Relative thickness of the cerebellar molecular layer in Klhl1 knock-out mice at 6, 12, and 24 weeks of age. Both homozygous and heterozygous show no significant difference when they are at the age of 6 or 12 weeks old compared with the wild-type controls. However, at the age of 24 weeks, the thickness of the molecular layer of homozygous Klhl1Δ/Δ and heterozygous Klhl1Δ/+mice were significantly decreased (mean ± SD, 123.5 ± 11.9, p = 0.015, n = 6; 128.4 ± 10.6, p = 0.048, n = 6; and 125.1 ± 1.8, p = 0.035, n = 2) compared with the wild-type Klhl1+/+controls (142.2 ± 7.3; n = 3). Error bars in F indicate SEM.
Discussion
We have shown that the KLHL1 protein encoded at the SCA8 ataxia locus is located exclusively in the cell bodies and dendrites of Purkinje cells and other neurons (Fig. 1). We knocked out the Klhl1 gene in mouse and find that loss of expression from even a single allele causes abnormal gait, progressive loss of motor coordination, and Purkinje cell dendritic deficits (Figs. 2, 3, 5). This degeneration occurs more quickly in mice in which both alleles have been deleted (Fig. 3). Mice with Klhl1 specifically deleted in only Purkinje cells have the same phenotype (Figs. 4, 5), indicating that the most critical function of Klhl1 is maintaining normal Purkinje cell functions and normal motor coordination. We hypothesize from these data that pathogenic SCA8 CTG expansions may cause ataxia in humans through a mechanism involving the loss of KLHL1 activity in Purkinje cells and that loss of proper expression from even a single KLHL1 allele would be sufficient to cause disease. Because the cerebellar dysfunction in our Klhl1 knock-out models is caused by Purkinje cell dysfunction rather than cell loss, we will be able to perform detailed molecular analyses on these mice to determine the precise molecular mechanisms that cause this dysfunction.
Diseases caused by expanded trinucleotide repeat mutations (Gatchel and Zoghbi, 2005) can involve either gain-of-function mechanisms, as is the case for the polyglutamine diseases and myotonic dystrophy, or loss-of-function mechanisms, as has been found in Friedreich ataxia and fragile X syndrome. In general, the dominantly inherited trinucleotide repeat diseases are caused by a gain-of-function mechanism, and the recessive or X-linked diseases are caused by loss-of-function mutations. None of the other dominantly inherited trinucleotide repeat diseases has as yet been found to be caused by loss-of-function mutations at a single allele, but examples of dominantly inherited human diseases caused by halpoinsufficiency are well documented (De Paepe, 1998; Santarosa and Ashworth, 2004; Zephir et al., 2005). Often the genes involved in this type of disease mechanism encode structural proteins that must form complexes with other proteins in proper stoichiometric ratios or compete for binding sites with other proteins.
The subcellular localization of Klhl1 indicates that this protein is involved in neuronal functions or structures associated with dendrites. The biochemical properties of the KLHL1 protein are consistent with those of known actin-organizing proteins, and the primary location of actin fibers in dendrites is in postsynaptic spines (Rao and Craig, 2000; Ethell and Pasquale, 2005; Lippman and Dunaevsky, 2005). We hypothesize from this that KLHL1 may be involved in cross-linking the actin cytoskeleton that determines the shape and dynamics of the dendritic spine head. Alternatively, KLHL1 could be involved in anchoring other associated proteins essential for postsynaptic functions, such as ion channels, to the actin network of the spine (Shirao and Sekino, 2001). Because disruptions of ion channel activity are known causes of Purkinje cell dysfunction and ataxia (Nahm et al., 2005; Walter et al., 2006), we find this latter possibility particularly intriguing.
The Klhl1 deletion used in the knock-out models we generated eliminate expression of both the Klhl1 protein product as well as the Klhl1as transcript. We cannot, therefore, formally exclude the possibility that some of the phenotype observed in these mice is attributable to the loss of Klhl1as RNA rather than the Klhl1 protein. The transcriptional organization of the Klhl1/Klhl1as locus in mice, however, appears to indicate the Klhl1as RNA is unlikely to be involved in activities other than regulating Klhl1 expression (Benzow and Koob, 2002). Whereas the human KLHL1AS RNA is a complex and multiply spliced RNA, the mouse antisense RNA is unspliced, is simply transcribed across the first Klhl1 exon, and terminates at a polyadenylation site in the Klhl1 promoter region (Fig. 1). The more complex human antisense RNA, however, may have additional activities not present in the mouse RNA. We found, for instance, that the KLHL1AS RNA specifically binds to staufen when overexpressed in Drosophila photoreceptor neurons (Mutsuddi et al., 2004), but the alternative 3′ terminal exon of this RNA containing the staufen binding site and the CTG repeat are not present in the mouse transcript. The staufen protein is known to mediate mRNA transport to dendritic spines (Miki et al., 2005), suggesting the possibility that KLHL1AS RNA may be transported to spines. We can speculate that interactions between the sense and antisense KLHL1 RNAs could in turn result in transport of the KLHL1 mRNA to the dendritic spines, as are numerous other proteins known to be essential for spine postsynaptic functions.
The data from our knock-out models demonstrate that Klhl1 is critical for maintaining normal motor coordination in mice and suggest that SCA8 could be caused by a partial loss of KLHL1 activity in Purkinje cells. These models were not, however, designed to directly test the pathogenic effects of the SCA8 expansion mutation itself, and we fully expect that the pathogenic cascade initiated by these mutations will prove to be significantly more complicated than those of simple deletion mutations. We found, for instance, that over-expression of the KLHLAS transcript in Drosophila photoreceptor neurons results in neurodegeneration, presumably as a consequence of titration of some of the RNA-binding proteins that interact with this RNA (Mutsuddi et al., 2004). Although this “toxic RNA” effect is seen with KLHLAS transcripts encoding unexpanded CTG repeats, the effect is stronger for transcripts containing expanded repeat tracts. Quite recently, Moseley et al. (2006) also reported that they can generate essentially pure polyglutamine peptides from expression constructs containing the SCA8 repeat expansion and hypothesized that polyglutamine toxicity may be part of the pathogenic mechanism of this disease. For obvious reasons, we have had very limited opportunities to directly study brain tissue samples from SCA8 patients and so, unfortunately, do not yet know if any, all, or none of these possible mechanisms play a significant role in the underlying pathophysiology of SCA8.
We can hypothesize a number of possible mechanisms through which KLHL1 activity could be reduced in Purkinje cells containing expanded SCA8 repeat tracts. For instance, an expanded CTG tract could have a direct negative impact on KLHL1 expression through interference with essential KLHL1 promoter elements. This direct mechanism, however, would not easily explain the incomplete penetrance of the SCA8 mutation or the observation that extremely large SCA8 repeat expansions do not appear to cause disease (Juvonen et al., 2000, 2005; Vincent et al., 2000; Sobrido et al., 2001; Corral et al., 2005). For this reason, we currently believe that mechanisms that require that the expansion be encoded in a transcript are more likely to be correct, because this provides a plausible explanation for the reduced penetrance associated with the SCA8 expansions (e.g., very large repeats may not be transcribed or spliced efficiently and would therefore not generate pathogenic RNA molecules). Possible RNA-mediated mechanisms include (1) loss of positive enhancement of KLHL1 expression normally provided by the antisense RNA, (2) RNA-initiated epigenetic silencing mediated by transcripts containing the expanded repeat (i.e., similar to the Fragile X repeat) (Jin et al., 2004), and (3) mislocalization of antisense and/or sense transcripts within the Purkinje cell by altering the protein binding characteristics of the KLHL1AS transcript, as suggested by our Drosophila KLHL1AS-overexpresion model (Mutsuddi et al., 2004).
Footnotes
This work was supported by the National Institute of Neurological Disorders and Stroke Grant NS36282.
References
- Albagli O, Dhordain P, Deweindt C, Lecocq G, Leprince D. The BTB/POZ domain: a new protein-protein interaction motif common to DNA- and actin-binding proteins. Cell Growth Differ. 1995;6:1193–1198. [PubMed] [Google Scholar]
- Bardwell VJ, Treisman R. The POZ domain: a conserved protein-protein interaction motif. Genes Dev. 1994;8:1664–1677. doi: 10.1101/gad.8.14.1664. [DOI] [PubMed] [Google Scholar]
- Benzow KA, Koob MD. The KLHL1-antisense transcript (KLHL1AS) is evolutionarily conserved. Mamm Genome. 2002;13:134–141. doi: 10.1007/s00335-001-2105-2. [DOI] [PubMed] [Google Scholar]
- Bomont P, Cavalier L, Blondeau F, Ben Hamida C, Belal S, Tazir M, Demir E, Topaloglu H, Korinthenberg R, Tuysuz B, Landrieu P, Hentati F, Koenig M. The gene encoding gigaxonin, a new member of the cytoskeletal BTB/kelch repeat family, is mutated in giant axonal neuropathy. Nat Genet. 2000;26:370–374. doi: 10.1038/81701. [DOI] [PubMed] [Google Scholar]
- Chen Y, Li M. Interactions between CAP70 and actinfilin are important for integrity of actin cytoskeleton structures in neurons. Neuropharmacology. 2005;49:1026–1041. doi: 10.1016/j.neuropharm.2005.05.022. [DOI] [PubMed] [Google Scholar]
- Clark HB, Burright EN, Yunis WS, Larson S, Wilcox C, Hartman B, Matilla A, Zoghbi HY, Orr HT. Purkinje cell expression of a mutant allele of SCA1 in transgenic mice leads to disparate effects on motor behaviors, followed by a progressive cerebellar dysfunction and histological alterations. J Neurosci. 1997;17:7385–7395. doi: 10.1523/JNEUROSCI.17-19-07385.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corral J, Genis D, Banchs I, San Nicolas H, Armstrong J, Volpini V. Giant SCA8 alleles in nine children whose mother has two moderately large ones. Ann Neurol. 2005;57:549–553. doi: 10.1002/ana.20421. [DOI] [PubMed] [Google Scholar]
- De Paepe A. Heritable collagen disorders: from phenotype to genotype. Verh K Acad Geneeskd Belg. 1998;60:463–482. discussion 482–484. [PubMed] [Google Scholar]
- Ethell IM, Pasquale EB. Molecular mechanisms of dendritic spine development and remodeling. Prog Neurobiol. 2005;75:161–205. doi: 10.1016/j.pneurobio.2005.02.003. [DOI] [PubMed] [Google Scholar]
- Gatchel JR, Zoghbi HY. Diseases of unstable repeat expansion: mechanisms and common principles. Nat Rev Genet. 2005;6:743–755. doi: 10.1038/nrg1691. [DOI] [PubMed] [Google Scholar]
- Jiang S, Avraham HK, Park SY, Kim TA, Bu X, Seng S, Avraham S. Process elongation of oligodendrocytes is promoted by the Kelch-related actin-binding protein Mayven. J Neurochem. 2005;92:1191–1203. doi: 10.1111/j.1471-4159.2004.02946.x. [DOI] [PubMed] [Google Scholar]
- Jin P, Alisch RS, Warren ST. RNA and microRNAs in fragile X mental retardation. Nat Cell Biol. 2004;6:1048–1053. doi: 10.1038/ncb1104-1048. [DOI] [PubMed] [Google Scholar]
- Juvonen V, Hietala M, Paivarinta M, Rantamaki M, Hakamies L, Kaakkola S, Vierimaa O, Penttinen M, Savontaus ML. Clinical and genetic findings in Finnish ataxia patients with the spinocerebellar ataxia 8 repeat expansion. Ann Neurol. 2000;48:354–361. [PubMed] [Google Scholar]
- Juvonen V, Hietala M, Kairisto V, Savontaus ML. The occurrence of dominant spinocerebellar ataxias among 251 Finnish ataxia patients and the role of predisposing large normal alleles in a genetically isolated population. Acta Neurol Scand. 2005;111:154–162. doi: 10.1111/j.1600-0404.2005.00349.x. [DOI] [PubMed] [Google Scholar]
- Klement IA, Skinner PJ, Kaytor MD, Yi H, Hersch SM, Clark HB, Zoghbi HY, Orr HT. Ataxin-1 nuclear localization and aggregation: role in polyglutamine-induced disease in SCA1 transgenic mice. Cell. 1998;95:41–53. doi: 10.1016/s0092-8674(00)81781-x. [DOI] [PubMed] [Google Scholar]
- Koob MD. Spinocerebellar Ataxia 8 (SCA8) In: Pulst SM, editor. Genetics of movement disorders. New York: Academic; 2003. pp. 95–102. [Google Scholar]
- Koob MD, Moseley ML, Schut LJ, Benzow KA, Bird TD, Day JW, Ranum LP. An untranslated CTG expansion causes a novel form of spinocerebellar ataxia (SCA8) Nat Genet. 1999;21:379–384. doi: 10.1038/7710. [DOI] [PubMed] [Google Scholar]
- Lippman J, Dunaevsky A. Dendritic spine morphogenesis and plasticity. J Neurobiol. 2005;64:47–57. doi: 10.1002/neu.20149. [DOI] [PubMed] [Google Scholar]
- Miki T, Takano K, Yoneda Y. The role of mammalian Staufen on mRNA traffic: a view from its nucleocytoplasmic shuttling function. Cell Struct Funct. 2005;30:51–56. doi: 10.1247/csf.30.51. [DOI] [PubMed] [Google Scholar]
- Moseley ML, Zu T, Ikeda Y, Gao W, Mosemiller AK, Daughters RS, Chen G, Weatherspoon MR, Clark HB, Ebner TJ, Day JW, Ranum LP. Bidirectional expression of CUG and CAG expansion transcripts and intranuclear polyglutamine inclusions in spinocerebellar ataxia type 8. Nat Genet. 2006;38:758–769. doi: 10.1038/ng1827. [DOI] [PubMed] [Google Scholar]
- Mutsuddi M, Marshall CM, Benzow KA, Koob MD, Rebay I. The spinocerebellar ataxia 8 noncoding RNA causes neurodegeneration and associates with staufen in Drosophila. Curr Biol. 2004;14:302–308. doi: 10.1016/j.cub.2004.01.034. [DOI] [PubMed] [Google Scholar]
- Nahm SS, Jung KY, Enger MK, Griffith WH, Abbott LC. Differential expression of T-type calcium channels in P/Q-type calcium channel mutant mice with ataxia and absence epilepsy. J Neurobiol. 2005;62:352–360. doi: 10.1002/neu.20107. [DOI] [PubMed] [Google Scholar]
- Nelson PN, Reynolds GM, Waldron EE, Ward E, Giannopoulos K, Murray PG. Monoclonal antibodies. Mol Pathol. 2000;53:111–117. doi: 10.1136/mp.53.3.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemes JP, Benzow KA, Moseley ML, Ranum LP, Koob MD. The SCA8 transcript is an antisense RNA to a brain-specific transcript encoding a novel actin-binding protein (KLHL1) Hum Mol Genet. 2000;9:1543–1551. doi: 10.1093/hmg/9.10.1543. [DOI] [PubMed] [Google Scholar]
- Prag S, Adams JC. Molecular phylogeny of the kelch-repeat superfamily reveals an expansion of BTB/kelch proteins in animals. BMC Bioinformatics. 2003;4:42. doi: 10.1186/1471-2105-4-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao A, Craig AM. Signaling between the actin cytoskeleton and the postsynaptic density of dendritic spines. Hippocampus. 2000;10:527–541. doi: 10.1002/1098-1063(2000)10:5<527::AID-HIPO3>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Robinson DN, Cooley L. Drosophila kelch is an oligomeric ring canal actin organizer. J Cell Biol. 1997;138:799–810. doi: 10.1083/jcb.138.4.799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santarosa M, Ashworth A. Haploinsufficiency for tumour suppressor genes: when you don't need to go all the way. Biochim Biophys Acta. 2004;1654:105–122. doi: 10.1016/j.bbcan.2004.01.001. [DOI] [PubMed] [Google Scholar]
- Shirao T, Sekino Y. Clustering and anchoring mechanisms of molecular constituents of postsynaptic scaffolds in dendritic spines. Neurosci Res. 2001;40:1–7. doi: 10.1016/s0168-0102(01)00209-7. [DOI] [PubMed] [Google Scholar]
- Sobrido MJ, Cholfin JA, Perlman S, Pulst SM, Geschwind DH. SCA8 repeat expansions in ataxia: a controversial association. Neurology. 2001;57:1310–1312. doi: 10.1212/wnl.57.7.1310. [DOI] [PubMed] [Google Scholar]
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- Vandaele S, Nordquist DT, Feddersen RM, Tretjakoff I, Peterson AC, Orr HT. Purkinje cell protein-2 regulatory regions and transgene expression in cerebellar compartments. Genes Dev. 1991;5:1136–1148. doi: 10.1101/gad.5.7.1136. [DOI] [PubMed] [Google Scholar]
- Vincent JB, Neves-Pereira ML, Paterson AD, Yamamoto E, Parikh SV, Macciardi F, Gurling HM, Potkin SG, Pato CN, Macedo A, Kovacs M, Davies M, Lieberman JA, Meltzer HY, Petronis A, Kennedy JL. An unstable trinucleotide-repeat region on chromosome 13 implicated in spinocerebellar ataxia: a common expansion locus. Am J Hum Genet. 2000;66:819–829. doi: 10.1086/302803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter JT, Alvina K, Womack MD, Chevez C, Khodakhah K. Decreases in the precision of Purkinje cell pacemaking cause cerebellar dysfunction and ataxia. Nat Neurosci. 2006;9:389–397. doi: 10.1038/nn1648. [DOI] [PubMed] [Google Scholar]
- Wooley CM, Sher RB, Kale A, Frankel WN, Cox GA, Seburn KL. Gait analysis detects early changes in transgenic SOD1(G93A) mice. Muscle Nerve. 2005;32:43–50. doi: 10.1002/mus.20228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zephir H, Stojkovic T, Latour P, Hurtevent JF, Blankaert F, Vermersch P. A family with a novel frameshift mutation in the PMP22 gene (c. 433_434insC) causing a phenotype of hereditary neuropathy with liability to pressure palsies. Neuromuscul Disord. 2005;15:493–497. doi: 10.1016/j.nmd.2005.04.007. [DOI] [PubMed] [Google Scholar]
- Zu T, Duvick LA, Kaytor MD, Berlinger MS, Zoghbi HY, Clark HB, Orr HT. Recovery from polyglutamine-induced neurodegeneration in conditional SCA1 transgenic mice. J Neurosci. 2004;24:8853–8861. doi: 10.1523/JNEUROSCI.2978-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]