

Abstract
The enormous cellular complexity of the brain is a major obstacle for gene expression profiling of neurological disease models, because physiologically relevant changes of transcription in a specific neuronal subset are likely to be lost in the presence of other neurons and glia. We solved this problem in transgenic mice by labeling genetically defined cells with a nuclear variant of GFP. When combined with laser-directed microdissection, intact RNA from unfixed, freeze-dried sections can be isolated, which is a prerequisite for high-quality global transcriptome analysis. Here, we compared gene expression profiles between pyramidal motor neurons and pyramidal somatosensory neurons captured from layer V of the adult neocortex. One striking feature of motor neurons is the elevated expression of ribosomal genes and genes involved in ATP synthesis. This suggests a molecular adaptation of the upper motor neurons to longer axonal projections and higher electrical activity. These molecular signatures were not detected when cortical layers and microareas were analyzed in toto.
Additionally, we used microarrays to determine the global mRNA expression profiles of microdissected Purkinje cells and cellularly complex cerebellar cortex microregions. In summary, our analysis shows that cellularly complex targets lead to averaged gene expression profiles that lack substantial amounts of cell type-specific information. Thus, cell type-restricted sampling strategies are mandatory in the CNS. The combined use of a genetic label with laser-microdissection offers an unbiased approach to map patterns of gene expression onto practically any cell type of the brain.
Keywords: cortex, cerebellum, microarrays, transcriptome, microdissection, GFP
Introduction
The mammalian brain is the most complex organ, comprising hundreds of neuronal subtypes that are intermingled with various glial cell types and non-neural cells, all of which differ in gene expression and responsiveness to particular stimuli (Stevens, 1998; Serafini, 1999). Unfortunately, standard gene expression profiles that are obtained from entire brain or subregions dissected in toto are not as helpful as expected, because they lack the spatial resolution that is necessary to address functional questions. In addition, the complexity of brain mRNA exceeds that of other tissues (Sutcliffe, 1988; Serafini, 1999), which raises the issue of sensitivity of hybridization-based microarray analyses. Indeed, in a pioneering study, Barlow and colleagues detected more cerebellum-expressed transcripts than neocortical transcripts (Sandberg et al., 2000), although the underlying complexity of mRNA is likely to be the reverse (Geschwind, 2000). Results of such global expression analyses are also difficult to interpret without localization of individual transcripts at the cellular level, typically by labor-intensive in situ hybridization (Zirlinger et al., 2001; Lein et al., 2004). Most importantly, functionally and clinically relevant changes of gene expression in the adult brain are rather subtle (Middleton et al., 2002; Panda et al., 2002; McClung and Nestler, 2003; Cirelli et al., 2004; Prabakaran et al., 2004), and dilution effects become a major issue, because relevant transcripts are likely to be missed by expression profiling of complex tissue samples. Finally, the metabolism of neurons in vivo is coupled to that of glial cells, and any attempt to study neuronal metabolism by transcriptional profiling of dissociated neurons in vitro is likely to lead to artificial results (Pellerin and Magistretti, 1994; Kasischke et al., 2004).
For all of these reasons, there is a strong need to develop better cell sampling strategies of the brain before applying global gene expression analysis (Serafini, 1999; Lockhart and Barlow, 2001; Henry et al., 2003).
However, there have been major technical limitations. Cellular gene expression profiles have been obtained from cultured neuronal precursor cells (Tietjen et al., 2003), but these in vitro profiles are most likely different from the in vivo situation. Another strategy has been the labeling of defined cell types in vivo for manual or automated fluorescent-activated cell sorting (Zhang et al., 2002; Buchstaller et al., 2004; Tumbar et al., 2004; Arlotta et al., 2005; Sugino et al., 2006). Here, the major limitations are the applicability to adult and aged brain tissue and a reduced spatial precision of isolation. Moreover, because prolonged tissue processing ex vivo is essential, an effect on gene expression and mRNA turnover cannot formally be excluded. Laser-directed microdissection (LDM) allows the isolation of very small regions and even single cells from fixed and stained histological sections (Schutze and Lahr, 1998; Simone et al., 1998). A combination of LDM sampling strategies with microarray analyses has been applied for Nissl-stained brain material (Luo et al., 1999; Kamme et al., 2003; Tietjen et al., 2003). Although this approach offers high spatial resolution, cell sampling relies solely on morphological criteria. Moreover, tissue fixation with cross-linking agents (e.g., formaldehyde) are generally not compatible with high-quality RNA isolation, and even ethanol fixation affects the reliability of subsequent microarray analysis (Karsten et al., 2002; Van Deerlin et al., 2002).
To overcome these limitations, we devised a strategy that combines a genetic labeling of neuronal cell types, using a nuclearly targeted green fluorescent protein (GFP) derivative, with fluorescence-guided LDM and microarray analysis. Importantly, this method does not require fixation or histological staining and is applicable in the adult brain. A small number of single neurons are sufficient to obtain RNA of high quality for global gene expression profiling.
In the present proof-of-principle, we used genechip analysis to study the gene expression profile of genetically labeled projection neurons of layer V of the neocortex and to compare these neurons between motor cortex and somatosensory cortex. We demonstrate that cortical motor neurons can be distinguished from corresponding somatosensory projection neurons by increased expression levels of genes that are involved in energy metabolism and protein synthesis. Moreover, cortical motor neurons increase the expression of enzymes that metabolize lactate, thought to be provided by associated astrocytes, but have unaltered expression of the core enzymes for glycolysis and the tricarboxylic acid (TCA) cycle. These in vivo observations support the existence of an astrocyte-neuron “lactate shuttle” that has recently been postulated based on cell culture data (Pellerin and Magistretti, 1994, 2004). We suggest that the molecular signatures may reflect an adaptation of motor neurons to higher electrical activity and increased projection distance (axonal length). Importantly, such cell type-specific signatures were not detected when the respective cortical layers were microdissected in toto for RNA analysis. We anticipate that expression profiling of individually collected neurons will become an important tool to monitor normal and pathological neuronal gene expression in a variety of neurological disease models.
Materials and Methods
Cell culture and transfections.
COS7 and HeLa cells were cultured in DMEM supplemented with 10% fetal calf serum. Cells were transfected with plasmids coding for GFP variants (BD Biosciences, Heidelberg, Germany) using Lipofectamin 2000 (Invitrogen, Karlsruhe, Germany) according to the instructions given by the manufacturer. Thirty-six to 48 h after transfection, GFP expression was checked; subsequently, cells were fixed, frozen, and reanalyzed for fluorescence.
Generation of transgenic mice.
All experiments were performed in accordance with the federal law and on approval of the animal care committee of the Bezirksregierung Braunschweig, Germany. Mice were housed under a 12 h light/dark cycle (lights on at 5:00 A.M.) with food and drinking water available ad libitum. Transgenic mice were generated by injection of a linearized Thy-1 minigene cassette into C57BL/6 oocytes (Caroni, 1997; Feng et al., 2000) and implantation into foster animals following standard procedures. In the Thy-1 minigene, the Thy-1 ORF was replaced by an enhance yellow fluorescent protein (EYFP) open reading frame fused to three nuclear localization signals derived from the SV40 large T antigen. Transgenic founder animals were bred with C57BL/6 mice to generate F1 offspring, which were analyzed for EYFP expression; one line called TYNC was used throughout this study. Transgenic mice were identified by PCR using the following primers: EFPs: CGGCTACGGCCTGCAGTGCT, EFPas: GGCGGATCTTGAAGTTCACCTTGATG.
Histological procedures.
For immunohistochemistry, mice were perfused with 4% buffered paraformaldehyde (PFA), and brains were removed and postfixed for 2 h at 4°C and then sunk in a 30% sucrose/0.1 m phosphate buffer (PB) solution overnight. Floating sections (50 μm) were prepared using a vibratome (Leica, Wetzlar, Germany), and tissue was rinsed in 0.1 m PB before immunostaining. Sections were then incubated in 0.1 m PB with 0.4% Triton X-100 for 30 min and blocked in PBS-T (PBS with 0.2% Triton X-100) with 4% horse serum for 1 h. Primary antibodies used were anti-GFAP (1:200; Novocastra, Newcastle upon Tyne, UK), anti- neuronal-specific nuclear protein (NeuN) (1:100; Chemicon, Hampshire, UK), anti-parvalbumin (1:500; Sigma, Hamburg, Germany), anti-calbindin (1:600; Sigma), anti-GAD67 (1:1000; Chemicon), and anti-GFP (1:1000; Invitrogen). Antibodies were diluted in PBS-T with 1% horse serum, and sections were incubated overnight at 4°C in this solution. Sections were washed three times with PBS-T and then incubated in a Cy3-conjugated secondary antibody (Cy3 anti-rabbit; Jackson ImmunoResearch, West Grove, PA) at a 1:2000 dilution in blocking solution. Sections were then washed three times in PBS-T before being mounted in Aqua-Poly/Mount (Polysciences, Warrington, PA) on slides.
Laser-directed microdissection.
Young adult TYNC males (2 months of age; ±30 g) were killed at 11:00 ± 1:00 A.M. under deep anesthesia. The brains were removed from the skull and were rapidly frozen on dry ice, sealed with Parafilm and stored at −80°C until further use. Serial coronal brain sections (8 μm) at the level of the striatum (bregma, +1.2 to +1.0), hippocampus (bregma, −1.7 to −2.0), and cerebellum (bregma, −6.5 to −7.0) were prepared using a cryostat (Leica) at −18°C and were mounted on POL-membrane slides (Leica). After sectioning, the slides were rapidly dried and stored in sealed boxes with silica at −80°C until further use. Selected sections from each series were Nissl-stained and kept as references. On the day of LDM, the slides were taken from −80°C and were subjected to LDM within the next hours. Single cells and cortical microregions were isolated from alternating sections to avoid any bias caused by handling time or slight regional differences. The EYFP-positive cells were microdissected from the anterior motor and somatosensory cortices from six slides in total (maximum of 20 cells from each hemisphere) and pooled; the corresponding layer V and layer I-VI microregions were microdissected from three immediately adjacent and intersected slides and were also pooled. For the microdissection, a Leica AS LDM was used. The isolation of YFP-positive cells was done by identifying single bright fluorescent cells in the fluorescence mode, switching to bright-field mode for microdissection and collection. Cells were only collected if no adjacent nuclei were detected in close proximity. Cutting and collection steps were subsequently examined in bright-field and fluorescent mode. Microdissected samples were collected in 100 μl of RNA lysis buffer (Qiagen, Hilden, Germany) and stored at room temperature until further processing.
RNA isolation and linear amplification.
RNA preparation was always performed at the day of microdissection using the RNeasy Micro Kit (Qiagen) according to the manufacturer instructions without adding any carrier. RNA quality and quantity of control samples was checked by analyzing 1 μl of RNA on the Agilent 2100 Bioanalyser using the RNA 6000 Pico LabChip kit (Agilent Technologies, Boeblingen, Germany). Total RNAs of all sample groups were stored at −80°C and were processed in parallel. Immediately before amplification, RNA was precipitated by adding Na-acetate [pH 5.2, 0.3 m, final concentration (f.c.)], 2 μl of PelletPaint (Calbiochem, La Jolla, CA; Novabiochem, Bad Soden, Germany), and ethanol. Total RNA of pooled single cells or microregions was resuspended with pretested T7-tagged dT21V oligonucleotides. Two-round T7-RNA polymerase-mediated linear amplification was performed according to optimized protocols for low-input RNA amounts (see Small Sample Target Labeling Assay Version II; Affymetrix). Biotin-labeled second-round aRNA was generated with a NTP-mix containing Biotin-11-CTP and Biotin-16-UTP (PerkinElmer, Boston, MA) (2 mm f.c.). Biotin-labeled amplified RNA (aRNA) size distribution and quantity was analyzed with the Agilent 2100 Bioanalyser using the RNA 6000 Nano LabChip kit (Agilent Technologies, Boeblingen, Germany). Samples with lower size compressed RNA products were discarded.
Microarray hybridization.
At least 10 μg of labeled cRNA was fragmented by heating the sample to 95°C for 35 min in a volume of 20 μl containing 40 mm Tris acetate, pH 8.1, 100 mm KOAc, and 30 mm MgOAc. Fragmentation was checked by alkaline agarose electrophoresis. Hybridization, washing, staining, and scanning were performed under standard conditions as described by the manufacturer. MOE430A genechips were used that contain over 22,600 probe sets representing transcripts and variants from over 14,000 well-characterized mouse genes.
Microarray data analysis.
Data analysis was initially performed using Microarray Suite software version 5.0 (MAS5.0; Affymetrix, Santa Clara, CA) to obtain signal intensities, detection calls, and detection p values. All signal intensities were scaled to a target value of 500 (TGT500). Microarray data were further analyzed with the Data Mining Tool 3.0 and NetAFFX (Affymetrix), dChip1.3 (http://www.dChip.org/), Rosetta Luminator (Rosetta Biosoftware, Seattle, WA), and Microsoft Excel and Microsoft Access (Microsoft, Seattle, WA). Confidence intervals for all replicate groups were determined with Excel for a p value of 0.05 (95% confidence) (see supplemental Methods, available at www.jneurosci.org as supplemental material). To obtain similarly scaled values for upregulated and downregulated genes in pair-wise comparisons, normalized signal intensities were log2-transformed (Signal-log2), and regulation factors were given as Signal-Log2-Ratios (SLRs).
Gene set analysis and statistics.
Selected probe set lists were downloaded using NetAffx (Liu et al., 2003) and were initially analyzed with the DAVID (Database for Annotation, Visualization, and Integrated Discovery) pathfinder tool [http://apps1.niaid.nih.gov/david/ (Dennis et al., 2003)]. The identification of coordinated changes in a priori defined sets of functionally grouped genes was performed using nonparametric tests and in parallel with the Gene Set Enrichment Analysis (GSEA) approach that was described recently by (Mootha et al., 2003). First, the summed average signal intensity differences of selected probe set groups (gene sets) for a given comparison were calculated with dChip1.3 (PM/MM difference model, model based expression normalization). The data were log2 converted and nonparametric Mann–Whitney U and Kolmogorov–Smirnov statistical tests were performed with Statistica 6.0 (StatSoft, Tulsa, OK). Gene sets were considered as regulated when the means (“all” vs “selected gene set”) of the summed log2 signal differences (Y + MCx − Y + MCx or MCx I-VI − SSCx I-VI) were scored as significant in both nonparametric tests with a p value <5 × 10−2. The GSEA software package was downloaded (http://www.broad.mit.edu/personal/aravind/GSEA/index.html) and used with default parameters (permutations set to 1000) and the selected gene sets as suggested.
Reverse Transcription-PCR.
For reverse transcription (RT)-PCR characterization of marker gene expression in EYFP-positive cells, total RNA was T7 amplified for one round (see above), aRNA was reverse transcribed using random primers, and PCR was performed with primer pairs corresponding to the following: (gene, accession number) Cyc (cyclophilin) (NM_008907), Thy-1 (NM_009382), CamKIIa (AK083245), GFAP (AK079577), and Otx-1 (NM_011023) (primer sequences are available in the supplemental Methods, available at www.jneurosci.org as supplemental material).
Quantitative RT-PCR.
Quantitative RT-PCR (qRT-PCR) was either performed using the LightCycler system (Roche, Mannheim, Germany) as described previously (Schneider et al., 2004) or with SybrGreen fluorescence analyzed with an ABI PRISM 7700 detection system (PerkinElmer, Rodgau, Germany). cDNAs were serially diluted to obtain three to four concentration points for quantification. The relative concentrations of mRNAs of interest in different cDNA samples were determined using the threshold cycle method (Ct) for each dilution and were normalized to levels of murine glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Reactions were performed in 25 μl using SYBR green PCR master mix (ABgene, Hamburg, Germany) according to the protocol of the manufacturer. Cycling was done for 15 min at 95°C, followed by 40 cycles of 95°C, 30 s, 60–65°C, 45 s 72°C, 45 s. PCR conditions for each primer pair were initially optimized with a melting curve analysis; the absence of primer by-products and correct product size was verified with agarose gel electrophoresis. The following gene products were analyzed (gene, probe set ID): Gapdh (AFFX-GAPDHMUR), Rps24 (1436064_X_AT), Rps10 (1438723_a_at), Cox7a2 (1416970_a_at), and Ndufb9 (1452184_AT) (primer sequences are available in supplemental Methods, available at www.jneurosci.org as supplemental material).
Results
The experimental approach that we developed to monitor global gene expression profiles of defined neuronal cell populations in the adult brain is based on the transgenic expression of microdissection- and cryosectioning-compatible fluorescent proteins (the general strategy is summarized in Fig. 1).
Figure 1.

Experimental strategy to monitor global gene expression patterns of defined neuronal cells. 1, The isolation of defined neuronal cell types with laser-mediated microdissection is based on the transgenic expression of fluorescent proteins that are compatible with the tissue-sectioning procedures indispensable for the isolation of intact RNA. The use of characterized promoters allows for the genetic labeling of cell types of choice. With this approach, no additional staining procedure is needed. 2, In freeze-dried cryosections, the fluorescent labeling as well as cellular RNAs are conserved for long periods of time. 3, The fluorescent label directs the precise isolation of defined cells with an appropriately equipped laser microdissection microscope. 4, High-quality RNA can be extracted, amplified, and labeled to obtain sufficient material to (5) snapshot the transcriptome of defined cells with high-density microarrays or related methods.
In a first attempt to combine transgenic labeling of neurons with LDM, we analyzed mice that express cytoplasmic enhanced GFP (EGFP) (or EYFP) in neurons and glial cells (Krestel et al., 2001; Meyer et al., 2002; Hirrlinger et al., 2004). Because formalin-fixed tissue is not compatible with the isolation of intact RNA, cryosectioning is mandatory (Van Deerlin et al., 2002). However, in all transgenic lines, such fluorescent signals faded rapidly or were completely lost, although the cells appeared morphologically intact (Fig. 2A). We determined that freezing itself did not reduce the activity of GFP, although cryoprotective substances appeared to stabilize fluorescence during repeated freeze-thaw cycles (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material). Thus, leakage from cells was the most likely cause for the loss of GFP from cryosections and could not be prevented by perfusion of mice with cryoprotective substances (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material) (data not shown).
Figure 2.
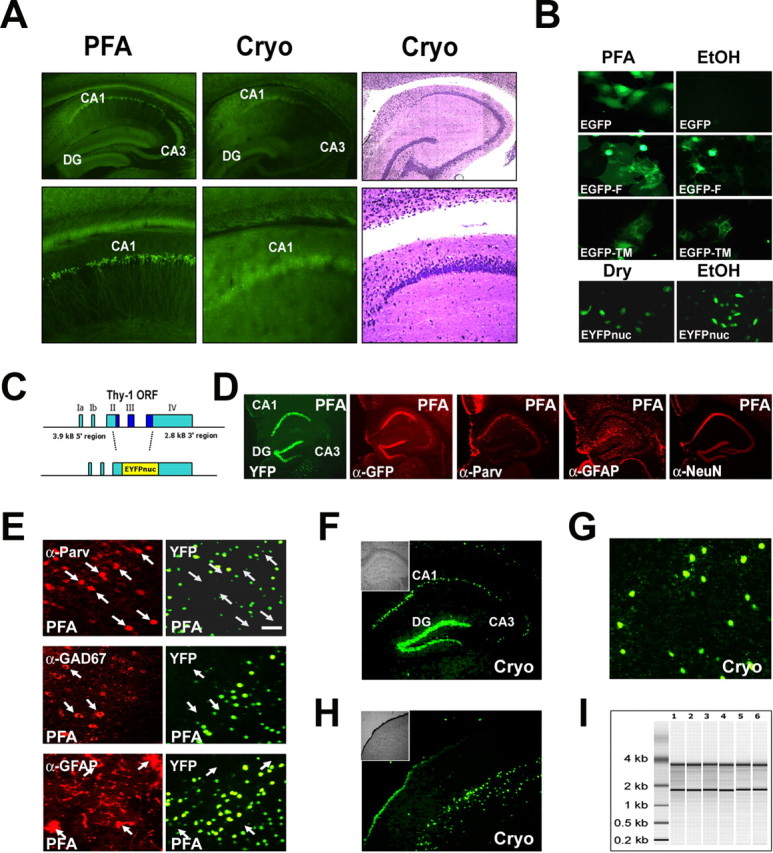
Conventional GFP derivatives are not compatible with laser-directed microdissection. The cellular signal of a nuclear-targeted EYFP (EYFPnuc) is preserved in freeze-dried cryosections obtained from transgenic mice. RNA isolated from cryosections is intact. A, The cellular resolution of EYFP expressed in transgenic mice is lost after cryosectioning. Sagittal sections of mouse brains from mice expressing cytoplasmic localized EYFP under the control of the Thy-1 promoter were either fixed with 4% buffered PFA or immediately frozen on dry ice. Vibratome sections (PFA, left) obtained from PFA fixed tissue (30 μm) and cryosections (15 μm; Cryo, middle) from frozen tissue, were analyzed for EYFP fluorescence. After cryosectioning, the cellular resolution of the EYFP fluorescent signal is lost. The EYFP signal is retained but appears to be diffuse and faded. Staining of the cryosection with thionin (Cryo, right) reveals that the cellular integrity of the tissue specimen is intact. B, Nuclear targeted fluorescent proteins appear to be promising candidates for a combined in vivo labeling and microdissection approach. COS7 or HeLa cells were transiently transfected with expression constructs coding for GFP derivatives localized to different cellular compartments: EGFP (cytoplasmic localization), EGFP carrying a farnesylation signal (EGFP-F, attached to membranes), EGFP fused to the N terminus of a synthetic transmembrane protein (TM-EGFP), and EYFP fused to three nuclear localization signals (EYFPnuc). Forty-eight hours after transfection, COS7 cells were either fixed with a PBS-buffered 4% PFA or fixed with 70% ethanol (EtOH) and subsequently dry mounted. HeLa cells transfected with EYFPnuc were either dry mounted (DRY) or fixed with 70% ethanol (EtOH) and subsequently frozen at −80°C before analysis. C, Transgenic mice were generated that express a nuclear-targeted EYFP (EYFPnuc) under control of the Thy-1 Promoter (TYNC mice). The Thy-1 minigene was modified by replacing the Thy-1 ORF with sequences coding for EYFPnuc. D, E, Vibratome sections (30 μm) from brains of PFA-fixed TYNC mice were analyzed for YFP fluorescence and for cellular marker gene expression with immunohistochemistry. D, Within the hippocampus, the YFP expression is most prominent in pyramidal neurons of the CA1 field (CA1) and in the granular cell layer of the dentate gyrus (DG). In the CA3 field (CA3), fewer cells are YFP positive. The YFP fluorescence appears to be strictly overlapping with the indirect GFP immune fluorescence analysis (α-GFP), although it is less sensitive. The pan-neuronal marker NeuN (α-NeuN) colocalizes with the nuclear YFP signal. Staining with GFAP (α-GFAP) and parvalbumin (α-Parv) does not reveal YFP overlapping signals. E, Higher-magnification photographs obtained from vibratome sections at the level of the primary motor and sensory cortex. YFP-positive nuclei (YFP) do not colocalize with the interneuronal marker parvalbumin (α-Parv) and GAD67 (α-GAD67) and also not with the astrocyte marker GFAP (α-GFAP). Arrows mark parvalbumin, GAD67, or GFAP-positive cells stained in red in the left panel. No overlap with YFP fluorescence can be detected in the right panel with arrows pointing to identical positions as in the left panel. Scale bar, 50 μm. F–H, Cryosections (8 μm; Cryo) were mounted on PEN foils (Leica) and analyzed for YFP fluorescence. F, In the hippocampus, YFP-positive nuclei can be detected in the granular cell layer (DG) and in the CA1 field of the pyramidal cell layer (CA1). In the CA3 field (CA3), fewer cells are YFP positive. The inset shows the morphology of the hippocampus in bright field. G, Higher magnification of YFP-positive nuclei in the somatosensory cortex. Scale bar, 50 μm. H, In the cortex, cells located in deeper layers show a nuclear YFP signal. The inset shows the morphology of the section at the level of the somatosensory cortex in bright field. I, Total RNA analyzed with a Bioanalyzer (Agilent); each lane represents half of the RNA isolated from one coronal. brain section. Adjacent 8-μ-thick sections were cryomounted on PEN foil slides, dried, and treated as follows: (1) stored for 2 h at room temperature, (2) stored for 8 h at room temperature, (3) stored for 24 h at room temperature, (4) stored for 48 h at room temperature, (5) frozen at −80°C and then kept at room temperature for 2 h, and (6) microdissected regions pooled from eight sections (total area size isolated comparable with one section). The ratio of the 28S versus the 18S rRNA bands was determined with the Bioanalyzer software and for all samples was 1.3± 0.1. The amount of RNA isolated from one coronal 8 μ brain cryosection was ∼50 ng.
To compare the feasibility of “anchored” forms of GFP, we tested cultured cells after transfection with various GFP expression constructs and subsequent fixation (with ethanol, or freezing and drying). In these experiments, all GFP variants that were targeted to membranous organelles survived the procedure (Fig. 2B) (supplemental Fig. 1C, available at www.jneurosci.org as supplemental material). Moreover, the nuclear localization of EYFP, achieved with three copies of the SV40 nuclear targeting sequence, appeared optimal for combining in vivo labeling and microdissection of individual neurons, because the fluorescent signal intensity was almost unaffected by subsequent freezing and drying (Fig. 2B).
Genetic labeling of neuronal cell nuclei
To obtain nuclear localization of EYFP in vivo, transgenic mice were generated by DNA microinjection of fertilized mouse oocytes (C57BL/6). The transgene encoding nuclear EYFP was driven by the neuron-specific Thy-1.2 minigene (Caroni, 1997; Feng et al., 2000) (Fig. 2C), which was chosen for this proof-of-principle study because of its proven efficiency to drive transgenic gene expression in subsets of principal neurons in forebrain structures. From four transgenic lines, we selected one for additional analysis that exhibited strong nuclear EYFP expression, primarily restricted to layer V of the neocortex, the CA fields, and the dentate gyrus (Fig. 2D,E). This line (termed TYNC) was chosen for all subsequent experiments. The overall distribution of transgene expression in the forebrain was in agreement with previous reports of Thy-1 gene-driven transgenes (Feng et al., 2000) (i.e., mosaic in genetically defined neuronal subsets. Expression of EYFP in projection neurons was verified by immunohistochemistry of PFA-fixed vibratome sections, using additional neuronal and glial markers (Fig. 2D,E). In the hippocampus, EYFP was primarily restricted to pyramidal cells of the CA1 field and to granule cells of the dentate (Fig. 2D). In the neocortex, colabeling of cell nuclei with 4′,6′-diamidino-2-phenylindole revealed that EYFP marked ∼30% of the large nuclei-bearing cells (i.e., principal neurons) throughout cortical layer V (data not shown). By hematoxylin-eosin staining, these cells were pyramidal neurons. Small neuronal or glial cell nuclei were never EYFP positive (data not shown). Similarly, the interneuronal marker (parvalbumin, GAD67) and astroglial marker (GFAP) did not overlap (Fig. 2E). Thus, nuclear EYFP labeling could be restricted to a specific subset of projection neurons, defined by the spatiotemporal expression pattern of a neuronal expressed transgene.
To test the compatibility of nuclear EYFP expression with LDM, we mounted unfixed cryosections (8 μm) onto membrane slides (see Materials and Methods for details). The distribution of EYFP-labeled neurons was identical to that observed in PFA-fixed vibratome sections, with bright fluorescent signals clearly localized to the nucleus (Fig. 2F–H). Sections were either stored at room temperature or frozen at −80°C. Interestingly, there was very little loss of fluorescence, even after prolonged storage at room temperature (data not shown). RNA isolated from single brain cryosections was well preserved (Fig. 2I)
Precision of fluorescence-directed laser microdissection
In the brain, the relevant neuronal and glial cells are typically defined by their location and by cell type-specific markers. Thus, we aimed to obtain gene expression profiles at the level of genetically defined, regionally restricted, and individually isolated cells. We used pools of 100 individually isolated EYFP-labeled neurons that yielded sufficient target for a microarray analysis under standard conditions (10 μg of aRNA). Such amplification products were analyzed with an Agilent Bioanalyzer and showed a broad peak from 0.3 to 1.0 kb, as shown in Figigure 3D. We also determined the efficiency of LDM by collecting 22 single cells for single-cell PCR. When RNA from individual cells was amplified in one round and analyzed by RT-PCR for two known targets (GAPDH and cyclophilin), PCR products were successfully obtained from 17 of 22 cells (data not shown).
Figure 3.

Fluorescence-directed laser microdissection enables the isolation of neuronal cell types with high precision. A, Fluorescent micrographs taken from an 8 μm TYNC brain cryosection mounted on a POL foil slide at the level of the striatum (St) (bregma, 1 mm) were manually assembled to an overview picture. The inset shows an adjacent section stained with thionin. For better orientation, the boundaries of the corpus callosum (CC) and of the lateral ventricle (LV) were marked in the thionin section, and the label was transferred to the fluorescent picture. In the cortex, at the level of the primary motor and somatosensory areas (MCx and SSCx), mainly cells of the deeper layers are EYFP positive (layer V and few cells in layer VI). In the upper layers II, III, and IV, a low number of dispersed cells were EYFP positive, usually with a weaker EYFP signal intensity. Samples isolated with laser microdissection were chosen from the primary motor and the somatosensory regions. B, Fluorescence-directed laser microdissection of a single YFP-positive cortical neuron. 1, In the fluorescent mode, a single cell was located and marked by an appropriate painting tool (Leica LDM software); only strong EYFP-positive cells in the plane of section were chosen for isolation. The laser-cutting line was drawn around the nucleus at a distance of half the diameter of the nucleus at the most. 2, The microscope was subsequently switched to bright-field mode; here, the corresponding nucleus is visible as a bright round structure (arrowhead). Cells with other nuclei in close proximity were not isolated. Depicted are two nuclei (arrows) at a distance where contamination was considered to be low. Laser cutting follows a manually or automatically drawn circle; using the 150× objective (Leica), the laser cut is ∼1 μm. 3, 4, A successful cut was controlled in the bright field and fluorescent mode. Scale bar, 20 μm. C, The accuracy of isolation was analyzed by RT-PCR using primers specific for the ubiquitously expressed cyclophilin (cyc), EYFP, the neuronal marker Thy-1, and CamKIIα. Otx-1 was chosen as a marker for cortical layer V projection neurons and GFAP as an astrocytic marker. All primers were located near the 3′ terminus. The cDNA template was generated from one round of linear amplified RNA. Total RNA was isolated from a pool of 100 EYFP-positive (Y) or 100 randomly selected EYFP-negative (N) cells from one TYNC cortex section not restricted to layer V. Lanes labeled with — are controls without cDNA. EYFP and Otx-1 mRNAs were detected in the YFP-positive pool of cells. The mRNA of markers for principal neurons Thy-1 and CamKIIα could be detected in the Y and N pools. GFAP expression was only detected in the EYFP-negative pool, suggesting that the contamination with cell material derived from astrocytes in the EYFP fraction is low. D, Two rounds amplified biotin-labeled RNA analyzed with the Bioanalyzer (Agilent). Shown are three independent samples of pools of 100 EYFP-positive cells isolated from layer V of the motor cortex (Y+MCx) and the somatosensory cortex (SSCx). The relative size distribution range is from 0.2 to >1 kB.
The nuclear YFP labeling allowed us to capture each neuron with highest precision, including the nucleus and an ∼5-μm-wide region containing neuronal cytoplasm (Fig. 3B). Unlabeled cells, even in close proximity, were easy to avoid, because all nuclei were also identified in bright-field mode (Fig. 3B-2,3). However, processes of neighboring cells remained a potential source for contaminating mRNA (Steward and Schuman, 2003). Thus, we tested the precision of LDM by amplifying different cDNAs from 100 pooled YFP-positive cells (and 100 nonfluorescent cells) from motor and somatosensory cortex. Whereas the ubiquitously expressed cyclophilin mRNA and the neuronal markers Thy-1 and calcium calmodulin-dependent protein kinase II-α (CaMKII-α) were detected in both samples, EYFP mRNA was only found in the YFP-positive fraction. The absence of GFAP mRNA in the YFP pool suggested that astrocytic processes were not a major source of unwanted RNA (Fig. 3C). In contrast, we amplified Otx1 transcripts, a marker for subcortically projecting layer V neurons (Weimann et al., 1999), selectively in the YFP-labeled neuronal pool. Moreover, we could not detect mRNA transcripts of the interneuron marker genes parvalbumin and calbindin in microdissected YFP-positive cell samples with qRT-PCR (data not shown). In concordance with the RT-PCR data, cluster analysis of microarray data from EYFP-positive cells (see below) showed an increased level of“′glutamatergic” and a decreased level of“′GABAergic” markers (supplemental Figs. 2, 3, available at www.jneurosci.org as supplemental material).
Microarray analyses after LDM sampling
To provide proof-of-principle of this method, we aimed to identify gene expression differences thought to exist between morphologically indistinguishable neuronal subtypes. We isolated three independent replicates of 100 YFP-positive pyramidal neurons from layer V of the motor cortex (the pool was termed Y+MCx in the following; n = 3) and as reference 100 pyramidal cells from layer V of the somatosensory cortex (Y+SSCx; n = 3) (Fig. 3A). To control for the benefits of cell type-restricted analysis, we additionally isolated (under microscopic control) the corresponding cortical microregions [i.e., entire layer V of the motor cortex (MCx V; n = 2) and layer V of the sensory cortex (SSCx V; n = 2), or all cortical layers but without white matter (MCx I-VI and SSCx I-VI; n = 3 for both)]. As an independent neuronal cell type, we used 400 pooled Purkinje cells (PC400; n = 3 for each) and the corresponding microdissected area of the cerebellar cortex (Cbx; n = 3) including the granular, Purkinje, and molecular cell layer excluding cerebellar white matter tracts for the analysis. Finally, we also prepared RNA from whole brain and kidney (n = 2 each) to compare the LDM samples to RNA isolated with standard procedures. All targets were generated after a two-round amplification protocol (see Materials and Methods) (supplemental Table 1, available at www.jneurosci.org as supplemental material). We obtained at least 10 μg labeled aRNA, and considerably more from the less demanding samples (supplemental Table 1, available at www.jneurosci.org as supplemental material). All amplified targets were hybridized under standard conditions and at identical concentrations to Affymetrix MOE430A gene expression arrays (see Materials and Methods).
We used several internal control criteria and confidence tests to assess the overall performance of targets obtained by LDM, compared with targets obtained by standard preparations (Fig. 3A). The number of expressed mRNAs (“present called probe sets”) ranged from >44% for the cortical projection neurons up to >60% for total brain, representing 10,000 and 13,000 different probe sets, respectively (supplemental Table 1, available at www.jneurosci.org as supplemental material). The dynamic range of expression (the relative distribution of weak, medium, and strong expressed mRNAs) was comparable for all samples, including the cell type-restricted targets (Fig. 2A–C). In summary, gene chip analyses performed with pooled single cells or microregions (obtained with LDM) worked equally well as the analysis with tissue-derived RNA, when compared with equal amounts of input RNA (see supplemental Results, available at www.jneurosci.org as supplemental material). Analyzing all quality control parameters (3′5′ ratios of the control probe sets b-actin and GAPDH, slope of all probe sets) and taking the replicate signal intensity-based statistics into account (Fig. 4C), we can order targets into “input groups” (from the least to the most demanding samples): (1) the brain and kidney samples, (2) LMD isolated microregions (Cbx, M/SSCx V, M/SSCx I-VI), and (3) the single isolated cells (PC400, Y+M/SSCx). We conclude that a maximally “bias-free” comparison is only possible within groups of similar input amount and quality; optimally when prepared in one batch (see supplemental Results, available at www.jneurosci.org as supplemental material).
Figure 4.

The GeneChip performance of amplified targets obtained from neuronal cell types isolated with laser-mediated microdissection compared with control preparations. A–C, Variable bin histogram for the samples isolated from primary motor and somatosensory cortex and cerebellum compared with total brain. The number of probe sets (on the y-axis) was plotted against grouped signal intensities (bins) (on the x-axis). The dynamic range of genes detected at low, middle, or high-expression levels comparing LMD isolated samples with the total brain sample is very similar, a technically introduced bias in the LMD isolated samples is therefore unlikely. D–I, Scatter plots of relative signal intensities of replicate arrays with absent called probe sets (gray dots) and present called probe sets (black dots). All replicate samples show a highly similar dynamic range of signal intensities over three orders of magnitude. The scatter does not increase with single-cell targets. D, Scatter plot of signal intensities of independent replicates of 100 pooled single isolated cells from somatosensory cortex (Y+SSCx 1 vs 2). E, Scatter plot of layer V isolated areas from somatosensory cortex (SSCx V 1 vs 2). F, Scatter plot of cortical layers I-VI isolated from somatosensory cortex (SSCx I-VI 1 vs 2). G, Scatter plot of 400 pooled single isolated Purkinje cells isolated from cerebellum (PC400 1 vs 2). H, Scatter plot of cerebellar cortex samples (Cbx 1 vs 2). I, Scatter plot of signal intensities of independent replicates of total brain targets amplified from 50 ng of input RNA (Brain 1 and 2). All errors are given as SD.
Complex cellular targets lack informative gene expression profiles
The pair-wise correlation of the motor and sensory cortical probes (Fig. 5A) and the determination of regulated genes at stringent cutoffs revealed that all gene expression profiles tend to average out with increasing cellular complexity (Fig. 5B,C). This became most obvious when the expression profiles of pyramidal projection neurons (motor and somatosensory neurons, samples collectively termed Y+Cx) were compared with the profiles of the entire microregions (motor and somatosensory samples collectively termed Cx V and Cx I-VI), which are cellularly complex. The number of genes detected as “regulated” by a signal-log2-ratio (SLR) of more than or equal to ±1.4 between the Y+Cx cells and the cellularly complex cortical samples is 264 (Cx V) and 476 (Cx I-VI), respectively. In contrast, only six probe sets were found to be differentially detected between the layer V and all layers containing cortical samples (Cx V vs Cx I-VI) (Fig. 5B). Indeed, their pair-wise correlation coefficient was high with 0.98 (Fig. 5A). Thus, even microdissected samples comprising cortical layer V yield in a highly averaged expression profile, which is caused by the high cell type complexity. This approach is therefore not suitable to unravel cell type- or layer-specific gene expression in depth. More importantly, when the layer V projection neurons (Y+Cx) were compared with cerebellar Purkinje cells (PC400) (representing the same input group; see above), we detected 396 probe sets as differentially expressed at a very stringent SLR of at least ±3.0 (196 up in Y+Cx, 200 up in PC400) (Fig. 5C) (supplemental Results, available at www.jneurosci.org as supplemental material). In contrast, when the corresponding microregions (Cx V vs Cbx) were compared, only 294 probe sets were detected at this stringency (104 up in Cx V and 90 in Cbx) (Fig. 5C). Importantly, when entire microregions were compared, only 62 (of 196) and 45 (of 200) cell type-specific genes were identified, suggesting that the majority of differentially expressed genes remain undetected in cellularly complex samples (Fig. 5C) (for a more detailed discussion, see supplemental Results, available at www.jneurosci.org as supplemental material).
Figure 5.

Statistical analysis of GeneChip data obtained with amplified targets isolated with laser-mediated microdissection. A, Average pair-wise Pearson's correlation coefficients (r) determined for selected comparisons of GeneChip data obtained from complex and cell type-specific samples. B, Numbers of regulated probe sets comparing EYFP-positive cells from layer V (Y+Cx) and layer V microregions (Cx V) with all layers comprising samples (Cx I-VI). The number of regulated genes was determined with MAS 5.0 with a cutoff at 66% change calls and a SLR of at least ± 1.4. The number of probe sets found to be differentially detected between Y+Cx and CxV (264) increases almost twofold in the more cellularly complex Cx I-VI comparison (476). Comparing both cellularly complex samples (Cx V and Cx I-VI) reveals only six probe sets to be differentially detected at the identical selection cutoff. C, Venn diagrams showing overlapping and differentially expressed mRNAs comparing (I) genetically defined pyramidal neurons from neocortex (Y+Cx) and cerebellar Purkinje cells (PC400) (left) and (II) the corresponding microregions in the cortex (Cx V) and in the cerebellum (Cbx). The number of mRNAs detected in both pyramidal neurons and Purkinje cells is 9113 (40.2% of all probe sets); the number of coexpressed genes in the corresponding microregions (Cx V and Cbx) is 10659 (47.0% of all probe sets). Highly differentially expressed genes were defined by an SLR ≥3.0/less than or equal to −3.0 (i.e., regulated ≥9-fold). The number of these genes is 196 and 200 for singly isolated cells and 104 and 90 for the corresponding microregions. Importantly, when entire microregions were compared, only 62 (of 196) and 45 (of 200) cell type-specific genes were identified, suggesting that the majority of differentially expressed genes remain undetected in cellularly complex samples.
Gene expression in motor and somatosensory cortex
The major aim of our study was to develop a protocol for obtaining informative gene expression profiles of genetically defined neuronal subtypes, unbiased by the overall tissue complexity. The analysis of morphologically indistinguishable layer V pyramidal neurons, isolated either from motor cortex or somatosensory cortex of adult mice, should provide proof-of-principle. Individually isolated YFP-positive layer V neurons from these regions corresponded to the same class of projection neurons, at the same hierarchical position (main cortical output neurons) of different modalities.
At first approximation, the gene expression profiles obtained from motor and somatosensory projection neurons (Y+MCx vs Y+SSCX and MCx I-VI vs SSCx I-VI) were very similar, as evidenced by correlation values of >0.99 (Fig. 5A). Indeed, we were expecting only subtle gene expression differences and performed first comparisons using nonstringent cutoff criteria (>66% change calls for all pair-wise comparisons) (McClung and Nestler, 2003). Here, we obtained 103 and 77 genes scored as differentially expressed between Y+MCx and Y+SSCx samples, 29 and 82 genes for MCx V versus SSCx V, and 68 and 31 genes for MCx I-VI versus SSCx I-VI (supplemental Table 3 and additional material, available at www.jneurosci.org as supplemental material). The SLRs of the vast majority of genes showed a regional gene expression difference in the range of 0.5–1 (i.e., 1.5- to 2-fold changes) in all comparisons. These rather subtle gene expression differences are in agreement with previous studies of larger brain regions (Panda et al., 2002; McClung and Nestler, 2003; Cirelli et al., 2004) and have been confirmed here by quantitative RT-PCR of independent samples (see further below) (Fig. 6D–G). Moreover, we found that cell type-specific and regional differences in gene expression detected with our approach are in many cases in accordance with published data (for a detailed discussion, see supplemental Results, available at www.jneurosci.org as supplemental material). In situ hybridization data confirming qualitatively the expression of most genes in mouse brain are available on-line at the Allen brain atlas (www.brain-map.org). For ∼80% of the genes detected in our comparison of the Y+MCx versus the Y+SSCx samples, mRNA expression can be detected in the adult mouse motor and somatosensory cortices (supplemental Table 3, available at www.jneurosci.org as supplemental material).
Figure 6.

The detection of coordinated gene expression changes in closely related neuronal cell populations is lost when comparing the corresponding motor or somatosensory cortical microareas. A, Biochemical pathways (KEGG chart, selected with the DAVID pathfinder tool) plotted for the probe sets found as upregulated in the Y+MCx samples. The cutoff for classification was set at 3. With these parameters, 42 (38%) of the 103 genes found upregulated in Y+MCx could be grouped into three biochemical pathways: (I) ribosomal function (27), (II) oxidative phosphorylation (12) and (III) ATP synthesis (see full data set in the supplemental Table 3, available at www.jneurosci.org as supplemental material). No other functional cluster could be generated for any other comparison at the given cutoff (see full data set in supplemental Table 3, available at www.jneurosci.org as supplemental material). B, C, Plotted are the SLRs for functionally grouped genes with a ribosomal function (b) or with a function in mitochondrial respiration (oxidative phosphorylation and ATP synthesis) (c) for all three MCx versus SSCx comparisons. The solid line at an SLR of 0.3 marks the 95% confidence interval for the average signal intensities obtained for the Cx V and Cx I-VI genechips (Fig. 4B) (see Materials and Methods). The upregulation is restricted to the Y+MCx samples when comparing the single-cell isolates. The higher variability in the SLRs observed in the layer V samples is likely to be attributable to the lower number of replicates in these samples (n = 2). D–G, Plots of the relative signal intensities obtained for all functional probe sets in the genechip analysis and two independent GAPDH normalized qRT-PCR measurements for Rps24 (D), Rps10 (E), Cox7a2 (F), and Ndufb9 (G). All values were normalized with the expression level found in the respective somatosensory sample (set to 1). All error bars are depicted as SD.
Coordinated gene expression changes in cortical projection neurons
We noticed that a substantial fraction (42 of 103) of transcripts enriched in motor cortex-derived layer V projection neurons were associated with a ribosomal or a mitochondrial function (supplemental Table 3, available at www.jneurosci.org as supplemental material). To further validate this finding and to search for other functionally grouped gene sets, we used the DAVID pathfinder tool (http://apps1.niaid.nih.gov/david/) (Dennis et al., 2003). The only apparent clusters (GoCharts for “Molecular Function” and “Cellular Component”) were scored in the Y+MCx upregulated gene list; with most hits confined to components of the cytosolic ribosome and to mitochondria (data not shown). Scanning for biochemical pathways [based on the KEGG annotations (www.genome.ad.jp/kegg/)], 27 hits were annotated for a ribosomal function and 15 with a function in mitochondrial respiration (12 scored for oxidative phosphorylation, 3 hits for ATP synthesis) (Fig. 6a). To corroborate this finding, additional software tools (dChip, Rosetta Luminator, R-project) were used to select for differentially expressed genes (Schadt et al., 2001). Although the independently generated lists of regulated genes were not identical, the enrichment of genes with a ribosomal or mitochondrial function was similarly apparent (data not shown).
Next, we plotted the log2-converted expression differences for ribosomal and mitochondrial clusters in all three comparisons (motor vs somatosensory cortex) side by side (Fig. 6B,C). Whereas the SLRs for the selected probe sets were all shifted toward a higher expression in the Y+MCx sample, they were close to zero in comparisons involving the entire layer V and all cortical layers (Fig. 6B,C). We verified the Y+MCx enriched expression of mRNAs coding for the ribosomal proteins Rps24 and Rps10, the cytochrome c-oxidase subunit VIIa 2 (Cox7a2), and the nicotinamide adenine dinucleotide phosphate (NADH) dehydrogenase 1-β subcomplex 9 (Ndufb9) with qRT-PCR (Fig. 6D–G). We plotted the normalized signal intensities for all probe sets (Y+MCx vs Y+SSCx, MCx V vs SSCx V, and MCx I-VI vs SSCx I-VI) as a reference and compared these to the GAPDH normalized qRT-PCR values. The overall fold induction measured with qRT-PCR (∼1.5-fold for all mRNAs examined) was close to that obtained with the genechips (Fig. 6D–G).
Metabolic and ribosomal gene sets in cortical motor neurons
It is well established that functionally related genes within metabolic pathways are coregulated by environmental changes (Eisen et al., 1998; Spellman et al., 1998). Recently, a strategy termed Gene Set Enrichment Analysis was described that allows detecting coordinated changes in a priori defined sets of functionally grouped genes (Mootha et al., 2003). This type of gene expression profiling allows recognizing potentially coregulated genes with high sensitivity, making use of the statistical power originating from increased numbers of data points. Because of the importance of energy balance for neuronal function, we focused on biochemical pathways important for ATP synthesis (Calabrese et al., 2001; Nicholls, 2002; Prabakaran et al., 2004). This knowledge might help to understand how cortical layer V motor neurons adapt, at the level of gene transcription, to their specific tasks in vivo. We manually selected or downloaded probe lists for biological pathways or cellular components (Fig. 7C) (probe sets used are available on-line, see supplemental note, available at www.jneurosci.org as supplemental material). Next, we used dChip to obtain the average signal intensities for all selected gene lists and determined the summed averaged intensity differences. We focused the analysis to the comparisons Y+MCx versus Y+SSCx and MCx I-VI versus SSCx I-VI, using the following groups of a priori defined gene sets: (1) all probe sets annotated for mitochondria (all-mito), (2) mitochondrial respiratory chain complexes I-IV (mito-resp), (3) mitochondrial F0-ATPases (complex V)(mito-ATP), (4) mitochondrial localized Krebs TCA cycle (mito-TCA), (5) all probe sets restricted to the cytoplasmic ribosome (ribo-cyto), (6) probe sets annotated for a function in the mitochondrial ribosome (mito-ribo), (7) enzymes linked to glycolysis, and (8) the hypothesized astroglia-neuron coupling lactate shuttle (lactate-shuttle) (Fig. 7C). In addition, we selected gene sets unrelated to energy balance, corresponding (9) to cholesterol biosynthesis and metabolism (cholesterol), and probe sets for genes linked to other subcellular functions (10–13) (peroxisome, proteasome, ubiquitin, apoptosis) (Fig. 7C) (supplemental Table 4, available at www.jneurosci.org as supplemental material).
Figure 7.

Coordinated upregulation of genes involved in neuronal ATP synthesis and ribosomal function in layer V projection neurons of the motor cortex. A, B, Histogram of the normalized mean gene expression level differences between Y+MCx and Y+SSCx samples (A) and between the MCx I-VI and SSCx I-VI samples (B) for all probe sets (gray bars), those corresponding to genes with a function in mitochondrial respiration (blue bars), and those corresponding to mRNAs coding for proteins of the cytoplasmic ribosome (red bars). C, Plot of the mean log2 transformed normalized gene expression level differences for all genes or defined groups of genes as indicated (for detailed description of gene sets, see supplemental Tables 4 and 5, available at www.jneurosci.org as supplemental material) showing the overall shift toward either motor or somatosensory cortex-derived samples. Black bars correspond to the Y+ comparisons, and gray bars correspond to the Cx I-VI comparisons; the mean average difference values were normalized to all Y+ and I-VI probe sets, respectively (set to 0). Significantly shifted gene sets are marked by asterisks (Mann–Whitney U and Kolmogorov–Smirnov tests; p < 0.05) (for a detailed description of the pathway sampling and detailed statistical analysis, see supplemental Table 4, available at www.jneurosci.org as supplemental material). D, Schematic drawing of the principal metabolic units involved in neuronal ATP synthesis. The flow chart summarizes the findings from the gene set analysis indicating unregulated and Y+MCx upregulated (gray) functionally grouped genes. The drawing illustrates the “lactate-bypass” hypothesis to fuel directly the respiratory chain complexes (RC I-IV) and oxidative phosphorylation machinery (F0F1-ATPase) with NADH, bypassing the glycolysis and at least partially the TCA cycle.
Plotting the intensity differences for all probe sets in bins, we obtained, as expected, a Gaussian distribution of approximately zero for the Y+ and Cx I-VI comparisons (Fig. 7A,B). However, when we analyzed groups of probe lists for distinct biochemical pathways or cellular functions, a coordinated difference in gene expression became obvious (Fig. 7A–C). Most prominently shifted toward higher expression in the genetically labeled motor neurons were genes of the mitochondrial respiratory chain complexes I-IV (mito-resp; n = 105; p < 10−10) and those coding for proteins of the cytoplasmic ribosome (cyto-ribo; n = 105; p < 10−12) (Fig. 7A–C) (for details, see supplemental Table 4, available at www.jneurosci.org as supplemental material). The coordinated change in groups of genes playing a role in mitochondrial energy metabolism, mito-resp, and mito-ATP (F0F1-ATPase subunits) and in the cytosolic ribosome (ribo-cyto) was expected, because we had already detected candidates from each group in the gene-by-gene analysis. However, we found additional gene groups significantly shifted for which no probe set had reached significance with DMT or dChip. Mitochondria-encoded ribosomal genes were also found slightly upregulated in the Y+MCx samples (mito-ribo; n = 48; p < 0.01). In contrast, the gene sets corresponding to the core enzymes of the mitochondrial Krebs tricarboxylic acid cycle (mito-TCA) and core enzymes of the glycolysis were not upregulated in cortical layer V motor neurons. It was therefore intriguing to find the lactate-shuttle gene set upregulated in the Y+MCx samples (n = 14; p < 0.05). These genes correspond to the hypothetical pathway of lactate as an additional source of carbohydrate for neurons provided by glial cells. This pathway was not identified in the corresponding cortical microareas (Fig. 7C,D) (supplemental Table 4, available at www.jneurosci.org as supplemental material).
The Na+/K+-ATPase requires >90% of the neuronal ATP (Nicholls, 2002). A lactate shuttle has been hypothesized to serve these high demands by a specialized energy coupling between neurons and neighboring astrocytes (Pellerin and Magistretti, 1994; Bouzier-Sore et al., 2003). However, direct evidence for a lactate shuttle in vivo has been lacking. Indirectly, two-photon microscopic visualization of NADH turnover (in slice preparations) has shown that activity-dependent rapid increases in neuronal energy demand are followed by a delayed NADH turnover in adjacent astrocytes (Kasischke et al., 2004). Lactate is thought to instantaneously replenish neuronal energy needs by generating NADH (Pellerin and Magistretti, 2004). Our molecular data, showing the increase of transcripts in cortical motor neurons for enzymes needed to metabolize lactate, provide in vivo evidence for the lactate shuttle hypothesis. We speculate that the lactate metabolism in neuronal processes, which we observe here at the level of coordinated nuclear gene transcription, helps to cope with long-lasting periods of high energy consumption.
Discussion
We have shown the feasibility to “snapshot” gene expression profiles of genetically defined neuronal subtypes in vivo and to compare morphologically similar neurons at a given time in a specific position. We obtained the critical informational gain by performing the transcriptional profiling at the cellular level, compared with the analysis of an entire cortical layer (comprising different cell types), or complex subregions of the brain. The data analysis suggests that such cell type-restricted gene expression analysis is mandatory to unravel physiological or pathological changes in complex tissues, such as the CNS.
Although single genes have been linked to many neurodegenerative diseases, the reasons for the remarkable cell type selectivity remain unknown (Cleveland and Rothstein, 2001). Cell type-specific gene expression differences are likely to contribute to the enhanced disease susceptibility in neurodegenerative diseases. We believe that truly cellular expression profiles in the nervous system will improve our understanding of normal development and mechanisms of neurodegeneration. In animal models of neurological and psychiatric diseases, we anticipate that appropriate molecular signatures will allow to better design and monitor experimental treatments. It will also be possible to follow gene expression changes at an early disease state, focusing on cells known to be susceptible to toxic insults or a genetic predisposition. Moreover, with our method, there are no technical restrictions with respect to tissue processing, and, importantly, our approach will enable us to study specific cell types in adult and aged animals, a prerequisite for neurodegenerative disease models.
Recently, a series of bacterial artificial chromosome-derived EGFP reporter constructs has been characterized in the mouse, all of which appear applicable to direct the expression of microdissection-compatible reporter proteins (Gong et al., 2003). Moreover, marker genes for cortical subtypes have been identified, which allow us to transgenically label these neurons (Arlotta et al., 2005). Transgenic mice expressing nuclear GFP variants under control of activity-regulated promoters will be useful to even map patterns of gene expression onto neuronal activity at the circuit level (Barth et al., 2004). Nonetheless, the generation of “universal” reporter mice is in progress, where microdissection-compatible GFP variants can be activated in a cell type-specific manner simply by crossing to already characterized Cre-recombinase expressing mouse lines. This may, in many cases, circumvent the necessity to establish novel reporter mouse lines. Although the present study has focused on gene expression, we expect that our cellular-restricted sampling strategy can also be applied to other “global-scaled” technologies that are currently developed in other model systems (Encode Consortium, 2004).
Footnotes
This work was supported by the Max-Planck-Society, a Bundesministerium für Bildung und Forschung Grant (FKZ 0312748), and a European Community Grant (NeuroproMiSe LSHM-CT-2005-018637). We acknowledge M. Pruess (Affymetrix), A. Schneider, M. Haubrock, and M. Kränzle for their help on microarray analysis; A. Rosenberger for assistance with the statistical analysis; S. Herberger for excellent technical assistance with sectioning and microdissection; K. Chowdhury for his support with the Affymetrix genechip instrument system; F. Kirchhoff for hGFAP–EGFP mice; H. Monyer and A. Meyer for Parvalbumin–EGFP mice; R. Sprengel and H. Krestel for mice expressing GFP in principal neurons; and A. Fahrenholz for help with immunohistochemistry. We thank T. Fischer and M. Pruess (Affymetrix) for critical reading of previous versions of this manuscript.
References
- Arlotta P, Molyneaux BJ, Chen J, Inoue J, Kominami R, Macklis JD. Neuronal subtype-specific genes that control corticospinal motor neuron development in vivo. Neuron. 2005;45:207–221. doi: 10.1016/j.neuron.2004.12.036. [DOI] [PubMed] [Google Scholar]
- Barth AL, Gerkin RC, Dean KL. Alteration of neuronal firing properties after in vivo experience in a FosGFP transgenic mouse. J Neurosci. 2004;24:6466–6475. doi: 10.1523/JNEUROSCI.4737-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouzier-Sore AK, Voisin P, Canioni P, Magistretti PJ, Pellerin L. Lactate is a preferential oxidative energy substrate over glucose for neurons in culture. J Cereb Blood Flow Metab. 2003;23:1298–1306. doi: 10.1097/01.WCB.0000091761.61714.25. [DOI] [PubMed] [Google Scholar]
- Buchstaller J, Sommer L, Bodmer M, Hoffmann R, Suter U, Mantei N. Efficient isolation and gene expression profiling of small numbers of neural crest stem cells and developing Schwann cells. J Neurosci. 2004;24:2357–2365. doi: 10.1523/JNEUROSCI.4083-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese V, Scapagnini G, Giuffrida Stella AM, Bates TE, Clark JB. Mitochondrial involvement in brain function and dysfunction: relevance to aging, neurodegenerative disorders and longevity. Neurochem Res. 2001;26:739–764. doi: 10.1023/a:1010955807739. [DOI] [PubMed] [Google Scholar]
- Caroni P. Overexpression of growth-associated proteins in the neurons of adult transgenic mice. J Neurosci Methods. 1997;71:3–9. doi: 10.1016/s0165-0270(96)00121-5. [DOI] [PubMed] [Google Scholar]
- Cirelli C, Gutierrez CM, Tononi G. Extensive and divergent effects of sleep and wakefulness on brain gene expression. Neuron. 2004;41:35–43. doi: 10.1016/s0896-6273(03)00814-6. [DOI] [PubMed] [Google Scholar]
- Cleveland DW, Rothstein JD. From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Nat Rev Neurosci. 2001;2:806–819. doi: 10.1038/35097565. [DOI] [PubMed] [Google Scholar]
- Dennis G, Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4:P3. [PubMed] [Google Scholar]
- Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci USA. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, Nerbonne JM, Lichtman JW, Sanes JR. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000;28:41–51. doi: 10.1016/s0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- Geschwind DH. Mice, microarrays, and the genetic diversity of the brain. Proc Natl Acad Sci USA. 2000;97:10676–10678. doi: 10.1073/pnas.97.20.10676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong S, Zheng C, Doughty ML, Losos K, Didkovsky N, Schambra UB, Nowak NJ, Joyner A, Leblanc G, Hatten ME, Heintz N. A gene expression atlas of the central nervous system based on bacterial artificial chromosomes. Nature. 2003;425:917–925. doi: 10.1038/nature02033. [DOI] [PubMed] [Google Scholar]
- Henry GL, Zito K, Dubnau J. Chipping away at brain function: mining for insights with microarrays. Curr Opin Neurobiol. 2003;13:570–576. doi: 10.1016/s0959-4388(03)00107-7. [DOI] [PubMed] [Google Scholar]
- Hirrlinger J, Hulsmann S, Kirchhoff F. Astroglial processes show spontaneous motility at active synaptic terminals in situ. Eur J Neurosci. 2004;20:2235–2239. doi: 10.1111/j.1460-9568.2004.03689.x. [DOI] [PubMed] [Google Scholar]
- Kamme F, Salunga R, Yu J, Tran DT, Zhu J, Luo L, Bittner A, Guo HQ, Miller N, Wan J, Erlander M. Single-cell microarray analysis in hippocampus CA1: demonstration and validation of cellular heterogeneity. J Neurosci. 2003;23:3607–3615. doi: 10.1523/JNEUROSCI.23-09-03607.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karsten SL, Van Deerlin VM, Sabatti C, Gill LH, Geschwind DH. An evaluation of tyramide signal amplification and archived fixed and frozen tissue in microarray gene expression analysis. Nucleic Acids Res. 2002;30:E4. doi: 10.1093/nar/30.2.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasischke KA, Vishwasrao HD, Fisher PJ, Zipfel WR, Webb WW. Neural activity triggers neuronal oxidative metabolism followed by astrocytic glycolysis. Science. 2004;305:99–103. doi: 10.1126/science.1096485. [DOI] [PubMed] [Google Scholar]
- Krestel HE, Mayford M, Seeburg PH, Sprengel R. A GFP-equipped bidirectional expression module well suited for monitoring tetracycline-regulated gene expression in mouse. Nucleic Acids Res. 2001;29:E39. doi: 10.1093/nar/29.7.e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lein ES, Zhao X, Gage FH. Defining a molecular atlas of the hippocampus using DNA microarrays and high-throughput in situ hybridization. J Neurosci. 2004;24:3879–3889. doi: 10.1523/JNEUROSCI.4710-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Loraine AE, Shigeta R, Cline M, Cheng J, Valmeekam V, Sun S, Kulp D, Siani-Rose MA. NetAffx: Affymetrix probe sets and annotations. Nucleic Acids Res. 2003;31:82–86. doi: 10.1093/nar/gkg121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockhart DJ, Barlow C. Expressing what's on your mind: DNA arrays and the brain. Nat Rev Neurosci. 2001;2:63–68. doi: 10.1038/35049070. [DOI] [PubMed] [Google Scholar]
- Luo L, Salunga RC, Guo H, Bittner A, Joy KC, Galindo JE, Xiao H, Rogers KE, Wan JS, Jackson MR, Erlander MG. Gene expression profiles of laser-captured adjacent neuronal subtypes. Nat Med. 1999;5:117–122. doi: 10.1038/4806. [DOI] [PubMed] [Google Scholar]
- McClung CA, Nestler EJ. Regulation of gene expression and cocaine reward by CREB and DeltaFosB. Nat Neurosci. 2003;6:1208–1215. doi: 10.1038/nn1143. [DOI] [PubMed] [Google Scholar]
- Meyer AH, Katona I, Blatow M, Rozov A, Monyer H. in vivo labeling of parvalbumin-positive interneurons and analysis of electrical coupling in identified neurons. J Neurosci. 2002;22:7055–7064. doi: 10.1523/JNEUROSCI.22-16-07055.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middleton FA, Mirnics K, Pierri JN, Lewis DA, Levitt P. Gene expression profiling reveals alterations of specific metabolic pathways in schizophrenia. J Neurosci. 2002;22:2718–2729. doi: 10.1523/JNEUROSCI.22-07-02718.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstrale M, Laurila E, Houstis N, Daly MJ, Patterson N, Mesirov JP, Golub TR, Tamayo P, Spiegelman B, Lander ES, Hirschhorn JN, Altshuler D, Groop LC. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- Nicholls D. Mitochondrial bioenergetics, aging, and aging-related disease. Sci Aging Knowledge Environ. 2002;2002:pe12. doi: 10.1126/sageke.2002.31.pe12. [DOI] [PubMed] [Google Scholar]
- Panda S, Antoch MP, Miller BH, Su AI, Schook AB, Straume M, Schultz PG, Kay SA, Takahashi JS, Hogenesch JB. Coordinated transcription of key pathways in the mouse by the circadian clock. Cell. 2002;109:307–320. doi: 10.1016/s0092-8674(02)00722-5. [DOI] [PubMed] [Google Scholar]
- Pellerin L, Magistretti PJ. Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proc Natl Acad Sci USA. 1994;91:10625–10629. doi: 10.1073/pnas.91.22.10625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellerin L, Magistretti PJ. Neuroscience Let there be (NADH) light. Science. 2004;305:50–52. doi: 10.1126/science.1100428. [DOI] [PubMed] [Google Scholar]
- Prabakaran S, Swatton JE, Ryan MM, Huffaker SJ, Huang JT, Griffin JL, Wayland M, Freeman T, Dudbridge F, Lilley KS, Karp NA, Hester S, Tkachev D, Mimmack ML, Yolken RH, Webster MJ, Torrey EF, Bahn S. Mitochondrial dysfunction in schizophrenia: evidence for compromised brain metabolism and oxidative stress. Mol Psychiatry. 2004;9:643. doi: 10.1038/sj.mp.4001511. [DOI] [PubMed] [Google Scholar]
- Sandberg R, Yasuda R, Pankratz DG, Carter TA, Del Rio JA, Wodicka L, Mayford M, Lockhart DJ, Barlow C. Regional and strain-specific gene expression mapping in the adult mouse brain. Proc Natl Acad Sci USA. 2000;97:11038–11043. doi: 10.1073/pnas.97.20.11038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schadt EE, Li C, Ellis B, Wong WH. Feature extraction and normalization algorithms for high-density oligonucleotide gene expression array data. J Cell Biochem. 2001;37(Suppl):120–125. doi: 10.1002/jcb.10073. [DOI] [PubMed] [Google Scholar]
- Schneider A, Laage R, von Ahsen O, Fischer A, Rossner M, Scheek S, Grunewald S, Kuner R, Weber D, Kruger C, Klaussner B, Gotz B, Hiemisch H, Newrzella D, Martin-Villalba A, Bach A, Schwaninger M. Identification of regulated genes during permanent focal cerebral ischaemia: characterization of the protein kinase 9b5/MARKL1/MARK4. J Neurochem. 2004;88:1114–1126. doi: 10.1046/j.1471-4159.2003.02228.x. [DOI] [PubMed] [Google Scholar]
- Schutze K, Lahr G. Identification of expressed genes by laser-mediated manipulation of single cells. Nat Biotechnol. 1998;16:737–742. doi: 10.1038/nbt0898-737. [DOI] [PubMed] [Google Scholar]
- Serafini T. Of neurons and gene chips. Curr Opin Neurobiol. 1999;9:641–644. doi: 10.1016/s0959-4388(99)00022-7. [DOI] [PubMed] [Google Scholar]
- Simone NL, Bonner RF, Gillespie JW, Emmert-Buck MR, Liotta LA. Laser-capture microdissection: opening the microscopic frontier to molecular analysis. Trends Genet. 1998;14:272–276. doi: 10.1016/s0168-9525(98)01489-9. [DOI] [PubMed] [Google Scholar]
- Spellman PT, Sherlock G, Zhang MQ, Iyer VR, Anders K, Eisen MB, Brown PO, Botstein D, Futcher B. Comprehensive identification of cell cycle-regulated genes of the yeast Saccharomyces cerevisiae by microarray hybridization. Mol Biol Cell. 1998;9:3273–3297. doi: 10.1091/mbc.9.12.3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens CF. Neuronal diversity: too many cell types for comfort? Curr Biol. 1998;8:R708–R710. doi: 10.1016/s0960-9822(98)70454-3. [DOI] [PubMed] [Google Scholar]
- Steward O, Schuman EM. Compartmentalized synthesis and degradation of proteins in neurons. Neuron. 2003;40:347–359. doi: 10.1016/s0896-6273(03)00635-4. [DOI] [PubMed] [Google Scholar]
- Sugino K, Hempel CM, Miller MN, Hattox AM, Shapiro P, Wu C, Huang ZJ, Nelson SB. Molecular taxonomy of major neuronal classes in the adult mouse forebrain. Nat Neurosci. 2006;9:99–107. doi: 10.1038/nn1618. [DOI] [PubMed] [Google Scholar]
- Sutcliffe JG. mRNA in the mammalian central nervous system. Annu Rev Neurosci. 1988;11:157–198. doi: 10.1146/annurev.ne.11.030188.001105. [DOI] [PubMed] [Google Scholar]
- Tietjen I, Rihel JM, Cao Y, Koentges G, Zakhary L, Dulac C. Single-cell transcriptional analysis of neuronal progenitors. Neuron. 2003;38:161–175. doi: 10.1016/s0896-6273(03)00229-0. [DOI] [PubMed] [Google Scholar]
- Tumbar T, Guasch G, Greco V, Blanpain C, Lowry WE, Rendl M, Fuchs E. Defining the epithelial stem cell niche in skin. Science. 2004;303:359–363. doi: 10.1126/science.1092436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Deerlin VM, Gill LH, Nelson PT. Optimizing gene expression analysis in archival brain tissue. Neurochem Res. 2002;27:993–1003. doi: 10.1023/a:1020996519419. [DOI] [PubMed] [Google Scholar]
- Weimann JM, Zhang YA, Levin ME, Devine WP, Brulet P, McConnell SK. Cortical neurons require Otx1 for the refinement of exuberant axonal projections to subcortical targets. Neuron. 1999;24:819–831. doi: 10.1016/s0896-6273(00)81030-2. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Ma C, Delohery T, Nasipak B, Foat BC, Bounoutas A, Bussemaker HJ, Kim SK, Chalfie M. Identification of genes expressed in C. elegans touch receptor neurons. Nature. 2002;418:331–335. doi: 10.1038/nature00891. [DOI] [PubMed] [Google Scholar]
- Zirlinger M, Kreiman G, Anderson DJ. Amygdala-enriched genes identified by microarray technology are restricted to specific amygdaloid subnuclei. Proc Natl Acad Sci USA. 2001;98:5270–5275. doi: 10.1073/pnas.091094698. [DOI] [PMC free article] [PubMed] [Google Scholar]