

Abstract
The β-amyloid (Aβ) precursor protein (APP) is cleaved sequentially by β-site of APP-cleaving enzyme (BACE) and γ-secretase to release the Aβ peptides that accumulate in plaques in Alzheimer's disease (AD). GGA1, a member of the Golgi-localized γ-ear-containing ARF-binding (GGA) protein family, interacts with BACE and influences its subcellular distribution. We now report that overexpression of GGA1 in cells increased the APP C-terminal fragment resulting from β-cleavage but surprisingly reduced Aβ. GGA1 confined APP to the Golgi, in which fluorescence resonance energy transfer analyses suggest that the proteins come into close proximity. GGA1 blunted only APP but not notch intracellular domain release. These results suggest that GGA1 prevented APP β-cleavage products from becoming substrates for γ-secretase. Direct binding of GGA1 to BACE was not required for these effects, but the integrity of the GAT (GGA1 and TOM) domain of GGA1 was. GGA1 may act as a specific spatial switch influencing APP trafficking and processing, so that APP–GGA1 interactions may have pathophysiological relevance in AD.
Keywords: Alzheimer's disease, amyloid precursor protein, Golgi-localized γ-ear-containing ARF binding protein 1, GGA1, BACE, trafficking, amyloid beta
Introduction
Metabolism of the amyloid precursor protein (APP) into amyloid β (Aβ) peptides is a central process in the development of senile plaques in Alzheimer disease (AD) (Selkoe, 2001). β-Site of APP-cleaving enzyme (BACE) is a type I membrane-associated aspartyl protease that cleaves APP (Hussain et al., 1999; Sinha et al., 1999; Vassar et al., 1999; Yan et al., 1999). Cleavage of APP by BACE releases soluble ectodomain of APP (sAPPβ) and generates a membrane bound C-terminal fragment (CTFβ or C99), which is then cleaved by γ-secretase to release Aβ and the APP intracellular domain (AICD). An alternative cleavage beginning with α-secretase is non-amyloidogenic. The precise subcellular locations for APP cleavages have not been established; Aβ appears to be generated in the Golgi, endoplasmic reticulum (ER), or near the cell surface under different experimental conditions (Kinoshita et al., 2003; Tarassishin et al., 2004; Chyung et al., 2005). Only a small fraction of APP produces the secreted Aβ that is deposited in plaques. The amount of Aβ generated thus appears to depend on several factors: (1) the balance between α-cleavage and β-cleavage of APP (Nitsch et al., 1996), (2) the amount and subcellular location of BACE cleavage (Billings et al., 2005; Lee et al., 2005), and (3) the extent to which BACE-processed APP undergoes γ-cleavage to produce Aβ.
In a series of studies examining APP and BACE interactions and trafficking, we demonstrated that APP and BACE interact and cotraffic through the secretory pathway to the cell surface and are cointernalized into early endosomes (Kinoshita et al., 2003). We also demonstrated that BACE trafficking through the secretory pathway depends in part on interactions with the sorting protein Golgi-localized γ-ear-containing ARF-binding protein (GGA), because C-terminal DISLL sequence of BACE binds the VHS (Vps-27, Hrs, and STAM) domain of GGA1 in the Golgi (He et al., 2002; T. Shiba et al., 2004; von Arnim et al., 2004; He et al., 2005); serine phosphorylation of the BACE motif modulates this interaction (von Arnim et al., 2004; He et al., 2005).
In the current study, we test the hypothesis that GGA also impacts APP trafficking and metabolism. GGA family proteins are believed to be important sorting adaptors required for the formation of clathrin-coated vesicles in the trans-Golgi network (TGN) (for review, see Bonifacino, 2004). GGA1 is a modular protein with several distinct domains: In addition to the VHS domain, a GAT (GGA1 and TOM) domain interacts with ADP-ribosylation factors (Arf), ubiquitin (Puertollano and Bonifacino, 2004; Scott et al., 2004; Y. Shiba et al., 2004), TSG101, and Rabaptin-5. The interaction of the GAT domain with Arf is necessary and sufficient for the recruitment of GGA to the TGN. The hinge and GAE (γ-adapting ear) domains of GGA bind clathrin. The GAE domain also interacts with adaptor protein-1, α-synergin, Rabaptin-5, and other potential regulators of vesicle coat assembly. Our current study shows that GGA1, via its GAT domain, acts as a “spatial switch,” confining APP to the Golgi and modulating its access to both BACE and γ-secretase, thereby impacting Aβ generation.
Materials and Methods
Plasmid constructs.
GGA1–myc, Fe65–myc, low-density lipoprotein receptor (LDLR)–green fluorescent protein (GFP), very low-density lipoprotein receptor (VLDLR)–GFP, leukocyte common antigen-related receptor (LARR), nicastrin, notch-Δ- extracellular domain (EC), notch intracellular domain (NICD), APP695–myc, APP770–myc, APP695–GFP, and APP C99–myc, as well as APP695 N-terminally fused to secreted alkaline phosphatase (SEAP–APP) and BACE–V5 constructs have been described previously (Kopan et al., 1996; Kinoshita et al., 2001, 2003; Lichtenthaler et al., 2003; von Arnim et al., 2004; Dunah et al., 2005). APP695 fused to the yeast transcription factor Gal4 (APP695-Gal4), Gal4-dependent luciferase reporter (pG5E1B–luc), and β-galactosidase (β-gal) reporter were gifts from Dr. T. Südhof (University of Texas Southwestern Medical Center, Dallas, TX) (Cao and Sudhof, 2001). To create deletion constructs, GGA1 ΔVHS (base pairs 467–1939), GGA1 ΔVHS/GAT (base pairs 973–1939), GGA1 ΔGAT (base pairs 1–467Δ973–1939), and GGA1 ΔGAE (base pairs 35–1357; GI 7021538) were put into pcDNA3.1–myc (Invitrogen, Carlsbad, CA) after amplification by PCR. The dileucine mutant of BACE was generated by substitution of Leu (499/500) to Ala. Authenticity of the constructs was confirmed by DNA sequencing. Constructs are summarized in Figure 1.
Figure 1.
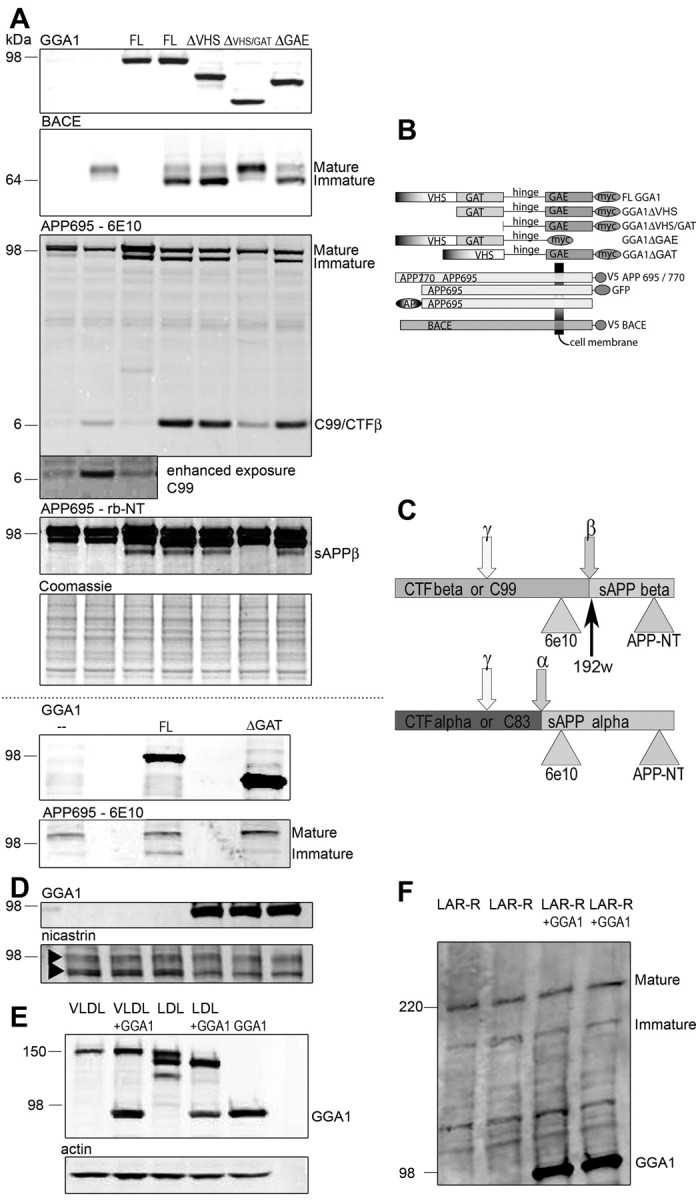
A, APP and BACE maturation during GGA1 coexpression. Cotransfection of APP695 (lanes 1–7) in N2a cells with BACE (lanes 2, 4–7), GGA1 [full length (FL); lanes 3, 4] or GGA1 deletion mutants (ΔVHS, lane 5; ΔVHS/GAT, lane 6; ΔGAE, lane 7; constructs are described in B) was performed as indicated. After lysis, cell samples were loaded onto a 10–20% Tris–glycine gel under reducing and denaturing conditions and were analyzed by immunoblotting with anti-myc Ab to detect GGA1 (row 1), mAb anti-V5 to detect BACE (row 2), and monoclonal APP Ab 6E10 (row 3; enhanced exposure in inlay) to detect maturation of APP and CTFβ/sAPPα or rabbit anti-APP N terminus Ab (row 4) to detect intracellular sAPPα and sAPPβ as indicated in C. C, Recognition of the sAPP band at ∼80 kDa only by rabbit anti-APP N terminal Ab (rb-NT) but not 6E10 indicates that this is sAPPβ. During overexposure of the CTFβ band, the increase of CTFβ during GGA1 transfection (lanes 1, 3, without BACE cotransfection) can be seen. Equal protein load is shown by Coomassie blue staining (A, row 5). In rows 6 and 7, cotransfection of APP695 (lanes 1–3) in N2a cells with BACE (lanes 1–3) and GGA1 ΔGAT was performed as indicated. D, Nicastrin maturation during GGA1 coexpression. Cotransfection of nicastrin in N2a cells GGA1 (lanes 3–6) was performed as indicated. Samples were loaded onto a 10–20% Tris–glycine gel under reducing and denaturing conditions and were analyzed by immunoblotting with anti-myc Ab to detect GGA1 (top row) or anti-nicastrin Ab to detect nicastrin (bottom row). No influence of GGA1 coexpression during nicastrin maturation is shown. E, VLDL receptor (VLDL) and LDL receptor (LDL) maturation during GGA1 coexpression. Cotransfection of VLDL (lanes 1, 2) or LDL (lanes 3, 4) in N2a cells with GGA1 (lanes 2, 4) or GGA1 alone (lane 5) was performed as indicated. Samples were loaded onto a 4–12% Tris–glycine gel under reducing and denaturing conditions and were analyzed by immunoblotting with anti-myc Ab to detect GGA1 or anti-GFP Ab to detect VLDL and LDL (as indicated). Equal protein load is shown by actin staining (F, bottom row). No change in VLDLR glycosylation can be detected during GGA1 coexpression, but a shift from mature to immature forms of LDLR can be observed during GGA1 coexpression. F, LAR receptor (LAR-R) maturation during GGA1 coexpression. Cotransfection of LAR-R (lanes 1–4) in N2a cells with GGA1 (lanes 3, 4) was performed as indicated. Samples were loaded onto a 7% Tris–glycine gel under reducing and denaturing conditions and were analyzed by immunoblotting with anti-myc Ab to detect GGA1 or anti-LARR Ab to detect LARR (as indicated). No change of LARR maturation is observed during GGA1 coexpression.
Cell culture conditions and transient transfection.
Mouse neuroblastoma N2a cells were cultured in OPTI-MEM I with 5% FBS and HEK293 cells in DMEM and 10% FBS. Transient transfection of the cells was performed using FuGene 6 (Roche, Indianapolis, IN).
Immunocytochemistry and antibodies.
Cells were fixed and immunostained as described previously (von Arnim et al., 2004). Antibodies (Abs) to the Golgi organelle marker GM130 and the early endosome antigen 1 (EEA1) marker were from BD Biosciences Transduction Laboratories (San Diego, CA). Rabbit anti-myc was from Upstate Biotechnology (Lake Placid, NY), anti-V5 monoclonal Ab (mAb) was from Sigma (St. Louis, MO), mAb 9E10 anti-myc was from Invitrogen, and mAb 6E10 against APP (see Fig. 1C) was obtained from Signet (Dedham, MA). mAbs against the APP C terminus (13G8) and APP N terminus (8E5) as well as a specific polyclonal Ab against the C terminus of sAPPβ (192wt) (see Fig. 1C) were gifts from Elan Pharmaceuticals (Gainesville, GA); rabbit anti-APP N terminus and actin were from Sigma. A specific rabbit anti-human GGA1 N terminus was from Abgent (San Diego, CA). GFP Abs were from Abcam (Cambridge, MA). Rabbit polyclonal antibody against LARR was a gift from A. Dunah (Harvard Medical School, Charlestown, MA). mAb against nicastrin was obtained from Chemicon (Temecula, CA). Secondary Abs were labeled with Alexa488 (Invitrogen) and cyanine 3 (Cy3) and Cy5 (Jackson ImmunoResearch, West Grove, PA). Immunostained cells were observed with the appropriate filters on a Bio-Rad (Hercules, CA) 1024 confocal three-channel microscope.
Fluorescence resonance energy transfer measurements using fluorescence lifetime imaging microscopy.
We applied a validated fluorescence lifetime imaging microscopy (FLIM) technique that can quantitate protein–protein interactions (Berezovska et al., 2003). Briefly, donor fluorophores (Alexa488) were excited on a two-photon system at 800 nm by a ∼100 fs pulse every ∼12.5 ns from a mode-locked titanium—sapphire laser (Spectra Physics, San Jose, CA). Images were acquired using a Bio-Rad Radiance 2000 multiphoton microscope. A high-speed Hamamatsu (Ichinocho, Japan) MCP detector (MCP5900) and hardware/software (SPC830, SpcImage2.60) from Becker and Hickl (Berlin, Germany) allowed measurement of lifetimes on a pixel-by-pixel basis. Donor fluorophore lifetimes were fit to two-exponential decay curves. The “non-FRETing” (fluorescence resonance energy transfer) population was represented as t2. The “FRETing” population (t1) is represented in the figures. Statistical analysis was by ANOVA and Fisher's PLSD post hoc test.
APP ectodomain shedding assay.
HEK293 cells were transfected in triplicate with β-gal, SEAP–APP, and constructs as indicated. Media was changed 24 h later and collected after another 24 h. SEAP activity in the conditioned media was measured in triplicate by chemiluminescent assay (Roche) and normalized to β-gal (Promega, Madison, WI). Transfection was confirmed by immunoblotting.
Aβ ELISA.
Media from transfected HEK293 cells was changed 48 h after transfection and collected after another 24 h. Cells were harvested in 70% formic acid. The samples were neutralized with Tris base and assayed for formic acid-extractable Aβ40 by a well characterized sandwich ELISA, sensitive to 1 pm Aβ (Fukumoto et al., 2002; Irizarry et al., 2004), using BNT77 (anti-Aβ11–28) as the capture Ab and horseradish peroxidase-linked BA27 as detection Abs (Takeda Chemical Company, Osaka, Japan). Purified Aβ40 (Bachem, King of Prussia, PA) served as standard.
Western blotting.
N2a cells were transfected as indicated and lysed the next day in 1% Triton X-100, and proteins were separated on 7%, 4–20% Tris–glycine, or 10% NuPAGE Novex Bis–Tris PAGE (Novex, San Diego, CA) under denaturing and reducing conditions. Proteins transferred to polyvinylidene difluoride (PVDF) membranes were detected on a LI-COR (Lincoln, NE) Odyssey IR detection system.
The Massachusetts Alzheimer Disease Research Center (MADRC) Brain Bank provided temporal cortex from eight control (77.6 ± 6.1 years old) and eight AD (78.1 ± 8.3 years old) subjects. All control brains were neuropathologically normal. Brain tissue was homogenized in 1:10 vol (w/v) of lysis buffer (10 mm NaF, 1 mm NaVO4, 2 mm EGTA, 0.2% SDS, 1× PBS, and Roche Complete protease inhibitor). Protein (50 μg) was loaded onto 4–20% Novex Tris–glycine gels and transferred to PVDF membranes. GGA1 and actin were visualized by chemiluminescence. Total protein was quantified after staining with Coomassie blue using the Odyssey IR system.
Fe65-dependent APP transactivation assay.
Transactivation assays were performed in HEK293 cells as described previously (Cao and Sudhof, 2001). Cells were cotransfected in triplicate with (1) APP695–Gal4, (2) Fe65–myc, (3) PG5E1B–luc, (4) β-gal, and (5) GGA1–myc or empty vector. After 48 h, luciferase activity was measured in triplicate and normalized to β-gal. Transfection was confirmed by immunoblotting.
Quantitative PCR.
To assess GGA mRNA levels, quantitative, real-time PCR (qPCR) was performed as described previously (Cantuti-Castelvetri et al., 2005). The MADRC Brain Bank provided temporal cortex from 20 control (82.4 ± 8.3 years old) and 35 AD (79.9 ± 7.9 years old) subjects. The mean postmortem interval was 14.4 ± 5.6 h in controls and 12.3 ± 6.2 h in AD subjects. Briefly, 30–40 mg of temporal cortex was homogenized in Trizol, and total RNA was prepared according to the instructions of the manufacturer (Invitrogen). All samples were controlled for integrity of 18S and 28S rRNA by microcapillary electrophoresis (RNA 6000 Nano Assay; Agilent Technologies, Palo Alto, CA). Reverse transcription was performed (Superscript II; Invitrogen).
Primers against GGA, the house-keeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH), and neuron-specific enolase 2 (ENO2) as a marker for neuronal cells and glial fibrillary acidic protein (GFAP) as a marker for astrocytes were developed. The sequences of the primers used were as follows: for GGA, 5′- CATCAAACCCAGCAACATC-3′ and 5′-GCAGCTTCACCTTCATAACC-3′; for GAPDH, 5′-GGTCTCCTCTGACTTCAACA-3′ and 5′-GTGAGGGTCTCTC-TCTTCCT-3′; for ENO2, 5′-CGCCACTACCACCGTCTG-3′ and 5′-TAGAGGCTCCACTGGGCACTG-3′; and for GFAP, 5′- GATCAACTCACCGCCAACAGC-3′ and 5′-CTCCTCCTCCAGCGACTCAATCT-3′. qPCR was performed on the iCycler (Bio-Rad) according to the SYBR green method (Applied Biosystems, Foster City, CA). For each reaction, 3 ng of cDNA was used in a 25 μl reaction. qPCR amplification for all of the targets proceeded to preincubate at 94°C for 6 min, followed by 50 cycles at 94°C for 30 s, at the primer-specific annealing temperature (at 60°C for GGA, at 55°C for GAPDH, at 62°C for ENO2, and at 58°C for GFAP) for 30 s and at 72°C for 45 s. The specificity of each candidate PCR amplicon was evaluated by direct sequencing. All primer pairs amplified a single peak of fluorescence by melt curve analysis. The transcripts of interest were quantified using a modification of the difference in cycle threshold method (Fink et al., 1998). Statistical analyses between AD and control brains were compared by Mann–Whitney U test because mRNA and protein levels studied here were not normally distributed. qPCR results can be expressed in terms of absolute number of molecules per microgram of RNA starting material or normalized to mRNA levels of a housekeeping gene (e.g., GAPDH) to correct for systematic differences in sample preparation or storage conditions. Because GAPDH levels tend to be lower in AD brains than controls (AD vs controls, 245,196.3 ± 161,923.4 vs. 599,132.2 ± 456,922.5 molecules per microgram of RNA; p = 0.0003), the latter correction might influence apparent levels of comparable mRNAs. Therefore, both approaches to normalizing the data are presented. Statistical significance was set at p < 0.05.
Results
GGA1 affects APP and BACE maturation and glycosylation
Given the role of GGA1 in trafficking, we asked whether GGA1 affects the maturation of BACE and APP. BACE was present as two bands that correspond to immature and fully modified mature protein (Capell et al., 2000). Cotransfection of GGA1 with BACE shifted the equilibrium from mainly mature to mainly immature. To determine whether this effect was attributable to a direct interaction, we used GGA1 deletion mutants (Fig. 1A). Surprisingly, eliminating the VHS domain of GGA1 and consequently preventing its direct interaction with BACE did not prevent the effect of GGA1 on BACE glycosylation. When the mutant lacking the GAT domain in addition to the VHS domain (ΔVHS/GAT domain) was cotransfected, however, the ratio of mature to immature BACE returned to normal (Fig. 1A, row 2, lane 6).
GGA1 also influenced APP maturation. In transfected cells, APP is present in multiple bands representing immature and fully glycosylated mature species (Caporaso et al., 1992). As with BACE, after coexpression of GGA1 with APP695, we observed an increase in immature APP dependent on the integrity of the GAT domain of GGA1 [Fig. 1A, row 3, lane 6 (ΔVHS/GAT) and row 7, lane 3 (ΔGAT)]. This demonstrates that the GAT domain of GGA1 affects BACE and APP maturation independent of its VHS domain-dependent interaction with BACE.
To test whether this was a general effect of GGA1 during trafficking, we evaluated the effect of GGA on the maturation of four other proteins. We coexpressed two secretory glycoproteins closely related to Alzheimer's disease because they are known apolipoprotein E receptors, VLDLR and LDLR (Herz, 2001). For VLDLR, we could not detect an unglycosylated band (Fig. 1E). However during cotransfection of LDLR with GGA1, we were able to detect an immature band analogous to those seen with APP and BACE (Fig. 1E). However, the overexpression of GGA1 did not change the ratio of immature to mature nicastrin (Fig. 1D) or leukocyte common antigen-related receptor (Fig. 1F). In addition, no effect on endogenous presenilin (PS) was observed because of GGA1 overexpression (data not shown). Nicastrin is a single transmembrane domain protein that is glycosylated. Nicastrin and presenilin are both obligate members of the γ-secretase complex. LARR is a secretory glycoprotein that is implicated in axon guidance and not known to interfere with APP trafficking (Dunah et al., 2005). Therefore, GGA1 overexpression affects APP, BACE, and LDLR glycosylation (Fig. 1A,E) but not all proteins that traffic through the secretory pathway. To examine whether this change in maturation of some proteins reflects a nonspecific effect on cell metabolism by GGA1 overexpression, we also measured toxicity during GGA1 overexpression by a lactate dehydrogenase assay. No GGA1-dependent toxicity was observed (data not shown).
APP shifts from cell surface to colocalize with GGA1
Because GGA1 traffics with and alters the localization of BACE by restricting it to the TGN (von Arnim et al., 2004; He et al., 2005), we asked whether GGA1 might affect APP localization as well. We cotransfected N2a cells with APP770 and GGA1 and observed the colocalization of these two proteins with Golgi and endosome markers (Fig. 2). Golgi formation was not disrupted under these conditions, as shown by GM130 staining. During GGA1 overexpression, APP shifted from compartments distal to the TGN (Fig. 2A) and showed complete colocalization with GGA1 and GM130. Although there is normally some colocalization of APP with EEA1, this was nearly completely abolished during cotransfection with GGA1 (Fig. 2B). To confirm that GGA1 reduced APP presentation on the cell surface, we transfected with APP695–GFP and performed cell surface staining with an antibody against the APP N terminus (Fig. 2C). To quantify the ratio of cell-surface to total APP, we calculated ratios of surface staining to total GFP intensity (Table 1), showing ∼35% decrease of cell surface APP (p < 0.01) on GGA1 cotransfection. Thus, GGA1 expression dramatically alters APP subcellular localization.
Figure 2.
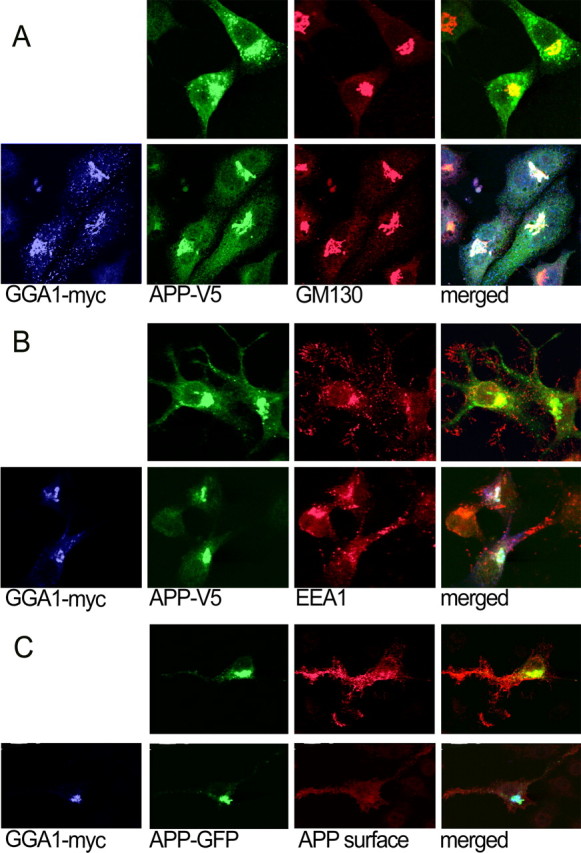
Localization of APP during cotransfection with GGA1. A, N2a cells cotransfected with APP770–V5 and GGA1–myc or empty vector were immunostained for APP (Alexa488; green), GGA1 (Cy5; blue), and the Golgi marker GM130 (Cy3; red). B, The same transfection was stained for the endosomal marker EEA1 (Cy3). C, To assess cell-surface localization, APP695–GFP and GGA1–myc or empty vector were cotransfected and then immunostained with an Ab to the APP ectodomain on ice without permeabilization (Cy3). Cells were then fixed, permeabilized, and stained for GGA1 (Cy5).
Table 1.
Cell surface staining was performed on cells transfected with APP695-GFP and empty vector or GGA1-myc
| APP695–GFP | APP695–GFP + GGA1–myc | |
|---|---|---|
| Ratio (cell surface/GFP intensity) | 1.32 ± 0.44 | 0.88 ± 0.06 |
The ratio of intensity of cell surface staining to intensity of GFP is shown (p < 0.01)
FLIM reveals close proximity of GGA1 and APP
Because increased GGA1 expression altered APP localization and trafficking independent of its direct interaction with BACE, we hypothesized that GGA1 might interact with APP itself. We therefore used a technique to probe GGA1/APP proximity that allowed us to confirm this interaction and identify where in the cell it occurred. FLIM is a morphology-based FRET technique that can reveal protein–protein proximity (<10 nm) in intact cells. Fluorescence lifetime of a donor fluorophore is shortened if a FRET acceptor is in proximity of the donor to an extent that often indicates direct interaction but can also occur within multiprotein complexes. The degree of lifetime shortening is concentration independent and can be displayed with high spatial resolution in a color-coded image. Therefore, lifetimes of fluorophores involving even a diminutive subpopulation of a protein in a unique subcellular locale can be identified. We measured changes in the lifetime of the donor fluorophore (Alexa488) as described previously (von Arnim et al., 2004). GGA1 and APP were cotransfected in N2a cells. If only GGA1 was immunostained as a negative control, the lifetime of Alexa488 was ∼2100 ps (Table 2). The lifetime of Alexa488 attached to the C terminus of GGA1 became significantly shorter when the coexpressed APP770 was labeled C terminally with the FRET acceptor Cy3, indicating FRET between the two fluorophores (Table 2). Close proximity between the two proteins was confirmed when the acceptor and donor fluorophores were exchanged and was not disrupted if APP695, an isoform lacking the Kunitz protease inhibitor domain, was used. Plotting the mean lifetimes calculated for each pixel shows that Alexa488 exhibits a single population of lifetimes in the absence of an acceptor fluorophore, but the close vicinity of APP with GGA1, and consequently their respective fluorophores, creates a second population with a shorter lifetime (Fig. 3A,B). By creating an image that assigns different colors to pixels belonging to each of these two populations, we were able to show that close proximity of APP with GGA1 occurs only in juxtanuclear compartments (Fig. 3). To confirm the idea that FRET observed in the APP/GGA1 FLIM assay was attributable to close vicinity (<10 nm), we performed an additional control. In this experiment, APP was labeled by Cy3 attached to 8E5, an antibody that binds the APP extracellular domain. Although there was still striking colocalization with Alexa488-labeled GGA1 on the opposite side of the membrane, the two fluorophores were too distant from one another to support FRET, and Alexa488 underwent no significant lifetime change (Table 2, Fig. 3C). This experiment demonstrates the specificity of this FLIM-based method. These results show that APP and GGA are in very close proximity but may not be directly interacting because repeated attempts to coimmunoprecipitate APP and GGA did not succeed.
Table 2.
If there is no interaction, donor fluorophore lifetimes are approximately ∼2000–2150 ps, as seen in the absence of the acceptor fluorophore
| Donor | Acceptor | Lifetime (ps) (mean ± SD) | n (cells) | Significance (vs control) |
|---|---|---|---|---|
| GGA1–myc (Alexa488) | None | 2133 ± 45 | 21 | |
| GGA1–myc (Alexa488) | APP695–V5 (Cy3) | 1947 ± 103 | 26 | p < 0.001 |
| GGA1–myc (Alexa488) | APP770–V5 (Cy3, C terminus) | 1951 ± 83 | 20 | p < 0.001 |
| GGA1–myc (Alexa488) | APP770–V5 (Cy3, N terminus) | 2088 ± 42 | 20 | NS |
| APP770–V5 (Alexa488) | None | 2030 ± 57 | 13 | |
| APP770–V5 (Alexa488) | GGA1–myc (Cy3) | 1799 ± 194 | 10 | p < 0.001 |
Statistically shorter lifetimes result from FRET between fluorophores bound to the GGA1 and APP C termini. However, if the fluorophore is located at the N terminus of APP, donor lifetime does not change, despite complete colocalization at the light level because the distance across the membrane (the N terminus compared with the C terminus) is too far to support a FRET interaction. FRET still occurs if the fluorophores are exchanged.
Figure 3.

FLIM analysis of APP and GGA1 proximity within cells. N2a cells were cotransfected with GGA1–myc and APP695–V5 (A) or APP770-V5 (B, C) [labeled by Cy3 on the C terminus (A, B) or on the N terminus (C)]. The confocal images show the previously observed immunostaining pattern. The color-coded FLIM image shows the lifetimes (in picoseconds) of Alexa488. The shorter lifetimes reflecting proximity between GGA1 and APP appear only in juxtanuclear compartments. C, The negative control, in which APP has been labeled on its N terminus (across the membrane), exhibits a single population of lifetimes despite complete overlay at the light microscopic level, reflecting the sensitivity of this method.
GGA1 reduces both sAPPα and sAPPβ secretion
Together with the changes in intracellular localization and maturation of APP attributable to GGA1, our FLIM data suggest a functional interaction between the proteins. We first tested the impact of GGA1 on secretion of sAPP using Western blot analysis. Cells transfected with APP and either empty vector or GGA1 were tested for sAPP secretion in the media by the sAPPβ-specific Ab 192wt and for sAPPα by 6E10 Ab (Fig. 1C). The analysis revealed reduced sAPPα and sAPPβ secretion during GGA1 overexpression (Fig. 4B). To investigate the influence of GGA1 on β-cleavage in more detail, we also looked for sAPP secretion when BACE is overexpressed. Remarkably, when BACE is cotransfected, GGA1 mainly reduces sAPPβ secretion, whereas sAPPα secretion is hardly detectable. Together, these results suggest that GGA1 blocks both APP α-secretion and β-secretion.
Figure 4.
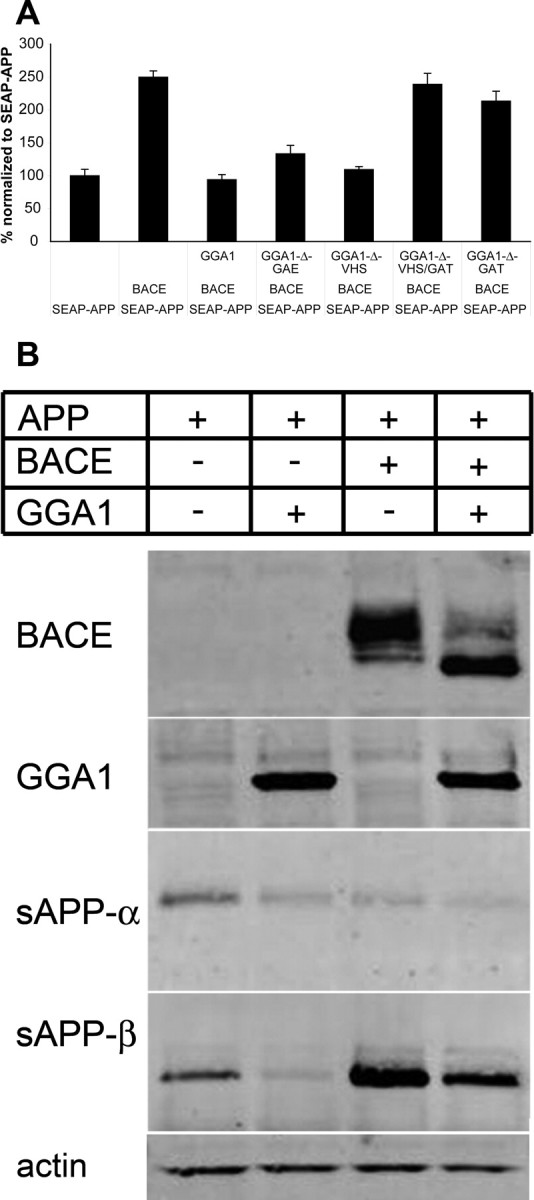
APP ectodomain shedding. A, HEK293 cells were transfected with SEAP–APP, β-gal, WT-GGA1, or GGA1 deletion mutants and empty vector as indicated. Shown is percentage ± SD of baseline alkaline phosphatase activity normalized to β-gal activity. Results from one of at least three independent assays are shown. B, HEK293 cells were transfected with APP695–myc, GGA1–myc, and BACE–V5 as indicated. Samples were loaded onto a 10–20% Tris–glycine gel under reducing and denaturing conditions. Immunoblotting was performed with anti-V5 (BACE), anti-myc (GGA1), mAb 6E10 (sAPP-α), 192wt (sAPP-β), and mAb anti-actin (actin). A reduction of both sAPPα and sAPPβ was observed during GGA1 cotransfection.
GGA1 influences on APP cleavage are GAT domain dependent
To determine which domain of GGA1 was inhibiting release of sAPP, we used an SEAP–APP assay (Lichtenthaler et al., 2003) After cotransfection of HEK293 cells with APP, BACE, GGA1, or GGA1 deletion mutants (Fig. 4), SEAP activity was measured. Eliminating the VHS domain responsible for the interaction of GGA1 with BACE did not impede the ability of the protein to reduce SEAP activity; however, after additional deletion of the GAT domain (ΔVHS/GAT or ΔGAT), the decrease in SEAP activity was reversed (Fig. 4A). We were therefore able to show that overexpression of GGA1 leads to less secretion of the BACE-cleaved ectodomain of APP independent of direct GGA1/BACE interaction but again dependent on the GAT domain of GGA1.
APP β-cleavage products accumulate intracellularly with GGA1
To test whether the GGA1-induced decrease in sAPPβ secretion resulted from a reduction in β-cleavage of APP, we cotransfected APP695, BACE, and GGA1 vectors into N2a cells. Western blots were probed with an antibody (6E10) that recognizes full-length APP, sAPPα, and CTFβ. As shown previously, BACE overexpression increased CTFβ. Surprisingly, coexpression of GGA1 resulted in an even larger increase of CTFβ. GGA1 alone, in the absence of transfected BACE, also increased the amount of CTFβ. Substituting the GGA1 ΔVHS/GAT or ΔGAT domain deletion construct, but not the ΔVHS or ΔGAE variants, for wild-type (WT)-GGA1 reversed this. An N-terminal antibody against APP revealed an additional band just below the immature APP (Fig. 1A, row 4, lanes 3–7). The inability of 6E10 to recognize this band allowed its identification as intracellular sAPPβ (Fig. 1A, row 3, lanes 3–7, C). Overexpression of GGA1 with an intact GAT domain significantly increased the intensity of this band. These data indicate that GGA1 does not inhibit proteolysis of APP by BACE but instead prevents secretion of the cleavage products and leads to their accumulation within the cell.
GGA1 lowers global Aβ40 production
We next investigated the effect of GGA1 on the production and secretion of Aβ40. Cotransfection of HEK293 cells with APP695 and BACE significantly enhanced Aβ40 secretion (Fig. 5A). In the absence of overexpressed BACE and GGA1 transfection, we did not observe a change in Aβ40 secretion, which was already at the lowest level detectable, but GGA1 did inhibit the increase in Aβ40 in the media induced by BACE overexpression. Once again, this effect was not dependent on the integrity of the VHS domain but was dependent on the GAT domain. In line with the observation that secreted Aβ40 was reduced when BACE is overexpressed, intracellular Aβ40 levels were also decreased under these conditions (Fig. 5B). This supports a hypothesis wherein APP remains in an intracellular compartment with BACE, with the resulting CTFβ prevented by GGA1 from being transported to another cell compartment in which γ-secretase could convert it to Aβ.
Figure 5.

GGA1 inhibits Aβ40 secretion and production. HEK293 cells were transfected in triplicate with APP, BACE–V5, and WT-GGA1 or GGA1 deletion mutants or empty vector as indicated. The levels of Aβ40 secreted into the media (A) and accumulated within the cell (B) were determined by ELISA in duplicate (n = 6). The mean Aβ concentration is shown as mean ± SD shown as percentage of Aβ levels in cells transfected with APP alone. Results from at least three independent assays are shown. C, GGA1 inhibits γ-secretase-dependent transactivation. HEK293 cells were transfected in triplicate with (1) APP695–Gal4, (2) Fe65–myc, (3) pG5E1B–luc, (4) β-gal, and (5) GGA1–myc or empty vector. The normalized average ± SD luciferase activity of three measurements of each of the three transfections (n = 9) is shown. Results from one of at least three independent assays are shown. D, GGA1 does not affect γ-secretase-dependent notch cleavage. HEK293 cells were transfected with (1) notch-Δ-EC–myc, (2) GGA1–myc with notch-Δ-EC—myc, (3) notch-Δ-EC–myc, and (4) NICD–myc. Samples were loaded onto a 4–12% Tris–glycine gel under reducing and denaturing conditions. Immunoblotting with 9E10 anti-myc Ab detected notch-Δ-EC and NICD during overexpression of notch-Δ-EC–myc (lane 1) as indicated. Coexpression of GGA1–myc with notch-Δ-EC–myc did not alter NICD generation by γ-secretase (lane 2). NICD generation during overexpression of notch-Δ-EC–myc was blunted by the γ-secretase inhibitor N-[N-(3,5-difluorophenacetyl)-l-alanyl]-S-phenylglycine-t-butylester (lane 3). NICD–myc as control for NICD size (lane 4).
GGA1 decreases access of APP to γ-secretase
γ-Secretase cleaves APP in the inner leaflet of the cell membrane to free the AICD (Cao and Sudhof, 2001). We postulated that, if GGA1 inhibits access of APP or CTFβ recycling to a compartment that contains γ-secretase, we may observe reduced γ-secretase-mediated AICD generation after transfection with GGA1. In HEK293 cells cotransfected with a C-terminal APP695–Gal4 fusion and a Gal4-dependent luciferase reporter plasmid, AICD production correlates with the amount of luciferase activity observed. Fe65 cotransfection, as expected, increased transcription of the luciferase reporter (Fig. 5C). GGA1 attenuated this effect. To further confirm that this is attributable to APP trafficking and not attributable to a decrease in γ-secretase activity, e.g., by altered trafficking of the γ-secretase components, we examined cleavage of another γ-substrate, notch. During cotransfection of notch-Δ-EC with GGA1, no change in generation of NICD was observed (Fig. 5D). In addition, we could not see any impact of overexpressed GGA1 on PS1 endoproteolytical processing to the PS1 N- and C-terminal fragments (data not shown). We interpret this result to suggest that GGA1 inhibits access of the γ-substrate CTFβ to γ-secretase, because there appears to be an alteration of assays measuring release of both Aβ and release of AICD.
GGA1 is present in human brain
Finally, to test the plausibility that GGA1 interaction with APP represents a physiologically relevant interaction in the context of AD, we confirmed that GGA1 is present in human brain tissue (Table 3). A GGA1-specific antibody showed several immunoreactive bands and recognized two bands centered at 70 kDa in brain samples (Ghosh and Kornfeld, 2003) (supplemental data A, available at www.jneurosci.org as supplemental material), possessing slightly greater mobility than myc-tagged GGA1 as expected (data not shown). To confirm that these bands were GGA1, we performed qPCR reactions that showed that GGA1 mRNA is present in human brain tissue (supplemental data B, available at www.jneurosci.org as supplemental material). When normalizing GGA1 to GAPDH as a marker of RNA integrity, we saw a significant decrease of GGA1 (p = 0.026) in AD. Additional correlation of GGA1 with the neuronal marker ENO2 and GFAP as a marker for astrocytes indicated that GGA1 reduction in AD brain can be mainly attributed to neuronal loss in AD.
Table 3.
mRNA levels of GGA1by qPCR in AD and control brains
| AD | Controls | p valuea | |
|---|---|---|---|
| mRNA levels | |||
| GAPDH, number of molecules/μg RNA | 24,5196 ± 161,923 | 599,132 ± 456,922 | 0.0003 |
| GGA, number of molecules/μg RNA | 31,638 ± 10,580 | 46,257 ± 30,831 | 0.205 |
| Standardized by GAPDH | 0.196 ± 0.143 | 0.116 ± 0.086 | 0.026 |
| ENO2, number of molecules/μg RNA | 49,603 ± 111,468 | 156,232 ± 175,201 | <0.0001 |
| Standardized by GAPDH | 0.147 ± 0.171 | 0.263 ± 0.171 | 0.001 |
| GFAP, number of molecules/μg RNA | 2,402,811 ± 2,300,672 | 1,244,964 ± 1,048,990 | 0.011 |
| Standardized by GAPDH | 12.50 ± 11.12 | 2.77 ± 2.66 | <0.0001 |
Values are mean ± SD
aMann–Whitney U test.
Discussion
Previous studies identifying GGA1 as a BACE-interacting protein suggested it might be relevant in AD. Because APP and BACE cotraffic, we now examined the functional significance of GGA1 on APP. We found that GGA overexpression leads to an increase in coexpressed, immature BACE and APP species, possibly by confining both proteins to the Golgi and preventing modifications that occur after export from this compartment. This does not seem to be an exclusive effect because other proteins such as LDLR undergo altered glycosylation as well. However, it is not a general effect because nicastrin, VLDLR, and LARR maturation are not affected. BACE (Capell et al., 2000) and APP (Caporaso et al., 1992) are subject to glycosylation, before APP is then cleaved in the secretory pathway in cultured cells (Tomita et al., 1998) or after it has entered into the fast axonal transport pathway in neurons (Koo et al., 1990; Amaratunga and Fine, 1995; Buxbaum et al., 1998; Kamal et al., 2001). By altering BACE and APP trafficking and maturation, GGA1 might therefore be expected to have implications for APP processing. Interestingly, inhibition of BACE maturation by GGA1 did not affect the ability of BACE to cleave APP. Despite incomplete BACE maturation with GGA1, we saw increases in the BACE cleavage products sAPPβ and CTFβ intracellularly. This is consistent with several reports showing that BACE in the TGN, although immature, is fully active (Haass et al., 1995; Thinakaran et al., 1996). Although cleavage of APP by BACE activity can occur in endosomes consistent with its acidic pH optimum (Kinoshita et al., 2003), we found here that restricting APP to the Golgi, which is also a rather acidic compartment, enhanced β-cleavage.
Importantly, trafficking of APP to the cell surface was impeded in our system, because sAPPα production on the cell surface was reduced and trafficking of sAPPβ to the cell surface for secretion was reduced. If we increased the amount of β-cleavage products of APP within the cell, we saw a decrease in the γ-secretase cleavage products Aβ and AICD attributable to GGA1 overexpression despite fully active γ-secretase as indicated by unchanged notch cleavage. Because our manipulation actually increased the total amount of the γ-substrate CTFβ within cells, these data indicate that CTFβ is being prevented from reaching compartments in which γ-secretase is most active. These results are reminiscent of recent data (Lee et al., 2005) showing that high levels of BACE expression lead to mistrafficking of CTFβ to the Golgi and diminished Aβ cleavage. Although the subcellular location of γ-cleavage has not been established definitively, some studies have suggested that it occurs in the ER, a pre-Golgi compartment, in which most presenilin is localized (De Strooper et al., 1997). Our data, under these experimental conditions, instead support other findings that γ-cleavage occurs primarily post-Golgi, perhaps on the way to the cell surface (Khvotchev and Sudhof, 2004; Tarassishin et al., 2004) or after endocytosis (Koo and Squazzo, 1994; Refolo et al., 1995; Berezovska et al., 2003). Our observation that GGA1 both reduces APP on the cell surface and prevents APP processing to sAPPα and sAPPβ particularly bolsters the theory that γ-cleavage of APP, like cleavage of notch, occurs on or near the cell surface (Berezovska et al., 2003; Ehehalt et al., 2003; Ramdya et al., 2003; Chyung et al., 2005; Kaether et al., 2006). We propose a model in which GGA1 acts as a sorting adaptor that regulates shuttling of APP between the Golgi and endosomes/lysosomes, consequently shifting APP away from peripheral compartments and therefore to compartments less favorable for α- and γ-cleavage. Of note, several studies suggest that CTFβ is elevated in AD brain (Yang et al., 2003); this observation has been somewhat paradoxical because β cleavage is viewed as the rate-limiting step in APP metabolism, and CTFβ accumulates in experimental systems primarily after inhibition of γ-secretase function.
Neither BACE phosphorylation (Walter et al., 2001) nor mutation of the BACE dileucine motif (Pastorino et al., 2002), modifications that affect the BACE/GGA1 interaction, affect Aβ production and BACE-dependent APP shedding. It is not surprising, therefore, that mutating the domain of GGA1 that interacts with BACE, the VHS domain, did not abolish the effect of GGA1 on BACE or APP maturation, processing, and transport under these conditions. However, the effects we observed depended on the GAT domain of GGA1. Rabaptin-5 represents a candidate to mediate the impact of GGA1 on APP because it interacts with the GAT domain, and upregulation of its effector, Rab5, leads to increased Aβ (Grbovic et al., 2003).
Endogenous GGA1 localizes predominantly to the TGN. APP resides predominantly in Golgi and endosomal compartments but can also be observed on the cell surface. Coexpression with GGA1 confines APP to the Golgi, diminishing its presence in endosomes and at the cell surface. As a general rule, however, colocalization is insufficient to prove actual protein–protein interactions. To detect proximity of an order indicating formation of APP/GGA complexes, we measured FRET by means of FLIM (Berezovska et al., 2003). Our FLIM data suggest that APP and GGA1 come into very close proximity primarily in the TGN. In indirect immunofluorescence (i.e., accounting for the size of the ∼7 nm antibody molecules), the greatest distance between two epitopes that have detectable FRET must be <30–40 nm (Chin et al., 2000; Siegel et al., 2000; Cho et al., 2001).
The quantitative change in lifetime is not as strong as we observed in some cases of direct protein–protein interaction and leaves open the possibility that this is an indirect interaction mediated by an adaptor protein. For instance, we observed FRET between the C termini of APP and LDL-related protein, although they are linked by the scaffolding protein Fe65 (Kinoshita et al., 2001). Additional research should examine the question of whether the close proximity of GGA1 and APP reflects direct or indirect interaction; other adapter proteins, including the munc 18 interacting proteins such as X11, can bind Arfs directly and, via interactions with proteins such as GGAs, have been implicated in trafficking of APP to vesicles and the cell surface (Mueller et al., 2000; Hill et al., 2003).
Our findings have identified GGA1 not only as a BACE-interacting protein but also as a sorting protein that affects APP and influences its trafficking and access to γ proteolytic processing. Thus, GGA1 acts as a spatial switch modulating to which compartments APP can traffic. It further acts as a switch for substrate specificity of γ-secretase because notch cleavage is, in contrast to APP γ-cleavage, not affected. It has been shown recently that other proteins involved in protein transport, including syntaxin1a (Khvotchev and Sudhof, 2004) and LR11/sorLA (Scherzer et al., 2004; Andersen et al., 2005; Spoelgen et al., 2006), can impact APP processing. Recent data from BACE transgenic mice also support the idea that the location of BACE, even more so than the amount of BACE in a cell, influences Aβ generation (Lee et al., 2005). Although our initial experiments were based on the observation that BACE and APP cotrafficked and that GGA influenced BACE trafficking, surprisingly in the experimental conditions applied, we observe a strong additional influence of GGA1 expression during APP processing independent of the GGA1/BACE interaction itself. Thus, altered expression or function of GGA1, perhaps in association with changes in proteins important in APP sorting and trafficking such as sorLA/LR11, X11, and Fe65, might potentially modify APP trafficking and ultimately be a risk for AD. Moreover, by pharmaceutically targeting APP trafficking, notch-associated problems of in vivo administration of γ-secretase inhibitors may be preventable, making γ-secretase a feasible therapeutic target for AD treatment.
Footnotes
This research was supported by National Institutes of Health Grant AG 12406, Deutsche Forschungsgemeinschaft Grant AR 379/1-1 (C.A.F.v.A.), and an American Federation for Aging Research Beeson award (M.C.I.). We thank M. S. Robinson for the GGA construct, T. Südhof for the APP–Gal4 construct, R. Kopan for the notch constructs, and A. Dunah for the LAR-R antibody. We also thank J. Walter for sharing results before submission of this manuscript.
References
- Amaratunga A, Fine RE. Generation of amyloidogenic C-terminal fragments during rapid axonal transport in vivo of [IMAGE]-amyloid precursor protein in the optic nerve. J Biol Chem. 1995;270:17268–17272. doi: 10.1074/jbc.270.29.17268. [DOI] [PubMed] [Google Scholar]
- Andersen OM, Reiche J, Schmidt V, Gotthardt M, Spoelgen R, Behlke J, von Arnim CA, Breiderhoff T, Jansen P, Wu X, Bales KR, Cappai R, Masters CL, Gliemann J, Mufson EJ, Hyman BT, Paul SM, Nykjaer A, Willnow TE. Neuronal sorting protein-related receptor sorLA/LR11 regulates processing of the amyloid precursor protein. Proc Natl Acad Sci USA. 2005;102:13461–13466. doi: 10.1073/pnas.0503689102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berezovska O, Ramdya P, Skoch J, Wolfe MS, Bacskai BJ, Hyman BT. Amyloid precursor protein associates with a nicastrin-dependent docking site on the presenilin 1–γ-secretase complex in cells demonstrated by fluorescence lifetime imaging. J Neurosci. 2003;23:4560–4566. doi: 10.1523/JNEUROSCI.23-11-04560.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billings LM, Oddo S, Green KN, McGaugh JL, Laferla FM. Intraneuronal abeta causes the onset of early Alzheimer's disease-related cognitive deficits in transgenic mice. Neuron. 2005;45:675–688. doi: 10.1016/j.neuron.2005.01.040. [DOI] [PubMed] [Google Scholar]
- Bonifacino JS. The GGA proteins: adaptors on the move. Nat Rev Mol Cell Biol. 2004;5:23–32. doi: 10.1038/nrm1279. [DOI] [PubMed] [Google Scholar]
- Buxbaum JD, Thinakaran G, Koliatsos V, O'Callahan J, Slunt HH, Price DL, Sisodia SS. Alzheimer amyloid protein precursor in the rat hippocampus: transport and processing through the perforant path. J Neurosci. 1998;18:9629–9637. doi: 10.1523/JNEUROSCI.18-23-09629.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantuti-Castelvetri I, Klucken J, Ingelsson M, Ramasamy K, McLean PJ, Frosch MP, Hyman BT, Standaert DG. Alpha-synuclein and chaperones in dementia with Lewy bodies. J Neuropathol Exp Neurol. 2005;64:1058–1066. doi: 10.1097/01.jnen.0000190063.90440.69. [DOI] [PubMed] [Google Scholar]
- Cao X, Sudhof TC. A transcriptionally [correction of transcriptively] active complex of APP with Fe65 and histone acetyltransferase Tip60. Science. 2001;293:115–120. doi: 10.1126/science.1058783. [DOI] [PubMed] [Google Scholar]
- Capell A, Steiner H, Willem M, Kaiser H, Meyer C, Walter J, Lammich S, Multhaup G, Haass C. Maturation and pro-peptide cleavage of β-secretase. J Biol Chem. 2000;275:30849–30854. doi: 10.1074/jbc.M003202200. [DOI] [PubMed] [Google Scholar]
- Caporaso GL, Gandy SE, Buxbaum JD, Greengard P. Chloroquine inhibits intracellular degradation but not secretion of Alzheimer β/A4 amyloid precursor protein. Proc Natl Acad Sci USA. 1992;89:2252–2256. doi: 10.1073/pnas.89.6.2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin JY, Knowles RB, Schneider A, Drewes G, Mandelkow EM, Hyman BT. Microtubule-affinity regulating kinase (MAPK) is tightly associated with neurofibrillary tangle in Alzheimer brain: a fluorescence resonance energy transfer study. J Neuropathol Exp Neurol. 2000;59:966–971. doi: 10.1093/jnen/59.11.966. [DOI] [PubMed] [Google Scholar]
- Chyung JH, Raper DM, Selkoe DJ. γ-Secretase exists on the plasma membrane as an intact complex that accepts substrates and effects intramembrane cleavage. J Biol Chem. 2005;280:4383–4392. doi: 10.1074/jbc.M409272200. [DOI] [PubMed] [Google Scholar]
- De Strooper B, Beullens M, Contreras B, Levesque L, Craessaerts K, Cordell B, Moechars D, Bollen M, Fraser P, George-Hyslop PS, Van Leuven F. Phosphorylation, subcellular localization, and membrane orientation of the Alzheimer's disease-associated presenilins. J Biol Chem. 1997;272:3590–3598. doi: 10.1074/jbc.272.6.3590. [DOI] [PubMed] [Google Scholar]
- Dunah AW, Hueske E, Wyszynski M, Hoogenraad CC, Jaworski J, Pak DT, Simonetta A, Liu G, Sheng M. LAR receptor protein tyrosine phosphatases in the development and maintenance of excitatory synapses. Nat Neurosci. 2005;8:458–467. doi: 10.1038/nn1416. [DOI] [PubMed] [Google Scholar]
- Ehehalt R, Keller P, Haass C, Thiele C, Simons K. Amyloidogenic processing of the Alzheimer β-amyloid precursor protein depends on lipid rafts. J Cell Biol. 2003;160:113–123. doi: 10.1083/jcb.200207113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink L, Seeger W, Ermert L, Hanze J, Stahl U, Grimminger F, Kummer W, Bohle RM. Real-time quantitative RT-PCR after laser-assisted cell picking. Nat Med. 1998;4:1329–1333. doi: 10.1038/3327. [DOI] [PubMed] [Google Scholar]
- Fukumoto H, Cheung BS, Hyman BT, Irizarry MC. Beta-secretase protein and activity are increased in the neocortex in Alzheimer disease. Arch Neurol. 2002;59:1381–1389. doi: 10.1001/archneur.59.9.1381. [DOI] [PubMed] [Google Scholar]
- Ghosh P, Kornfeld S. Phosphorylation-induced conformational changes regulate GGAs 1 and 3 function at the trans-Golgi network. J Biol Chem. 2003;278:14543–14549. doi: 10.1074/jbc.M212543200. [DOI] [PubMed] [Google Scholar]
- Grbovic OM, Mathews PM, Jiang Y, Schmidt SD, Dinakar R, Summers-Terio NB, Ceresa BP, Nixon RA, Cataldo AM. Rab5-stimulated up-regulation of the endocytic pathway increases intracellular β-cleaved amyloid precursor protein carboxyl-terminal fragment levels and Aβ production. J Biol Chem. 2003;278:31261–31268. doi: 10.1074/jbc.M304122200. [DOI] [PubMed] [Google Scholar]
- Haass C, Lemere CA, Capell A, Citron M, Seubert P, Schenk D, Lannfelt L, Selkoe DJ. The Swedish mutation causes early-onset Alzheimer's disease by beta-secretase cleavage within the secretory pathway. Nat Med. 1995;1:1291–1296. doi: 10.1038/nm1295-1291. [DOI] [PubMed] [Google Scholar]
- He X, Chang WP, Koelsch G, Tang J. Memapsin 2 (beta-secretase) cytosolic domain binds to the VHS domains of GGA1 and GGA2: implications on the endocytosis mechanism of memapsin 2. FEBS Lett. 2002;524:183–187. doi: 10.1016/s0014-5793(02)03052-1. [DOI] [PubMed] [Google Scholar]
- He X, Li F, Chang WP, Tang J. GGA proteins mediate the recycling pathway of memapsin 2 (BACE) J Biol Chem. 2005;280:11696–11703. doi: 10.1074/jbc.M411296200. [DOI] [PubMed] [Google Scholar]
- Herz J. The LDL receptor gene family: (un)expected signal transducers in the brain. Neuron. 2001;29:571–581. doi: 10.1016/s0896-6273(01)00234-3. [DOI] [PubMed] [Google Scholar]
- Hill K, Li Y, Bennett M, McKay M, Zhu X, Shern J, Torre E, Lah JJ, Levey AI, Kahn RA. Munc18 interacting proteins: ADP-ribosylation factor-dependent coat proteins that regulate the traffic of β-Alzheimer's precursor protein. J Biol Chem. 2003;278:36032–36040. doi: 10.1074/jbc.M301632200. [DOI] [PubMed] [Google Scholar]
- Hussain I, Powell D, Howlett DR, Tew DG, Meek TD, Chapman C, Gloger IS, Murphy KE, Southan CD, Ryan DM, Smith TS, Simmons DL, Walsh FS, Dingwall C, Christie G. Identification of a novel aspartic protease (Asp 2) as beta-secretase. Mol Cell Neurosci. 1999;14:419–427. doi: 10.1006/mcne.1999.0811. [DOI] [PubMed] [Google Scholar]
- Cho HS, Hyman BT, Greenberg SM, Rebeck GW. Quantitation of apoE domains in Alzheimer disease brain suggests a role for apoE in AB aggregation. J Neuropathol Exp Neurol. 2001;60:342–349. doi: 10.1093/jnen/60.4.342. [DOI] [PubMed] [Google Scholar]
- Irizarry MC, Deng A, Lleo A, Berezovska O, Von Arnim CA, Martin-Rehrmann M, Manelli A, LaDu MJ, Hyman BT, Rebeck GW. Apolipoprotein E modulates gamma-secretase cleavage of the amyloid precursor protein. J Neurochem. 2004;90:1132–1143. doi: 10.1111/j.1471-4159.2004.02581.x. [DOI] [PubMed] [Google Scholar]
- Kaether C, Schmitt S, Willem M, Haass C. Amyloid precursor protein and notch intracellular domains are generated after transport of their precursors to the cell surface. Traffic. 2006;7:408–415. doi: 10.1111/j.1600-0854.2006.00396.x. [DOI] [PubMed] [Google Scholar]
- Kamal A, Almenar-Queralt A, LeBlanc JF, Roberts EA, Goldstein LSB. Kinesin-mediated axonal transport of a membrane compartment containing β-secretase and presenilin-1 requires APP. Nature. 2001;414:643–648. doi: 10.1038/414643a. [DOI] [PubMed] [Google Scholar]
- Khvotchev M, Sudhof TC. Proteolytic processing of amyloid-beta precursor protein by secretases does not require cell surface transport. J Biol Chem. 2004;279:47101–47108. doi: 10.1074/jbc.M408474200. [DOI] [PubMed] [Google Scholar]
- Kinoshita A, Whelan CM, Smith CJ, Mikhailenko I, Rebeck GW, Strickland DK, Hyman BT. Demonstration by fluorescence resonance energy transfer of two sites of interaction between the low-density lipoprotein receptor-related protein and the amyloid precursor protein: role of the intracellular adapter protein Fe65. J Neurosci. 2001;21:8354–8361. doi: 10.1523/JNEUROSCI.21-21-08354.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita A, Fukumoto H, Shah T, Whelan CM, Irizarry MC, Hyman BT. Demonstration by FRET of BACE interaction with the amyloid precursor protein at the cell surface and in early endosomes. J Cell Sci. 2003;116:3339–3346. doi: 10.1242/jcs.00643. [DOI] [PubMed] [Google Scholar]
- Koo EH, Sisodia S, Archer D, Martin L, Weidemann A, Beyreuther K, Fischer P, Masters C, Price D. Precursor of amyloid protein in Alzheimer disease undergoes fast anterograde axonal transport. Proc Natl Acad Sci USA. 1990;87:1561–1565. doi: 10.1073/pnas.87.4.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo EH, Squazzo SL. Evidence that production and release of amyloid β-protein involves the endocytic pathway. J Biol Chem. 1994;269:17386–17389. [PubMed] [Google Scholar]
- Kopan R, Schroeter EH, Weintraub H, Nye JS. Signal transduction by activated mNotch: importance of proteolytic processing and its regulation by the extracellular domain. Proc Natl Acad Sci USA. 1996;93:1683–1688. doi: 10.1073/pnas.93.4.1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EB, Zhang B, Liu K, Greenbaum EA, Doms RW, Trojanowski JQ, Lee VM. BACE overexpression alters the subcellular processing of APP and inhibits Aβ deposition in vivo. J Cell Biol. 2005;168:291–302. doi: 10.1083/jcb.200407070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtenthaler SF, Dominguez DI, Westmeyer GG, Reiss K, Haass C, Saftig P, De Strooper B, Seed B. The cell adhesion protein P-selectin glycoprotein ligand-1 is a substrate for the aspartyl protease BACE1. J Biol Chem. 2003;278:48713–48719. doi: 10.1074/jbc.M303861200. [DOI] [PubMed] [Google Scholar]
- Mueller HT, Borg JP, Margolis B, Turner RS. Modulation of amyloid precursor protein metabolism by X11α/Mint-1. A deletion analysis of protein-protein interaction domains. J Biol Chem. 2000;275:39302–39306. doi: 10.1074/jbc.M008453200. [DOI] [PubMed] [Google Scholar]
- Nitsch RM, Wurtman RJ, Growdon JH. Regulation of APP processing. Potential for the therapeutical reduction of brain amyloid burden. Ann NY Acad Sci. 1996;777:175–182. doi: 10.1111/j.1749-6632.1996.tb34416.x. [DOI] [PubMed] [Google Scholar]
- Pastorino L, Ikin AF, Nairn AC, Pursnani A, Buxbaum JD. The carboxyl-terminus of BACE contains a sorting signal that regulates BACE trafficking but not the formation of total Aβ. Mol Cell Neurosci. 2002;19:175–185. doi: 10.1006/mcne.2001.1065. [DOI] [PubMed] [Google Scholar]
- Puertollano R, Bonifacino JS. Interactions of GGA3 with the ubiquitin sorting machinery. Nat Cell Biol. 2004;6:244–251. doi: 10.1038/ncb1106. [DOI] [PubMed] [Google Scholar]
- Ramdya P, Skoch J, Bacskai BJ, Hyman BT, Berezovska O. Activated Notch1 associates with a presenilin-1/gamma-secretase docking site. J Neurochem. 2003;87:843–850. doi: 10.1046/j.1471-4159.2003.02030.x. [DOI] [PubMed] [Google Scholar]
- Refolo LM, Sambamurti K, Efthimiopoulos S, Pappolla MA, Robakis NK. Evidence that secretase cleavage of cell surface Alzheimer amyloid precursor occurs after normal endocytic internalization. J Neurosci Res. 1995;40:694–706. doi: 10.1002/jnr.490400515. [DOI] [PubMed] [Google Scholar]
- Scherzer CR, Offe K, Gearing M, Rees HD, Fang G, Heilman CJ, Schaller C, Bujo H, Levey AI, Lah JJ. Loss of apolipoprotein E receptor LR11 in Alzheimer disease. Arch Neurol. 2004;61:1200–1205. doi: 10.1001/archneur.61.8.1200. [DOI] [PubMed] [Google Scholar]
- Scott PM, Bilodeau PS, Zhdankina O, Winistorfer SC, Hauglund MJ, Allaman MM, Kearney WR, Robertson AD, Boman AL, Piper RC. GGA proteins bind ubiquitin to facilitate sorting at the trans-Golgi network. Nat Cell Biol. 2004;6:252–259. doi: 10.1038/ncb1107. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- Shiba T, Kametaka S, Kawasaki M, Shibata M, Waguri S, Uchiyama Y, Wakatsuki S. Insights into the phosphoregulation of beta-secretase sorting signal by the VHS domain of GGA1. Traffic. 2004;5:437–448. doi: 10.1111/j.1600-0854.2004.00188.x. [DOI] [PubMed] [Google Scholar]
- Shiba Y, Katoh Y, Shiba T, Yoshino K, Takatsu H, Kobayashi H, Shin HW, Wakatsuki S, Nakayama K. GAT (GGA and Tom1) domain responsible for ubiquitin binding and ubiquitination. J Biol Chem. 2004;279:7105–7111. doi: 10.1074/jbc.M311702200. [DOI] [PubMed] [Google Scholar]
- Siegel RM, Frederikson JK, Zacharias DA, Chan FK, Johnson M, Lynch D, Tsien RY, Lenardo MJ. Fas preassociation required for apoptosis signaling and dominant inhibition by pathogenic mutations. Science. 2000;288:2354–2357. doi: 10.1126/science.288.5475.2354. [DOI] [PubMed] [Google Scholar]
- Sinha S, Anderson JP, Barbour R, Basi GS, Caccavello R, Davis D, Doan M, Dovey HF, Frigon N, Hong J, Jacobson-Croak K, Jewett N, Keim P, Knops J, Lieberburg I, Power M, Tan H, Tatsuno G, Tung J, Schenk D, et al. Purification and cloning of amyloid precursor protein beta-secretase from human brain. Nature. 1999;402:537–540. doi: 10.1038/990114. [DOI] [PubMed] [Google Scholar]
- Spoelgen R, von Arnim CA, Thomas AV, Peltan ID, Koker M, Deng A, Irizarry MC, Andersen OM, Willnow TE, Hyman BT. Interaction of the cytosolic domains of sorLA/LR11 with the amyloid precursor protein (APP) and β-secretase β-site APP-cleaving enzyme. J Neurosci. 2006;26:418–428. doi: 10.1523/JNEUROSCI.3882-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarassishin L, Yin YI, Bassit B, Li Y-M. Processing of Notch and amyloid precursor protein by γ-secretase is spatially distinct. Proc Natl Acad Sci USA. 2004;101:17050–17055. doi: 10.1073/pnas.0408007101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thinakaran G, Teplow DB, Siman R, Greenberg B, Sisodia SS. Metabolism of the “Swedish” amyloid precursor protein variant in Neuro2a (N2a) cells. J Biol Chem. 1996;271:9390–9397. doi: 10.1074/jbc.271.16.9390. [DOI] [PubMed] [Google Scholar]
- Tomita S, Kirino Y, Suzuki T. Cleavage of Alzheimer's amyloid precursor protein (APP) by secretases occurs after O-glycosylation of APP in the protein secretory pathway. Identification of intracellular compartments in which APP cleavage occurs without using toxic agents that interfere with protein metabolism. J Biol Chem. 1998;273:6277–6284. doi: 10.1074/jbc.273.11.6277. [DOI] [PubMed] [Google Scholar]
- Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, Teplow DB, Ross S, Amarante P, Loeloff R, Luo Y, Fisher S, Fuller J, Edenson S, Lile J, Jarosinski MA, Biere AL, Curran E, Burgess T, Louis JC, et al. Beta-secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286:735–741. doi: 10.1126/science.286.5440.735. [DOI] [PubMed] [Google Scholar]
- von Arnim CA, Tangredi MM, Peltan ID, Lee BM, Irizarry MC, Kinoshita A, Hyman BT. Demonstration of BACE (beta-secretase) phosphorylation and its interaction with GGA1 in cells by fluorescence-lifetime imaging microscopy. J Cell Sci. 2004;117:5437–5445. doi: 10.1242/jcs.01422. [DOI] [PubMed] [Google Scholar]
- Walter J, Fluhrer R, Hartung B, Willem M, Kaether C, Capell A, Lammich S, Multhaup G, Haass C. Phosphorylation regulates intracellular trafficking of β-secretase. J Biol Chem. 2001;276:14634–14641. doi: 10.1074/jbc.M011116200. [DOI] [PubMed] [Google Scholar]
- Yan R, Bienkowski MJ, Shuck ME, Miao H, Tory MC, Pauley AM, Brashier JR, Stratman NC, Mathews WR, Buhl AE, Carter DB, Tomasselli AG, Parodi LA, Heinrikson RL, Gurney ME. Membrane-anchored aspartyl protease with Alzheimer's disease beta-secretase activity. Nature. 1999;402:533–537. doi: 10.1038/990107. [DOI] [PubMed] [Google Scholar]
- Yang LB, Lindholm K, Yan R, Citron M, Xia W, Yang XL, Beach T, Sue L, Wong P, Price D, Li R, Shen Y. Elevated beta-secretase expression and enzymatic activity detected in sporadic Alzheimer disease. Nat Med. 2003;9:3–4. doi: 10.1038/nm0103-3. [DOI] [PubMed] [Google Scholar]