

Abstract
Odorant receptors comprise the biggest subfamily of G-protein-coupled receptors. Although the endocytic mechanisms of other G-protein-coupled receptors have been characterized extensively, almost nothing is known about the intracellular trafficking of odorant receptors. The present study describes the endocytic pathway of mammalian odorant receptors, which bind β-arrestin2 with high affinity and are internalized via a clathrin-dependent mechanism. After prolonged odorant exposure, receptors are not targeted to lysosomal degradation but accumulate in recycling endosomes. Odorant-induced odorant receptor desensitization is promoted by cAMP-dependent protein kinase A phosphorylation and is dependent on serine and threonine residues within the third intracellular loop of the receptor. Moreover, β-arrestin2 is redistributed into the dendritic knobs of mouse olfactory receptor neurons after treatment with a complex odorant mixture. Prolonged odorant exposure resulted in accumulation of β-arrestin2 in intracellular vesicles. Adaptation of olfactory receptor neurons to odorants can be abolished by the inhibition of clathrin-mediated endocytosis, showing the physiological relevance of the here described mechanism of odorant receptor desensitization. A better understanding of odorant receptor trafficking and additional insight into the molecular determinants underlying the interactions of odorant receptors with β-arrestin2 and other trafficking proteins will therefore be important to fully understand the mechanisms of adaptation and sensitization in the olfactory epithelium.
Keywords: olfactory receptor, arrestin, olfactory epithelium, endocytosis, desensitization, receptor trafficking
Introduction
Detection of odor chemicals is initiated by activation of G-protein-coupled mammalian odorant receptors (ORs) that are expressed in olfactory receptor neurons (ORNs) of the nose (Buck and Axel, 1991; Buck, 1992; Mombaerts, 2004). ORs signal through stimulation of Gαolf, which leads to activation of type III adenylyl cyclase and opening of cAMP-gated cation channels (Reed, 1992; Zufall et al., 1994). Previous research has provided considerable insight into the molecular mechanisms underlying adaptation in the olfactory system, including Ca2+- and phosphoinositide-dependent cyclic nucleotide-gated channel modulation (Zufall et al., 1991; Kurahashi and Menini, 1997; Munger et al., 2001; Bradley et al., 2004; Zhainazarov et al., 2004), Ca2+/calmodulin kinase II-dependent attenuation of adenylyl cyclase (Leinders-Zufall et al., 1999), and the modulatory activities of nitric oxide, carbon monoxide, and cGMP (Zufall and Leinders-Zufall, 2000). Odor adaptation also involves stimulus-dependent phosphorylation of elements of the signal transduction cascade, because stimulation of the olfactory epithelium causes significant phosphorylation of cilia proteins (Boekhoff et al., 1992). A sequential interplay of either second-messenger-dependent or receptor-specific kinases [G-protein-coupled receptor kinases (GRKs)] has been suggested because inhibition of both results in complete abolition of the olfactory desensitization process (Dawson et al., 1993; Schleicher et al., 1993; Peppel et al., 1997). The cAMP-dependent protein kinase (PKA) was further shown to modulate voltage-gated ion channels (Wetzel et al., 2001).
However, the fate of the stimulated receptor molecule is far less clear, and the pathways and proteins involved in intracellular trafficking of ORs after stimulation remain elusive. Clathrin-dependent internalization pathways have been shown in ORNs of channel catfish (Rankin et al., 1999). G-protein-coupled receptors (GPCRs) are typically internalized after ligand binding, and the mechanisms involved in agonist-induced receptor endocytosis have been well described (Seachrist and Ferguson, 2003; von Zastrow, 2003; Lefkowitz, 2004). After agonist binding and activation of the heterotrimeric G-protein, signaling is turned off by phosphorylation of serine and threonine residues within the C terminus and/or the third intracellular loop of the receptors, followed by the recruitment of arrestins (Oakley et al., 2001; Lefkowitz, 2004). Arrestin binding not only interrupts coupling between the receptor and its cognate G-protein but also mediates the activation of “novel” signaling pathways and targeting of the receptors to clathrin-coated pits with subsequent internalization (Ferguson, 2001). Two distinct patterns of β-arrestin trafficking have been delineated, leading to the division of GPCRs into two classes. Class A receptors show preferential binding to β-arrestin2 compared with β-arrestin1, and arrestin binding is only transient. Class B receptors display equal affinity for both arrestins and build stable β-arrestin complexes, which remain in endosomal compartments for extended periods (Shenoy and Lefkowitz, 2003).
For non-olfactory GPCRs, the basic mechanisms of intracellular trafficking have been characterized successfully in heterologous cell systems. To learn about potential OR endocytosis, we investigated the endocytic pathway of recombinant ORs and compared our findings with observations in odor-activated ORNs. We show that stimulated ORs stably interact with β-arrestin and undergo clathrin-dependent endocytosis, resulting in their accumulation in endocytic vesicles for extended periods of time. In ORNs, inhibition of β-arrestin2-mediated endocytosis affects sensory adaptation to repetitive odor stimulation.
Materials and Methods
Antibodies.
The following primary antibodies were used: (1) mouse polyclonal antibody against cathepsin D (University of Texas Southwestern CBI Antibody Core, Southwestern Medical Center, University of Texas, Dallas, TX); (2) mouse monoclonal against β-arrestin2 (Santa Cruz Biotechnology, Santa Cruz, CA); and (3) rabbit monoclonal antibody against the phospho-(Ser/Thr) PKA substrate recognizing the R-X-X-S/T-X-X motif (Cell Signaling Technology, Beverly, MA). For immunofluorescence, a secondary goat anti-mouse or goat anti-rabbit antibodies conjugated to Alexa 568 or Alexa 660 were used (Invitrogen, Carlsbad, CA).
Cell culture and transfection.
Hana3A cells (Saito et al., 2004) were kindly provided by Dr. Hiroaki Matsunami (Department of Molecular Genetics and Microbiology, Duke University Medical Center, Durham, NC). Experiments using heterologous expression system were performed in HEK293 and Hana3A cells, which were maintained under standard conditions in MEM supplemented with 10% FBS, 100 U/ml penicillin and streptomycin, and 2 mm l-glutamine. Transfections were done using a calcium phosphate precipitation technique.
DNA constructs and plasmids.
Human β-arrestin2 was amplified from RNA obtained from human testis tissue by reverse transcription-PCR using specific primers that amplify the complete open reading frame and contain restriction sites for additional subcloning, and the PCR product was cloned into pcDNA3. OR2AG1 (olfactory receptor, family 2, subfamily AG, member 1) was amplified from genomic DNA, which was isolated from blood using a commercial kit (Qiagen, Hilden, Germany). PCR reactions were done with 100 ng genomic DNA and specific primers for OR2AG1. OR2AG1–green fluorescent protein (GFP) and β-arrestin2–GFP fusion constructs were generated by cloning the PCR products into a linearized GFP containing plasmid with restriction sites for the generation of N-terminal fusion constructs (Neuhaus et al., 2005). OR2AG1-RLuc plasmid was obtained by subcloning the coding sequences of Renilla luciferase (RLuc) from the plasmid pGFP2–multiple cloning site–RLuc(h) (Packard BioScience, Meriden, CT) into the ClaI-linearized, odorant-receptor-containing plasmid. The OR2AG1–GFP and OR2AG1-RLuc fusion proteins were tested for functionality by Ca2+ imaging using amylbutyrat (Neuhaus et al., 2006).
Site mutations in OR2AG1 were introduced at the C terminus (positions 314S-A and 315T-A) and the third intracellular loop (positions 224T-A, 240T-A, 230S-A, 242S-A, and 243S-A).
The C-terminus mutant of OR2AG1 was obtained by PCR from the pcDNA3 vector containing the full-length receptor using specific downstream primer with S mutated to A at position 314 and T mutated to A at position 315 (GCC-TCG-AGT-TAA-TCG-ATA-TCG-AGC-GCG-GCG-TGT-GCT-GG). To obtain the third loop phosphorylation mutant, the N-terminal part of the OR2AG1 sequence was amplified up to the end of the third intracellular loop by PCR with a specific downstream primer mutating T to A at positions 224 and 240 and S to A at positions 230, 242, and 243. This PCR product (700 bp) was cloned into pcDNA3 vector. The second PCR was done to amplify the rest of the receptor sequence. To obtain full-length mutated ORAG1 construct, this PCR product (270 bp) was cloned into pcDNA3 containing the N-terminal part of the receptor sequence using specific restriction sites. All generated plasmids were verified by sequencing.
Immunohistochemistry.
Cells were grown on poly-lysine-coated coverslips (80–100 μm; Menzel Gläser, Braunschweig, Germany). After the treatment as indicated in the figure legends, coverslips were washed and fixed in 3% paraformaldehyde in Ringer’s solution containing 10 mm glucose (30 min at room temperature). Cells were permeabilized with 0.1% Triton X-100 in PBS containing 1% cold-water fish skin gelatin (Sigma, St. Louis, MO) and incubated with primary antibodies in PBS/gelatin/Triton X-100. After washing, coverslips were incubated with fluorescently labeled secondary antibodies and mounted in ProLong Antifade (Invitrogen). All fluorescence images were obtained with a confocal microscope (LSM510 Meta; Zeiss, Oberkochen, Germany) using a 40×, 1.4 numerical aperture objective (pinhole set to one Airey unit) and further processed with Photoshop (Adobe Systems, San Jose, CA).
Translocation assays.
For β-arrestin–GFP and OR2AG1 translocation assays, cells were transfected with a 5:1 ratio of the OR2AG1–GFP construct and β-arrestin2–GFP. Two days after transfection, growth media were removed and replaced with standard Ringer’s solution; 500 μm amylbutyrat was added at room temperature. In some cases, cells were preincubated with 10 μg/ml Texas Red-conjugated transferring (Tfn) (Invitrogen). All images were obtained with a confocal microscope (LSM510 Meta; Zeiss). Typically, cells were investigated for 60 min, and pictures were taken every 10 s. Quantification of the fluorescence intensities of the OR2AG1–GFP in the membrane compared with cytosol was performed with the original data. Images were further processed with Photoshop (Adobe Systems).
Ca2+ imaging in heterologous cells.
Cells were incubated (45 min) in Ringer’s solution containing 7.5 μm fura-2 AM (Invitrogen) at room temperature. Inhibitors were applied 30–60 min before amylbutyrat application at concentrations of 0.25 mg/ml concanavalinA (Sigma), 10 μm phenylarsine oxide (Sigma), 5 μg/ml filipin III (Sigma), 10 mm β-cyclodextrin (Sigma), or 500 nm of water-soluble PKA inhibitor H-89 (N-[2-(p-bromo-cinnamylamino)-ethyl]-5-isoquinoline-sulfon-amide 2HCl) (Calbiochem, La Jolla, CA). Ratiofluorometric Ca2+ imaging was performed as described previously (Spehr et al., 2003) using a Zeiss inverted microscope equipped for ratiometric imaging. Images were acquired from up to 15 cells in a randomly selected field of view, and integrated fluorescence ratios (F340/F380 ratio) were measured. Cells were exposed to either 100 or 500 μm amylbutyrat, which are typical concentrations used for heterologously expressed ORs (Saito et al., 2004; Spehr et al., 2004; Katada et al., 2005). Exposure to amylbutyrat was accomplished using a specialized microcapillary application system. The time after which the Ca2+ concentration returned to the preexposure level was used to determine the response duration in the different experiments.
Bioluminescence resonance energy transfer assay.
Hana3A cells grown in 35 mm dishes were transiently transfected with 0.5 μg of the β-arrestin–GFP and 2.5 μg of the hOR2AG1–RLuc fusion construct or the hOR2AG1–RLuc plasmid alone using ExGen500 transfection reagent (Fermentas, Hanover, MD). At 48 h after transfection ∼60,000 cells per well were distributed in a 96-well microplate (white Optiplate; Packard BioScience) in the presence or absence of amylbutyrat. The bioluminescence resonance energy transfer (BRET) effect was measured using DeepBlueC reagent (BioSignal Packard, Meriden, CT) according to the instructions of the manufacturer.
Mice odorant exposure and tissue preparation.
Fifteen-day-old mice were held in standard cages at an animal facility at room temperature (22°C). Each cage was housed in a customized hood designed to deliver an isolated air stream, and the “exhaust” air was extracted to a fume cupboard. The apparatus was designed to ensure that the vapor phase concentration of the odorant remained consistent throughout the experiment. All tubes were silicon coated to minimized odorant absorbance, and the hood was constructed from Perspex glass. A complex mixture of 100 compounds (Henkel 100), including aromatic and short-chain aliphatic hydrocarbons (Wetzel et al., 1999; Spehr et al., 2003), was applied in a 1:10,000 dilution for the time periods indicated in the figure legend.
Mice were killed after the time of odorant exposure indicated in each particular experiment, and snouts were fixed in 3% paraformaldehyde in Ringer’s solution containing 10 mm glucose. Fixed tissue was cryoprotected in 30% sucrose in Ringer’s solution before embedding and freezing in Tissue-Tek OCT (Leica, Nussloch, Germany). Immunostaining was done on 10 μm coronal cryosections on Superfrost slides (Menzel Gläser). All fluorescence images were obtained with a confocal microscope (LSM510 Meta; Zeiss) using a 40×, 1.4 numerical aperture objective (pinhole set to one Airey unit); fluorescence intensities were quantified using the LSM510 software.
Electro-olfactogram recordings.
12 mice were pre-treated with the Henkel 100 mixture in a 1:10,000 dilution for 2 h, 11 mice did not receive any treatment (control group). After treatment mice were killed by decapitation, the septal bone with the intact olfactory epithelium was dissected from the head and used intact for electro-olfactogram (EOG) recordings as described previously (Spehr et al., 2002). Briefly, the septal bone and its associated epithelium was placed in a recording chamber continually perfused with oxygenated Ringer’s buffer. Field potentials were recorded using glass pipettes (tips filled with extracellular solution in 1% agar), which were placed on the air face of the epithelium. Electrophysiological signals were transduced by a unipolar silver/silver chloride (Ag/AgCl) electrode “housed” in the glass pipette; the direct current output was differentially amplified (P18C; Grass Instruments, Quincy, MA) and referenced to an identical electrode (Ag/AgCl) grounding the bath. Increasing concentrations of Henkel 100 mixture were applied in the vapor phase for 2 s, with 1 min interval. The odorant was injected into a constant stream of humidified air that continuously superfused the epithelium. Signals were recorded on a chart recorder, and the peak amplitudes were measured manually.
Endocytosis of horseradish peroxidase.
Mice were killed, and olfactory epithelium was dissected from the septal bone as described previously (Spehr at al., 2002) and incubated in Ringer’s solution containing 0.25 mg/ml horseradish peroxidase (HRP) for indicated periods of time at room temperature. An odorant mixture (Henkel 100) was added at 1:10,000 dilution. After that, the epithelium was placed on ice, washed three times with PBS, placed in PBS containing 0.5% Triton X-100, and homogenized with the tight-fitting pestle. Lysates were cleared by centrifugation at 10,000 × g for 10 min at 4°C, and equal aliquots were removed for peroxidase assay. 2,2′-Azino-bis(3-ethylbenzthiazoline-6-sulfonic acid (ABTS) (Sigma) was used as a chromogen peroxidase substrate for the detection of HRP activity; the ABTS solution was prepared according to the instructions of the manufacturer. The green color of the substrate was determined by measuring absorption at 405 nm with a spectrophotometer (Helios Gamma; Thermo Electron Corporation, Waltham, MA). Negative controls were performed with lysis buffer including ABTS and subtracted from each value. The HRP activity was normalized to the total protein determined using a colorimetric assay (Bio-Rad, Hercules, CA).
Ca2+ imaging on single mouse ORNs.
Ca2+-imaging recordings from acutely dissociated ORNs was performed as described previously (Spehr et al., 2002). Briefly, mice were killed, and the sensory epithelium was carefully dissected from both the septal bone and endoturbinates and placed for 10 min in papain (Sigma) solution of low divalent cation concentration (in mm: 140 NaCl, 5 KCl, 0.5 CaCl2, 5 EGTA, 10 HEPES, and 10 glucose). Next, the tissue was transferred to Ringer’s solution containing an AM ester derivative of the Ca2+-sensitive reporter dye fura-2. Tissue pieces were gently triturated with a fire-polished Pasteur pipette. The solution was then filtered using a 70 μm nylon cell strainer (Becton Dickinson, Mountain View, CA), and dissociated cells were collected in coated glass-bottom dishes (World Precision Instruments, Sarasota, FL) and allowed to settle and attach. After a loading period of 30 min, the fura-2 AM solution was carefully replaced with the nonsupplemented Ringer’s solution. The dish was transferred to the stage of an inverted microscope equipped for ratiometric live-cell imaging (model IX71; Olympus Optical, Tokyo, Japan) and viewed at 63× magnification using an oil-immersion objective. ORNs were identified based on their characteristic morphology (elliptical soma, apical dendrite, and knob) and illuminated sequentially at 340 and 380 nm (10 ms each, 1000 ms cycle time). The average pixel intensity within the ORN dendritic knobs (user-selected regions of interest) was digitized and stored on a personal computer. The Ca2+-dependent fluorescence signal at 510 nm was expressed as the F340/F380 ratio. Odorant mixtures (Henkel 50, individual odorants at 10 μm) were applied for 5 s at various intervals using a custom-made pressure-driven multibarrel perfusion system that allows for instantaneous solution change. To study a potential role of receptor internalization in olfactory adaptation, ORNs were exposed for 5 min to dynamin inhibitory peptide (50 μm).
Results
Activated ORs are internalized and accumulate in recycling endosomes
Acute stimulation of neurons with ligands for GPCRs induces endocytosis of the receptor protein, which occurs mainly through the formation of clathrin-coated pits. To investigate whether ORs also undergo regulated internalization from the cell surface, trafficking of recombinant OR2AG1–GFP fusion protein was analyzed in heterologous cells by confocal microscopy. Treatment with its cognate ligand amylbutyrat (Neuhaus et al., 2006) resulted in redistribution of OR2AG1–GFP from the plasma membrane to intracellular vesicles (Fig. 1A,B). To further characterize the fate of the internalized OR, localization of OR2AG1 was compared with that of the Tfn receptor, a membrane protein with a well described endocytic trafficking route. Cells expressing OR2AG1–GFP were incubated with amylbutyrat in the presence of fluorescently labeled Tfn (Fig. 1C). In the absence of a ligand, the receptor predominantly resided at the cell surface, whereas after prolonged ligand exposure (1 h), the majority of OR was present in intracellular, transferrin-positive vesicles. We observed only marginal colocalization between the internalized OR and the lysosomal protease cathepsin D (Fig. 1D), demonstrating that at least after 1 h of agonist stimulation the receptor is not targeted for lysosomal degradation. Together, recombinant ORs accumulate in an endocytic recycling compartment after stimulation.
Figure 1.

OR2AG1 undergoes endocytosis during stimulation. A, Confocal microscopy of cells transfected with OR2AG1–GFP before (0) and after (30 min) amylbutyrat treatment. OR2AG1–GFP is redistributed from the cell membrane to intracellular vesicles during stimulation. B, Quantification of the OR2AG1–GFP fluorescence intensity in the membrane relative to the intensity in the cytosol, measured in single confocal sections before (contr) and after the amylbutyrat (amyl) application. The averages of at least 15 cells demonstrate a ∼4.5 fold decrease in membrane GFP fluorescence intensity after stimulation. C, Confocal microscopy of HEK293 cells expressing OR2AG1–GFP before (0) and after (1 h) amylbutyrat stimulation. OR2AG1–GFP-containing vesicles (green) show substantial colocalization with transferrin (red, top). After 1 h of amylbutyrat stimulation (yellow, top), only marginal colocalization was observed with the lysosomal protease cathepsin D (red, bottom).
Endocytosis of OR occurs via a clathrin-mediated mechanism
At least two distinct pathways of GPCR internalization have been described, clathrin-coated pits and caveolae. Because of the fact that activation of transiently expressed ORs in HEK293 cells leads to an IP3-mediated increase in the cytosolic Ca2+ concentration, ratiofluorometric imaging techniques can be used to detect OR activation (Wetzel et al., 1999; Spehr et al., 2003). We asked which endocytosis mechanism is used by ORs and performed Ca2+-imaging experiments in OR2AG1-expressing HEK293 cells after selective inhibition of different endocytic pathways (Fig. 2A). In untreated (control) cells, responses to short amylbutyrat pulses decreased within 20–30 s. Pretreatment with both filipin (+filipinIII), a sterol binding agent inhibiting the caveolae pathway, and β-methylcyclodextrin (+cycloD), which causes the disruption of caveolae by cholesterol depletion, did not affect response duration or amplitude (Fig. 2A). In marked contrast, preincubation with described inhibitors of clathrin-mediated endocytosis, such as phenylarsine oxide (+PAO) and concanavalinA (+conA), evoked significantly prolonged responses (Fig. 2A,B). After recruitment of clathrin to forming pits at the plasma membrane, detachment of these pits from the membrane is induced by the GTPase dynamin. Using a cell-permeable peptide that functions as an inhibitor of the GTPase dynamin by competitively blocking the binding of dynamin to amphiphysin also resulted in significantly prolonged responses compared with control cells (Fig. 2A,B). Together, these findings indicate that OR activation directs the receptor to internalize via a clathrin-dependent mechanism.
Figure 2.

Endocytosis of OR2AG1 is mediated by a clathrin-dependent mechanism. A, Representative ratiofluorometric recordings of the individual cells. The cytosolic Ca2+ level of fura-2-loaded cells is depicted as fluorescence ratio (F340/F380) and viewed as a function of time. Cells were preincubated with inhibitors of the clathrin pathway [phenylarsine oxide (+PAO), concanavalinA (+conA), and dynamin inhibitory peptide (+DIP)] or the caveolae pathway [filipin III (+filipin III) and β-cyclodextrin (+cycloD)] as described in Materials and Methods. B, Preincubation of the cells with clathrin inhibitors resulted in significantly prolonged responses compared with the control, although response amplitude was only slightly increased. The duration of the Ca2+ response was measured from the onset of the stimulus to the decrease to the basal level. Results are the mean from at least 10 cells. Error bars represent SEM; **p < 0.01. Amylbutyrat (100 μm) was applied for 5 s in each case.
Activation of ORs induces β-arrestin2–GFP redistribution into endosomes
Because targeting of GPCRs to clathrin-coated pits via β-arrestin2 is well established, we investigated the role of β-arrestin2 in OR signaling. We first examined a potential interaction of ORs and β-arrestin2 in a heterologous cell system. We visualized redistribution of GFP-tagged β-arrestin2 in response to OR activation. In the absence of agonist, β-arrestin2–GFP was diffusely localized throughout the cytoplasm (Fig. 3A). In response to OR2AG1 activation by amylbutyrat (500 μm), β-arrestin2–GFP was redistributed over time into intracellular vesicular structures (Fig. 3A). The same phenomenon also occurred with other recombinant ORs tested (data not shown). Translocation of β-arrestin2–GFP was specifically dependent on OR2AG1 activation, because it occurred after neither application of odorants that do not activate this OR2AG1 (data not shown) nor amylbutyrat application to untransfected HEK293 cells (data not shown).
Figure 3.
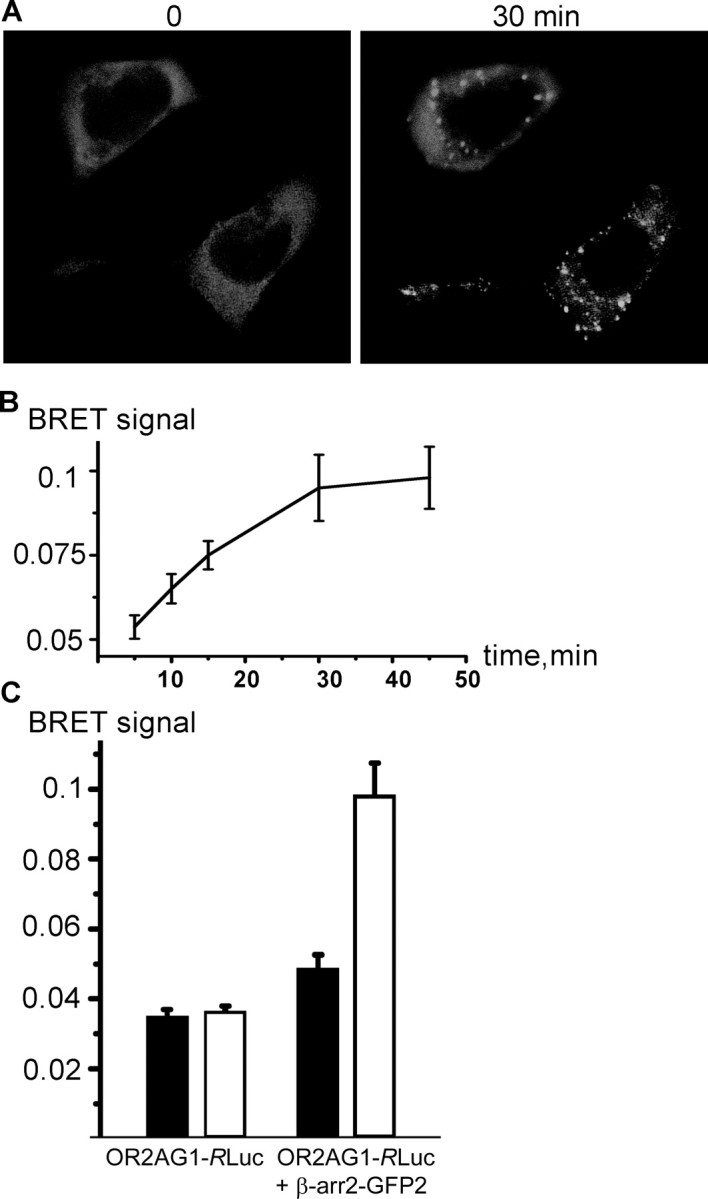
β-arrestin2 interacts with olfactory receptors during ligand binding. A, Confocal microscopy of HEK293 cells cotransfected with OR2AG1 and β-arrestin2–GFP. Amylbutyrat treatment (30 min) causes translocation of β-arrestin2 into intracellular vesicular structures. B, Cells were cotransfected with β-arrestin–GFP and OR2AG1–RLuc fusion constructs, and the BRET ratio was measured at different time points of amylbutyrat stimulation 48 h after transfection. The BRET signal increased in a time-dependent manner during addition of the agonist. Data are the mean from five independent experiments. C, Cells were cotransfected with β-arrestin–GFP and OR2AG1–RLuc fusion constructs or with OR2AG1–RLuc alone. The BRET ratio was determined 48 h after transfection on amylbutyrat-stimulated (white bars) and nonstimulated (black bars) cells. No significant change of the BRET ratio during addition of amylbutyrat is observed in cells expressing OR2AG1–RLuc alone. In contrast, addition of the agonist to cells expressing the OR2AG1–RLuc fusion construct together with β-arrestin–GFP leads to an increase of the BRET signal. Error bars represent SEM.
ORs form high-affinity complexes with β-arrestin2
To show the direct interaction of OR with β-arrestin2, we used a BRET assay, which has been successfully used to measure the GPCR/β-arrestin2 interaction in living cells (Heding, 2004). BRET is a noninvasive, sensitive technique involving the proximity-dependent transfer of energy from RLuc, which releases energy during oxidization of the substrate coelenterazine, to an energy acceptor (GFP), resulting in the emission of light. BRET involves the use of fusion proteins; in our case, the OR is fused to RLuc and GFP is fused to β-arrestin2. In control cells expressing OR2AG1--RLuc fusion protein alone, the BRET signal did not significantly change during amylbutyrat application (Fig. 3B). In contrast, in cells coexpressing OR2AG1-RLuc and β-arrestin2–GFP fusion proteins, the BRET signal increased in a time-dependent manner during addition of the agonist (Fig. 3B,C), providing a clear indication that β-arrestin2 binds to the activated receptor. The BRET assay can therefore be a useful tool to monitor agonist-promoted interactions occurring during OR activation. The stability of the interaction between β-arrestin2 and ORs over time together with the fact that β-arrestin2 accumulates in endocytic vesicles after receptor activation suggests that ORs belong to class B GPCRs.
Phosphorylation of the third intracellular loop is required for OR desensitization
To directly assess whether agonist-mediated receptor phosphorylation is essential for arrestin binding and desensitization (Gainetdinov et al., 2004), we next aimed to identify phosphorylation sites important for β-arrestin2 binding and OR desensitization. We created constructs in which potential phosphorylation sites within the C terminus and the third intracellular loop of OR2AG1 were mutated (Fig. 4A). Intracellular loop 3 mutants failed to induce β-arrestin2–GFP translocation to endosomes in response to ligand stimulation. In contrast, C-terminus mutants showed normal β-arrestin2–GFP translocation (Fig. 4B).
Figure 4.
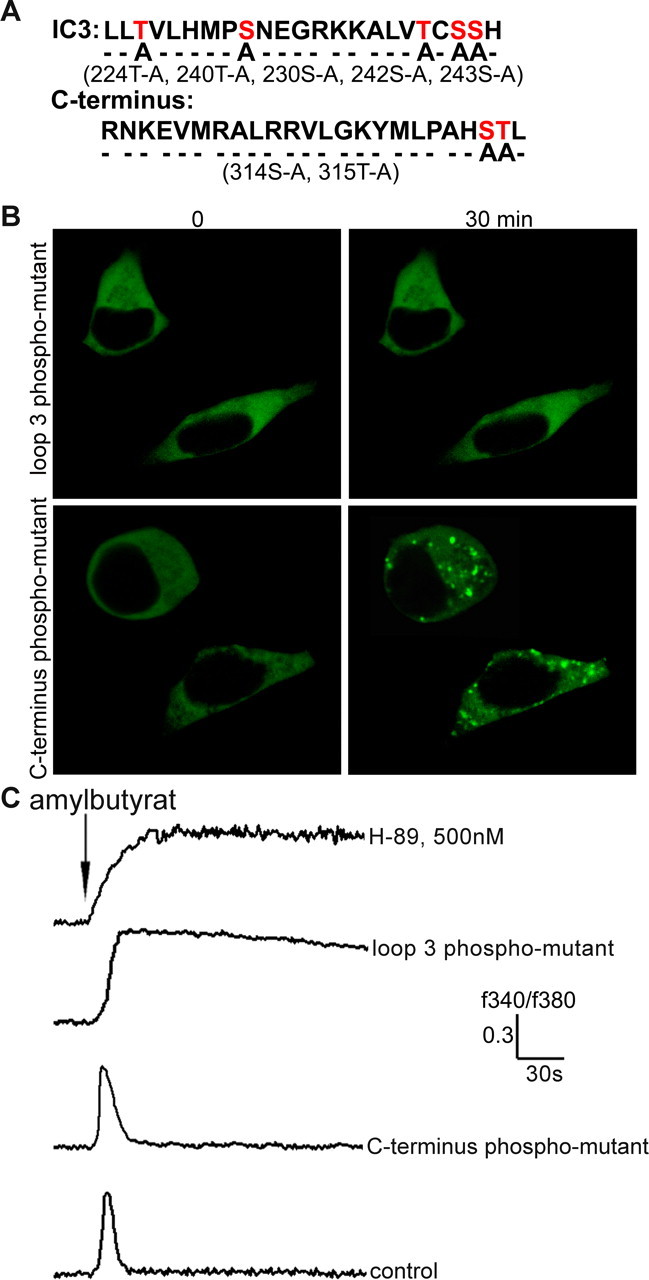
Agonist promoted OR phosphorylation is essential for OR desensitization and β-arrestin2 binding. A, Site-directed mutagenesis experiments were performed to delete the potential phosphorylation sites of OR2AG1; the scheme shows mutated phosphorylation sites within the third intracellular loop (positions 224T-A, 240T-A, 230S-A, 242S-A, and 243S-A) and the C terminus (positions 314S-A and 315T-A). B, OR2AG1 with mutated intracellular loop 3 failed to induce β-arrestin2–GFP translocation, whereas OR2AG1 with mutated C terminus shows wild-type-like β-arrestin2–GFP translocation into endosomes in response to amylbutyrat stimulation. C, Ca2+ responses elicited by the activation of the C-terminus phosphorylation mutant (C terminus phospho-mutant) in HEK293 cells were undistinguishable from the control. In contrast, a strong delay in desensitization was found for the third loop phosphorylation mutant (loop 3 phospho-mutant). OR2AG1 desensitization is promoted by PKA phosphorylation, as reflected by a strong delay in desensitization in the presence of the PKA inhibitor H-89 (500 nm). These delays in desensitization were observed in at east 10 independent experiments. Amylbutyrat (100 μm) was applied for 5 s in each case.
The heterologous expressed recombinant OR in HEK293 cells activates an endogenous G-protein, which triggers a phosphatidylinositol-mediated rise in intracellular Ca2+. To visualize this rise in the Ca2+ concentration, Ca2+-imaging experiments were performed. Activation of the C-terminus phosphorylation mutant (C-terminus phospho-mutant) was indistinguishable from that of the wild-type receptor (Fig. 4C). In contrast, prolonged Ca2+ signals were observed for the third loop phosphorylation mutant (loop 3 phospho-mutant), indicating that phosphorylation of residues T224, S230, T240, S242, and S243 within the third intracellular loop is important for OR desensitization. The observed delay in the decay of the Ca2+ signal is somewhat similar to the prolonged signal caused by the PKA inhibitor H-89 (Fig. 4C). The fact that the Ca2+ signal does not increase continuously, although the receptor cannot be inactivated, can be explained by the activation of calcium pumps in the plasma membrane (Ca2+-ATPase and Na+/Ca2+ exchangers) and in the endoplasmic reticulum (sarcoendoplasmic reticulum Ca2+-ATPases) that act to reestablish low resting Ca2+ concentrations.
OR induced β-arrestin2 redistribution in vivo
Having established that β-arrestin2 interacts with ORs in a heterologous expression system, we traced β-arrestin2 redistribution during OR activation in vivo. We took advantage of the olfactory marker protein (OMP)–GFP transgenic mouse line, which expresses GFP in all mature ORNs (Potter et al., 2001), thus allowing the visualization of neurons in olfactory epithelium without applying additional immunohistochemical staining. Two groups of mice (postnatal day 15) were exposed to a complex odorant mixture (Henkel 100) for either 15 min or 2 h.
In untreated mice (control), β-arrestin2 was homogeneously distributed throughout the cytoplasm of ORNs (Fig. 5A). Short time exposure (15 min) to the odorant mixture (each odorant ∼10−6 m) caused translocation of β-arrestin2 to ORN dendritic knobs (Fig. 5B, arrows) in which, along with olfactory cilia, the initial events of olfactory signal transduction are known to occur (Menini et al., 2004). Arrestin accumulation in the knobs occurred in 5–10% of the ORNs, similar to the fraction of dissociated ORNs responding to the odorant mixture with a Ca2+ response (Spehr et al., 2002). A 10-fold increase in individual odorant concentration (each odorant ∼10−5 m) increased the number of knobs with strong β-arrestin2 staining (Fig. 5C). After 2 h of odorant treatment, β-arrestin2 was no longer detected in the dendritic knobs but instead showed a vesicular-like staining in the cytoplasm (Fig. 5G, arrows). β-arrestin2-containing vesicles were also present in the ORN axons (higher magnification in Fig. 5F). Quantifying the number of β-arrestin-labeled knobs before and after odorant treatment (15 min), we found a concentration-dependent increase in β-arrestin2 recruitment after odorant exposure (Fig. 5D). Moreover, we quantified the increase in arrestin immunoreactivity in the knobs relative to the OMP signal, which remained unchanged throughout the experiment, and found an approximately fourfold increase in the β-arrestin2 staining intensity (Fig. 5E).
Figure 5.
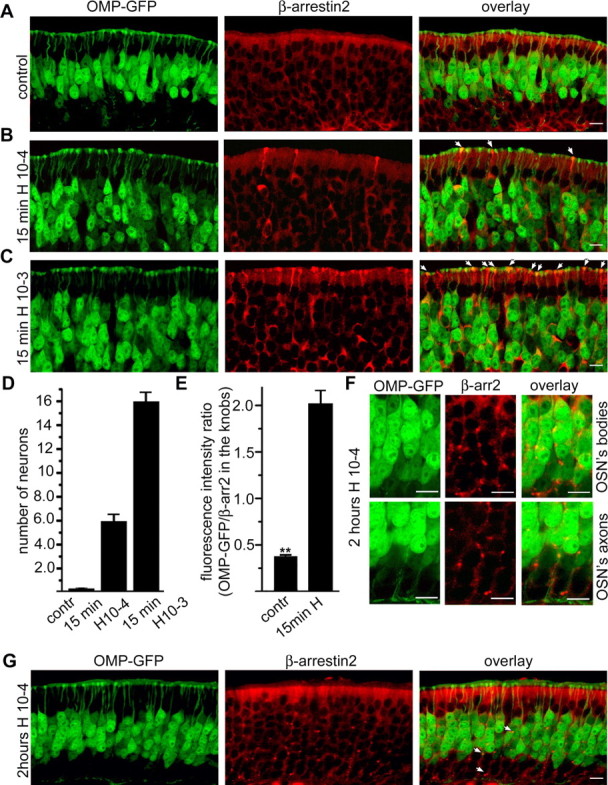
β-Arrestin2 is redistributed in ORNs during odorant treatment. A–C, β-Arrestin2 redistribution in mouse ORNs after odorant exposure. Fifteen-day-old OMP–GFP mice were exposed or not (control) to a complex odorant mixture of 100 compounds (Henkel 100) for 15 min. Cryosections through the mouse olfactory epithelium were probed with antibodies against β-arrestin2. A, In untreated mice (control), β-arrestin2 was homogeneously distributed throughout the cytoplasm of all cells in the olfactory epithelium, including ORNs (yellow on the overlay). B, Fifteen minutes of exposure to the Henkel 100 mixture (diluted 1:10,000) caused translocation of β-arrestin2 to the dendritic knobs of ORNs as indicated by arrows. C, Treatment of mice with higher concentration of Henkel 100 mixture (1:1000) resulted in increased number of ORNs with β-arrestin2 accumulation in the dendritic knob. D, E, Quantification of the observed effects; three mice from the control and three mice from the exposed group were investigated (4 slices with complete olfactory epithelium per mouse). D, The number of neurons showing strong anti-β-arrestin2 reactivity in the dendritic knob was counted over a 500 μm segment of a 10-μm-thick cryosection from the olfactory epithelium. Error bars represent SEM. E, Quantification of the anti-β-arrestin2 fluorescence intensity in the dendritic knobs of ORNs relative to the fluorescence intensity from OMP–GFP. The comparison of the fluorescence intensities in the dendritic knobs from the ORNs from the control group and the intensities from the group exposed to the lower Henkel 100 concentration demonstrates an approximate sixfold increase. At least 70 neurons from each group were investigated. Error bars represent SD; **p < 0.01. F, Higher-magnification pictures show a redistribution of β-arrestin2 to vesicular structures in the cell bodies (OSN‘s bodies) and in the axons (OSN‘s axons) of ORNs after 2 h odorant treatment. G, After 2 h of odorant treatment (Henkel 100 mixture, 2 h Henkel), β-arrestin2 showed vesicular-like staining in the ORNs as indicated by arrows. Scale bars, 10 μm.
β-Arrestin2 recruitment to the knobs is accompanied by PKA phosphorylation
Multiple studies on GPCRs have shown that agonist-mediated receptor phosphorylation is essential for arrestin binding and receptor desensitization (Gainetdinov et al., 2004). Treatment of ORNs for 15 min with the odorant mixture treatment resulted in labeling of dendritic knobs with antibodies detecting PKA phosphorylated substrates (Fig. 6A). Without odorant application, such immunoreactivity could not be detected (Fig. 6C). Antibody labeling was specifically found in those knobs that also showed accumulation of β-arrestin2 (Fig. 6A,B). Approximately 70% of the knobs that showed staining of the PKA phosphorylated substrates also showed accumulation of β-arrestin2, and nearly all knobs in which β-arrestin2 accumulated contained PKA phosphorylated substrates. Moreover, pretreatment of the olfactory epithelium with the PKA inhibitor H-89 resulted in a reduced number of β-arrestin2-positive dendritic knobs (Fig. 6D). These results indicate that OR phosphorylation is required for stimulus-induced recruitment of β-arrestin2 to the dendritic knob.
Figure 6.
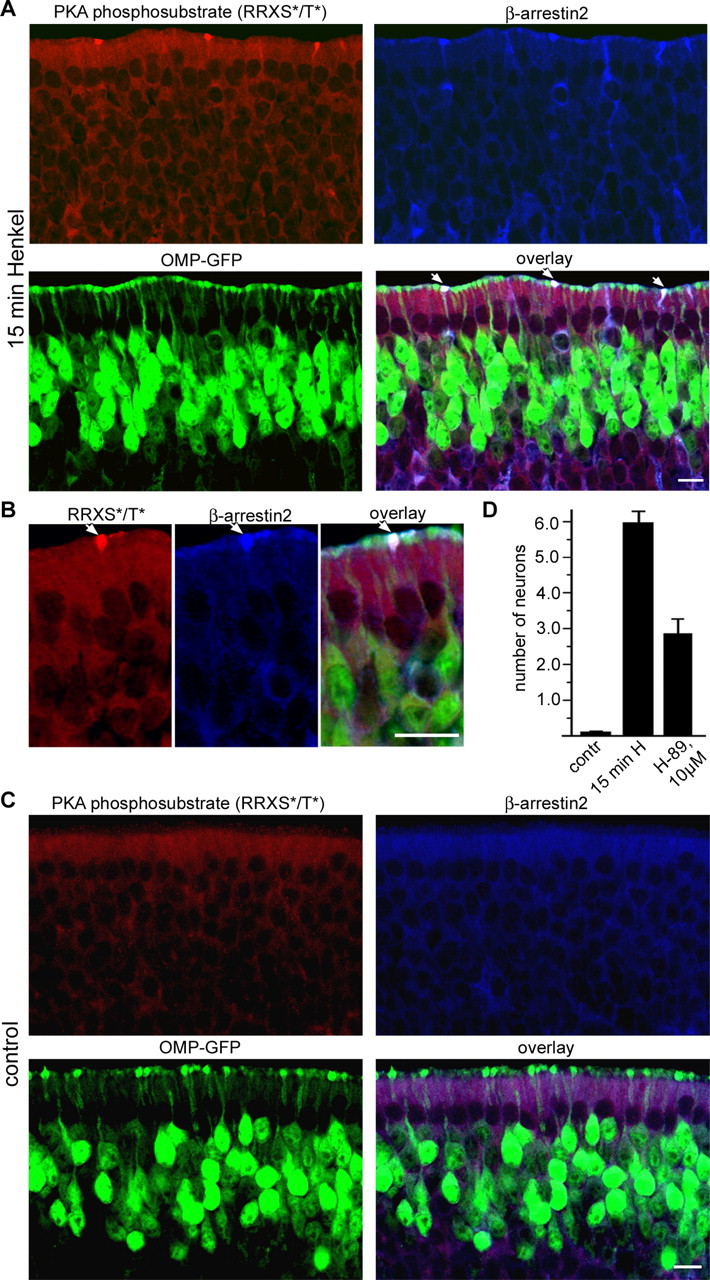
PKA is involved in β-arrestin2 accumulation in the dendritic knobs. A, Fifteen-day-old OMP–GFP mice were exposed to a complex odorant mixture of 100 compounds (Henkel 100) for 15 min, and cryosections through the olfactory epithelium were costained with antibodies against β-arrestin2 (blue) and antibodies that detect PKA phosphorylated proteins, motif RRXS*/T* (red). Strong anti-PKA phosphosubstrate immunoreactivity was observed in the cytoplasm and dendritic knobs of ORNs after odorant treatment (top left). In addition, ∼70% of the PKA phosphosubstrate-positive knobs also show strong anti-β-arrestin2 immunoreactivity (indicated by arrows on the overlay). B, Higher-magnification pictures show colocalization of PKA phosphorylated proteins (RRXS*/T*) and β-arrestin2 in the dendritic knobs of ORNs after 15 min of Henkel 100 treatment. Scale bars, 10 μm. C, Without odorant application, β-arrestin2 accumulation in the dendritic knobs and staining with the anti-PKA phosphosubstrate was not observed. D, Pretreatment of olfactory epithelium with 10 μm of the PKA inhibitor H-89 decreases the number of β-arrestin2-positive dendritic knobs after 15 min of Henkel 100 mixture treatment. Septal bone with the intact olfactory epithelium was dissected from the head and preincubated for 15 min in the large volume of Ringer’s solution (control, 15 min H) or in Ringer’s solution containing 10 μm of the PKA inhibitor H-89 (10 μm). After that, the olfactory epithelium was placed in 1:10,000 diluted Henkel 100 mixture (15 min H; H-89, 10 μm) or in Ringer’s solution (control) for 15 min and fixed. Cryosections from olfactory epithelium were stained with anti-β-arrestin2 antibodies. The number of neurons showing strong anti-β-arrestin2 reactivity in the dendritic knob was counted per 500 μm of olfactory epithelium surface. Error bars represent SEM. Scale bars, 10 μm.
Clathrin-dependent endocytosis is involved in desensitization of the odorant response
It is well known that ORNs adapt to prolonged and/or repeated odorant exposure (Zufall and Leinders-Zufall, 2000). Accordingly, mice that have been exposed to Henkel 100 for 2 h, a time when vesicular-like β-arrestin2 staining in the cytoplasm is observed, showed significantly reduced odorant responses in EOG recordings (Fig. 7A). To test whether odorant-stimulated endocytosis takes place in ORNs, we incubated olfactory epithelia with a fluid phase marker (HRP). After stimulation, the amount of internalized HRP was detected using ABTS as an HRP substrate (Fig. 7B). Odorant application increased the amount of fluid that is taken up by the cells, indicating that endocytosis can be stimulated by OR activation.
Figure 7.

Endocytosis is important for adaptation of ORNs. A, A 2 h pretreatment of mice with Henkel 100 mixture results in significantly reduced amplitude of EOG responses. Twelve mice were pretreated with Henkel 100 mixture for 2 h, and 11 mice did not receive any treatment (control group). The septal bone with the intact olfactory epithelium was dissected from the head and used intact for EOG recording as described in Materials and Methods. Increasing concentrations of Henkel 100 mixture were applied in the vapor phase for 2 s, with 1 min interval. Bars are the mean amplitude of the EOG responses from the control group (shown in white) and Henkel 100 pretreated mice (shown in black). Factorial ANOVA was performed. Error bars represent SEM; *p < 0.001. B, Treatment of mouse olfactory epithelium with Henkel 100 mixture enhances uptake of fluid phase marker HRP. Olfactory epithelium was dissected from the septal bone and incubated in solution containing 0.25 mg/ml HRP in Ringer’s solution (control; squares) or in Ringer’s solution containing Henkel 100 mixture at 1:10,000 dilution (circles) for indicated periods of time at room temperature. After the incubation, the olfactory epithelium was washed and lysed, and HRP activity was determined as described in Material and Methods. Each point is the mean from three independent experiments. Error bars represent SEM. C, D, Inhibition of clathrin-mediated endocytic pathway affects sensory adaptation in ORNs. C, A representative ratiofluorometric recording of the cytosolic Ca2+ level (fluorescence ratio, F340/F380, viewed as a function of time) shows the effect of dynamin inhibitory peptide (5 min incubation; DIP), resulting in a significant loss of adaptation. Arrows indicate repeated stimulation with a complex odorant mixture (Henkel 50 B, 5 s each) at changing intervals. D, Fluorescence image of an acutely dissociated ORN loaded with the ratiometric Ca2+ dye fura-2. Neurons are easily identified based on their characteristic morphology. Changes in Ca2+ concentration were measured in the dendritic knob region (depicted by the circle). E, Quantification of the observed effects. Bars represent the average residual response normalized to an initial odorant application as a function of the prestimulation interval. Overall, 17 individual ORNs were included in the analysis. Under control conditions, all of these neurons responded with decreasing signal amplitudes to repeated stimulation. At interstimulus intervals, ≤40 s residual response amplitudes were 57.65 ± 2.14% (control; n = 10) and 78.48 ± 8.85% (treatment; n = 6), whereas at intervals, ≤60 s residual amplitudes were 81.77 ± 4.63% (control; n = 5) and 104.13 ± 5.51% (treatment; n = 5). Response amplitudes before and after pharmacological treatment were statistically analyzed using a homoscedastic Student’s t test. Error bars represent SEM; * p < 0.1; **p < 0.01.
To investigate whether ORN desensitization is affected by clathrin-dependent internalization events, we performed Ca2+ imaging in freshly dissociated neurons and used the dynamin inhibitory peptide to selectively block the clathrin-mediated endocytosis. ORNs were identified by their characteristic morphology, and Ca2+ responses were measured selectively in individual dendritic knobs (Fig. 7D). In all ORNs tested, repeated odorant application (Henkel 50; 5 s pulses) evoked decreasing Ca2+ signals, whereas extended interstimulus intervals (∼2 min) restored the Ca2+ signal to the original level (Fig. 7C). Preincubation with the inhibitor diminished the decrease of the signal during repeated application of the odorant (Fig. 7C). When residual responses were normalized to the corresponding initial signal and plotted as a function of the interstimulus interval (Fig. 7E), we find a significantly smaller decrease of the response after pharmacological inhibition of clathrin-mediated endocytosis. These results suggest a physiological role of clathrin-dependent OR internalization plays in ORN sensory adaptation.
Discussion
ORs are expressed in the ciliary and dendritic compartments of ORNs, which have the ability to adapt to odorant stimuli in a very short time, but also can optimize their sensitivity to frequently encountered odorants. Whether and how receptor desensitization processes contribute to this cellular habituation is still unclear, partly because of a lack of knowledge about receptor endocytosis and the fate of the activated OR. In the present study, we tried to understand the basic principles of OR trafficking and desensitization in both a heterologous expression system and individual mouse ORNs and showed that ORs undergo arrestin-mediated endocytosis via a clathrin-dependent pathway.
Agonist-evoked pathways for removal of GPCRs from the cell surface, receptor endocytosis, resensitization, and degradation have successfully been revealed in cells expressing recombinant GPCRs (Seachrist and Ferguson, 2003; von Zastrow, 2003; Lefkowitz, 2004). Although receptor internalization is a common process in GPCR signaling, the fate of the receptor after agonist stimulation strongly depends on the interaction with regulatory proteins such as arrestins, which control the rate and extent of agonist-elicited turnover, as well as the fate (recycling/degradation) after endocytosis. To better understand the principles of OR desensitization and trafficking, initial experiments were performed in a heterologous expression system. By measuring the translocation of GFP-tagged β-arrestin2 during receptor activation, we found that ORs stably interact with β-arrestin2 and accumulate in endocytic vesicles, implying that ORs belong to class B of GPCRs. No differences were observed when the experiments were performed in either HEK293 or Hana3A cells (Saito et al., 2004), indicating that RTP1 (receptor transporting protein 1) and REEP1 (receptor expression enhancing protein 1), proteins involved in translocation of ORs to the plasma membrane, are not involved in receptor desensitization.
Using a bioluminescence resonance energy transfer-based β-arrestin2 recruitment assay, we also detected a sustained interaction of ORs with arrestin that steadily increased over time. Activation of class A receptors normally results in a rapid increase in agonist-promoted BRET, which peaks at ∼5 min (vasopressin receptor 1A) to ∼10 min (β2-adrenergic receptor) and then gradually declines (Hamdan et al., 2005). The sustained interaction of ORs and β-arrestin may allow monitoring of OR/arrestin complex formation in BRET assays and could thus be useful for high-throughput ligand screening of at least some members of this largest GPCR family. The nature of GPCR/arrestin interactions also determines the receptor recycling rate, and stably associated GPCRs appear to be less efficiently dephosphorylated and more slowly recycled.
During activation, GPCRs become phosphorylated by both second messenger-activated and GPCR kinases on serine and threonine residues located in the third intracellular loop and/or C-terminal tail of the receptors (Ferguson, 2001). A conserved motif consisting of clustered serine and threonine residues in the C-terminal tail of GPCRs was described to be responsible for the agonist-dependent formation of high-affinity β-arrestin complexes by binding of the phosphorylation recognition domain (arrestin) to the GRK-phosphorylated C terminus (receptor) (Gurevich and Benovic, 1997; Oakley et al., 2001). ORs lack a phosphorylation cluster in C-terminal tail but contain a putative phosphorylation cluster composed of a highly conserved triplet serine/threonine motif in their third intracellular loop. We mutated these potential phosphorylation sites (T224, S230, T240, S242, and S243 in the case of OR2AG1) and could correlate their phosphorylation with receptor desensitization. Because the mutant receptor also fails to induce endosomal accumulation of β-arrestin2–GFP, this defect in desensitization is likely caused by impaired arrestin binding. In the present study, we moreover found that PKA phosphorylation can promote OR desensitization, as reflected by a strong delay in the receptor desensitization in the presence of the PKA inhibitor H-89. Furthermore, treatment of mice with a complex odorant mixture resulted in increased PKA-mediated protein phosphorylation in ORN dendritic knobs, additional in vivo evidence of the role of PKA in olfactory signal transduction. Clearly, an important goal for future studies will be to further elucidate the mechanisms that control stable β-arrestin2/OR interaction as well as phosphorylation and ubiquitination events in OR desensitization.
In general, GPCRs can internalize via two distinct machineries, clathrin-coated pits and caveolae. Our Ca2+-imaging results provide a clear indication that ORs are internalized via clathrin coated-pits both in the heterologous expression system and in vivo in ORNs. Both, β-arrestin2 (Menco, 2005) and ORs (Barnea et al., 2004; Strotmann et al., 2004) are localized in the olfactory cilia and dendritic knobs of ORNs. We found that β-arrestin2 seems to be involved in OR signaling in vivo, because it accumulated into dendritic knobs of ORNs during treatment of mice with a complex odorant mixture. In rod photoreceptors, arrestin relocalization from the inner to the outer segment is believed to play a role in light and dark adaptation, with phosphorylated rhodopsin acting as a sink for passively diffusing arrestin molecules (Arshavsky, 2003; Nair et al., 2005). In our experiments, we do not see whether there is also redistribution of β-arrestin2 to the “outer parts” of the ORNs, the cilia of the dendritic knob. Redistribution of β-arrestin2 to the cilia could precede its accumulation in the dendritic knob, depending on where cilia or dendritic knob arrestin binds to the activated OR. Because coated vesicle-mediated transport of ORs to the base of the cilium is likely compromised because of the limited space between axonemal microtubules and the ciliary membrane, internalization of the receptor–arrestin complex via clathrin-coated vesicles could only occur in the dendritic knob, in which coated pits invaginate and numerous endocytic vesicles exist (Bannister and Dodson, 1992). Interestingly, although endocytosis has not been described as a main pathway to shut off phototransduction in photoreceptor outer segments, endocytic compartments and activity have been described in the inner segments (Cotter, 1989), and light induces internalization of rhodopsin and phosphorylated arrestin into rod cells bodies (Reme et al., 1999; Chuang et al., 2004).
Recent electrophysiological work has suggested that the termination of the activity of the receptor is simply caused by unbinding of the odorant molecule (Bhandawat et al., 2005). Our data now suggest that, at least during prolonged and/or intense stimulation of ORs, desensitization involves phosphorylation and recruitment of arrestins. Prolonged exposure of mice to an odorant mixture resulted in reduced EOG amplitudes. Because the residual responses observed in field potential recordings result from integrated effects of various adaptation mechanisms on all levels of olfactory signal transduction, they cannot be explained solely by receptor endocytosis, and the extent of the contribution of receptor internalization to the observed reduced EOG responses remains unclear. Long odorant exposure likely results in a lowered density of ORs at the membrane, resulting in reduced signal strength but not necessarily in a complete surface removal of all receptors. In addition, some odorants in the mixture might activate ORs that are not internalized or are recycled fast. Although we observed stable β-arrestin2 binding for different ORs, we cannot rule out that some ORs show a different behavior. Ca2+-imaging recordings of repetitive odorant responses in single ORNs reveal a significant decrease in signal adaptation when clathrin-dependent endocytosis is inhibited. As derived from data quantification (Fig. 7E), this inhibitory effect, however, does not abolish signal adaptation in ORNs but rather adds another molecular mechanism to the complex biochemical interplay that governs peripheral response adaptation in the olfactory system.
The fact that, after prolonged receptor stimulation, the majority of the receptor protein colocalized with transferrin and only marginally with a lysosomal marker indicates that recombinant ORs mimic to some extent the trafficking route of typical class B GPCRs, as for example V2 receptors, which accumulate in the perinuclear recycling compartment (Innamorati et al., 2001). Interestingly, our finding that ORs are not targeted for degradation even after relatively long odorant exposure implies that the internalized receptor could return to the cell surface and reengage in signaling, likely via an indirect “long cycle” traversing the perinuclear recycling endosomes (Innamorati et al., 2001). After prolonged treatment of ORNs with odorants, β-arrestin2 accumulated in intracellular vesicular structures, which may represent an endocytic compartment. Considering the fact that recombinant ORs stably interact with β-arrestin2 and accumulated in an endocytic compartment, one could speculate that the receptor might also be present in these vesicular structures. The stability of the GPCR/β-arrestin complex regulates the receptor resensitization kinetics (Oakley et al., 1999; Pan et al., 2003) but is also involved in the initiation and/or regulation of other signaling pathways. β-Arrestin functions as a molecular scaffold that recruits components of mitogen-activated protein kinase (MAPK) cascade (Shenoy and Lefkowitz, 2005), and prolonged association between receptor and β-arrestin ensures the proper localization and/or duration of the MAPK response. Phosphorylated MAPK accumulated in the cytosol of ORNs after prolonged agonist stimulation (data not shown), which may be related to the presence of arrestin in the endosomal membranes.
This report may help to understand the role of OR trafficking as a background for signaling events in the olfactory epithelium. Insight into the molecular determinants underlying high-affinity OR/β-arrestin2 interactions may help to fully understand the mechanisms and consequences of OR activation in ORNs. Finally, it seems that ORs have to be added to the long list of GPCRs that internalize via clathrin-dependent pathway using β-arrestin2.
Footnotes
This study was supported by grants from the Deutsche Forschungsgemeinschaft Sonderforschungsbereich 642 (E.M.N., H.H.) and SP724/2-1 (M.S.), and the International Max-Planck Research School for Chemical Biology. We thank W. Grabowski, H. Bartel, and J. Gerkrath for excellent technical assistance, R. Dooley for critical reading of this manuscript, H. Matsunami (Duke University Medical Center, Durham, NC) for donation of Hana3A cells, and P. Mombaerts (Rockefeller University, New York, NY) for donation of the transgenic OMP/GFP-mice.
References
- Arshavsky VY. Protein translocation in photoreceptor light adaptation: a common theme in vertebrate and invertebrate vision. Sci STKE. 2003;2003:E43. doi: 10.1126/stke.2003.204.pe43. [DOI] [PubMed] [Google Scholar]
- Bannister LH, Dodson HC. Endocytic pathways in the olfactory and vomeronasal epithelia of the mouse: ultrastructure and uptake of tracers. Microsc Res Tech. 1992;23:128–141. doi: 10.1002/jemt.1070230204. [DOI] [PubMed] [Google Scholar]
- Barnea G, O’Donnell S, Mancia F, Sun X, Nemes A, Mendelsohn M, Axel R. Odorant receptors on axon termini in the brain. Science. 2004;304:1468. doi: 10.1126/science.1096146. [DOI] [PubMed] [Google Scholar]
- Bhandawat V, Reisert J, Yau KW. Elementary response of olfactory receptor neurons to odorants. Science. 2005;308:1931–1934. doi: 10.1126/science.1109886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boekhoff I, Schleicher S, Strotmann J, Breer H. Odor-induced phosphorylation of olfactory cilia proteins. Proc Natl Acad Sci USA. 1992;89:11983–11987. doi: 10.1073/pnas.89.24.11983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley J, Bonigk W, Yau KW, Frings S. Calmodulin permanently associates with rat olfactory CNG channels under native conditions. Nat Neurosci. 2004;7:705–710. doi: 10.1038/nn1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck L, Axel R. A novel multigene family may encode odorant receptors: a molecular basis for odor recognition. Cell. 1991;65:175–187. doi: 10.1016/0092-8674(91)90418-x. [DOI] [PubMed] [Google Scholar]
- Buck LB. The olfactory multigene family. Curr Opin Neurobiol. 1992;2:282–288. doi: 10.1016/0959-4388(92)90116-3. [DOI] [PubMed] [Google Scholar]
- Chuang JZ, Vega C, Jun W, Sung CH. Structural and functional impairment of endocytic pathways by retinitis pigmentosa mutant rhodopsin-arrestin complexes. J Clin Invest. 2004;114:131–140. doi: 10.1172/JCI21136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotter RJ. Endocytosis of cationized ferritin by rat photoreceptors. Neurosci Lett. 1989;106:65–70. doi: 10.1016/0304-3940(89)90203-6. [DOI] [PubMed] [Google Scholar]
- Dawson TM, Arriza JL, Jaworsky DE, Borisy FF, Attramadal H, Lefkowitz RJ, Ronnett GV. Beta-adrenergic receptor kinase-2 and beta-arrestin-2 as mediators of odorant-induced desensitization. Science. 1993;259:825–829. doi: 10.1126/science.8381559. [DOI] [PubMed] [Google Scholar]
- Ferguson SS. Evolving concepts in G protein-coupled receptor endocytosis: the role in receptor desensitization and signaling. Pharmacol Rev. 2001;53:1–24. [PubMed] [Google Scholar]
- Gainetdinov RR, Premont RT, Bohn LM, Lefkowitz RJ, Caron MG. Desensitization of G protein-coupled receptors and neuronal functions. Annu Rev Neurosci. 2004;27:107–144. doi: 10.1146/annurev.neuro.27.070203.144206. [DOI] [PubMed] [Google Scholar]
- Gurevich VV, Benovic JL. Mechanism of phosphorylation-recognition by visual arrestin and the transition of arrestin into a high affinity binding state. Mol Pharmacol. 1997;51:161–169. doi: 10.1124/mol.51.1.161. [DOI] [PubMed] [Google Scholar]
- Hamdan FF, Audet M, Garneau P, Pelletier J, Bouvier M. High-throughput screening of G protein-coupled receptor antagonists using a bioluminescence resonance energy transfer 1-based beta-arrestin2 recruitment assay. J Biomol Screen. 2005;10:463–475. doi: 10.1177/1087057105275344. [DOI] [PubMed] [Google Scholar]
- Heding A. Use of the BRET 7TM receptor/beta-arrestin assay in drug discovery and screening. Expert Rev Mol Diagn. 2004;4:403–411. doi: 10.1586/14737159.4.3.403. [DOI] [PubMed] [Google Scholar]
- Innamorati G, Le Gouill C, Balamotis M, Birnbaumer M. The long and the short cycle. Alternative intracellular routes for trafficking of G-protein-coupled receptors. J Biol Chem. 2001;276:13096–13103. doi: 10.1074/jbc.M009780200. [DOI] [PubMed] [Google Scholar]
- Katada S, Hirokawa T, Oka Y, Suwa M, Touhara K. Structural basis for a broad but selective ligand spectrum of a mouse olfactory receptor: mapping the odorant-binding site. J Neurosci. 2005;25:1806–1815. doi: 10.1523/JNEUROSCI.4723-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurahashi T, Menini A. Mechanism of odorant adaptation in the olfactory receptor cell. Nature. 1997;385:725–729. doi: 10.1038/385725a0. [DOI] [PubMed] [Google Scholar]
- Lefkowitz RJ. Historical review: a brief history and personal retrospective of seven-transmembrane receptors. Trends Pharmacol Sci. 2004;25:413–422. doi: 10.1016/j.tips.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Leinders-Zufall T, Ma M, Zufall F. Impaired odor adaptation in olfactory receptor neurons after inhibition of Ca2+/calmodulin kinase II. J Neurosci. 1999;19(RC19):1–6. doi: 10.1523/JNEUROSCI.19-14-j0005.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menco BP. The fine-structural distribution of G-protein receptor kinase 3, beta-arrestin-2, Ca2+/calmodulin-dependent protein kinase II and phosphodiesterase PDE1C2, and a Cl−-cotransporter in rodent olfactory epithelia. J Neurocytol. 2005;34:11–36. doi: 10.1007/s11068-005-5045-9. [DOI] [PubMed] [Google Scholar]
- Menini A, Lagostena L, Boccaccio A. Olfaction: from odorant molecules to the olfactory cortex. News Physiol Sci. 2004;19:101–104. doi: 10.1152/nips.1507.2003. [DOI] [PubMed] [Google Scholar]
- Mombaerts P. Genes and ligands for odorant, vomeronasal and taste receptors. Nat Rev Neurosci. 2004;5:263–278. doi: 10.1038/nrn1365. [DOI] [PubMed] [Google Scholar]
- Munger SD, Lane AP, Zhong H, Leinders-Zufall T, Yau KW, Zufall F, Reed RR. Central role of the CNGA4 channel subunit in Ca2+-calmodulin-dependent odor adaptation. Science. 2001;294:2172–2175. doi: 10.1126/science.1063224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair KS, Hanson SM, Mendez A, Gurevich EV, Kennedy MJ, Shestopalov VI, Vishnivetskiy SA, Chen J, Hurley JB, Gurevich VV, Slepak VZ. Light-dependent redistribution of arrestin in vertebrate rods is an energy-independent process governed by protein-protein interactions. Neuron. 2005;46:555–567. doi: 10.1016/j.neuron.2005.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuhaus EM, Gisselmann G, Zhang W, Dooley R, Stortkuhl K, Hatt H. Odorant receptor heterodimerization in the olfactory system of Drosophila melanogaster. Nat Neurosci. 2005;8:15–17. doi: 10.1038/nn1371. [DOI] [PubMed] [Google Scholar]
- Neuhaus EM, Mashukova A, Zhang W, Barbour J, Wolters D, Hatt H. A specific heat shock protein enhances the expression of mammalian olfactory receptor proteins. Chem Senses. 2006;31:445–452. doi: 10.1093/chemse/bjj049. [DOI] [PubMed] [Google Scholar]
- Oakley RH, Laporte SA, Holt JA, Barak LS, Caron MG. Association of beta-arrestin with G protein-coupled receptors during clathrin-mediated endocytosis dictates the profile of receptor resensitization. J Biol Chem. 1999;274:32248–32257. doi: 10.1074/jbc.274.45.32248. [DOI] [PubMed] [Google Scholar]
- Oakley RH, Laporte SA, Holt JA, Barak LS, Caron MG. Molecular determinants underlying the formation of stable intracellular G protein-coupled receptor-beta-arrestin complexes after receptor endocytosis*. J Biol Chem. 2001;276:19452–19460. doi: 10.1074/jbc.M101450200. [DOI] [PubMed] [Google Scholar]
- Pan L, Gurevich EV, Gurevich VV. The nature of the arrestin x receptor complex determines the ultimate fate of the internalized receptor. J Biol Chem. 2003;278:11623–11632. doi: 10.1074/jbc.M209532200. [DOI] [PubMed] [Google Scholar]
- Peppel K, Boekhoff I, McDonald P, Breer H, Caron MG, Lefkowitz RJ. G protein-coupled receptor kinase 3 (GRK3) gene disruption leads to loss of odorant receptor desensitization. J Biol Chem. 1997;272:25425–25428. doi: 10.1074/jbc.272.41.25425. [DOI] [PubMed] [Google Scholar]
- Potter SM, Zheng C, Koos DS, Feinstein P, Fraser SE, Mombaerts P. Structure and emergence of specific olfactory glomeruli in the mouse. J Neurosci. 2001;21:9713–9723. doi: 10.1523/JNEUROSCI.21-24-09713.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rankin ML, Alvania RS, Gleason EL, Bruch RC. Internalization of G protein-coupled receptors in single olfactory receptor neurons. J Neurochem. 1999;72:541–548. doi: 10.1046/j.1471-4159.1999.0720541.x. [DOI] [PubMed] [Google Scholar]
- Reed RR. Signaling pathways in odorant detection. Neuron. 1992;8:205–209. doi: 10.1016/0896-6273(92)90287-n. [DOI] [PubMed] [Google Scholar]
- Reme CE, Wolfrum U, Imsand C, Hafezi F, Williams TP. Photoreceptor autophagy: effects of light history on number and opsin content of degradative vacuoles. Invest Ophthalmol Vis Sci. 1999;40:2398–2404. [PubMed] [Google Scholar]
- Saito H, Kubota M, Roberts RW, Chi Q, Matsunami H. RTP family members induce functional expression of mammalian odorant receptors. Cell. 2004;119:679–691. doi: 10.1016/j.cell.2004.11.021. [DOI] [PubMed] [Google Scholar]
- Schleicher S, Boekhoff I, Arriza J, Lefkowitz RJ, Breer H. A beta-adrenergic receptor kinase-like enzyme is involved in olfactory signal termination. Proc Natl Acad Sci USA. 1993;90:1420–1424. doi: 10.1073/pnas.90.4.1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seachrist JL, Ferguson SS. Regulation of G protein-coupled receptor endocytosis and trafficking by Rab GTPases. Life Sci. 2003;74:225–235. doi: 10.1016/j.lfs.2003.09.009. [DOI] [PubMed] [Google Scholar]
- Shenoy SK, Lefkowitz RJ. Multifaceted roles of beta-arrestins in the regulation of seven-membrane-spanning receptor trafficking and signalling. Biochem J. 2003;375:503–515. doi: 10.1042/BJ20031076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenoy SK, Lefkowitz RJ. Seven-transmembrane receptor signaling through beta-arrestin. Sci STKE. 2005;2005:cm10. doi: 10.1126/stke.2005/308/cm10. [DOI] [PubMed] [Google Scholar]
- Spehr M, Wetzel CH, Hatt H, Ache BW. 3-phosphoinositides modulate cyclic nucleotide signaling in olfactory receptor neurons. Neuron. 2002;33:731–739. doi: 10.1016/s0896-6273(02)00610-4. [DOI] [PubMed] [Google Scholar]
- Spehr M, Gisselmann G, Poplawski A, Riffell JA, Wetzel CH, Zimmer RK, Hatt H. Identification of a testicular odorant receptor mediating human sperm chemotaxis. Science. 2003;299:2054–2058. doi: 10.1126/science.1080376. [DOI] [PubMed] [Google Scholar]
- Spehr M, Schwane K, Riffell JA, Barbour J, Zimmer RK, Neuhaus EM, Hatt H. Particulate adenylate cyclase plays a key role in human sperm olfactory receptor-mediated chemotaxis. J Biol Chem. 2004;279:40194–40203. doi: 10.1074/jbc.M403913200. [DOI] [PubMed] [Google Scholar]
- Strotmann J, Levai O, Fleischer J, Schwarzenbacher K, Breer H. Olfactory receptor proteins in axonal processes of chemosensory neurons. J Neurosci. 2004;24:7754–7761. doi: 10.1523/JNEUROSCI.2588-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Zastrow M. Mechanisms regulating membrane trafficking of G protein-coupled receptors in the endocytic pathway. Life Sci. 2003;74:217–224. doi: 10.1016/j.lfs.2003.09.008. [DOI] [PubMed] [Google Scholar]
- Wetzel CH, Oles M, Wellerdieck C, Kuczkowiak M, Gisselmann G, Hatt H. Specificity and sensitivity of a human olfactory receptor functionally expressed in human embryonic kidney 293 cells and Xenopus laevis oocytes. J Neurosci. 1999;19:7426–7433. doi: 10.1523/JNEUROSCI.19-17-07426.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wetzel CH, Spehr M, Hatt H. Phosphorylation of voltage-gated ion channels in rat olfactory receptor neurons. Eur J Neurosci. 2001;14:1056–1064. doi: 10.1046/j.0953-816x.2001.01722.x. [DOI] [PubMed] [Google Scholar]
- Zhainazarov AB, Spehr M, Wetzel CH, Hatt H, Ache BW. Modulation of the olfactory CNG channel by Ptdlns(3,4,5)P3. J Membr Biol. 2004;201:51–57. doi: 10.1007/s00232-004-0707-4. [DOI] [PubMed] [Google Scholar]
- Zufall F, Leinders-Zufall T. The cellular and molecular basis of odor adaptation. Chem Senses. 2000;25:473–481. doi: 10.1093/chemse/25.4.473. [DOI] [PubMed] [Google Scholar]
- Zufall F, Shepherd GM, Firestein S. Inhibition of the olfactory cyclic nucleotide gated ion channel by intracellular calcium. Proc Biol Sci; 1991. pp. 225–230. [DOI] [PubMed] [Google Scholar]
- Zufall F, Firestein S, Shepherd GM. Cyclic nucleotide-gated ion channels and sensory transduction in olfactory receptor neurons. Annu Rev Biophys Biomol Struct. 1994;23:577–607. doi: 10.1146/annurev.bb.23.060194.003045. [DOI] [PubMed] [Google Scholar]