

Abstract
Aberrant aggregation of α-synuclein (α-syn) to form fibrils and insoluble aggregates has been implicated in the pathogenic processes of many neurodegenerative diseases. Despite the dramatic effects of dopamine in inhibiting the formation of α-syn fibrils by stabilization of oligomeric intermediates in cell-free systems, no studies have examined the effects of intracellular dopamine on α-syn aggregation. To study this process and its association with neurodegeneration, intracellular catechol levels were increased to various levels by expressing different forms of tyrosine hydroxylase, in cells induced to form α-syn aggregates. The increase in the steady-state dopamine levels inhibited the formation of α-syn aggregates and induced the formation of innocuous oligomeric intermediates. Analysis of transgenic mice expressing the disease-associated A53T mutant α-syn revealed the presence of oligomeric α-syn in nondegenerating dopaminergic neurons that do contain insoluble α-syn. These data indicate that intraneuronal dopamine levels can be a major modulator of α-syn aggregation and inclusion formation, with important implications on the selective degeneration of these neurons in Parkinson's disease.
Keywords: α-synuclein, catecholamines, neurodegeneration, Parkinson's disease, tyrosine hydroxylase, protein aggregation
Introduction
α-Synuclein (α-syn; NACP, synelfin) was first cloned from the electric lobe of Torpedo californica, where it was found concentrated around vesicles in synaptic terminals (Maroteaux et al., 1988). Primary structure analysis of α-syn revealed seven recurring imperfect repeats in the N terminus, which facilitates folding into amphipathic α-helices, resembling the lipid-binding domain of exchangeable apolipoproteins (George et al., 1995). Subsequent studies revealed that α-syn indeed assumes an α-helical structure after binding to phospholipids (Davidson et al., 1998) and functions to modulate synaptic transmission through maintenance of reserve pool vesicles (Murphy et al., 2000; Cabin et al., 2002), as well as contributes to the maintenance of SNARE (soluble N-ethylmaleimide-sensitive factor attachment protein receptor) protein complexes by chaperone-like functions (Chandra et al., 2005). In support of a chaperone function, α-syn shows functional homology to 14-3-3 proteins and can inhibit thermally or chemically induced protein aggregation (Ostrerova et al., 1999; Souza et al., 2000a).
Pathologically, intraneuronal inclusions composed of α-syn fibrils have been detected in a variety of neurodegenerative diseases including Parkinson's disease (PD), dementia with Lewy bodies, Lewy body variant of Alzheimer's disease, and multiple system atrophy, collectively termed synucleinopathies (Spillantini et al., 1997; Trojanowski and Lee, 2003). Additionally, modifications in the α-syn gene (SNCA), including point mutations (A53T, A30P, and E46K), are responsible for the familial forms of PD (Polymeropoulos et al., 1997; Kruger et al., 1998; Zarranz et al., 2004); SNCA locus duplication and triplication have also linked α-syn to neurodegenerative mechanisms (Singleton and Gwinn-Hardy, 2004). The A53T and E46K mutations accelerate the rate of α-syn aggregation (Giasson et al., 1999; Greenbaum et al., 2005). Along with the lack of disease phenotype in α-syn knock-out mice (Abeliovich et al., 2000), this suggests that a gain in toxic function through aberrant α-syn aggregation is responsible for α-syn-mediated neuronal dysfunction. Factors or compounds that inhibit the process of aggregation may thus be useful to treat a variety of synucleinopathies.
The biochemical and biophysical parameters that govern the organized assembly of α-syn into fibers are not known, but several proposals have been advanced. These include an increase in α-syn concentration (e.g., by gene triplication) to a critical threshold that could overcome the thermodynamic requirements for the initiation of fibril formation (Giasson et al., 1999; Singleton and Gwinn-Hardy, 2004); an inappropriate interaction with other proteins, such as tau, that leads to an ordered coassembly of the two proteins primarily to homopolymeric fibrils (Giasson et al., 2003); post-translational modifications such as phosphorylation at Ser 129 (Fujiwara et al., 2002; Chen and Feany, 2005; Smith et al., 2005); partial or inappropriate cleavage of the flexible and highly negatively charged C terminus (Murray et al., 2003; Li et al., 2005; Mishizen-Eberz et al., 2005; Tofaris et al., 2006); and oxidative post-translational modifications, which result in the formation of oxidized and nitrated α-syn (Giasson et al., 2000b; Norris et al., 2003; Hodara et al., 2004).
One of the most potentially important oxidation-dependent events is the interaction of dopamine (DA) with α-syn. This interaction studied exclusively in vitro has been shown to inhibit the formation of fibrils by kinetically stabilizing oligomeric α-syn intermediates (Conway et al., 2001; J. Li et al., 2004; Cappai et al., 2005; Norris et al., 2005). Despite the profound implications for the pathogenesis of PD and related synucleinopathies, the interaction of α-syn with DA in vivo has not been evaluated. To that end, we have developed cellular model systems to molecularly manipulate the intracellular steady-state levels of DA to evaluate the effect on α-syn aggregation.
Materials and Methods
Cell culture and transfection to generate α-syn-expressing stable cell lines.
SH-SY5Y cells (catalog #CRL-2266; American Type Culture Collection, Manassas, VA) were maintained in DMEM/F-12 medium (Invitrogen, Carlsbad, CA) supplemented with 10% heat-inactivated fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin (complete medium) at 37°C with 5% CO2. Cells were transfected using Lipofectamine 2000 (Invitrogen) with the empty pcDNA3.1 vector (control) or pcDNA3.1 expression vectors in which the full-length human wild-type (wt) or A53T α-syn cDNAs were cloned in the polylinker at the KpnI and ApaI sites. Stable transfected cell lines were selected and maintained in complete medium containing 300 μg/ml G418 (Invitrogen).
Generation of tyrosine hydroxylase-containing lentiviral vectors and cell infection.
Generation of human tyrosine hydroxylase-1 (hTH-1) construct containing the R37G/R38G (RR-GG) or R37E/R38E (RR-EE) mutations have been described previously (Nakashima et al., 2002). Coding sequences of wt, RR-GG, or RR-EE mutants of hTH-1 were removed from pcDNA3.1 vectors and subcloned into the pTY linker lentiviral expression vector at the PmeI site. Lentivirus was prepared according to the optimized protocol described by Karolewski et al. (2003). Briefly, HEK 293T cells were seeded at 8 × 106 cells/10 cm poly-d-lysine-coated dish and transfected with calcium-phosphate N,N-bis(2-hydroxyethyl)-2-aminoethanesulfonic acid–DNA precipitates at 37°C with 3% CO2 overnight. The following amounts of plasmids per plate were used for the tranfections: JS86-VSV-G (1.25 μg), CMVΔ82 (3.75 μg), and pTY-linker expression vector (5 μg). Forty milliliters of culture medium containing virus were collected every 24 h (up to 72 h), filtered through 0.45 μm membranes, concentrated by ultracentrifugation at 50,000 × g for 2 h at 4°C, and resuspended in 1 ml of cold complete DMEM/F-12 medium to make a 40× concentrated virus stock. The virus was titered by TH immunostaining of SH-SY5Y target cells. Equal viral titers were verified by TH Western blot analysis (see Fig. 2 a).
Figure 2.
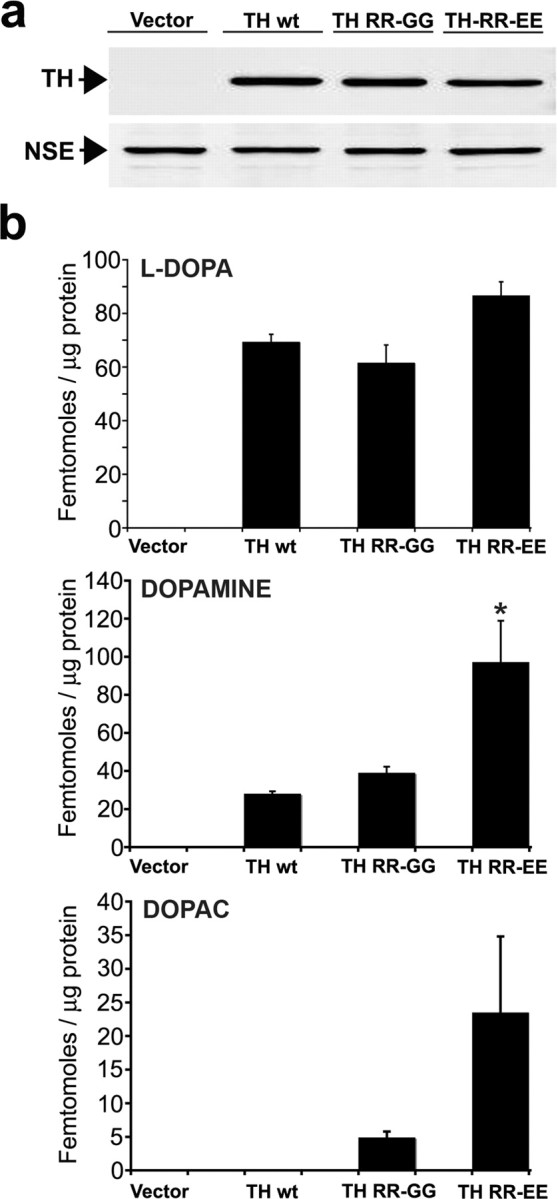
Manipulation of intracellular catechol levels in SH-SY5Y cells expressing A53T α-syn by infection with lentivirus-containing TH expression plasmids. A53T α-syn-expressing cells were infected with lentivirus generated with empty vector plasmid (Vector), or vector expressing wt TH (TH wt), 37,38 RR-GG TH (TH RR-GG), or 37,38 RR-EE TH (TH RR-EE). The cells were infected using a multiplicity of infection that transduced ∼50% of total cells and cultured for 5 d in the presence of 20 μm RA. a, Western blot analysis of TH-infected cells shows similar expression of each TH protein. NSE was used as a loading control. b, Quantification of intracellular catechol levels HPLC with on-line electrochemical detection. Similar results were obtained when wt α-syn-expressing cells were infected with TH-containing plasmids (data not shown). Values are the mean ± SEM (n = 3). *p < 0.05 relative to TH wt and TH RR-GG.
For each experiment, cells were transduced using a final concentration of 5× concentrated viral supernatant (average infectious units per milliliter are 3 × 106), which corresponds to ∼40–50% total naive cell transduction (or a multiplicity of infection of 10). For all experiments with SH-SY5Y cells stably expressing wt or A53T α-syn, cells were seeded at 5–6.5 × 104 cells/cm2 in culture dishes, allowed to recover for 2 d, infected with lentivirus containing empty vector or TH expression plasmids, and differentiated with 20 μm trans-retinoic acid (RA) for 5 d.
Catechol quantification by HPLC with electrochemical detection.
After infection with lentivirus-containing TH expression plasmids and differentiation with RA, cells were washed with cold Dulbecco's PBS without calcium and magnesium, pH 7.4 (D-PBS), and harvested in 100 μl of 0.1 m perchloric acid containing 100 nm 3,4 dihydroxybenzylamine (DHBA). Cell lysates were sonicated and centrifuged at 16,000 × g for 10 min at 4°C. Supernatants were filtered through 0.22 μm membranes, and 50 μl was injected on the HPLC for catechol analysis using an Agilent (Palo Alto, CA) 1100 series HPLC controlled by ChemStation software (Agilent). Catechols were resolved on a reverse-phase C18 Luna column (150 × 4.6 mm, 5 μm; Phenomenex, Torrance, CA) at a flow rate of 1 ml/min in a mobile phase consisting of 72 mm citric acid, 28 mm sodium phosphate, 2% methanol, and 0.04% sodium azide, pH 2.8, with a 2–10% methanol gradient, and detected using a Coularray detector (ESA Biosciences, Chelmsford, MA) with the following working potentials (in mV): −200, +200, +300, and +400. Catechol quantification was done by comparing the peak areas of a known amount of standards and using Chemstation software (version 1.04; ESA Biosciences).
Protein pellets were solubilized in 2% SDS and 50 mm Tris-Cl, pH 7.4, containing protease inhibitors (Sigma, St. Louis, MO), and 50 μg of total lysate was analyzed by Western blot using an anti-TH monoclonal antibody (1:5000; EMD Biosciences, La Jolla, CA) and a polyclonal antibody specific for neural-specific enolase (NSE; 1:2000; Polysciences, Warrington, PA). Western blotting was performed as described in below (see Sequential protein extraction and Western blot analysis). Protein concentration determination was done using the micro BCA kit (Pierce, Rockford, IL) using BSA as a standard. Intracellular catechol levels were normalized to both DHBA and protein concentration and expressed as femtomoles per microgram of protein.
Sequential protein extraction and Western blot analysis.
Cells were washed twice with cold D-PBS and scraped in 200 μl of lysis buffer [1% Triton X-100, 20 mm HEPES, 150 mm NaCl, 10% glycerol, 1 mm EGTA, 1.5 mm MgCl2, 1 mm PMSF, 10 mm Na pyruvate, 50 mm NaF, 2 mm Na orthovanadate, 1 μm lactacystin, and a protease inhibitor mixture (Sigma), pH 7.4]. Lysates were homogenized with a pestle and subjected to four successive freeze–thaw cycles to disrupt membranes. Triton-soluble fractions were obtained by ultracentifugation at 100,000 × g for 30 min at 4°C. Triton-insoluble pellets were extracted in 2% SDS, 50 mm Tris, pH 7.4, and a protease inhibitor mixture by sonication and boiling for 20 min and ultracentrifuged at 110,000 × g for 30 min at 25°C. The supernatants were saved as the SDS-soluble fractions, and remaining pellets were extracted in 70% formic acid by sonication and incubation at 37°C for 20 min. Protein concentration from Triton and SDS-soluble fractions was determined by the BCA assay as described above. Formic acid fractions were normalized according to the protein concentration in the corresponding Triton-soluble fractions. Samples were boiled in 1× sample buffer (20 mm Tris, 1% glycerol, 180 mm β-mercaptoethanol, 0.003% bromophenol blue, and 2% SDS, pH 6.8), loaded on 12% SDS-PAGE gels, and transferred to polyvinylidene difluoride membranes (0.45-μm-pore immobilon FL; Millipore, Billerica, MA). The blots were blocked in 1× TBS-T (20 mm Tris, pH 7.4, 150 mm NaCl, and 0.2% Tween 20) containing 5% milk for 1 h and incubated with the following primary antibodies in blocking solution at the indicated dilutions for 2 h at 25°C or overnight at 4°C: monoclonal Syn 211 and LB509 (1:1000), monoclonal TH (1:5000; EMD Biosciences), polyclonal NSE (1:2000; Polysciences), and monoclonal vimentin (1:800; BD PharMingen, San Diego, CA). LB509 recognizes amino acids 115–122 and has been described previously and characterized (Baba et al., 1998; Jakes et al., 1999). Syn 211 recognizes amino acids 120–125 (Giasson et al., 2000a). Primary antibodies were detected by 1 h incubation with either goat anti-mouse IgG Alexa Fluor 680 (1:5000; Invitrogen, Eugene, OR) or goat anti-rabbbit IgG IRDye 800 (1:5000; Rockland, Gilbertsville, PA)-conjugated secondary antibodies in blocking buffer and scanned at intensity level 2 with an Odessey Infrared Imaging System (Li-Cor, Lincoln, NE).
Gel filtration analysis.
After infection with lentivirus-containing TH expression plasmids and differentiation with RA, cells were harvested as described above. One milligram of total Triton-soluble protein in a total volume of 250 μl was resolved onto a Superdex 200 HR10/30 column (GE Healthcare Bio-Sciences, Uppsala, Sweden) for size exclusion chromatography (SEC) analysis. Western blot analysis for NSE and UV absorbance at 210 and 280 nm was used to verify that equal amounts of protein were used. Protein extracts were resolved at a flow rate of 0.3 ml/min in 25 mm HEPES, pH 7.25, and 150 mm NaCl using an Agilent 1100 series HPLC system. Fractions (0.5 ml) were collected and concentrated with 5000 NMWL Ultrafree-MC filters (Millipore). Fractions 2–5 (99–63 Å), 6–9 (63–48 Å), 18–20 (25–20 Å), and 21–24 (20–11 Å) were combined and loaded into one lane on SDS-PAGE gels to enhance the signals. Western blotting was performed as described above using Syn 211, stripped, and reprobed with LB509. The gel filtration column was calibrated using the following mixture of globular proteins standards: thyroglobulin (669 kDa, 85 Å), ferritin (440 kDa, 61 Å), catalase (232 kDa, 52.2 Å), aldolase (158 kDa, 48 Å), albumin (67 kDa, 35.5 Å), ovalbumin (43 kDa, 30.5 Å), chymotrypsin (25 kDa, 20.9 Å), and ribonuclease A (13.7 kDa, 16.4 Å). The void volume was determined from the elution migration of blue dextran (2000 kDa, 99 Å).
Immunofluorescence analysis.
Cells were washed twice with cold D-PBS and fixed with cold methanol for 20 min at −20°C, followed by incubation in a 1:1 mix of cold methanol/acetone for 5 min at −20°C. The cells were washed with cold PBS-T (50 mm phosphate, 50 mm NaCl, pH 7.2, and 0.3% Triton) and incubated overnight at 4°C in PBS-T. The next day, the cells were blocked in blocking buffer (5% BSA and 10% normal goat serum in PBS-T) for 30 min, washed in PBS-T, and incubated with the following primary antibodies in blocking buffer overnight at 4°C: Syn 211, LB509, and SNL-1 (all 1:700); and monoclonal or polyclonal anti-TH (1:1000; EMD Biosciences). The cells were washed three times for 10 min in PBS-T, and the following secondary antibodies were incubated for 1 h at room temperature: Cy3-conjugated anti-mouse or anti-rabbit IgG (1:100; Jackson ImmunoResearch, West Grove, PA), Alexa 488-conjugated anti-mouse or anti-rabbit IgG (1:300; Invitrogen), and 7-amino-4-methylcoumarin-3-acetic acid (AMCA)-conjugated anti-rabbit IgG (1:50; Vector Laboratories, Burlingame, CA). To detect amyloidogenic aggregates, 0.05% thioflavin S (Sigma) in 50% ethanol was added to fixed cells for 15 min at room temperature, after antibody incubations. The cells were washed two times for 20 min in 50% ethanol, followed by one 20 min wash in 80% ethanol. Ethanol was removed by briefly washing in PBS-T. Nuclei were visualized by staining with 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI) at 200 ng/ml for 15 min. Cells were visualized using an inverted IX70 microscope using the 20× objective with an IX-FLA fluorescence observation attachment (Olympus Optical, Tokyo, Japan). Fluorophores were excited at the following wavelengths (in nm): AMCA and DAPI, 350; Cy3, 550; Alexa 488 and thioflavin S, 485. Cell images were captured with a Hamamatsu (Hamamatsu City, Japan) digital camera using MetaMorph software (version 6.0; Universal Imaging Corporation, West Chester, PA). For quantitation of cells containing aggregates, 10 different fields of view were randomly selected for each condition, using C-terminal recognition monoclonal antibodies Syn 211 and LB509 and polyclonal antibody SNL-1 (epitope amino acids 104–119). Between 100 and 300 cells were counted for each condition per antibody. For Figure 3 c, transduced cells were clearly identified by TH immunofluoresence, and the number of TH-positive cells containing α-syn aggregates was quantitated as described above. For qualitative data presented in Figure 1 c and supplemental Figure S2 (available at www.jneurosci.org as supplemental material), representative large juxtanuclear inclusions, thioflavin S positive, and small punctate aggregates (see Fig. 1 c) were used as a standard.
Figure 3.
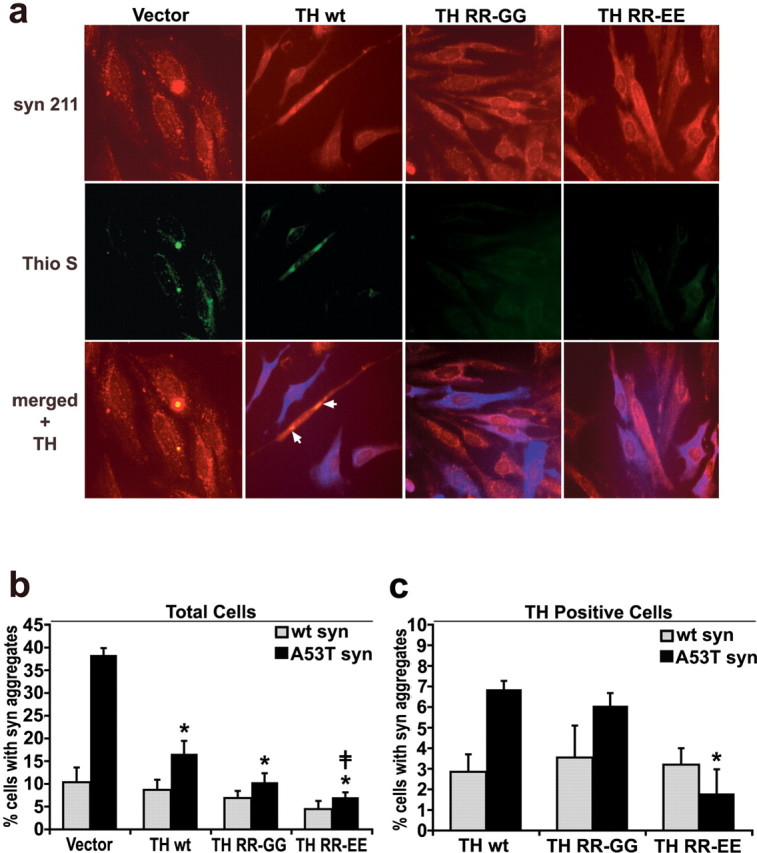
Increasing TH protein expression and intracellular catechol levels decreases the number of α-syn aggregates in A53T α-syn-expressing cells. wt α-syn- and A53T α-syn-expressing cells were infected with lentivirus containing TH expression plasmids and cultured for 5 d in 20 μm RA. a, A53T α-syn cells were stained with Syn 211 (red), 0.05% thioflavin S (Thio S; green) to visualize amyloidogenic aggregates, and anti-TH antibodies (blue) (20× magnification). The arrows show that thioflavin S-positive structures were also detected in the cells treated with TH lentivirus, although this was observed mostly in cells that had not been transduced. b, Quantification of total cells containing α-syn punctate structures using both LB509 (n = 2) and Syn 211 (n = 3) antibodies. Values are the mean ± SEM (n = 5). *p < 0.05 relative to vector control; ‡ p < 0.05 relative to TH wt. c, Quantification of only the TH-positive cells containing α-syn punctate structures using LB509 and Syn 211 shows that the observed decrease in α-syn aggregates correlates with increased catechol production by the TH RR-EE mutant rather than TH protein levels. Values are the mean ± SEM (n = 3). *p < 0.05 relative to TH wt and p < 0.05 relative to TH RR-GG.
Figure 1.

Differentiation of SH-SY5Y cells expressing A53T α-syn induces the formation of amyloidogenic, Triton-insoluble aggregates. SH-SY5Y cells were stably transfected with empty vector, wt Syn, or A53T α-syn (A53T Syn) expression plasmids and cultured with or without 20 μm RA. a, Cells were harvested after 5 d of culture with either DMSO (vehicle) or 20 μm RA. Triton-soluble lysates (40 μg of protein/lane) were analyzed by Western blot analysis using monoclonal antibody Syn 211, and NSE was used as a loading control. b, wt α-syn cells were stained with the anti-α-syn monoclonal antibody Syn 211 (red) and 0.05% thioflavin S to detect amyloidogenic aggregates (Thio S; green) (20× magnification). The majority of naive cells show a diffuse staining pattern for α-syn, and differentiation by RA treatment for 5 d does not change the distribution patterns of α-syn. c, A53T α-syn cells were analyzed as in b, showing the induction of α-syn aggregation by RA differentiation. Both large juxtanuclear aggregates (middle row) and small punctate aggregates (bottom row) were observed. d, Quantification of the number of cells containing α-syn-positive punctate structures. Values are the mean ± SEM (n = 3). *p = 0.01 for total aggregates relative to A53T α-syn naive condition. e, wt or A53T α-syn cells were subjected to sequential biochemical extraction in 1% Triton-X (T-sol), 2% SDS (SDS-sol), and 70% formic acid (FA-sol), followed by Western blot analysis using LB509. Similar results were obtained when the blot was stripped and reprobed with Syn 211. To monitor the efficiency of the extractions and protein loading, NSE was used for Triton-soluble fractions, whereas the Triton-insoluble intermediate filament protein vimentin (Vim) was used for SDS- and FA-soluble fractions. The molecular weight marker located at the left side of the blot indicates the protein size in kilodaltons. Although differentiated A53T α-syn cells contain Triton-insoluble α-syn, wt-syn cells do not form detectable amounts of Triton-insoluble materials after 5 d in culture.
Assessment of cell viability.
Cells were infected with lentivirus-containing TH expression plasmids, and differentiation with RA was as described above. The cell culture medium was changed at days 2 and 4 after the addition of RA. At day 5, the cell culture supernatant was collected and combined with two washes in warm D-PBS. Cells were trypsinized, combined with culture supernatant/D-PBS washes, and collected by centrifugation at 200 × g for 5 min at room temperature. After two washes in cold D-PBS, cells were either resuspended in annexin-binding buffer (10 mm HEPES, 140 mm NaCl, and 2.5 mm CaCl2, pH 7.4) for flow cytometry analysis or resuspended in complete medium for measurement of cell viability through trypan blue exclusion (0.4% trypan blue). For flow cytometry analysis, apoptotic and necrotic-associated events [phosphatidylserine translocation through annexin V binding and propidium iodide (PI) fluorescence, respectively] were measured. FITC-conjugated annexin V and PI (100 μg/ml) were added to resuspended cells according to the manufacturer's protocol (Vybrant apoptosis assay kit #3; Invitrogen). The percentage of cells with FITC and/or PI fluorescence was determined by flow cytometry. Control experiments comparing RA-differentiated cells with lentiviral-transduced cells revealed that lentiviral infection did not significantly alter cell viability of cells stably expressing α-syn or empty vector containing cell lines.
Pharmacological inhibition of catechol-synthesizing enzymes in A53T α-syn cells.
A53T α-syn cells were pretreated for 24 h with the following compounds (final concentrations) (Sigma): 0.5 mm α-methyl-p-tyrosine dissolved in 2 μm HCl (TH inhibitor) and 0.2 mm 3-hydroxybenzylhydrazine dihydrochloride (NSD 1015) dissolved in D-PBS (aromatic amino acid decarboxylase inhibitor). Cells were then infected with lentivirus-containing TH RR-EE expression plamids and differentiated with RA for 5 d. The medium containing fresh inhibitor was changed every 24 h. Intracellular catechol levels, biochemical analyses, and densitometric quantitation were performed as described.
Analysis of A53T transgenic mice.
A53T transgenic (tg) mice (M83 line) between 9 and 17 months of age, clearly displaying motor deficits as described previously (Giasson et al., 2002), were used for biochemical analysis. After the mice were killed and brains were rapidly dissected, the entire brain was mounted and sliced into 1-mm-thick coronal sections using a vibroslice (World Precision Instruments, Sarasota, FL). The substantia nigra (SN) and cortex (Ctx) were microdissected using a Stemi DRC (Zeiss, Oberkochen, Germany) microscope from slices corresponding to −2.5 to −3.88 mm relative to the bregma point. Tissues were homogenized with pestles in 10 vol of lysis buffer, incubated on ice for 10 min, and ultracentrifuged at 100,000 × g at 4°C for 30 min. The remaining pellets were then re-extracted with 10 vol of lysis buffer and ultracentrifuged as before, and the Triton-soluble supernatants of both extractions were combined. The remaining pellets were extracted in 5 vol of 2% SDS/50 mm Tris-Cl, pH 7.4, with a protease inhibitor mixture (Sigma) by sonication and boiling for 20 min and ultracentrifuged at 110,000 × g for 30 min at 25°C. The supernatants were saved as the Triton-insoluble fractions. Western blot analysis was performed as described above. The following antibodies were used: Syn 211, LB509, Syn 303, Syn 506, and Syn 514 (all 1:1000); monoclonal TH (1:5000); polyclonal NSE (1:2000); and monoclonal vimentin (1:800). Syn 303, Syn 506, and Syn 514 have been described previously (Duda et al., 2002). Gel filtration analysis was performed as described, using 500 μg of total Triton-soluble lysate injected onto the column in a volume of 250 μl.
For catechol measurements, cortical (bregma, −2.5 to −3.88 mm) or striatal (bregma, +0.66 to −0.34 mm) tissues were homogenized in 10 vol of 0.1 m perchloric acid containing 1 μm DHBA, then processed and analyzed as described above. Twenty microliters of filtered lysate were injected on the HPLC.
Densitometric analysis.
Bands from Western blot analyses were quantified by densitometric analysis using Infrared Imaging System software (version 1.2; Odyssey, Lincoln, NE). Samples used for comparison of α-syn levels were quantified from the same blot and scanned at intensity level 2. All Triton-soluble fractions were normalized to NSE levels, whereas Triton-insoluble levels were normalized to vimentin. To determine the percentage of insoluble α-syn in A53T mice, normalized integrated lane intensities from LB509 Western blots were divided by total α-syn (quantified from both Triton-soluble and Triton-insoluble fractions). To estimate the α-syn oligomer:monomer ratios in Figure 6 b, integrated lane intensities from 99 to 63 Å size fractions were quantified and divided by the lane intensity of the monomeric fraction (32–35 Å).
Figure 6.

Decreased Triton-insoluble α-syn and an increased proportion of 63–99 Å α-syn oligomers in nondegenerating SN of diseased A53T tg mice. a, Ctx and SN from A53T mice displaying motor impairments were dissected, sequentially extracted (see Materials and Methods), and analyzed by Western blot using a panel of α-syn antibodies (50 μg of total protein/lane). SN tissue contains TH as expected, whereas it is not detected in the Ctx. Western blot for NSE was used to monitor extraction and loading for Triton-soluble (T-sol) fractions, whereas vimentin (Vim) was used for Triton-insoluble (T-in) fractions. The molecular weight marker indicates the protein size in kilodaltons. b, T-soluble fractions were analyzed by SEC, followed by Western blot using the monoclonal antibody Syn 211 (500 μg of total protein injected on column). The horizontal marker indicates the molecular size in angstroms. c, The level of Triton-insoluble α-syn detected by LB509 was quantified by densitometry and normalized to vimentin (percentage of insoluble α-syn: SN, 6 ± 1; Ctx, 17 ± 3). Values are the mean ± SEM (n = 3). *p < 0.05. Although Ctx tissue contains α-syn-positive perikaryal inclusions and dystrophic neurites, cells of the SN are completely devoid of α-syn pathology in this mouse model (Giasson et al., 2002).
Statistical analysis.
Data were analyzed using SigmaStat software (version 2.03; SPSS, Chicago, IL) and are expressed as the mean ± SEM. One-way ANOVA followed by Tukey's post hoc test was used to determine whether groups were statistically different. p values <0.05 were considered significant.
Results
Human neuroblastoma cells, SH-SY5Y, were stably transfected with either empty vector plasmid, wt, or A53T α-syn expression plasmids and differentiated into a neuron-like phenotype with RA for up to 5 d. As expected, increased expression of α-syn was observed after transfection with wt or A53T α-syn compared with empty vector-transfected control cells, and differentiation with RA increased expression in all three cell lines (Fig. 1 a and supplemental Fig. S1, available at www.jneurosci.org as supplemental material). To determine the effect of overexpression and differentiation on α-syn cellular distribution, cells were analyzed by immunofluorescence using anti-α-syn antibodies. Figure 1 b–d shows that differentiation of A53T, but not wt α-syn-expressing cells, increases the number of cells containing α-syn aggregates. Both small punctate aggregates and large juxtanuclear inclusions (Fig. 1 c) were observed by 5 d in culture, whereas naive cells displayed a less punctate, more diffuse cytoplasmic distribution. The large juxtanuclear α-syn-positive aggregates colocalized with thioflavin S, a fluorescent dye that specifically intercalates within structures rich in β-pleated sheet conformation, indicating the presence of amyloid structures (Fig. 1 c). To determine the effect of RA differentiation on α-syn solubility, we sequentially extracted A53T and wt α-syn cell lysates with buffers of increasing protein solubility. As expected from the presence of α-syn inclusions in Figure 1 c, intensely labeled α-syn-positive smears were observed in the SDS and formic acid fractions of A53T but not the wt α-syn extracts (Fig. 1 e). These results demonstrate that RA differentiation of A53T α-syn-expressing SH-SY5Y cells induces the formation of amyloidogenic, α-syn-positive aggregates.
To determine the steady-state levels of catechols in α-syn-transfected SH-SY5Y cell lines, HPLC with electrochemical detection was used. Analysis of differentiated empty vector, wt α-syn, and A53T α-syn cell lines revealed that steady-state catechol levels were below the limit of detection (data not shown). Furthermore, protein levels of tyrosine hydroxylase (TH), the catecholamine synthesis rate-limiting enzyme, were undetectable in cell lysates assessed by Western blot analysis (Fig. 2 a). Therefore, to increase steady-state levels of intracellular catechols, we took advantage of the fact that SH-SY5Y cells contain no detectable TH but sufficient amounts of other enzymes involved in DA synthesis and processing. Using a recombinant lentiviral plasmid expressing hTH-1, Figure 2 shows that transduction of differentiated A53T α-syn cells increases intracellular levels of the DA precursor dihydroxyphenylalanine (l-DOPA), DA, and the DA metabolite 3,4-dihydroxyphenylacetic acid (DOPAC). To further increase intracellular catechol levels without increasing the amount of total TH protein, we used TH mutants that lack the catecholamine feedback inhibition binding site by replacing arginine residues at positions 37 and 38 with either glycine (RR-GG) or glutamate (RR-EE). Transduction of cells with the RR-GG and RR-EE TH mutants significantly increased intracellular steady-state levels of DA and DOPAC (Fig. 2 b).
To determine the effects of altered catechol levels on the process of α-syn aggregation, wt and A53T α-syn-expressing cells were infected with lentivirus-containing TH expression plasmids and differentiated with RA for 5 d. Immunofluorescence analysis revealed a decrease in the total number of A53T α-syn-expressing cells with α-syn-positive amyloidogenic aggregates in cells transduced to express TH (Fig. 3 and supplemental Fig. S2, available at www.jneurosci.org as supplemental material). Importantly, cells transduced with mutant TH RR-EE contained significantly fewer aggregates compared with TH wt transduced cells, suggesting that catechols alone can modulate α-syn aggregation. By counting only the TH-positive cells containing α-syn aggregates, we observed a similar correlation when comparing TH wt with RR-EE mutant transduced cells (Fig. 3 c). The TH RR-EE-transduced cells were almost completely devoid of large juxtanuclear inclusions (see supplemental Fig. S2, available at www.jneurosci.org as supplemental material), and only occasionally small punctate aggregates were noted. Although a small percentage of wt α-syn cells containing aggregates were detected, increasing TH expression and intracellular catechols had no effect (Fig. 3 b,c). These data indicate that increasing TH expression and intracellular steady-state levels of catechols in A53T α-syn-expressing cells inhibits the formation of α-syn inclusions.
To evaluate the effect of increasing TH expression and catechol levels on the solubility of α-syn, sequential extractions of A53T and wt α-syn cell lysates were analyzed by Western blot analysis. Transduction of A53T α-syn-expressing cells with lentivirus-containing wt or mutant TH expression plasmids resulted in a decrease in the total amount of Triton-insoluble α-syn, whereas the amount of α-syn in the Triton-soluble fraction did not change (Fig. 4 c). The same treatment in wt α-syn cells had no effect, because they do not demonstrate detectable Triton-insoluble α-syn (Fig. 4 a). These data indicate that an increase in TH protein and steady-state intracellular catechols inhibit the formation of Triton-insoluble α-syn.
Figure 4.

Increasing intracellular catechol levels decreases Triton-insoluble A53T α-syn with a concomitant accumulation of soluble oligomers. a, wt α-syn-expressing SH-SY5Y cells were transduced with lentivirus constructed with the empty vector or lentivirus containing wt, RR-GG, or RR-EE TH expression plasmids and cultured for 5 d in the presence of 20 μm RA. Cell lysates were subjected to sequential extraction and analyzed by Western blot using LB509. Western blot for TH verifies similar expression of each TH variant. NSE was used to monitor extraction and loading for Triton-soluble fractions, and vimentin was used for Triton-insoluble fractions. b, Triton-soluble fractions from wt α-syn cells were analyzed by SEC using a superdex 200 HR 10/30 gel filtration column. Fractions were collected and analyzed by Western blot analysis using antibody Syn 211. The horizontal marker indicates the molecular size in angstroms, whereas the vertical marker indicates the molecular weight in kilodaltons. A value of 99 Å corresponds to the column void volume (∼2000 kDa), and the α-syn monomer elutes off at a peak between 32 and 35 Å (or ∼57 kDa). c, A53T α-syn cells were infected, differentiated, and analyzed as in a. d, Triton-soluble fractions from A53T α-syn cells were analyzed by SEC as in b. SDS/heat-stable dimers, trimers, and oligomers are not detected in 50 μg of total cell protein (c) but were detected when 1 mg of total cell protein is analyzed by SEC. e, Naive A53T α-syn-expressing cells were infected with lentivirus containing TH RR-EE plasmid and cultured without RA for 5 d. SEC/Western blot analysis of the Triton-soluble fraction using antibody Syn 211 reveals the absence of α-syn eluting at the void volume. f, A53T α-syn cells were infected with lentivirus containing empty vector plasmid and cultured for 5 d with RA. The addition of 0.5 μm DA in the lysis buffer (threefold excess of the concentration detected in TH RR-EE-infected cells) did not dramatically shift the elution profile of α-syn, suggesting that catechol-induced oligomers are formed by active intracellular processes. The data are representative of three independent experiments. V, Vector; RG, RR-GG; RE, RR-EE; Vim, vimentin; T-sol, Triton-soluble fractions; T-insol, Triton-insoluble fractions.
It has been demonstrated previously that inhibition of α-syn aggregation by catecholamines occurs in vitro, through the kinetic stabilization of oligomeric intermediates (Conway et al., 2001). Therefore, we sought to determine whether these species exist in differentiated α-syn cells transduced with TH expression plasmids by SEC/Western blot analysis of Triton-soluble fractions. In empty vector-transduced A53T α-syn-expressing cells, the majority of monomeric α-syn eluted between 32 and 35 Å, which indicate a protein with a molecular size of ∼57 kDa (Fig. 4 d). This higher gel filtration migration pattern of α-syn, relative to the expected molecular size of 18 kDa, is attributable to the natively unfolded, elongated structure of this protein and has been observed previously (Weinreb et al., 1996). In A53T α-syn-expressing cells transduced with TH wt, no change in the elution profile was observed (Fig. 4 d). Analysis of cells transduced to express TH variants (RR-GG and RR-EE), however, revealed an increase in the amount of α-syn eluting at the void volume (between 48 and 99 Å, or ∼158 and 2000 kDa) (Fig. 4 d). These high-molecular-size species appeared to be composed of the 18 kDa α-syn monomer and heat/SDS-stable dimers (36 kDa), trimers (54 kDa), and oligomers (Fig. 4 d). Conversely, when TH RR-EE was expressed in either naive A53T α-syn cells or RA-differentiated wt α-syn cells (cells that do not contain biochemically detectable amounts of Triton-insoluble α-syn aggregates), no change in the elution profile of α-syn was observed (Fig. 4 b,e). This suggests that elevating steady-state catechols prevents the formation of Triton-insoluble aggregates at the level of soluble oligomers, and this occurs only if α-syn is in the process of polymerizing. Moreover, the addition of DA to differentiated empty vector-infected A53T α-syn-expressing cells during cell lysis did not increase the α-syn eluting at the void volume, indicating that catechol-induced α-syn oligomer stabilization occurs as a cellular process (Fig. 4 f).
To exclude the possibility that the TH RR-EE protein facilitates the formation of soluble α-syn oligomers rather than catechols, A53T α-syn cells were pretreated with the TH inhibitor α-methyl-p-tyrosine, or the aromatic amino acid inhibitor NSD 1015, infected with lentivirus-containing TH RR-EE expression plasmid, and differentiated with RA for 5 d. Consistent with the data in Figure 4, inhibition of DA synthesis increased the amount of Triton-insoluble α-syn with a concomitant decrease in Triton-soluble oligomers compared with vehicle-treated TH RR-EE-expressing cells (supplemental Fig. S3, available at www.jneurosci.org as supplemental material).
We next sought to determine the viability of α-syn-expressing SH-SY5Y cells by trypan blue exclusion. After 5 d of RA differentiation, expression of both wt and A53T α-syn enhanced cellular degeneration, with A53T-expressing cells exhibiting the highest amount of degenerating cells (percentage of cell death: empty vector, 4.5 ± 0.3; wt α-syn, 13.8 ± 1.4*; A53T α-syn, 28.5 ± 1.9*; mean ± SEM; n = 3; *p < 0.05). To evaluate the effect of modulating α-syn aggregation through increasing intracellular catechol levels, viability was measured in cells transduced to express TH by FITC-conjugated annexin labeling and PI fluorescence, followed by flow cytometry. A53T α-syn-expressing cells transduced with empty vector degenerated similarly to the TH RR-EE-transduced cells (Fig. 5 b), suggesting that decreasing Triton-insoluble α-syn along with increasing levels of soluble α-syn oligomers does not alter toxicity. The same treatment in wt α-syn cells also had no effect on cell viability (Fig. 5 a).
Figure 5.

Accumulation of catechol-induced α-syn oligomers does not accelerate A53T α-syn-mediated cell death in SH-SY5Y cells. wt and A53T α-syn cells were infected with lentivirus generated with empty vector or TH-containing plasmids, and the percentage of FITC-conjugated annexin V- and PI-positive cells was measured after 5 d in culture with RA by flow cytometry. Although A53T α-syn-expressing cells degenerate more rapidly compared with wt α-syn cells, increasing catechol levels does not change cell viability in either wt or A53T α-syn-expressing cell lines. Values are the mean ± SEM (n = 4–6). Similar results were obtained when measuring cell viability by trypan blue exclusion (data not shown).
To further evaluate the presence of α-syn oligomers in dopaminergic neurons in vivo, brain tissue was analyzed from diseased A53T tg mice displaying motor impairment [tg line M83 described by Giasson et al. (2002)]. Previous histopathological characterization of this mouse model revealed age-dependent neurodegeneration and α-syn inclusion formation in a wide range of brain regions such as the Ctx, but not in neurons of the SN, which are rich in DA. Using a panel of α-syn antibodies, data in Figure 6, a and c, verified that SN tissue contains less Triton-insoluble α-syn compared with cortical tissue, consistent with the presence of α-syn inclusions in the Ctx but not in the SN. Although monoclonal antibody LB509 appeared to show the most dramatic difference of Triton-insoluble α-syn levels between the SN and Ctx, monoclonal antibodies (Syn 303, Syn 506, Syn 514) that preferentially detect oxidized and potentially pathological α-syn (Duda et al. 2002) also showed a clear increase of Triton-insoluble α-syn in the Ctx (Fig. 6 a). To assess the presence of α-syn oligomers, SEC/Western blot analysis was used to analyze Triton-soluble lysates. In SN lysates, the majority of α-syn eluted off at the void volume (63–99 Å or ∼440–2000 kDa) with α-syn dimers and trimers eluting later (40–63 Å or ∼116–440 kDa) (Fig. 6 b). Unexpectedly, almost no monomeric α-syn eluting between 32 and 35 Å was detected. Conversely in cortical tissue, the α-syn monomer was detected at 32 Å as well as dimers and trimers between 40 and 99 Å (Fig. 6 b). Although the intensity of the high molecular weight smear detected in the 63–99 Å fraction of the Ctx appeared to be similar to that of the α-syn detected in the same fraction of the SN, dopaminergic neurons appear to contain a higher proportion of large molecular-size α-syn oligomers compared with non-dopaminergic cortical cells (oligomer/monomer ratio quantified by densitometry is 19 for SN and 2.6 for Ctx). Moreover, we verified that SN neurons in diseased A53T mice were still able to produce DA by measuring catechol levels in the striatum (l-DOPA, 206 ± 54 fmol/μg protein; DA, 861 ± 89 fmol/μg protein; DOPAC, 197 ± 49 fmol/μg protein; mean ± SEM; n = 3). As expected, these levels were significantly higher than the catechol levels in the Ctx of the same mice (l-DOPA, 8 ± 4 fmol/μg protein; DA, 3 ± 1 fmol/μg protein; no detectable DOPAC; mean ± SEM; n = 3). Collectively, these data suggest that the absence of α-syn inclusions in SN neurons of A53T tg mice may occur through the accumulation of catechol-induced oligomeric intermediates.
Discussion
Previous findings in cell-free systems indicated that catechols, including DA, interact with α-syn to stabilize soluble oligomeric species provided new clues into the pathogenic mechanism of PD (Conway et al., 2001). Despite the limitations of studying the formation and properties of these species exclusively in vitro, the data collectively generated new, testable hypotheses to explain the selective vulnerability of DA neurons in PD. These oligomeric α-syn species, products of the interaction of oxidized catechols with α-syn, can assume spherical structures (Norris et al., 2005) and may represent the species responsible for the demise of these neurons. This hypothesis was further supported by recent findings demonstrating that α-syn fibrils and inclusions may not be required for α-syn-mediated toxicity (Chen and Feany, 2005) despite the sound documentation that Lewy bodies and inclusions are composed of α-syn fibrils. Therefore, by using biochemical and molecular approaches, we generated a cellular model system to evaluate the effect of intracellular catechol levels on α-syn aggregation and cellular degeneration.
The data document for the first time that TH expression and intracellular catechols such as DA and DOPAC have the ability to modulate α-syn aggregation in cultured human cells and possibly in mouse brain. Using TH variants RR-GG and RR-EE, which exhibit increased enzymatic production of catechols, the data revealed that elevated cytosolic catechols alone have the ability to alter α-syn aggregation. As predicted from the in vitro data, the increase in cytosolic catechol levels was associated with the decline in the formation of inclusions that contain amyloidogenic α-syn (Fig. 3). The amount of α-syn accumulating in Triton-insoluble fractions also declined as a result of increased cytosolic catechols (Fig. 4 c) resulting from TH expression. Expression of TH RR-GG and TH-RR-EE resulted in a concomitant accumulation of soluble α-syn oligomers (between 48 and 99 Å in size) observed by SEC analysis in differentiated A53T α-syn-expressing cells (Fig. 4 d). In TH RR-EE-expressing cells, these oligomers were also predominantly heat and SDS resistant compared with those in cells expressing TH RR-GG, which could be partially dissociated into monomers by SDS. This difference could be a result of the higher levels of catechols in cells expressing TH RR-EE (Fig. 2), which may promote oxidative cross-linking of α-syn associated with production of reactive species generated during the intracellular oxidation of catechols (Souza et al., 2000b; Norris et al., 2003). Expression of wt TH prevented the accumulation of Triton-insoluble α-syn, albeit less efficiently than the two TH mutants, without significant formation of α-syn oligomers. These results are likely attributable to the lower levels of catechols in the wt TH-infected cells, which determine the kinetics of catechol oxidation, and thus the ability to stabilize α-syn oligomers as suggested by in vitro studies (Cappai et al., 2005; Norris et al., 2005). Together, the data demonstrate that intracellular levels of catechols can modulate the α-syn aggregation process.
The specific mechanism by which catechols prevent α-syn aggregation in transduced SH-SY5Y cells was not established. Uniformly, all of the previous publications in cell-free systems have indicated that DA oxidation is required for the kinetic stabilization of the oligomeric species (Conway et al., 2001; W. Li et al., 2004; Norris et al., 2005). This kinetic stabilization appears to be primarily dependent on the noncovalent interaction of the oxidized DA with amino acids Tyr125-Glu-Met-Pro-Ser129 (Norris et al., 2005). Covalent interactions of oxidized forms of DA with α-syn have not been documented in the cell-free systems because cysteine residues, the nucleophile that reacts primarily with the electron-poor oxidized forms of DA, are not present in α-syn. Previous work also explored the possibility of a histidine adduct formation with oxidized forms of DA. However, mutation of the His 50 in α-syn did not prevent the effect of DA inhibiting the fibril formation (Norris et al., 2005). Therefore, the noncovalent interaction between the oxidized products of catechols and α-syn induces reversible conformational alterations in α-syn, which are not permissive for fibril formation (Norris et al., 2005). Although a variety of compounds containing a catechol ring can inhibit α-syn in vitro (Conway et al., 2001), we speculate that intracellular DA has the greatest effect in vivo because of its ability to auto-oxidize more rapidly than other catechols at physiological pH to generate reactive electrophiles and reactive oxygen species (Graham, 1978). This notion is partly corroborated by the pharmacological data provided in supplemental Figure S3 (available at www.jneurosci.org as supplemental material), which showed that increased intracellular l-DOPA levels by inhibition of aromatic amino acid decarboxylase failed to inhibit the formation of Triton-insoluble synuclein and did not induce the formation of Triton-soluble oligomers.
In the cell model system used here, increased expression of both wt and A53T α-syn enhances cellular degeneration compared with empty vector-transfected cells, consistent with previous reports (Stefanis et al., 2001; Xu et al., 2002; Zhou et al., 2006). In other cellular model systems, inhibition of TH with α-methyl-p-tyrosine prevented α-syn-mediated toxicity (Xu et al., 2002; Zhou et al., 2006), indicating a dependency on endogenous catechol levels. Based on these observations, it was expected that the increase in the intracellular catechol levels produced by TH RR-GG and TH RR-EE mutants would further augment α-syn-mediated cellular degeneration. However, increasing intracellular catechol levels through TH expression did not further augment cellular degeneration in α-syn-expressing cells despite altering the aggregation of the protein. It is possible that a significant portion of the intracellular catechols, primarily DA, that undergo oxidation to generate electron-deficient oxidized DA species and reactive oxygen species interacted with the overexpressed α-syn preventing cellular injury. Preventing the aggregation of α-syn suggested that, indeed, the oxidized catechols interacted with α-syn. Moreover, the presence of heat- and SDS-stable oligomers of α-syn in the TH RR-EE-infected cells also supports this suggestion because in vitro data has clearly implicated reactive species-mediated oxidative cross-linking as the mechanisms responsible for the formation of these stable α-syn oligomers (Souza et al., 2000b; Norris et al., 2003).
The data presented here suggest that the formation of soluble oligomeric species induced by catechols does not lead to toxicity. However, it is possible that the structure of soluble oligomeric species documented in this study are not identical to the potentially toxic species shown in vitro (Volles et al., 2001), despite similarities in the SEC profiles. A53T α-syn-expressing cells, which contain Triton-insoluble aggregates, degenerate significantly more than wt α-syn cells. Although this suggests that formation of Triton-insoluble α-syn may partly contribute to degeneration, we found that decreasing Triton-insoluble α-syn had no effect on cell viability. Therefore, the data indicate that α-syn-mediated cell death in SH-SY5Y cells may occur by mechanisms that do not solely include the formation of Triton-insoluble aggregates, possibly through elevated levels of soluble, monomeric forms of α-syn. Previous reports have indicated that expression of α-syn induces cellular degeneration that can be attributed to a multitude of events, such as lysosomal and proteosomal dysfunction (Stefanis et al., 2001; Cuervo et al., 2005), sequestration of anti-apoptotic proteins during conditions of oxidative stress (Xu et al., 2002), or alterations of signal transduction pathways (Ostrerova et al., 1999; Hashimoto et al., 2002)
Consistent with the data in SH-SY5Y cell, the studies in A53T α-syn tg mice also support the notion that catechols may inhibit α-syn inclusions by blocking α-syn polymerization at the stage of soluble oligomers. The data in these mice also indicate that catechols may promote the formation of these oligomers, but without any apparent toxicity as documented in dopaminergic SN neurons. Several tg mouse lines expressing A53T α-syn have been generated and characterized, and the lack of pathology in the SN appears to be a consistent feature (Matsuoka et al., 2001; Giasson et al., 2002; Martin et al., 2006) despite pathology in many other brain regions. In support of the view that DA may be a factor in preventing α-syn aggregation, the only tg mouse model demonstrating the presence of nigral α-syn inclusions was generated by expressing a truncated form of wt α-syn (1–120) driven by the TH promoter (Tofaris et al., 2006), and this truncated mutant lacks the putative catechol-interaction site in α-syn (Norris et al., 2005).
Overall the data suggest that the intracellular levels of catechols are critical chemical modifiers that may participate in the development of PD and related disorders. Previous data indicated that an early event in PD pathophysiology is the decrease in catecholamine levels (Nagatsu, 1990), which could occur through oxidative or nitrative damage to TH causing a decrease in catalytic activity (Ara et al., 1998; Kuhn et al., 1999). The decline in catecholamines may lead to the formation of insoluble α-syn fibrils, a process that could induce toxicity, or additionally sequester α-syn, preventing its association with phospholipids and proteins, as well as eliminating putative molecular chaperone function.
Footnotes
This work was supported by National Institutes of Health Grants AG09215 (B.I.G.), NS45986 (D.R.L.), and AG13966 (H.I.). We thank Drs. Michael B. Robinson and John H. Wolfe for providing the viral vectors, Li-Bin Li and Brian A. Karolewski for assistance in lentiviral generation, Elpida Tsika for technical assistance in mouse brain dissection, and Eric Riedel for assistance with flow cytometry.
References
- Abeliovich A, Schmitz Y, Farinas I, Choi-Lundberg D, Ho WH, Catillo PE, Shinsky N, Verdugo JM, Armanini M, Ryan A, Hynes M, Phillips H, Sulzer D, Rosenthal A. Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron. 2000;25:239–252. doi: 10.1016/s0896-6273(00)80886-7. [DOI] [PubMed] [Google Scholar]
- Ara J, Przedborski S, Naini AB, Jackson-Lewis V, Trifiletti RR, Horwitz J, Ischiropoulos H. Inactivation of tyrosine hydroxylase by nitration following exposure to peroxynitrite and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) Proc Natl Acad Sci USA. 1998;95:7659–7663. doi: 10.1073/pnas.95.13.7659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba M, Nakajo S, Tu P, Tomita T, Nakaya K, Lee VM, Trojanowski JQ, Iwatsubo T. Aggregation of alpha-synuclein in Lewy bodies of sporadic Parkinson's disease and dementia with Lewy bodies. Am J Pathol. 1998;152:879–884. [PMC free article] [PubMed] [Google Scholar]
- Cabin DE, Shimazu K, Murphy DD, Cole NB, Gottschalk W, McIIwain KL, Orrison B, Chen A, Ellis CE, Paylor R, Lu B, Nussbaum RL. Synaptic vesicle depletion correlates with attenuated synaptic responses to prolonged repetitive stimulation in mice lacking α-synuclein. J Neurosci. 2002;22:8797–8807. doi: 10.1523/JNEUROSCI.22-20-08797.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cappai R, Leck SL, Tew DJ, Williamson NA, Smith DP, Galatis D, Sharples RA, Curtain CC, Ali FE, Cherny RA, Culvenor JG, Bottomley SP, Masters CL, Barnham KJ, Hill AF. Dopamine promotes alpha-synuclein aggregation into SDS-resistant soluble oligomers via a distinct folding pathway. FASEB J. 2005;19:1377–1379. doi: 10.1096/fj.04-3437fje. [DOI] [PubMed] [Google Scholar]
- Chandra S, Gallardo G, Fernandez-Chacon R, Schluter OM, Sudhof TC. Alpha-synuclein cooperates with CSP-alpha in preventing neurodegeneration. Cell. 2005;123:383–396. doi: 10.1016/j.cell.2005.09.028. [DOI] [PubMed] [Google Scholar]
- Chen L, Feany MB. Alpha-synuclein phosphorylation controls neurotoxicity and inclusion formation in a Drosophila model of Parkinson disease. Nat Neurosci. 2005;8:657–663. doi: 10.1038/nn1443. [DOI] [PubMed] [Google Scholar]
- Conway KA, Rochet JC, Bieganski RM, Lansbury PT., Jr Kinetic stabilization of the alpha-synuclein protofibril by a dopamine-alpha-synuclein adduct. Science. 2001;294:1346–1349. doi: 10.1126/science.1063522. [DOI] [PubMed] [Google Scholar]
- Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2005;305:1292–1295. doi: 10.1126/science.1101738. [DOI] [PubMed] [Google Scholar]
- Davidson WS, Jonas A, Clayton DF, George JM. Stabilization of alpha-synuclein secondary structure upon binding to synthetic membranes. J Biol Chem. 1998;273:9443–9449. doi: 10.1074/jbc.273.16.9443. [DOI] [PubMed] [Google Scholar]
- Duda JE, Giasson BI, Mabon ME, Lee VM, Trojanowski JQ. Novel antibodies to synuclein show abundant striatal pathology in Lewy body diseases. Ann Neurol. 2002;52:205–210. doi: 10.1002/ana.10279. [DOI] [PubMed] [Google Scholar]
- Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS, Shen J, Takio K, Iwatsubo T. alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol. 2002;4:160–164. doi: 10.1038/ncb748. [DOI] [PubMed] [Google Scholar]
- George JM, Jin H, Woods WS, Clayton DF. Characterization of a novel protein regulated during the critical period for song learning in the zebra finch. Neuron. 1995;15:361–372. doi: 10.1016/0896-6273(95)90040-3. [DOI] [PubMed] [Google Scholar]
- Giasson BI, Uryu K, Trojanowski JQ, Lee VM. Mutant and wild type human alpha-synucleins assemble into elongated filaments with distinct morphologies in vitro. J Biol Chem. 1999;274:7619–7622. doi: 10.1074/jbc.274.12.7619. [DOI] [PubMed] [Google Scholar]
- Giasson BI, Jakes R, Goedert M, Duda JE, Leight S, Trojanowski JQ, Lee VM. A panel of epitope-specific antibodies detects protein domains distributed throughout human alpha-synuclein in Lewy bodies of Parkinson's disease. J Neurosci Res. 2000a;59:528–533. doi: 10.1002/(SICI)1097-4547(20000215)59:4<528::AID-JNR8>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Giasson BI, Duda JE, Murray IV, Chen Q, Souza JM, Hurtig HI, Ischiropoulos H, Trojanowski JQ, Lee VM. Oxidative damage linked to neurodegeneration by selective alpha-synuclein nitration in synucleinopathy lesions. Science. 2000b;290:985–989. doi: 10.1126/science.290.5493.985. [DOI] [PubMed] [Google Scholar]
- Giasson BI, Duda JE, Quinn SM, Zhang B, Trojanowski JQ, Lee VM. Neuronal alpha-synucleinopathy with severe movement disorder in mice expressing A53T human alpha-synuclein. Neuron. 2002;34:521–533. doi: 10.1016/s0896-6273(02)00682-7. [DOI] [PubMed] [Google Scholar]
- Giasson BI, Forman MS, Higuchi M, Golbe LI, Graves CL, Kotzbauer PT, Trojanowski JQ, Lee VM. Initiation and synergistic fibrillization of tau and alpha-synuclein. Science. 2003;300:636–640. doi: 10.1126/science.1082324. [DOI] [PubMed] [Google Scholar]
- Graham DG. Oxidative pathways for catecholamines in the genesis of neuromelanin and cytotoxic quinones. Mol Pharmacol. 1978;14:633–643. [PubMed] [Google Scholar]
- Greenbaum EA, Graves CL, Mishizen-Eberz AJ, Lupoli MA, Lynch DR, Englander SW, Axelsen PH, Giasson BI. The E46K mutation in alpha-synuclein increases amyloid fibril formation. J Biol Chem. 2005;280:7800–7807. doi: 10.1074/jbc.M411638200. [DOI] [PubMed] [Google Scholar]
- Hashimoto M, Hsu LJ, Rockenstein E, Takenouchi T, Mallory M, Masliah E. Alpha-Synuclein protects against oxidative stress via inactivation of the c-Jun N-terminal kinase stress-signaling pathway in neuronal cells. J Biol Chem. 2002;277:11465–11472. doi: 10.1074/jbc.M111428200. [DOI] [PubMed] [Google Scholar]
- Hodara R, Norris EH, Giasson BI, Mishizen-Eberz AJ, Lynch DR, Lee VM-Y, Ischiropoulos H. Functional consequences of α-synuclein tyrosine nitration: diminished binding to lipid vesicles and increased fibril formation. J Biol Chem. 2004;279:47746–47753. doi: 10.1074/jbc.M408906200. [DOI] [PubMed] [Google Scholar]
- Jakes R, Crowther A, Lee VM, Trojanowski JQ, Iwatsubo T, Goedert M. Epitope mapping of LB509, a monoclonal antibody directed against human alpha-synuclein. Neurosci Lett. 1999;269:13–16. doi: 10.1016/s0304-3940(99)00411-5. [DOI] [PubMed] [Google Scholar]
- Karolewski BA, Watson DJ, Parente MK, Wolfe JH. Comparison of transfection conditions for a lentivirus vector produced in large volumes. Hum Gene Ther. 2003;14:1287–1296. doi: 10.1089/104303403322319372. [DOI] [PubMed] [Google Scholar]
- Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, Przuntek H, Epplen JT, Schols L, Riess O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson's disease. Nat Genet. 1998;18:106–108. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- Kuhn DM, Aretha CW, Geddes TJ. Peroxynitrite inactivation of tyrosine hydroxylase: mediation by sulfhydryl oxidation, not tyrosine nitration. J Neurosci. 1999;19:10289–10294. doi: 10.1523/JNEUROSCI.19-23-10289.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Zhu M, Manning-Bog AB, Di Monte DA, Fink AL. Dopamine and L-dopa disaggregate amyloid fibrils: implications for Parkinson's and Alzheimer's disease. FASEB J. 2004;18:962–964. doi: 10.1096/fj.03-0770fje. [DOI] [PubMed] [Google Scholar]
- Li W, Lesuisse C, Xu Y, Troncoso JC, Price DL, Lee MK. Stabilization of α-synuclein protein with aging and familial Parkinson's disease-linked A53T mutation. J Neurosci. 2004;24:7400–7409. doi: 10.1523/JNEUROSCI.1370-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, West N, Colla E, Pletnikova O, Troncoso JC, Marsh L, Dawson TM, Jakala P, Hartmann T, Price DL, Lee MK. Aggregation promoting C-terminal truncation of alpha-synuclein is a normal cellular process and is enhanced by the familial Parkinson's disease-linked mutations. Proc Natl Acad Sci USA. 2005;102:2162–2167. doi: 10.1073/pnas.0406976102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroteaux L, Campanelli JT, Scheller RH. Synuclein: a neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J Neurosci. 1988;8:2804–2815. doi: 10.1523/JNEUROSCI.08-08-02804.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin LJ, Pan Y, Price AC, Sterling W, Copeland NG, Jenkins NA, Price DL, Lee MK. Parkinson's disease α-synuclein transgenic mice develop neuronal mitochondrial degeneration and cell death. J Neurosci. 2006;26:41–50. doi: 10.1523/JNEUROSCI.4308-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka Y, Vila M, Lincoln S, McCormack A, Picciano M, LaFroncois J, Yu X, Dickson D, Langston WJ, McGown E, Farrer M, Hardy J, Duff K, Przedborski S, Di Monte DA. Lack of nigral pathology in transgenic mice expressing human alpha-synuclein driven by the tyrosine hydroxylase promoter. Neurobiol Dis. 2001;8:535–539. doi: 10.1006/nbdi.2001.0392. [DOI] [PubMed] [Google Scholar]
- Mishizen-Eberz AJ, Norris EH, Giasson BI, Hodara R, Ischiropoulos H, Lee VM, Trojanowski JQ, Lynch DR. Cleavage of alpha-synuclein by calpain: potential role in degradation of fibrillized and nitrated species of alpha-synuclein. Biochemistry. 2005;44:7818–7829. doi: 10.1021/bi047846q. [DOI] [PubMed] [Google Scholar]
- Murphy DD, Rueter SM, Trojanowski JQ, Lee VM. Synucleins are developmentally expressed, and α-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. J Neurosci. 2000;20:3214–3220. doi: 10.1523/JNEUROSCI.20-09-03214.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray IV, Giasson BI, Quinn SM, Koppaka V, Axelsen PH, Ischiropoulos H, Trojanowski JQ, Lee VM. Role of alpha-synuclein carboxy-terminus on fibril formation in vitro. Biochemistry. 2003;42:8530–8540. doi: 10.1021/bi027363r. [DOI] [PubMed] [Google Scholar]
- Nagatsu T. Change of tyrosine hydroxylase in the Parkinsonian brain and in the brain of MPTP-treated mice as revealed by homospecific activity. Neurochem Res. 1990;15:425–429. doi: 10.1007/BF00969928. [DOI] [PubMed] [Google Scholar]
- Nakashima A, Kaneko YS, Mori K, Fujiwara K, Tsugu T, Suzuki T, Nagatsu T, Ota A. The mutation of two amino acid residues in the N-terminus of tyrosine hydroxylase (TH) dramatically enhances the catalytic activity in neuroendocrine AtT-20 cells. J Neurochem. 2002;82:202–206. doi: 10.1046/j.1471-4159.2002.00921.x. [DOI] [PubMed] [Google Scholar]
- Norris EH, Giasson BI, Ischiropoulos H, Lee VM. Effects of oxidative and nitrative challenges on alpha-synuclein fibrillogenesis involve distinct mechanisms of protein modifications. J Biol Chem. 2003;278:27230–27240. doi: 10.1074/jbc.M212436200. [DOI] [PubMed] [Google Scholar]
- Norris EH, Giasson BI, Hodara R, Xu S, Trojanowski JQ, Ischiropoulos H, Lee VM. Reversible inhibition of alpha-synuclein fibrillization by dopaminochrome-mediated conformational alterations. J Biol Chem. 2005;280:21212–21219. doi: 10.1074/jbc.M412621200. [DOI] [PubMed] [Google Scholar]
- Ostrerova N, Petrucelli L, Farrer M, Mehta N, Choi P, Hardy J, Wolozin B. α-Synuclein shares physical and functional homology with 14-3-3 proteins. J Neurosci. 1999;19:5782–5791. doi: 10.1523/JNEUROSCI.19-14-05782.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- Singleton A, Gwinn-Hardy K. Parkinson's disease and dementia with Lewy bodies: a difference in dose? Lancet. 2004;364:1105–1107. doi: 10.1016/S0140-6736(04)17117-1. [DOI] [PubMed] [Google Scholar]
- Smith WW, Margolis RL, Li X, Troncoso JC, Lee MK, Dawson VL, Dawson TM, Iwatsubo T, Ross CA. α-Synuclein phosphorylation enhances eosinophilic cytoplasmic inclusion formation in SH-SY5Y cells. J Neurosci. 2005;25:5544–5552. doi: 10.1523/JNEUROSCI.0482-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souza JM, Giasson BI, Lee VM, Ischiropoulos H. Chaperone-like activity of synucleins. FEBS Lett. 2000a;474:116–119. doi: 10.1016/s0014-5793(00)01563-5. [DOI] [PubMed] [Google Scholar]
- Souza JM, Giasson BI, Chen Q, Lee VM-Y, Ischiropoulos H. Dityrosine cross-linking promotes formation of stable a-synuclein polymers: implication of nitrative and oxidative stress in the pathogenesis of neurodegenerative synucleinopathies. J Biol Chem. 2000b;275:18344–18349. doi: 10.1074/jbc.M000206200. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- Stefanis L, Larsen KE, Rideout HJ, Sulzer D, Greene LA. Expression of A53T mutant but not wild-type α-synuclein in PC12 cells induces alterations of the ubiquitin-dependent degradation system, loss of dopamine release, and autophagic cell death. J Neurosci. 2001;21:9549–9560. doi: 10.1523/JNEUROSCI.21-24-09549.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tofaris GK, Reitbock PG, Humby T, Lambourne SL, O'Connell M, Ghetti B, Gossage H, Emson PC, Wilkinson LS, Goedert M, Spillantini MG. Pathological changes in dopaminergic nerve cells of the substantia nigra and olfactory bulb in mice transgenic for truncated human α-synuclein(1–120): implications for Lewy body disorders. J Neurosci. 2006;26:3942–3950. doi: 10.1523/JNEUROSCI.4965-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trojanowski JQ, Lee VM. Parkinson's disease and related alpha-synucleinopathies are brain amyloidoses. Ann NY Acad Sci. 2003;991:107–110. doi: 10.1111/j.1749-6632.2003.tb07468.x. [DOI] [PubMed] [Google Scholar]
- Volles MJ, Lee S-J, Rochet J-C, Shtilerman MD, Ding TT, Kessler JC, Lansbury PT. Vesicle permeabilization by protofibrillar alpha-synuclein: implications for the pathogenesis and treatment of Parkinson's disease. Biochemistry. 2001;40:7812–7819. doi: 10.1021/bi0102398. [DOI] [PubMed] [Google Scholar]
- Weinreb PH, Zhen W, Poon AW, Conway KA, Lansbury PT., Jr NACP, a protein implicated in Alzheimer's disease and learning, is natively unfolded. Biochemistry. 1996;35:13709–13715. doi: 10.1021/bi961799n. [DOI] [PubMed] [Google Scholar]
- Xu J, Kao SY, Lee FJ, Song W, Jin LW, Yankner BA. Dopamine-dependent neurotoxicity of alpha-synuclein: a mechanism for selective neurodegeneration in Parkinson disease. Nat Med. 2002;8:600–606. doi: 10.1038/nm0602-600. [DOI] [PubMed] [Google Scholar]
- Zarranz JJ, Alegre J, Gomez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atares B, Llorens V, Gomez Tortosa E, del Ser T, Munoz DG, de Yebenes JG. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol. 2004;55:153–156. doi: 10.1002/ana.10795. [DOI] [PubMed] [Google Scholar]
- Zhou ZD, Yap BP., Gung AY, Leong SM, Ang ST, Lim TM. Dopamine-related and caspase-independent apoptosis in dopaminergic neurons induced by overexpression of human wild type or mutant alpha-synuclein. Exp Cell Res. 2006;312:156–170. doi: 10.1016/j.yexcr.2005.10.012. [DOI] [PubMed] [Google Scholar]