

Abstract
Sporadic Parkinson's disease (sPD) is a common neurodegenerative disorder, characterized by selective degeneration of dopaminergic neurons in the substantia nigra. Although the pathogenesis of the disease remains undetermined, phosphorylation of α-synuclein and its oligomer formation seem to play a key role. However, the protein kinase(s) involved in the phosphorylation in the pathogenesis of sPD has not been identified. Here, we found that G-protein-coupled receptor kinase 5 (GRK5) accumulated in Lewy bodies and colocalized with α-synuclein in the pathological structures of the brains of sPD patients. In cotransfected cells, GRK5 phosphorylated Ser-129 of α-synuclein at the plasma membrane and induced translocation of phosphorylated α-synuclein to the perikaryal area. GRK5-catalyzed phosphorylation also promoted the formation of soluble oligomers and aggregates of α-synuclein. Genetic association study revealed haplotypic association of the GRK5 gene with susceptibility to sPD. The haplotype contained two functional single-nucleotide polymorphisms, m22.1 and m24, in introns of the GRK5 gene, which bound to YY1 (Yin Yang-1) and CREB-1 (cAMP response element-binding protein 1), respectively, and increased transcriptional activity of the reporter gene. The results suggest that phosphorylation of α-synuclein by GRK5 plays a crucial role in the pathogenesis of sPD.
Keywords: α-synuclein, GRK5, haplotype, Lewy body, phosphorylation, SNP
Introduction
Parkinson's disease (PD) is a common neurodegenerative disease that usually occurs after middle age. Neurologically, PD is characterized by progressive deterioration of tremor, rigidity, bradykinesia/akinesia, gait disturbance, and postural instability. Most PD patients eventually become bedridden and die in an average of 13 years after diagnosis. The neuropathological features of the disease are progressive degeneration of dopaminergic neurons in the substantia nigra and the formation of the characteristic intracytoplasmic inclusion bodies, called Lewy bodies (LBs). Because the mechanism of dopaminergic neuron death is not fully understood, none of the currently available therapies for PD is able to hold or reverse the disease process (Dawson and Dawson, 2002).
Causal genes for subtypes of familial PD, which is inherited in a Mendelian manner, include α-synuclein (αS), parkin, PINK1 [PTEN (phosphatase and tensin homolog deleted on chromosome 10)-induced putative kinase protein 1], DJ-1, and LRRK2 (leucine-rich repeat kinase 2) (Polymeropoulos et al., 1997; Kruger et al., 1998; Dawson and Dawson, 2003a; Paisan-Ruiz et al., 2004; Vila and Przedborski, 2004; Zimprich et al., 2004). However, the etiology or risk factors for sporadic PD (sPD), which comprises the majority of PD, remain undetermined. sPD is considered a multifactorial disease, in which both genetic and environmental factors may be involved (Chung et al., 2003; Dawson and Dawson, 2003b; Siderowf and Stern, 2003; Moore et al., 2005). The genetic role in the pathogenesis of sPD was indicated by familial clustering of sPD patients and a twin study using positron emission tomography scans (Foltynie et al., 2002). Indeed, many candidate-gene approaches were made to find sPD-associated genes (Gasser, 2001; Martin et al., 2001; Foltynie et al., 2002).
αS is a soluble protein with 140 aa residues and localizes primarily at presynaptic terminals in the CNS (Dev et al., 2003; Recchia et al., 2004). αS is not only a causative gene for a minor type of familial PD but was also implicated in the pathogenesis of sPD, because αS is deposited in LBs of sPD (Spillantini et al., 1997, 1998; Baba et al., 1998). Phosphorylation of αS at Ser-129 (pS129–αS) was identified as a major modification of αS in LBs in sPD brains (Fujiwara et al., 2002). Substitution of Ser-129 of αS to alanine (S129A) to abolish phosphorylation at this site decreased its interaction with synphilin-1, another component of LBs (Smith et al., 2005). Quite recently, phosphorylation at Ser-129 of αS has been shown to be essential for its toxicity to dopaminergic neurons in a Drosophila model of PD (Chen and Feany, 2005). Thus, αS phosphorylation seems to be involved in the pathogenesis of sPD. Up to the present time, it has been reported that several protein kinases, such as casein kinase 1 (CK1), CK2, and a family of G-protein-coupled receptor kinases (GRKs), are able to phosphorylate αS (Okochi et al., 2000; Pronin et al., 2000). However, it remains undetermined which protein kinase is really involved in the pathogenesis of human sPD. Here, we report immunohistochemical, biochemical, and genetic evidence that GRK5 has a crucial role in the pathogenesis of sPD.
Materials and Methods
Expression constructs.
A full-length wild-type human αS cDNA (IMAGE clone 3604532) was subcloned into the pcDNA3.1 vector (Invitrogen, Carlsbad, CA). Human GRK5 cDNA was generated by PCR from reverse-transcribed human brain RNA. GRK5 cDNA fused with a C-terminal FLAG tag was subcloned into the pcDNA3.1 vector. Substitution mutant αS cDNA with serine-129 changed to alanine and GRK5 cDNA with lysine-215 changed to arginine were generated by using the Quick Change Site-Directed Mutagenesis kit (Stratagene, La Jolla, CA). DNA sequences of all cDNA constructs were confirmed.
Cell culture and transfection.
Human embryonic kidney 293 (HEK293) cells were maintained in DMEM supplemented with 10% fetal bovine serum. Neuroblastoma SH-SY5Y cells were maintained in DMEM/F-12 medium with 10% fetal bovine serum at 37°C in a humidified 5% CO2 atmosphere. Cells were transfected using Lipofectamine Plus reagents (Invitrogen), according to the manufacturer's protocols. Primary cortical neurons were obtained from embryos of Institute of Cancer Research mice on embryonic day 18 and cultured in Neurobasal medium with B-27 supplement and 0.5 mm l-glutamine (Invitrogen). Primary neurons were transiently cotransfected with wild-type αS and GRK5 cDNAs using Lipofectamine 2000 reagents (Invitrogen). HEK293 cell lines stably expressing wild-type αS (HEK293–αS) were generated by selection for neomycin resistance using 1 mg/ml G418 (Invitrogen).
αS phosphorylation in cells and immunoblotting.
For detection of phosphorylation of αS in HEK293 or SH-SY5Y cells, okadaic acid (OA; Wako Chemicals, Osaka, Japan) in the indicated concentrations was added to the conditioned medium at 30 h after transfection, and the cells were incubated for 16 h. Collected cell pellets were lysed on ice with PBS containing 1% Nonidet P-40, 1× protease inhibitor cocktail (Roche Diagnostics, Indianapolis, IN), and 1× phosphatase inhibitor cocktail (Sigma, St. Louis, MO). After centrifugation at 12,000 × g for 30 min, supernatants were collected. For immunoblot analysis, all proteins were denatured by boiling for 5 min in Laemmli's sample buffer containing 2.5% 2-mercaptoethanol, subjected to 12.5% polyacrylamide gels, and transferred to a polyvinylidene difluoride membrane (Millipore, Billerica, MA). Membrane was probed with antibodies specific for αS (Syn-1, monoclonal IgG; BD Biosciences, San Jose, CA), pS129–αS (psyn#64, monoclonal IgG) (Saito et al., 2003), FLAG tag (M2; Sigma), β-actin (AC-15; Sigma), superoxide dismutase 1 (SOD1; Stressgen Biotechnologies, San Diego, CA), or GRK5 (H-64, polyclonal; Santa Cruz Biotechnology, Santa Cruz, CA) overnight at 4°C. Bands were visualized by ECL or ECL Plus (GE Healthcare, Piscataway, NJ). The relative intensity of the band was scanned and quantified with the Scion image program, version 4.02 (Scion, Frederick, MD). Detailed procedures were described previously by Arawaka et al. (2002).
Chemical cross-linking and immunoprecipitation.
Transfected HEK293 cells in 100 mm dishes were rinsed twice with ice-cold PBS, resuspended in 6 ml of cross-linking buffer [PBS containing 10 mm HEPES, pH 7.4, and 2.5 mm of dithiobis(succinimidylpropionate) (DSP; Pierce Biotechnology, Rockford, IL)], and incubated for 30 min at room temperature (RT). The cross-linking buffer was removed, and the cells were extracted with 1 ml of ice-cold lysis buffer (50 mm Tris-HCl, pH 7.6, 150 mm NaCl, 1 mm EDTA, 10% glycerol, 1% Nonidet P-40, and 1× protease inhibitor cocktail). After centrifugation at 12,000 × g for 30 min, the supernatants were incubated with one of the indicated antibodies overnight at 4°C and then incubated with protein G or A agarose beads for 2 h. Beads were washed three times with ice-cold lysis buffer, and immunoprecipitates were dissolved in Laemmli's sample buffer. Equivalent amounts of samples were analyzed by immunoblotting.
For experiments using human brain, frozen tissue of the temporal cortex was homogenized in PBS containing protease inhibitors with a Teflon Potter homogenizer, and the homogenate was centrifuged at 800 × g for 5 min. DSP was added to the supernatant at a final concentration of 2.5 mm and incubated for 30 min at RT with gentle rotation for cross-linking. After the reaction was quenched with 50 mm of Tris, pH 7.6, the brain homogenate was centrifuged at 100,000 × g for 30 min and the resultant pellets were extracted with 10 passages through a 27 gauge needle in 1:5 (w/v) ice-cold lysis buffer. The lysate was centrifuged again at 100,000 × g for 30 min, and the supernatant was used for immunoprecipitation as described above.
Protein fractionation and size exclusion chromatography.
For fatty acid-induced αS oligomerization, HEK293–αS cells were incubated for 24 h after transfection with GRK5 cDNA. The cells were resuspended in fresh medium containing 20 nm OA. After incubation for 8 h [for size exclusion chromatography (SEC) experiments] or 16 h (for time course analysis of αS oligomerization), the medium was discarded, and the cells were incubated with serum-free medium containing 500 μm α-linolenic acid and 100 μm BSA (Sharon et al., 2001, 2003) and OA for the indicated time.
The ultracentrifuged cytosols (S370 cytosols) of HEK293–αS cells were fractionated based on the previously published protocol (Sharon et al., 2001, 2003), with slight modifications. All procedures were performed at 4°C. Collected cells were suspended in homogenization buffer (20 mm HEPES, pH 7.4, 1 mm MgCl2, 0.32 m sucrose, 43 mm 2-mercaptoethanol, 1× protease inhibitor cocktail, and 1× phosphatase inhibitor cocktail) and disrupted by a Teflon Potter homogenizer. The homogenate was centrifuged at 200 × g for 10 min, and the supernatant was centrifuged at 8000 × g for 15 min. The resultant supernatant was finally centrifuged at 370,000 × g for 1 h, and the supernatant was subjected to immunoblotting or SEC experiments. Protein concentration was measured by the Bradford method (Bio-Rad, Hercules, CA).
For SEC, 300 μg of the S370 cytosol of the HEK293–αS cells transfected with GRK5 cDNA or a set of molecular weight standards were loaded onto a Superdex 75 10/300 column (bed volumes, 24 ml; GE Healthcare). Elution was performed at a linear flow rate of 6 ml/h in 50 mm ammonium acetate, pH 7.4, containing 20 nm OA using the FPLC system (GE Healthcare Bio-Sciences), and 0.5 ml of fractions were collected. A 0.25 ml volume of the indicated fractions was lyophilized, followed by resolving in the homogenization buffer.
For lipid removal, the S370 cytosols or SEC samples were treated at 65°C for 16 h (Sharon et al., 2001, 2003) and then resuspended in Laemmli's sample buffer containing 2.5% 2-mercaptoethanol for immunoblotting.
In vitro αS oligomerization.
Human αS cDNA was subcloned into the pGEX-4T-1 vector (GE Healthcare) to generate glutathione S-transferase fusion protein. αS was expressed in Escherichia coli and purified using glutathione–Sepharose 4 Fast Flow beads (GE Healthcare) and thrombin. The purity of the protein was >95%, as determined on the gel stained with Coomassie brilliant blue R-250 after PAGE. Recombinant αS protein (5 μm) was incubated with α-linolenic acid (100 μm) in PBS at 37°C for 16 h and then incubated with Lipidex 1000 (10% w/v; Sigma) at 37°C with rocking for 1 h. For the analysis of fatty acid–protein complexes, the reaction mixture was centrifuged at 15,000 × g, and the resultant pellet was collected. The pellet was boiled for 5 min in Laemmli's sample buffer, and the sample was analyzed by immunoblotting.
Immunocytochemistry and immunohistochemistry.
HEK293–αS cells were grown on the four-chamber slides (Nalge Nunc, Rochester, NY) and were transiently transfected with either wild-type GRK5–FLAG or mutant GRK5–K215R–FLAG cDNA. Cells were fixed with PBS containing 4% paraformaldehyde at RT for 15 min, blocked with PBS containing 5% skim milk and 0.05% Triton X-100 for 1 h, and then incubated with a mixture of H-64 and either LB509 (monoclonal anti-αS antibody; Zymed Laboratories, South San Francisco, CA) or psyn#64 overnight at 4°C. After washing, cells were incubated with a mixture of Alexa 488 anti-mouse IgG (green) and Alexa 568 anti-rabbit IgG (red) (Invitrogen) for 1 h at 37°C. Images were acquired on an LSM 510 Meta laser-scanning confocal microscope (Zeiss, Jena, Germany).
For immunohistochemistry, formalin-fixed, paraffin-embedded tissue sections (6 μm thickness) of the brain from six autopsied patients with sPD were deparaffinized. Tissue sections of the brains from patients with dementia with LBs (DLB) (three cases) and multiple system atrophy (MSA) (two cases) were also used for our immunohistochemical study. Some sections were heated in a citrate solution, pH 6.0, using a microwave oven for antigen retrieval. The sections were incubated with 1% H2O2 in distilled water for 10 min to block intrinsic peroxidase activity and then with 5% normal goat serum in PBS containing 0.03% Triton X-100 for 30 min to block nonspecific binding sites. The sections were then incubated overnight at 4°C with polyclonal antibody against human GRK5 (H-64), monoclonal anti-αS antibody (LB509), or the polyclonal antibodies listed in Table 1 and then with appropriate biotinylated secondary antibodies, followed by incubation with avidin–biotin–peroxidase conjugate. Color was developed with 3,3′-diaminobenzidine tetrahydrochloride, with or without nickel ammonium sulfate. To obtain a direct correlation between hematoxylin and eosin (H&E)-stained and immunostained LBs in the same preparations, some sections were first stained with H&E, photographed, and finally immunostained with anti-GRK5 (Kato et al., 1989). The specificity of the commercial antibodies against each one of the GRK and CK families (Table 1) was confirmed by using the lysates of HEK293 cells overexpressing the respective proteins, and the results are shown in supplemental Figure 1 (available at www.jneurosci.org as supplemental material).
Table 1.
Primary antibodies used for immunohistochemistry
| Antibodya | Isotype | Dilution | Staining of Lewy bodies |
|---|---|---|---|
| GRK1 (H-160) | Rabbit polyclonal IgG | 1:100 | No |
| GRK2 (C-15) | Rabbit polyclonal IgG | 1:100 | No |
| GRK3 (E-15) | Goat polyclonal IgG | 1:100 | No |
| GRK4 (I-20) | Rabbit polyclonal IgG | 1:100 | No |
| GRK5 (H-64) | Rabbit polyclonal IgG | 1:100 | Yes |
| GRK6 (C-20) | Rabbit polyclonal IgG | 1:100 | No |
| CK Iα (C-19) | Goat polyclonal IgG | 1:100 | No |
| CK Iγ1 (N-14) | Goat polyclonal IgG | 1:30 | No |
| CK Iγ2 (C-20) | Goat polyclonal IgG | 1:30 | No |
| CK Iϵ (H-60) | Rabbit polyclonal IgG | 1:30 | No |
| CK Iδ (N-19) | Goat polyclonal IgG | 1:30 | No |
| CK IIα (C-18) | Goat polyclonal IgG | 1:30 | No |
| CK IIβ (FL-215) | Rabbit polyclonal IgG | 1:30 | No |
a All antibodies were obtained from Santa Cruz Biotechnology.
Electrophoretic mobility shift assays.
Nuclear extract was prepared from SH-SY5Y cells and stored in aliquots at −80°C until use. Protein concentration was measured using the Bradford method. Double-stranded oligonucleotides specific for m24 T or C allele and m22.1 A or G allele were labeled with 5′-32P using T4 polynucleotide kinase. 32P-labeled probes were incubated with nuclear extracts for 30 min at RT, and these were then separated by electrophoresis on a 5% nondenaturing polyacrylamide gel with 0.5-fold Tris-borate-EDTA running buffer. DNA–protein complexes were detected by autoradiography. For the supershift assays, nuclear extracts were preincubated with 2 μg of antibodies against cAMP response element-binding protein 1 (CREB-1) (X-12), c-Jun (D), or c-Fos (K-25; Santa Cruz Biotechnology) for m24 and 10 μg of antibody against Yin Yang-1 (YY1) (H414; Santa Cruz Biotechnology) for m22.1 for 10 min at 4°C before addition of 32P-labeled probes. Competition assays were conducted by adding 200- or 400-fold excess of nonlabeled probes.
Luciferase assay.
Luciferase reporter plasmids were constructed by cloning single or three-tandem copies of DNA fragments that consist of the 21 nt bearing m24 or m22.1 in the center into the pGL3-promoter vector (Promega, Madison, WI) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). The inserted portions of all constructs were verified by sequencing. SH-SY5Y cells (8 × 105) were transfected with 0.1 μg of the reporter constructs and with 0.002 μg of pRL-TK Renilla luciferase vector (Promega) with Lipofectamine Plus reagents. The Renilla luciferase vector served as an internal control for transfection efficiency. At 18 h after transfection, the cells were treated with or without 1 mm cAMP (Calbiochem, La Jolla, CA). After 6 h, the cells were harvested, and luciferase activity was measured with the Dual-Luciferase Reporter Assay System (Promega).
Chromatin immunoprecipitation assay.
Chromatin immunoprecipitation (ChIP) was performed according to the manufacturer's protocols (Millipore). SH-SY5Y cells were fixed with 1% formaldehyde for 10 min at 37°C. Cells were sonicated on ice for generating soluble chromatin complexes with DNA fragment length <2 kb. Samples containing the equivalent of 1 × 106 cells were immunoprecipitated with 5 μg of anti-CREB-1 antibody (C-21) or 5 μg of anti-YY1 antibody (C-20; Santa Cruz Biotechnology). After removing proteins, DNA was amplified by PCR using the following primer sets: 5′-tgccggagttcgactggtcag-3′ and 5′-tagcagctcaatggctttgtc-3′ for the DNA sequence including m24; 5′-aaagcctcaaccttctaatcc-3′ and 5′-tccttggaatttctgagtaata-3′ for the DNA sequences including m22.1.
Subjects.
Patients with sPD were collected through the Yamagata Study Group of PD, which surveyed all of the 782 hospitals and clinics located in the Yamagata prefecture, which has a population of 1.2 million (Kimura et al., 2002). A total of 1477 patients diagnosed as having PD were registered during the period of September and October, 2000, and of these, 963 patients were selected for additional critical evaluation for sPD. Diagnosis was made by experienced neurologists and/or neurosurgeons who gave careful interviews and medical and neurological examinations, and brain magnetic resonance imaging (MRI) was examined for all patients. Among these, DNA samples were obtained from 317 patients with written informed consent, and 286 patients whose age of onset was >50 years were enrolled in this study. We used the diagnostic criteria recommended by “Multicentric Research Study on the Skill and Indication of Surgical Therapy for PD,” a research group supported by the Japanese government. In brief, sPD was defined as the presence of all of the following five items: (1) insidious onset after 20 years of age, (2) resting tremor of 4–6 Hz or cogwheel rigidity with akinesia or small-step gait, (3) apparent improvement of parkinsonism by l-dopa, a dopamine receptor agonist, or an anticholinergic agent, (4) no history of administration of drugs known to cause parkinsonism, and (5) exclusion of symptomatic parkinsonism, such as vascular parkinsonism, multiple system atrophy, progressive supranuclear palsy, or normal-pressure hydrocephalus. This careful sampling enabled acquisition of “clinically high-quality” samples of sPD by exclusion of secondary parkinsonism as strictly as possible. Age (mean ± SD) in PD patients at the time of examination was 71.3 ± 7.3 years, and age (mean ± SD) at the onset of PD was 65.0 ± 7.4 years. The PD group consisted of 62.4% women and 37.6% men. This gender proportion corresponds to that in Japanese sPD patients. Quite different from European countries and the United States of America, a female preponderance of sPD has been observed in Japan (Kimura et al., 2002).
The control samples were obtained from residents of the same prefecture through a publically financed health examination for 60- and 70-year-olds. In addition to the routine medical examinations, all participants underwent neurological examinations and brain MRIs for exclusion of neurological diseases, including parkinsonism. DNA samples were obtained with written informed consent from 496 individuals who had no neurological abnormalities. Age (mean ± SD) of the controls was 67.0 ± 5.3 years. The overall samples consisted of 59.2% women and 40.8% men; there was no difference in sex ratio between the cases and controls (p = 0.290; Fisher's test).
Single-nucleotide polymorphism genotyping.
Single-nucleotide polymorphisms (SNPs) were genotyped by the TaqMan assay (Livak, 1999). SNPs showing minor allele frequency <0.1 or a typing failure rate >0.05 were omitted for our analysis. SNPs showing Hardy–Weinberg equilibrium p value <0.05 in the control samples were also omitted.
Haplotype trend regression test.
We used a “sliding window approach” in the haplotype trend regression (HTR) test. HTR is the permutation-based haplotype assessment program for case–control association study using a linear multiple-regression model that was constructed with all inferred haplotype probabilities in each unrelated individual as the dependent variable and the phenotypic response (0 or 1) as the independent variable. Inference of haplotypes in HTR was performed by using an expectation maximization algorithm. Empirical p values of both overall and individual haplotype association tests were obtained by testing this regression model by a permutation procedure that randomly shuffled the affection status among individuals. The empirical p value was the proportion of times when shuffled data produced a value equal to or larger than the statistic of the original data set. In a sliding window approach, an SNP window with fixed numbers of contiguous SNPs shifts one marker at a time (Zaykin et al., 2002). A triangular matrix of pair-wise linkage disequilibriums (LDs) between scanned SNPs was calculated to confirm that each sliding block had an adequate degree of LD for haplotypic inference (Table 2).
Table 2.
Pair-wise LD matrix, showing a triangular matrix of pair-wise LD parameters D' and p value in the GRK5 gene

Pair-wise LD coefficients (D') and the log likelihood ratio p values are shown. D' > 0.3 and p < 0.05 are shaded.
Evaluation of population stratification.
Overall genomic control χ2 test was performed using 25 unlinked SNPs. This test revealed the absence of stratification (p = 0.919).
Results
Immunohistochemical identification of GRK5 in LBs of sPD
We performed immunohistochemical examination of sPD brains for detection of any protein kinase known to phosphorylate αS (CK1, CK2, and a family of GRKs shown in Table 1). Among the kinases tested, positive staining was found only when anti-GRK5 antibody was used (Table 1). GRK5 accumulated in LBs in dopaminergic neurons of the substantia nigra and in neurons of the locus ceruleus (Fig. 1A,B,E–G). It was noted that GRK5 and αS colocalized in LBs (Fig. 1E–G). Both anti-αS and anti-GRK5 antibodies bound to pale bodies (Fig. 1C,D). Although the anti-GRK5 antibody did not bind to typical Lewy neurites in the neuropil, it bound to “filamentous” or “rod-like” structures in the neurites of nigral neurons in sPD brains (Fig. 1H–K). The immunostaining with the anti-GRK5 antibody was completely abolished by the addition of recombinant human GRK5 to the primary antibody solution (Fig. 1N–Q). None of the antibodies specific for CK1, CK2, GRK1, GRK2, GRK3, GRK4, or GRK6 bound to LBs, Lewy neurites, or pale bodies under the experimental conditions shown in Table 1. GRK5 was not detected in the cortical-type LBs of DLB (Fig. 1L,M) nor in the αS-positive glial cytoplasmic inclusions of MSA (data not shown). The accumulation of GRK5 distinctly in LBs and pale bodies of sPD brains suggested its specific role in the pathogenesis of sPD.
Figure 1.

Anti-GRK5 antibody immunostains Lewy bodies in the brainstem of sPD patients. A, B, The same section of the locus ceruleus stained with H&E (A) and anti-GRK5 (H-64) (B). C, D, Pale bodies in the locus ceruleus (C, H&E; D, H-64) were also positive for GRK5 staining. E–G, Confocal microscopic images of Lewy body in a nigral neuron using H-64 (E) and anti-αS antibodies (LB509; F). A merged image of E and F is shown in G. H, Filamentous or rod-like structure in the substantia nigra stained with H-64. I–K, Confocal microscopic images of filamentous or rod-like structures in the substantia nigra using H-64 (I) and LB509 (J). A merged image of I and J is shown in K. L, M, No immunostaining for GRK5 was found in cortical-type Lewy bodies (L, LB509; M, H-64). N–Q, The immunostaining with H-64 (N, P) was completely abolished by the addition of recombinant human GRK5 to the primary antibody solution (O, Q). The boxed areas in N and O are shown in P and Q, respectively. The arrows in N indicate Lewy bodies. L–O are the serial sections, and the asterisks indicate the same blood vessels. L–Q are counterstained with neutral fast red. Scale bars, 20 μm.
GRK5 phosphorylates αS at Ser-129
Colocalization of GRK5 and αS in LBs prompted us to investigate biochemical interaction between the two molecules. For this purpose, we established HEK293 cell lines stably expressing human wild-type αS. These cells were transfected either with GRK5–FLAG cDNA or with an empty vector. Because pS129–αS is easily dephosphorylated (Fujiwara et al., 2002), we treated the cells with OA, a protein phosphatase 1 (PP1)/PP2A-specific inhibitor (Okochi et al., 2000). Immunoblot analysis of cell lysates showed that coexpression of αS and GRK5 in HEK293–αS cells resulted in an increase in pS129–αS, which was detected by psyn#64 antibody to pS129–αS (Saito et al., 2003), in a manner dependent on the dose of OA (Fig. 2A, top). The level of pS129–αS also increased in a manner dependent on incubation time with OA (Fig. 2A, bottom). A faint signal of pS129–αS was observed in the cells transfected with empty vector at 24 h incubation with OA (Fig. 2A, bottom), suggesting that background phosphorylation of αS occurred with endogenous undetermined kinase(s). When HEK293–αS cells were transfected with increasing doses of GRK5 cDNA, the amount of pS129–αS increased in parallel to that of GRK5 (Fig. 2B, top). To confirm the role of GRK5 in the phosphorylation of Ser-129 of αS, we made substitution mutants at the phosphorylated site of αS (αS–S129A) and the catalytic site of GRK5 (GRK5–K215R). The mutant αS–S129A was not phosphorylated by GRK5, whereas the catalytic inactive mutant, GRK5–K215R, failed to phosphorylate αS (Fig. 2B, bottom). GRK5-catalyzed αS phosphorylation was detected in neuroblastoma SH-SY5Y cells as well (data not shown).
Figure 2.
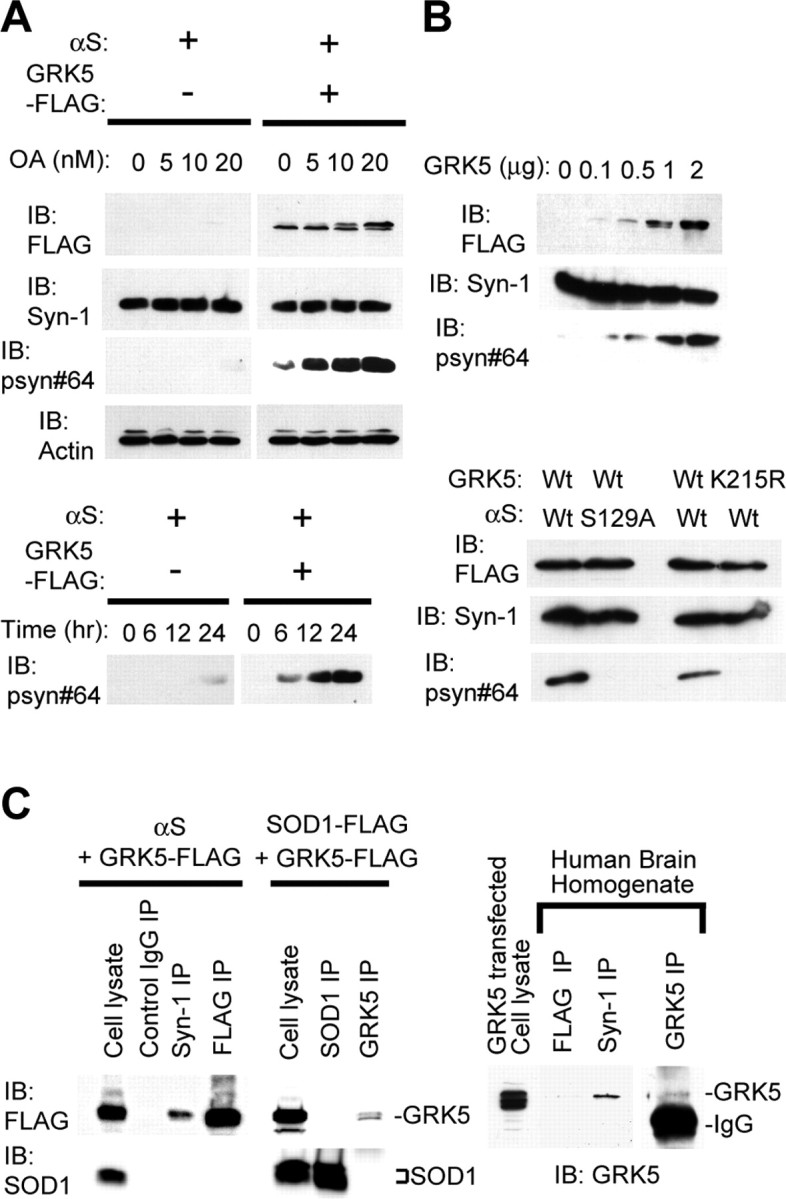
GRK5 phosphorylates αS at Ser-129. A, Immunoblots (IB) using one of the antibodies for GRK5–FLAG (FLAG), total αS (Syn-1), pS129–αS (psyn#64), and β-actin (AC-15). HEK293–αS cells transfected with or without GRK5–FLAG cDNA were treated with OA at concentrations of 0, 5, 10, and 20 nm (top). They were incubated for 0, 6, 12, and 24 h in the presence of 20 nm OA in medium (bottom). In HEK293–αS cells expressing GRK5, pS129–αS was detected in OA dose- and incubation time-dependent manners. The levels of total αS and actin were constant (top). B, Phosphorylation of αS at Ser-129 through GRK5 enzymatic activity. Top, HEK293–αS cells were transfected with increasing amounts of GRK5–FLAG cDNA, and cell lysates were subjected to immunoblot analysis. Although the expression levels of total αS were approximately equal, the pS129–αS levels increased in parallel to the level of GRK5. Bottom, Immunoblotting of cell lysates expressing the combination of wild-type (Wt) αS or αS–S129A and wild-type GRK5 or GRK5–K215R. pS129–αS was seen only in the cells expressing both wild-type GRK5 and wild-type αS. The cells expressing either αS–S129A and GRK5 or αS and GRK5–K215R did not produce pS129–αS. C, Physical association of GRK5 with αS in cultured cells and human brain tissue. A cross-linker, DSP, was used in all experiments. Left and middle, HEK293 cells were transfected with αS and GRK5–FLAG cDNAs, and cell lysate was immunoprecipitated (IP) with control IgG, Syn-1, and anti-FLAG antibody (left). HEK293 cells were also transfected with SOD1–FLAG and GRK5–FLAG, and the cell lysates were immunoprecipitated with anti-SOD1 or anti-GRK5 (H-64) antibodies (middle). Immunoprecipitates were analyzed by immunoblotting with anti-FLAG antibody (top) or with anti-SOD1 antibody (bottom). Binding of GRK5 to αS was detected (left), whereas nonspecific binding of GRK5 to endogenous/overexpressed SOD1 was not (left and middle). Right, Tissue extracts from human temporal cortex were immunoprecipitated with anti-FLAG (negative control), Syn-1, or anti-GRK5 (H-64), followed by immunoblotting with H-64. Binding of GRK5 and αS was detected in the extracts.
To prove a physical association between GRK5 and αS, we performed an immunoprecipitation study. HEK293 cells were transfected with αS and GRK5–FLAG cDNAs. The cells were then treated with DSP, a membrane-permeable cross-linker. Cell lysates were incubated with anti-αS antibody, and immunoprecipitates were analyzed by SDS-PAGE and immunoblotting with anti-FLAG. As shown in Figure 2C (left), coprecipitation of GRK5 with αS was detected only after treatment of the cells with DSP, suggesting a close, but weak or transient, association between the two molecules. To rule out the possibility that the use of DSP might have caused nonspecific protein–protein coupling, we examined whether GRK5 might be coimmunoprecipitated with endogenous and overexpressed SOD1, a ubiquitous cytosolic protein, in HEK293 cells. As shown in Figure 2C (middle), anti-SOD1 antibody failed to coimmunoprecipitate GRK5 in the presence of DSP. We further examined the interaction between GRK5 and αS by using recombinant proteins in the cell-free system. The results showed that Ser-129 of the recombinant αS was phosphorylated in the presence, but not in the absence, of recombinant GRK5, indicating the interaction between the two proteins (data not shown). Coprecipitation of endogenous GRK5 and αS using anti-αS was also observed in human cerebral cortex tissue when the tissue was treated with DSP (Fig. 2C, right).
αS colocalizes with GRK5 at the plasma membrane
To confirm the association between GRK5 and αS morphologically, HEK293–αS cells were transiently transfected with either wild-type GRK5 or GRK5–K215R and incubated in the presence or absence of OA. The cells were fixed and processed for direct immunofluorescence with polyclonal anti-GRK5 antibody and either monoclonal anti-αS antibody or monoclonal anti-pSer129–αS antibody. Examination of the cells by confocal microscopy revealed that the wild-type GRK5 localized predominantly in the area of plasma membrane and a little in the perikaryal area (Fig. 3A,B, top). The distribution of wild-type GRK5 was in agreement with previous reports (Pronin et al., 1998; Thiyagarajan et al., 2004). αS localized mainly in the cytoplasm, but a portion of αS colocalized with GRK5 in the area of the plasma membrane (Fig. 3A, top). In the cells expressing wild-type GRK5, phosphorylated αS was distributed in the areas of perikarya and plasma membrane (Fig. 3B, top). In the cells expressing mutant GRK5–K215R, there was no fluorescence of pS129–αS, although the intracellular distribution of GRK5–K215R was similar to that of wild-type GRK5 (Fig. 3B, bottom). Phosphorylation of αS by GRK5 was also observed in double-transfected primary neurons from the cerebral cortex of fetal mice (Fig. 3C).
Figure 3.
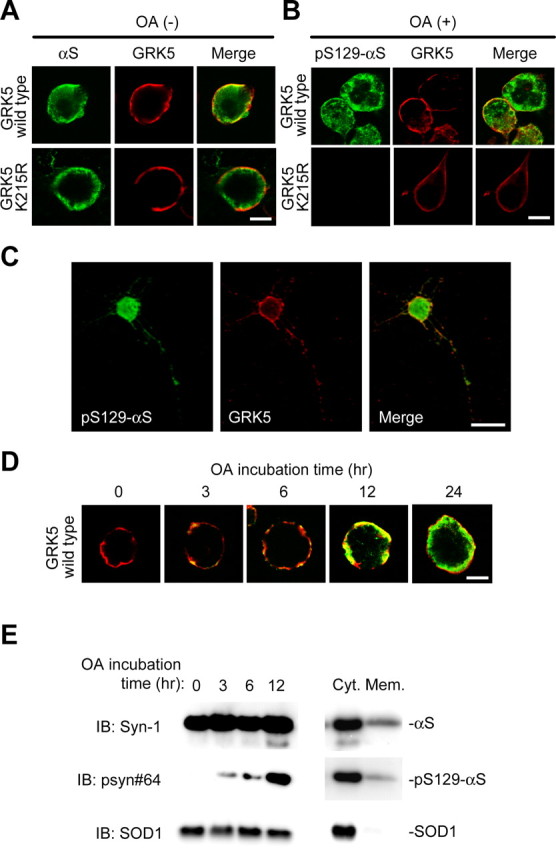
αS colocalizes with GRK5 at the plasma membrane of HEK293 cells. A, B, HEK293–αS cells were transiently transfected with either wild-type GRK5–FLAG (top) or GRK5–K215R-FLAG (bottom) and incubated in the presence (B) or absence (A) of OA. Fixed cells were processed for direct immunofluorescence with polyclonal anti-GRK5 antibody (H-64) (A and B, red channel) and either monoclonal anti-αS antibody (LB509; A, green channel) or monoclonal anti-pS129–αS antibody (psyn#64; B, green channel). In HEK293–αS cells expressing GRK5, αS and GRK5 colocalized in the area of the plasma membrane (A, top), and pS129–αS was distributed in the areas of the perikarya and plasma membrane (B, top). In the cells expressing mutant GRK5–K215R, there was no fluorescence of pS129-αS, although the distribution of GRK5–K215R was similar to that of wild-type GRK5 (B, bottom). Scale bars, 10 μm. C, Phosphorylation of αS by GRK5 in primary neurons from the cerebral cortex of fetal mice. The primary cortical neurons were cotransfected with wild-type αS and GRK5 cDNAs, treated with 10 nm OA for 12 h, and immunostained with anti-pS129–αS (psyn#64; left) and anti-GRK5 (H-64; middle) antibodies. A merged image is shown at right. Scale bar, 20 μm. D, Translocation of pS129–αS catalyzed by GRK5. HEK293–αS cells were transiently transfected with wild-type GRK5–FLAG cDNA, and the cells were incubated with 20 nm OA for various time periods. Fixed cells were immunostained with anti-GRK5 (H-64; red) and anti-pS129–αS (psyn#64; green) antibodies. After 3 h of incubation, pS129–αS was detected in the area of the plasma membrane. With time, pS129–αS was gradually translocated from the plasma membrane to the perikaryal area. Scale bar, 10 μm. E, Immunoblotting (IB) of the cytosol and membrane fractions of HEK293–αS cells transiently transfected with wild-type GRK5 cDNA. After incubation of the cells with 20 nm OA for various time periods, the cells were disrupted, and the homogenate was sequentially centrifuged at 800 × g for 10 min and at 100,000 × g for 30 min. The fractions were tested for the presence of pS129–αS by immunoblotting. In the presence of OA, the amount of pS129–αS in the cytosol fraction gradually increased with time, although the expression levels of the total αS and SOD1 were constant (left). In the same preparation of cell homogenate at 12 h of OA incubation, the amount of pS129–αS in the cytosol fraction (Cyt.) was much larger than that in the membrane fraction (Mem.; right). Abundant SOD1 was detectable in the cytosol fraction.
To observe possible changes in the distribution of pS129–αS in the cells with time, we performed a time course experiment: HEK293–αS cells were transiently transfected with GRK5 cDNA. At 24 h after transfection, these cells were incubated with OA for 0, 3, 6, 12, or 24 h, and fixed cells were immunostained with anti-GRK5 and anti-pSer129–αS (psyn#64) antibodies. As shown in Figure 3D, pS129–αS was first generated at the plasma membrane, at which GRK5 and αS colocalized, and then gradually translocated to the perikaryal area. This finding was confirmed by fractionation of the homogenate of the HEK293–αS cells transiently transfected with GRK5 cDNA, followed by immunoblotting of the cytosol and membrane fractions. As shown in Figure 3E (left), pS129–αS became detectable in the cytosol 3–6 h after the addition of OA to the culture medium, and the amount of pS129–αS in the cytosol fraction gradually increased with time. At 12 h of OA incubation, pS129–αS accumulated much more abundantly in the cytosol fraction than in the membrane fraction (Fig. 3E, right). The colocalization of GRK5 and αS at the plasma membrane was compatible with the in vitro observation that phospholipids promote phosphorylation of recombinant αS by GRK5 (Pronin et al., 2000).
GRK5 promotes α-linolenic acid-induced oligomerization of αS
αS shares biophysical properties with a family of fatty acid-binding proteins (FABPs) (Sharon et al., 2001). Sharon et al. (2003) reported that the formation of highly soluble oligomers of αS is induced by polyunsaturated fatty acids (PUFAs), and this process is known to be enhanced in the brains of αS-transgenic mice and in human PD. Selkoe (2004) suggested that these oligomers may be a pathogenic species of αS in PD.
Because the detection of SDS-stable, soluble αS oligomers by immunoblotting requires delipidation procedures, we conducted heat treatment to remove lipids from αS oligomers to make their bands detectable by anti-αS (Sharon et al., 2001, 2003). High-speed (370,000 × g) soluble fraction from cell lysates was heat treated (65°C for 16 h) and subjected to immunoblot analysis (Sharon et al., 2001). In HEK293–αS cells, soluble oligomers of transfected αS were formed in the presence of α-linolenic acid (Fig. 4A, left and middle). OA, which was added in the culture medium to inhibit dephosphorylation of αS, did not have any effect on the oligomerization of αS by itself (Fig. 4A, left and middle). To test whether self-association of αS occurs in the presence of PUFA, recombinant αS was mixed with α-linolenic acid and immunoblotted. As shown in Figure 4A (right), αS monomer, dimer, trimer, and tetramer were detected. To exclude the possibility that oligomerization of αS might have been induced by heat treatment, the experiment using size exclusion chromatography was performed, in which fractionation was conducted under nondenatured conditions before heat treatment. A large amount of pS129–αS oligomers was recovered in the fractions of high molecular weights (45–70 kDa) (Fig. 4B). The results suggested that the formation of αS oligomers occurred in the living cells.
Figure 4.
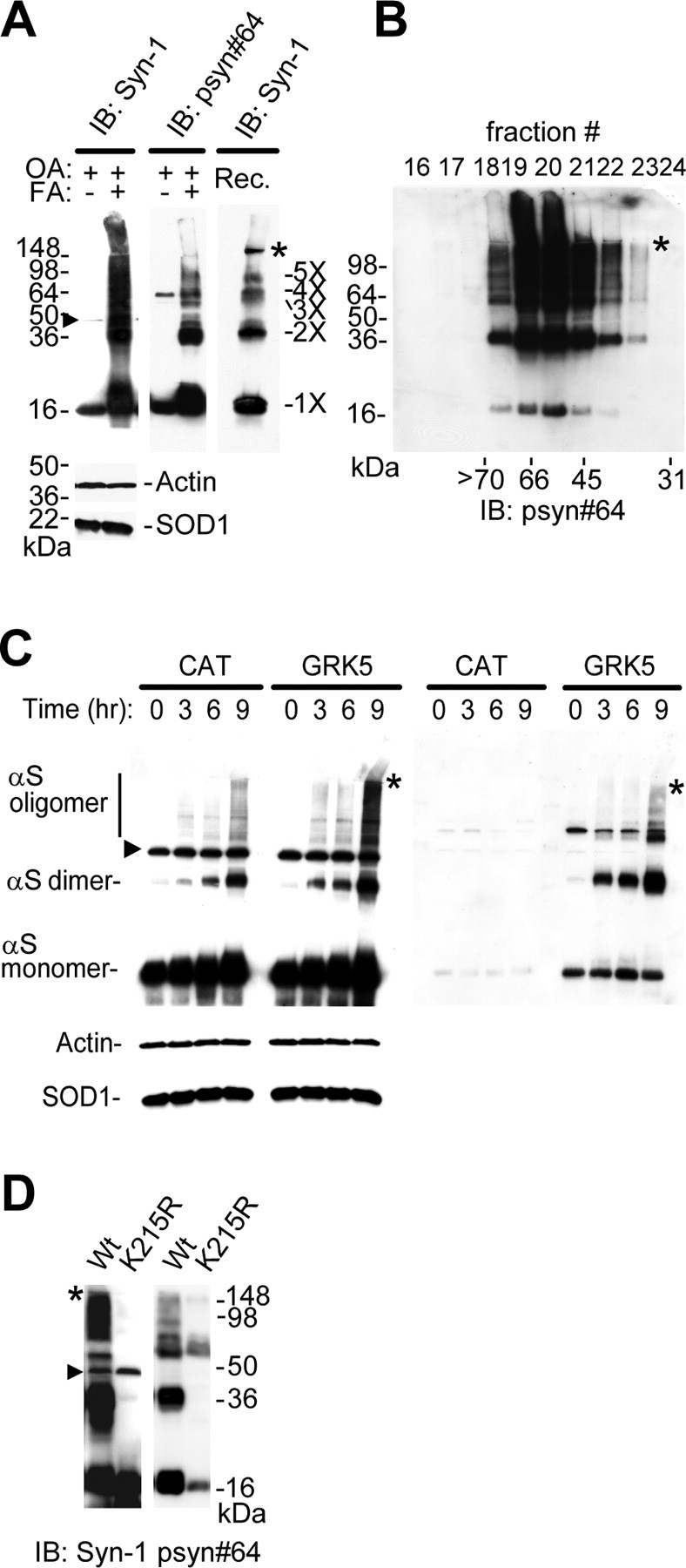
GRK5 promotes α-linolenic acid-induced oligomerization of αS. A, To test the effect of fatty acid (FA) on αS oligomerization, HEK293–αS cells expressing GRK5–FLAG were preincubated with 20 nm OA for 8 h and then conditioned with or without α-linolenic acid for 16 h along with OA. High-speed soluble cytosol fractions (30 μg of protein) from cell lysates were heat treated (65°C for 16 h) to remove lipids before gel loading and were immunoblotted (IB) with Syn-1 (top left) or psyn#64 (top middle). For loading control, the same amounts of non-heat-treated samples were immunoblotted with anti-actin and anti-SOD1 antibodies (bottom left). As the molecular weight standards, αS oligomers formed in vitro by the treatment of recombinant αS (Rec.) with α-linolenic acid (see Materials and Methods) are shown at right. B, High-speed soluble cytosol from cells expressing αS and GRK5 was loaded onto a Superdex 75 column and eluted with ammonium acetate buffer. A large amount of pS129–αS oligomers was eluted into the fractions of high molecular weights (45–70 kDa), indicating that the formation of pS129–αS oligomers occurred under nondenatured conditions. Immunoblotting was made with psyn#64 after heat treatment. C, HEK293–αS cells expressing either GRK5–FLAG or CAT were incubated with 20 nm OA for 16 h and conditioned with α-linolenic acid for 0, 3, 6, and 9 h along with OA. High-speed soluble cytosol fractions (30 μg of protein) from cell lysates were heat treated (65°C for 16 h) to remove lipids before gel loading and immunoblotted with Syn-1 (left) or psyn#64 (right). Although the levels of αS monomers and oligomers dominantly containing nonphosphorylated species increased within 3 h in CAT-expressing cells, αS containing phosphorylated species yielded more oligomers in GRK5-expressing cells at 9 h. The smear pattern in the background and the gel-excluded immunoreactive material, which seemed to represent aggregated αS species, were also seen at 9 h after incubation of α-linolenic acid in the presence of GRK5. For loading control, the same amounts of non-heat-treated samples were immunoblotted with anti-actin and anti-SOD1 antibodies (bottom left). D, Expression of the enzymatic-inactive mutant GRK5 (K215R) in HEK293–αS cells, instead of wild-type (Wt) GRK5, failed to promote the oligomerization of αS. The cells transfected with either wild-type GRK5 or GRK5 K215R cDNA were preincubated with 20 nm OA for 16 h and incubated for 8 h with α-linolenic acid along with OA. The high-speed soluble cytosol fractions of the cells were analyzed by immunoblotting. Asterisks (A–D) indicate the interface of resolving/stacking gel, and the arrowheads (A, C, D) point to a nonspecific band (∼47 kDa) by Syn-1 antibody (Perrin et al., 2003).
To explore the effect of GRK5 on the oligomerization of αS, HEK293–αS cells with or without transfection of GRK5 cDNA were conditioned with α-linolenic acid for 0, 3, 6, or 9 h. As shown in Figure 4C [left, chloramphenicol acetyltransferase (CAT)], α-linolenic acid induced the formation of soluble αS oligomers in a time-dependent manner. It was found that expression of GRK5 promoted the oligomerization of αS (Fig. 4C, left, GRK5). The “smear pattern” in the background and the gel-excluded immunoreactive material were also seen at 9 h after incubation of HEK293–αS cells expressing GRK5 with α-linolenic acid (Fig. 4C, left, GRK5). The smear pattern may represent aggregated αS species that are partially SDS soluble. The gel-excluded material may be the species that are no longer SDS soluble. The levels of αS dimers and oligomers (>50 kDa) are significantly higher in GRK5-expressing cells than in CAT-expressing cells. The relative ratios of the 37 kDa dimer to the 17 kDa monomer, determined by densitometry, were 0.009 ± 0.006 in CAT-expressing cells and 0.059 ± 0.024 in GRK5-expressing cells (mean ± SD; p = 0.0246) at 6 h after administration of α-linolenic acid. The relative ratio of the >50 kDa oligomer to the monomer in CAT-expressing cells was 0.058 ± 0.026, whereas it was 0.179 ± 0.06 in GRK5-expressing cells (p = 0.0326; Student's t test). The αS dimers and oligomers in GRK5-expressing cells contained a large amount of pS129–αS, compared with control gene (CAT) expression (Fig. 4C, right). We also performed the experiments of αS oligomerization by α-linolenic acid in the absence or the presence of different concentrations of OA, and we found that the addition of OA increased oligomerization of αS in a dose-dependent manner (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). Furthermore, the expression of the enzymatic-inactive mutant GRK5 (K215R), instead of wild-type GRK5, in the HEK293–αS cells failed to promote αS oligomerization by α-linolenic acid and OA (Fig. 4D).
Genetic association of the GRK5 gene with sPD
To examine whether GRK5 is associated with sPD, we conducted a haplotype-based association study for the GRK5 gene using a clinically high-quality case–control setting (286 cases of sPD and 496 control subjects, all of whom were residents of Yamagata prefecture, Japan) (see Materials and Methods). An HTR analysis (Zaykin et al., 2002) detected a low empirical p value of 0.0002 (10,000 permutations) in a haplotype G-A-C in the m22-m23-m24 block of the GRK5 gene (Fig. 5A,B). It should be noted that this p value was corrected by the number of estimated haplotypes because it was empirically determined by permutation test. Population stratification was not observed by overall χ2 test (p = 0.919). The haplotype G-A-C was a low-frequency haplotype, which increased sPD risk twofold [9.3% in cases vs 5.1% in controls; odds ratio (OR) = 1.91; 95% confidence interval (CI) = 1.25–2.79] (Fig. 5B). The empirical p value for overall haplotype association test for m22-m23-m24 block was 0.0152. No nonsynonymous SNP in the GRK5 gene was found in 31 individuals except for SNP m32 located in exon 9. Among 15 SNPs identified in our analysis of the GRK5 gene, only a single SNP, m24, gave a marginal sign of association with sPD under the dominant model (p = 0.031; OR = 1.39; 95% CI = 1.04–1.86). Genetic analysis for the variations of the other protein kinases known to phosphorylate αS in vitro did not reveal any significant association with sPD (supplemental Table 1, available at www.jneurosci.org as supplemental material).
Figure 5.

Haplotype analysis of the GRK5 gene. A, Names and positions of genotyped SNPs in the GRK5 gene (m19, rs10886424; m20, rs11198856; m21, rs1473799; m22, rs871196; m22.1, rs2420616; m23, rs7069375; m24, rs4752293; m25, rs884970; m26, rs291979; m27, rs2275036; m28, rs11198907; m29, rs3740564; m30, rs933048; m31, rs11198922; m32, rs12718341; m33, rs1999628). B–D, The results of HTR analysis in the m22-m23-m24 (B), m22.1-m23-m24 (C), and m22-m22.1-m23-m24 (D) blocks are shown. Haplotypes showing significant empirical p value (10,000 permutations) are shown in bold. ORs were calculated by mean response values (case, 1; control, 0). Overall haplotype empirical p values (10,000 permutations) were 0.0152, 0.0053, and 0.0127 for the blocks m22-m23-m24, m22.1-m22-m24, and m22-m22.1-m23-m24, respectively.
The C allele of SNP m24 increases transcriptional activity
Electrophoretic mobility shift assay (EMSA) and reporter assay were performed to identify functional SNPs around this at-risk haplotype G-A-C (m22-m23-m24). Because a cAMP-responsive element overlaps with m24 site, we investigated a DNA segment containing m24 C or T allele. Oligonucleotide probe derived from the C allele (m24C) had a higher affinity to a nuclear protein than the T allele (m24T) (Fig. 6A). Binding of CREB-1 was confirmed by a supershift assay using anti-CREB-1 antibody. Shifted bands were more intense in m24C than in m24T (Fig. 6B), indicating that CREB-1 binding to m24C was more tight than to m24T. Allelic effect on transcriptional activity was examined by luciferase reporter assay. In the presence of cAMP, C allele showed a 1.5-fold increase in luciferase expression in single-copy assays and a 9.7-fold increase in assays with three-concatenated copies, compared with T allele (Fig. 6C; supplemental Fig. 2, available at www.jneurosci.org as supplemental material). ChIP assay confirmed the binding of CREB-1 to the m24 region of the GRK5 gene within intact SH-SY5Y cells (Fig. 6D).
Figure 6.

Allelic difference in SNP m24 (T/C). A, EMSA. Double-stranded oligonucleotide with the sequence of SNP m24 T allele (m24T, 5′cagaagactcTgtcatcaggc; lanes 1–4) or m24 C allele (m24C, 5′cagaagactcCgtcatcaggc; lanes 5–8) was 32P labeled and incubated with nuclear extract from SH-SY5Y cells with (lanes 2–4, 6–8) or without (lanes 1, 5) 200-fold excess of nonlabeled competitors. Shift bands (arrowhead) were seen in lanes 1 and 5, and the intensity was higher in m24C (lane 5) than in m24T (lane 1), indicating that m24C had a higher affinity to nuclear protein(s) than m24T. The higher affinity of m24C than m24T was supported by a competition assay, which showed that shift bands were more efficiently inhibited by nonlabeled m24C (lanes 4, 8) than by nonlabeled m24T (lanes 3, 7). An unrelated control sequence did not inhibit shift bands (lanes 2, 6). The asterisk indicates nonspecific shifted bands. B, Supershift assay. 32P-labeled m24T (lanes 1–5) or m24C (lanes 6–10) was incubated with nuclear extract from SH-SY5Y cells with (lanes 2–5, 7–10) or without (lanes 1, 6) antibodies. Anti-CREB-1 antibody diminished the intensity of shift bands (arrowhead; lanes 5, 10) and produced two supershifted bands (double arrowhead; lanes 5, 10). Anti-c-Jun antibody also produced a supershifted band, albeit weaker than CREB-1 (triple arrowhead; lanes 3, 8). The intensity of supershifted bands was higher in m24C (lanes 8, 10) than in m24T (lanes 3, 5). A control anti-STAT-1 antibody did not affect the intensity of shift bands, nor did it produce supershifted bands (lanes 2, 7). C, Luciferase assay. A DNA fragment corresponding to m24T or m24C was inserted into pGL-3 promoter vector to produce reporter with single m24T or m24C and three-tandem m24T or m24C (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). The reporters were transfected into SH-SY5Y cells, and relative luciferase activity (RLA) was assayed. No difference in RLA was seen among the five constructs in the absence of cAMP. In the presence of cAMP, m24C showed a 1.5-fold increase in RLA compared with m24T (*p < 0.05), and m24C × 3 showed a 9.7-fold increase compared with m24T × 3 (**p < 0.0001). D, ChIP assay. DNA–protein complexes were immunoprecipitated with no antibody (lane 1), unrelated antibody (lane 2), or anti-CREB-1 antibody (lane 3). Lane 3 shows the binding of CREB-1 to the m24 region of the GRK5 gene within intact SH-SY5Y cells. Lane 4 shows input DNA. Experiments were done in duplicate.
Replication of the results of association using genotype data of two functional SNPs
A search for functional SNPs using dbSNP (National Center for Biotechnology Information) and the TRANSFAC database identified a novel SNP, m22.1, which was located in intron 1 between m22 and m23 (Fig. 5A), as one of the candidates to make a potential allelic difference in cis-acting regulatory transcription factors. Additional investigation showed that this SNP comprised allelic difference in YY1 binding and transcriptional activity, in which YY1 bound to the A allele (Fig. 7A,B) and repressed its transcriptional activity, compared with the G allele (Fig. 7C; supplemental Fig. 2, available at www.jneurosci.org as supplemental material). ChIP assay confirmed the binding of YY1 to the m22.1 region of the GRK5 gene within intact SH-SY5Y cells (Fig. 7D). To examine the effect of a combination of two SNPs on the expression of the reporter gene, we performed the experiments using the constructs with tandem repeats of m22.1 and m24 sequences and found that the increased activity of luciferase by m24C was suppressed by the addition of m22.1A (Fig. 7E; supplemental Fig. 2, available at www.jneurosci.org as supplemental material).
Figure 7.

Allelic difference in SNP m22.1 (A/G). A, Left, EMSA. Double-stranded oligonucleotide with the sequence of SNP m22.1 A allele (m22.1A, 5′taaactcagatAtggcttcaggg; lanes 1–3) or m22.1 G allele (m22.1G, 5′taaactcagatGtggcttcaggg; lanes 4–6) was 32P labeled and incubated with nuclear extract from SH-SY5Y cells with (lanes 2, 3, 5, 6) or without (lanes 1, 4) 400-fold excess of nonlabeled competitors. Shift bands (single and double arrowheads) were seen in m22.1A (lanes 1–3) but not in m22.1G (lanes 4, 5). The intensity of shift bands was diminished by addition of competitor m22.1A (lane 2) but not by m22.1G (lane 3), suggesting the binding of nuclear protein(s) to m22.1A. The asterisks indicate nonspecific shifted bands. Right, Competition assays using oligonucleotide with YY1-binding motif. 32P-labeled m22.1A oligonucleotide (5′taaactcagatAtggcttcaggg), which contained a core consensus sequence of YY1-binding site, was incubated with nuclear extract from SH-SY5Y cells with (lanes 2–6) or without (lane 1) 400-fold excess of nonlabeled competitors. The shift band (double arrowhead) was markedly abolished by addition of nonlabeled representative YY1-binding consensus sequence (YY1co; 5′cagccgccaagatggccggggag; lane 5) but not by nonlabeled mutated YY1-binding consensus sequence (YY1mu; 5′cagccgccaagataatcgcggag; lane 6) or by nonlabeled mutated m22.1A (m22.1Amu, 5′taaactcagatAcaccttcaggg; lane 3). B, Supershift assay. m22.1A (lanes 1, 2), m22.1Amu (lanes 3, 4), m22.1G (lanes 5, 6), YY1co (lanes 7, 8), or YY1mu (lanes 9, 10) oligonucleotide labeled with 32P was incubated with nuclear extract from SH-SY5Y cells with (lanes 2, 4, 6, 8, 10) or without (lanes 1, 3, 5, 7, 9) antibody against YY1. The shift band (double arrowhead) was produced by m22.1A (lane 1) or YY1co (lane 7) oligonucleotide labeled with 32P; this band was markedly abolished by the antibody (lanes 2, 8), suggesting the binding of YY1 to m22.1A as well as YY1co. C, Luciferase assay. Single- or three-concatenated oligonucleotide with m22.1A or m22.1G was inserted into pGL-3 promoter vector (supplemental Fig. 2, available at www.jneurosci.org as supplemental material), and relative luciferase activity (RLA) was assayed. m22.1A × 3 showed a 42% decrease in RLA compared with m22.1G × 3 (**p < 0.0001), indicating a repressed transcriptional activity by YY1. D, ChIP assay. DNA–protein complexes were immunoprecipitated with either unrelated antibody (lane 1) or anti-YY1 antibody (lanes 2, 3). Lanes 2 and 3 show the binding of YY1 to the m22.1 region of the GRK5 gene within intact SH-SY5Y cells. Lane 4 shows input DNA. Experiments were done in duplicate. E, A combination effect of two SNPs (supplemental Fig. 2, available at www.jneurosci.org as supplemental material) on the expression of the reporter gene, showing that the increased activity of luciferase by m24C × 3 was suppressed by the addition of m22.1A × 3 (**p < 0.0001). Experiments were performed in triplicate.
This SNP showed no allelic association with sPD in a single-SNP analysis. To test the possibility that allelic combination of the two functional SNPs, G at m22.1 and C at m24, is in the same at-risk haplotype, HTR analysis was performed for the haplotype blocks, including a newly found SNP, m22.1. The tests replicated the results of the haplotypic association shown above. An analysis of the m22.1-m23-m24 block revealed a low empirical p value of 0.0002 in 10,000 permutations for G-A-C haplotype (4.0% in cases and 1.2% in controls; OR = 3.45; 95% CI = 1.67–6.86) (Fig. 5C). The empirical overall p value for this block was 0.0053 (10,000 permutations). It should be noted that both overall and individual p values were obtained by a single HTR test but not by a sliding window approach. Furthermore, analysis of the m22-m22.1-m23-m24 block revealed a low empirical p value of 0 in 10,000 permutations for haplotype G-G-A-C (OR = 3.94; 95% CI = 1.68–8.30) (Fig. 5D). The empirical overall p value for this block was 0.0127 (10,000 permutations). As expected by these results, the SNP m22.1 was found to be in tight LD (D′ = 0.89; Δ = 0.88) with m24 (Table 2). Each allele at m22.1 and m24 (G at m22.1 and C at m24) on the at-risk haplotypes (G-A-C in m22.1-m22-m23 and G-G-A-C in m22-m22.1-m23-m24) conferred higher transcription activity of the reporter gene (Figs. 6C, 7C).
To compare the contents of GRK5 protein in the brains, we performed Western blotting of the protein using the frontal cortex tissues from four autopsy cases of sPD, one case of DLB, and nine controls. No significant difference in the intensity of the GRK5 band was found between the sPD and control groups (supplemental Fig. 4, left, available at www.jneurosci.org as supplemental material). Next, we divided the samples into three groups on the basis of the genotypes of SNP m24 (T/T, C/T, and C/C). However, difference in the intensity of GRK5 protein was not statistically significant among the three groups (supplemental Fig. 4, middle, available at www.jneurosci.org as supplemental material). Differences in the expression of GRK5 in the three genotypes of m22.1 (A/A, G/A, and G/G) were not significant either (supplemental Fig. 4, right, available at www.jneurosci.org as supplemental material). We need more samples to increase the power of statistical analysis. However, the possibility could not be excluded that the results of the frontal cortex may not reflect the changes of GRK5 in the substantia nigra of sPD patients.
Discussion
In the present study, we demonstrated that GRK5 is accumulated in LBs in the brains of sPD patients and colocalized with αS in the pathological structures of the substantia nigra (Fig. 1). However, GRK5 was not detected in the cortical-type LBs of DLB, nor in the glial cytoplasmic inclusions of MSA, which are known to contain phosphorylated αS (Fujiwara et al., 2002). It appears that αS in cortical neurons of DLB and glial cells of MSA is phosphorylated by protein kinase(s) other than GRK5. In the brain tissues and in HEK293 cells transfected with GRK5 and αS, GRK5 was closely associated with αS (Fig. 2C). Pronin et al. (2000) reported biochemical evidence that GRK5 phosphorylates αS at Ser-129. In the present study, we established the cell line stably expressing αS and showed that phosphorylation of αS at Ser-129 depends on the expression levels of GRK5 and its kinase activity (Fig. 2B). The phosphorylation occurred at the area of plasma membrane, at which GRK5 and αS colocalized, and the phosphorylated αS was translocated to the perikaryal area (Fig. 3). The translocation of pS129–αS from the plasma membrane to the perikaryal area was consistent with decreased affinity of pS129–αS for phospholipids (Pronin et al., 2000). Evidence was also obtained indicating that GRK5 promoted the formation of soluble oligomers and aggregates of αS by phosphorylation of αS (Fig. 4).
Formation and deposition of proteinaceous fibrillar aggregates, such as the amyloid β-protein in Alzheimer's disease, the prion protein in Creutzfeldt–Jacob disease, and the huntingtin protein in Huntington's disease, seem to be related to the pathogenesis of many neurodegenerative diseases. αS seems to be such a protein in sPD. These proteinaceous fibrils are generated from the monomeric forms through the formation of intermediate species (Walsh and Selkoe, 2004). It has been shown that the intermediate species bind to synthetic phospholipid vesicles with high affinity and transiently permeabilize the vesicles (Volles et al., 2001), and the intermediate species were thought to be more cytotoxic than the mature fibrils (Bucciantini et al., 2002; Volles and Lansbury, 2003; Walsh and Selkoe, 2004). Soluble forms of αS were also identified, which are described as “soluble protein complexes” (Xu et al., 2002) and “soluble oligomers” (Sharon et al., 2003; Walsh and Selkoe, 2004) based on their biochemical features. Because of a motif at the N and C termini of αS that is homologous to an FABP signature and its actual binding to fatty acids, αS was regarded as a brain-type FABP (Sharon et al., 2001). The addition of fatty acids, especially PUFAs, to culture medium of the cells overexpressing αS promotes the formation of soluble αS oligomers (Sharon et al., 2003). In the αS-transgenic mice, an increased amount of soluble oligomers was observed in the brains of the old mice, and these soluble oligomers seem to ultimately convert to highly insoluble aggregates and mature fibrils (Sharon et al., 2003; Walsh and Selkoe, 2004). Accumulation of soluble oligomers was also observed in human PD brains (Sharon et al., 2003). In the present study, we found that phosphorylation of αS by GRK5 promoted the formation of soluble oligomers and aggregates of αS in the presence of α-linolenic acid, a PUFA. We suspect that such biochemical processes are involved in the formation and deposition of proteinaceous aggregates in sPD. However, the significance of this process in the pathogenesis of sPD remains unknown. It is possible that αS aggregates detected in sPD brains may be formed at a later stage of neurodegeneration.
Previous studies suggested neurotoxicity of high-molecular-weight species of αS, such as soluble oligomers and intermediate/prefibrillar species (Dawson and Dawson, 2003; Walsh and Selkoe, 2004). Quite recently, Chen and Feany (2005) reported definitive evidence in the Drosophila model of PD that phosphorylation at Ser-129 of αS is essential for its toxicity to dopaminergic neurons and that overexpression of Drosophila GRK2 increases phosphorylation of αS at Ser-129 and enhances αS neurotoxicity. In our study, no genetic association was found between GRK2 and sPD (supplemental Table 1, available at www.jneurosci.org as supplemental material), and GRK2 was not detectable in LBs of sPD brains (Table 1). Alternatively, we found that GRK5 colocalized with αS both in double-transfected cells and in LBs of sPD and that a haplotype of SNPs in the GRK5 gene was associated with susceptibility to sPD. In our more recent studies, which will be published separately, we observed that co-overexpression of human αS and wild-type GRK5 to dopaminergic neurons in C. elegans resulted in phosphorylation of αS and produced a noxious effect on dopaminergic neurons, the function of which was evaluated by “food-sensing behavior assay (bending assay).” Overexpression of either human αS or GRK5 alone, or co-overexpression of human αS and catalytic-inactive GRK5 (GRK5–K215R) failed to induce such a noxious effect on dopaminergic neurons in C. elegans (C.-H. Ren, T. Kuwahara, S. Koyama, S. Arawaka, S. Mitani, T. Kato, and T. Iwatsubo, unpublished observations). These observations strongly support the hypothesis that αS phosphorylated by GRK5 has a toxic effect on the dopaminergic neurons.
To connect GRK5 to an sPD phenotype, we performed an association study of sPD patients and found that a haplotype G-A-C in the m22-m23-m24 block of the GRK5 gene was associated with susceptibility to sPD. Moreover, within the genomic region of this haplotype block, we identified two functional SNPs, m22.1 and m24, in introns 1 and 2 of the GRK5 gene. Additional haplotype analysis demonstrated that a combination of these alleles (G at m22.1 and C at m24) on the same chromosome increased the sPD risk. We calculated the exact empirical p value by permutation procedures. This procedure created the null distribution of p values of simulated data sets by random labeling of affection status in all of the samples. Consequently, both individual and overall p values described in this article are corrected for the number of estimated haplotypes in each block. Multiple testing for the number of tests in a sliding approach might be necessary. However, because tests of overlapping windows are not independent from each other, simple Bonferroni's correction using the number of sliding tests is inappropriate for this statistical analysis. However, it should be noted that the empirical p value of the at-risk haplotype in a sliding window approach (m22-m23-m24 block) have survived a conservative Bonferroni's correction, which uses the exact number of sliding windows, 13 (a corrected p value was 0.0026). Confirmation of genetic association in an independent population will further strengthen our finding.
In the gel-shift assays, an allelic difference of m22.1 (A/G) and m24 (T/C) in the affinity for nuclear protein(s) was observed (Figs. 6A, 7A). The supershift assays demonstrated that m22.1 and m24 sequences bound to YY1 and CREB-1, respectively (Figs. 6B, 7B). ChIP assays also confirmed the binding of YY1 and CREB-1 to the endogenous DNA sequences of m22.1 and m24, respectively, of the GRK5 gene in SH-SY5Y cells (Figs. 6D, 7D). In the luciferase assays, the m24 C allele on the at-risk haplotype did increase the transcriptional activity of the reporter gene when the cells were stimulated with cAMP (Fig. 6C). In the absence of cAMP, however, the m24 C allele did not affect the transcription of the gene (Fig. 6C). In m22.1, the G allele on the at-risk haplotype had no apparent effect on the transcriptional activity compared with the control, whereas the A allele suppressed its activity (Fig. 7C). The A allele of m22.1 also diminished the enhanced transcriptional activity by the m24 C allele in the presence of cAMP (Fig. 7E), suggesting that the function of m22.1 is a modulator of m24. The results of the luciferase assays suggest that the functional SNPs of the GRK5 gene exert the effect on GRK5 transcription by internal and/or external stimuli that activate the cAMP pathway. Such stimuli might induce a repeated, transient increase of GRK5 expression in nigral dopaminergic neurons, which phosphorylates αS to cause accumulation of oligomerized αS. Lack of a significant difference in the “steady-state” expression level of GRK5 protein in the absence of any specific stimulus in the postmortem tissues of the frontal cortex between sPD patients and controls (supplemental Fig. 4, available at www.jneurosci.org as supplemental material) does not necessarily exclude a possible contribution of GRK5 to the pathogenesis of sPD.
It should be noted that only a minor subgroup (9.3%) of sPD patients had the at-risk haplotype of the GRK5 gene and therefore that this genetic polymorphism cannot fully explain the pathogenesis in the majority of sPD patients. In fact, intense immunostaining of phosphorylated αS in LBs was observed in all six cases of sPD examined, but all of the cases, including those with intense GRK5 immunostaining in LBs, had no at-risk haplotype of the GRK5 gene (all six cases had the m24T/T homozygote). The results suggest that an increase in phosphorylated αS in nigral dopaminergic neurons and GRK5 accumulation in LBs could occur in the sPD patients who do not have the at-risk haplotype of the GRK5 gene. Among the protein kinases that are known to phosphorylate αS, such as those in CK and GRK families, however, only GRK5 was detected in LBs of sPD patients (Table 1). Genetic analysis did not support that the kinases, other than GRK5, are involved in the pathogenesis of sPD (supplemental Table 1, available at www.jneurosci.org as supplemental material). One may speculate that the accumulation of GRK5 in LBs could be induced by several different mechanisms and that the polymorphism of the GRK5 gene represents one of these factors.
Because phosphorylation seems to be a common mechanism controlling the neurotoxicity of aggregation-prone proteins in some neurodegenerative diseases, it is crucial to identify pathophysiologically relevant protein kinases and/or phosphatases for focusing on therapeutic targets of these diseases. One may expect that GRK5 is one of the promising targets for the development of a novel drug therapy for sPD.
Footnotes
This work was supported in part by the 21st Century Center of Excellence Program (F03), Japan. We thank the members of the Yamagata Study Group of Parkinson's Disease for clinical assistance.
References
- Arawaka S, Hasegawa H, Tandon A, Janus C, Chen F, Yu G, Kikuchi K, Koyama S, Kato T, Fraser PE, St George-Hyslop P. The levels of mature glycosylated nicastrin are regulated and correlate with gamma-secretase processing of amyloid beta-precursor protein. J Neurochem. 2002;83:1065–1071. doi: 10.1046/j.1471-4159.2002.01207.x. [DOI] [PubMed] [Google Scholar]
- Baba M, Nakajo S, Tu PH, Tomita T, Nakaya K, Lee VM, Trojanowski JQ, Iwatsubo T. Aggregation of alpha-synuclein in Lewy bodies of sporadic Parkinson's disease and dementia with Lewy bodies. Am J Pathol. 1998;152:879–884. [PMC free article] [PubMed] [Google Scholar]
- Bucciantini M, Giannoni E, Chiti F, Baroni F, Formigli L, Zurdo J, Taddei N, Ramponi G, Dobson CM, Stefani M. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature. 2002;416:507–511. doi: 10.1038/416507a. [DOI] [PubMed] [Google Scholar]
- Chen L, Feany MB. Alpha-synuclein phosphorylation controls neurotoxicity and inclusion formation in a Drosophila model of Parkinson disease. Nat Neurosci. 2005;8:657–663. doi: 10.1038/nn1443. [DOI] [PubMed] [Google Scholar]
- Chung KK, Dawson VL, Dawson TM. New insights into Parkinson's disease. J Neurol. 2003;250([Suppl 3]):III15–III24. doi: 10.1007/s00415-003-1304-9. [DOI] [PubMed] [Google Scholar]
- Dawson TM, Dawson VL. Neuroprotective and neurorestorative strategies for Parkinson's disease. Nat Neurosci. 2002;5:1058–1061. doi: 10.1038/nn941. [DOI] [PubMed] [Google Scholar]
- Dawson TM, Dawson VL. Rare genetic mutations shed light on the pathogenesis of Parkinson's disease. J Clin Invest. 2003a;111:145–151. doi: 10.1172/JCI17575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson TM, Dawson VL. Molecular pathways of neurodegeneration in Parkinson's disease. Science. 2003b;302:819–822. doi: 10.1126/science.1087753. [DOI] [PubMed] [Google Scholar]
- Dev KK, Hofele K, Barbieri S, Buchman VL, van der Putten H. Part II: alpha-synuclein and its molecular pathophysiological role in neurodegenerative disease. Neuropharmacology. 2003;45:14–44. doi: 10.1016/s0028-3908(03)00140-0. [DOI] [PubMed] [Google Scholar]
- Foltynie T, Sawcer S, Brayne C, Barker RA. The genetic basis of Parkinson's disease. J Neurol Neurosurg Psychiatry. 2002;73:363–370. doi: 10.1136/jnnp.73.4.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS, Shen J, Takio K, Iwatsubo T. Alpha-synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol. 2002;4:160–164. doi: 10.1038/ncb748. [DOI] [PubMed] [Google Scholar]
- Gasser T. Genetics of Parkinson's disease. J Neurol. 2001;248:833–840. doi: 10.1007/s004150170066. [DOI] [PubMed] [Google Scholar]
- Kato T, Katagiri T, Hirano A, Kawanami T, Sasaki H. Lewy body-like hyaline inclusions in sporadic motor neuron disease are ubiquitinated. Acta Neuropathol (Berl) 1989;77:391–396. doi: 10.1007/BF00687373. [DOI] [PubMed] [Google Scholar]
- Kimura H, Kurimura M, Wada M, Kawanami T, Kurita K, Suzuki Y, Katagiri T, Daimon M, Kayama T, Kato T. Female preponderance of Parkinson's disease in Japan. Neuroepidemiology. 2002;21:292–296. doi: 10.1159/000065527. [DOI] [PubMed] [Google Scholar]
- Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, Przuntek H, Epplen JT, Schols L, Riess O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson's disease. Nat Genet. 1998;18:106–108. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- Livak KJ. Allelic discrimination using fluorogenic probes and the 5′ nuclease assay. Genet Anal. 1999;14:143–149. doi: 10.1016/s1050-3862(98)00019-9. [DOI] [PubMed] [Google Scholar]
- Martin ER, Scott WK, Nance MA, Watts RL, Hubble JP, Koller WC, Lyons K, Pahwa R, Stern MB, Colcher A, Hiner BC, Jankovic J, Ondo WG, Allen FH, Jr, Goetz CG, Small GW, Masterman D, Mastaglia F, Laing NG, Stajich JM, Ribble RC, et al. Association of single-nucleotide polymorphisms of the tau gene with late-onset Parkinson disease. JAMA. 2001;286:2245–2250. doi: 10.1001/jama.286.18.2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore DJ, West AB, Dawson VL, Dawson TM. Molecular pathophysiology of Parkinson's disease. Annu Rev Neurosci. 2005;28:57–87. doi: 10.1146/annurev.neuro.28.061604.135718. [DOI] [PubMed] [Google Scholar]
- Okochi M, Walter J, Koyama A, Nakajo S, Baba M, Iwatsubo T, Meijer L, Kahle PJ, Haass C. Constitutive phosphorylation of the Parkinson's disease associated alpha-synuclein. J Biol Chem. 2000;275:390–397. doi: 10.1074/jbc.275.1.390. [DOI] [PubMed] [Google Scholar]
- Paisan-Ruiz C, Jain S, Evans EW, Gilks WP, Simon J, van der Brug M, Lopez de Munain A, Aparicio S, Gil AM, Khan N, Johnson J, Martinez JR, Nicholl D, Carrera IM, Pena AS, de Silva R, Lees A, Marti-Masso JF, Perez-Tur J, Wood NW, et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson's disease. Neuron. 2004;44:595–600. doi: 10.1016/j.neuron.2004.10.023. [DOI] [PubMed] [Google Scholar]
- Perrin RJ, Payton JE, Barnett DH, Wraight CL, Woods WS, Ye L, George JM. Epitope mapping and specificity of the anti-alpha-synuclein monoclonal antibody Syn-1 in mouse brain and cultured cell lines. Neurosci Lett. 2003;349:133–135. doi: 10.1016/s0304-3940(03)00781-x. [DOI] [PubMed] [Google Scholar]
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- Pronin AN, Carman CV, Benovic JL. Structure-function analysis of G protein-coupled receptor kinase-5. Role of the carboxyl terminus in kinase regulation. J Biol Chem. 1998;273:31510–31518. doi: 10.1074/jbc.273.47.31510. [DOI] [PubMed] [Google Scholar]
- Pronin AN, Morris AJ, Surguchov A, Benovic JL. Synucleins are a novel class of substrates for G protein-coupled receptor kinases. J Biol Chem. 2000;275:26515–26522. doi: 10.1074/jbc.M003542200. [DOI] [PubMed] [Google Scholar]
- Recchia A, Debetto P, Negro A, Guidolin D, Skaper SD, Giusti P. Alpha-synuclein and Parkinson's disease. FASEB J. 2004;18:617–626. doi: 10.1096/fj.03-0338rev. [DOI] [PubMed] [Google Scholar]
- Saito Y, Kawashima A, Ruberu NN, Fujiwara H, Koyama S, Sawabe M, Arai T, Nagura H, Yamanouchi H, Hasegawa M, Iwatsubo T, Murayama S. Accumulation of phosphorylated alpha-synuclein in aging human brain. J Neuropathol Exp Neurol. 2003;62:644–654. doi: 10.1093/jnen/62.6.644. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Cell biology of protein misfolding: the examples of Alzheimer's and Parkinson's diseases. Nat Cell Biol. 2004;6:1054–1061. doi: 10.1038/ncb1104-1054. [DOI] [PubMed] [Google Scholar]
- Sharon R, Goldberg MS, Bar-Josef I, Betensky RA, Shen J, Selkoe DJ. Alpha-synuclein occurs in lipid-rich high molecular weight complexes, binds fatty acids, and shows homology to the fatty acid-binding proteins. Proc Natl Acad Sci USA. 2001;98:9110–9115. doi: 10.1073/pnas.171300598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharon R, Bar-Joseph I, Frosch MP, Walsh DM, Hamilton JA, Selkoe DJ. The formation of highly soluble oligomers of alpha-synuclein is regulated by fatty acids and enhanced in Parkinson's disease. Neuron. 2003;37:583–595. doi: 10.1016/s0896-6273(03)00024-2. [DOI] [PubMed] [Google Scholar]
- Siderowf A, Stern M. Update on Parkinson disease. Ann Intern Med. 2003;138:651–658. doi: 10.7326/0003-4819-138-8-200304150-00013. [DOI] [PubMed] [Google Scholar]
- Smith WW, Margolis RL, Li X, Troncoso JC, Lee MK, Dawson VL, Dawson TM, Iwatsubo T, Ross CA. α-Synuclein phosphorylation enhances eosinophilic cytoplasmic inclusion formation in SH-SY5Y cells. J Neurosci. 2005;25:5544–5552. doi: 10.1523/JNEUROSCI.0482-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M. Alpha-synuclein in filamentous inclusions of Lewy bodies from Parkinson's disease and dementia with Lewy bodies. Proc Natl Acad Sci USA. 1998;95:6469–6473. doi: 10.1073/pnas.95.11.6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiyagarajan MM, Stracquatanio RP, Pronin AN, Evanko DS, Benovic JL, Wedegaertner PB. A predicted amphipathic helix mediates plasma membrane localization of GRK5. J Biol Chem. 2004;279:17989–17995. doi: 10.1074/jbc.M310738200. [DOI] [PubMed] [Google Scholar]
- Vila M, Przedborski S. Genetic clues to the pathogenesis of Parkinson's disease. Nat Med. 2004;10([Suppl]):S58–S62. doi: 10.1038/nm1068. [DOI] [PubMed] [Google Scholar]
- Volles MJ, Lansbury PT., Jr Zeroing in on the pathogenic form of alpha-synuclein and its mechanism of neurotoxicity in Parkinson's disease. Biochemistry. 2003;42:7871–7878. doi: 10.1021/bi030086j. [DOI] [PubMed] [Google Scholar]
- Volles MJ, Lee SJ, Rochet JC, Shtilerman MD, Ding TT, Kessler JC, Lansbury PT., Jr Vesicle permeabilization by protofibrillar alpha-synuclein: implications for the pathogenesis and treatment of Parkinson's disease. Biochemistry. 2001;40:7812–7819. doi: 10.1021/bi0102398. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Selkoe DJ. Oligomers on the brain: the emerging role of soluble protein aggregates in neurodegeneration. Protein Pept Lett. 2004;11:213–228. doi: 10.2174/0929866043407174. [DOI] [PubMed] [Google Scholar]
- Xu J, Kao SY, Lee FJ, Song W, Jin LW, Yankner BA. Dopamine-dependent neurotoxicity of alpha-synuclein: a mechanism for selective neurodegeneration in Parkinson disease. Nat Med. 2002;8:600–606. doi: 10.1038/nm0602-600. [DOI] [PubMed] [Google Scholar]
- Zaykin DV, Westfall PH, Young SS, Karnoub MA, Wagner MJ, Ehm MG. Testing association of statistically inferred haplotypes with discrete and continuous traits in samples of unrelated individuals. Hum Hered. 2002;53:79–91. doi: 10.1159/000057986. [DOI] [PubMed] [Google Scholar]
- Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, Kachergus J, Hulihan M, Uitti RJ, Calne DB, Stoessl AJ, Pfeiffer RF, Patenge N, Carbajal IC, Vieregge P, Asmus F, Muller-Myhsok B, Dickson DW, Meitinger T, Strom TM, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44:601–607. doi: 10.1016/j.neuron.2004.11.005. [DOI] [PubMed] [Google Scholar]