

Abstract
The release of glutamate from astrocytes activates synchronous slow inward currents (SICs) in hippocampal pyramidal neurons, which are mediated by the NMDA receptor and represent a nonsynaptic mechanism to promote the synchronization of neuronal activity. Two recent studies demonstrate that SICs generate neuronal paroxysmal depolarizations resembling those typical of interictal epileptiform activity and proposed that there could be an astrocytic basis of epilepsy (Kang et al., 2005; Tian et al., 2005). We tested this hypothesis using two in vitro models of epileptiform activity in hippocampal slices. Removal of extracellular Mg2+ and application of picrotoxin or perfusion with 0.5 mm Mg2+ and 8.5 mm K+-containing saline result mainly in neuronal ictal- and interictal-like epileptiform activity, respectively. Although both models trigger epileptiform activity, astrocytic Ca2+ oscillations were increased only after slice perfusion with 0 mm Mg2+ and picrotoxin. The activation of astrocytic Ca2+ signaling correlates with an increased frequency of SICs, and, when paired neurons were within 100 μm of one another with synchronous neuronal Ca2+ elevations, the generation of synchronous neuronal depolarizations and action potential discharges. TTX blocked both ictal- and interictal-like epileptiform activity without affecting SICs or SIC-mediated neuronal synchronization. In contrast, NMDA receptor antagonists, which block SICs, did not prevent the generation of either ictal- or interictal-like events. Based on this clear-cut pharmacology, our data demonstrate that nonsynaptic glutamate release from astrocytes is not necessary for the generation of epileptiform activity in vitro, although we cannot exclude the possibility that it may modulate the strength of the ictal (seizure)-like event.
Keywords: glia, glutamate release, epileptiform activity, epilepsy, NMDA receptor, synchrony
Introduction
Seizures are associated with intense neuronal discharges that are generated by the “hyper-”synchronous activity of a large number of neurons in different brain regions, including the cortex and the hippocampus. How such a large population of neurons becomes synchronized is not known, but it is believed to result from an imbalance between excitatory and inhibitory inputs to the network (Jefferys, 1994; McNamara, 1994, 1999; McCormick and Contreras, 2001). Indeed, in slice preparations, increased neuronal excitability obtained by relieving the Mg2+ block of the NMDA receptors (NMDARs) (Anderson et al., 1986; Walther et al., 1986; Jones and Heinemann, 1988; Wilson et al., 1988; Dreier and Heinemann, 1991) or by blockade of the interneuron network by application of GABAA antagonists (Schwartzkroin and Prince, 1980; Swann and Brady, 1984) results in abnormal neuronal activities resembling those recorded in vivo (Bragin et al., 1999). Although this abnormal epileptiform activity is generally considered to be generated exclusively in neurons, there have been suggestions of a nonsynaptic source of excitation contributing to these events (Konnerth et al., 1986; Jefferys, 1995).
Astrocytes, the major glial cell type in the CNS, have long been considered to be a silent partner of neurons in the brain providing only structural and metabolic support. However, many recent studies demonstrate that astrocytes are a non-neuronal source of many neuroactive molecules that can modulate neuronal excitability, including the excitatory neurotransmitter glutamate (Parpura et al., 1994; Bezzi et al., 1998; Haydon, 2001; Volterra and Meldolesi, 2005). Astrocytes can detect the synaptic release of neurotransmitters and display Ca2+ oscillations (Porter and McCarthy, 1996; Pasti et al., 1997). Ca2+ signaling in astrocytes, in turn, induces the release from these cells of chemical transmitters, suggesting that astrocytes can respond to synaptic activation and provide reciprocal signals to neurons (Haydon, 2001). Ca2+ elevations in astrocytes, either produced by receptor-evoked Ca2+ release from internal stores or experimentally induced photolytic elevation of Ca2+, induce the exocytotic release of glutamate from astrocytes (Bezzi et al., 1998, 2004; Parpura and Haydon, 2000; Pasti et al., 2001; Fellin et al., 2004). Glutamate released from astrocytes has been shown to generate slow inward currents (SICs) in hippocampal pyramidal neurons, which are attributable to the selective activation of neuronal NMDA receptors (Angulo et al., 2004; Fellin et al., 2004; Perea and Araque, 2005). Importantly, SICs have been shown to occur with a high degree of synchrony among different neurons (Angulo et al., 2004; Fellin et al., 2004). Thus, by releasing the excitatory neurotransmitter glutamate and by acting as a nonsynaptic mechanism of neuronal synchrony, astrocytes have the potential to control neuronal excitability (Fellin et al., 2006) and to generate the widespread, synchronous neuronal activity typical of seizures (Carmignoto and Fellin, 2006). Indeed, two recent studies (Kang et al., 2005; Tian et al., 2005) have shown that astrocytic glutamate can generate paroxysmal depolarization shifts (PDSs) in neurons, suggesting that astrocytes play a key role in interictal events, in which neurons are known to exhibit PDSs, and possibly in seizures (Fellin and Haydon, 2005; Rogawski, 2005; Seifert et al., 2006). However, an experimental test of the requirement for astrocytes in the generation of interictal activity has not been performed.
Here we investigated the contribution of astrocytic glutamate to the generation of abnormal neuronal activity in two in vitro models of epileptiform activity in hippocampal slices. We initially confirmed some of the results by Tian et al. (2005) by showing that astrocytes may be activated during a period of epileptiform activity and that their activation may result in increased glutamate release from these cells and, consequently, in an increased NMDA receptor-dependent SIC frequency in hippocampal pyramidal neurons. Then we extended their studies by asking whether SICs contribute to either ictal- or interictal-like epileptiform activity. Although SICs can trigger synchronous depolarizations and action potential firing in small groups of hippocampal neurons, these events do not play any central role in the genesis of interictal-like activity, but they may modulate the strength of the ictal-like event. Indeed, NMDA receptor antagonists, which block SICs, do not prevent interictal-like epileptiform activity but they shorten significantly the duration of ictal, seizure-like events. In contrast, TTX, which does not affect nonsynaptic SICs, completely blocks epileptiform activity. These results demonstrate that astrocytic glutamate is not necessary for the initiation of epileptiform activity, although by acting on NMDARs, it might modulate the duration of ictal, seizure-like events.
Materials and Methods
All procedures were in strict accordance with either the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the University of Pennsylvania Institutional Animal Care and Use Committee or the Italian and European Union regulations on animal welfare and were authorized by the Italian Ministry of Health.
Slice preparation.
Coronal cortical–hippocampal slices (400–500 μm) were obtained from C57BL/6 and FVBN/J mice (The Jackson Laboratory, Bar Harbor, ME) at postnatal days 11–24 as described previously (Edwards et al., 1989; Pasti et al., 1997). Slices from Wistar rats of the same age were used for the experiments shown in Figure 2 and some experiments shown in Figures 3 and 4. No differences in SIC properties between slices from rat and mouse were observed, as observed previously (Fellin et al., 2004). In brief, the mice were anesthetized with halothane, and, after cervical dislocation, the brain was rapidly removed and put in an ice-cold cutting solution containing the following (in mm): 120 NaCl, 3.2 KCl, 1 NaH2PO4, 26 NaHCO3, 2 MgCl2, 1 CaCl2, 2.8 glucose, 2 Na-pyruvate, and 0.6 ascorbic acid at pH 7.4 (with O2 95%, CO2 5%). After removal of the cerebellum, the brain was glued and transverse slices were cut using a vibratome (VT1000S; Leica, Mannheim, Germany). Before recording, slices were incubated at 37°C for a recovery period of 1 h. For Ca2+ imaging experiments, slices were loaded with the Ca2+ indicator Indo-1/AM or Oregon Green BAPTA-1 AM (Invitrogen, Carlsbad, CA) and 0.02% pluronic under mild stirring at 37°C for 50 or 90 min, respectively. After recovery, slices were put in the recording chamber (Warner Instruments, Hamden, CT) and continuously perfused with normal artificial CSF (ACSF) containing the following (in mm): 120 NaCl, 3.2 KCl, 1 NaH2PO4, 26 NaHCO3, 1 MgCl2, 2 CaCl2, and 2.8 glucose at pH 7.4 (with O2 95%, CO2 5%).
Figure 2.
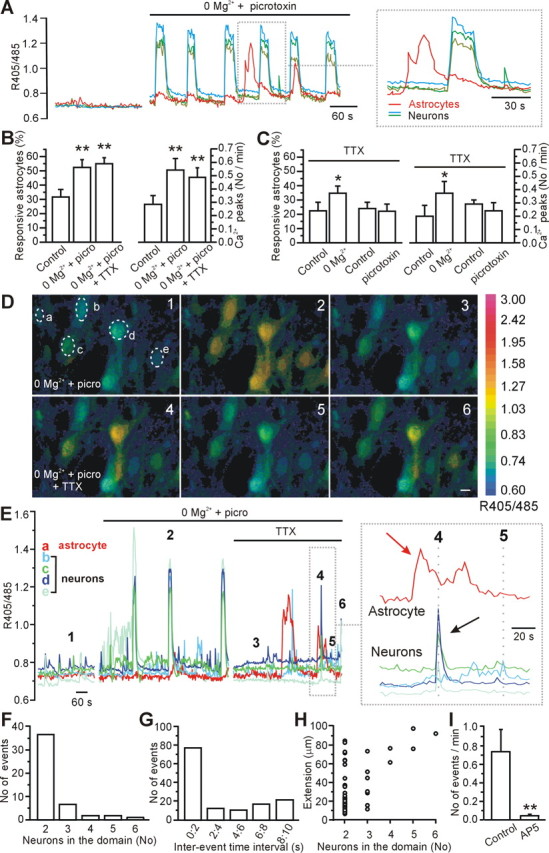
Mg2+ (0 mm) and picrotoxin-induced epileptiform activity triggers Ca2+ oscillations in and glutamate release from astrocytes in the CA3 region. A, Representative example showing synchronous Ca2+ responses in pyramidal neurons as well as Ca2+ oscillations in one astrocyte (red line) after slice perfusion with 0 mm Mg2+ and 50 μm picrotoxin. B, Average values of responsive astrocytes (left) and average frequency of Ca2+ peaks in astrocytes (right) under the different experimental conditions. Once activated by 0 mm Mg2+ and picrotoxin, astrocytic Ca2+ signaling is not reduced by subsequent blockade of neuronal activity with 0.5–1.0 μm TTX (n = 173 astrocytes analyzed from 22 experiments). In this as well as in the following figures, *p < 0.05 and **p < 0.01. C, When neuronal activity is blocked by the continuous presence of TTX, 0 Mg2+ but not picrotoxin causes a small increase in Ca2+ oscillations in astrocytes (n = 78 astrocytes analyzed from 4 experiments for 0 mm Mg2+ and 55 from 3 experiments for picrotoxin). D, Pseudocolor images showing astrocytic (cell a) and neuronal (cells b–e) responses after slice perfusion with 0 mm Mg2+ and 50 μm picrotoxin (images 1–3). Neuronal Ca2+ events generally occur synchronously in all pyramidal neurons in the field (99.7 ± 0.11%; n = 127, 12 experiments). After TTX application, neuronal Ca2+ elevations are observed to occur only in small groups of closely spaced neurons (images 4–6). Scale bar, 10 μm. E, Time course of the 405/485 nm ratio changes in neurons and in the astrocyte marked in D, at rest, after slice perfusion with 0 mm Mg2+ and 50 μm picrotoxin, and after a subsequent TTX application (1 μm). The numbers 1–6 mark the timing for the images shown in D. The inset reports the 405/485 nm ratio changes at an expanded timescale. F, Frequency distribution histogram displaying the number of coactive neuronal Ca2+ responses within a domain. G, Interevent time interval histogram for the Ca2+ elevations in neurons (bin, 2 s). H, Maximal spatial extension of domains recorded in 0 Mg2+, picrotoxin, and TTX as a function of the number of neurons in the domain. I, Average frequency of Ca2+ elevations recorded in each responsive CA3 pyramidal neuron (n = 22 from 4 experiments) in the absence and presence of 100 μm d-AP-5.
Figure 3.

Mg2+ (0 mm) and picrotoxin-induced epileptiform activity in brain slices stimulates astrocyte-evoked neuronally detected NMDAR-dependent currents. A, Paired patch-clamp recording from two CA1 pyramidal neurons under control conditions (a), during interictal-like epileptiform activity triggered by 0 Mg2+ and picrotoxin (100 μm; b, picro), and after subsequent application of TTX (1 μm; c). Note the increased frequency of SICs in cells 1 and 2 (c) compared with the basal condition (a) after epileptiform-like activity induced by 0 Mg2+ and picrotoxin. SICs are not synchronous in the two cells because the neuronal somata are >100 μm apart. B, TTX application (1 μm) blocks the epileptiform events and unmasks the presence of SICs. C, Average frequency of SICs is shown during a basal period recorded in ACSF (Basal, gray bars) before perfusion with 0 Mg2+ and picrotoxin. After epileptiform activity was initiated by perfusing 0 Mg2+ and picrotoxin, TTX (1 μm) was added to this saline to block neuronal activity. Thereafter, SIC frequency was again measured (black bars) in the 9 of 12 CA1, 7 of 14 CA3, and 7 of 18 cortical neurons displaying SICs. D, A similar experiment to that described in C was performed except that the initiation of epileptiform activity was prevented by preincubation with TTX (white bars). Perfusion of 0 Mg2+ and picrotoxin in TTX did not significantly increase SIC frequency. The average SIC frequency (black bars) in 5 of 7 CA1, 3 of 6 CA3, and 3 of 7 cortical neurons displaying SICs. E, Superimposition of a typical interictal event (black trace) and an SIC (gray trace) recorded from the same cell showing the distinct rise times of these two events. F, Average frequency of SICs before and after the epileptiform activity in the presence of TTX and CNQX (25 μm) in the 8 of 19 cortical neurons that displayed SICs. G, Left, SICs recorded in TTX and induced by 0 Mg2+ and picrotoxin are reversibly blocked by d-AP-5 (50–100 μm). Right, Mean number of SICs per minute under the different experimental conditions (n = 4).
Figure 4.

Glutamate release from astrocytes leads to action potential (AP) firing and TTX-insensitive neuronal synchronization. A, Paired recording from two closely spaced CA1 neurons showing spontaneous SIC activity in voltage clamp (VC, first panel on the left) and current clamp (CC, second panel) in the absence of extracellular Mg2+. SIC-mediated membrane depolarizations lead to action potential firing in cell 1 (for expanded traces, see inset). Subsequent application of TTX (1 μm) blocks action potential generation but not the membrane depolarizations (third panel) or SICs (last panel on the right). SICs measured in voltage clamp before and after TTX application share similar kinetics: rise time, 71.2 ± 5.7 and 75.0 ± 10.9 ms (p > 0.8); decay time, 441.1 ± 64.6 and 399.2 ± 49.4 ms (n = 21 and 31; p > 0.5), respectively. The paired recording experiment shown in this portion of this figure has an unusual high frequency of spontaneous SICs with respect to the average (Fellin et al., 2004) and is shown here because this frequency allowed a sequence of manipulations to be performed in the same experiment. Other recordings with SIC frequency similar to the average value showed similar properties. SICs and SIC-mediated depolarization frequency in each portion of the experiment shown in the figure was as follows (in SICs/min): voltage clamp, 7 ± 1; current clamp, 13 ± 5; current clamp plus TTX, 11 ± 3; voltage clamp plus TTX, 7 ± 1. B, Left, Average value of the SIC-mediated membrane depolarization in the absence (n = 36 from 5 cells) and presence (1 μm; n = 49) of TTX. Right, Average number of action potentials per SIC in the absence (n = 36 from 5 cells) and presence (n = 49) of TTX. C, Mean rise and decay time of SIC-mediated membrane depolarization under control conditions (n = 36) and in the presence of TTX (n = 49; p > 0.4). D, Paired recording from two closely spaced CA3 neurons in the presence of TTX (1 μm), showing synchronous SICs (bottom traces in voltage clamp) and membrane depolarizations (top traces in current clamp). E, The event highlighted in D is shown at an expanded timescale. F, Average membrane depolarization (left) and average amplitude (right) for SIC-mediated depolarization and for SICs in CA3 neurons (n = 8 and 16, respectively). G, Kinetics of SICs recorded from CA3 neurons in current (CC) and voltage clamp (VC) configuration (n = 8 and 16, respectively).
Confocal microscopy of brain slices.
A Nikon (Tokyo, Japan) RCM8000 and a Leica TCS SP2 RS confocal microscope were used for [Ca2+]i imaging as described previously (Pasti et al., 1997). Slices were continuously perfused with normal ACSF (as above) with sulfinpyrazone at 0.2 mm. Laser emissions at 351 and 488 nm were used for excitation of Indo-1 and Oregon Green BAPTA, respectively. Indo-1 fluorescence was separated into two components (405 and 485 nm) by a dichroic mirror. The ratio of the signal collected at the two wavelengths (405/485 nm) is displayed in a pseudocolor scale. Confocal images were acquired every 2 or 4 s. Experiments in Figure 2 were performed at either room temperature or 35°C, and experiments in Figure 8, C and D, were performed at 35°C. Neurons and astrocytes were distinguished on the basis of the distinct kinetics of their Ca2+ response to high K+ stimulation (40 mm), which was always performed at the end of the experiment as discussed previously (Pasti et al., 1997). The interevent time interval of [Ca2+]i elevations in neurons was estimated as by Fellin et al. (2004).
Figure 8.

The 8.5 mm K+ and 0.5 mm Mg2+ model of epileptiform activity, which generates pure interictal-like activity, does not trigger activation of astrocytic Ca2+ signaling. A, Representative example of epileptiform activity generated by bath application of 0.5 mm Mg2+ and 8.5 mm K+-containing saline recorded extracellularly (left) and intracellularly (right) in the CA3 region. B, Average duration (left) and frequency (right) of 447 interictal-like events recorded from 17 slices after perfusion of 0.5 mm Mg2+ and 8.5 mm K+-containing saline. C, Example of the Ca2+ response of three different CA3 astrocytes under basal conditions and in the presence of 0.5 mm Mg2+ and 8.5 mm K+-containing saline. D, Mean number of astrocytes displaying Ca2+ oscillations (left) and mean frequency of Ca2+ peaks (right) in 46 CA3 astrocytes from four experiments (p > 0.39) under the different experimental conditions.
Extracellular recordings.
Slices were visualized with a 4× objective in an upright Olympus Optical (Tokyo, Japan) BX51WI microscope. Glass electrodes for extracellular recording were filled with normal ACSF. Electrodes were positioned in the CA3 and CA1 pyramidal cells layers. Electrophysiological signals were amplified and filtered at 0.1 Hz and 1 kHz by a MultiClamp 700 B (Molecular Devices, Palo Alto, CA) or 1800 microelectrode (A-M Systems, Sequim, WA) amplifier, digitized at 5 kHz with Digidata 1320 (Molecular Devices), and stored in the computer with Clampex 9.2 (Molecular Devices). Recording were performed at 30–35°C. Epileptiform events were determined with a spike detection algorithm (Mini Analysis software; Synaptosoft, Decatur, GA) and verified visually. Spikes with amplitude greater than two times the peak-to-peak noise level (∼200 μV) were accepted. The duration of an epileptiform event was measured as the time interval between the first and the last population spike present in each epileptiform event. Events with duration <1 s were defined as interictal-like events (see Fig. 1) and events with duration >2 s as ictal-like events (see Fig. 1) according to previous reports (McNamara, 1994; McCormick and Contreras, 2001; Avoli et al., 2002). Interictal-like epileptiform events are composed of a primary population spike and subsequent, secondary afterdischarges as described previously (Dzhala and Staley, 2003) and occur at high frequency (>3 events/min). Ictal (seizure)-like epileptiform events occur at lower frequency (∼1 event/min) and consist usually of an initial sustained phase (tonic) and a subsequent phase characterized by intermittent pattern of population discharges (clonic) as described previously (Dzhala and Staley, 2003). The delay between epileptiform events recorded from the different hippocampal regions was measured as the time interval between the onsets of the first population spike in each event.
Figure 1.

Epileptiform-like activity triggered in hippocampal slices by removing extracellular Mg2+ and by blocking GABAA receptors. A–E, Extracellular (A, D) and patch-clamp (B, E) recordings in the CA3 region showing ictal-like (A, B) and interictal-like (D, E) events triggered by bath application of 0 Mg2+ ACSF and 50–100 μm picrotoxin. C, F, Average values of event duration (left) and frequency (right) for ictal-like events (C; n = 344 events from 35 experiments) and interictal-like events (F; n = 274 events from 9 experiments).
Patch-clamp recordings and analysis.
Pipette resistance was 3–4 MΩ. Intrapipette solution contained the following (in mm): 145 K-gluconate, 2 MgCl2, 5 EGTA, 2 Na2ATP, 0.2 NaGTP, and 10 HEPES to pH 7.2 with KOH. Pipette liquid junction potential was approximately −15 mV. This value should be added to all voltages to obtain the correct value of the membrane potential in whole-cell configuration. Patch-clamp recordings were performed using standard procedures (Hamill et al., 1981) using Axopatch-200B/multiclamp 700B amplifiers (Molecular Devices) or 2400 patch-clamp amplifiers (A-M Systems). Data were filtered at 1 kHz and sampled at 5 kHz with a Digidata 1200 or 1320 interface and pClamp software (Molecular Devices). Experiments were performed at either room temperature or 35°C. Neurons were voltage clamped at −60 mV. Recordings from cortical neurons were performed from the somatosensory, visual, and auditory cortex. Most of these recordings were conducted from cells located within 100 μm from the surface of the brain. Data analysis and fitting were performed with Clampfit 9.2 (Molecular Devices), Origin 7.5 (Microcal Software), and SigmaPlot 8.0 (SPSS, Chicago, IL) software. During epileptiform activity, unclamped action potentials were occasionally detected. For presentation purposes, and to allow resolution of the underling currents, the current caused by unclamped action potentials has been truncated. Slow inward currents with an amplitude greater than −20 pA and a rise time slower than 10 ms [more then five times greater than miniature EPSC (supplemental Tables 1, 2, available at www.jneurosci.org as supplemental material)] are classified as SICs as described previously (Fellin et al., 2004). SIC amplitude was measured at the peak; rise time was calculated with the 20–80% criterion and the decay time as the time constant of a single exponential fit. Cellular input resistance was calculated from hyperpolarizing current injections (100–200 pA). Analysis of miniature PSC was performed using Mini Analysis software (Synaptosoft). Data are expressed as mean ± SEM. Student’s t test and Mann–Whitney’s nonparametric test were used to evaluate statistical significance.
Results
Because astrocytes can sense synaptically released glutamate (Porter and McCarthy, 1996), we asked whether chemically induced seizure-like or epileptiform activity in brain slice preparations activates Ca2+ signaling in these glial cells. Removal of external Mg2+ from the ACSF and blockade of inhibitory synapses with picrotoxin (50–100 μm) evoked synchronous epileptiform activity in CA3 pyramidal neurons that was detected in field recordings (Fig. 1A,D) as well as in patch-clamp recordings (Fig. 1B,E). The histogram of the duration of the epileptiform events recorded extracellularly shows a biphasic distribution (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material), reflecting two major patterns of activity: in most of the slices (80%; n = 44), we predominantly observed long-duration (>2 s) (Fig. 1A–C) events occurring at low frequency (<1 event/min), which are usually referred as ictal-like events (McNamara, 1994; McCormick and Contreras, 2001; Avoli et al., 2002). In the remaining slices (20%; n = 44), perfusion with 0 Mg2+ and picrotoxin resulted in shorter-duration events (<1 s) (Fig. 1D–F) occurring at a higher frequency, which are usually referred to as interictal-like events (McNamara, 1994; McCormick and Contreras, 2001; Avoli et al., 2002). Based on previous reports (Traynelis and Dingledine, 1988; Dzhala and Staley, 2003; Sayin and Rutecki, 2003) and the clear bimodal distribution of event duration (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material), events of <1 s in duration will be referred to as interictal-like events and those of duration >2 s as ictal-like events. Under these experimental conditions, Ca2+ oscillations in CA3 astrocytes were significantly increased with respect to control (n = 22 slices): both the number of astrocytes displaying Ca2+ oscillations and the frequency of oscillations almost doubled (Fig. 2A,B). On average, the amplitude of the Ca2+ peaks in astrocytes was unchanged in the presence of 0 Mg2+ and picrotoxin before and after TTX (0.5–1.0 μm) application (405/485 nm ratio, 0.22 ± 0.02 vs 0.21 ± 0.03, respectively; p > 0.6). In the presence of TTX (0.5–1.0 μm) to prevent the generation of the epileptiform activity, application of picrotoxin alone did not affect astrocyte Ca2+ signaling (p > 0.23), although removal of extracellular Mg2+ in the presence of TTX resulted in a small increase in astrocytic Ca2+ excitability (Fig. 2C).
Addition of TTX (0.5–1.0 μm) to suppress action potential activity blocks, in all of the experiments performed (n = 22), the ongoing synchronous epileptiform events that involve all neurons in the field of view (Fig. 2D,E). However, when neuronal activity is blocked, sporadic Ca2+ transients were still observed in astrocytes and in some neurons (Fig. 2D, images 4–6, E, right). These TTX-insensitive neuronal Ca2+ oscillations resemble those induced by astrocyte-evoked neuronal excitation (Fellin et al., 2004) because they occur synchronously in groups of closely spaced neurons with a maximal extension of ∼100 μm (Fig. 2D,H) (supplemental movie S1, available at www.jneurosci.org as supplemental material). Astrocytic Ca2+ oscillations that are initiated by epileptiform activity are not blocked by subsequent application of TTX (Fig. 2B,E), whereas TTX-insensitive neuronal Ca2+ oscillations are prevented by the NMDAR antagonist d-AP-5 (n = 22 neurons from 4 slices; 100 μm) (Fig. 2I), which is known to block astrocyte-evoked neuronal excitation (Angulo et al., 2004; Fellin et al., 2004; Perea and Araque, 2005). In Figure 2E, we show a correlated example of a Ca2+ elevation in an astrocyte and neuron that were in the same optical plane. In this type of imaging experiment in which one is correlating Ca2+ signals in two cell types, it is never possible to know which astrocyte is responsible for inducing NMDA receptor-dependent neuronal excitation. Thus, whether the ∼10 s delay between the astrocytic Ca2+ signal and the neuronal event is a true reflection of the latency in this experiment is unknown. However, previous photolysis studies do show that such prolonged delays can occur because neuronal SICs were detected >10 s after photolytic elevation of Ca2+ in the astrocytic cell body. In such examples, the delay resulted from the time required for the Ca2+ signal to spread within the astrocytic processes to sites in which it induces glutamate release (Fellin et al., 2004). Nonetheless, these data demonstrate that epileptiform activity in 0 Mg2+ and picrotoxin triggers Ca2+ signaling in astrocytes, that these oscillations persist beyond the period of neuronal activity, and that NMDAR-mediated neuronal Ca2+ signals share similarity to previously identified astrocyte-evoked neuronal Ca2+ signals.
Glutamate release from astrocytes is increased after a period of 0 Mg2+ and picrotoxin-induced epileptiform activity
Ca2+ oscillations in area CA1 astrocytes are known to evoke glial glutamate release and, as a consequence, NMDAR-mediated SICs in adjacent pyramidal neurons (Angulo et al., 2004; Fellin et al., 2004; Perea and Araque, 2005). We therefore asked whether neuronally detected SICs are activated by epileptiform activity. Astrocyte-evoked SICs can be identified based on four properties: their slow kinetics, insensitivity to TTX, insensitivity to CNQX, and blockade by d-AP-5 (Angulo et al., 2004; Fellin et al., 2004). Indeed, spontaneous and induced SICs with these properties were detected in area CA3 pyramidal neurons (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Moreover, a transient episode of epileptiform activity triggered a persistent increase in SIC frequency that was detected after neuronal activity was blocked by TTX (1 μm) (Fig. 3A,B). The ability of epileptiform activity to stimulate SICs is a widespread phenomenon that was detected in area CA1, CA3, and also in the cortex (Fig. 3C). Preincubation and maintenance of slices in TTX (1 μm) before perfusion with 0 Mg2+ and picrotoxin ACSF to prevent epileptiform activity did not lead to a significant increase in SIC frequency, demonstrating that in 0 mm Mg2+ and picrotoxin epileptiform activity is required for the activation of astrocytes and the generation of SICs (Fig. 3D). These epileptiform-evoked slow currents share kinetics typical of previously identified SICs (Angulo et al., 2004; Fellin et al., 2004; Perea and Araque, 2005), which are orders of magnitude slower than synaptic-evoked currents (supplemental Tables 1, 2, available at www.jneurosci.org as supplemental material) and slower than epileptiform events (Fig. 3E).
Epileptiform activity-induced SICs fulfill the remaining pharmacological criteria to assign their origin to the astrocyte: they are mediated by the selective activation of NMDARs because they are reversibly blocked by d-AP-5 (n = 4 cells; 50–100 μm) (Fig. 3G), whereas CNQX (25 μm) does not change the number of cells displaying SICs (43% control, 42% CNQX; n = 14 and 19 cells, respectively), the increase in SIC frequency (Fig. 3F), or the amplitude of SICs (supplemental Table 1, available at www.jneurosci.org as supplemental material). Because SICs detected after epileptiform activity exhibit the kinetic and pharmacological hallmarks of previously described astrocyte-evoked SICs, we conclude that prolonged astrocytic Ca2+ signaling induced by a transient period of epileptiform activity in 0 Mg2+ and picrotoxin (Fig. 2A,B) results in a persistent increase in the excitation of hippocampal and cortical neurons mediated by glial glutamate acting on NMDA receptors.
Glutamate-dependent gliotransmission evokes neuronal depolarization and action potential generation
Typically, SICs have a mean amplitude of >100 pA but can reach amplitudes of several hundreds of picoamperes (supplemental Table 1, available at www.jneurosci.org as supplemental material) (Angulo et al., 2004; Fellin et al., 2004). Currents of such an amplitude can depolarize the neuronal membrane potential close to the threshold for action potential firing (supplemental Table 3, available at www.jneurosci.org as supplemental material). Figure 4A shows a paired recording in the absence of extracellular Mg2+ from two adjacent CA1 neurons that display a high frequency of spontaneous SIC activity, as judged from the recording in voltage-clamp configuration (left panel). (Note that, because recordings were made from paired cells <100 μm apart, some SICs are detected synchronously in these neurons.) In current clamp, SICs cause a depolarization in the neuronal membrane, which can lead to action potential firing (second panel and inset). The average membrane depolarization and the average number of action potentials evoked by SICs in five neurons are shown in Figure 4B. SICs can evoke action potentials in 53% of the examples (n = 36 events from 5 cells). Perfusion of the slice with TTX (1 μm) abolished action potential generation but does not modify the depolarization induced by SICs (Fig. 4A,B). Accordingly, the slow kinetics of the membrane depolarization (Fig. 4C), which resemble those of SICs in voltage-clamp configuration (legend of Fig. 4), are not affected by TTX. In two of these five cells, SICs were characterized pharmacologically with d-AP-5 (100 μm; data not shown) and shown to be sensitive to this NMDAR antagonist. Importantly, the same TTX-insensitive synchronous depolarizations were observed in four pairs of closely spaced (<60 μm) neurons from the CA3 area (Fig. 4D–G). In conclusion, these data clearly show that SICs generated by glutamate released from astrocytes can significantly affect neuronal excitability and lead to the generation of action potentials in small groups of closely spaced hippocampal neurons.
Astrocytic glutamate is not necessary for the generation of epileptiform activity
Because astrocytic glutamate provides an excitatory drive to small groups of pyramidal neurons, it is possible that this nonsynaptic source of excitation is required for epileptiform activity. To test this possibility, we monitored epileptiform activity using four extracellular recordings positioned in the CA3 and CA1 regions of the hippocampus and performed pharmacological manipulations that target neuronal and glial signaling pathways. Despite the fact that each electrode is separated by >150 μm, epileptiform activity is synchronous in all of the four electrodes within a time window of <30 ms (n = 9 slices) (Fig. 5A,B). Analysis of the delay between the onsets of the epileptiform activity recorded from the different electrodes is consistent with previous findings (Korn et al., 1987; Colom and Saggau, 1994; Dzhala and Staley, 2003), suggesting that this hypersynchronous activity occurs first in the CA3a-b region and then spreads to the other hippocampal regions (Fig. 5B). In agreement with an essential neuronal role in the spread of epileptiform activity, the application of TTX (1 μm) to block action potential generation blocks extracellularly detected ictal (n = 8) (Fig. 5C,D,G) and interictal (n = 3) (Fig. 5E–G; but see also Fig. 3A,B) epileptiform activity.
Figure 5.

Epileptiform-like activity is synchronous within the whole hippocampus and is blocked by TTX. A, Extracellular recording from four different electrodes positioned in the CA1 and CA3 pyramidal cell layers as shown in the inset [left, CA1 proximal (CA1 prox); CA1 distal (CA1 dist)]. The electrodes were separated by >150 μm. Inset, Right, A typical ictal-like event seen at an expanded timescale reveals synchronization of neuronal activity in the whole hippocampus. B, Average values of the delay between the onset of the signal in the electrode positioned in the CA3b region and the ones positioned in the other areas of the hippocampus (n = 9). C, Ictal-like activity is blocked by TTX. D, Normalized histograms for the signals recorded from CA1 distal (top) and CA3c (bottom) under control conditions (gray), 0 Mg2+ and picrotoxin (picro; black), and 0 Mg2+, picrotoxin, and TTX (gray; picrotoxin at 100 μm, TTX at 1 μm). E, F, Same as in C and D for a representative experiment showing interictal-like activity. G, Average frequency of ictal-like (n = 8) and interictal-like (n = 3) events in control conditions, in the presence of picrotoxin and 0 extracellular Mg2+, and in the presence of picrotoxin, 0 extracellular Mg2+, and TTX (1 μm).
Because astrocyte-evoked neuronal SICs are mediated exclusively through NMDARs (Angulo et al., 2004; Fellin et al., 2004; Perea and Araque, 2005), we asked whether these receptors are necessary for epileptiform activity (Figs. 6, 7). After initiating epileptiform activity with slice perfusion of 0 Mg2+ and picrotoxin, we separated the experiments into two groups, ictal like (Fig. 6A) and interictal like (Fig. 6D), according to the predominant pattern of activity during this initial phase of the experiment. We then applied the NMDA receptor antagonists d-AP-5 (50 μm) or MK-801 [(5S,10R)-(+)-5-methyl-10,11-dihydro-5H-dibenzo [a,d]cyclohepten-5,10-imine maleate] (10–20 μm) and determined the actions of NMDAR blockade on ongoing epileptiform activity. NMDAR antagonists never blocked ongoing epileptiform activity as described previously (Stasheff et al., 1989). In slices displaying initial ictal-like activity (Fig. 6A), the duration of the epileptiform events was, however, significantly shortened during d-AP-5 application (n = 6). The frequency of the epileptiform events was not changed in the presence of d-AP-5 in five of the six experiments performed, although in one it was clearly increased. As a consequence, the average frequency of the epileptiform activity is slightly, but not significantly, increased (Fig. 6B). Similar results were obtained with MK-801 (n = 5) (Fig. 6C). In slices displaying interictal-like activity, the frequency of the interictal events was unchanged, although the duration of individual events was also significantly shortened by either d-AP-5 (n = 6) or MK-801 (n = 5) application (Fig. 6D–F).
Figure 6.

Block of the NMDA receptor does not prevent the initiation or maintenance of epileptiform activity but modulates its duration. A–F, Experiments were separated into ictal like (A–C) and interictal like (D–F) according to the type of activity displayed after the initial application of 0 Mg2+ and picrotoxin. A, Representative traces of slices displaying ictal-like activity recorded extracellularly in 0 Mg2+ and picrotoxin in the CA3 region under control conditions. Subsequent application of d-AP-5 (50 μm) shortens the epileptiform event duration, which fully recovers after d-AP-5 washout. B, Average duration (left) and frequency (right) for the epileptiform events recorded in 0 Mg2+ and picrotoxin under the experimental conditions shown in A in six slices initially displaying ictal-like activity. Note that the average frequency of epileptiform activity tends to be increased during d-AP-5 as a consequence of a switch from ictal- to interictal-like activity in one of the six experiments performed. Statistical significance in this as well as in C was calculated using the Mann–Whitney’s test. C, Same as in B using the open channel NMDA blocker MK-801 (10–20 μm; n = 5). D, Representative extracellular recording from the CA3 region showing interictal-like activity during the initial slice perfusion with 0 Mg2+ and picrotoxin. Application of d-AP-5 results in a reversible decrease of the epileptiform event duration. E, Average duration (left) and frequency (right) for the epileptiform events recorded in 0 Mg2+ and picrotoxin under the different experimental conditions shown in D in six slices initially displaying interictal-like activity. F, Same as in E using the NMDA blocker MK-801 (10–20 μm; n = 5). G, Block of the NMDA receptor does not prevent the initiation of ictal and interictal epileptiform activity. Extracellular recording showing interictal-like (left) and ictal-like (right) activity arising in the slice after perfusion with 0 Mg2+ and picrotoxin-containing saline in the presence of d-AP-5 (50 μm). d-AP-5 was preincubated in the slice at least 5 min before the application of 0 Mg2+ and picrotoxin (100 μm). d-AP-5 washout can result in a switch from interictal-like to an ictal-like activity (left). H, Average duration (left) and frequency (right) of the epileptiform events recorded in the different experimental conditions shown in G (n = 7). picro, Picrotoxin. Data in this figure are obtained from 12- to 20-d-old animals.
Figure 7.

Astrocyte-evoked SICs and epileptiform activity are two distinct electrophysiological events. A–C, Simultaneous recording from a pyramidal cell in the CA1 region (middle trace in each panel) and two distal extracellular electrodes placed in the CA3b and CA3c regions (top and bottom traces in each panel, respectively). When the slice is challenged with 0 Mg2+ and picrotoxin (A), synchronous ictal-like events are recorded from all of the electrodes. In contrast, locally generated SICs (asterisk) are recorded only by the whole-cell patch electrode and not by the extracellular electrode. For an example of ictal-like event and SIC displayed at an expanded timescale, see inset. In the same experiment shown in A, application of d-AP-5 (B; 50 μm) blocks SIC but not epileptiform activity. Note that, in this experiment, application of d-AP-5 results in a switch of the duration of epileptiform activity from ictal- to interictal-like, as described previously (see legend of Fig. 6). Subsequent washout of d-AP-5 results in the recovery of the SIC activity (C). D, Average SIC (left) and epileptiform event (right) frequency under the different experimental conditions shown in A–C (n = 4 experiments).
To simultaneously and unambiguously monitor SICs and epileptiform activity, we use a combination of whole-cell patch-clamp and extracellular field recordings (Fig. 7) (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). As shown in Figure 7, epileptiform ictal-like activity is recorded synchronously in all electrodes positioned in different regions of the hippocampus when the slice is perfused with 0 Mg2+ and picrotoxin. In contrast, SICs are recorded exclusively in the patch electrode. Of utmost importance is the fact that the occurrence of an SIC in the pyramidal neuron does not result in the generation of epileptiform events that can be detected by the extracellular electrodes (Fig. 7A) (supplemental Fig. 3A, available at www.jneurosci.org as supplemental material). These data demonstrate the inability of SICs to generate a widespread epileptiform event. Based on the data described previously (Figs. 3–6), SICs and epileptiform activity can be distinguished on the basis of a clear-cut pharmacology: SICs are TTX insensitive and blocked by d-AP-5, whereas epileptiform activity is TTX sensitive and is not prevented by d-AP-5 application. With the multielectrode approach shown in Figure 7, we could test this distinct pharmacology in the same experiment. Indeed, when TTX (1 μm) is applied in the bath, epileptiform activity ceases, although SICs are unaffected (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). When d-AP-5 (50 μm) is applied to the slice, SICs are reversibly blocked, although epileptiform activity is still present (Fig. 7B–D).
Although these data demonstrate that the activation of the target of astrocytic glutamate, i.e., the NMDA receptor, is not necessary for the maintenance of epileptiform activity in the slice, the generation of synchronous astrocyte-evoked neuronal excitation in a small group of hippocampal neurons could in principle initiate the hypersynchronous neuronal activity typical of seizures. To test this possibility, we incubated slices in NMDAR antagonists to block astrocyte-evoked neuronal excitation, before switching to pro-epileptiform conditions. Preincubation of hippocampal slices in d-AP-5 (50 μm; n = 7) before the application of low Mg2+ and picrotoxin prevented the initiation of neither interictal-like nor ictal-like epileptiform activity (Fig. 6G, left and right, respectively). After epileptiform activity had been initiated, the washout of d-AP-5 resulted in an increased frequency and duration of the epileptiform events, and, in the cases in which interictal activity was present in d-AP-5 (n = 3), a switch to ictal-like activity was observed (Fig. 6G,H). Together, these data demonstrate that, although the NMDA receptor is important for the modulation of the duration of the ictal and interictal events, these receptors, and thus gliotransmission, are not necessary for the generation of epileptiform activity.
A different model of epileptiform activity does not activate astrocytes
We made use of an additional in vitro model of epileptiform activity (Rutecki et al., 1985; Korn et al., 1987) in which perfusion with a modified ACSF containing elevated (8.5 mm) K+ and reduced Mg2+ (0.5 mm) resulted in 17 of 20 slices (85%) in the generation of an epileptiform activity characterized, exclusively, by fast, high-frequency interictal-like events (Fig. 8A,B) (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material). Under these experimental conditions, astrocytic Ca2+ signaling (Fig. 8C,D) and SIC frequency in CA3 pyramidal cells (0.09 ± 0.02 SICs/min) were unchanged with respect to controls (0.04 ± 0.02 SICs/min; p > 0.2). Although at first sight this might seem at odds with the ability of epileptiform activity in the 0 mm Mg2+ and picrotoxin model to stimulate astrocytic Ca2+ oscillations and SICs, it should be noted that the 8.5 mm K+ and 0.5 mm Mg2+ model exclusively supports interictal activity, whereas the 0 Mg2+ and picrotoxin model predominantly supports ictal activity, raising the possibility that astrocytes are more effectively activated by ictal activity. Despite the fact that astrocytes are not activated, interictal-like epileptiform activity in 8.5 mm K+ and 0.5 mm Mg2+ is observed to be synchronous over large distances in the hippocampus and to be dependent on action potential-mediated synaptic transmission, being completely blocked by TTX (1 μm) (Fig. 9A,B). Application of d-AP-5 (50 μm) either before or after the induction of the epileptiform activity modifies neither the frequency nor the duration of the interictal events (Fig. 9C–F).
Figure 9.

Interictal-like activity, generated by 0.5 mm Mg2+ and 8.5 mm K+, is synchronized within the hippocampus, and its generation is not dependent on the activation of the NMDA receptors. A, Simultaneous recording from two pyramidal neurons recorded in the CA3c region in voltage clamp and two extracellular electrodes placed in CA3b and CA3c regions under control conditions (left), during slice perfusion with 0.5 mm Mg2+ and 8.5 mm K+-containing saline (middle, High K+), and with 0.5 mm Mg2+ and 8.5 mm K+-containing saline in the presence of TTX (1 μm; right, High K+ + TTX). B, Average frequency of epileptiform events under the different experimental conditions shown in A (n = 7). C, Extracellular recording showing interictal-like activity generated, in the CA3 region, by slice perfusion with 0.5 mm Mg2+ and 8.5 mm K+-containing saline before (top), during (middle), and after (bottom) d-AP-5 (50 μm) application. The insets show interictal-like events under each condition at an expanded timescale. D, Average frequency (top) and duration (bottom) of interictal-like events under basal condition (n = 31 events), in the presence of d-AP-5 (50 μm; n = 42), and after d-AP-5 washout (n = 46) from four different experiments. E, F, Representative traces (left) and average frequency and duration of interictal-like events (right) during d-AP-5 application, in the presence of d-AP-5 and 0.5 mm Mg2+ and 8.5 mm K+-containing saline (High K+ + D-AP5; n = 30 events), and after d-AP-5 washout (High K+; n = 25) in four different experiments.
Discussion
Although astrocytes have been shown capable of releasing glutamate in response to elevations of internal Ca2+, the physiological role of this gliotransmitter in astrocyte-to-neuron communication is still under investigation. However, glial glutamate generates synchronous excitation in small groups of closely spaced hippocampal neurons (Angulo et al., 2004; Fellin et al., 2004), which can lead to the generation of action potentials (Fig. 4A). Moreover, recent work has proposed that these astrocyte-evoked currents generate depolarizations resembling the paroxysmal depolarization shifts that characterize interictal epileptiform events (Kang et al., 2005; Tian et al., 2005). On the basis of this result, an intriguing hypothesis has been proposed concerning an astrocytic basis of epilepsy (Tian et al., 2005). Indirect corroboration for this hypothesis is provided by the observation that some proconvulsants evoke Ca2+ signaling in astrocytes (Chuang et al., 2001; Rutecki et al., 2002; Sayin and Rutecki, 2003; Tian et al., 2005) and that anticonvulsants can attenuate astrocytic Ca2+ signaling (Tian et al., 2005). However, a direct test of the role of astrocytes in generating ictal and interictal epileptiform activity has been elusive because of an absence of gliotransmission-selective pharmacology. In this study, we exploited the observation that glutamate-dependent gliotransmission is exclusively mediated by NMDA receptors (Angulo et al., 2004; Fellin et al., 2004; Perea and Araque, 2005), whereas synaptic transmission is mediated by AMPA and NMDA receptors. Thus, by using NMDA receptor antagonists, we could begin to probe the involvement of gliotransmission in epileptiform activity. Although we demonstrate that astrocytes do excite pyramidal neurons in area CA3 (Fig. 3) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material), an area of epileptiform generation, blockade of this nonsynaptic excitation using NMDAR antagonists (Figs. 3, 7) did not prevent the initiation of epileptiform activity (Fig. 6). We therefore conclude that glutamate-mediated gliotransmission is not necessary for epileptiform activity.
Our results are at variance, at least in part, with those recently reported by Tian et al. (2005) and Kang et al. (2005) that lead to the proposal for a predominant role of astrocytic glutamate in generating interictal activity. Indeed, although we confirmed that Ca2+ signaling in astrocytes is activated during 0 Mg2+ and picrotoxin-induced epileptiform activity, we failed to detect TTX-insensitive interictal events. Indeed, in agreement with a bulk of previous data (Perreault and Avoli, 1991; Mattia et al., 1993; Stasheff et al., 1993; Stringer, 1994; Siniscalchi et al., 1997; Graber and Prince, 2004), we found that TTX is highly effective in either preventing or blocking, depending on the time of application, both the slow depolarization with the overriding discharge of several action potentials typical of interictal events and the sustained depolarization with the prolonged action potential firing typical of ictal events. It thus appears that the discharge of action potentials in neurons, rather than the astrocytic glutamate, is a conditio sine qua non for the genesis of epileptiform activity. Additional support for a neuronal basis of the interictal-like activity is that elevated K+ and decreased Mg2+ failed to stimulate Ca2+ oscillations in astrocytes and failed to increase SIC frequency despite triggering interictal-like epileptiform events.
Although not necessary for epileptiform activity, we do not discount the possibility that astrocytic glutamate either amplifies or modulates synaptic actions during this process. Because Ca2+ oscillations within astrocytes may be initiated during epileptiform activity, the resultant Ca2+-dependent release of glutamate has the potential to contribute excitation during periods of neuronal activity. Indeed, NMDAR antagonists reduce the duration of ictal-like discharges, raising the potential for gliotransmission contributing to the prolonged duration of these events. Interestingly, the observation that SICs occur, on average, at a low frequency, matching that of ictal events, renders more likely the possibility that glutamate released from astrocytes contributes specifically to the prolonged duration of ictal events rather than to that of interictal events.
Because the NMDAR antagonists d-AP-5 and MK-801 do not select between synaptic- and gliotransmission-activated NMDA receptors and because ifenprodil, an NR2B subunit-selective antagonist only partially attenuates gliotransmission (Fellin et al., 2004), it has not been possible to determine the relative role for synaptic and glial NMDA currents in modulating the duration of ictal- and interictal-like events. An examination of this potential modulatory role for astrocytic glutamate awaits the development of astrocyte-selective tools to prevent gliotransmission.
The ability of gliotransmission to be activated for a period outlasting the original stimulus has been reported in previous studies. In the current study, the blockade of ongoing epileptiform activity with TTX revealed a persistent elevation of astrocytic Ca2+ signals and SIC frequency lasting for minutes (Figs. 2, 3). Previously, we have shown that stimulation of the Schaffer collaterals evokes gliotransmission, as monitored by SICs recorded from CA1 pyramidal neurons that were detected for several minutes after the termination of the stimulus (Fellin et al., 2004, their Fig. 1). Although there is no mechanistic understanding of this persistent activation of gliotransmission, astrocytes have been shown capable of maintaining a memory of previous stimuli. Repeated application of the metabotropic glutamate receptor agonist (±)-1-aminocyclopentrane-trans-1,3-dicarboxylic acid to astrocytes evoked Ca2+ oscillations whose frequency increased on subsequent delivery of the same stimulus (Pasti et al., 1997).
Although the results of our experimental test of the hypothesis of Tian et al. do not support an astrocytic basis of chemically induced seizures, it will be important in future studies to determine whether the excitatory contribution of the astrocyte is significantly altered in tissue isolated from epileptic animals. After traumatic brain injury, or status epilepticus, astrocytes become reactive and animals become epileptic. Do reactive astrocytes release larger quantities of glutamate that is sufficient to initiate an epileptic discharge? Astrocytes isolated from human epileptic foci exhibit elevated Ca2+ oscillation frequency (Manning and Sontheimer, 1997). Additionally, of great interest is the observation that synaptotagmin IV, a vesicle protein essential for glutamate release from astrocytes (Zhang et al., 2004), is upregulated after seizures (Tocco et al., 1996). Is the expression of this protein within astrocytes altered to cause an enhanced release of glutamate?
In conclusion, we demonstrate that epileptiform activity can initiate astrocytic Ca2+ signaling and glutamate-mediated gliotransmission, which provides a powerful excitation to neurons leading to increased excitability and action potential discharges. However, because blockade of NMDA receptors, the target of gliotransmission, does not prevent the initiation or the maintenance of interictal and ictal activity, we conclude that gliotransmission is not required for epileptiform activity. Because NMDA receptor antagonists do reduce the duration of ictal activity, our results support the possibility that this non-neuronal source of excitation could regulate the intensity of ictal events.
Footnotes
This work was supported by the Epilepsy Foundation (T.F.), Italian University and Health Ministries (Fondo per gli Investimenti della Ricerca di Base) Grant RBNE01RHZM_003 (G.C.), and National Institutes of Health Grants R01NS043142, R37NS037585, and P20MH071705 (P.G.H.). We thank Dr. D. Coulter for helpful comments on this manuscript and the members of the laboratory for discussion during the course of this work.
References
- Anderson WW, Lewis DV, Swartzwelder HS, Wilson WA. Magnesium-free medium activates seizure-like events in the rat hippocampal slice. Brain Res. 1986;398:215–219. doi: 10.1016/0006-8993(86)91274-6. [DOI] [PubMed] [Google Scholar]
- Angulo MC, Kozlov AS, Charpak S, Audinat E. Glutamate released from glial cells synchronizes neuronal activity in the hippocampus. J Neurosci. 2004;24:6920–6927. doi: 10.1523/JNEUROSCI.0473-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avoli M, D’Antuono M, Louvel J, Kohling R, Biagini G, Pumain R, D’Arcangelo G, Tancredi V. Network and pharmacological mechanisms leading to epileptiform synchronization in the limbic system in vitro. Prog Neurobiol. 2002;68:167–207. doi: 10.1016/s0301-0082(02)00077-1. [DOI] [PubMed] [Google Scholar]
- Bezzi P, Carmignoto G, Pasti L, Vesce S, Rossi D, Rizzini BL, Pozzan T, Volterra A. Prostaglandins stimulate calcium-dependent glutamate release in astrocytes. Nature. 1998;391:281–285. doi: 10.1038/34651. [DOI] [PubMed] [Google Scholar]
- Bezzi P, Gundersen V, Galbete JL, Seifert G, Steinhauser C, Pilati E, Volterra A. Astrocytes contain a vesicular compartment that is competent for regulated exocytosis of glutamate. Nat Neurosci. 2004;7:613–620. doi: 10.1038/nn1246. [DOI] [PubMed] [Google Scholar]
- Bragin A, Engel J, Wilson CL, Vizentin E, Mathern GW. Electrophysiologic analysis of a chronic seizure model after unilateral hippocampal KA injection. Epilepsia. 1999;40:1210–1221. doi: 10.1111/j.1528-1157.1999.tb00849.x. [DOI] [PubMed] [Google Scholar]
- Carmignoto G, Fellin T. Glutamate release from astrocytes as a non-synaptic mechanism for neuronal synchronization in the hippocampus. J Physiol (Paris) 2006;99:98–102. doi: 10.1016/j.jphysparis.2005.12.008. [DOI] [PubMed] [Google Scholar]
- Chuang SC, Bianchi R, Kim D, Shin HS, Wong RKS. Group I metabotropic glutamate receptors elicit epileptiform discharges in the hippocampus through PLCβ1 signaling. J Neurosci. 2001;21:6387–6394. doi: 10.1523/JNEUROSCI.21-16-06387.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colom LV, Saggau P. Spontaneous interictal-like activity originates in multiple areas of the Ca2-Ca3 region of hippocampal slices. J Neurophysiol. 1994;71:1574–1585. doi: 10.1152/jn.1994.71.4.1574. [DOI] [PubMed] [Google Scholar]
- Dreier JP, Heinemann U. Regional and time-dependent variations of low Mg2+ induced epileptiform activity in rat temporal cortex slices. Exp Res. 1991;87:581–596. doi: 10.1007/BF00227083. [DOI] [PubMed] [Google Scholar]
- Dzhala VI, Staley KJ. Transition from interictal to ictal activity in limbic networks in vitro. J Neurosci. 2003;23:7873–7880. doi: 10.1523/JNEUROSCI.23-21-07873.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards FA, Konnerth A, Sakmann B, Takahashi T. A thin slice preparation for patch clamp recordings from neurons of the mammalian central nervous system. Pflügers Arch. 1989;414:600–612. doi: 10.1007/BF00580998. [DOI] [PubMed] [Google Scholar]
- Fellin T, Haydon PG. Do astrocytes contribute to excitation underlying seizures? Trends Mol Med. 2005;11:530–533. doi: 10.1016/j.molmed.2005.10.007. [DOI] [PubMed] [Google Scholar]
- Fellin T, Pascual O, Gobbo S, Pozzan T, Haydon PG, Carmignoto G. Neuronal synchrony mediated by astrocytic glutamate through activation of extrasynaptic NMDA receptors. Neuron. 2004;43:729–743. doi: 10.1016/j.neuron.2004.08.011. [DOI] [PubMed] [Google Scholar]
- Fellin T, Pascual O, Haydon PG. Astrocytes coordinate synaptic networks: balanced excitation and inhibition. Physiology. 2006;21:208–215. doi: 10.1152/physiol.00161.2005. [DOI] [PubMed] [Google Scholar]
- Graber KD, Prince DA. A critical period for prevention of posttraumatic neocortical hyperexcitability in rats. Ann Neurol. 2004;55:860–870. doi: 10.1002/ana.20124. [DOI] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Haydon PG. GLIA: listening and talking to the synapse. Nat Rev Neurosci. 2001;2:185–193. doi: 10.1038/35058528. [DOI] [PubMed] [Google Scholar]
- Jefferys JGR. Experimental neurobiology of epilepsies. Curr Opin Neurol. 1994;7:113–122. doi: 10.1097/00019052-199404000-00007. [DOI] [PubMed] [Google Scholar]
- Jefferys JGR. Nonsynaptic modulation of neuronal activity in the brain: electric currents and extracellular ions. Physiol Rev. 1995;75:689–723. doi: 10.1152/physrev.1995.75.4.689. [DOI] [PubMed] [Google Scholar]
- Jones RSG, Heinemann U. Synaptic and intrinsic responses of medial entorhinal cortical cells in normal and magnesium-free medium in vitro. J Neurophysiol. 1988;59:1476–1496. doi: 10.1152/jn.1988.59.5.1476. [DOI] [PubMed] [Google Scholar]
- Kang N, Xu J, Xu Q, Nedergaard M, Kang J. Astrocytic glutamate release-induced transient depolarization and epileptiform discharges in hippocampal CA1 pyramidal neurons. J Neurophysiol. 2005;94:4121–4130. doi: 10.1152/jn.00448.2005. [DOI] [PubMed] [Google Scholar]
- Konnerth A, Heinemann U, Yaari Y. Nonsynaptic epileptogenesis in the mammalian hippocampus in vitro. I. Development of seizurelike activity in low extracellular calcium. J Neurophysiol. 1986;56:409–423. doi: 10.1152/jn.1986.56.2.409. [DOI] [PubMed] [Google Scholar]
- Korn SJ, Giacchino JL, Chamberlin NL, Dingledine R. Epileptiform burst activity induced by potassium in the hippocampus and its Regulation by GABA-mediated inhibition. J Neurophysiol. 1987;57:325–340. doi: 10.1152/jn.1987.57.1.325. [DOI] [PubMed] [Google Scholar]
- Manning TJ, Sontheimer H. Spontaneous intracellular calcium oscillations in cortical astrocytes from a patient with intractable childhood epilepsy (Rasmussen’s encephalitis) Glia. 1997;21:332–337. [PubMed] [Google Scholar]
- Mattia D, Hwa GGC, Avoli M. Epileptiform activity-induced by 4-aminopyridine in guinea-pig and rat neocortices. Neurosci Lett. 1993;154:157–160. doi: 10.1016/0304-3940(93)90195-q. [DOI] [PubMed] [Google Scholar]
- McCormick DA, Contreras D. On the cellular and network bases of epileptic seizures. Annu Rev Physiol. 2001;63:815–846. doi: 10.1146/annurev.physiol.63.1.815. [DOI] [PubMed] [Google Scholar]
- McNamara JO. Cellular and molecular-basis of epilepsy. J Neurosci. 1994;14:3413–3425. doi: 10.1523/JNEUROSCI.14-06-03413.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamara JO. Emerging insights into the genesis of epilepsy. Nature. 1999;399:A15–A22. doi: 10.1038/399a015. [DOI] [PubMed] [Google Scholar]
- Parpura V, Haydon PG. Physiological astrocytic calcium levels stimulate glutamate release to modulate adjacent neurons. Proc Natl Acad Sci USA. 2000;97:8629–8634. doi: 10.1073/pnas.97.15.8629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parpura V, Basarsky TA, Liu F, Jeftinija K, Jeftinija S, Haydon PG. Glutamate-mediated astrocyte-neuron signalling. Nature. 1994;369:744–747. doi: 10.1038/369744a0. [DOI] [PubMed] [Google Scholar]
- Pasti L, Volterra A, Pozzan T, Carmignoto G. Intracellular calcium oscillations in astrocytes: a highly plastic, bidirectional form of communication between neurons and astrocytes in situ. J Neurosci. 1997;17:7817–7830. doi: 10.1523/JNEUROSCI.17-20-07817.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasti L, Zonta M, Pozzan T, Vicini S, Carmignoto G. Cytosolic calcium oscillations in astrocytes may regulate exocytotic release of glutamate. J Neurosci. 2001;21:477–484. doi: 10.1523/JNEUROSCI.21-02-00477.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perea G, Araque A. Properties of synaptically evoked astrocyte calcium signal reveal synaptic information processing by astrocytes. J Neurosci. 2005;25:2192–2203. doi: 10.1523/JNEUROSCI.3965-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perreault P, Avoli M. Physiology and pharmacology of epileptiform activity induced by 4-aminopyridine in rat hippocampal slices. J Neurophysiol. 1991;65:771–785. doi: 10.1152/jn.1991.65.4.771. [DOI] [PubMed] [Google Scholar]
- Porter JT, McCarthy KD. Hippocampal astrocytes in situ respond to glutamate released from synaptic terminals. J Neurosci. 1996;16:5073–5081. doi: 10.1523/JNEUROSCI.16-16-05073.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogawski MA. Astrocytes get in the act in epilepsy. Nat Med. 2005;11:919–920. doi: 10.1038/nm0905-919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutecki PA, Lebeda FJ, Johnston D. Epileptiform activity induced by changes in extracellular potassium in hippocampus. J Neurophysiol. 1985;54:1363–1374. doi: 10.1152/jn.1985.54.5.1363. [DOI] [PubMed] [Google Scholar]
- Rutecki PA, Sayin U, Yang Y, Hadar E. Determinants of ictal epileptiform patterns in the hippocampal slice. Epilepsia. 2002;43:179–183. doi: 10.1046/j.1528-1157.43.s.5.34.x. [DOI] [PubMed] [Google Scholar]
- Sayin U, Rutecki PA. Group I metabotropic glutamate receptor activation produces prolonged epileptiform neuronal synchronization and alters evoked population responses in the hippocampus. Epilepsy Res. 2003;53:186–195. doi: 10.1016/s0920-1211(03)00020-2. [DOI] [PubMed] [Google Scholar]
- Schwartzkroin PA, Prince DA. Changes in excitatory and inhibitory synaptic potentials leading to epileptogenic activity. Brain Res. 1980;183:61–77. doi: 10.1016/0006-8993(80)90119-5. [DOI] [PubMed] [Google Scholar]
- Seifert G, Schilling K, Steinhauser C. Astrocyte dysfunction in neurological disorders: a molecular perspective. Nat Rev Neurosci. 2006;7:194–206. doi: 10.1038/nrn1870. [DOI] [PubMed] [Google Scholar]
- Siniscalchi A, Calabresi P, Mercuri NB, Bernardi G. Epileptiform discharge induced by 4-aminopyridine in magnesium-free medium in neocortical neurons: physiological and pharmacological characterization. Neuroscience. 1997;81:189–197. doi: 10.1016/s0306-4522(97)00178-4. [DOI] [PubMed] [Google Scholar]
- Stasheff SF, Anderson WW, Clark S, Wilson WA. NMDA antagonists differentiate epileptogenesis from seizure expression in an in vitro model. Science. 1989;245:648–651. doi: 10.1126/science.2569762. [DOI] [PubMed] [Google Scholar]
- Stasheff SF, Hines M, Wilson WA. Axon terminal hyperexcitability associated with epileptogenesis in vitro. 1. Origin of ectopic spikes. J Neurophysiol. 1993;70:961–975. doi: 10.1152/jn.1993.70.3.961. [DOI] [PubMed] [Google Scholar]
- Stringer JL. Pentylenetetrazol elicits epileptiform activity in the dentate gyrus of the urethane-anesthetized rat by activation of the entorhinal cortex. Brain Res. 1994;636:221–226. doi: 10.1016/0006-8993(94)91020-0. [DOI] [PubMed] [Google Scholar]
- Swann JW, Brady RJ. Penicillin-induced epileptogenesis in immature rat CA3 hippocampal pyramidal cells. Dev Brain Res. 1984;12:243–254. doi: 10.1016/0165-3806(84)90046-4. [DOI] [PubMed] [Google Scholar]
- Tian GF, Azmi H, Takano T, Xu Q, Peng W, Lin J, Oberheim N, Lou N, Wang X, Zielke HR, Kang J, Nedergaard M. An astrocytic basis of epilepsy. Nat Med. 2005;11:973–981. doi: 10.1038/nm1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tocco G, Bi XN, Vician L, Lim IK, Herschman H, Baudry M. Two synaptotagmin genes, Syt1 and Syt4, are differentially regulated in adult brain and during postnatal development following kainic acid-induced seizures. Mol Brain Res. 1996;40:229–239. doi: 10.1016/0169-328x(96)00055-1. [DOI] [PubMed] [Google Scholar]
- Traynelis SF, Dingledine R. Potassium-induced spontaneous electrographic seizures in the rat hippocampal slice. J Neurophysiol. 1988;59:259–276. doi: 10.1152/jn.1988.59.1.259. [DOI] [PubMed] [Google Scholar]
- Volterra A, Meldolesi J. Astrocytes, from brain glue to communication elements: the revolution continues. Nat Rev Neurosci. 2005;6:626–640. doi: 10.1038/nrn1722. [DOI] [PubMed] [Google Scholar]
- Walther H, Lambert JDC, Jones RSG, Heinemann U, Hamon B. Epileptiform activity in combined slices of the hippocampus, subiculum and entorhinal cortex during perfusion with low magnesium medium. Neurosci Lett. 1986;69:156–161. doi: 10.1016/0304-3940(86)90595-1. [DOI] [PubMed] [Google Scholar]
- Wilson WA, Swartzwelder HS, Anderson WW, Lewis DV. Seizure activity in vitro: a dual focus model. Epilepsy Res. 1988;2:289–293. doi: 10.1016/0920-1211(88)90036-8. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Fukuda M, Van Bockstaele E, Pascual O, Haydon PG. Synaptotagmin IV regulates glial glutamate release. Proc Natl Acad Sci USA. 2004;101:9441–9446. doi: 10.1073/pnas.0401960101. [DOI] [PMC free article] [PubMed] [Google Scholar]