

Abstract
The excitability of a neuron is regulated by the balance of excitatory and inhibitory inputs that impinge on it. Such modulation can occur either presynaptically or postsynaptically. Here, we show that an excitatory transmitter can increase the release of an inhibitory transmitter and thus paradoxically produces a long-lasting enhancement of inhibitory synaptic transmission. This occurs at a near-physiological temperature. These findings from cerebellar stellate neurons reveal a novel form of long-term potentiation that is induced by the activation of NMDA-type glutamate receptors and that requires both glutamate and glycine. Our results indicate that Ca2+ entry into the presynaptic terminals during the activation of presynaptic NMDARs is necessary to induce the potentiation. This presynaptic modulation provides a mechanism by which an excitatory transmitter can induce a long-term increase in the release of an inhibitory transmitter and thus modify the activity of a simple neuronal circuit.
Keywords: NMDA receptors, cerebellar stellate cell, synaptic plasticity, GABA release, presynaptic LTP, glycine
Introduction
The activity of a neuronal network depends on the balance between its excitatory and inhibitory inputs. How excitatory transmission interacts with inhibitory transmission will have an impact on the activity of the circuit as a whole. One potential mechanism that may underlie lasting changes in excitability involves the regulation of inhibitory transmission by glutamate.
There is evidence that glutamate can activate NMDA receptors (NMDARs) in the postsynaptic membrane of inhibitory synapses and alter their response to GABA (Gaiarsa et al., 2002). In addition to these postsynaptic changes, lasting suppression of the release of inhibitory transmitter can be induced by the activation of metabotropic glutamate receptors in the postsynaptic pyramidal cell in the hippocampus, an effect that is mediated by cannabinoids (Chevaleyre and Castillo, 2003). However, glutamate receptors, including NMDARs, have also been found on the presynaptic terminals of GABAergic neurons (Belan and Kostyuk, 2002). Activation of these glutamate receptors can transiently enhance or suppress the release of GABA from inhibitory interneurons (Engelman and MacDermott, 2004). Whether glutamate can produce a lasting enhancement of the release of transmitters from inhibitory neurons remains mostly unknown.
GABAergic interneurons in the cerebellum, stellate/basket cells, form inhibitory synapses onto Purkinje cells and control the timing and firing frequency of Purkinje cells (Hausser and Clark, 1997). Stellate cells also innervate themselves by GABAergic autaptic connections (Pouzat and Marty, 1998), which may enhance the precision of spike-timing in these neurons, as demonstrated in neocortical inhibitory interneurons (Bacci and Huguenard, 2006). Several lines of evidence strongly indicate the presence of NMDARs on the presynaptic terminals of cerebellar stellate cells (Glitsch and Marty, 1999; Duguid and Smart, 2004; Fiszman et al., 2005). First, immunostaining of NMDAR subunits has been found on the axonal terminals of cerebellar stellate/basket cells in vitro. Second, functional NMDARs have been detected in stellate/basket cell axon terminals. Third, activation of NMDARs in stellate/basket cells can transiently increase the release of GABA from these cells. Could activation of NMDARs at the presynaptic terminals of these inhibitory neurons produce a long-lasting increase in the secretion of GABA?
Here, we investigate whether glutamate released from parallel fibers (axons of granule cells) can induce a lasting increase in the release of GABA from stellate cells. The evoked secretion of GABA from the axon of a single neuron was monitored via an autaptic synapse. Using this preparation, we describe a new form of synaptic plasticity in which a lasting increase in the release of GABA is induced by the activation of excitatory NMDARs. Our results additionally suggest that Ca2+ entry into the presynaptic terminals during the activation of presynaptic NMDARs is required for the induction of this lasting increase in spontaneous GABA release.
Materials and Methods
Slice preparation.
Saggital and horizontal slices (250 mm) of the cerebellum from postnatal day 14 (P14) to P23 C57BL/6J mice were cut with a Leica (Nussloch, Germany) VT1000S vibrating microslicer as previously described (Liu and Cull-Candy, 2000; Liu and Cull-Candy, 2005). Slices from P14–P20 and P18–P23 mice were used for recordings of autaptic currents and spontaneous/miniature IPSCs, respectively.
Electrophysiology.
Voltage-clamp recordings were made with an Axopatch 700A amplifier (Molecular Devices, Foster City, CA) at room temperature. The extracellular solution (in mm: 125 NaCl, 2.5 KCl, 2 CaCl2, 1 MgCl2, 1.25 NaH2PO4, 26 NaHCO3, and 25 glucose, pH 7.4) was continuously bubbled with 95% O2 and 5% CO2. Electrode resistance was 4–8 MΩ. Inhibitory synaptic currents were filtered at 2 or 10 kHz and digitized at 10 kHz. Stellate cells were identified by their location in the outer two-thirds of the molecular layer and by the presence of spontaneous synaptic currents.
Synaptic currents.
Autoreceptor/autaptic currents were evoked by a depolarizing step from −70 to 0 mV for 1 ms and recorded at −70 mV in the perforated patch configuration. Electrodes were filled with a pipette solution (in mm: 135 KCl, 4.6 MgCl2, 0.1 CaCl2, 10 HEPES, 1 EGTA, 4 Na-ATP, and 0.4 Na-GTP, pH 7.4) with the addition of amphotericin B (300 μg/ml). Series resistance was monitored throughout the experiment, and the experiment was terminated if this changed by >20%. Autaptic currents were recorded at a room temperature before and after parallel fiber stimulation or application of NMDA/glycine.
Stimulation of parallel fibers.
A parallel bipolar electrode (150 μm spacing) was placed across the molecular layer ∼200 μm from the recording electrode to stimulate parallel fibers (PFs) in horizontal slices (P18 and P19). The stimulus intensity ranged from 15 to 50 V with duration of 0.1–0.2 ms. The stimulation protocol was a train of four pulses at 100 Hz, which was repeated 150 times at 1 Hz. During this time, the cell was voltage-clamped at −70 mV and depolarized to 0 mV for 1 ms (1 ms after each burst stimulation), in external solution containing 5 μm 2,3-dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7-sulfonamide (NBQX), 20 μm glycine, 10 μm SCH 50911 ((+)-5,5-dimethyl-2-morpholineacetic acid), and 1 μm strychnine.
Chemical induction of inhibitory-long-term potentiation.
NMDA (15 μm) or NMDA plus glycine (20 or 100 μm) was applied for 2.5 min in extracellular solution containing strychnine (1 μm) and NBQX (10 μm). AMPA (0.3 μm) was applied for 10 min. The cell was transiently depolarized at 1 and 0.2 Hz during the NMDA/glycine and AMPA application, respectively.
Miniature spontaneous IPSCs (mIPSCs) were recorded at −70 mV before and after a 5 min application of 30 μm NMDA and 20 μm glycine in the whole-cell configuration at room temperature. During NMDA/glycine application, the postsynaptic cell was held at −70 mV. TTX (0.3 μm), NBQX (5 μm), and strychnine (1 μm) were present throughout the experiment. A Cs-based pipette solution (in mm: 135 CsCl, 4.6 MgCl2, 0.1 CaCl2, 10 HEPES, 10 EGTA, 4 Na-ATP, and 0.4 Na-GTP, pH 7.4) was used in a few experiments. No difference in the NMDA/glycine-induced increase in mIPSC frequency was found between this and the K-internal solution described above, and therefore the data were pooled. In some experiments, a pipette solution [which contained the following (in mm): 115 KCl or CsCl, 4.6 MgCl2, 0.1 CaCl2, 10 HEPES, 10 or 20 BAPTA, 4 Na-ATP, and 0.4 Na-GTP, pH 7.4] was used to buffer a Ca2+ rise in the postsynaptic cell.
Spontaneous and miniature IPSCs were recorded at 32°C in some experiments. To determine the reversal potential for spontaneous IPSCs, gramicidin D (50 μg/ml) was added to the pipette solution and spontaneous IPSCs were recorded in a perforated patch configuration at several holding potentials (from −120 mV to −40 mV with 10 mV increments). The effect of PF stimulation on the frequency of mIPSCs were determined using a pipette solution (in mm: 135 K gluconate and 2.9 MgCl2, 0.1 CaCl2, 10 HEPES, 1 EGTA, 4 Na-ATP, and 0.4 Na-GTP, pH 7.4), which set the reversal potential for Cl to −80.5 mV. mIPSCs were recorded at −30 mV in a whole-cell configuration and detected as outward currents in the presence of TTX. The slice was then washed with artificial CSF (ACSF) for 25 min to remove TTX before PF stimulation. The PFs were stimulated with a train of four pulses at 100 Hz, repeated 60 times at 1 Hz, while the postsynaptic cell was voltage-clamped at −70 mV, in an external solution that contained 5 μm NBQX, 20 μm glycine, and 1 μm strychnine. The stimulus intensity ranged from 5 to 30 V with duration of 0.1–0.2 ms.
Data analysis.
Average autaptic IPSCs (aIPSCs) were constructed from 10 consecutive sweeps. Each mean paired-pulse response was obtained from 50 to 70 events. Events with a nontypical shape and latency were rejected (these were likely to be spontaneous IPSCs). The paired pulse ratio (PPR) was calculated as mean aIPSC2 amplitude/mean aIPSC1 amplitude. The coefficient of variation (CV) of synaptic transmission was calculated from the peak amplitude of at least 70 events. Spontaneous and miniature IPSC events were identified using Clampfit (version 9.0). Data are expressed as mean ± SEM. A two-tailed Student’s t test was used to assess statistical significance.
Results
Autaptic/autoreceptor currents were inhibited by 6-imino-3-(4-methoxyphenyl)-1(6H)-pyridazinebutanine acid hydrobromide (SR-95531) and thus mediated by GABAA receptors (aIPSCs) (Fig. 1A). aIPSCs recorded in the perforated patch-clamp configuration were stable for 1–2 h (Fig. 1B).
Figure 1.
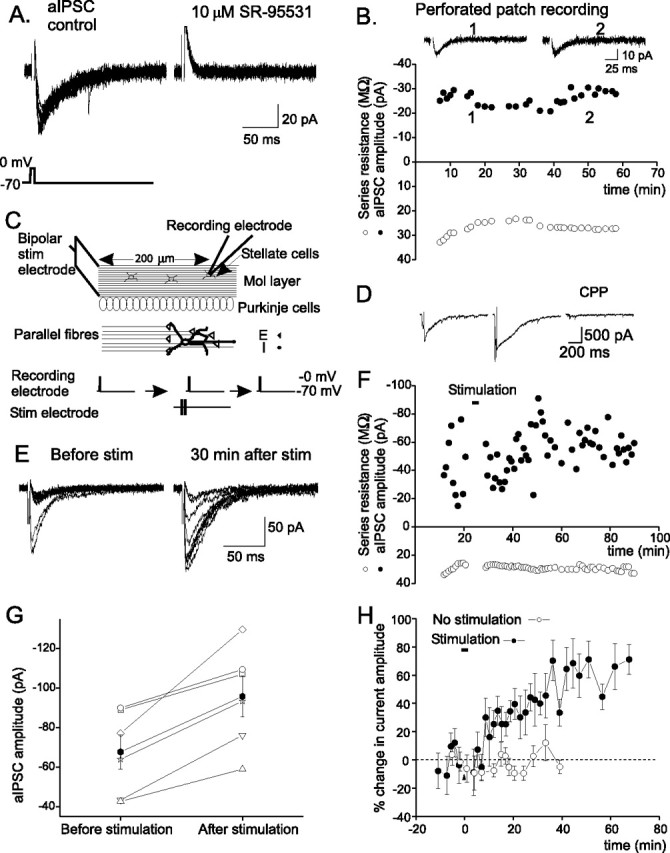
Stimulation of parallel fiber inputs induces a lasting increase in the autaptic current in stellate cells. A, Autaptic currents evoked by a brief depolarizing pulse, which was blocked by SR-95531, a GABAA receptor blocker. B, The aIPSC amplitude versus time. C, Schematic diagram of the experimental protocol. D, Currents evoked by PF stimulation. Representative current traces (middle and right for experiments described in Fig. 2B,C) are shown. E, Ten consecutive aIPSC traces. F, aIPSC amplitude (average of 10 consecutive traces) versus time. G, Averaged aIPSC amplitudes of individual stellate cells (open symbols) before and 15–60 min after PF stimulation. The average current amplitude (filled symbols) increased after stimulation (n = 6; p < 0.02). H, Plot of percentage change in mean aIPSC amplitude versus time (n = 6). Bar indicates the time of PF stimulation. Error bars indicate SEM.
Stimulation of parallel fibers induces a long-lasting increase in the amplitude of aIPSCs
Stellate cells receive glutermatergic excitatory inputs from PFs, and project their axons in the molecular layer along a horizontal course. In vivo recordings of cerebellar granule cells show that sensory stimulation evokes a burst of action potentials at ∼80 Hz (Chadderton et al., 2004). Such burst PF stimulation increases glutamate release, and the spillover of glutamate activates extrasynaptic NMDARs in the postsynaptic cell and in other stellate cells with which the activated parallel fibers do not directly form synaptic contacts (Carter and Regehr, 2000; Clark and Cull-Candy, 2002). We therefore stimulated PFs with bursts of four depolarizations at 100 Hz, as illustrated in Figure 1C, to determine how glutamate released from the PFs modulates the release of GABA from stellate cells. Glycine is a coagonist for NMDARs, and because the extracellular basal glycine concentration in the cerebellum determined by in vivo microdialysis is ∼20 μm (Matsui et al., 1995), 20 μm glycine was added to ACSF during the stimulation. This stimulation protocol evoked an NMDAR-mediated current of ∼500 pA (n = 6) (Figs. 1D, 2A) at −70 mV, which is comparable with the current activated by PF stimulation with a monopolar electrode (Carter and Regehr, 2000).
Figure 2.
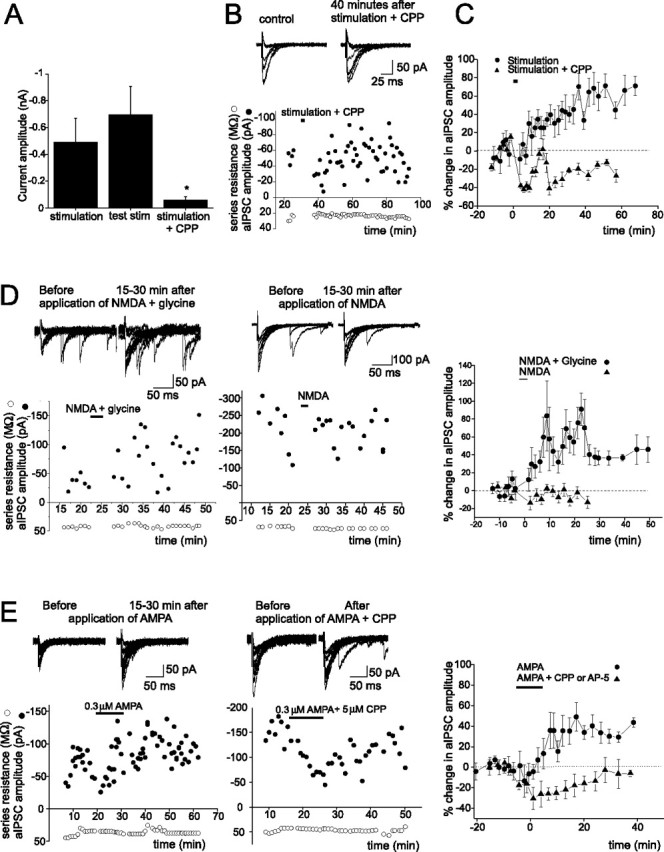
PF stimulation-induced increase in aIPSC amplitude requires activation of NMDARs. A, Group data of currents evoked by PF stimulation, before (middle; currents were evoked by a single sweep of testing stimulation) and during the application of CPP (right; n = 5, *p < 0.04), and without CPP (left; in experiments described in Fig. 1). B, aIPSC traces (top) and aIPSC amplitude before and after PF stimulation in the presence of an NMDAR blocker (CPP). C, CPP blocks the PF stimulation-induced potentiation of aIPSC amplitudes (n = 6; p < 0.03). D, E, aIPSC traces (top) and the plot of aIPSC amplitude versus time. A plot of percentage change in mean aIPSC amplitudes versus time (right; NMDA plus glycine, n = 7; NMDA, n = 5; p < 0.007; AMPA, n = 7; AMPA plus NMDAR blocker, n = 4; p < 0.03) is shown. Error bars indicate SEM.
As shown in Figure 1, E–H, the amplitude of autaptic currents increased after the PF stimulation in all cells recorded. The increase in the current amplitude lasted for at least 1 h. On average, the autaptic current increased by ∼50% (n = 6; p < 0.02). Thus, the PF stimulation induced a lasting enhancement of the efficacy of inhibitory synaptic transmission [inhibitory-long-term potentiation (I-LTP)].
Long-term potentiation of aIPSCs is induced by activation of NMDARs and requires glutamate and glycine
To determine whether activation of NMDARs was required for the induction of I-LTP, PFs were stimulated in the presence of a NMDAR blocker, 3-(CR-2-carboxypiperazin-4-yl)-propyl-1-phosphonic acid (CPP) (10–20 μm). The duration and amplitude of the PF stimulation were first adjusted such that it evoked a slow NMDAR current with an amplitude that was comparable with that generated for the induction of I-LTP in the previous section. As illustrated in Figure 2, A–C, CPP blocked the slow NMDAR current and the PF stimulation-induced I-LTP, indicating that activation of NMDARs is necessary to induce the lasting potentiation of aIPSCs in stellate cells.
Is NMDAR activation sufficient to induce I-LTP in stellate cells? To address this question, exogenous NMDA and glycine were coapplied to selectively activate NMDARs. This induced an increase in the aIPSC amplitude, which lasted for 30–60 min (Fig. 2D). A sustained potentiation of the aIPSC was consistently observed (seven of eight cells). On average, the current amplitude increased by ∼60% (from −31 ± 4 to −50 ± 8 pA; n = 8; p < 0.02). Thus, activation of NMDARs is sufficient to induce the lasting enhancement of the aIPSC. Unexpectedly, however, the coapplication of both NMDA and glycine was required to induce I-LTP, because the application of NMDA or glycine (data not shown) alone did not alter aIPSC amplitude.
The question then arises whether endogenously released glycine and glutamate can induce the I-LTP by activating NMDARs in stellate cells. Bergmann glial cells can release glycine when the membrane is depolarized (Huang et al., 2004). Activation of Ca2+-permeable AMPARs in these cells reduces the resting K conductance (Muller et al., 1992), which would depolarize the membrane potential and stimulate the release of glycine from these cells. To test this possibility, AMPA was applied to the cerebellar slice for 10 min. A lasting increase in the amplitude of aIPSCs was subsequently observed in five of seven cells (Fig. 2E). On average, the aIPSC amplitude increased by ∼37% (n = 7; p < 0.05). When AP-5 (50 μm) or CPP (5 μm) was included in the extracellular solution during AMPA application, AMPA no longer enhanced aIPSC amplitude. This result suggests that the induction of I-LTP by AMPA involves the activation of NMDARs on stellate cells presumably by endogenously released glycine and glutamate.
Exogenously applied glycine potentiates extrasynaptic NMDAR currents in stellate cells
Glycine may be required to potentiate the glutamate-activated current via NMDARs, which induces I-LTP in stellate cells. If the concentration of endogenous glycine in cerebellar slices is <20 μm (the extracellular glycine concentration in vivo) and glycine binding sites are not saturated, one would predict that exogenously applied glycine should enhance the NMDAR current.
Stimulation of PFs with a train of four stimulations at 100 Hz evoked a slow outward current at +40 mV, which was blocked by 10 μm CPP or 50 μm d-AP-5. Bath application of 20 μm glycine increased the current amplitude and charge transfer by ∼40% (n = 5; p < 0.02) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material), which is expected to enhance Ca2+ entry. Therefore, glycine sites on NMDARs are not saturated in cerebellar slices by endogenously released glycine.
The NMDAR-mediated increase of aIPSCs occurs via a presynaptic mechanism
We examined whether enhancing the presynaptic release of GABA from stellate cells contributes to the lasting increase in aIPSC amplitude after PF stimulation. If this were the case, then a decrease in the PPR and failure rate and a change in the variance of the synaptic current should be associated with the potentiation.
To measure the PPR, two depolarizing pulses separated by 20 ms were delivered and the amplitude of the aIPSC evoked by the second pulse was divided by that of the first pulse (A2/A1). As shown in Figure 3, A–C, the PPR decreased 15–60 min after PF stimulation in all cells (from 0.92 ± 0.10 to 0.72 ± 0.08; n = 6; p < 0.01). The frequency of successfully evoked aIPSCs increased and the failure rate decreased from 36 ± 10% to 24.3 ± 7% (n = 6; p < 0.05) (Fig. 4D–F). These results suggest that the PF stimulation enhanced the presynaptic release of GABA from stellate cells.
Figure 3.

PF stimulation induces a lasting increase in the presynaptic release of GABA from stellate cells. A, Average aIPSC traces. B, The PPR (=A2/A1) of individual cells before and 15–60 min after the stimulation. C, PPR decreased after stimulation (n = 6; *p < 0.01). D, Plots of aIPSC amplitude distribution from one cell. E, The failure rate of individual cells. F, Average failure rate decreased after the stimulation (n = 6; *p < 0.03). G, Comparison of the value of 1/CV2 of individual cells before and 15–60 min after the stimulation. H, Average 1/CV2 increased after stimulation (n = 6; *p < 0.03). Error bars indicate SEM.
Figure 4.
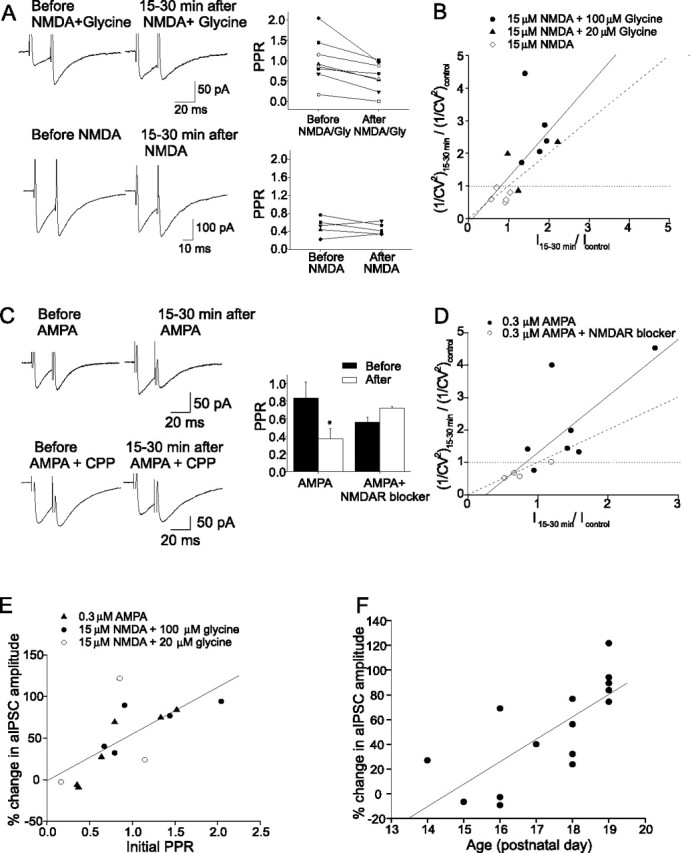
Presynaptic expression of chemically induced I-LTP. A, aIPSC traces (left) and the PPR of individual cells (right) before and after application of NMDA plus glycine (top) and NMDA (bottom). B, Plot of 1/CV2 versus current amplitude (I). Values of 1/CV2 and I were normalized to their control values before the application of NMDA plus glycine or NMDA. Each symbol represents a separate experiment. There was a significant correlation between the change in 1/CV2 and I (solid line) (r = 0.83; p < 0.003). C, aIPSC traces (left) and average PPR. Mean PPR was significantly reduced after AMPA application (n = 7; *p < 0.05; AMPA plus NMDAR blocker, n = 4). Error bars indicate SEM. D, The AMPA-induced potentiation of aIPSCs was correlated with an increase in 1/CV2 (r = 0.76; p < 0.007). E, Correlation between changes in the aIPSC amplitude 15–30 min after activation of NMDARs and the initial PPR (r = 0.70; p < 0.006). F, Largest potentiation of aIPSC amplitude (with NMDA plus glycine or AMPA) occurred in cells from older animals (r = 0.74; p < 0.002).
In additional support of a presynaptic mechanism of action, the I-LTP was found to be associated with a change in the CV of the aIPSCs. 1/CV2 (=I2/s2, where I is the aIPSC amplitude and s2 is the variance about the peak of the aIPSCs) was found to increase after PF stimulation (n = 6; p < 0.03) (Fig. 3G,H).
No changes in PPR, failure rate, or CV were observed when PF stimulation occurred in the presence of CPP (Fig. 3). The observation that I-LTP was associated with a decrease in PPR and failure rate, and with an increase in 1/(CV)2, strongly suggests that PF stimulation induces a lasting increase in the release of GABA from stellate cells via a presynaptic mechanism.
Because the induction of I-LTP requires both glycine and glutamate/NMDA, one would predict that the increased release could be induced by NMDA/glycine, but not by NMDA alone. As illustrated in Figure 4A, the PPR consistently decreased after the application of NMDA/glycine (in NMDA/20 μm glycine, from 0.72 ± 0.27 to 0.48 ± 0.23, n = 3, p < 0.03; in NMDA/100 μm glycine, from 1.17 ± 0.25 to 0.86 ± 0.15, n = 5, p < 0.04). The percentage failure rate also changed from 30 ± 8 to 19 ± 6% (p < 0.005), and the coefficient of variation (1/CV2) increased after NMDAR activation by NMDA/glycine (r = 0.83; p < 0.003) (Fig. 4B). These results indicate that an increase in the presynaptic release of GABA has occurred. The fact that NMDA alone failed to alter the PPR, failure rate, and 1/CV2 suggests that the NMDAR-mediated increase in the presynaptic release of GABA requires glycine.
Finally, consistent with a presynaptic expression of the NMDAR-mediated I-LTP, the potentiation of aIPSCs by AMPA application was accompanied by a decrease in the PPR from 0.84 ± 0.19 to 0.38 ± 0.11 (n = 7; p < 0.03), which was prevented by NMDAR blockers (Fig. 4C). As illustrated in Figure 4D, the ratio of 1/CV2 increased as the AMPA-induced enhancement of current amplitude became more pronounced (r = 0.78; p < 0.003). Thus, the site of expression of the AMPA-induced I-LTP also appears to be presynaptic.
A modulation of GABA release by glutamate is developmentally regulated
One of the predictions of the presynaptic expression of I-LTP is that the potentiation is more likely to be induced in stellate cells that have an initially low release probability. To address this point, the long-term change in the aIPSC amplitude was plotted against the associated PPR before the activation of NMDARs. As predicted, the degree of potentiation strongly correlates with the initial PPR (r = 0.7; p < 0.006) (Fig. 4E). That is, there is a greater potentiation of aIPSCs in cells that had a high initial PPR (low release probability).
The initial PPR was higher in P18–P19 mice (1.12 ± 0.19; n = 9) than younger mice (P14–P17, 0.5 ± 0.1; n = 6; p < 0.03). Thus, the probability of GABA release from stellate cells declines as the neurons become more mature. Is I-LTP more likely to be induced in mature neurons than in immature neurons? As expected, the potentiation occurs more frequently and becomes larger in magnitude in older animals (r = 0.74; p < 0.002) (Fig. 4F).
The NMDAR-induced sustained increase in miniature IPSC frequency requires presynaptic Ca2+ entry
To assess the extent of potentiation of presynaptic GABA release and possible alterations in the responsiveness of postsynaptic GABAA receptors, mIPSCs were recorded from a stellate cell before and after coapplication of NMDA and glycine. Because stellate cells form inhibitory synapses onto each other, an enhancement in the frequency of mIPSCs would indicate an increase in the spontaneous release of GABA, whereas a change in the amplitude of mIPSCs would reflect a postsynaptic modification. Spontaneous synaptic currents in the presence of 5 μm NBQX and 0.3 μm TTX at −70 mV were blocked by SR-95531 (supplemental Fig. 2, available at www.jneurosci.org as supplemental material) and thus mediated by GABAA receptors. As shown in Figure 5, A–C and G, NMDA/glycine application induced a long-lasting increase in mIPSC frequency by ∼77% (n = 12; p < 0.03) without a change in the amplitude of mIPSCs. This is consistent with the idea that I-LTP is associated with an increase in spontaneous GABA release, and is not mediated by a postsynaptic mechanism. However, the possibility that NMDAR activation may increase the number of functional inhibitory synapses (Kilman et al., 2002) cannot be ruled out.
Figure 5.

Ca entry into presynaptic terminals during NMDAR activation triggers I-LTP. A, mIPSCs before and after application of NMDA/glycine. B, Plot of percentage change in mean mIPSC frequency versus time (n = 12 without CPP; n = 6 with CPP). C, Plot of percentage change in mean mIPSC amplitude versus time (n = 12; except for the first two data points, n = 2). D, A brief application of NMDA alone induced a transient but not sustained increase in mIPSC frequency (n = 5). E, Application of NMDA/glycine in a Ca2+-free ACSF did not induce a lasting change in mIPSC frequency (n = 8). F, Inclusion of 10 or 20 mm BAPTA in the pipette solution did not prevent NMDAR-induced increase in mIPSC frequency (n = 8). G, Percentage change in mean mIPSC frequency of individual cells (open triangles; BAPTA: 10 mm, open triangles, and 20 mm, filled triangles) and average data (filled circles; *p < 0.02). Error bars indicate SEM.
The NMDA/glycine-induced increase in mIPSC frequency was completely blocked by 10 μm CPP, an NMDAR blocker (Fig. 5B). Thus, the activation of NMDARs is required. Furthermore, glycine is necessary for the I-LTP induction because NMDA alone failed to induce the potentiation (Fig. 5D).
To address the question of whether NMDAR-induced I-LTP requires Ca2+ entry, we applied NMDA and glycine in a Ca2+-free ACSF (0 mm Ca, 200 μm EGTA, and 2 mm MgCl2). Under these conditions, NMDA/glycine failed to induce a lasting increase in mIPSC frequency (Fig. 5E,G). Thus, Ca2+ entry is needed for the I-LTP induction.
NMDARs are present extrasynaptically in the postsynaptic stellate cell and at the axon terminals of presynaptic cells (Carter and Regehr, 2000; Clark and Cull-Candy, 2002; Duguid and Smart, 2004). If I-LTP induction requires activation of presynaptic NMDARs, preventing a Ca2+ increase in the postsynaptic cell is not expected to block the NMDAR-induced I-LTP. Indeed, when 10 or 20 mm BAPTA was included in the pipette solution to buffer the Ca2+ rise in the postsynaptic cell, NMDA/glycine application still induced a sustained increase in mIPSC frequency (n = 8; p < 0.001) (Fig. 5F,G). The fact that BAPTA in the postsynaptic cell failed to abolish I-LTP indicates that Ca2+ entry via NMDARs in the postsynaptic cell is not necessary for the presynaptic I-LTP. This is consistent with the idea that activation of presynaptic NMDARs is sufficient to induce I-LTP.
I-LTP can be induced at a physiological temperature
The activity of glutamate transporters increases with temperature (Asztely et al., 1997). Thus, we tested whether I-LTP can also be induced at a physiological-like temperature. First, we measured spontaneous IPSCs at various holding potentials at 32°C and found that the reversal potential for Cl− in stellate cells was 81 ± 6 mV (n = 6), consistent with a previous report (Carter and Regehr, 2002).
Using a low Cl− pipette solution (which set the reversal potential for Cl− to −81 mV), we observed outward spontaneous synaptic currents in the presence of 5 μm NBQX and 0.3 μm TTX at −30 mV (Fig. 6A). These currents were blocked by 20 μm SR-95531 and are thus mediated by GABAA receptors. We then determined mIPSC frequency at 32°C before and after PF stimulation (Fig. 6B). The stimulation of PFs induced an NMDAR current (49 ± 12 pA; n = 5), which was blocked by 10 μm CPP (Fig. 6C). After PF stimulation, mIPSC frequency increased by 110 ± 27% (n = 5; p < 0.02) (Fig. 6D,E) without a change in mIPSC amplitude (Fig. 6F), indicating an increase in the spontaneous release of GABA via a presynaptic mechanism. This activity-induced potentiation of mIPSC frequency was prevented by the presence of 10 μm CPP during PF stimulation. Therefore, burst activity of PFs can activate NMDARs, and produces a lasting increase in the spontaneous release of GABA at a physiological-like temperature.
Figure 6.

PF stimulation at 32°C induces a lasting increase in the frequency of mIPSCs. A, Miniature synaptic currents recorded at −30 mV in the presence NBQX. B, Schematic diagram of the experimental protocol. C, Representative average current traces evoked by PF stimulation, without CPP (top two traces), before (third; the current was evoked by a single sweep of testing stimulation) and during the application of CPP. D, mIPSCs before and 30 min after PF stimulation. E, Plot of percentage change in mean mIPSC frequency versus time (n = 5). F, Plot of percentage change in mean mIPSC amplitude versus time (n = 5).
Discussion
Although many excitatory synapses show LTP, much less is known about the lasting potentiation of inhibitory transmission. In particular, what physiological transmitters trigger an enduring increase in the presynaptic release of inhibitory transmitters is far from clear. The present study examines this question and describes a novel type of long-term potentiation of inhibitory synaptic transmission that occurs in cerebellar stellate cells. It is unusual in that it is paradoxically induced by excitatory transmitters that transiently activate NMDARs and requires the presence of glycine. The evidence suggests that the lasting potentiation is induced and expressed presynaptically and that it involves an increase in the release of GABA. This modulation of the release of GABA by glutamate presumably allows fine control in the balance that exists between excitation and inhibition within a cerebellar circuit.
Curiously, although NMDA alone will transiently enhance the release of GABA from stellate/basket cells (Glitsch and Marty, 1999; Duguid and Smart, 2004), glycine, a coagonist for NMDARs, is required for the induction of the lasting increase in the presynaptic release of GABA and a sustained enhancement in mIPSC frequency. One possible mechanism for this requirement is that glycine potentiates NMDAR-mediated responses by reducing receptor desensitization (Kleckner and Dingledine, 1988) and enhancing Ca2+ entry through the receptors. Indeed, glycine binding to NMDARs has been shown to facilitate the induction of LTP at excitatory synapses (Martina et al., 2004). In the cerebellum, the extracellular glycine concentration is ∼20 mm (Matsui et al., 1995), at which concentration stimulation of PFs is sufficient to induce the I-LTP. This suggests that endogenous glutamate and glycine should be able to induce this form of plasticity in the cerebellum under physiological conditions. In addition, the discrepancy could also be attributable to the difference in the age of the animals. The present study shows that I-LTP is more likely to be induced in mature neurons, whereas Duguid and Smart (2004) used younger animals (P6–P14 rats) for their experiments.
NMDARs are found on the presynaptic stellate/basket cell terminals (Duguid and Smart, 2004; Fiszman et al., 2005). Because our results show that activation of NMDARs and Ca2+ entry into the presynaptic terminal (but not the postsynaptic cell) are required to induce the presynaptic I-LTP, the lasting increase in GABA release described in this study most likely involves the activation of these presynaptic NMDARs. However, the possibility that activation of extrasynaptic NMDARs on other stellate cells may trigger the release of retrograde signals, such as NO (Shin and Linden, 2005), and thus alter the release of GABA, cannot be ruled out. It would be of interest to determine whether activation of presynaptic NMDARs in other GABAergic neurons (Paquet and Smith, 2000) can also modulate the release of GABA.
What are the possible sources for the endogenous glycine? Both glutamate and glycine appear to be released during AMPA application, perhaps from Bergmann glial cells or cerebellar neurons. These transmitters then induced I-LTP by activating NMDARs. Glycine can be released from Bergmann glial cells via the glycine transporter when the membrane is depolarized (Huang et al., 2004). Ca2+ entry through Ca2+-permeable AMPARs during stimulation of parallel and climbing fibers (Grosche et al., 1999; Matsui and Jahr, 2003) is expected to depolarize the membrane potential (Muller et al., 1992) and stimulate the release of glycine from these cells. Glycine could also originate from Golgi and Lugaro cells (Zeilhofer et al., 2005).
Several lines of evidence strongly suggest that the type of I-LTP described in the present work is associated with an enduring increase in the release probability of GABA from stellate cells. First, the paired pulse ratio of aIPSCs decreases after the activation of NMDARs. Second, the failure rate of evoked autaptic currents is also reduced. Third, the potentiation of aIPSCs is accompanied by an increase in 1/(CV)2. Fourth, activation of NMDARs could only induce a lasting increase in GABA release from cells that had a low initial release probability. Fifth, the frequency of mIPSCs increases. The molecular mechanisms underlying presynaptic LTP at mossy fiber synapses in the hippocampus and at the parallel fiber to Purkinje cell synapses in the cerebellum involve Ca2+-induced Ca2+ release from intracellular stores, presynaptic cAMP-dependent protein kinase, phosphorylation of RIM1α by PKA and Rab (Weisskopf et al., 1994; Salin et al., 1996; Castillo et al., 1997, 2002; Storm et al., 1998; Linden and Ahn, 1999; Lauri et al., 2003; Lonart et al., 2003). Because PKA and Ca2+ release from intracellular stores are involved in triggering a transient increase in GABA release from stellate cells (Kondo and Marty, 1997; Duguid and Smart, 2004), it would be interesting to determine whether these molecular events are also responsible for the induction of presynaptic LTP at inhibitory synapses.
In conclusion, these results demonstrate that glutamate can directly enhance the release of an inhibitory transmitter, producing a lasting potentiation in the efficacy of inhibitory synaptic transmission. This mechanism may be particularly important in the cerebellar cortex where motor learning is associated with a reduction in the activity of Purkinje cells (Thompson, 1990). Simultaneously enhancing the release of an inhibitory transmitter from the presynaptic stellate/basket cells (this study) and increasing the postsynaptic responsiveness of the Purkinje cells to GABA (Kano et al., 1992), is likely to produce a powerful suppression of the activity of Purkinje cells. Thus, the lasting enhancement of the release of GABA from stellate/basket cells is positioned to play an important role in cerebellar motor learning.
Footnotes
This work was supported by National Science Foundation Grant IBN-0344559. We thank Richard Ordway and Matthew Whim for helpful discussions and comments on this manuscript.
References
- Asztely F, Erdemli G, Kullmann DM. Extrasynaptic glutamate spillover in the hippocampus: dependence on temperature and the role of active glutamate uptake. Neuron. 1997;18:281–293. doi: 10.1016/s0896-6273(00)80268-8. [DOI] [PubMed] [Google Scholar]
- Bacci A, Huguenard JR. Enhancement of spike-timing precision by autaptic transmission in neocortical inhibitory interneurons. Neuron. 2006;49:119–130. doi: 10.1016/j.neuron.2005.12.014. [DOI] [PubMed] [Google Scholar]
- Belan PV, Kostyuk PG. Glutamate-receptor-induced modulation of GABAergic synaptic transmission in the hippocampus. Pflügers Arch. 2002;444:26–37. doi: 10.1007/s00424-002-0783-3. [DOI] [PubMed] [Google Scholar]
- Carter AG, Regehr WG. Prolonged synaptic currents and glutamate spillover at the parallel fiber to stellate cell synapse. J Neurosci. 2000;20:4423–4434. doi: 10.1523/JNEUROSCI.20-12-04423.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter AG, Regehr WG. Quantal events shape cerebellar interneuron firing. Nat Neurosci. 2002;5:1309–1318. doi: 10.1038/nn970. [DOI] [PubMed] [Google Scholar]
- Castillo PE, Janz R, Sudhof TC, Tzounopoulos T, Malenka RC, Nicoll RA. Rab3A is essential for mossy fibre long-term potentiation in the hippocampus. Nature. 1997;388:590–593. doi: 10.1038/41574. [DOI] [PubMed] [Google Scholar]
- Castillo PE, Schoch S, Schmitz F, Sudhof TC, Malenka RC. RIM1α is required for presynaptic long-term potentiation. Nature. 2002;415:327–330. doi: 10.1038/415327a. [DOI] [PubMed] [Google Scholar]
- Chadderton P, Margrie TW, Hausser M. Integration of quanta in cerebellar granule cells during sensory processing. Nature. 2004;428:856–860. doi: 10.1038/nature02442. [DOI] [PubMed] [Google Scholar]
- Chevaleyre V, Castillo PE. Heterosynaptic LTD of hippocampal GABAergic synapses: a novel role of endocannabinoids in regulating excitability. Neuron. 2003;38:461–472. doi: 10.1016/s0896-6273(03)00235-6. [DOI] [PubMed] [Google Scholar]
- Clark BA, Cull-Candy SG. Activity-dependent recruitment of extrasynaptic NMDA receptor activation at an AMPA receptor-only synapse. J Neurosci. 2002;22:4428–4436. doi: 10.1523/JNEUROSCI.22-11-04428.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duguid IC, Smart TG. Retrograde activation of presynaptic NMDA receptors enhances GABA release at cerebellar interneuron-Purkinje cell synapses. Nat Neurosci. 2004;7:525–533. doi: 10.1038/nn1227. [DOI] [PubMed] [Google Scholar]
- Engelman HS, MacDermott AB. Presynaptic ionotropic receptors and control of transmitter release. Nat Rev Neurosci. 2004;5:135–145. doi: 10.1038/nrn1297. [DOI] [PubMed] [Google Scholar]
- Fiszman ML, Barberis A, Lu C, Fu Z, Erdelyi F, Szabo G, Vicini S. NMDA receptors increase the size of GABAergic terminals and enhance GABA release. J Neurosci. 2005;25:2024–2031. doi: 10.1523/JNEUROSCI.4980-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaiarsa JL, Caillard O, Ben-Ari Y. Long-term plasticity at GABAergic and glycinergic synapses: mechanisms and functional significance. Trends Neurosci. 2002;25:564–570. doi: 10.1016/s0166-2236(02)02269-5. [DOI] [PubMed] [Google Scholar]
- Glitsch M, Marty A. Presynaptic effects of NMDA in cerebellar Purkinje cells and interneurons. J Neurosci. 1999;19:511–519. doi: 10.1523/JNEUROSCI.19-02-00511.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosche J, Matyash V, Moller T, Verkhratsky A, Reichenbach A, Kettenmann H. Microdomains for neuron-glia interaction: parallel fiber signaling to Bergmann glial cells. Nat Neurosci. 1999;2:139–143. doi: 10.1038/5692. [DOI] [PubMed] [Google Scholar]
- Hausser M, Clark BA. Tonic synaptic inhibition modulates neuronal output pattern and spatiotemporal synaptic integration. Neuron. 1997;19:665–678. doi: 10.1016/s0896-6273(00)80379-7. [DOI] [PubMed] [Google Scholar]
- Huang H, Barakat L, Wang D, Bordey A. Bergmann glial GlyT1 mediates glycine uptake and release in mouse cerebellar slices. J Physiol (Lond) 2004;560:721–736. doi: 10.1113/jphysiol.2004.067801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kano M, Rexhausen U, Dreessen J, Konnerth A. Synaptic excitation produces a long-lasting rebound potentiation of inhibitory synaptic signals in cerebellar Purkinje cells. Nature. 1992;356:601–604. doi: 10.1038/356601a0. [DOI] [PubMed] [Google Scholar]
- Kilman V, van Rossum MC, Turrigiano GG. Activity deprivation reduces miniature IPSC amplitude by decreasing the number of postsynaptic GABAA receptors clustered at neocortical synapses. J Neurosci. 2002;22:1328–1337. doi: 10.1523/JNEUROSCI.22-04-01328.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleckner NW, Dingledine R. Requirement for glycine in activation of NMDA-receptors expressed in Xenopus oocytes. Science. 1988;241:835–837. doi: 10.1126/science.2841759. [DOI] [PubMed] [Google Scholar]
- Kondo S, Marty A. Protein kinase A-mediated enhancement of miniature IPSC frequency by noradrenaline in rat cerebellar stellate cells. J Physiol (Lond) 1997;498:165–176. doi: 10.1113/jphysiol.1997.sp021849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauri SE, Bortolotto ZA, Nistico R, Bleakman D, Ornstein PL, Lodge D, Isaac JT, Collingridge GL. A role for Ca2+ stores in kainate receptor-dependent synaptic facilitation and LTP at mossy fiber synapses in the hippocampus. Neuron. 2003;39:327–341. doi: 10.1016/s0896-6273(03)00369-6. [DOI] [PubMed] [Google Scholar]
- Linden DJ, Ahn S. Activation of presynaptic cAMP-dependent protein kinase is required for induction of cerebellar long-term potentiation. J Neurosci. 1999;19:10221–10227. doi: 10.1523/JNEUROSCI.19-23-10221.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SJ, Cull-Candy SG. Subunit interaction with PICK and GRIP controls Ca2+ permeability of AMPARs at cerebellar synapses. Nat Neurosci. 2005;8:768–775. doi: 10.1038/nn1468. [DOI] [PubMed] [Google Scholar]
- Liu SQ, Cull-Candy SG. Synaptic activity at calcium-permeable AMPA receptors induces a switch in receptor subtype. Nature. 2000;405:454–458. doi: 10.1038/35013064. [DOI] [PubMed] [Google Scholar]
- Lonart G, Schoch S, Kaeser PS, Larkin CJ, Sudhof TC, Linden DJ. Phosphorylation of RIM1α by PKA triggers presynaptic long-term potentiation at cerebellar parallel fiber synapses. Cell. 2003;115:49–60. doi: 10.1016/s0092-8674(03)00727-x. [DOI] [PubMed] [Google Scholar]
- Martina M, Gorfinkel Y, Halman S, Lowe JA, Periyalwar P, Schmidt CJ, Bergeron R. Glycine transporter type 1 blockade changes NMDA receptor-mediated responses and LTP in hippocampal CA1 pyramidal cells by altering extracellular glycine levels. J Physiol (Lond) 2004;557:489–500. doi: 10.1113/jphysiol.2004.063321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui K, Jahr CE. Ectopic release of synaptic vesicles. Neuron. 2003;40:1173–1183. doi: 10.1016/s0896-6273(03)00788-8. [DOI] [PubMed] [Google Scholar]
- Matsui T, Sekiguchi M, Hashimoto A, Tomita U, Nishikawa T, Wada K. Functional comparison of d-serine and glycine in rodents: the effect on cloned NMDA receptors and the extracellular concentration. J Neurochem. 1995;65:454–458. doi: 10.1046/j.1471-4159.1995.65010454.x. [DOI] [PubMed] [Google Scholar]
- Muller T, Moller T, Berger T, Schnitzer J, Kettenmann H. Calcium entry through kainate receptors and resulting potassium-channel blockade in Bergmann glial cells. Science. 1992;256:1563–1566. doi: 10.1126/science.1317969. [DOI] [PubMed] [Google Scholar]
- Paquet M, Smith Y. Presynaptic NMDA receptor subunit immunoreactivity in GABAergic terminals in rat brain. J Comp Neurol. 2000;423:330–347. doi: 10.1002/1096-9861(20000724)423:2<330::aid-cne10>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- Pouzat C, Marty A. Autaptic inhibitory currents recorded from interneurones in rat cerebellar slices. J Physiol (Lond) 1998;509:777–783. doi: 10.1111/j.1469-7793.1998.777bm.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salin PA, Malenka RC, Nicoll RA. Cyclic AMP mediates a presynaptic form of LTP at cerebellar parallel fiber synapses. Neuron. 1996;16:797–803. doi: 10.1016/s0896-6273(00)80099-9. [DOI] [PubMed] [Google Scholar]
- Shin JH, Linden DJ. An NMDA receptor/nitric oxide cascade is involved in cerebellar LTD but is not localized to the parallel fiber terminal. J Neurophysiol. 2005;94:4281–4289. doi: 10.1152/jn.00661.2005. [DOI] [PubMed] [Google Scholar]
- Storm DR, Hansel C, Hacker B, Parent A, Linden DJ. Impaired cerebellar long-term potentiation in type I adenylyl cyclase mutant mice. Neuron. 1998;20:1199–1210. doi: 10.1016/s0896-6273(00)80500-0. [DOI] [PubMed] [Google Scholar]
- Thompson RF. Neural mechanisms of classical conditioning in mammals. Philos Trans R Soc Lond B Biol Sci. 1990;329:161–170. doi: 10.1098/rstb.1990.0161. [DOI] [PubMed] [Google Scholar]
- Weisskopf MG, Castillo PE, Zalutsky RA, Nicoll RA. Mediation of hippocampal mossy fiber long-term potentiation by cyclic AMP. Science. 1994;265:1878–1882. doi: 10.1126/science.7916482. [DOI] [PubMed] [Google Scholar]
- Zeilhofer HU, Studler B, Arabadzisz D, Schweizer C, Ahmadi S, Layh B, Bosl MR, Fritschy JM. Glycinergic neurons expressing enhanced green fluorescent protein in bacterial artificial chromosome transgenic mice. J Comp Neurol. 2005;482:123–141. doi: 10.1002/cne.20349. [DOI] [PubMed] [Google Scholar]