

Abstract
The death of motor neurons in amyotrophic lateral sclerosis (ALS) is thought to result from the interaction of a variety of factors including excitotoxicity, accumulation of toxic proteins, and abnormal axonal transport. Previously, we found that the susceptibility of motor neurons to excitotoxic insults can be limited by inhibiting signals evoked by brain-derived neurotrophic factor (BDNF) activation of the receptor tyrosine kinase B (TrkB). Here we show that this can be achieved by direct kinase inhibition or by blockade of a transactivation pathway that uses adenosine A2a receptors and src-family kinases (SFKs). Downstream signaling cascades (such as mitogen-activated protein kinase and phosphatidylinositol-3 kinase) are inhibited by these blockers. In addition to protecting motor neurons from excitotoxic insult, these agents also prevent toxicity that follows from the expression of mutant proteins (G85R superoxide dismutase 1; G59S p150glued) that cause familial motor neuron disease. TrkB, adenosine A2a receptors, and SFKs associate into complexes in lipid raft and nonlipid raft membranes and the signaling from lipids rafts may be particularly important because their disruption by cholesterol depletion blocks the ability of BDNF to render motor neurons vulnerable to insult. The neuroprotective versatility of Trk antagonism suggests that it may have broad utility in the treatment of ALS patients.
Keywords: adenosine A2a receptor, brain-derived neurotrophic factor, Trk receptor, transactivation, amyotrophic lateral sclerosis, motoneuron
Introduction
Although the majority of cases of amyotrophic lateral sclerosis (ALS) are sporadic, motor neuron disease can also result from mutation of single genes (i.e., Cu2+/Zn2+ superoxide dismutase 1, p150glued subunit of dynactin) (Bruijn et al., 2004). One of the most effective ways to slow the progression of motor neuron death in model systems of ALS is to modulate excitatory glutamatergic neurotransmission (Rothstein et al., 1993, 2005; Estevez et al., 1995; Kwak and Nakamura, 1995; Carriedo et al., 1996; Roy et al., 1998; Kruman et al., 1999; Van Damme et al., 2003).
Previous work demonstrated that the vulnerability of motor neurons to excitotoxic insult in vitro is promoted by the peptide growth factor BDNF and is mediated by the receptor tyrosine kinase B (TrkB) (Fryer et al., 2000; Hu and Kalb, 2003). Although this is counter-intuitive, because BDNF-TrkB signaling is essential for survival of populations of developing neurons (Jones et al., 1994; Ernfors et al., 1995; Liu et al., 1995; Schwartz et al., 1997; Silos-Santiago et al., 1997), an additional prodeath activity of BDNF has also been observed in a variety of in vitro stressed-neuron paradigms (Koh et al., 1995; Ishikawa et al., 2000; Kim et al., 2003). These antipodal actions may be related to the complexity of BDNF–TrkB signaling and the maturity of the neurons under study (Kalb, 2005).
Two sets of in vivo observations provide additional support for the notion that BDNF might have an adverse action in ALS. First, a trial of recombinant BDNF treatment for ALS was conducted from February, 1998, to December, 1999 (BDNF 970278; French Centres, V. Meininger, unpublished observations). Patients were randomized to receive intrathecal BDNF, 25 μg/d or 150 μg/d, or vehicle for 18 months. The primary endpoint was survival free of ventilatory support. In the two active treatment groups, the endpoint was reached faster than in the placebo group with a dose effect (worse in the 150 μg/d vs the 25 μg/d group). Because of the clinical course of subjects in the active treatment groups versus controls, the study was discontinued before the log rank tests reached statistical significance. Second, in a study of the expression of neurotrophin family trophic factors, Küst et al. (2002) found a selective increase in the abundance of BDNF message and protein in muscle of ALS patients and this was most pronounced early in the disease. This collection of in vitro and in vivo observations raises the possibility that pharmacological manipulations that inhibit TrkB signaling might be motor neuron protective.
In addition to the canonical pathway of TrkB activation by extracellular BDNF, Trk receptors can be transactivated by the adenosine A2a G-protein-coupled receptor (GPCR) (Lee and Chao, 2001; Lee et al., 2002a,b). We inquired whether manipulation of adenosine A2a receptor activation influenced Trk signaling in spinal cord neurons and if this provided neuroprotection against excitotoxic injury as well as the baleful actions of mutant proteins shown previously to cause familial forms of motor neuron disease.
Materials and Methods
Sources of reagents.
CEP4416 and CEP701 were obtained from Cephalon (Frazer, PA), insulin-like growth factor 1 (IGF1) was from Bachem (King of Prussia, PA), CM-DiI was from Invitrogen (Eugene, OR), SQ 22536 was from Biomol (Plymouth Meeting, PA), H89 was from CalBiochem (La Jolla, CA), and PP1 was from Pfizer (Groton, CT). All other chemicals were obtained from Sigma (St. Louis, MO) and were of the highest possible grade. The source of tissue culture materials has been described previously (Hu and Kalb, 2003). KW6002 was a gift from Jacques Petzer (University of Witwatersrand, Johannesburg, South Africa).
Preparation of spinal cord cultures.
Embryonic Sprague Dawley rat spinal cord neurons were grown on previously established cortical astrocyte monolayers as described previously (Hu and Kalb, 2003). Briefly, the cerebral cortex of postnatal day 1–3 (P1–P3) rat pups was dissociated and plated on tissue culture plastic (Primaria, Falcon, Becton Dickinson) or laminin/polylysine-coated glass coverslips and maintained in minimal essential media (10% horse serum, 10% fetal bovine serum) and 10 μm AraC was added for 1 d to arrest additional proliferation when confluent. Under these conditions, no cortical neurons are present in these cultures. Spinal cord from embryonic day 15 (E15) rats was dissociated and plated on the astrocytes at a density of 1 spinal cord/4 ml of media conditioned previously over astrocytes with 10 ng/ml of trophic factors added [ciliary neurotrophic factor, cardiotrophin-1 (CT1), BDNF, neurotrophin 4, and glial-derived neurotrophic factor, all from Alomone Labs (Jerusalem, Israel)]. Unless otherwise specified, BDNF was added to all cultures. Approximately 50% of media was replaced every third day with fresh media until cultures were used after 14 d in vitro (DIV) for biochemistry or toxicity assays. Purified motor neuron cultures were prepared as described previously using metrizamide gradient and immunopanning techniques (Fryer et al., 1999, 2000). Dye labeling of motor neurons was accomplished by incubating purified motor neurons with chloromethyl DiI (2 μm) for 5 min at 37°C, then 20 min at 4°C, washing three times in culture media, and adding these cells to the dissociated nonmotor neuron spinal cord neurons at the time of plating.
In studies using CEP4416 and CEP701, the vehicle solution to which neurons was exposed contained dimethyl sulfoxide (DMSO) at a final concentration of 0.2%. A recent study showed that DMSO at concentrations as low as 0.02% can potentiate NMDA and non-NMDA excitatory postsynaptic potentials (Tsvyetlynska et al., 2005). To control for any confounding effect of DMSO, all experiments included controls exposed to vehicle with a final DMSO concentration of 0.2%.
Live labeling of cell surface TrkB.
Twenty-fours hours after plating, chicken anti-N-terminal TrkB was added to purified motor neuron cultures with a final dilution of 1:500. After 1 h at 37°C, cultures were washed twice in room temperature PBS, fixed in 4% paraformaldehyde for 20 min, and then washed three times in PBS. Identical results were obtained by incubating with the anti-TrkB antibody at 4°C.
Western blots and immunoprecipitation.
Cells were lysed in 1% Nonidet P-40 (NP-40) lysis buffers containing protease inhibitors, subjected to SDS-PAGE, and immunoblotted as described previously (Fryer et al., 1999, 2000). For immunoprecipitation, lysates from 60 mm dishes were precleared by incubation with protein A beads (Santa Cruz Biotechnology, Santa Cruz, CA), incubated with primary antibody (4 μg/ml) for 2 h, and then 50 μl of beads were added and incubated overnight with gentle agitation at 4°C. Beads were pelleted, washed five times with 10 mm Tris, pH 7.4, plus 0.5 mm EGTA plus 0.5 mm EDTA plus 0.1% NP-40 before elution in 1× Laemmeli sample buffer. Antibodies were from the following sources: TrkB and mitogen-activated protein kinase (MAPK) used at 1:500, Transduction Laboratories (Lexington, KY); Akt 1:1000, phosphoTrk (tyr490) 1:1000, phospho-p42/44 MAP Kinase (thr 202/tyr204) 1:1000, phosphoAkt (ser473) 1:500, and IGF1 receptor antibody (tyr 1131) 1:500, Cell Signaling (Danvers, MA); adenosine A2a 1:1000, Millipore (Bedford, MA); actin 1:200, Sigma; active src (clone 28) 1:200, Biosource (Camarillo, CA).
Preparation of lipid rafts.
We used previously published methods for the isolation of lipid rafts (Kawabuchi et al., 2000; Suzuki et al., 2004). Briefly, neonatal rat spinal cords (0.25 g) were homogenized in Triton X-100 lysis buffer (50 mm Tris-HCl, pH 8.0, 10 mm MgCl2, 0.15 mm NaCl, 1% Triton X, 5% glycerol, 20 mm NaF, 1 mm Na3VO4, 5 mm β-mercaptoethanol, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 1 mm PMSF) at a 8:1 ratio of buffer to tissue. Two milliliters of lysate were combined with 2 ml of 80% sucrose in buffer A and layered over 5 ml of 30% sucrose and 1 ml of 5% sucrose (buffer A: 500 mm NaCl, 10 mm MgCl2, 50 mm Tris-HCl, pH 7.5, 1 mm Na3VO4). Using an SW-40 rotor, the samples were spun 12 h at 200,000 × g. Ten 1 ml samples were removed from the top and labeled 1–10 and used in subsequent analysis.
Excitotoxicity assays.
The media was removed (and saved), cells were exposed to 100 μm kainic acid or vehicle in Locke’s buffer for 1 h, washed 3× in Locke’s buffer not containing kainic acid, and the original media restored to each dish (Hu and Kalb, 2003). Twenty-four hours later, dishes were fixed in 4% paraformaldehyde, washed extensively, and immunostained. Coverslips were incubated with primary antibody overnight, washed, and incubated with species-specific secondary antibodies. When double labeling using two spectrally distinct chromophores, control incubations omitting either primary antibody were used to confirm staining specificity. To quantify the number of stained cells, we counted the number of labeled cells in three nonoverlapping fields using a 5× objective on a Zeiss (Oberkochen, Germany) upright Axioskop microscope and the value from one coverslip was averaged. The results from 3–5 independently treated coverslips were the basis of the means and variance used in subsequent statistical analysis. When cell body diameter was assessed in select experiments, a reticule was introduced into an eye piece and the length of the long axis of a neuron was noted. Absolute values were made using a micrometer calibration slide (Swift Optics, San Jose, CA). Antibodies were from the following sources: SMI-32 1:1000, Sternberger Monoclonals (Lutherville, MD); choline acetyl transferase (ChAT) 1:500, peripherin 1:200, Millipore; islet 1/2, undiluted 4D5 supernatant, Developmental Studies Hybridoma Bank (Iowa City, IA). Alexa 488 and 594 conjugated species-specific secondary antibodies were obtained from Invitrogen and used at 1:200.
Determination of glutamate concentration.
Amino acids were derivatized with fluoraldehyde o-phthaldehyde (Pierce, Rockford, IL) and subjected to liquid chromatography (Varicon 9010, Varian Analytical Instruments, Lake Forest, CA).
Determination of cholesterol content.
Two hundred thousand motor neurons were plated in wells of a 96-well dish and after 24 h treated with β-methylcyclodextrin (βMCD) or vehicle. Thirty minutes later, cholesterol content was determined using the Amplex Red Cholesterol Assay kit (Invitrogen) according to the manufacturers instructions. All determinations were made in quintuplicate and normalized to protein content (determined using Protein Assay kit; Bio-Rad, Hercules, CA).
Determination of BDNF concentration.
We measured the concentration of BDNF in culture media using ELISA (BDNF Emax immunoassay; Promega, Madison, WI) according to the manufacturers instructions.
Recombinant HSV.
Various cDNAs were cloned into the PrpUC amplicon plasmid and were used to generate recombinant herpes simplex viruses (HSVs) as described previously (Neve et al., 1997). The titer of viruses used in these studies were routinely 3–5 ×107 plaque-forming units/ml. The cDNA for wild-type (WT)/mutant superoxide dismutase (SOD) and wild-type/mutant p150glued were gifts from D. Borchelt (University of Florida) and E. Holzbaur (University of Pennsylvania), respectively. The K295R mutant version of chick src was a gift from J. Brugge (Harvard University).
Cell imaging.
Images of motor neurons were obtained on an Olympus (Tokyo, Japan) BX51 microscope equipped with an FV300 laser confocal optical microscope using the Fluoview version 4.3 software. Images were obtained with a 40× 1.00 numerical aperture oil UPlan Apo objective at room temperature. Abode (San Jose, CA) Photoshop CS version 8.0 for Apple computers was used to crop and assemble images into figures, but no alterations in contrast, brightness, hue or gamma setting were undertaken.
Statistical analysis.
The results from at least four independent cell survival or biochemistry experiments are reported here. Every observation within any individual cell survival experiment was obtained in triplicate. Means and SDs are reported.
When two groups of observations were compared, Student’s t test was used; when three or more groups of observations were compared, ANOVA was used. Repeated-measures ANOVA (RMANOVA) was used to examine the association between the transgene expressed (wild-type and mutant forms of SOD and p150glued) and the number of surviving motor neurons over time.
Results
Many previous in vitro investigations have identified motor neurons in mixed spinal cord cell culture by immunostaining for nonphosphorylated neurofilaments (or peripherin) and restricting analysis to cells with a soma diameter of 25 μm or greater (Carriedo et al., 2000; Hu and Kalb, 2003). We wanted to confirm the validity of this approach using an independent method for determining whether a given cell is a motor neuron. To specifically identify motor neurons in our cultures, we purified these cells by metrizamide gradient and immunopanning techniques (Henderson et al., 1993; Fryer et al., 1999), live-labeled them with CM-DiI, and added these fluorescently tagged, bona fide motor neurons to mixed cell cultures at the time of initial plating. All CM-DiI-labeled cells stained for motor neuron markers such as nonphosphorylated neurofilament H (using antibody, SMI-32), peripherin, ChAT, and islet 1/2 (Fig. 1a). We then subjected spinal cord cultures to an excitotoxic insult and determined motor neuron survival using multiple methods for identifying motor neurons. Regardless of how we quantify motor neuron number (immunostaining for SMI-32, peripherin, ChAT, or CM-DiI), we found that an excitotoxic insult led to the loss of ∼50% of motor neurons (Fig. 1b). Because all SMI-32(+) cells with a cell body diameter of 25 μm or greater were CM-DiI(+) and all CM-DiI(+) cells were SMI-32(+), for the remainder of the studies herein, we could confidently study motor neuron death in vitro by counting SMI-32(+) cells with a cell body diameter of 25 μm or greater.
Figure 1.
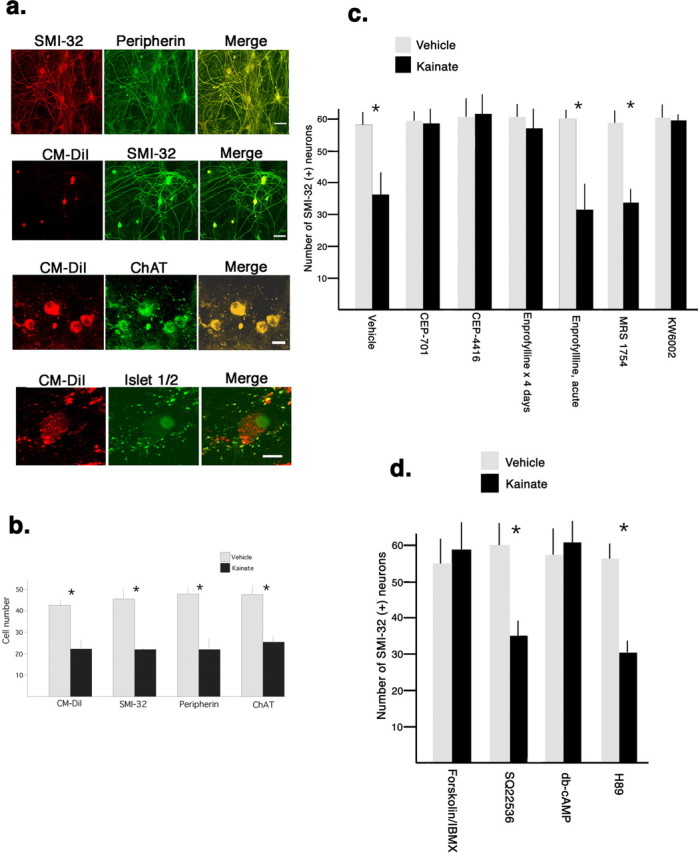
Chronic but not acute Adenosine A2a receptor antagonism protects motor neurons from excitotoxic insult in a manner not mimicked by manipulating cAMP levels. a, Immunochemical methods were used to identify motor neurons in mixed spinal cord cultures. Two previously used markers of motor neurons (Carriedo et al., 1996), nonphosphorylated neurofilament H and peripherin, stained a subset of neurons in spinal cord cultures with large cell bodies and extensive neurites. Virtually all (>98 ± 1%) SMI-32(+) neurons were peripherin(+) and vice versa (merge image). Motor neurons, purified by metrizamide gradient and immunopanning, were live labeled by incubation with CM-DiI and washed motor neurons were plated with the remaining population of spinal cord neurons. Immunostaining of cultures revealed that 100% of CM-DiI labeled cells were immunostained with SMI-32(+), anti-choline acetyl transferase, and anti-islet 1/2. CM-DiI labeled cells are seen with conventional rhodamine optics, immunostaining was seen with fluorescein optics, and the merged image demonstrates colocalization. Scale bars: (from top to bottom): 50 μm, 50 μm, 20 μm, 10 μm. b, After 14–16 DIV, the excitotoxicity assay was performed: spinal cord neurons were exposed to 100 μm kainic acid (kainate) or vehicle (saline) for 1 h in Locke’s Buffer, washed, and 24 h later the number of CM-DiI-labeled cells or cells immunostained with SMI-32, peripherin, or ChAT, and cell body diameter >20 μm was determined. Kainate treatment caused the loss of ∼45% of motor neurons regardless of the method of cell identification (Student’s t test, *p < 0.01). c, After 14 DIV, cultures were exposed to CEP701, CEP4416, enprofylline, MRS1754, or KW6002 for 4 d before the excitotoxicity assay. Acute exposure to enprofylline was also studied. Excitotoxic insult lead to a loss of ∼40% of SMI(+) neurons in cultures treated with vehicle treated, MRS1754, or acute enprofylline treated cultures, whereas 4 d exposure to CEP701, CEP4416, enprofylline, or KW6002 completely blocked the loss of SMI(+) neurons (Student’s t test, *p < 0.01). d, Fourteen DIV cultures were exposed to forskolin/IBMX, SQ 22536, db-cAMP, or H89 for 4 d before the excitotoxicity assay. No excitotoxic cell death occurred in forskolin/IBMX or db-cAMP treated cultures whereas ∼40% of SMI(+) neurons were killed in cultures treated with SQ 22536 or H89 (Student’s t test, *p < 0.01). Error bars indicate SE.
In previous work (using function blocking anti-TrkB antibodies or by expressing a dominant-negative TrkB construct), we showed that antagonism of TrkB activation protected motor neurons from excitotoxic insult (Hu and Kalb, 2003). To be able to translate this work into therapeutics, we wanted to identify small molecules that either directly or indirectly inhibit TrkB activation. We examined the neuroprotective efficacy of derivatives of the Trk antagonist K252a that have in vivo activity and have been used in humans (Evans et al., 1999, 2001; Smith et al., 2004). CEP4416 (30 nm), CEP701 (30 nm), or vehicle (0.2% DMSO) was added to spinal cord cultures 4 and 2 d before an excitotoxic insult with Kainic acid (100 μm). No motor neuron death occurred in cultures that were treated with the Trk antagonists whereas ∼50% of motor neurons were killed in vehicle pretreated cultures (Fig. 1c). Thus, direct pharmacological inhibition of Trk activation protects motor neurons from excitotoxic insult.
Stimulation of adenosine A2a receptors causes Trk receptor activation (monitored by receptor phosphorylation and increases in downstream signaling), a process referred to as transactivation (Lee and Chao, 2001; Rajagopal et al., 2004). Because adenosine receptors are typically tonically active in vitro [because of ambient adenosine in the serum of growth media and the efficient generation of adenosine from extracellular ATP by exonucleases (Hirschhorn et al., 1981; Dunwiddie et al., 1997)], we asked whether they were transactivating TrkB in our culture system and contributing to the TrkB-induced vulnerability of motor neurons to excitotoxic insult. One attraction of adenosine receptor antagonists is that they are small molecules in clinical use for human disease (Kase et al., 2003). We began to study this issue by treating spinal cord cultures (grown in the presence of a cocktail of trophic factors including 10 ng/ml BDNF) 4 and 2 d before excitotoxic insult with enprofylline. This antagonist of adenosine A1, A2a, and A2b receptors (relative potency A2b > A2a ≫ A1) has good bioavailability, is commercially available, and is 10–100 times less expensive than other adenosine receptor antagonists (Robeva et al., 1996). We also studied the selective antagonist of A2a receptors (KW6002, 1 μm) and A2b receptors (MRS1754, 50 nm). There was no excitotoxic motor neuron death in cultures treated with enprofylline or KW6002 and ∼50% of motor neurons were killed in cultures pretreated with MRS1754 or vehicle (Fig. 1c). These results complement our previous observations in which we found that no excitotoxic cell death occurred in cultures pretreated with ZM241385 (A2a antagonist) but death occurred when cultures were pretreated with DPCPX (A1 antagonist) (Mojsilovic-Petrovic et al., 2005). Thus, three distinct compounds that antagonize adenosine A2a receptor were neuroprotective in this experimental paradigm. For most of the subsequent experiments we used enprofylline and KW6002.
The mechanism by which adenosine A2a receptors transactivate Trk receptors is not understood at a cell biological level. One possibility is that active adenosine A2a receptors stimulate the release of BDNF from cells, which then binds to and activates TrkB. The transactivation of vascular smooth muscle cell epidermal growth factor receptors by angiotensin II has been shown to operate in such an autocrine/paracrine loop (Eguchi et al., 2001). To see whether a similar phenomenon was occurring in our cultures, we measured the level of BDNF in the medium of cultures (not provided with exogenous BDNF) treated for two days with enprofylline or vehicle and found no statistically significant drug effect (enprofylline, 33 ± 1.2 pg/ml vs vehicle, 34 ± 4.1 pg/ml; n = 6; 2 separate experiments; Students t test, p = 0.724). These results argue against the notion that adenosine A2a receptor antagonism modulates TrkB signaling by diminishing the ambient levels of BDNF in our culture system.
Adenosine A2a receptors are known to have rapid modulatory actions on neurotransmission (Dunwiddie and Masino, 2001; Fredholm et al., 2001), raising the possibility that the neuroprotective action of enprofylline is mediated by influencing excitatory neurotransmission among neurons in our cultures. If so, brief pretreatment of cultures with enprofylline might also be neuroprotective. To test this, we grew cultures as described above and added enprofylline to the media 30 min before the excitotoxic challenge (Fig. 1c). We counted the number of motor neurons 24 h later and found that in this paradigm, enprofylline was not neuroprotective (percent motor neuron death in short term enprofylline group, −46 ± 4 vs percent motor neuron death in short term vehicle group, −43 ± 5; t test, p > 0.40). In addition, analysis of ambient glutamate concentrations in the media of cultures treated with enprofylline or vehicle for 24 h also revealed no differences between the groups (2.2 ± 0.7 vs 1.7 ± 0.4 μm; t test, p = 0.47). These findings argue against the idea that the neuroprotection afforded by A2a antagonism is mediated by rapid alteration of excitability by the drug or changes in extracellular concentration of glutamate.
Transactivation of some receptor tyrosine kinases by GPCRs occurs in a G-protein-dependent manner (Oak et al., 2001) and, thus, we wanted to examine this possibility in our system. Adenosine A2a receptors are GPCRs that, after ligand binding, lead to Gα loading with GTP, dissociation from Gβγ, and the activation of adenylate cyclase (Dunwiddie and Masino, 2001; Fredholm et al., 2001). Adenosine A2a receptor antagonists would be expected to reduce adenylate cyclase activation and result in a reduction in the intracellular level of cAMP. There is plausibility to the idea that reductions in cAMP levels might be neuroprotective because stress resistance in nondividing yeast Saccharomyces cerevisiae is enhanced by reductions in the CYR1 gene, an adenylate cyclase (Fabrizio et al., 2001). If neuroprotection was mediated by a reduction in cAMP levels, then agents that reduce cAMP levels (such as the adenylate cyclase inhibitor SQ 22536) or reduce the activity of cAMP-dependent kinase PKA (using H89) ought to mimic the neuroprotective actions of adenosine A2a receptor antagonism. Pretreatment of cultures with the adenylate cyclase inhibitor SQ 22536 (10 μm) for 4 d did not adversely affect the basal survival of motor neurons and did not protect against excitotoxic challenge (42 ± 4% cell death) (Fig. 1d). Similarly treating cultures with H89 (2 μm) did not protect against excitotoxic challenge (53 ± 5% cell death) (Fig. 1d). These results argue that the neuroprotective activity of adenosine A2a receptor antagonism is not mimicked by maneuvers that reduce cAMP levels or downstream signaling via PKA.
For two reasons we wanted to study the effects of raising cAMP levels. First, it would allow us to ask whether the neuroprotection afforded by adenosine A2a receptor antagonism is tied to its ability to reduce cAMP levels. If so, maneuvers that raise cAMP levels (i.e., IBMX plus forskolin) ought to erase the neuroprotective activity of adenosine A2a receptor antagonism. Second, at higher concentrations enprofylline (but not KW6002) has phosphodiesterase inhibitory activity and, thus, (at least theoretically) could lead to a rise in cAMP levels. Would elevation of cAMP levels be neuroprotective? Four day pretreatment of cultures with IBMX (100 μm) plus forskolin (10 μm) had no adverse effect on motor neuron survival and this pretreatment itself protected motor neurons from excitotoxic insult (−3 ± 4% cell death) (Fig. 1d). Similarly, treating cultures with the cell-permeable, nonhydrolyzable cAMP analog, dibutryl-cAMP (db-cAMP) at 1 mm, 4 and 2 d before the excitotoxic insult, was neuroprotective (−2 ± 3% cell death) (Fig. 1d). These findings are consistent with previous work demonstrating that manipulations that raise cAMP levels strongly support the long-term survival of motor neurons (Hanson et al., 1998). Because IBMX plus forskolin and db-cAMP were neuroprotective, we could not investigate the potential interaction of adenosine A2a antagonists with elevated cAMP levels. In addition, we could not dissociate elevation of cAMP levels from the neuroprotective action of enprofylline and, thus, exclude its potential inhibitory action as a phosphodiesterase antagonist. In sum, the neuroprotective activity of adenosine A2a receptor block is at least partially dissociable from an effect on cAMP levels and suggests that it occurs in a G-protein-independent process. We cannot rule out the possibility that the neuroprotective activity of enprofylline (specifically) is mediated, in part, by increases in cAMP levels. These findings are consistent with the view, however, that blockade of adenosine A2a receptors protects neurons from insult by virtue of another biochemical activity(ies).
We next asked about the effect of the adenosine A2 receptor antagonism on Trk signaling. Cultures of spinal cord cells (grown in the presence of a cocktail of trophic factors including 10 ng/ml BDNF) received KW6002 (1 μm), MRS1754 (50 nm), or vehicle (DMSO, 0.1%) and 24 h later lysates were prepared for immunoblots for Trk and the MAP kinase downstream signaling module (Fig. 2a). Immunoblots for phosphoTrk and phosphoMAPK were used to monitor the state of activation of these proteins. KW6002 led to a substantial reduction in the abundance of phosphoTrk and phosphoMAPK without altering the abundance of the unphosphorylated species. The effects of MRS1754 were indistinguishable from the vehicle control. These results indicate that antagonism of adenosine A2a, but not A2b, receptors inhibits activation of Trk and downstream signaling, even when cells are provided with enough extracellular BDNF to ordinarily activate its receptor.
Figure 2.
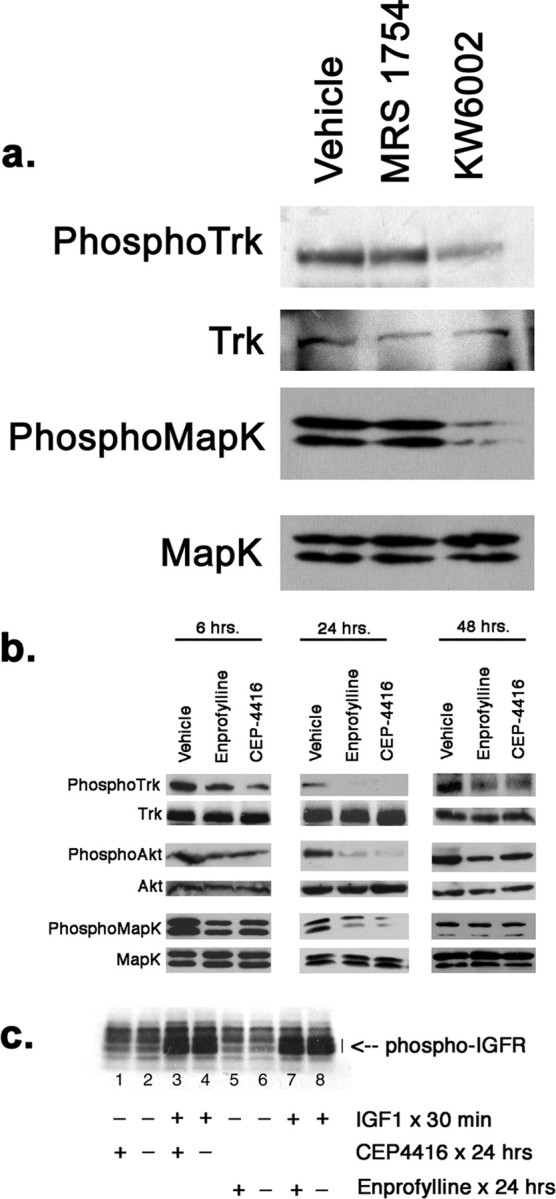
Biochemical analysis of the effects of adenosine or Trk receptor antagonists on cultures of spinal cord neurons. a, Fourteen DIV cultures of embryonic rat spinal cord neurons grown on astrocyte feeder layer were incubated with MRS1754 (10 nm), KW6002 (1 μm), or vehicle (DMSO, final concentration 0.1%) for 24 h and lysates were prepared in radioimmunoprecipitation assay (RIPA) buffer and subjected to immunoblot analysis. KW6002, but not MRS1754 or vehicle, led to a decrease in the abundance of phosphoTrk and phosphoMAPK without influencing the abundance unphosphorylated species. b, Fourteen DIV cultures of embryonic rat spinal cord neurons grown on astrocyte feeder layer were incubated with 100 μm enprofylline, 30 nm CEP4416, or vehicle (DMSO, final concentration 0.1%) for 6, 24, or 48 h and lysates were prepared in RIPA buffer and subjected to immunoblot analysis. Enprofylline and CEP4416 led to a decrease in the abundance of phosphoTrk at all time points with the most profound effect seen at 24 h. Similarly, enprofylline and CEP4416 led to a decrease in the abundance of phosphoAKT and phosphoMAPK at 6 and 24 h with the most profound effect seen at 24 h. PhosphoMAPK returned to drug treatment had no effect on the unphosphorylated species. c, Cultures were prepared as above and immunoblots of cell lysates were probed for phosphoIGFR after treatment with CEP4416 or vehicle for 24 h (lane 1 vs 2), or enprofylline or vehicle for 24 h (lane 5 vs 6). Neither drug led to an alteration in the abundance in the phosphorylated species. An additional set of cultures were treated for 24 h with CEP4416 or vehicle (lane 3 vs 4) or enprofylline or vehicle (lane 7 vs 8) and then acutely treated with IGF1 for 30 min. Immunoblots revealed robust induction of phosphoIGFR after acute stimulation with IGF and neither CEP4416 nor enprofylline blunted the acute response.
We followed up these observations by determining the effect of enprofylline on Trk signaling and determined the extent to which the resultant biochemical changes mimic the effects of direct pharmacological Trk antagonism with CEP4416. Six hours after drug administration, we regularly detected decreases in the phosphorylation of Trk, MAP kinase, and Akt (Fig. 2b). By 24 h after drug administration, there was a robust reduction in phosphoTrk, phosphoMAPK, and phosphoAkt without any alteration in the overall abundance of the unphosphorylated forms of these proteins. The effect of drug treatment on Akt was particularly marked. Forty-eight hours after drug administration, decrease in phosphoTrk could still be detected in the drug-treated cells although it was less pronounced that seen 24 h earlier. The abundance of phosphoAkt remained depressed and this was particularly evident in the enprofylline treated cells; phosphorylation of MAPK had largely returned to baseline by this time point. To determine the specificity of these agents, we examined their effect on IGF1 signaling through the IGF receptor tyrosine kinase (IGFR). Twenty-four hour treatment of spinal cord cultures with CEP4416 or enprofylline had no effect on the basal state of IGFR phosphorylation (Fig. 2c, lane 1 vs 2, 5 vs 6). In addition, the acute administration of IGF1 (5 nm) to spinal cord cultures led to a strong increase in IGFR phosphorylation and this was undiminished by 24 h pretreatment with CEP4416 or enprofylline (Fig. 2c, lane 3 vs 4, 7 vs 8). In sum, both enprofylline and CEP4416 lead to similar reductions in the activation of Trk and the actions of these drugs appear specific, to the extent that they do not interfere with the activation of the IGF1 receptor. The inhibition of signaling downstream of Trk was most pronounced 24 h after treatment and the blockade of adenosine A2a receptors appears to have a longer-lasting effect on the phosphatidylinositol-3 kinase–Akt pathway. It is unlikely that phosphodiesterase activity of enprofylline can explain these observations as increased cAMP levels enhance, not suppress, TrkB activation (Boulanger and Poo, 1999; Ji et al., 2005).
The simplest formulation posits that the modulation of Trk signaling by adenosine receptors occurs on (or within) motor neurons themselves. This is supported by two lines of evidence. First, immunostaining of purified motor neurons demonstrated that all motor neurons are colabeled with antibodies to Trk and the A2a receptor. In addition to cell body staining, colocalized puncta of Trk and A2a receptor immunoreactivity are evidence on dendritic shafts. The same observation was made on mixed spinal cord cultures where we found that all SMI-32-positive motor neurons were immunoreactive for Trk and A2a (Fig. 3a). These studies were performed using an antibody to the extracellular N terminus of the protein in a live labeling protocol and, thus, reflect the expression of cell surface TrkB with adenosine A2a receptors. We complemented these observations by subjecting lysates from spinal cord cultures to coimmunoprecipitation analysis. Anti-Trk-coated beads immunoprecipitated both Trk and adenosine A2a receptors and conversely anti-adenosine A2a-coated beads immunoprecipitated both Trk and adenosine A2a receptors (Fig. 3b). Beads not coated with primary antibody immunoprecipitated neither Trk nor adenosine A2a receptors. Similar results were obtained with homogenates of spinal cord tissue (data not shown). Thus, adenosine A2a and TrkB receptors appear to be components of a macromolecular complex, at least a portion of which is on the plasma membrane. Second, we asked whether adenosine A2a antagonists (or CEP4416) protected cultures of purified motor neurons from excitotoxic injury. One day after isolation, purified motor neurons grown in the presence of BDNF plus CT1 were incubated with enprofylline, CEP4416, or vehicle for 24 h and then underwent excitotoxic challenge (Fig. 3c). Quantification of motor neuron numbers in the various groups revealed that both drugs had no adverse effects on basal survival. Whereas the excitotoxic insult caused the death of 65 ± 4% of vehicle-treated motor neurons, there was no motor neuron death in the cultures pretreated with enprofylline (3 ± 1%) or CEP4416 (−2 ± 1%). Thus, while a non-cell-autonomous interaction between Trk and A2a receptors remains possible, our findings are consistent with adenosine A2a receptor modulation of Trk function at the level of the motor neuron itself.
Figure 3.

Adenosine A2a and TrkB receptors are colocalized on motor neurons and antagonism of either receptor protects against excitotoxic insult. a, Purified motor neurons were live labeled with chick anti-TrkB, washed, and fixed before incubation with mouse anti-adenosine A2a receptor and species-specific secondary antibodies. All motor neurons were immunostained with both antibodies. The anti-A2a antibody stained an intracellular pool of antigen as well as discrete punta associated with dendrites. The TrkB antibody only stained the cell surface of neurons in both a diffuse and punctate pattern on dendrites. In the merge image several puncta are immunostained for both antigens and inset at higher power shows colocalization of puncta (denoted with arrowheads). b, Lysates of 14 DIV spinal cord cultures underwent immunoprecipitation with anti-Trk or anti-adenosine A2a-coated beads and immunoblotted with anti-Trk, anti-adenosine A2a, or anti-actin antibodies. Coimmunoprecipitation of Trk with adenosine A2a receptors is observed. Actin is not part of this complex. c, Bar graph shows the results from the excitotoxicity assay performed on purified motor neurons pretreated for 24 h with enprofylline, CEP4416, or vehicle. Kainic acid treatment caused the death of ∼50% of motor neurons in the vehicle-treated group (*statistically significantly different, Student’s t test, p < 0.01; n = 4). No death of motor neurons occurred in the groups pretreated with enprofylline or CEP4416. Error bars indicate SE.
Mutations in SOD or the p150glued subunit of dynactin are known to account for a familial form of ALS in a subpopulation of individuals (Rosen et al., 1993; Puls et al., 2003; Bruijn et al., 2004). We inquired whether expression the mutant forms of SOD or p150glued caused motor neuron death in vitro and whether Trk antagonism was neuroprotective. Recombinant HSVs were generated that expressed wild-type or mutant forms of SOD (G85R) and wild-type or mutant p150glued (G59S). Viruses engineered to express wild-type or mutant proteins expressed transgenes at similar levels when assayed by Western blot (data not shown). Spinal cord cultures were infected with virus at 14 DIV and the number of motor neurons was determined 2, 4, 6, or 8 d later (Fig. 4a). No decrement in motor neuron number occurred over 8 d in cultures uninfected with virus, or infected with HSV-LacZ, HSV-WT-SOD, or HSV-WT-p150glued. In contrast, infecting cultures with HSV-G85R-SOD or HSV-G59S-p150glued led to a progressive loss of motor neurons that differed in a statistically significant manner when compared with controls 4, 6, and 8 d after infection [F = 13.757; p < 0.001 (multivariate analysis, linear model) repeated measure ANOVA, followed by post hoc Scheffé’s test with significance set at p < 0.05].
Figure 4.
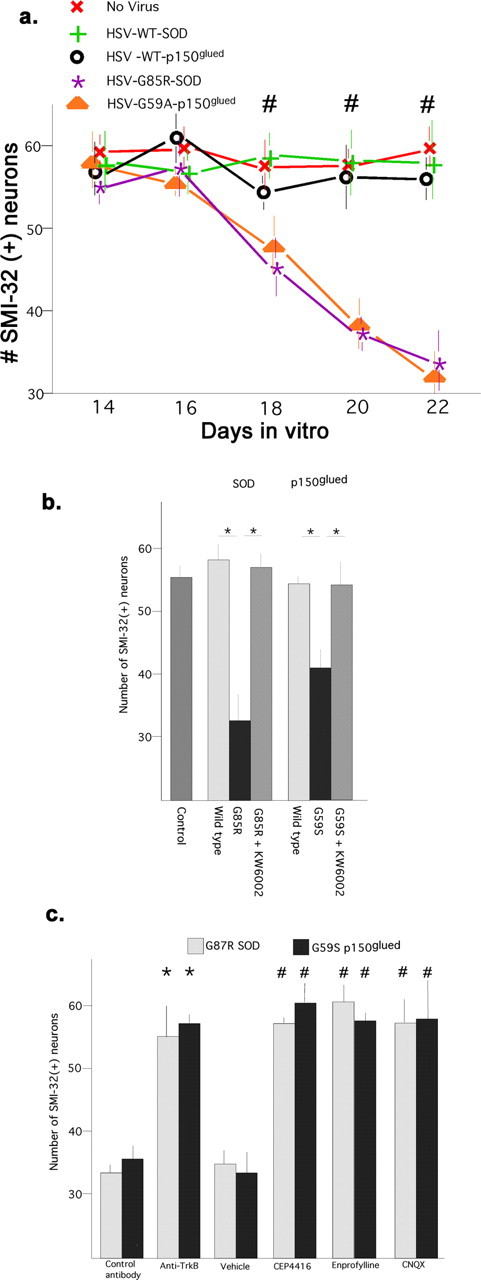
Time-dependent death of motor neurons triggered by expression of mutant forms of SOD1 and p150glued: neuroprotection afforded by antagonists of Trk, adenosine A2a, or AMPA receptors. a, Fourteen DIV spinal cord cultures were infected with recombinant HSV or vehicle (2 μl/ml of culture media) and the number of motor neurons was determined at 2 d intervals thereafter. RMANOVA was used to examine the association between the transgene expressed (wild-type and mutant forms of SOD and p150glued) and the number of surviving motor neurons over time. For this analysis, the four transgene-expressing groups and no-virus control represent the between-group factor (5 levels) and the survival over the subsequent 8 d (16, 18, 20, and 22 DIV) represents the within-group factor (4 levels). The effects of interest are the group by days interaction and F = 13.757, p < 0.001 (multivariate analysis, linear model) indicates that survival over time is significantly different as a function of transgene expression. Post hoc comparisons between groups using Scheffé’s F test with significance set at p < 0.05 showed that the G85R SOD mutant and the G59S p150glued mutant differed significantly from the other groups (denoted with #). No differences in survival were found between the no-virus versus the WT SOD versus the WT p150glued groups. b, Survival of motor neurons 6 d after infection with HSV expressing the wild-type or mutant versions of SOD or p150glued; effects of KW6002. Expression of the mutant versions of SOD or p150glued led to motor neuron death that could be completely reversed by 6 d pretreatment with KW6002. (Student’s t test, *p < 0.01). c, One set of cultures received function-blocking anti-TrkB (monoclonal; Transduction Laboratories) or a control mouse antibody (10A8, anti human apolipoprotein A1; ICN Pharmaceuticals, Costa Mesa, CA), 5 μg/ml daily for 5 d. There were significantly more motor neurons in the anti-TrkB-treated cultures compared with the control (denoted with asterisks, Student’s t test, p < 0.01; n = 6). This experimental paradigm was repeated with HSV G59S p150glued and the same neuroprotective effects seen. Another set of cultures was infected with viruses and received CEP4416 (every other day), enprofylline (every other day), CNQX (daily), or vehicle. There was a significantly greater number of motor neurons in each of the drug-treated groups in comparison with the controls both for the HSV-G85R-SOD cultures (F(7,25) = 8.697; p < 0.0001) and HSV-G59S-p150glued cultures (F(7,25) = 16.93; p < 0.0001). Multiple comparisons were made with ANOVA with Scheffé’s post hoc tests with significance set at p < 0.05 (denoted by #). Error bars indicate SE.
We next determined whether KW6002 could protect motor neurons from the toxic effects of mutant proteins. Spinal cord cultures were infected with viruses expressing wild-type or mutant versions of SOD or p150glued at 14 DIV, then treated with drug or vehicle for 4 d (Fig. 4b). The number of motor neurons in cultures uninfected with virus (control) or wild-type versions of SOD or p150glued were not significantly different (55 ± 2 vs 58 ± 3 vs 54 ± 4). Expression of the mutant versions of SOD or p150glued led to the loss of ∼40% of motor neurons and KW6002 completely prevented this (F(6,14) = 67.10; p < 0.0001, ANOVA). Post hoc analysis (with significant set at p < 0.05) showed that the number of motor neurons in the mutant protein expressing cultures was significantly less than motor neurons expressing wild-type proteins. KW6002 led to a significant abrogation of the toxic effect of the mutant proteins. These results indicate that, in vitro, KW6002 inhibits motor neuron death caused by the expression of mutant versions of SOD or p150glued.
Under normal growth conditions, BDNF is endogenously produced in spinal cord cultures and introduction of function blocking anti-TrkB antibodies daily for 4 d will reduce the abundance of phosphorylated Trk (Hu and Kalb, 2003). To determine whether motor neuron death caused by G85R-SOD or G59S-p150glued required intact BDNF-TrkB signaling, cultures were infected with the respective viral vectors and were subsequently treated (daily) with function blocking anti-TrkB antibodies or a control antibody (Fig. 4c). Of note, the neurotrophic factor CT1 was included in all incubations. When we assessed motor neuron survival 6 d postinfection, we found that function blocking anti-TrkB antibodies, but not the control antibody, eliminated the toxicity of the mutant protein (number of motor neurons: 56 ± 2 vs 28 ± 4, anti-TrkB vs control antibody, G85R-SOD1 groups, p < 0.01, t test; 58 ± 2 vs 32 ± 5, anti-TrkB vs control antibody, G59S-p150glued groups, p < 0.01, t test). Simply treating naive cultures with the function blocking anti-TrkB antibodies had no effect on motor neuron survival. We next asked whether enprofylline or CEP4416 (administered every other day for 6 d) or CNQX, an AMPA receptor antagonist, affected the survival of motor neurons in cultures infected with HSV-G85R-SOD or HSV-G59S-p150glued and found that all three drugs completely prevented the toxicity of mutant protein expression (Fig. 4c). All three drug treatments led to a statistically significant neuroprotective effect against the toxic action of G85R SOD (F(7,25) = 8.697; p < 0.0001, ANOVA). Similarly, all drug treatments led to a statistically significant neuroprotective effect against the toxic action of G59S p150glued (F(7,25) = 16.930; p < 0.0001, ANOVA). These results indicate that (1) antagonism of Trk signaling with function-blocking antibodies or CEP4416 protects motor neurons from the toxicity associated with mutant proteins known to underlie familial ALS, (2) antagonism of adenosine A2a receptor displays similar neuroprotective activity, and (3) basal excitatory neurotransmission ongoing in our culture system provides a necessary substrate for mutant protein toxicity of motor neurons.
The precise mechanism by which G-protein-coupled receptors transactivate receptor tyrosine kinases is complex and to some extent receptor-subtype specific (Luttrell et al., 1999; Downward, 2003; Piiper and Zeuzem, 2004; Waters et al., 2004). Some evidence indicates that src-family kinases (SFKs) participate in adenosine A2a receptors transactivation of Trk (Lee and Chao, 2001). If this pathway was operative in spinal cord neurons, then antagonism of adenosine A2a receptors might lead to reduced src activation and antagonism of SFKs might be neuroprotective. Src is negatively regulated by phosphorylation of tyrosine 529 (refers to the human src sequence) and an antibody that recognizes a dephospho-epitope in this region (“clone 28”) is a reliable measure of active src and SFKs (Kawakatsu et al., 1996). We began by asking whether adenosine A2a receptors and active SFKs are present in the same cells and, if so, for their subcellular distribution. Pure motor neurons grown in the presence of BDNF were immunostained with a rabbit anti-adenosine A2a receptor and the clone 28 antibody. One hundred percent of motor neurons expressed both antigens and on dendrites the immunoreactivity formed discrete fine puncta (Fig. 5a). Many, but not all, puncta were stained with both antibodies. To study the participation of SFKs in A2a antagonism action, cultures were exposed to KW6002, MRS1754, or vehicle for 24 h and lysates probed with the clone 28 antibody. KW6002, but not MRS1754, caused a reduction in the abundance of active SFKs when compared with vehicle treated cultures (Fig. 5b). We also exposed cultures to enprofylline or vehicle for 24 h and probed lysates with the clone 28 antibody. Enprofylline also caused a reduction in the abundance of active SFKs when compared with vehicle-treated cultures (Fig. 5c). Pharmacological block of SFKs can be achieved with PP1 (Hanke et al., 1996) and we found that pretreatment of spinal cord cultures with PP1 prevented motor neuron death induced by excitotoxic challenge (percent motor neuron survival, 100 ± 2 vs 62 ± 3, PP1 vs vehicle, respectively; p < 0.01, t test) (Fig. 5c). We used a second method for inhibiting SFK activity by engineering HSV to express a dominant-negative form of src (K295R) in neurons (Mukhopadhyay et al., 1995). Spinal cord cultures were infected with the HSV-K295R-src or HSV-LacZ and 24 h later the excitotoxicity assay was performed. Expression of the dominant-negative src (but not β-galactosidase) rescued motor neurons from the excitotoxic challenge (percent motor neuron survival, 99 ± 3 vs 59 ± 2, src-K295R vs LacZ, respectively; p < 0.01, t test) (Fig. 5c). These experiments reveal that (1) some adenosine A2a receptors reside in close proximity to active SFKs, (2) SFK activation is downstream of adenosine A2a receptors, and (3) the neuroprotective action of adenosine A2a receptor antagonists uses an SFK pathway.
Figure 5.
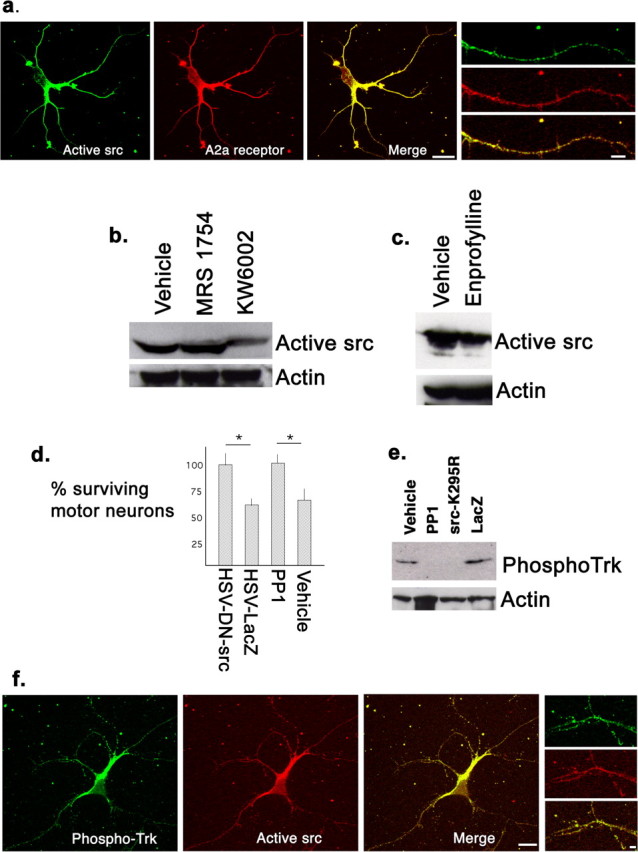
Relationship between the activation of SFKs and adenosine A2a receptors in Trk activation and neuroprotection. a, A motor neuron immunostained for active SFKs (using the clone 28 monoclonal antibody) and adenosine A2a receptors (using a rabbit serum). Immunoreactivity with both antibodies is evident at the cell body and dendrites and much of it colocalizes (yellow in “merge” image; scale bar, 20 μm). Examination at a higher power (images of a single dendrite at right) shows that immunoreactivity is mainly puncta and many (but not all) puncta are immunopositive for both antigens. Scale bar, 7 μm. b, Immunoblots for active SFKs (clone 28) after 24 h treatment of spinal cord cultures with MRS1754, KW6002, or vehicle. The abundance of active SFK is only reduced in cultures treated with KW6002 treatment. Protein loading levels (monitored by actin blots) were equivalent. c, Immunoblots for active src family kinases (clone 28) after 24 h treatment of spinal cord cultures with enprofylline or vehicle. The abundance of active SFKs is reduced in the enprofylline-treated cultures. Protein loading levels (monitored by actin blots) were equivalent. d, The number of motor neurons surviving excitotoxic challenge after treatment with two different methods of inhibiting SFK activation: expression of dominant-negative src versus LacZ or treatment with PP1 versus vehicle. Expression of the dominant-negative src led to a statistically significant protection when compared with expression of LacZ (asterisk denotes statistically significantly different, Student’s t test, p < 0.01; n = 5). Similarly, there was a statistically significant greater number of motor neurons after excitotoxic insult in cultures treated with PP1 versus vehicle (Student’s t test, p < 0.01; n = 4). e, Immunoblots for phosphoTrk after treating cultures for 24 h with dominant-negative src versus LacZ or PP1 versus vehicle. Both methods for SFK inhibition led to a reduction in the abundance of phosphorylated Trk. Protein loading levels (monitored by actin blots) were equivalent. f, A motor neuron immunostained for active, phosphoTrk (using a rabbit serum) and active SFKs (using the clone 28 monoclonal antibody). Immunoreactivity with both antibodies is evident at the cell body and dendrites and, as in a, much of it colocalizes (yellow in merge image; scale bar, 17 μm). Examination at a higher power (images of a dendrite at right) shows that immunoreactivity is largely puncta and many (but not all) puncta are immunopositive for both antigens. Scale bar, 3 μm. Error bars indicate SE.
We next inquired about the relationship between SFKs and Trk receptors. When pure motor neurons grown in the presence of BDNF were immunostained with a rabbit anti-phosphoTrk receptor antibody and the clone 28 antibody we found that 100% of motor neurons expressed both antigens at the cell body and on dendrites (Fig. 5f). As noted above for adenosine A2a and active SFKs, the immunoreactivity formed discrete fine puncta and in many instances there was colocalization. To determine whether this close spatial relationship between active Trk receptors and active SFKs reflected a functional relationship, we monitored the state of phosphorylation of Trk after blocking SFK activity with PP1 or HSV-src-K295R. We found that maneuvers that caused antagonism of SFK activation, led to a reduction in the abundance of phosphoTrk (Fig. 5e). These findings suggest that active adenosine A2a receptors contribute to SFK activation, and src (or src-family) kinases are upstream of TrkB activation.
Lipid rafts are cholesterol/sphingolipid rich microdomains of plasma membrane with specialized signaling capacity and recent work demonstrates that TrkB receptors are recruited into lipid rafts with activation with BDNF (Suzuki et al., 2004). Because SFKs are residents of lipid rafts (by virtue of their lipid modification by myristate) we wondered whether Trk and A2A receptors were found in the same membrane compartments and whether they physically associate. The yield of detergent insoluble membrane fractions (i.e., lipid rafts) is generally low and, therefore, to be certain we would have enough material for analysis, in these studies we used homogenates of neonatal spinal cord tissue. After homogenization in cold 0.5% Triton X-100, centrifugation through a discontinuous sucrose gradient, and fraction collection, we recovered lipid raft marker Thy-1 immunoreactivity within fraction 2 (by convention “lipid rafts”). Trk receptors, adenosine A2a receptors and active SFK were found in both lipid raft and nonlipid raft fractions (Fig. 6a). Using the clone 28 antibody that recognizes four active SFK members (src, fyn, yes, and fgr) that differ in their migration through SDS-PAGE gel, we found a predominance of the slower migrating SFK(s) in the lipid raft fraction and a predominance of the faster migrating SFK(s) in the nonlipid raft fractions. Coimmunoprecipitation analysis using beads coated with anti-Trk or anti-phosphoTrk demonstrated that adenosine A2a receptors associated with Trk receptors in both lipid raft and nonlipid raft fractions. Because we loaded the same amount of protein in each lane of the Western blot, and the bands were more intense in the lipid raft fractions, it is likely the association of these proteins is enriched in this subcellular fraction. Coimmunoprecipitation analysis using beads coated with anti-adenosine A2a or anti-phosphoTrk receptors brought down SFK members in the lipid raft and nonlipid raft fractions. In both immunoprecipitates, it was the slower migrating SFK member that associated with adenosine A2a or phosphoTrk receptors in the lipid raft fraction and the faster migrating SFK member associated with adenosine A2a or phosphoTrk receptors in the nonlipid raft fraction (Fig. 6a). These results indicate that in vivo SFKs are part of a physical complex with adenosine A2a receptors and active Trk receptors, who themselves are physically associated. Whether a heterotrimeric complex exists (active Trk receptors plus active SFKs plus adenosine A2a receptors), or multiple binary complexes, is not known. At the very least, a series of binary complexes exist and those in the lipid raft fraction that include SFKs are molecularly distinct from those in the nonlipid raft fraction. A model of the relationship between adenosine A2a receptors, SFKs, and Trk receptors is shown in supplemental Figure 1 (available at www.jneurosci.org as supplemental material).
Figure 6.
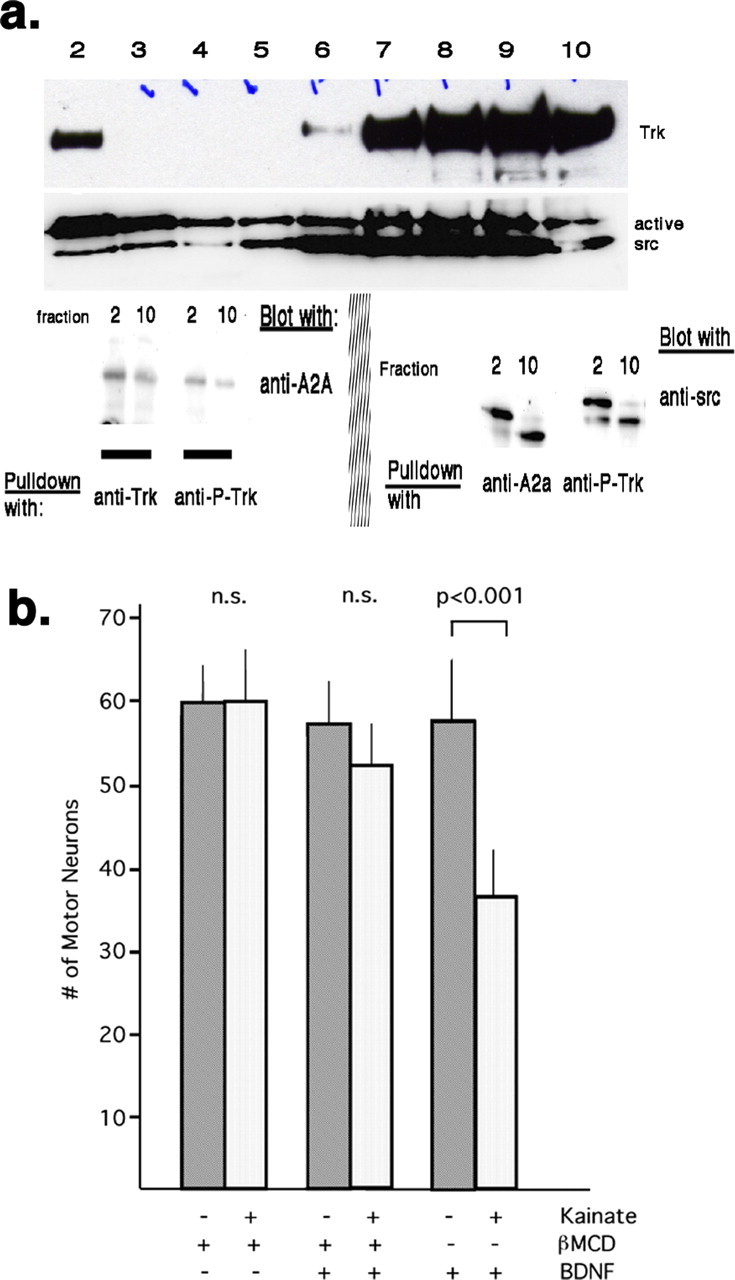
Trk receptors, adenosine A2a receptors, and SFKs are present in lipid rafts and nonlipid rafts. a, Discontinuous sucrose gradients were used to separate rafts from nonlipid rafts and aliquots from 1 ml fractions of the gradient were subjected to Western blots for Trk and active SFKs. The lipid raft fraction (2) contains all three proteins. Trk and active SFKs are also present in nonraft fractions. Lower panels (left) show coimmunoprecipitation (pull down with anti-Trk beads, blot with anti-adenosine A2a receptor) of A2a with Trk in lipid raft fractions (2) and nonraft fractions (10). The adenosine A2a receptor also associates with P-Trk in both the lipid raft fractions and nonraft fractions (pull down with anti-P-Trk beads, blot with anti-adenosine A2a receptor). Active SFKs can be coimmunoprecipitated from rafts and nonraft fractions with anti-A2a or anti-P-Trk (lower right). In the lipid raft fractions, the slower migrating SFK preferentially associates with Trk and adenosine A2a receptors. In the nonlipid raft fractions, the faster migrating SFK preferentially associates with Trk and adenosine A2a receptors. b, Purified motor neurons were grown in the presence of CT1 and then acutely exposed to βMCD or vehicle followed by BDNF or vehicle. The excitotoxicity assay was performed (exposure kainate or vehicle) and the number of motor neurons assessed one day later. Excitotoxic cell death occurred in motor neurons exposed to BDNF, but did not occur if cells were pretreated with βMCD before BDNF. βMCD itself was nontoxic. Error bars indicate SE.
Finally, to begin to determine the biological relevance of the lipid raft-signaling complex, we asked whether their disruption with cholesterol-depleting agents would affect the capacity of BDNF to confer excitotoxic sensitivity on motor neurons. Pure motor neurons were grown in the presence of CT1 but in the absence of BDNF for 24 h, at which point the cholesterol-depleting agent βMCD or vehicle was added to the cultures. Thirty minutes later, BDNF or vehicle was added to the culture and 3 h later the excitotoxicity assay was performed (Fig. 6b). We determined the amount of cholesterol in our pure motor neuron cultures after 30 min of βMCD treatment and found an ∼25% reduction in comparison with vehicle treatment (βMCD vs vehicle; 404 ± 31 μg/μg of protein vs 550 ± 16 μg/μg of protein; n = 5 samples; t test, p < 0.001). In terms of cell death, a statistically significant difference between experimental groups was identified by ANOVA (F(5,42) = 32.797; p < 0.001) and a post hoc analysis (Scheffé with level of significance set at p < 0.05) indicated excitotoxic death only occurred in the (+)BDNF/(−)βMCD treatment group (number of motor neurons exposed to vehicle or kainate; 57 ± 7 vs 40 ± 4). No excitotoxic motor neuron death occurred in the (−)BDNF/(+)βMCD treatment group (number of motor neurons exposed to vehicle or kainate; 60 ± 3 vs 60 ± 5) or in the (+)BDNF/(+)βMCD treatment group (number of motor neurons exposed to vehicle or kainate; 58 ± 4 vs 51 ± 5). These results suggest that disruption of lipid rafts by cholesterol depletion interferes with the ability of BDNF signaling to evoke excitotoxic sensitivity.
Discussion
One approach to developing effective therapy for ALS aims to identify the primary pathophysiological process(es) that initiate cell dysfunction and ultimately death. Despite intensive effort and enormous progress, this remains an elusive goal. Another approach focuses on the identification of physiological processes that render motor neurons vulnerable to insult. Adopting this strategy we found that the activation of TrkB by BDNF induces a state of susceptibility within motor neurons to insult. Here, we show that blocking TrkB activation by a variety of means prevents excitotoxic motor neuron death as well as death caused by the expression of ALS-causing mutant proteins. This unexpectedly broad neuroprotective action of TrkB antagonism drives the inquiry into the various ways cells activate TrkB and regulate downstream signaling cascades.
Many receptor tyrosine kinases (RTKs) undergo transactivation by agonists of GPCRs (Daub et al., 1997; Lee and Chao, 2001; Oak et al., 2001; Lee et al., 2002b; Fischer et al., 2004; Zahradka et al., 2004) and our observations that antagonism of adenosine A2a receptors in spinal cord cultures leads to a reduction in TrkB activation complement these results. They imply that under physiological conditions, adenosine A2a and TrkB receptors endogenously interact and active adenosine A2a receptors contribute to the basal level of TrkB activation. Work from the Chao lab shows that TrkA receptor transactivation involves transcription/translation and is restricted to intracellular membranes (probably the Golgi apparatus) (Rajagopal et al., 2004). We identify an additional niche for the TrkB and adenosine receptor interaction: at the plasma membrane concentrated in hot spots. In addition we show that adenosine A2a and the TrkB receptor physically interact (either directly or as part of a larger multiprotein complex), an observation, to our knowledge, not reported previously. The colocalization of the receptors might allow for dynamic, local changes in TrkB activation as a function of adenosinergic neurotransmission. The present work indicates that the reduction in TrkB activation that follows from adenosine A2a blockade underlies, at least in part, its neuroprotective properties. We do not establish that this is the exclusive molecular mechanism and other changes in cellular neurochemistry evoked by adenosine A2a blockade have the potential to be healthful for motor neurons.
SFKs participate in GPCR-mediated transactivation of several RTKs (Keely et al., 2000; Lee and Chao, 2001; Krieg et al., 2002; Zahradka et al., 2004). Although intermediates in the pathway, we do not know whether SFKs directly phosphorylate these receptor tyrosine kinases or if the effect is indirect. Similarly, although it is parsimonious to posit that the neuroprotective action of blocking SFKs is attributable to their ability to decrease TrkB activation, the plethora of SFK substrates (in particular MAPK) raise other possibilities (Murray et al., 1998; Runden et al., 1998; Grewal et al., 1999; Ishikawa et al., 2000; Kaplan and Miller, 2000; Kim et al., 2003; Bromann et al., 2004; Choi et al., 2004; Luttrell and Luttrell, 2004). Regardless of its precise mechanism of action, increases in SFK activity can adversely affect neuronal health (or exacerbate toxic insult, i.e., Aβ peptides) and inhibition of SFK activity can protect neurons from insult (Lambert et al., 1998; Chin et al., 2004, 2005; Lennmyr et al., 2004).
We and others have detected RTKs, transactivating GPCRs and SFKs in lipid rafts and their colocalization bolsters the case for the physiological relevance of this membrane compartment as a platform for signal transduction integration (supplemental Fig. 1, available at www.jneurosci.org as supplemental material) (Ushio-Fukai et al., 2001; Hur et al., 2004). These specialized membrane domains are dynamic: proteins enter into and egress from lipid rafts after ligand binding and lipid rafts themselves may associate to bring new signaling capacities to liganded receptors (Simons and Toomre, 2000; Tansey et al., 2000; Paratcha and Ibanez, 2002; Ma et al., 2003; Golub et al., 2004). For example, in cortical neuron cultures, BDNF can recruit a subpopulation of TrkB receptors into lipid rafts and whereas BDNF-stimulated TrkB receptors preferentially activate MAP kinase in lipid rafts, they activate Akt in nonlipid raft membrane (Suzuki et al., 2004). A subtype of lipid rafts are caveolae, flask-shaped membrane invaginations that contain the cholesterol-binding protein caveolin as well as a diversity of receptors and signaling molecules (Krajewska and Maslowska, 2004). Dissolution of lipid rafts by cholesterol depletion can block transactivation of the epidermal growth factor RTK by the angiotensin II GPCR type 1 in a process that involves caveolin (Ushio-Fukai et al., 2001). These findings might be relevant to our current observations (Fig. 6) because Trk receptors have been shown to reside in caveolae (at least in PC12 cells) and physically associate with caveolin (Bilderback et al., 1999; Peiro et al., 2000). Future work will be needed to explore the differences in signals propagating from TrkB receptors activated by BDNF versus those transactivated by adenosine A2a receptors as a function of lipid raft integrity.
For two reasons we believe the neuroprotective activity of adenosine A2a receptor antagonism is operating in motor neurons themselves. First, motor neurons express both A2a receptors and TrkB receptors and they are colocalized into distinct subcellular domains (punta) suggesting a privileged signaling capacity. Second, A2a receptor antagonists protect purified motor neurons from insult indicating the lack of necessity of nonmotor neurons and glial cells in the biological effect. Thus, although ALS is increasingly being viewed a disorder that involves the participation of multiple cell types (i.e., astrocytes, muscle, microglia), the present results highlight motor neuron-specific processes (Clement et al., 2003; Dupuis et al., 2003, 2004; Barbeito et al., 2004; Pehar et al., 2004; Cassina et al., 2005; Schutz et al., 2005).
Several investigators have found that a pathological rise in intracellular calcium is an essential component of the mechanism of excitotoxic insult to motor neurons (Carriedo et al., 1996) and interestingly, the toxicity of mutant forms of SOD in vitro are abrogated by maneuvers that antagonize AMPA receptor activation and limit rises in intracellular calcium (Roy et al., 1998). Because activation of Trk receptors or adenosine A2a receptors can modulate intracellular calcium levels, our current studies in mixed cell cultures might reflect effects on modulation of intracellular calcium levels, particularly in specific subcellular domains. In a variety of neuronal preparations, application of BDNF leads to a rise in intracellular calcium in a tyrosine kinase-dependent manner (Mizoguchi et al., 2002; Lamb and Bielefeldt, 2003; Mizoguchi and Nabekura, 2003). With regard to adenosine A2a receptors, the situation is more complex; their blockade on presynaptic terminals prevents stimulus-evoked rises in intraterminal calcium levels (Correia-de-Sa et al., 2000; Li and Wong, 2000) whereas their activation on the cell soma blocks hypoxia-induced rises in intracellular calcium (Kobayashi et al., 1998). Given these observations, reduction in TrkB activation on the cell soma or adenosine A2a receptors on presynaptic terminals might have neuroprotective effects by blunting rises in intracellular calcium that follow noxious insult.
We showed previously that agonist-evoked rises in intracellular calcium are not higher in purified motor neurons grown in the presence of BDNF versus those grown in the presence of other trophic factors (Fryer et al., 2000). Thus, although synaptic activity can be enhanced by BDNF-TrkB signaling (Poo, 2001), it is unlikely that BDNF makes motor neurons vulnerable to excitotoxic insult simply by potentiating the capacity of glutamate to excite neurons. Our results indicate that activity-dependent rise in intracellular calcium is necessary (but not sufficient) for motor neuron death (Fryer et al., 1999) and that the effects of BDNF, in our experimental paradigms, are downstream of this rise in intracellular calcium.
One of the particular attractions of the present observations is the potential ability to translate them into human therapeutics. Trk antagonists related to CEP4416 (Cephalon) are in clinical use currently for the treatment of leukemia and solid tumors (Smith et al., 2004; Undevia et al., 2004). After peripheral administration, CEP4416 is detectable in CNS tissues and causes a reduction in the abundance of activated Trk (data not shown). The long-term utility of Trk antagonists may be limited by their effects on the maintenance of neuromuscular junction integrity (Gonzalez et al., 1999) and learning and memory (Korte et al., 1996; Messaoudi et al., 2002; Pang et al., 2004). Adenosine A2a receptor antagonists have been shown to be neuroprotective agents in animal models of parkinsonism (Shiozaki et al., 1999; Koga et al., 2000; Ikeda et al., 2002; Fink et al., 2004). Potential adverse effects on inflammation (Ohta and Sitkovsky, 2001; Thiel et al., 2005) and coronary artery vasoregulation (Belardinelli et al., 1998) have not limited their safe use in humans with Parkinson’s disease (Bara-Jimenez et al., 2003; Kase et al., 2003). Seven transmembrane domain receptors (such as the adenosine A2a receptors) are the most common target of therapeutic drugs (Lefkowitz and Shenoy, 2005), raising the possibility that future agents may be developed that antagonize the pathway by which A2a receptors transactivate Trk receptors. Although in vitro observations on ALS therapeutics do not invariably translate into in vivo efficacy (compare Gurney et al., 1996; Li et al., 2000 with Groeneveld et al., 2003), this combination of favorable characteristic could hasten the evaluation of adenosine A2a or Trk antagonists for the treatment of ALS.
Footnotes
This work was supported by the Amyotrophic Lateral Sclerosis Association (ALSA), the Philadelphia Chapter of the ALSA, the Institutional Development Fund of the Children’s Hospital of Philadelphia (CHOP), and United States Public Health Service–National Institutes of Health Grants NS 29837 and NS 52325. We thank Rachael Neve (Harvard University, Boston, MA) for packaging the recombinant HSV, Ilana B. Nissim and Marc Yudkoff (CHOP) for help with the liquid chromatography, Louis Reichardt (University of California, San Francisco, CA) for the chick anti-N-terminal TrkB antibody, Joel Linden for discussions on adenosine receptor pharmacology, and David Pleasure for critical review of this manuscript.
References
- Bara-Jimenez W, Sherzai A, Dimitrova T, Favit A, Bibbiani F, Gillespie M, Morris MJ, Mouradian MM, Chase TN. Adenosine A(2A) receptor antagonist treatment of Parkinson’s disease. Neurology. 2003;61:293–296. doi: 10.1212/01.wnl.0000073136.00548.d4. [DOI] [PubMed] [Google Scholar]
- Barbeito LH, Pehar M, Cassina P, Vargas MR, Peluffo H, Viera L, Estevez AG, Beckman JS. A role for astrocytes in motor neuron loss in amyotrophic lateral sclerosis. Brain Res Brain Res Rev. 2004;47:263–274. doi: 10.1016/j.brainresrev.2004.05.003. [DOI] [PubMed] [Google Scholar]
- Belardinelli L, Shryock JC, Snowdy S, Zhang Y, Monopoli A, Lozza G, Ongini E, Olsson RA, Dennis DM. The A2A adenosine receptor mediates coronary vasodilation. J Pharmacol Exp Ther. 1998;284:1066–1073. [PubMed] [Google Scholar]
- Bilderback TR, Gazula VR, Lisanti MP, Dobrowsky RT. Caveolin interacts with Trk A and p75(NTR) and regulates neurotrophin signaling pathways. J Biol Chem. 1999;274:257–263. doi: 10.1074/jbc.274.1.257. [DOI] [PubMed] [Google Scholar]
- Boulanger L, Poo M. Gating of BDNF-induced synaptic potentiation by cAMP. Science. 1999;284:1982–1984. doi: 10.1126/science.284.5422.1982. [DOI] [PubMed] [Google Scholar]
- Bromann PA, Korkaya H, Courtneidge SA. The interplay between Src family kinases and receptor tyrosine kinases. Oncogene. 2004;23:7957–7968. doi: 10.1038/sj.onc.1208079. [DOI] [PubMed] [Google Scholar]
- Bruijn LI, Miller TM, Cleveland DW. Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu Rev Neurosci. 2004;27:723–749. doi: 10.1146/annurev.neuro.27.070203.144244. [DOI] [PubMed] [Google Scholar]
- Carriedo SG, Yin HZ, Weiss JH. Motor neurons are selectively vulnerable to AMPA/KA receptor-mediated injury in vitro. J Neurosci. 1996;16:4069–4079. doi: 10.1523/JNEUROSCI.16-13-04069.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carriedo SG, Sensi SL, Yin HZ, Weiss JH. AMPA exposures induce mitochondrial Ca2+ overload and ROS generation in spinal motor neurons in vitro. J Neurosci. 2000;20:240–250. doi: 10.1523/JNEUROSCI.20-01-00240.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassina P, Pehar M, Vargas MR, Castellanos R, Barbeito AG, Estevez AG, Thompson JA, Beckman JS, Barbeito L. Astrocyte activation by fibroblast growth factor-1 and motor neuron apoptosis: implications for amyotrophic lateral sclerosis. J Neurochem. 2005;93:38–46. doi: 10.1111/j.1471-4159.2004.02984.x. [DOI] [PubMed] [Google Scholar]
- Chin J, Palop JJ, Yu GQ, Kojima N, Masliah E, Mucke L. Fyn kinase modulates synaptotoxicity, but not aberrant sprouting, in human amyloid precursor protein transgenic mice. J Neurosci. 2004;24:4692–4697. doi: 10.1523/JNEUROSCI.0277-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin J, Palop JJ, Puolivali J, Massaro C, Bien-Ly N, Gerstein H, Scearce-Levie K, Masliah E, Mucke L. Fyn kinase induces synaptic and cognitive impairments in a transgenic mouse model of Alzheimer’s disease. J Neurosci. 2005;25:9694–9703. doi: 10.1523/JNEUROSCI.2980-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SY, Hwang JJ, Koh JY. NR2A induction and NMDA receptor-dependent neuronal death by neurotrophin-4/5 in cortical cell culture. J Neurochem. 2004;88:708–716. doi: 10.1046/j.1471-4159.2003.02187.x. [DOI] [PubMed] [Google Scholar]
- Clement AM, Nguyen MD, Roberts EA, Garcia ML, Boillee S, Rule M, McMahon AP, Doucette W, Siwek D, Ferrante RJ, Brown RH, Jr, Julien JP, Goldstein LS, Cleveland DW. Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science. 2003;302:113–117. doi: 10.1126/science.1086071. [DOI] [PubMed] [Google Scholar]
- Correia-de-Sa P, Timoteo MA, Ribeiro JA. A(2A) adenosine receptor facilitation of neuromuscular transmission: influence of stimulus paradigm on calcium mobilization. J Neurochem. 2000;74:2462–2469. doi: 10.1046/j.1471-4159.2000.0742462.x. [DOI] [PubMed] [Google Scholar]
- Daub H, Wallasch C, Lankenau A, Herrlich A, Ullrich A. Signal characteristics of G protein-transactivated EGF receptor. EMBO J. 1997;16:7032–7044. doi: 10.1093/emboj/16.23.7032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downward J. Role of receptor tyrosine kinases in G-protein-coupled receptor regulation of Ras: transactivation or parallel pathways? Biochem J. 2003;376:e9–e10. doi: 10.1042/BJ20031745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunwiddie TV, Masino SA. The role and regulation of adenosine in the central nervous system. Annu Rev Neurosci. 2001;24:31–55. doi: 10.1146/annurev.neuro.24.1.31. [DOI] [PubMed] [Google Scholar]
- Dunwiddie TV, Diao L, Proctor WR. Adenine nucleotides undergo rapid, quantitative conversion to adenosine in the extracellular space in rat hippocampus. J Neurosci. 1997;17:7673–7682. doi: 10.1523/JNEUROSCI.17-20-07673.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupuis L, di Scala F, Rene F, de Tapia M, Oudart H, Pradat PF, Meininger V, Loeffler JP. Up-regulation of mitochondrial uncoupling protein 3 reveals an early muscular metabolic defect in amyotrophic lateral sclerosis. Faseb J. 2003;17:2091–2093. doi: 10.1096/fj.02-1182fje. [DOI] [PubMed] [Google Scholar]
- Dupuis L, Oudart H, Rene F, Gonzalez de Aguilar JL, Loeffler JP. Evidence for defective energy homeostasis in amyotrophic lateral sclerosis: benefit of a high-energy diet in a transgenic mouse model. Proc Natl Acad Sci USA. 2004;101:11159–11164. doi: 10.1073/pnas.0402026101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eguchi S, Dempsey PJ, Frank GD, Motley ED, Inagami T. Activation of MAPKs by angiotensin II in vascular smooth muscle cells. Metalloprotease-dependent EGF receptor activation is required for activation of ERK and p38 MAPK but not for JNK. J Biol Chem. 2001;276:7957–7962. doi: 10.1074/jbc.M008570200. [DOI] [PubMed] [Google Scholar]
- Ernfors P, Lee KF, Jaenisch R. Mice lacking brain-derived neurotrophic factor develop with sensory deficits. Nature. 1995;368:147–150. doi: 10.1038/368147a0. [DOI] [PubMed] [Google Scholar]
- Estevez AG, Stutzmann JM, Barbeito L. Protective effect of riluzole on excitatory amino acid-mediated neurotoxicity in motoneuron-enriched cultures. Eur J Pharmacol. 1995;280:47–53. doi: 10.1016/0014-2999(95)00186-o. [DOI] [PubMed] [Google Scholar]
- Evans AE, Kisselbach KD, Yamashiro DJ, Ikegaki N, Camoratto AM, Dionne CA, Brodeur GM. Antitumor activity of CEP-751 (KT-6587) on human neuroblastoma and medulloblastoma xenografts. Clin Cancer Res. 1999;5:3594–3602. [PubMed] [Google Scholar]
- Evans AE, Kisselbach KD, Liu X, Eggert A, Ikegaki N, Camoratto AM, Dionne C, Brodeur GM. Effect of CEP-751 (KT-6587) on neuroblastoma xenografts expressing TrkB. Med Pediatr Oncol. 2001;36:181–184. doi: 10.1002/1096-911X(20010101)36:1<181::AID-MPO1043>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Fabrizio P, Pozza F, Pletcher SD, Gendron CM, Longo VD. Regulation of longevity and stress resistance by Sch9 in yeast. Science. 2001;292:288–290. doi: 10.1126/science.1059497. [DOI] [PubMed] [Google Scholar]
- Fink JS, Kalda A, Ryu H, Stack EC, Schwarzschild MA, Chen JF, Ferrante RJ. Genetic and pharmacological inactivation of the adenosine A2A receptor attenuates 3-nitropropionic acid-induced striatal damage. J Neurochem. 2004;88:538–544. doi: 10.1046/j.1471-4159.2003.02145.x. [DOI] [PubMed] [Google Scholar]
- Fischer OM, Giordano S, Comoglio PM, Ullrich A. Reactive oxygen species mediate Met receptor transactivation by G protein-coupled receptors and the epidermal growth factor receptor in human carcinoma cells. J Biol Chem. 2004;279:28970–28978. doi: 10.1074/jbc.M402508200. [DOI] [PubMed] [Google Scholar]
- Fredholm BB, Ijzerman AP, Jacobson KA, Klotz KN, Linden J. International union of pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol Rev. 2001;53:527–552. [PMC free article] [PubMed] [Google Scholar]
- Fryer HJ, Knox RJ, Strittmatter SM, Kalb RG. Excitotoxic death of a subset of embryonic rat motor neurons in vitro. J Neurochem. 1999;72:500–513. doi: 10.1046/j.1471-4159.1999.0720500.x. [DOI] [PubMed] [Google Scholar]
- Fryer HJ, Wolf DH, Knox RJ, Strittmatter SM, Pennica D, O’Leary RM, Russell DS, Kalb RG. Brain-derived neurotrophic factor induces excitotoxic sensitivity in cultured embryonic rat spinal motor neurons through activation of the phosphatidylinositol 3-kinase pathway. J Neurochem. 2000;74:582–595. doi: 10.1046/j.1471-4159.2000.740582.x. [DOI] [PubMed] [Google Scholar]
- Golub T, Wacha S, Caroni P. Spatial and temporal control of signaling through lipid rafts. Curr Opin Neurobiol. 2004;14:542–550. doi: 10.1016/j.conb.2004.08.003. [DOI] [PubMed] [Google Scholar]
- Gonzalez M, Ruggiero FP, Chang Q, Shi Y-J, Rich MM, Kraner S, Balice-Gordon RJ. Disruption of TrkB-mediated signaling induces disassembly of postsynaptic receptor clusters at the neuromuscular junction. Neuron. 1999;24:567–583. doi: 10.1016/s0896-6273(00)81113-7. [DOI] [PubMed] [Google Scholar]
- Grewal SS, York RD, Stork PJ. Extracellular-signal-regulated kinase signalling in neurons. Curr Opin Neurobiol. 1999;9:544–553. doi: 10.1016/S0959-4388(99)00010-0. [DOI] [PubMed] [Google Scholar]
- Groeneveld GJ, Veldink JH, van der Tweel I, Kalmijn S, Beijer C, de Visser M, Wokke JH, Franssen H, van den Berg LH. A randomized sequential trial of creatine in amyotrophic lateral sclerosis. Ann Neurol. 2003;53:437–445. doi: 10.1002/ana.10554. [DOI] [PubMed] [Google Scholar]
- Gurney ME, Cutting FB, Zhai P, Doble A, Taylor CP, Andrus PK, Hall ED. Benefit of vitamin E, riluzole, and gabapentin in a transgenic model of familial amyotrophic lateral sclerosis. Ann Neurol. 1996;39:147–157. doi: 10.1002/ana.410390203. [DOI] [PubMed] [Google Scholar]
- Hanke JH, Gardner JP, Dow RL, Changelian PS, Brissette WH, Weringer EJ, Pollok BA, Connelly PA. Discovery of a novel, potent, and Src family-selective tyrosine kinase inhibitor. Study of Lck- and FynT-dependent T cell activation. J Biol Chem. 1996;271:695–701. doi: 10.1074/jbc.271.2.695. [DOI] [PubMed] [Google Scholar]
- Hanson MG, Jr, Shen S, Wiemelt AP, McMorris FA, Barres BA. Cyclic AMP elevation is sufficient to promote the survival of spinal motor neurons in vitro. J Neurosci. 1998;18:7361–7371. doi: 10.1523/JNEUROSCI.18-18-07361.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson CE, Camu W, Mettling C, Gouin A, Poulsen K, Karihaloo M, Rullamas J, Evans T, McMahon SB, Armanini MP, Berkemeier L, Phillips HS, Rosenthal A. Neurotrophins promote motor neuron survival and are present in embryonic limb bud. Nature. 1993;363:266–270. doi: 10.1038/363266a0. [DOI] [PubMed] [Google Scholar]
- Hirschhorn R, Roegner-Maniscalco V, Kuritsky L, Rosen FS. Bone marrow transplantation only partially restores purine metabolites to normal in adenosine deaminase-deficient patients. J Clin Invest. 1981;68:1387–1393. doi: 10.1172/JCI110389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu P, Kalb RG. BDNF heightens the sensitivity of motor neurons to excitotoxic insults through activation of TrkB. J Neurochem. 2003;84:1421–1430. doi: 10.1046/j.1471-4159.2003.01599.x. [DOI] [PubMed] [Google Scholar]
- Hur EM, Park YS, Lee BD, Jang IH, Kim HS, Kim TD, Suh PG, Ryu SH, Kim KT. Sensitization of epidermal growth factor-induced signaling by bradykinin is mediated by c-Src. Implications for a role of lipid microdomains. J Biol Chem. 2004;279:5852–5860. doi: 10.1074/jbc.M311687200. [DOI] [PubMed] [Google Scholar]
- Ikeda K, Kurokawa M, Aoyama S, Kuwana Y. Neuroprotection by adenosine A2A receptor blockade in experimental models of Parkinson’s disease. J Neurochem. 2002;80:262–270. doi: 10.1046/j.0022-3042.2001.00694.x. [DOI] [PubMed] [Google Scholar]
- Ishikawa Y, Ikeuchi T, Hatanaka H. Brain-derived neurotrophic factor accelerates nitric oxide donor-induced apoptosis of cultured cortical neurons. J Neurochem. 2000;75:494–502. doi: 10.1046/j.1471-4159.2000.0750494.x. [DOI] [PubMed] [Google Scholar]
- Ji Y, Pang PT, Feng L, Lu B. Cyclic AMP controls BDNF-induced TrkB phosphorylation and dendritic spine formation in mature hippocampal neurons. Nat Neurosci. 2005;8:164–172. doi: 10.1038/nn1381. [DOI] [PubMed] [Google Scholar]
- Jones KR, Farinas I, Backus C, Reichardt LF. Targeted Disruption of the BDNF Gene Perturbs Brain and Sensory Neuron Development but not Motor Neuron Development. Cell. 1994;76:989–999. doi: 10.1016/0092-8674(94)90377-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalb R. The protean actions of neurotrophins and their receptors on the life and death of neurons. Trends Neurosci. 2005;28:5–11. doi: 10.1016/j.tins.2004.11.003. [DOI] [PubMed] [Google Scholar]
- Kaplan DR, Miller FD. Neurotrophin signal transduction in the nervous system. Curr Opin Neurobiol. 2000;10:381–391. doi: 10.1016/s0959-4388(00)00092-1. [DOI] [PubMed] [Google Scholar]
- Kase H, Aoyama S, Ichimura M, Ikeda K, Ishii A, Kanda T, Koga K, Koike N, Kurokawa M, Kuwana Y, Mori A, Nakamura J, Nonaka H, Ochi M, Saki M, Shimada J, Shindou T, Shiozaki S, Suzuki F, Takeda M, et al. Progress in pursuit of therapeutic A2A antagonists: the adenosine A2A receptor selective antagonist KW6002: research and development toward a novel nondopaminergic therapy for Parkinson’s disease. Neurology. 2003;61:S97–S100. doi: 10.1212/01.wnl.0000095219.22086.31. [DOI] [PubMed] [Google Scholar]
- Kawabuchi M, Satomi Y, Takao T, Shimonishi Y, Nada S, Nagai K, Tarakhovsky A, Okada M. Transmembrane phosphoprotein Cbp regulates the activities of Src-family tyrosine kinases. Nature. 2000;404:999–1003. doi: 10.1038/35010121. [DOI] [PubMed] [Google Scholar]
- Kawakatsu H, Sakai T, Takagaki Y, Shinoda Y, Saito M, Owada MK, Yano J. A new monoclonal antibody which selectively recognizes the active form of Src tyrosine kinase. J Biol Chem. 1996;271:5680–5685. doi: 10.1074/jbc.271.10.5680. [DOI] [PubMed] [Google Scholar]
- Keely SJ, Calandrella SO, Barrett KE. Carbachol-stimulated transactivation of epidermal growth factor receptor and mitogen-activated protein kinase in T(84) cells is mediated by intracellular Ca(2+), PYK-2, and p60(src) J Biol Chem. 2000;275:12619–12625. doi: 10.1074/jbc.275.17.12619. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Hwang JJ, Behrens MM, Snider BJ, Choi DW, Koh JY. TrkB mediates BDNF-induced potentiation of neuronal necrosis in cortical culture. Neurobiol Dis. 2003;14:110–119. doi: 10.1016/s0969-9961(03)00103-7. [DOI] [PubMed] [Google Scholar]
- Kobayashi S, Conforti L, Pun RY, Millhorn DE. Adenosine modulates hypoxia-induced responses in rat PC12 cells via the A2A receptor. J Physiol (Lond) 1998;508:95–107. doi: 10.1111/j.1469-7793.1998.095br.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koga K, Kurokawa M, Ochi M, Nakamura J, Kuwana Y. Adenosine A(2A) receptor antagonists KF17837 and KW-6002 potentiate rotation induced by dopaminergic drugs in hemi-Parkinsonian rats. Eur J Pharmacol. 2000;408:249–255. doi: 10.1016/s0014-2999(00)00745-7. [DOI] [PubMed] [Google Scholar]
- Koh JY, Gwag BJ, Lobner D, Choi DW. Potentiated necrosis of cultured cortical neurons by neurotrophins. Science. 1995;268:573–575. doi: 10.1126/science.7725105. [DOI] [PubMed] [Google Scholar]
- Korte M, Staiger V, Griesbeck O, Thoenen H, Bonhoeffer T. The involvement of brain-derived neurotrophic factor in hippocampal long-term potentiation revealed by gene targeting experiments. J Physiol (Paris) 1996;90:157–164. doi: 10.1016/s0928-4257(97)81415-5. [DOI] [PubMed] [Google Scholar]
- Krajewska WM, Maslowska I. Caveolins: structure and function in signal transduction. Cell Mol Biol Lett. 2004;9:195–220. [PubMed] [Google Scholar]
- Krieg T, Qin Q, McIntosh EC, Cohen MV, Downey JM. ACh and adenosine activate PI3-kinase in rabbit hearts through transactivation of receptor tyrosine kinases. Am J Physiol Heart Circ Physiol. 2002;283:H2322–H2330. doi: 10.1152/ajpheart.00474.2002. [DOI] [PubMed] [Google Scholar]
- Kruman II, Pedersen WA, Springer JE, Mattson MP. ALS-linked Cu/Zn-SOD mutation increases vulnerability of motor neurons to excitotoxicity by a mechanism involving increased oxidative stress and perturbed calcium homeostasis. Exp Neurol. 1999;160:28–39. doi: 10.1006/exnr.1999.7190. [DOI] [PubMed] [Google Scholar]
- Kust BM, Copray JC, Brouwer N, Troost D, Boddeke HW. Elevated levels of neurotrophins in human biceps brachii tissue of amyotrophic lateral sclerosis. Exp Neurol. 2002;177:419–427. doi: 10.1006/exnr.2002.8011. [DOI] [PubMed] [Google Scholar]
- Kwak S, Nakamura R. Acute and late neurotoxicity in the rat spinal cord in vivo induced by glutamate receptor agonists. J Neurol Sci. 1995;129([Suppl]):99–103. doi: 10.1016/0022-510x(95)00076-e. [DOI] [PubMed] [Google Scholar]
- Lamb K, Bielefeldt K. Rapid effects of neurotrophic factors on calcium homeostasis in rat visceral afferent neurons. Neurosci Lett. 2003;336:9–12. doi: 10.1016/s0304-3940(02)01223-5. [DOI] [PubMed] [Google Scholar]
- Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL. Diffusible, nonfibrillar ligands derived from Abeta1–42 are potent central nervous system neurotoxins. Proc Natl Acad Sci USA. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee FS, Chao MV. Activation of Trk neurotrophin receptors in the absence of neurotrophins. Proc Natl Acad Sci USA. 2001;98:3555–3560. doi: 10.1073/pnas.061020198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee FS, Rajagopal R, Chao MV. Distinctive features of Trk neurotrophin receptor transactivation by G protein-coupled receptors. Cytokine Growth Factor Rev. 2002a;13:11–17. doi: 10.1016/s1359-6101(01)00024-7. [DOI] [PubMed] [Google Scholar]
- Lee FS, Rajagopal R, Kim AH, Chang PC, Chao MV. Activation of Trk neurotrophin receptor signaling by pituitary adenylate cyclase-activating polypeptides. J Biol Chem. 2002b;277:9096–9102. doi: 10.1074/jbc.M107421200. [DOI] [PubMed] [Google Scholar]
- Lefkowitz RJ, Shenoy SK. Transduction of receptor signals by beta-arrestins. Science. 2005;308:512–517. doi: 10.1126/science.1109237. [DOI] [PubMed] [Google Scholar]
- Lennmyr F, Ericsson A, Gerwins P, Akterin S, Ahlstrom H, Terent A. Src family kinase-inhibitor PP2 reduces focal ischemic brain injury. Acta Neurol Scand. 2004;110:175–179. doi: 10.1111/j.1600-0404.2004.00306.x. [DOI] [PubMed] [Google Scholar]
- Li M, Ona VO, Guegan C, Chen M, Jackson-Lewis V, Andrews LJ, Olszewski AJ, Stieg PE, Lee JP, Przedborski S, Friedlander RM. Functional role of caspase-1 and caspase-3 in an ALS transgenic mouse model. Science. 2000;288:335–339. doi: 10.1126/science.288.5464.335. [DOI] [PubMed] [Google Scholar]
- Li SN, Wong PT. The adenosine receptor agonist, APNEA, increases calcium influx into rat cortical synaptosomes through N-type channels associated with A2a receptors. Neurochem Res. 2000;25:457–459. doi: 10.1023/a:1007503907823. [DOI] [PubMed] [Google Scholar]
- Liu X, Ernfors P, Wu H, Jaenisch R. Sensory but not motor neuron deficits in mice lacking NT4 and BDNF. Nature. 1995;375:238–241. doi: 10.1038/375238a0. [DOI] [PubMed] [Google Scholar]
- Luttrell DK, Luttrell LM. Not so strange bedfellows: G-protein-coupled receptors and Src family kinases. Oncogene. 2004;23:7969–7978. doi: 10.1038/sj.onc.1208162. [DOI] [PubMed] [Google Scholar]
- Luttrell LM, Daaka Y, Lefkowitz RJ. Regulation of tyrosine kinase cascades by G-protein-coupled receptors. Curr Opin Cell Biol. 1999;11:177–183. doi: 10.1016/s0955-0674(99)80023-4. [DOI] [PubMed] [Google Scholar]
- Ma L, Huang YZ, Pitcher GM, Valtschanoff JG, Ma YH, Feng LY, Lu B, Xiong WC, Salter MW, Weinberg RJ, Mei L. Ligand-dependent recruitment of the ErbB4 signaling complex into neuronal lipid rafts. J Neurosci. 2003;23:3164–3175. doi: 10.1523/JNEUROSCI.23-08-03164.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messaoudi E, Ying SW, Kanhema T, Croll SD, Bramham CR. Brain-derived neurotrophic factor triggers transcription-dependent, late phase long-term potentiation in vivo. J Neurosci. 2002;22:7453–7461. doi: 10.1523/JNEUROSCI.22-17-07453.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizoguchi Y, Nabekura J. Sustained intracellular Ca2+ elevation induced by a brief BDNF application in rat visual cortex neurons. NeuroReport. 2003;14:1481–1483. doi: 10.1097/00001756-200308060-00015. [DOI] [PubMed] [Google Scholar]
- Mizoguchi Y, Monji A, Nabekura J. Brain-derived neurotrophic factor induces long-lasting Ca2+-activated K+ currents in rat visual cortex neurons. Eur J Neurosci. 2002;16:1417–1424. doi: 10.1046/j.1460-9568.2002.02198.x. [DOI] [PubMed] [Google Scholar]
- Mojsilovic-Petrovic J, Arneja A, Kalb R. Enprofylline protects motor neurons from in vitro excitotoxic challenge. Neurodegener Dis. 2005;2:160–165. doi: 10.1159/000089621. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay D, Tsiokas L, Zhou XM, Foster D, Brugge JS, Sukhatme VP. Hypoxic induction of human vascular endothelial growth factor expression through c-Src activation. Nature. 1995;375:577–581. doi: 10.1038/375577a0. [DOI] [PubMed] [Google Scholar]
- Murray B, Alessandrini A, Cole AJ, Yee AG, Furshpan EJ. Inhibition of the p44/42 MAP kinase pathway protects hippocampal neurons in a cell-culture model of seizure activity. Proc Natl Acad Sci USA. 1998;95:11975–11980. doi: 10.1073/pnas.95.20.11975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neve RL, Howe JR, Hong S, Kalb RG. Introduction of the glutamate receptor subunit 1 into motor neurons in vitro and in vivo using a recombinant herpes simplex virus. Neuroscience. 1997;79:435–447. doi: 10.1016/s0306-4522(96)00645-8. [DOI] [PubMed] [Google Scholar]
- Oak JN, Lavine N, Van Tol HH. Dopamine D(4) and D(2L) receptor stimulation of the mitogen-activated protein kinase pathway is dependent on trans-activation of the platelet-derived growth factor receptor. Mol Pharmacol. 2001;60:92–103. doi: 10.1124/mol.60.1.92. [DOI] [PubMed] [Google Scholar]
- Ohta A, Sitkovsky M. Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature. 2001;414:916–920. doi: 10.1038/414916a. [DOI] [PubMed] [Google Scholar]
- Pang PT, Teng HK, Zaitsev E, Woo NT, Sakata K, Zhen S, Teng KK, Yung WH, Hempstead BL, Lu B. Cleavage of proBDNF by tPA/plasmin is essential for long-term hippocampal plasticity. Science. 2004;306:487–491. doi: 10.1126/science.1100135. [DOI] [PubMed] [Google Scholar]
- Paratcha G, Ibanez CF. Lipid rafts and the control of neurotrophic factor signaling in the nervous system: variations on a theme. Curr Opin Neurobiol. 2002;12:542–549. doi: 10.1016/s0959-4388(02)00363-x. [DOI] [PubMed] [Google Scholar]
- Pehar M, Cassina P, Vargas MR, Castellanos R, Viera L, Beckman JS, Estevez AG, Barbeito L. Astrocytic production of nerve growth factor in motor neuron apoptosis: implications for amyotrophic lateral sclerosis. J Neurochem. 2004;89:464–473. doi: 10.1111/j.1471-4159.2004.02357.x. [DOI] [PubMed] [Google Scholar]
- Peiro S, Comella JX, Enrich C, Martin-Zanca D, Rocamora N. PC12 cells have caveolae that contain TrkA. Caveolae-disrupting drugs inhibit nerve growth factor-induced, but not epidermal growth factor-induced, MAPK phosphorylation. J Biol Chem. 2000;275:37846–37852. doi: 10.1074/jbc.M000487200. [DOI] [PubMed] [Google Scholar]
- Piiper A, Zeuzem S. Receptor tyrosine kinases are signaling intermediates of G protein-coupled receptors. Curr Pharm Des. 2004;10:3539–3545. doi: 10.2174/1381612043382936. [DOI] [PubMed] [Google Scholar]
- Poo M. Neurotrophins as synaptic modulators. Nat Rev Neurosci. 2001;2:24–32. doi: 10.1038/35049004. [DOI] [PubMed] [Google Scholar]
- Puls I, Jonnakuty C, LaMonte BH, Holzbaur EL, Tokito M, Mann E, Floeter MK, Bidus K, Drayna D, Oh SJ, Brown RH, Ludlow CL, Fischbeck KH. Mutant dynactin in motor neuron disease. Nat Genet. 2003 doi: 10.1038/ng1123. [DOI] [PubMed] [Google Scholar]
- Rajagopal R, Chen ZY, Lee FS, Chao MV. Transactivation of Trk neurotrophin receptors by G-protein-coupled receptor ligands occurs on intracellular membranes. J Neurosci. 2004;24:6650–6658. doi: 10.1523/JNEUROSCI.0010-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robeva SA, Woodard RL, Jin X, Gao Z, Bhattacharya S, Taylor HE, Rosin DL, Linden J. Molelcular characterization of recombinant human adenosine receptors. Drug Dev Res. 1996;39:243–252. [Google Scholar]
- Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentatl A, Donaldson D, Goto J, O’Regan JP, Deng H-X, Rahmani Z, Krizus A, McKenna-Yasek D, Cayabyab A, Gaston S, Tanzi RE, Halperin JJ, Herzfeldt B, Van den Berg R, Hung W-Y, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Jin L, Dykes-Hoberg M, Kuncl RW. Chronic inhibition of glutamate uptake produces a model of slow neurotoxicity. Proc Natl Acad Sci USA. 1993;90:6591–6595. doi: 10.1073/pnas.90.14.6591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothstein JD, Patel S, Regan MR, Haenggeli C, Huang YH, Bergles DE, Jin L, Dykes Hoberg M, Vidensky S, Chung DS, Toan SV, Bruijn LI, Su ZZ, Gupta P, Fisher PB. Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature. 2005;433:73–77. doi: 10.1038/nature03180. [DOI] [PubMed] [Google Scholar]
- Roy J, Minotti S, Dong L, Figlewicz DA, Durham HD. Glutamate potentiates the toxicity of mutant Cu/Zn-superoxide dismutase in motor neurons by postsynaptic calcium-dependent mechanisms. J Neurosci. 1998;18:9673–9684. doi: 10.1523/JNEUROSCI.18-23-09673.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runden E, Seglen PO, Haug FM, Ottersen OP, Wieloch T, Shamloo M, Laake JH. Regional selective neuronal degeneration after protein phosphatase inhibition in hippocampal slice cultures: evidence for a MAP kinase-dependent mechanism. J Neurosci. 1998;18:7296–7305. doi: 10.1523/JNEUROSCI.18-18-07296.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schutz B, Reimann J, Dumitrescu-Ozimek L, Kappes-Horn K, Landreth GE, Schurmann B, Zimmer A, Heneka MT. The oral antidiabetic pioglitazone protects from neurodegeneration and amyotrophic lateral sclerosis-like symptoms in superoxide dismutase-G93A transgenic mice. J Neurosci. 2005;25:7805–7812. doi: 10.1523/JNEUROSCI.2038-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz PM, Borghesani PR, Levy RL, Pomeroy SL, Segal RA. Abnormal cerebellar development and foliation in BDNF−/− mice reveals a role for neurotrophins in CNS patterning. Neuron. 1997;19:269–281. doi: 10.1016/s0896-6273(00)80938-1. [DOI] [PubMed] [Google Scholar]
- Shiozaki S, Ichikawa S, Nakamura J, Kitamura S, Yamada K, Kuwana Y. Actions of adenosine A2A receptor antagonist KW-6002 on drug-induced catalepsy and hypokinesia caused by reserpine or MPTP. Psychopharmacology (Berl) 1999;147:90–95. doi: 10.1007/s002130051146. [DOI] [PubMed] [Google Scholar]
- Silos-Santiago I, Fagan AM, Garber M, Fritzsch B, Barbacid M. Severe sensory deficits but normal CNS development in newborn mice lacking TrkB and TrkC tyrosine protein kinase receptors. Eur J Neurosci. 1997;9:2045–2056. doi: 10.1111/j.1460-9568.1997.tb01372.x. [DOI] [PubMed] [Google Scholar]
- Simons K, Toomre D. Lipid rafts and signal transduction. Nat Rev Mol Cell Biol. 2000;1:31–39. doi: 10.1038/35036052. [DOI] [PubMed] [Google Scholar]
- Smith BD, Levis M, Beran M, Giles F, Kantarjian H, Berg K, Murphy K, Dauses T, Allebach J, Small D. Single agent CEP-701, a novel FLT3 inhibitor, shows biological and clinical activity in patients with relapsed or refractory acute myeloid leukemia. Blood. 2004;103:3669–3676. doi: 10.1182/blood-2003-11-3775. [DOI] [PubMed] [Google Scholar]
- Suzuki S, Numakawa T, Shimazu K, Koshimizu H, Hara T, Hatanaka H, Mei L, Lu B, Kojima M. BDNF-induced recruitment of TrkB receptor into neuronal lipid rafts: roles in synaptic modulation. J Cell Biol. 2004;167:1205–1215. doi: 10.1083/jcb.200404106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tansey MG, Baloh RH, Milbrandt J, Johnson EM., Jr GFRalpha-mediated localization of RET to lipid rafts is required for effective downstream signaling, differentiation, and neuronal survival. Neuron. 2000;25:611–623. doi: 10.1016/s0896-6273(00)81064-8. [DOI] [PubMed] [Google Scholar]
- Thiel M, Chouker A, Ohta A, Jackson E, Caldwell C, Smith P, Lukashev D, Bittmann I, Sitkovsky MV. Oxygenation inhibits the physiological tissue-protecting mechanism and thereby exacerbates acute inflammatory lung injury. PLoS Biol. 2005;3:e174. doi: 10.1371/journal.pbio.0030174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsvyetlynska NA, Hill RH, Grillner S. Role of AMPA receptor desensitization and the side effects of a DMSO vehicle on reticulospinal EPSPs and locomotor activity. J Neurophysiol. 2005;94:3951–3960. doi: 10.1152/jn.00201.2005. [DOI] [PubMed] [Google Scholar]
- Undevia SD, Vogelzang NJ, Mauer AM, Janisch L, Mani S, Ratain MJ. Phase I clinical trial of CEP-2563 dihydrochloride, a receptor tyrosine kinase inhibitor, in patients with refractory solid tumors. Invest New Drugs. 2004;22:449–458. doi: 10.1023/B:DRUG.0000036687.26604.8c. [DOI] [PubMed] [Google Scholar]
- Ushio-Fukai M, Hilenski L, Santanam N, Becker PL, Ma Y, Griendling KK, Alexander RW. Cholesterol depletion inhibits epidermal growth factor receptor transactivation by angiotensin II in vascular smooth muscle cells: role of cholesterol-rich microdomains and focal adhesions in angiotensin II signaling. J Biol Chem. 2001;276:48269–48275. doi: 10.1074/jbc.M105901200. [DOI] [PubMed] [Google Scholar]
- Van Damme P, Leyssen M, Callewaert G, Robberecht W, Van Den Bosch L. The AMPA receptor antagonist NBQX prolongs survival in a transgenic mouse model of amyotrophic lateral sclerosis. Neurosci Lett. 2003;343:81–84. doi: 10.1016/s0304-3940(03)00314-8. [DOI] [PubMed] [Google Scholar]
- Waters C, Pyne S, Pyne NJ. The role of G-protein coupled receptors and associated proteins in receptor tyrosine kinase signal transduction. Semin Cell Dev Biol. 2004;15:309–323. doi: 10.1016/j.semcdb.2003.12.020. [DOI] [PubMed] [Google Scholar]
- Zahradka P, Litchie B, Storie B, Helwer G. Transactivation of the insulin-like growth factor-I receptor by angiotensin II mediates downstream signaling from the angiotensin II type 1 receptor to phosphatidylinositol 3-kinase. Endocrinology. 2004;145:2978–2987. doi: 10.1210/en.2004-0029. [DOI] [PubMed] [Google Scholar]