

Abstract
Dysregulation of dopamine homeostasis and elevation of the cytosolic level of the transmitter have been suggested to underlie the vulnerability of catecholaminergic neurons in Parkinson’s disease. Because several known mutations in α-synuclein or overexpression of the wild-type (WT) protein causes familial forms of Parkinson’s disease, we investigated possible links between α-synuclein pathogenesis and dopamine homeostasis. Chromaffin cells isolated from transgenic mice that overexpress A30P α-synuclein displayed significantly increased cytosolic catecholamine levels as measured by intracellular patch electrochemistry, whereas cells overexpressing the WT protein and those from knock-out animals were not different from controls. Likewise, catechol concentrations were higher in l-DOPA-treated PC12 cells overexpressing A30P or A53T compared with those expressing WT α-synuclein, although the ability of cells to maintain a low cytosolic dopamine level after l-DOPA challenge was markedly inhibited by either protein. We also found that incubation with low-micromolar concentrations of WT, A30P, or A53T α-synuclein inhibited ATP-dependent maintenance of pH gradients in isolated chromaffin vesicles and that the WT protein was significantly less potent in inducing the proton leakage. In summary, we demonstrate that overexpression of different types of α-synuclein disrupts vesicular pH and leads to a marked increase in the levels of cytosolic catechol species, an effect that may in turn trigger cellular oxyradical damage. Although multiple molecular mechanisms may be responsible for the perturbation of cytosolic catecholamine homeostasis, this study provides critical evidence about how α-synuclein might exert its cytotoxicity and selectively damage catecholaminergic cells.
Keywords: synuclein, cytosol, catecholamine, IPE, PC12, chromaffin cell, vesicular pH
Introduction
Parkinson’s disease (PD) is a neurodegenerative disorder characterized by a loss of dopaminergic neurons in substantia nigra pars compacta, a loss of norepinephrinergic neurons in the locus ceruleus, and the presence of intraneuronal inclusions known as Lewy bodies (Dauer and Przedborski, 2003). The major component of Lewy bodies, α-synuclein, is linked to familial forms of PD (Vila and Przedborski, 2004), although the mechanism by which point mutations in or overexpression of this protein provoke the demise of catecholaminergic neurons in PD remains unknown (Dauer and Przedborski, 2003; Eriksen et al., 2005). Monomeric α-synuclein is mainly unfolded in solution (Eliezer et al., 2001); however, both WT and mutant α-synuclein can form oligomeric protofibrils and amyloid fibrils that resemble those in Lewy bodies (Conway et al., 1998; Giasson et al., 2003). Beyond the possible cytotoxicity of α-synuclein polymerization products (Conway et al., 2000a; Rochet et al., 2004), the protein may induce mitochondrial and Golgi deficits (Gosavi et al., 2002; Song et al., 2004; Moore et al., 2005) or an inhibition of protein degradation via chaperone-mediated autophagy (Cuervo et al., 2004) or the proteosome pathway (Stefanis et al., 2001; Tanaka et al., 2001; Petrucelli et al., 2002).
Several reports suggest that α-synuclein may affect neuronal catecholamine homeostasis, leading to a buildup of cytosolic dopamine and a consequent oxidative stress (Lotharius et al., 2002; Perez and Hastings, 2004; Sidhu et al., 2004). Cytosolic catecholamines are translocated into the vesicles by the vesicular monoamine transporter (VMAT), using the energy of a proton concentration gradient between the cytosol and the vesicle lumen generated by the vesicular H+-ATPase. The maintenance of the gradient relies on the integrity of vesicular membrane, and even minor changes in its permeability could lead to significant perturbation of the equilibrium proton and catecholamine concentrations. α-Synuclein interacts with synthetic phospholipids (Davidson et al., 1998; Jo et al., 2002), brain-derived vesicles (Jensen et al., 1998), and lipid rafts purified from native membranes (Fortin et al., 2004; Kubo et al., 2005). The ability of α-synuclein protofibrils to form annular structures reminiscent of bacterial pore-forming toxins could account for the in vitro permeabilization activity of α-synuclein (Volles et al., 2001; Lashuel et al., 2002). Additionally, α-synuclein protofibrils are stabilized by dopamine (Conway et al., 2000a; Rochet et al., 2004), which may underlie the selective vulnerability of catecholaminergic neurons in PD.
The aforementioned findings raise the possibility of a vicious pathogenic cycle: oligomerization of α-synuclein leads to the leakage of dopamine from synaptic vesicles into the cytosol, which could promote additional accumulation of the permeabilizing protofibrils. To investigate the proposed link between α-synuclein pathogenesis and intracellular dopamine homeostasis, we used intracellular patch electrochemistry (IPE) to determine cytosolic catecholamine and total catechol concentrations in adrenal chromaffin and PC12 cells stably overexpressing WT, A30P, or A53T α-synuclein. We report that overexpression of α-synuclein indeed elevated cytosolic catechols, likely because of the leakage of transmitter from the storage vesicles. Mutant α-synucleins were significantly more potent in both increasing the concentration of cytosolic catecholamines and inducing the leakage of protons from isolated chromaffin vesicles.
Materials and Methods
PC12 cell cultures.
The cells were grown on rat tail collagen-coated dishes in RPMI 1640 media supplemented with 5% fetal bovine serum, 10% heat-inactivated horse serum, 50 U/ml penicillin, and 50 μg/ml streptomycin as described previously (Greene and Tischler, 1976; Rukenstein et al., 1991). Cell lines overexpressing human WT or mutant A30P or A53T α-synuclein types were generated based on the TET-On system (Clontech, Mountain View, CA). The cloning of cDNA from WT or mutant alleles of α-synuclein inserts into a pcDNA3 vector (Invitrogen, San Diego, CA) has been described previously (Stefanis et al., 2001). To subclone genes into a pTRE vector (Clontech), a SacII site was added to the 5′ end of each DNA fragment by PCR using pcDNA3-based constructs as templates. The correctness of obtained fragments was verified by sequencing, and the PCR products were restriction digested with SacII/XbaI. PC12 cells were then cotransfected with a pTRE vector encoding WT, A30P, or A53T α-synuclein and a pTK-Hyg vector encoding hygromycin resistance, which allowed us to select stable transfectants. Two control cell lines were generated, TET-On cells transfected with empty pTRE vector and cells expressing enhanced green fluorescent protein (EGFP) from pBI-EGFP vector (Clontech). Clones were selected after hygromycin treatment (400 μg/ml; Roche Diagnostics, Indianapolis, IN), and monoclonal and polyclonal cell lines were generated. Screening of clones for α-synuclein overexpression was performed by Western blotting (mouse monoclonal anti-α-Syn-1; BD Biosciences Pharmingen, San Diego, CA). For this purpose, cells were cultured using Tet-free FBS (Clontech), and lysates were collected 24 h after exposure to the medium with or without doxycycline (2 ng/ml).
Multiple clonal cell lines cultured in the absence of doxycycline produced baseline α-synuclein expression that was stable yet insufficient for the induction of an autophagic stress response described previously for PC12 cells with high α-synuclein overexpression levels (Stefanis et al., 2001). Morphological characterization of PC12 cell lines revealed substantial variability in cell size, which did not seem to depend on the presence of overexpressed α-synuclein. Two cell lines (empty vector and A53T) with distinctly smaller cell sizes (cell diameters more than twofold smaller) also showed no cytosolic catecholamine response after l-DOPA treatment (see below) and were excluded from additional analysis.
Measurement of total dopamine and DOPAC levels by HPLC.
PC12 cells were grown to ∼70% confluency in 4 cm culture dishes as described above. Cells were treated with 100 μm l-DOPA at 37°C for 1 h before sample collection. The cells were rinsed twice with ice-cold PBS and collected by washing them off with a 1 ml pipette. After centrifugation (4000 × g for 2 min), the supernatant was removed, and the cells were resuspended in 100 μl of PBS. One aliquot (50 μl) of the cell suspension was centrifuged (4000 × g for 2 min), 40 μl of supernatant was discarded, and the remaining cell precipitate was mixed with 90 μl of lysis buffer containing 20 mm HEPES, pH 7.4, 25 mm KCl, 1 mm phenylmethanesulfonyl fluoride, and 0.5% Nonidet P-40. The suspension was mixed well and placed on ice for 10 min. After centrifugation (10,000 × g for 2 min), the protein concentration in the supernatant was determined using Bradford reagent (Bio-Rad, Hercules, CA) with BSA as the standard and used later for normalization of dopamine amounts in each sample. The second 50 μl aliquot of cell suspension was deproteinized by mixing with an equal amount of 0.5 m trichloric acid solution. After sonication at room temperature for 10 min, the mixture was centrifuged (10,000 × g for 2 min), and the supernatant was used for dopamine and DOPAC determination by HPLC with electrochemical detection as described previously (Pothos et al., 1996). Briefly, samples were diluted 100- to 1000-fold depending on the catechol content and separated on VeloSep RP-18, 3 μm, 100 × 3.2 mm column (Chrom Tech, Apple Valley, MN) using Agilent Technologies (Palo Alto, CA) 1100 series quaternary pump and mobile phase containing 45 mm NaH2PO4, pH 3.2, 0.2 mm EDTA, 1.4 mm heptanesulfonic acid, and 5% methanol. Compounds were detected on ESA (Chelmsford, MA) Coulochem II electrochemical detector at 350 mV oxidation potential. Catechol amounts were calculated from areas under HPLC peaks using calibration curves and normalized to protein concentrations in each sample.
Western blotting of PC12 cells.
PC12 cells were collected in saline [in mm: 10 HEPES, 10 KCl, 1.5 MgCl2, 1 DTT, and protease inhibitor mixture (Complete Mini; Roche Diagnostics)] and homogenized in a Dounce homogenizer. The homogenate was centrifuged for 10 min at 500 × g, the supernatant was collected, and protein concentrations were measured using the BCA assay (Pierce, Rockford, IL). Before loading onto the SDS-polyacrylamide gel, proteins were denatured by mixing the supernatant with an equal volume of 2× sample loading buffer (31 mm Tris-HCl, pH 6.8, 10% glycerol, 1% SDS, 2% β-mercaptoethanol, and 0.1% bromphenol blue) and by boiling for 5 min. Overnight incubation at 4°C was performed with the following primary antibodies: mouse monoclonal anti-α-synuclein (1:2000; BD Biosciences Pharmingen), rabbit polyclonal anti-tyrosine hydroxylase (TH; 1:10,000; Calbiochem, La Jolla, CA), rabbit polyclonal anti-aromatic acid decarboxylase (AADC; 1:1000; Abcam, Cambridge, MA), goat polyclonal anti-monoamine oxidase A (MAO-A; 1:200; Santa Cruz Biotechnology, Santa Cruz, CA), and sheep polyclonal anti-Cu/Zn superoxide dismutase (SOD1; 1:4000; Calbiochem). Anti-mouse and anti-rabbit horseradish-conjugated secondary antibodies (1:2000; GE Healthcare, Piscataway, NJ) were used respectively in the α-synuclein, TH, and AADC blots. For MAO-A and SOD1 blots, anti-goat horseradish-conjugated secondary antibodies were used (1:2000; Santa Cruz Biotechnology). Bands were quantified using the FluorChem 8800 imaging system (Alpha Innotech, San Leandro, CA) after incubating the nitrocellulose membranes in chemiluminescent substrate (SuperSignal Ultra; Pierce).
Transgenic mice.
Transgenic mice overexpressing WT or A30P α-synuclein were generated as described previously (Fortin et al., 2004). These mice express high levels of human α-synuclein throughout the brain and adrenal glands under the control of an expanded prion protein promoter. All mice were congenic (at least seven generations backcrossed) in the C57BL6/J background. α-Synuclein knock-out mice were from William Dauer (Columbia University, New York, NY) (Dauer et al., 2002).
Western blotting of adrenal glands.
Adult ∼1-year-old mice (30–35 g) were anesthetized with intraperitoneal pentobarbital injections and decapitated, and the adrenal glands were rapidly dissected and homogenized in ice-cold buffer containing the following: 4 mm HEPES, pH 7.4, 32 mm sucrose, 1 mm EGTA, and protease inhibitors (Complete Protease Inhibitor cocktail; Roche Diagnostics). Homogenates were centrifuged at 1000 × g for 10 min. Equal amounts of supernatant protein (15 or 20 μg) from transgenic and nontransgenic adrenal gland homogenates were separated in 12.5% denaturing acrylamide gel, transferred to nitrocellulose membrane, and immunoblotted with rat monoclonal anti-human α-synuclein (1:2000; 15G7; Alexis Biochemicals, San Diego, CA) or with mouse monoclonal anti-Syn-1 (1:1000; BD Biosciences Pharmingen) antibodies. α-Synuclein bands were detected using horseradish-conjugated secondary antibodies (1:2000; GE Healthcare) and enhanced chemiluminescence (Pierce).
Primary mouse chromaffin cell cultures.
Adrenal glands from 3- to 6-month-old transgenic mice and their nontransgenic littermates were dissected in ice-cold HBSS. The capsule and cortex of adrenal glands were removed, and the remaining medullas were cut into two pieces. After several washes with HBSS, the tissue was incubated with Ca2+-free collagenase IA (250 U/ml; Worthington Biochemical, Lakewood, NJ) in HBSS for 30 min at 30°C with stirring. The digested tissue was rinsed three times and triturated gently in HBSS containing 1% heat-inactivated bovine serum albumin and 0.02% DNase I. Dissociated cells were collected at 1000 × g for 2 min and resuspended in a culture medium composed of DMEM, 10% fetal bovine serum, 2 mm glutamine, 50 U/ml penicillin, and 50 μg/ml streptomycin. Cell suspension (100 μl) was plated onto poly-d-lysine and laminin-coated 1 cm2 glass wells in 50 mm dishes and, after 1–2 h, the dishes were flooded with the culture medium. Cells were maintained in a 7% CO2 incubator at 37°C. All measurements were conducted between days 2 and 4 after plating.
IPE.
Measurements of cytosolic catecholamine and total catechol concentrations were performed as described previously (Mosharov et al., 2003). Briefly, a polyethylene-coated 5 μm carbon fiber electrode (CFE) was placed inside the glass patch pipette and used in amperometric or cyclic voltammetric (CV) mode of detection using a subroutine locally written in IgorPro (WaveMetrics, Lake Oswego, OR). For the amperometric mode, the CFE potential was held at +700 mV. After achieving a seal between the cell and the patch pipette, the plasma membrane was ruptured by suction, and substances diffusing from the cytosol into the pipette were observed as a slow wave of oxidation current. After baseline subtraction, the total area of this oxidation current wave, the charge (Q), was used to estimate cytosolic catecholamine concentration (C) using the formula C = Q/(n × F × V), where n is the number of electrons transferred from a molecule to the CFE, which was assumed to equal 2 (Baur et al., 1988), F is Faraday’s constant, and V is the cytosolic volume estimated from photographs taken before each recording. Because amperometry has the same sensitivity for any molecule oxidizable at 700 mV, the signal was attributed to the sum of catechol species [dopamine, norepinephrine, epinephrine, l-DOPA, DOPAC, and 3,4-dihydroxyphenylglycol (DOPEG)].
For IPE in CV mode of detection, voltage ramps from −450 mV holding potential to +800 mV and back to −450 mV over 10 ms (scan rate of 250 mV/ms) were applied at 90 ms intervals. Subtraction voltammograms were generated, and catecholamine concentration at the maximum of the oxidation wave was calculated using calibration curves generated for CFEs with different detection surface areas (Mosharov et al., 2003). The initial dopamine concentration in cellular cytosol was calculated using the cell body volume and the volume of the pipette tip estimated from photographs taken before each recording. Because CFEs in CV mode are 15- to 20-fold more sensitive to catecholamine compared with other catechols, the majority of oxidation signal was attributable to dopamine alone (in PC12 cells) or together with norepinephrine and epinephrine (in chromaffin cells). No difference was found in the IPE signal recorded in intracellular and extracellular patch-pipette salines (Mosharov et al., 2003). We therefore used the same extracellular saline as the bath and the pipette solutions, which reduced the background CV current drifts because of the exchange of ions between the patch pipette and the bath. The saline contained the following (in mm): 10 HEPES-NaOH, pH 7.2, 128 NaCl, 2 KCl, 2 MgCl2, 1.2 CaCl2, 1 NaH2PO4, and 10 glucose.
The sensitivity of IPE to catecholamines depends on multiple factors, including the rms noise, the exposed area of the CFE, the geometry of the cell, and the proximity of the cell to the CFE (Mosharov et al., 2003). Typically, the detection limit of IPE in CV mode of detection for dopamine was ∼0.1 μm, which corresponds to an ∼50 pA catecholamine oxidation peak on the subtraction voltammogram. Because the concentration of cytosolic dopamine was below the detection limits in untreated PC12 cells, the cultures were incubated with 100 μm l-DOPA for 1 h before and during the recordings. The drug was present in the bath and in the patch pipette, which increased the baseline oxidation current but did not otherwise interfere with IPE measurements. Within each experiment, the same CFE was used for measurements from experimental and control groups of cells.
In vitro chromaffin granule acidification assay.
Bovine adrenal chromaffin granules were prepared as described previously (Nelson et al., 1988). α-Synuclein monomers containing <5% protofibrils were prepared using the Rochet technique (“flash void”) as described previously (Volles et al., 2001). For spectrofluorometric quantification of granule acidification, granules (∼100 μg) were resuspended in morpholinopropane sulfonic acid (MOPS) buffer (in mm: 20 MOPS, 200 sucrose, 100 KCl, 1 MgCl2, pH 7.4, buffered with Tris-base) in a cuvette with stirring. Acridine orange (AO) was added to a final concentration of 670 nm and baseline fluorescence (excitation, 496 nm; emission, 526 nm) was measured for 3–5 min before the addition of 1 mm Mg-ATP (Sigma, St. Louis, MO). α-Synuclein was added after ATP-dependent acidification had reached a maximum and the fluorescence signal was stable for at least 1 min. Once the fluorescence had stabilized after the addition of α-synuclein, carbonyl p-trifluoromethoxyphenylhydrazone (FCCP; 1 μm) was added to collapse the remaining concentration gradient. The V-type H+-ATPase inhibitor bafilomycin (10 nm) completely blocked ATP-dependent vesicle acidification (data not shown).
Statistical analysis.
Statistics were performed in Prism 4 (GraphPad Software, San Diego, CA). Nonparametric Kruskal–Wallis test was followed by Dunn’s multiple-comparison post hoc test.
Results
Increased catecholamine turnover in PC12 cells overexpressing α-synuclein
Rat pheochromocytoma-derived PC12 cells provide a convenient model system for the purposes of our study because although they have all the key enzymes of catecholamine homeostasis, they express very small levels of endogenous α-synuclein in their undifferentiated state (Stefanis et al., 2001). The majority of the protein detected by the Western blot was therefore attributable to the human exogenous α-synuclein (Fig. 1 A). The levels of protein overexpression were similar between the WT and A30P cell lines, whereas A53T cells expressed approximately one-half of these amounts (Fig. 1 B). Cells that overexpressed EGFP were used as controls.
Figure 1.
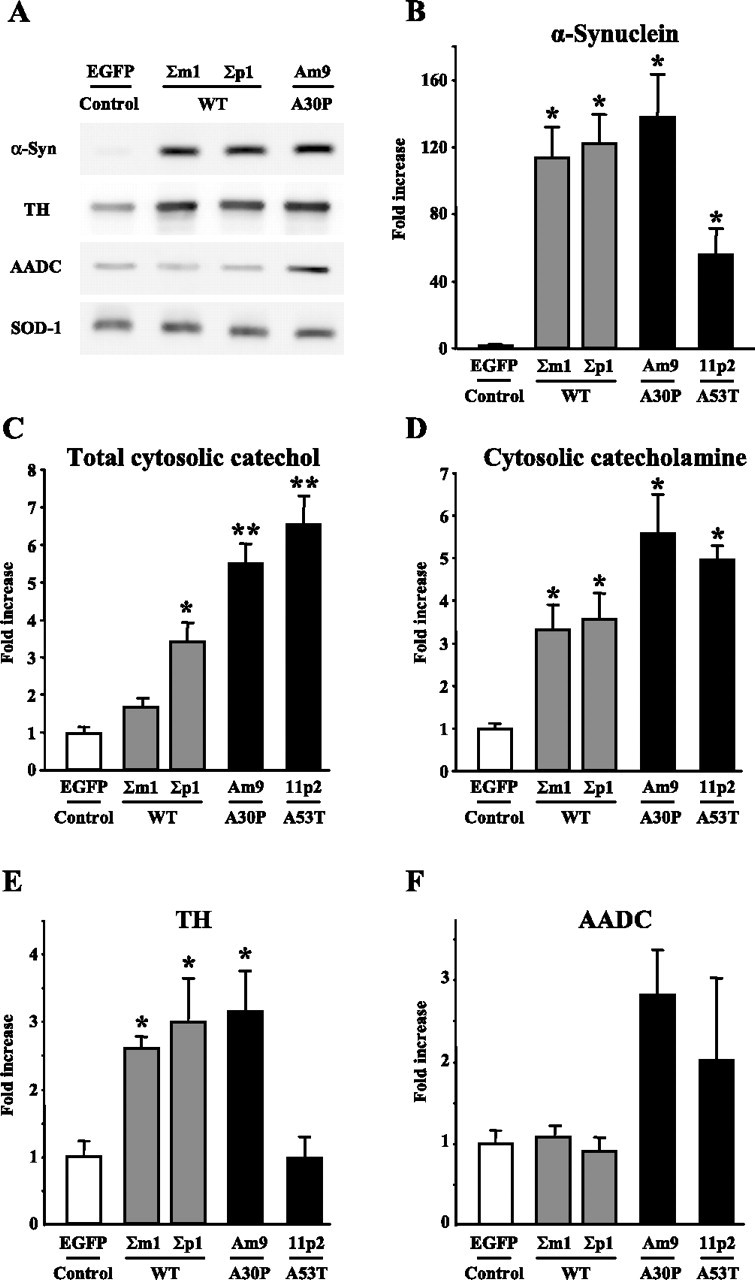
α-Synuclein overexpression increases cytosolic catechol concentrations in PC12 cells. A, Representative Western blots of α-synuclein (α-Syn), TH, AADC, and superoxide dismutase 1 (loading control) in PC12 cells extracts. B, E, F, Relative protein levels (fold increase over control) of α-synuclein (B), TH (E), and AADC (F) in PC12 cells stably overexpressing EGFP, WT α-synuclein, or mutated A30P or A53T α-synuclein (mean ± SEM; n = 4–10 gels). C, D, Cytosolic concentrations of all catechol metabolites (C) and catecholamines (D) measured by IPE in cells treated with 100 μm l-DOPA for 1 h (mean ± SEM; n = 20–54 cells in different groups). p < 0.05 versus *EGFP or versus **EGFP and WT groups by Kruskal–Wallis test.
Because cancer-derived cells are prone to random mutation, we first wanted to characterize the ability of the selected PC12 lines to synthesize and metabolize catecholamines (Table 1). We determined the concentrations of dopamine and its major intracellular catabolic product, DOPAC, in whole-cell extracts from untreated cells and those treated with dopamine precursor l-DOPA. Exposure of the control EGFP cell line to 100 μm l-DOPA for 1 h, which is known to augment the packaging of catecholamines into individual vesicles in PC12 cells (Pothos et al., 1996), also produced a large increase in the levels of total intracellular dopamine and DOPAC. Likewise, all α-synuclein-overexpressing lines displayed similar or higher degrees of catechol elevation after l-DOPA treatment, confirming the ability of these cells to both synthesize and break down dopamine. Although some cell lines showed levels of dopamine or DOPAC significantly different from control, these types of measurement do not provide information about the distribution of the transmitter between the intracellular pools and might also be biased by many factors, including possible differences in the number of available vesicles, cell size, and the amount of total protein.
Table 1.
Total cellular DA and DOPAC contents in PC12 cells before and after l-DOPA treatment
| Untreated |
l-DOPA |
l-DOPA/untreated |
||||||
|---|---|---|---|---|---|---|---|---|
| DA | DOPAC | DA/DOPAC | DA | DOPAC | DA/DOPAC | DA | DOPAC | |
| Control (EGFP) | 680 ± 120 | 13.3 ± 2.5 | 40 ± 4 | 8330 ± 1380 | 960 ± 70 | 8.7 ± 1.4 | 12 | 72 |
| WT synuclein (Σm1) | 150 ± 40* | 4.5 ± 0.3* | 34 ± 7 | 2540 ± 340 | 490 ± 145 | 8.7 ± 2.4 | 17 | 110 |
| WT synuclein (Σp1) | 300 ± 60 | 11.4 ± 1.7 | 33 ± 10 | 15060 ± 1040 | 1860 ± 380 | 9.7 ± 1.5 | 51 | 164 |
| A30P synuclein (Am9) | 650 ± 80 | 9.3 ± 0.5 | 51 ± 11 | 13360 ± 730 | 690 ± 60 | 19.5 ± 0.8* | 21 | 74 |
| A53T synuclein (11p2) | 760 ± 30 | 13.2 ± 0.5 | 58 ± 4 | 29450 ± 3050* | 1870 ± 210 | 15.8 ± 0.6* | 39 | 142 |
PC12 cells were treated with 100 μm l-DOPA for 1 hr at 37°C before harvesting and fixation (see Materials and Methods). Total intracellular levels of DA and DOPAC were determined by HPLC with electrochemical detection and normalized to protein concentration in each sample. Data are shown as mean ± SEM of metabolite concentrations in femtomoles per microgram of protein from three to six samples (culture dishes) for each cell line.
*p < 0.05, different from EGFP cells (Kruskal–Wallis test).
In contrast to the whole-cell HPLC measurements, IPE provides a means to specifically determine the concentration of cytosolic catecholamines, which constitute only a small fraction of the total intracellular transmitter content (Chien et al., 1990; Mosharov et al., 2003). However, IPE measurements in all selected PC12 cell lines revealed that the concentration of cytosolic catecholamine in untreated cells was below the detection limit of the technique (<0.1 μm; data not shown). In this respect, PC12 cells were similar to untreated cultured midbrain neurons, in which the cytosolic dopamine level is also below the detection threshold of IPE (E. V. Mosharov, unpublished data). We have shown previously that exposure to l-DOPA leads to a rapid accumulation of cytosolic catecholamines in cultured rat chromaffin cells (Mosharov et al., 2003). Similarly, l-DOPA treatment of PC12 cells (100 μm for 1 h) increased cytosolic levels of catecholamines (dopamine and norepinephrine) and total catechols (catecholamines plus l-DOPA, DOPAC, and DOPEG; see Materials and Methods) to detectable levels (Fig. 1 C,D). Cytosolic catecholamine concentration in l-DOPA-treated PC12 cells that expressed EGFP was 25.5 ± 2.4 μm (mean ± SEM; n = 54 cells), whereas total catechols were at 301.4 ± 72.9 μm (n = 20). The l-DOPA-induced elevation of cytosolic catechol metabolites was significantly greater in cells that overexpressed either WT or mutant α-synuclein. Moreover, total catechol levels were highest in cells expressing A30P or A53T mutant proteins.
Cytosolic catecholamine is increased in transgenic A30P α-synuclein chromaffin cells
Next, to rule out a possibility that the effect of α-synuclein on catecholamine intracellular distribution is only evident after l-DOPA treatment, we conducted IPE measurements in the cytosol of adrenal chromaffin cells derived from transgenic mice that overexpress either human WT or A30P α-synuclein under the control of prion promoter (Fig. 2 A). Similar to catecholamine levels in untreated rat chromaffin cells and in contrast to PC12 cells, cytosolic catecholamine concentration in untreated mouse chromaffin cells from nontransgenic animals was at a detectable level of 7.5 ± 2.2 μm (mean ± SEM of the medians from each batch of cells; n = 7 experiments, 106 cells). Overexpression of A30P α-synuclein led to an approximately twofold increase of the cytosolic catecholamine concentration (14.8 ± 4.5 μm; n = 8 experiments, 91 cells), whereas adrenal cells overexpressing WT protein or those from α-synuclein knock-out animals had cytosolic catecholamine levels comparable to matched nontransgenic controls (Fig. 2 B).
Figure 2.
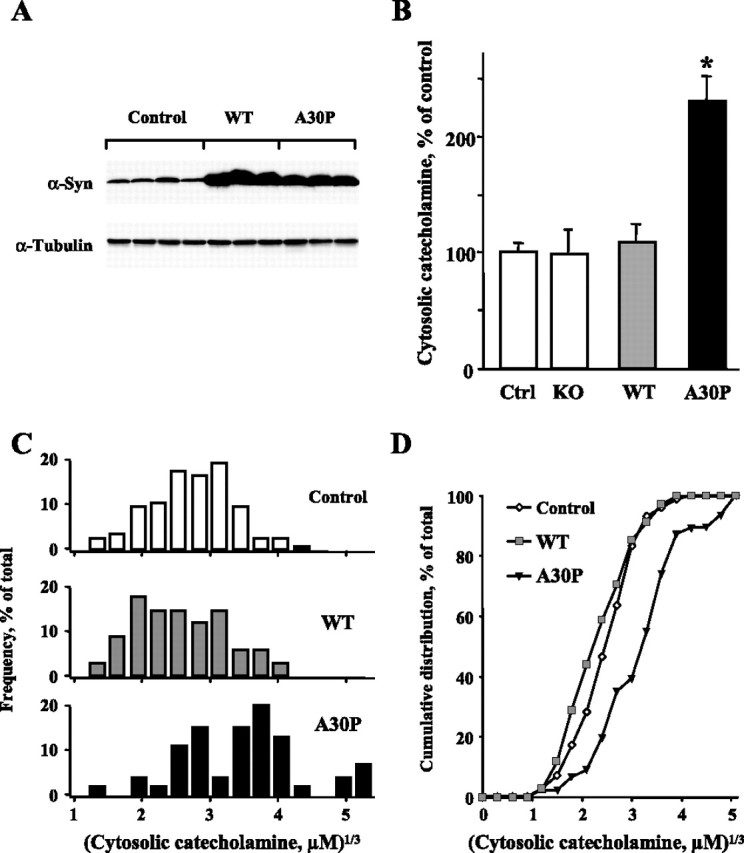
Cytosolic catecholamine concentration is increased in transgenic mice overexpressing A30P α-synuclein. A, Western blots of α-synuclein (α-syn) and α-tubulin (loading control) of the adrenal gland extract from WT and A30P α-synuclein mice and their nontransgenic littermates (control); multiple wells represent different animals. B, Cytosolic catecholamine levels in untreated cultured chromaffin cells from α-synuclein knock-out (KO) mice and those overexpressing WT or A30P α-synuclein. Within each batch of cell cultures, cytosolic catecholamine concentration in each cell was normalized to the average catecholamine level in the nontransgenic group (Ctrl). Data are presented as averages of normalized values ± SEM (n = 55–106 cells; *p < 0.001 by Kruskal–Wallis test). C, Distribution of cube root transformed cytosolic catecholamine concentrations from three representative experiments that displayed similar catecholamine levels in cells from nontransgenic animals. D, Cumulative distribution plot of the data sets presented in C. Cytosolic catecholamine concentrations in A30P cells are significantly different from those in nontransgenic cells by Mann–Whitney test (p < 0.001).
For detailed analysis of α-synuclein-induced changes in cytosolic catecholamine concentrations, we chose three experiments in which catecholamine levels in nontransgenic groups were not significantly different from each other by Mann–Whitney test (a total of 71 cells in nontransgenic, 34 cells in WT, and 46 cells in A30P α-synuclein groups). Statistical comparison of the distributions of cube root transformed catecholamine levels from individual cells confirmed the conclusions from the analyses of averaged medians from each experiment (see above) and of the IPE values normalized to matched controls (Fig. 2 B). Although the histograms of cytosolic catecholamine concentrations in cells from WT α-synuclein-overexpressing animals and their nontransgenic littermates did not differ, those from A30P α-synuclein mice exhibited a rightward shift of the entire population of values (Fig. 2 C,D), indicating that the mutated protein induced an elevation of the cytosolic catecholamines in all examined cells.
Status of catecholamine synthetic and catabolic enzymes
Cytosolic catecholamine concentration is determined by the balance between catecholamine synthesis, degradation, and vesicular sequestration. We therefore examined the expression levels of several key enzymes of dopamine homeostasis in chromaffin and PC12 cells overexpressing various α-synuclein types. In adrenal gland homogenates from α-synuclein transgenic animals, no changes in the expression of tyrosine hydroxylase and MAO, the rate-limiting enzyme for catecholamine synthesis and the major catecholamine catabolic enzyme, respectively, were observed (data not shown). Western blots of PC12 cell extracts confirmed that α-synuclein overexpression did not alter the levels of MAO (data not shown). In contrast, WT and A30P α-synuclein lines displayed increased levels of TH, although the amounts of this enzyme were unchanged in A53T cells (Fig. 1 E). Although changes in the activity of TH may affect dopamine steady state in the cytosol, these cannot explain the increased cytosolic catecholamine levels in PC12 cells in the presence of excessive amounts of l-DOPA, because it enters the catecholamine synthetic pathway downstream of TH. Alternatively, the combinatorial effects of l-DOPA and α-synuclein overexpression could be explained by an enhanced activity of AADC, the enzyme that converts l-DOPA to dopamine. Although Western blots of PC12 cell extracts showed twofold to threefold increased expression of this enzyme in A30P and A53T lines compared with both EGFP and the WT α-synuclein cells, these differences were not statistically significant by Kruskal–Wallis multiple-groups comparison test (Fig. 1 F).
WT and mutant α-synucleins permeabilize purified chromaffin vesicles
In addition to potential effects on the enzymes of catecholamine synthesis and degradation, α-synuclein might have interfered with the ability of secretory vesicles to sequester catecholamines, as suggested by effects on artificial vesicle membrane permeabilization. We tested the ability of isolated chromaffin granules from bovine adrenal glands to maintain the ATP-dependent pH gradient in the presence and in the absence of different types of purified α-synuclein using a previously developed technique that employs the amphiphilic vital dye AO (Sulzer and Rayport, 1990). Addition of ATP to the suspension of vesicles results in acidification of the vesicular lumen, protonation of lumenal AO, and intravesicular accumulation of lumenal AO. A stacking-dependent change in AO fluorescent emission from green to orange (530 to 655 nm) leads to a net decrease of green fluorescence (Nelson et al., 1988). This fluorescence quenching can be prevented by vesicular H+-ATPase inhibitor bafilomycin or the protonophore FCCP, both of which collapse vesicular pH gradients causing redistribution of AO from the vesicles to the media and a restoration of green fluorescent signal (Fig. 3 A). Similarly, all types of α-synuclein induced a leakage of protons from chromaffin granules in a dose-dependent manner (Fig. 3 B). However, WT protein was significantly less potent than both A30P and A53T mutants at low-micromolar concentrations (p < 0.05 by two-way ANOVA test); at 1 μm α-synuclein levels, the mutants produced an approximately twofold greater effect on vesicle permeability compared with the WT protein.
Figure 3.
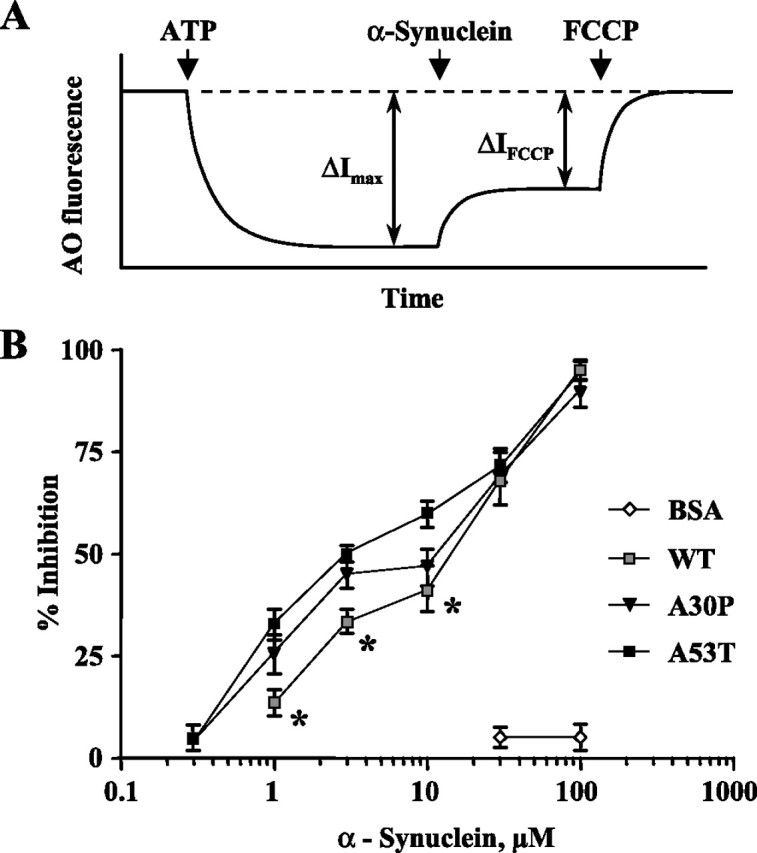
α-Synuclein induces a leakage of protons from isolated bovine chromaffin granules. A, Protocol used to measure the degree of proton leakage from chromaffin vesicles. H+-ATPase inhibitor bafilomycin (10 nm; dashed line) and the protonophore FCCP (1 μm) were used to completely collapse vesicular pH. Addition of ATP in the presence of AO resulted in fluorescence quenching caused by the dye uptake into the vesicles. ΔI max, maximal ATP-dependent vesicle acidification; ΔI FCCP, the change in fluorescence intensity as a result of FCCP. B, Concentration dependence of α-synuclein-induced proton leakage from the vesicles, which was calculated as (ΔI max − ΔI FCCP) × 100/ΔI max for each protein concentration. The curve for A53T α-synuclein is significantly different from WT (p < 0.0001), and the curve for A30P is significantly different from WT α-synuclein in the 0.3–10 μm range (*p < 0.05) by two-way ANOVA test.
Discussion
Dysregulation of dopamine homeostasis and elevation of the cytosolic level of the transmitter have been suggested to underlie the vulnerability of catecholaminergic neurons in PD (Stokes et al., 1999; Shastry, 2001) and may contribute to the neurotoxicity of methamphetamine (Cubells et al., 1994; LaVoie and Hastings, 1999; Larsen et al., 2002) and 1-methyl-4-phenylpyridinium (Liu et al., 1992; Lotharius and O’Malley, 2000). Such effects are proposed to occur once cytosolic catecholamines exceed the capacity of the neurons to buffer oxyradicals that result from dopamine oxidation (Hastings and Berman, 1999; Sulzer and Zecca, 2000). Additional cellular damage may arise from modifications of proteins, lipids, and nucleic acids by dopamine-derived quinones and semiquinones (Sulzer and Zecca, 2000). Until the recent development of IPE (Mosharov et al., 2003), however, it had not been possible to directly verify the assumed changes in the concentration of cytosolic catechol metabolites in cellular models of PD.
Direct measurements of cytosolic catecholamine concentrations in chromaffin cells and l-DOPA-treated PC12 cells showed that overexpression of different types of α-synuclein indeed disrupted the ability of the cells to maintain cytosolic catecholamines at a low level. These observations are consistent with previous reports that α-synuclein overexpression in human mesencephalic MESC2.10 cells enhances cytosolic immunofluorescence from an anti-dopamine antibody (Lotharius et al., 2002). Although α-synuclein could disturb catecholamine homeostasis via effects on synthesis, breakdown, or compartmentalization, apparently it acted downstream of TH, because in PC12 cells, catecholamines were elevated even after incubation with the TH product l-DOPA. An additional argument against increased TH activity as the sole source of disturbance of catecholamine homeostasis is that we did not observe any alteration in TH expression in A53T PC12 cells and in A30P chromaffin cells, both of which displayed significantly elevated cytosolic catecholamine concentrations. In contrast, increased expression of AADC in A30P and A53T PC12 cells might indeed have explained the difference between the WT and mutant α-synuclein cells observed with IPE (Fig. 1 C) and whole-cell HPLC measurements [Table 1, dopamine (DA)/DOPAC ratio]; however, this upregulation of DA-synthesizing enzyme did not reach the level of statistical significance. There was also no change in the expression of MAO, the major intracellular catabolic enzyme, and it is unlikely that changes in catecholamine uptake via plasma membrane transporter (Sidhu et al., 2004) played any significant role in sparsely plated chromaffin cells (cell density, <100 cells/cm2). Finally, possible changes in VMAT activity (Lotharius et al., 2002) are unlikely to account for the observed changes in cytosolic catecholamine in chromaffin cells, because (1) VMAT inhibitors alone fail to produce any detectable alteration of these levels in rat adrenal cells (Mosharov et al., 2003), and (2) amperometric measurements of stimulation-dependent catecholamine release from sister cultures of chromaffin cells used in our experiments show that the amount of transmitter release per individual granule was unchanged in cells from transgenic α-synuclein WT or A30P mice (Y. Schmitz, unpublished data). We therefore conclude that, although α-synuclein overexpression significantly altered the steady-state levels of several key proteins of catecholamine metabolism, these changes alone cannot fully explain the observed increase of cytosolic catecholamine concentration.
Another possible mechanism that might have affected the cytosolic catecholamine pool is based on the reports that α-synuclein protofibrils can interact with artificial vesicles and form pore-like structures within their membranes (Lashuel et al., 2002). If such α-synuclein-mediated permeabilization occurs with real secretory vesicles, the leakage of stored dopamine could affect cytosolic transmitter concentration, which normally constitutes only a miniscule fraction of the total intracellular catecholamine content. For chromaffin cells, in which vesicular and cytosolic catecholamine concentrations are known (Albillos et al., 1997; Mosharov et al., 2003), we estimated that the leakage of all transmitter molecules from a single vesicle would be sufficient to increase cytosolic catecholamine by 1.5- to 2-fold.
Using isolated bovine chromaffin granules, we found that exogenously added α-synuclein disrupts the ATP-dependent vesicular proton gradient, which has been previously demonstrated to result in a proportional loss of vesicular catecholamine (Carlsson et al., 1963; Johnson, 1988; Sulzer and Rayport, 1990). Moreover, we observed that at low-micromolar concentrations, WT α-synuclein was significantly less potent in diminishing vesicular pH than either the A30P or A53T mutants. Although α-synuclein is often said to be an abundant protein, there are no reliable data to our knowledge on its free cytosolic concentration in nerve terminals. The maximal difference in chromaffin vesicle proton gradients between wild-type and pathogenic mutant proteins was observed at ∼1 μm, a level substantially lower than that typically used in studies of α-synuclein fibrillization.
α-Synuclein can readily produce oligomeric and polymeric forms in solution (Conway et al., 2000b), making it difficult to ascertain the particular form of the protein responsible for pH gradient disruption under our assay conditions. However, the augmented ability of the A30P protein to permeabilize secretory vesicles might stem from its greater propensity to produce membrane-damaging protofibrils (Conway et al., 2000a; Lashuel et al., 2002). Analogous effects could also underlie the observation that an overexpression of mutant A53T, but not WT, α-synuclein in PC12 cells interferes with the maintenance of acidic organelles (Stefanis et al., 2001). The effect of α-synuclein on vesicular membrane permeability may not depend on the ability of the monomer to interact with phospholipid membranes (Eliezer et al., 2001), because A30P mutant α-synuclein, which is deficient in membrane binding (Jensen et al., 1998; Outeiro and Lindquist, 2003; Fortin et al., 2004), also induced leakage of protons from isolated vesicles. Finally, it is unclear whether α-synuclein induces a uniform leakage from all vesicles within a given cell or whether the process of vesicle permeabilization is discrete, resulting in subpopulations of depleted vesicles and those that have normal vesicular contents. Although the latter model may explain the observed lack of changes in the vesicular quantal size in chromaffin cells from transgenic α-synuclein A30P animals (Y. Schmitz, unpublished data), our data are insufficient to exclude a uniform leakage model, because compensatory changes in the activity of the enzymes of catecholamine homeostasis might result in seemingly unchanged amounts of vesicular catecholamine.
In summary, we measured the effects of different types of α-synuclein on cytosolic catecholamine concentrations in situ and on the proton gradient of isolated secretory vesicles. The effect of the WT and the mutant pathogenic α-synuclein on vesicular pH suggests that a buildup of this protein, whether through impaired degradation, as suggested for pathogenic mutants (Cuervo et al., 2004), or enhanced synthesis, as follows from familial Parkinson’s disease cases because of α-synuclein gene multiplication (Eriksen et al., 2005), may lead to excess cytosolic transmitter. Importantly, we observed a significantly greater ability of mutant α-synuclein to cause proton leakage from the vesicles and to augment cytosolic catecholamine concentration in both PC12 and chromaffin cells. Elevation of steady-state concentration of dopamine in cellular cytosol, even by a small degree, will provide a source of continuous oxyradical damage via dopamine–quinone formation and enhanced dopamine cleavage by MAO, which produces hydrogen peroxide as a byproduct. It may be that vesicular levels of other classical transmitters are also altered by α-synuclein; however, only monoamines are known to produce oxidative stress in the cytosol. Although additional experiments, possibly on cells overexpressing various α-synuclein levels, are required to determine the physiological relevance of observed effects of the protein on catecholamine homeostasis, our findings provide critical evidence for the mechanism that could explain how α-synuclein may execute its cytotoxicity and selectively damage dopaminergic neurons.
Footnotes
R.H.E. was supported by the National Institute of Mental Health, the National Institute of Neurological Disorders and Stroke (NINDS), and the National Parkinson Foundation; M.D.T. was supported by NINDS; S.P. was supported by the Muscular Dystrophy Association/Wings-over-Wall Street, NINDS, National Institutes of Health Grant AG 021191, and the United States Department of Defense; and D.S. was supported by grants from the National Institute on Drug Abuse, NINDS, and the Picower, Lowenstein, and Parkinson’s Disease Foundations. We thank Drs. Hardy J. Rideout and Leonidas Stefanis for PC12 cells, William Dauer for α-synuclein knock-out mice, and Michael J. Volles and Peter Lansbury for purified α-synuclein.
References
- Albillos A, Dernick G, Horstmann H, Almers W, Alvarez de Toledo G, Lindau M. The exocytic event in chromaffin cells revealed by patch amperometry. Nature. 1997;389:509–512. doi: 10.1038/39081. [DOI] [PubMed] [Google Scholar]
- Baur JE, Kristensen EW, May LJ, Wiedemann DJ, Wightman RM. Fast-scan voltammetry of biogenic amines. Anal Chem. 1988;60:1268–1272. doi: 10.1021/ac00164a006. [DOI] [PubMed] [Google Scholar]
- Carlsson A, Hillarp N, Waldeck B. Analysis of the Mg++-ATP dependent storage mechanism in the amine granules of the adrenal medulla. Acta Physiol Scand. 1963;59:215–238. [PubMed] [Google Scholar]
- Chien JB, Wallingford RA, Ewing AG. Estimation of free dopamine in the cytoplasm of the giant dopamine cell of Planorbis corneus by voltammetry and capillary electrophoresis. J Neurochem. 1990;54:633–638. doi: 10.1111/j.1471-4159.1990.tb01918.x. [DOI] [PubMed] [Google Scholar]
- Conway KA, Harper JD, Lansbury PT. Accelerated in vitro fibril formation by a mutant alpha-synuclein linked to early-onset Parkinson disease. Nat Med. 1998;4:1318–1320. doi: 10.1038/3311. [DOI] [PubMed] [Google Scholar]
- Conway KA, Lee SJ, Rochet JC, Ding TT, Williamson RE, Lansbury PT., Jr Acceleration of oligomerization, not fibrillization, is a shared property of both alpha-synuclein mutations linked to early-onset Parkinson’s disease: implications for pathogenesis and therapy. Proc Natl Acad Sci USA. 2000a;97:571–576. doi: 10.1073/pnas.97.2.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway KA, Harper JD, Lansbury PT., Jr Fibrils formed in vitro from alpha-synuclein and two mutant forms linked to Parkinson’s disease are typical amyloid. Biochemistry. 2000b;39:2552–2563. doi: 10.1021/bi991447r. [DOI] [PubMed] [Google Scholar]
- Cubells JF, Rayport S, Rajendran G, Sulzer D. Methamphetamine neurotoxicity involves vacuolation of endocytic organelles and dopamine-dependent intracellular oxidative stress. J Neurosci. 1994;14:2260–2271. doi: 10.1523/JNEUROSCI.14-04-02260.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2004;305:1292–1295. doi: 10.1126/science.1101738. [DOI] [PubMed] [Google Scholar]
- Dauer W, Przedborski S. Parkinson’s disease: mechanisms and models. Neuron. 2003;39:889–909. doi: 10.1016/s0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- Dauer W, Kholodilov N, Vila M, Trillat AC, Goodchild R, Larsen KE, Staal R, Tieu K, Schmitz Y, Yuan CA, Rocha M, Jackson-Lewis V, Hersch S, Sulzer D, Przedborski S, Burke R, Hen R. Resistance of alpha-synuclein null mice to the parkinsonian neurotoxin MPTP. Proc Natl Acad Sci USA. 2002;99:14524–14529. doi: 10.1073/pnas.172514599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson WS, Jonas A, Clayton DF, George JM. Stabilization of alpha-synuclein secondary structure upon binding to synthetic membranes. J Biol Chem. 1998;273:9443–9449. doi: 10.1074/jbc.273.16.9443. [DOI] [PubMed] [Google Scholar]
- Eliezer D, Kutluay E, Bussell R, Jr, Browne G. Conformational properties of alpha-synuclein in its free and lipid-associated states. J Mol Biol. 2001;307:1061–1073. doi: 10.1006/jmbi.2001.4538. [DOI] [PubMed] [Google Scholar]
- Eriksen JL, Przedborski S, Petrucelli L. Gene dosage and pathogenesis of Parkinson’s disease. Trends Mol Med. 2005;11:91–96. doi: 10.1016/j.molmed.2005.01.001. [DOI] [PubMed] [Google Scholar]
- Fortin DL, Troyer MD, Nakamura K, Kubo S, Anthony MD, Edwards RH. Lipid rafts mediate the synaptic localization of α-synuclein. J Neurosci. 2004;24:6715–6723. doi: 10.1523/JNEUROSCI.1594-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giasson BI, Forman MS, Higuchi M, Golbe LI, Graves CL, Kotzbauer PT, Trojanowski JQ, Lee VM. Initiation and synergistic fibrillization of tau and alpha-synuclein. Science. 2003;300:636–640. doi: 10.1126/science.1082324. [DOI] [PubMed] [Google Scholar]
- Gosavi N, Lee HJ, Lee JS, Patel S, Lee SJ. Golgi fragmentation occurs in the cells with prefibrillar alpha-synuclein aggregates and precedes the formation of fibrillar inclusion. J Biol Chem. 2002;277:48984–48992. doi: 10.1074/jbc.M208194200. [DOI] [PubMed] [Google Scholar]
- Greene LA, Tischler AS. Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc Natl Acad Sci USA. 1976;73:2424–2428. doi: 10.1073/pnas.73.7.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastings TG, Berman SB. Dopamine-induced toxicity and quinone modification of proteins: implications for Parkinson’s disease. In: Creveling CR, editor. Role of catechol quinone species in cellular toxicity. Johnson City, TN: Graham; 1999. pp. 69–89. [Google Scholar]
- Jensen PH, Nielsen MS, Jakes R, Dotti CG, Goedert M. Binding of alpha-synuclein to brain vesicles is abolished by familial Parkinson’s disease mutation. J Biol Chem. 1998;273:26292–26294. doi: 10.1074/jbc.273.41.26292. [DOI] [PubMed] [Google Scholar]
- Jo E, Fuller N, Rand RP, St George-Hyslop P, Fraser PE. Defective membrane interactions of familial Parkinson’s disease mutant A30P alpha-synuclein. J Mol Biol. 2002;315:799–807. doi: 10.1006/jmbi.2001.5269. [DOI] [PubMed] [Google Scholar]
- Johnson RG. Accumulation of biological amines into chromaffin granules: a model for hormone and neurotransmitter transport. Physiol Rev. 1988;68:232–307. doi: 10.1152/physrev.1988.68.1.232. [DOI] [PubMed] [Google Scholar]
- Kubo S, Nemani VM, Chalkley RJ, Anthony MD, Hattori N, Mizuno Y, Edwards RH, Fortin DL. A combinatorial code for the interaction of alpha-synuclein with membranes. J Biol Chem. 2005;280:31664–31672. doi: 10.1074/jbc.M504894200. [DOI] [PubMed] [Google Scholar]
- Larsen KE, Fon EA, Hastings TG, Edwards RH, Sulzer D. Methamphetamine-induced degeneration of dopaminergic neurons involves autophagy and upregulation of dopamine synthesis. J Neurosci. 2002;22:8951–8960. doi: 10.1523/JNEUROSCI.22-20-08951.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lashuel HA, Petre BM, Wall J, Simon M, Nowak RJ, Walz T, Lansbury PT., Jr Alpha-synuclein, especially the Parkinson’s disease-associated mutants, forms pore-like annular and tubular protofibrils. J Mol Biol. 2002;322:1089–1102. doi: 10.1016/s0022-2836(02)00735-0. [DOI] [PubMed] [Google Scholar]
- LaVoie MJ, Hastings TG. Dopamine quinone formation and protein modification associated with the striatal neurotoxicity of methamphetamine: evidence against a role for extracellular dopamine. J Neurosci. 1999;19:1484–1491. doi: 10.1523/JNEUROSCI.19-04-01484.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Peter D, Roghani A, Schuldiner S, Prive GG, Eisenberg D, Brecha N, Edwards RH. A cDNA that suppresses MPP+ toxicity encodes a vesicular amine transporter. Cell. 1992;70:539–551. doi: 10.1016/0092-8674(92)90425-c. [DOI] [PubMed] [Google Scholar]
- Lotharius J, O’Malley KL. The parkinsonism-inducing drug 1-methyl-4-phenylpyridinium (MPP+) triggers intracellular dopamine oxidation: a novel mechanism of toxicity. J Biol Chem. 2000;275:38581–38588. doi: 10.1074/jbc.M005385200. [DOI] [PubMed] [Google Scholar]
- Lotharius J, Barg S, Wiekop P, Lundberg C, Raymon HK, Brundin P. Effect of mutant alpha-synuclein on dopamine homeostasis in a new human mesencephalic cell line. J Biol Chem. 2002;277:38884–38894. doi: 10.1074/jbc.M205518200. [DOI] [PubMed] [Google Scholar]
- Moore DJ, West AB, Dawson VL, Dawson TM. Molecular pathophysiology of Parkinson’s disease. Annu Rev Neurosci. 2005;28:57–87. doi: 10.1146/annurev.neuro.28.061604.135718. [DOI] [PubMed] [Google Scholar]
- Mosharov EV, Gong LW, Khanna B, Sulzer D, Lindau M. Intracellular patch electrochemistry: regulation of cytosolic catecholamines in chromaffin cells. J Neurosci. 2003;23:5835–5845. doi: 10.1523/JNEUROSCI.23-13-05835.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson N, Cidon S, Moriyama Y. Chromaffin granule proton pump. Methods Enzymol. 1988;157:619–633. doi: 10.1016/0076-6879(88)57110-0. [DOI] [PubMed] [Google Scholar]
- Outeiro TF, Lindquist S. Yeast cells provide insight into alpha-synuclein biology and pathobiology. Science. 2003;302:1772–1775. doi: 10.1126/science.1090439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez RG, Hastings TG. Could a loss of alpha-synuclein function put dopaminergic neurons at risk? J Neurochem. 2004;89:1318–1324. doi: 10.1111/j.1471-4159.2004.02423.x. [DOI] [PubMed] [Google Scholar]
- Petrucelli L, O’Farrell C, Lockhart PJ, Baptista M, Kehoe K, Vink L, Choi P, Wolozin B, Farrer M, Hardy J, Cookson MR. Parkin protects against the toxicity associated with mutant alpha-synuclein: proteasome dysfunction selectively affects catecholaminergic neurons. Neuron. 2002;36:1007–1019. doi: 10.1016/s0896-6273(02)01125-x. [DOI] [PubMed] [Google Scholar]
- Pothos E, Desmond M, Sulzer D. l-3,4-Dihydroxyphenylalanine increases the quantal size of exocytic dopamine release in vitro. J Neurochem. 1996;66:629–636. doi: 10.1046/j.1471-4159.1996.66020629.x. [DOI] [PubMed] [Google Scholar]
- Rochet JC, Outeiro TF, Conway KA, Ding TT, Volles MJ, Lashuel HA, Bieganski RM, Lindquist SL, Lansbury PT. Interactions among alpha-synuclein, dopamine, and biomembranes: some clues for understanding neurodegeneration in Parkinson’s disease. J Mol Neurosci. 2004;23:23–34. doi: 10.1385/jmn:23:1-2:023. [DOI] [PubMed] [Google Scholar]
- Rukenstein A, Rydel RE, Greene LA. Multiple agents rescue PC12 cells from serum-free cell death by translation- and transcription-independent mechanisms. J Neurosci. 1991;11:2552–2563. doi: 10.1523/JNEUROSCI.11-08-02552.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shastry BS. Parkinson disease: etiology, pathogenesis and future of gene therapy. Neurosci Res. 2001;41:5–12. doi: 10.1016/s0168-0102(01)00254-1. [DOI] [PubMed] [Google Scholar]
- Sidhu A, Wersinger C, Vernier P. Alpha-synuclein regulation of the dopaminergic transporter: a possible role in the pathogenesis of Parkinson’s disease. FEBS Lett. 2004;565:1–5. doi: 10.1016/j.febslet.2004.03.063. [DOI] [PubMed] [Google Scholar]
- Song DD, Shults CW, Sisk A, Rockenstein E, Masliah E. Enhanced substantia nigra mitochondrial pathology in human alpha-synuclein transgenic mice after treatment with MPTP. Exp Neurol. 2004;186:158–172. doi: 10.1016/S0014-4886(03)00342-X. [DOI] [PubMed] [Google Scholar]
- Stefanis L, Larsen KE, Rideout HJ, Sulzer D, Greene LA. Expression of A53T mutant but not wild-type α-synuclein in PC12 cells induces alterations of the ubiquitin-dependent degradation system, loss of dopamine release, and autophagic cell death. J Neurosci. 2001;21:9549–9560. doi: 10.1523/JNEUROSCI.21-24-09549.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokes AH, Hastings TG, Vrana KE. Cytotoxic and genotoxic potential of dopamine. J Neurosci Res. 1999;55:659–665. doi: 10.1002/(SICI)1097-4547(19990315)55:6<659::AID-JNR1>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Sulzer D, Rayport S. Amphetamine and other psychostimulants reduce pH gradients in midbrain dopaminergic neurons and chromaffin granules: a mechanism of action. Neuron. 1990;5:797–808. doi: 10.1016/0896-6273(90)90339-h. [DOI] [PubMed] [Google Scholar]
- Sulzer D, Zecca L. Intraneuronal dopamine-quinone synthesis: a review. Neurotox Res. 2000;1:181–195. doi: 10.1007/BF03033289. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Engelender S, Igarashi S, Rao RK, Wanner T, Tanzi RE, Sawa A, Dawson VL, Dawson TM, Ross CA. Inducible expression of mutant alpha-synuclein decreases proteasome activity and increases sensitivity to mitochondria-dependent apoptosis. Hum Mol Genet. 2001;10:919–926. doi: 10.1093/hmg/10.9.919. [DOI] [PubMed] [Google Scholar]
- Vila M, Przedborski S. Genetic clues to the pathogenesis of Parkinson’s disease. Nat Med. 2004;10:S58–S62. doi: 10.1038/nm1068. [DOI] [PubMed] [Google Scholar]
- Volles MJ, Lee SJ, Rochet JC, Shtilerman MD, Ding TT, Kessler JC, Lansbury PT., Jr Vesicle permeabilization by protofibrillar alpha-synuclein: implications for the pathogenesis and treatment of Parkinson’s disease. Biochemistry. 2001;40:7812–7819. doi: 10.1021/bi0102398. [DOI] [PubMed] [Google Scholar]