

Abstract
The neurogenic potential of the postnatal neocortex has not been tested previously with a combination of both retroviral and bromodeoxyuridine (BrdU) labeling. Here we report that injections of enhanced green fluorescent protein (eGFP) retrovirus into 134 postnatal rats resulted in GFP labeling of 642 pyramidal neurons in neocortex. GFP-labeled neocortical pyramidal neurons, however, unlike GFP-labeled glia, did not incorporate BrdU. Closer inspection of retrovirally labeled neurons revealed microglia fused to the apical dendrites of labeled pyramidal neurons. Retroviral infection of mixed cultures of cortical neurons and glia confirmed the presence of specific neuronal–microglial fusions. Microglia did not fuse to other glial cell types, and cultures not treated with retrovirus lacked microglial–neuronal fusion. Furthermore, activation of microglia by lipopolysaccharide greatly increased the virally induced fusion of microglia to neurons in culture. These results indicate a novel form of specific cell fusion between neuronal dendrites and microglia and further illustrate the need for caution when interpreting evidence for neuronogenesis in the postnatal brain.
Keywords: cerebral cortex, neurogenesis, macrophage, inflammation, virus, bromodeoxyuridine, rat, pyramidal cell, dendrite, phagocytosis, cell fusion
Introduction
Determining the intrinsic neuronogenic potential of structures in the adult CNS has important implications for the regenerative capacities of the mammalian brain. Although the evidence for continual neurogenesis in the subventricular zone (SVZ) and the dentate gyrus is widely accepted, continued neuronal addition or replacement in the postnatal neocortex is unclear. Some studies have reported the generation of neocortical neurons during adulthood under physiological conditions (Kaplan, 1981; Gould et al., 1999, 2001; Dayer et al., 2005), whereas others have demonstrated a lack of evidence for continuing neocortical neurogenesis using similar bromodeoxyuridine (BrdU) labeling methodologies (Magavi et al., 2000; Kornack and Rakic, 2001; Ehninger and Kempermann, 2003; Koketsu et al., 2003). Recently, retrospective birthdating in humans using fallout from nuclear testing has also shown no support for large-scale neurogenesis in neocortex after birth (Spalding et al., 2005).
The primary method used to assess the generation of new cells has been the incorporation of the thymidine analog BrdU into the genome of dividing cells during S-phase of the cell cycle (Miller and Nowakowski, 1988). One concern over the use of BrdU to identify newly generated cells is that false positives may arise from aborted DNA synthesis known to occur in injured neurons (Katchanov et al., 2001; Yang et al., 2001; Kuan et al., 2004). In addition, such labeling can create difficulties in accurately determining the phenotype of newly generated neurons with double immunocytochemistry (Rakic, 2002).
Infection with certain classes of retrovirus is another method used routinely to label newly generated cells. Retroviral vectors based on C-type murine retroviruses can only integrate into the genome of dividing cells (Roe et al., 1993) during M-phase of the cell cycle, which eliminates the potential artifact of labeling DNA repair or aborted DNA synthesis that can confound BrdU methods. In addition, expression of an enhanced green fluorescent protein (eGFP) transgene can reveal the full morphology of a newly generated cell, making identification of cell types unequivocal (Okada et al., 1999). This method has been used effectively in postnatal brain to show that postnatally generated dentate granule cells and olfactory bulb (OB) interneurons integrate into existing neuronal circuits (van Praag et al., 2002; Belluzzi et al., 2003). Retroviral vectors have not been used previously to specifically assess neuronogenic potential in the postnatal neocortex.
A disadvantage of the retrovirus method is that, in general, it is less efficient in labeling dividing cells than BrdU. This arises because of limitations in viral titer and the requirement of direct injection into the population of dividing cells. We reasoned that this relative inefficiency of retroviral labeling could be circumvented by injecting a large number of brains to ensure that a large number of cells are labeled. We found that stereotaxic injection of an eGFP retrovirus into the cerebral cortex of 134 postnatal rats resulted in labeled pyramidal neurons in frontoparietal motor cortex and occasionally in Ammon's horn of hippocampus. These GFP-positive (GFP+) pyramidal neurons possessed a mature neuronal morphology as soon as GFP expression could be detected and never incorporated BrdU. The labeled neurons were often fused to GFP-positive secondary cells that expressed markers of microglia. This fusion could be reproduced in mixed microglial–neuronal cultures and potentiated by microglial activation. Thus, activated microglia can readily fuse to pyramidal neurons, and this fusion may create a false readout of new neurogenesis.
Materials and Methods
eGFP retrovirus preparation.
Replication-incompetent retrovirus encoding enhanced green fluorescent protein was synthesized using gapEGFPm4 plasmid (a gift from Dr. S. K. McConnell, Stanford University, Stanford, CA) and the 293GPG packaging cell line (Ory et al., 1996) (a gift from Dr. R. Mulligan, Harvard University, Boston, MA) as described previously (Okada et al., 1999). The eGFP retrovirus was harvested by filtering retroviral media from the 293GPG cell line with low-protein binding 0.45 μm syringe filters. Retrovirus was concentrated by ultracentrifugation in a SW-28 rotor at 4°C at 24,000 rpm. Concentrated retrovirus was resuspended in 0.1% HBSS, divided into 5 μl aliquots, and stored at −70°C. Titer of eGFP retrovirus was assayed by infection of 3T3 cells and counting the number of GFP-expressing clones 3 d after. Viral titer was determined to be 1–2 × 108 colony forming units (cfu)/ml.
Animals and stereotaxic surgery.
All animal procedures were performed according to protocols approved by the University of Connecticut Animal Care and Use Committee. Flathead rats were obtained from bred colonies at the University of Connecticut (Storrs, CT). Wild-type rats included normal littermates from the flathead colony or Wistar rats ordered from Charles River Laboratories (Wilmington, MA). Adult animals were aged postnatal day 28 (P28) (n = 2) or P68 and older (n = 9). Animals were kept under isoflurane inhalation anesthesia for the duration of surgery. Glass micropipettes (Drummond Scientific, Broomall, PA) were pulled to a diameter of ∼100 μm and loaded with 1 μl of eGFP retrovirus. Stereotaxic injection of retrovirus was performed either unilaterally or bilaterally in each animal. Injection of 1 μl of eGFP retrovirus into the lateral ventricle was performed slowly over the course of 3 min using an Eppendorf (Hamburg, Germany) Femptojet pressure injector. After surgery, animals were warmed on an isothermal pad, and, for postnatal pups, the animals were returned to the dam.
Rats were killed with halothane and perfused intracardially with PBS, followed by 4% paraformaldehyde. Brains were dissected out and postfixed overnight at 4°C in 4% paraformaldehyde, followed by blocking in 2% agarose. Brains were cut at 40 μm on a vibratome (VT-1000S; Leica, Nussloch, Germany), collected as free-floating sections in cryostorage solution (1% w/v polyvinylpyrrolidone, 30% w/v sucrose, and 30% v/v ethylene glycol in 0.2 m phosphate buffer, pH 7.4), and stored at −20°C until use.
BrdU injections and immunocytochemistry.
Postnatal rat pups received 40 mg/kg injections of BrdU intraperitoneally. The number of BrdU injections per animal ranged from one to four sequential injections spaced 2–4 h apart before or after stereotaxic surgery (see Fig. 2 a). BrdU immunocytochemistry was performed by rinsing free-floating brain sections in PBS, followed by denaturation in 2N HCl for 30 min at room temperature. Sections were neutralized in 0.1% sodium borate and rinsed in PBS before continuation with the standard immunocytochemistry protocol using donkey serum and donkey-raised secondaries. The standard immunocytochemistry protocol included blocking sections in 5% donkey serum (D9663; Sigma, St. Louis, MO) with 0.3% Triton X-100 in PBS for 1 h at room temperature, followed by incubation in primary antibodies overnight at 4°C. After rinsing for 1 h, sections were incubated in secondary antibodies for 1 h at room temperature, followed by a final rinse in PBS and mounted on glass slides. Slides were coverslipped in either Vectashield (Vector Laboratories, Burlingame, CA) or Prolong Antifade (Invitrogen, Carlsbad, CA) and stored at either 4 or −20°C. The primary antibodies used were as follows: rabbit anti-GFP (1:300, AB3080; Chemicon, Temecula, CA), rat anti-BrdU (1:100; OBT0030; Accurate Chemical and Scientific Corporation, Westbury, NY), mouse anti-neuronal-specific nuclear protein (NeuN) (1:1000; MAB377; Chemicon), mouse anti-microtubule-associated protein-2 (MAP-2) (1:200; catalog #05-346; Upstate Biotechnology, Lake Placid, NY), mouse anti-rat pyramidal cell (1:1000; catalog #345; Swant, Bellinzona, Switzerland), mouse anti-OX42 (1:200; MCA275G; Serotec, Oxford, UK), mouse anti-vimentin (1:200; MAB3400; Chemicon), mouse anti-GFAP (1:200; G3893; Sigma), and mouse anti-NG2 (1:200; a gift from A. Nishiyama, University of Connecticut, Storrs, CT). The secondary antibodies used were as follows: donkey anti-rabbit Alexa 488 (1:200; Invitrogen), donkey anti-rabbit Alexa 568 (1:200; Invitrogen), donkey anti-mouse Alexa 568 (1:200; Invitrogen), donkey anti-mouse Alexa 568 (1:200; Invitrogen), donkey anti-rat cyanine 3 (Cy3) (1:200; Jackson ImmunoResearch, West Grove, PA), donkey anti-mouse Alexa 647 (1:200; Invitrogen), and donkey anti-rat Cy5 (1:200; Jackson ImmunoResearch). Sections were counterstained with the nuclear stains 4′,6′-diamidino-2-phenylindole (DAPI) or TO-PRO-3 (Invitrogen) or with Neurotrace Nissl (Invitrogen).
Figure 2.
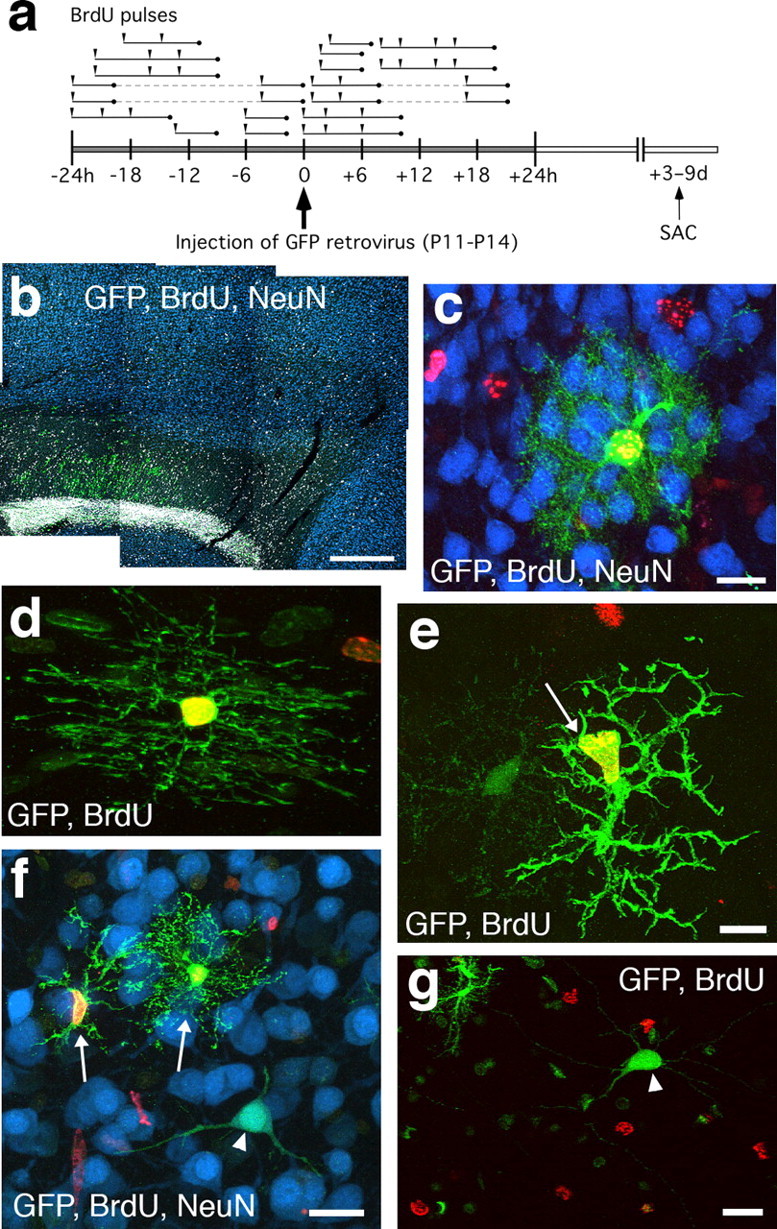
BrdU is not incorporated into virally labeled neurons in postnatal rat neocortex. a, BrdU colabeling scheme. Postnatal rats received one to four BrdU injections (arrowheads) within 24 h of stereotaxic eGFP retrovirus injection. Each line segment indicates the approximate total S-phase time covered by the BrdU pulses given to each animal (n = 18 animals). Animals received 3–9 d survivals after surgery (SAC). b, Sagittal section of frontal cortex from a P20 wild-type animal that received stereotaxic injection of eGFP retrovirus at P11. The animal had received one BrdU injection 1 h after surgery. GFP+ cells (green) are predominant in the RMS and the white matter, with few scattered in neocortex. No BrdU+ nuclei (gray) are colabeled with the neuronal marker NeuN (blue). c, GFP retrovirally labeled astrocyte (green) that is positive for BrdU (red) but not NeuN (blue). d, GFP retrovirally labeled oligodendrocyte in the corpus callosum that is BrdU positive (red). e, GFP retrovirally labeled cell in neocortex with the morphology of a microglia cell (arrow) that is BrdU positive. f, GFP-labeled neuron (arrowhead) that is BrdU negative (red) but NeuN positive (blue). Two nearby glial cells (arrows) are BrdU positive (red). g, GFP-labeled neuron (arrowhead) that is BrdU negative. Scale bars: b, 500 μm; c, f, g, 20 μm; d, e, 10 μm.
To stain microglia with isolectin-B4 (IB4), sections or coverslips were blocked as in the standard immunocytochemistry protocol with the exception that PBS++ (PBS with 0.1 mm CaCl2, 0.1 mm MnCl2, and 0.1 mm MgCl2) was used followed by a streptavidin/biotin blocking kit (Vector Laboratories). Tissue was then incubated in 5 μg/ml biotinylated Griffonia simplicifolia isolectin B4 (B-1205; Vector Laboratories), rinsed with PBS++, and revealed with streptavidin-568 (1:1000; S11226; Invitrogen).
Microscopy and statistical analysis.
Imaging was done using a laser scanning confocal microscope (TCS-SP2; Leica) with laser lines at 488, 543, and 633. Multichannel imaging was performed in sequential scan mode. Contrast, color adjustments, and an unsharp mask filter were applied to images in Adobe Photoshop (Adobe Systems, San Jose, CA). Statistical results are reported as means ± SEM. Differences in means was assessed using a two-tailed unpaired Student's t test. For the microglial activation experiments, differences in the mean microglial activation index (mean number of IB4+ microglia per culture divided by the average IB4+ population per culture for a particular dissociation date) and the mean number of GFP+ neurons per culture was assessed using the nonparametric Mann–Whitney U test. The level of significance was set at p < 0.05.
Dissociated cultures.
For long-term neuronal cultures, hippocampi were dissected from embryonic day 18 rat embryos as described previously (Brewer et al., 1993; Walikonis et al., 2000). Isolated hippocampal tissue was incubated with trypsin for 15 min at 37°C, and the cells were dissociated by trituration. Cells were plated at a density of 200 cells/mm2 on poly-dl-lysine- and laminin-coated coverslips in Neurobasal media supplemented with B27 (Invitrogen), 25 μm glutamate, and 500 μm glutamine. After 21–33 d in vitro (DIV), 1 μl of eGFP retrovirus or 1 μl of eGFP retrovirus plus lipopolysaccharide (LPS) (Sigma) were added to the cultures. The cells were fixed with 4% paraformaldehyde 2–14 d later and processed using the standard immunocytochemistry protocol. For the BrdU-fused cell colabeling experiment, BrdU was added to primary neuronal cultures at a concentration of 28 μm at 12 and 8 h before addition of 2 μl of eGFP retrovirus. Cultures were fixed and processed for BrdU immunocytochemistry 3.5 d after BrdU addition.
For the anti-mitotic experiments, primary neural cultures were established from the dissociated cerebral cortex of P9 wild-type and flathead rats. After 11 DIV, cytosine-β-d-arabinofuranoside (Ara-C) (Sigma) was added to the cultures at a concentration of 5 μm, followed by 1 μl of eGFP retrovirus 24 h later. Cultures were fixed and stained 2 d after virus addition.
Results
Retrovirally labeled neurons in the postnatal rat neocortex
To assess the neurogenic potential of the postnatal neocortex, we made stereotaxic injections of GFP retrovirus into the anterior lateral ventricle of 123 rats aged between P2 and P19 and 11 adult rats. Our assessment of neurogenic potential was performed in the cerebral cortex of normal Wistar rats (n = 75) as well as the neurological mutant rat flathead (n = 59). The flathead rat is a citron-kinase null mutant and has microcephaly and frequent seizures (Sarkisian et al., 2002). We reasoned that there would be a possibility that the epileptic or damaged brain may have increased or compensatory neurogenesis. The injections of GFP retrovirus-labeled cells throughout the SVZ and rostral migratory stream (RMS), corpus callosum, dorsal striatum, and ventral neocortex within 10 d after injection in wild-type and flathead mutant rats (Fig. 1 a,b). GFP-expressing cells had morphologies consisting of bipolar migratory cells, astrocytes, oligodendrocytes and NG2+ polydendrocytes, and microglia. After longer survival times (>2 weeks), granule neurons and periglomerular neurons in the OB were labeled. The morphology and location of GFP-expressing cells in flathead was similar to that seen in wild-type except that the flathead lacked GFP-expressing cells in the RMS (supplemental Fig. 1, available at www.jneurosci.org as supplemental material) and OB 10–14 d after surgery (n = 8 brains).
Figure 1.

Retrovirally labeled neurons in the postnatal rat neocortex. a, Wistar rats received a single stereotaxic injection of a solution containing GFP retrovirus into the anterior SVZ. b, Low-magnification sagittal montage showing GFP-expressing cells in the cerebral cortex 9 d after stereotaxic injection of eGFP retrovirus into P11 wild-type rat. c, A deep layer GFP+ neuron from a P17 rat that received stereotaxic retrovirus injection at P11. The column of GFP+ glial cells next to the GFP+ neuron (arrow) is near the injection tract. d, Higher-magnification confocal image showing the same GFP+ neuron from c with the nuclear label TO-PRO-3 (blue). Note the inverted-type pyramidal morphology of the cell. e–g, Examples of deep layer pyramidal neurons labeled with the GFP retrovirus from an animal that received a 3 d survival (d.s.) after virus injection at P11. h–j, GFP+ neurons labeled with the retrovirus at P11 express markers for mature neurons. h, A GFP+ neuron expressing NeuN (red) from an animal that had a 9 d.s. after stereotaxic virus injection. i, Higher-magnification confocal z-section of the same cell from h with orthogonal views. j, A GFP-labeled neuron in layer VI from a 3 d.s. animal that is positive for a marker found in rat pyramidal neurons (RT-PYR, red). k, Low-magnification sagittal montage showing GFP-expressing cells in both the upper and lower cortical layers. Arrows indicate GFP+ neurons. l, m, Examples of GFP-labeled neurons in layers II/III from an animal that received a 3 d.s. after stereotaxic injection at P14. n, A GFP-labeled pyramidal neuron in hippocampal layer CA3 from an animal that received a 3 d.s. after retrovirus injection at P14. In all images, dorsal is up and rostral is to the right. ctx, Neocortex; lv, lateral ventricle; cc, corpus callosum. Scale bars: b, k, 500 μm; c, e, 100 μm; d, f, h, l–n, 40 μm; g, 50 μm; i, 10 μm; j, 20 μm.
In addition to glial cells, we observed GFP+ pyramidal neurons in the neocortex of both wild-type (Fig. 1 c,d) and flathead (Fig. 1 e–g) rats. We observed these GFP+ neocortical neurons in 54% of animals in which the age of injection was between P2 and P19 and the length of survival was 2 weeks or less (n = 21 of 49 brains, 43% for wild type; n = 37 of 59 brains, 63% for flathead; mean GFP+ neurons per brain, 4.0 ± 1.6 for wild type; 7.6 ± 2.4 for flathead; p = 0.213, Student's t test). The GFP+ neocortical neurons included cells with either pyramidal or inverted pyramidal morphologies in the deep layers of frontoparietal neocortex (Fig. 1 e,f). Pyramidal neurons were found both proximal and distal to the injection site (Fig. 1 d,g). These GFP+ neurons were reactive for a fluorescent Nissl stain (Fig. 1 g) and expressed the neuronal markers NeuN (Fig. 1 h,i) and anti-rat pyramidal cell antigen (Fig. 1 j). Some stereotaxic injections resulted in strong labeling of cells along the pial surface (Fig. 1 k). In these cases, we found layer II/III pyramidal neurons labeled with GFP (Fig. 1 l,m) (n = 11 brains). Some surgeries also resulted in GFP-labeled cells along the posterior periventricular space and in the SVZ adjacent to Ammon's horn in hippocampus. In these brains, GFP-positive CA1 or CA3 pyramidal neurons were found (n = 5 brains; n = 7 total GFP+ pyramidal neurons) (Fig. 1 n). In addition, GFP+ neurons with the morphology of medium spiny neurons were observed in striatum (n = 8 brains) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material).
Labeled pyramidal neurons in all instances (n = 58 brains) were found in wild-type and flathead rats that had received a single stereotaxic injection of retrovirus between P2 and P19. GFP+ cortical neurons were observed as soon as 24–48 h after stereotaxic injection (n = 5 brains) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material) and were present as long as 2 weeks after injection (n = 6 brains). We did not observe retrovirally labeled neurons in the neocortex of wild-type animals that received stereotaxic injection between P2 and P19 and were then allowed to survive for >3 weeks (n = 15). In addition, no GFP+ neurons were observed in the neocortex of adult wild-type rats after survival periods of 1–3 weeks (n = 5) or >3 weeks after injection (n = 6) (Table 1).
Table 1.
GFP retroviral labeling of neurons in postnatal rat
| Age of injection, length of survival |
Number of brains |
Number of brains with labeled neurons in ctx |
Total number of labeled neurons in ctx |
Mean number of labeled neurons per brain |
|---|---|---|---|---|
| P2–P4, 3–14 d | 13 | 4 | 11 | 0.9 ± 0.4 |
| P2–P4, >21 d | 1 | 0 | ||
| P10–P19, 1–2 d | 19 | 5 | 29 | 1.5 ± 0.8 |
| P10–P19, 3–7 d | 58 | 37 | 537 | 9.3 ± 2.7 |
| P10–P19, 8–14 d | 18 | 12 | 65 | 3.6 ± 0.9 |
| P10–P19, >21 d | 14 | 0 | ||
| Adult, <21 d | 5 | 0 | ||
| Adult, >21 d | 6 | 0 | ||
| 134 | 58 | 642 | 5.9 ± 1.5 |
GFP+ cell labeling was analyzed in consecutive 40 μ m sagittal brain sections in frontoparietal cortex. Approximately 15–30 sections were examined per brain, spanning the mediolateral extent of the retrovirally labeled population. Means are reported as mean ± SEM. ctx, Neocortex.
Lack of BrdU incorporation into retrovirally labeled neurons in postnatal neocortex
To determine whether these retrovirally labeled neurons in the postnatal neocortex were generated by a cell division that occurred in the postnatal period, we attempted to colabel GFP+ neurons with BrdU (Fig. 2 a). Tissue from postnatal rats that had received one to four BrdU injections within 24 h before or after GFP retrovirus injection (P11-P14; n = 14 flathead; n = 4 wild type) was processed for BrdU, GFP, and NeuN immunocytochemistry within 3–9 d after stereotaxic surgery. In addition, because GFP-labeled neurons were found to express NeuN (Fig. 1 h) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material), we also searched for BrdU+/NeuN+ cells in the neocortex that were not GFP positive. Sections that were stained for the neuronal marker NeuN (n = 96 flathead; n = 11 wild type) were analyzed for BrdU incorporation in neuronal nuclei in frontoparietal motor cortex (Fig. 2 b), but no BrdU+/NeuN+ cells were found. In cases in which BrdU+ nuclei appeared to colocalize with NeuN+ cells, imaging under high magnification combined with confocal optical sectioning always revealed these cases to be instances of satellite cells closely apposed to neuronal somata as has been reported previously (Kornack and Rakic, 2001). GFP-labeled cells with clear glial morphologies in contrast (Fig. 2 c–e) were found to colabel with BrdU frequently (BrdU+/GFP+, 56.6 ± 7.3%; n = 255 cells). However, GFP+ neurons were never found to colabel with BrdU in flathead or wild type (n = 82 cells; n = 10 brains) (Fig. 2 f,g). This result indicates that the GFP-labeled pyramidal neurons had not undergone an S-phase in the postnatal brain. The remaining possibilities were that either the retrovirus is capable of infecting nondividing neurons or a dividing cell population is capable of fusing to neurons and delivering the GFP label. We were able to rule out the possibility of nondividing cells being infected by retrovirus by performing infections of mitotically inactive primary neural cell cultures that had been treated with the anti-mitotic agent Ara-C (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). In these experiments, we found no GFP+ cells, confirming multiple previous studies showing that C-type retrovirus can only successfully integrate into dividing cells (Roe et al., 1993; Lewis and Emerman, 1994).
Secondary cells fused to retrovirally labeled neurons in postnatal neocortex
Inspection of GFP+ neurons in the postnatal neocortex of both wild-type and flathead rats revealed the presence of GFP+ secondary cell bodies closely apposed to GFP+ neuronal dendrites (Fig. 3). GFP-labeled neurons in layers II/III of neocortex typically had regular pyramidal cell morphology but with an additional cell body attached to the apical dendrite (Fig. 3 a). Confocal optical sectioning of the main process emanating from the GFP+ secondary cell soma indicated that the apical dendrite of GFP+ neurons was continuous with the secondary cell (Fig. 3 b,c). The GFP+ secondary cells fused to the apical dendrites of layer II/III pyramidal neurons typically were present in layer I. The fused cell often had many short ramified processes emanating from its soma (Fig. 3 d,e) and was confirmed to have a nucleus with the DNA label TO-PRO-3 (Fig. 3 e). Retrovirally labeled neurons in layer VI of neocortex typically had inverted pyramidal or horizontally oriented morphologies, and the secondary fused cells were again located on the apical dendrite (Fig. 3 f–h).
Figure 3.
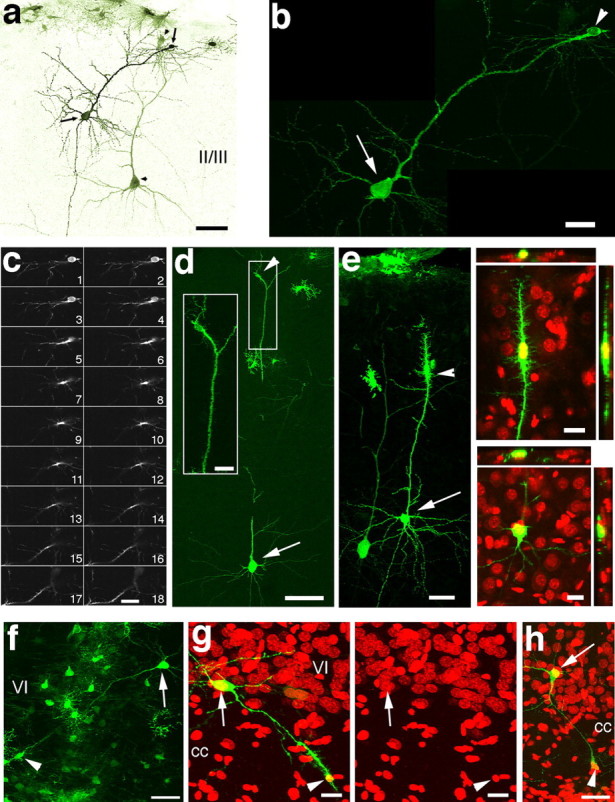
Fused neuronal–glial cells in postnatal rat neocortex. a, GFP+ layer II/III neurons from a P14 rat that received stereotaxic injection of eGFP retrovirus at P11. A secondary cell body (arrowheads) appears to be fused to the apical dendrite of each neuron (arrow and arrowhead pairs). The lookup table for the image is inversed and pseudocolored. b, Higher-magnification confocal image of one of the neurons from a. c, Series of 18 confocal z-sections (0.3 μm per optical section) of the cell indicated by the arrowhead in b. The primary process emanating from the small GFP+ secondary cell could not be distinguished from the apical dendrite of the GFP+ neuron. d, A labeled layer III neuron (arrow) with a secondary cell fused to its apical dendrite (arrowhead). The apical dendrite was followed in and out of a neighboring brain section. The inset shows the secondary cell body at higher magnification. e, GFP+ layer II/III neurons. One of the neurons (arrow) has a secondary cell fused to its apical dendrite (arrowhead). Panels at right show orthogonal views of the secondary cell body (top) and the neuronal cell body (bottom). The secondary cell is colabeled with TO-PRO-3 (red). f, GFP+ layer VI neuron with a secondary cell body (arrowhead) fused to one of the dendrites of the neuron (arrow). The column of GFP-expressing glial cells is near the injection site. g, A GFP+ layer VI neuron (arrow) that has a secondary cell fused to one of the dendrites in the corpus callosum (cc; arrowhead). Right panel is TO-PRO-3 (red) alone. h, Additional example of a labeled deep layer neuron with a secondary cell fused to a primary dendrite (TO-PRO-3, red). Scale bars: a, 50 μm; b, c, inset in d, orthogonal in e, 20 μm; d, 80 μm; e–h, 40 μm.
Labeled neurons in vitro are associated with GFP+ secondary fused-type cells
To test whether the GFP retrovirus labels mature neurons in vitro, we added virus to cultures of dissociated cortical neurons and glia without addition of mitotic inhibitors. After 33 d in vitro, we added 1 μl of GFP retrovirus (∼1 × 105 cfu/culture). GFP-positive cells with neuronal morphology were detected in the cultures within 2 d after addition of GFP retrovirus and could be found for at least up to 14 d after retrovirus addition (Fig. 4) (3.7 ± 1.8 total GFP+ neurons/culture; n = 9 cultures). GFP+ neurons were positive for the neuronal marker MAP-2 (Fig. 4 a–f,j–n). As in vivo, labeled neurons had secondary cell bodies associated with primary dendrites (Fig. 4 a–c) (n = 31 of 33 cells, 94%). The GFP+ secondary cells associated with GFP+ neuronal dendrites contained DAPI+ nuclei, and, as in vivo, the GFP+ cell body could not be distinguished from the GFP+ dendrite of the neuron in stacks of confocal optical sections (Fig. 4 d–i). The MAP-2+ immunoreactivity in the GFP+ dendrite enveloped the soma of the secondary cells, and the GFP+ cytoplasm of the secondary cells were weakly MAP-2+ (Fig. 4 g–i). GFP+ neurons that did not have GFP+ secondary cells associated with their dendrites (n = 2 cells) came from cultures that received the longest survivals (14 d after GFP retrovirus addition), and these two cells contained a second, small nucleus inside the soma of the neuron (supplemental Fig. 4, available at www.jneurosci.org as supplemental material). GFP+ neurons occasionally had more than one GFP+ secondary cell body fused to GFP+ neuronal dendrites (Fig. 4 j–l). The fused secondary cells were typically ameboid in appearance and often had irregular membrane and cytoplasmic protrusions resembling pseudopodia (Fig. 4 m,n). We also observed fusions between GFP-negative (GFP−) neurons and GFP− secondary cells in cultures that received GFP retrovirus (Fig. 4 o,p) (n = 5 cultures), indicating that fusion between neurons and secondary cells is not dependent on expression of the viral genome. In these GFP-negative neuronal-secondary cell fusions, the MAP-2 immunoreactivity in the dendrite was seen to envelop the DAPI+ secondary cell nucleus (Fig. 4 o,p) (supplemental Fig. 5, available at www.jneurosci.org as supplemental material) in a manner similar to that seen in GFP+ cell fusions (Fig. 4 h). In addition, we tested whether the fused-type cell incorporated BrdU by incubating the primary cultures with BrdU together with eGFP retrovirus. We found that both GFP-positive and GFP-negative fused cells were BrdU+ (supplemental Fig. 6, available at www.jneurosci.org as supplemental material). The nucleus of GFP labeled or nonlabeled neurons was never BrdU+.
Figure 4.
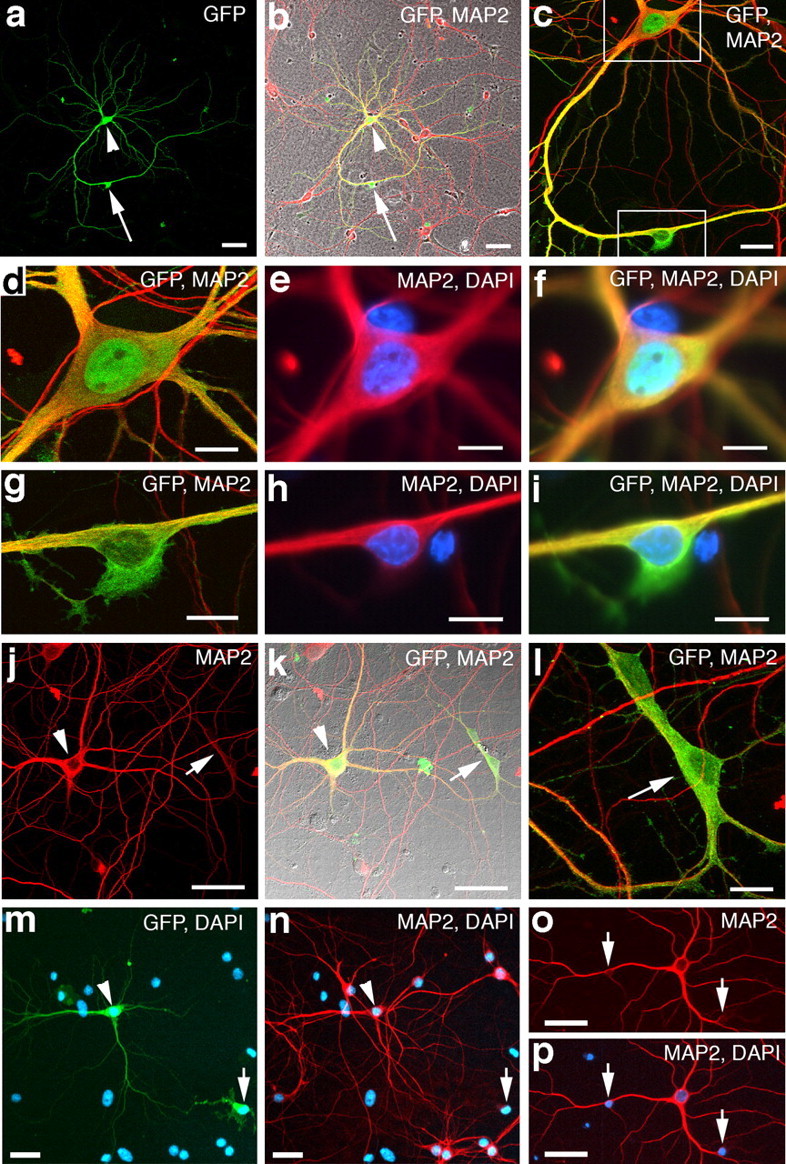
Cell fusion in primary cultures of cortical neurons. a, b, A GFP-labeled neuron from a primary cortical culture that received eGFP retrovirus at 33 DIV and fixed at 36 DIV. The GFP+ neuron (green; arrowhead) has a secondary cell body closely apposed to one of its dendrites (arrow). b, The GFP+ neuron is colabeled with the neuronal marker MAP-2 (red). Merged image is overlaid on a phase-contrast image of the culture. c, Higher magnification of same cells from b. Notice that the GFP+/MAP-2+ dendrite appears to course into the GFP+ secondary cell (bottom box). d–f, Higher-magnification image of the neuron from the top box in c. e–f, Wide-field epifluorescence images showing the nucleus of the neuron labeled with DAPI (blue). g–i, Higher-magnification images of the secondary cell from the bottom box in c. Notice that the GFP+ cytoplasm of the cell cannot be distinguished from the GFP+ neuronal dendrite (yellow). h, i, Wide-field epifluorescence images of the same cell in g showing the nucleus labeled with DAPI (blue). Notice that the GFP+ secondary cell contains weak MAP-2 reactivity (red) that cannot be distinguished from the MAP-2+ neuronal dendrite. j–l, Example of a labeled MAP-2+ neuron (arrowhead) that appears to have two secondary cells fused to one of its primary dendrites (arrow). m, n, Additional example of a labeled neuronal-secondary cell fusion (arrow, arrowhead). o, p, A GFP−/MAP-2+ neuron (red) from a primary culture that received eGFP retrovirus. Notice that the GFP-negative neuron has two DAPI-positive nuclei (arrows) that appear to be fused to two of its primary dendrites. Scale bars: a, b, o, p, 50 μm; c, m, n, 20 μm; d–i, l, 10 μm; j, k, 40 μm.
Secondary cells fused with cortical neurons are of the microglial/macrophage lineage
The morphology of cells fused to the dendrites of pyramidal neurons indicated that they may be microglia. To test for this more directly, we performed OX42 immunocytochemistry, which labels activated microglia, and IB4 histochemistry, which labels microglia. We found that GFP+ secondary cells fused to pyramidal neurons both in vivo and in vitro were OX42 and IB4 positive (Figs. 5 a–c, 6). The GFP+ fused cells were negative for the astrocyte marker GFAP (Figs. 5 d,e, 6 l) and for the polydendrocyte–oligodendrocyte progenitor marker NG2 (Fig. 5 f). GFP+ fused cells were also negative for the astrocyte/endothelial cell marker vimentin (data not shown), although previous work has shown that vimentin may occasionally be expressed in microglia (Graeber et al., 1988). In addition, GFP+ neuronal dendrites to which secondary cells were fused were IB4+ (Fig. 6 a–k), and the fused cells were MAP-2+ (Fig. 6 b–e). Nonfused IB4+ microglia in the cultures were never MAP-2+. Furthermore, we detected GFP−/IB4+ fused-type microglia with morphologies similar to GFP+/IB4+ fused-type microglia (Fig. 6 k) that were fused to GFP-negative neurons (supplemental Fig. 5, available at www.jneurosci.org as supplemental material) (30.8 ± 9.4 cells/culture; n = 9 cultures) in cultures that had received GFP retrovirus.
Figure 5.
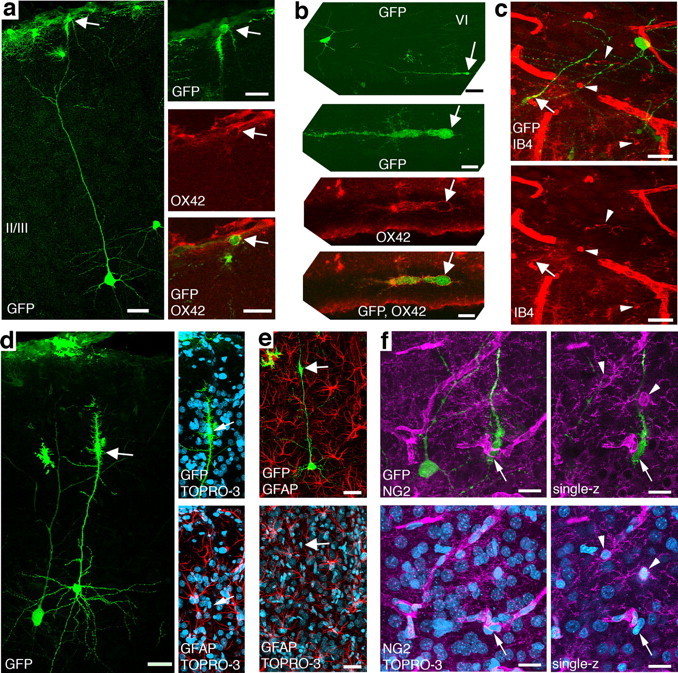
Neuron-fused glial cells are microglia. a, A GFP+ neuron in layer III, with a GFP+ cell fused to its apical dendrite (arrow). The right panel is a higher-magnification image of GFP+ cell indicated by arrow in the left panel. The cell is positive for the activated-microglia marker OX42 (red). The middle and bottom images are single optical sections. b, A GFP+ neuron in layer VI, with a GFP+ cell fused to dendrite in white matter (arrow). The dendrite of a neuron coursed out and back into the sagittal brain section. Middle and bottom are higher-magnification image of the GFP+ cell indicated by arrow in the top. The two bottom images are single optical sections. The cell is positive for OX42 (red). c, GFP+ fused-type glial cell (arrow) in white matter with GFP+ neuron in layer VI nearby. The fused cell is positive for the microglia marker IB4 (red). Examples of IB4+/GFP− microglia are indicated by arrowheads. d, GFP+ neuron in layer II/III with a GFP+ cell fused to its apical dendrite (arrow). The fused cell is not positive for GFAP (red). TOPRO-3 is in blue. e, Another GFP+ neuron in layer II/III with a GFP+ cell fused to its apical dendrite (arrow). This fused cell is also not GFAP positive (red). f, GFP+ fused-type glial cell (arrow) in layer VI with GFP+ neuron in nearby. The fused-type glial cell is not positive for NG2 (magenta). The right column of f are single optical sections. Examples of NG2+/GFP− cells are indicated by arrowheads. Scale bars: a, left, 40 μm; a, right, 20 μm; b, top, 40 μm; b, middle and bottom, 10 μm; c, d, e, 40 μm; f, 20 μm.
Figure 6.

Cells fused to neurons in vitro are microglia. a, b, A GFP+ neuron from a 33 DIV primary cortical culture that received eGFP retrovirus at 30 DIV. The GFP+ neuron has a GFP+ cell (arrow in box) fused to its primary dendrite (arrowheads) that is IB4+ (red). MAP-2 is blue, and DAPI is gray. c–e, Higher magnification of the fused cell from the box in a. The fused cell is positive for IB4 (red, d) and MAP-2 (blue, c). Notice that IB4 and MAP-2 colocalize along the dendrite of the labeled neuron (arrowheads) distal to the fused cell body. f–h, A GFP+ neuron (asterisk) from a 32 DIV primary cortical culture that received eGFP retrovirus at 30 DIV. The GFP+ neuron has a GFP+ cell fused to one of its primary dendrites (top arrow). The fused cell and the neuronal dendrite is positive for IB4 (red, g) (n = 92 of 114 cells; n = 8 cultures). MAP-2 is in blue. i, j, Higher magnification of the microglial cell indicated by arrows in f–h. Both the GFP+ neuronal dendrite and portions of the microglial cell body exhibit immunoreactivity for the neuronal microtubule marker MAP-2 (blue). The inset wide-field image in i shows the DAPI+ nucleus within the IB4+ (red) cell body. k, A GFP+ neuron (asterisk) with a GFP+ cell fused to a primary dendrite (arrows). The reactivity for the microglial marker IB4 (red) colocalizes with the GFP+ fused cell and extends down into the proximal portions of the GFP+ neuronal dendrite (arrows). Arrowheads indicate other GFP+/IB4+ microglia nearby. l, A GFP+ neuron (arrowhead) with a GFP+/IB4+ microglial cell fused to its primary dendrite (arrow). The GFP+/IB4+ fused cell is not GFAP positive (blue). Scale bars: a–e, f–h, k, l, 40 μm; i, j, 20 μm; i, inset, 10 μm.
Activation of microglia increases microglia–neuronal fusion
When activated, microglia increase in number and exhibit increased motility and phagocytic behavior. We hypothesized that an increase in microglia activation would increase the amount of cell fusion with neurons (Fig. 7 a). We added the bacterial membrane antigen LPS to cultures to activate microglia. After addition of LPS, IB4 labeling intensity increased: IB4+ cells underwent a morphological change (i.e., transformation to large ameboid shape) (Fig. 7 b), and there was a twofold increase in the number of IB4+ cells in culture (Fig. 7 b,d) (microglial reactivity index, 1.04 ± 0.22, n = 3 for LPS; 0.45 ± 0.18, n = 4 for control). Addition of LPS together with GFP retrovirus to dissociated cortical cultures resulted in an additional increase in microglial activation with respect to control cultures (Fig. 7 c,d) (microglial reactivity index, 1.41 ± 0.41, n = 5 for LPS plus virus; p = 0.01587, Mann–Whitney U test). Moreover, there was a >10-fold increase in the number of GFP+ pyramidal neurons in LPS-treated cultures relative to untreated cultures (Fig. 7 c–g) (mean GFP+ neurons/culture, 45.8 ± 23.6, n = 12 for LPS plus virus; 3.7 ± 1.8, n = 9 for virus only; p = 0.00375, Mann–Whitney U test).
Figure 7.
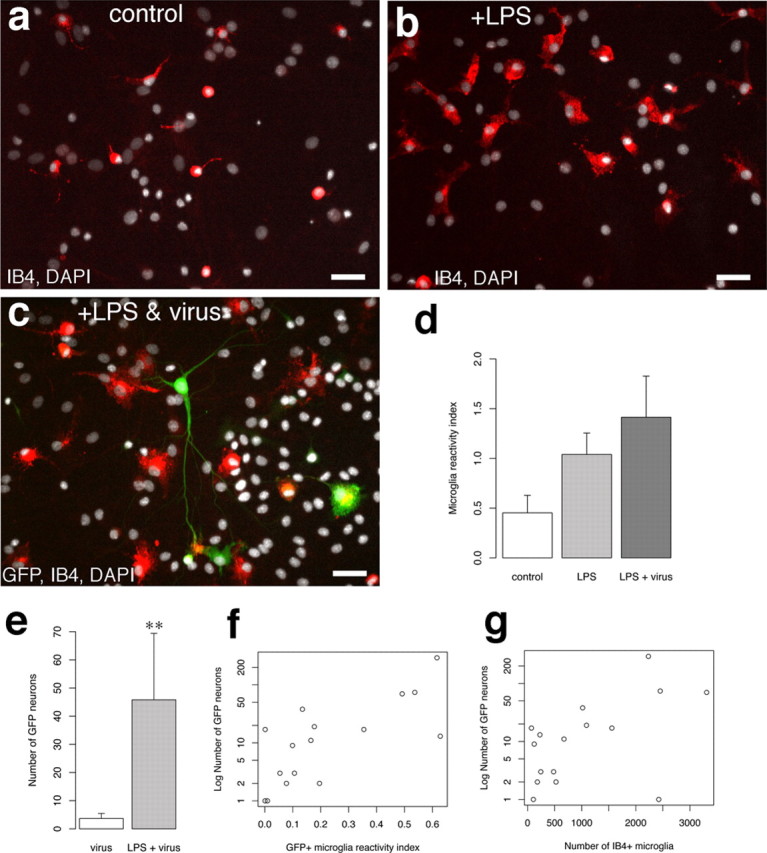
Activation of microglia increases the incidence of retrovirally labeled neurons and fusion. a, Image of IB4-positive microglia from an untreated primary cortical cell culture. Notice that the microglia cell bodies are small and round with occasional ramified processes extending from the soma. b, IB4-positive microglia from a culture that was incubated with the known microglia cell activator LPS (1 μg/ml). Notice that the IB4+ cells now have a large soma with ameboid appearance, a characteristic of activated microglia. c, A GFP+ neuron from a culture that received LPS and GFP retrovirus. The GFP+ neuron has a GFP+/IB4+ microglia fused to its primary dendrite. Notice the IB4+ microglia with large, ameboid cell bodies nearby. d, Index of IB4 labeling in LPS, LPS plus virus, and control cultures. The reactivity index is the average number of IB4+ cells for each condition expressed as a fraction of the total average IB4+ population for all conditions. e, Average number of GFP+ neurons in virus alone and LPS plus virus cultures. **p = 0.00375, Mann–Whitney U test. f, Plot of number of GFP neurons per culture (n = 16) with respect to the index of IB4+/GFP+ cell reactivity (number of IB4+/GFP+ microglia divided by number of total IB4+ cells). g, Plot of number of GFP neurons per culture with respect to the total number of IB4+ cells per culture. Scale bars: a–c, 40 μm.
Microglial–dendritic fusion in culture requires presence of retrovirus
The detection of fused cell profiles positive for MAP-2 and IB4 and negative for GFP indicated that expression of the integrated provirus was not required for cell–cell fusion (Fig. 4 o,p) (supplementary Fig. 5, available at www.jneurosci.org as supplemental material). The existence of this distinctive cellular profile, independent of eGFP expression, provided a means to address whether fusion was dependent on the addition of retrovirus to the cultures. In contrast to cultures that received retrovirus, cultures that did not receive retrovirus were never found to contain MAP-2+/DAPI+ secondary cells fused to dendrites or IB4+ secondary cells fused to dendrites (n = 22 cultures). This absence of fused cells was true for both LPS-treated cultures (n = 9) and cultures not treated with LPS (n = 13). Thus, microglial activation is not sufficient, and the presence of retroviral particles is necessary for fusion of microglia to dendrites.
Discussion
Although it is well known that gliogenesis can occur during physiological and pathological conditions in the adult cerebral cortex, the possibility that neocortical neuronogenesis extends beyond embryonic and fetal neuronogenic periods has remained uncertain. One source of concern in studies of postnatal or adult neocortical neurogenesis has been methodological complications that lead to false positives. Here, we demonstrate that a standard method for labeling newly generated cells, retroviral-mediated gene transfer, can mark nondividing neocortical neurons in vivo and in vitro. Labeled neurons had retrovirally labeled secondary cells syncytially fused to the primary dendrites, and we identified these fused cells to be microglia. Moreover, we found that activation of microglia greatly increased the incidence of microglial–neuronal fusion. The consistent location of the fusion to apical dendrites and lack of fusion to other neural cell types, both in vitro and in vivo, suggests a unique interaction that may exist between microglia and the dendrites of neurons. Furthermore, these results underscore cell–cell fusion as an important concern for studies of neurogenesis.
Labeling of cortical neurons with a retrovirus may have occurred from integration of provirus into a dividing progenitor and subsequent differentiation of a daughter cell into a neuron or by cell fusion of a dividing cell that fused to a mature pyramidal neuron. Four pieces of evidence argue for fusion and against generation of neurons from dividing progenitors in our study. First, GFP+ neurons in vivo and in culture had mature morphologies with extensive dendritic arbors and expressed mature neuronal markers as soon as the GFP labeling was evident (<2 d). Second, we did not detect NeuN+/BrdU+ cells in the neocortex. Third, unlike labeled glial cells that were also colabeled for BrdU, no GFP-positive neurons were labeled by BrdU injections. Fourth, GFP+ microglial cells were fused directly to the dendrites of pyramidal neurons in vivo and in culture.
Macrophages and cell fusion
Microglia are the resident tissue macrophages of the CNS. Macrophages originate from bone-marrow-derived (BMDCs) monocytes and play an important role during development and regeneration as one of the primary phagocytic and immune responsive cell types. In addition, physiologically significant fusion events occur between cells of the macrophage lineage. For example, fusion between cells of the macrophage lineage has been shown to have a physiological role in the formation of the multinucleate osteoclasts of the bone, which do not resorb bone as efficiently when fusion is prevented, and also in the formation of multinucleate giant cells observed in inflammatory reactions (Anderson, 2000; Vignery, 2000). Formation of multinucleate macrophages increases cell size and confers on macrophages the ability to degrade larger extracellular targets (Vignery, 2005). Furthermore, the ability of macrophages to cross intact capillary walls (diapedesis) and invade any tissue implicates the ability for macrophages to fuse with tumor cells in contributing to cancer metastasis (Pawelek, 2005; Vignery, 2005). This enhanced migratory and fusogenic ability in macrophages could make cell fusion a useful tool for delivery of therapeutic transgenes or reprogrammed nuclei into injured cells (Pomerantz and Blau, 2004; Chen and Olson, 2005). Because our results indicate that microglia are fusogenic in the brain and that this fusion is highly specific to the apical dendrite of neurons, elucidation of the mechanisms of cell fusion described in our experiments may help realize the use of macrophages to deliver therapeutic agents across the blood–brain barrier and into neurons.
Heterotypic cell fusion in the CNS has been reported previously for BMDCs with neurons in the cerebellum (Alvarez-Dolado et al., 2003; Weimann et al., 2003b). Although the phenotype of the fusogenic cell was not identified in these studies, the other primary cell type in the brain labeled with bone-marrow-derived reporter genes was microglia. Our results indicate that microglia have the potential to fuse to cortical neurons and suggest that microglial fusion with neuronal dendrites may be an important consideration when assessing transdifferentiation versus fusion events in the brain. For example, previous studies of cell fusion or BMDC transdifferentiation have focused on the appearance of heterokaryons and hybrid nuclei inside neuronal somata to assess fusion (Brazelton et al., 2000; Priller et al., 2001; Alvarez-Dolado et al., 2003; Weimann et al., 2003a,b). Our study indicates that distal fusion of microglia with neuronal dendrites may represent an additional type of cell–cell fusion that could be pertinent to studies of stem cell plasticity and may represent one of the early steps in fusion with neurons.
Virally induced cell fusion
In our experiments, fusion of microglia with neurons was detected only when retrovirus was present. Virus envelope glycoproteins from several families of virus are fusogenic and cause syncytium formation in cultured cells (Hernandez et al., 1996), and endogenous retroviral proteins have been implicated in physiological cell fusion in the placenta (Mi et al., 2000; Dupressoir et al., 2005). In our study, we used a retroviral vector that had been pseudotyped with the VSV-G envelope protein from a rhabdovirus, vesicular stomatitis virus. VSV-G is a 63 kDa surface membrane glycoprotein that inserts into target membranes and mediates fusion between viral particle and host cell (Florkiewicz and Rose, 1984; Pal et al., 1988). VSV-G contains a fusogenic domain (Zhang and Ghosh, 1994; Roche et al., 2006), and membrane fusion is believed to be triggered by a conformational change that occurs below pH 6.1 (Blumenthal et al., 1987; Doms et al., 1987; Puri et al., 1988). Because VSV-G is known to cause membrane fusion between neighboring cells in vitro (Blumenthal et al., 1987; Puri et al., 1988), it is possible that VSV-G-containing membranes in the hours after infection may mediate the cell fusion observed in our study. We did not observe fusion between cell types other than microglia and neurons in vivo or in dissociated cultures, and in fact the fusion was specific between apical neuronal dendrites and microglia. Therefore, the mechanism of virally induced fusion has a high degree of specificity. One potential possibility for this specificity would be through close association of microglia with neuronal dendrites combined with the lowering of extracellular pH by activated microglia (Eder and DeCoursey, 2001).
Our finding that microglia have the potential to fuse with cortical neurons has implications for studies of adult neurogenesis and stem cell-based plasticity. For example, previous work has used GFP retrovirus to demonstrate evidence for regeneration of pyramidal neurons in hippocampal layer CA1 after ischemic injury (Nakatomi et al., 2002). The results of our study do not preclude the use of retroviral labeling for marking newly generated cells, but these results do underscore the need for caution when interpreting results from experiments that assess the short-term lineage relationships of cells labeled with retrovirus. Additionally, our results suggest that viral infection and activation of microglia by tissue damage or inflammation may exacerbate the problem of cell fusion between microglia and neurons, causing a false readout of neurogenesis.
Footnotes
This work was supported by National Institutes of Health Grant MH056524 (J.J.L.). We thank Dr. S. K. McConnell for the gapEGFPm4 plasmid, Dr. R. Mulligan for the 293GPG packaging cell line, and Dr. A. Nishiyama for the OX-42 and NG2 antibodies.
References
- Alvarez-Dolado M, Pardal R, Garcia-Verdugo JM, Fike JR, Lee HO, Pfeffer K, Lois C, Morrison SJ, Alvarez-Buylla A. Fusion of bone-marrow-derived cells with Purkinje neurons, cardiomyocytes and hepatocytes. Nature. 2003;425:968–973. doi: 10.1038/nature02069. [DOI] [PubMed] [Google Scholar]
- Anderson JM. Multinucleated giant cells. Curr Opin Hematol. 2000;7:40–47. doi: 10.1097/00062752-200001000-00008. [DOI] [PubMed] [Google Scholar]
- Belluzzi O, Benedusi M, Ackman J, LoTurco JJ. Electrophysiological differentiation of new neurons in the olfactory bulb. J Neurosci. 2003;23:10411–10418. doi: 10.1523/JNEUROSCI.23-32-10411.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumenthal R, Bali-Puri A, Walter A, Covell D, Eidelman O. pH-dependent fusion of vesicular stomatitis virus with Vero cells. Measurement by dequenching of octadecyl rhodamine fluorescence. J Biol Chem. 1987;262:13614–13619. [PubMed] [Google Scholar]
- Brazelton TR, Rossi FM, Keshet GI, Blau HM. From marrow to brain: expression of neuronal phenotypes in adult mice. Science. 2000;290:1775–1779. doi: 10.1126/science.290.5497.1775. [DOI] [PubMed] [Google Scholar]
- Brewer GJ, Torricelli JR, Evege EK, Price PJ. Optimized survival of hippocampal neurons in B27-supplemented Neurobasal, a new serum-free medium combination. J Neurosci Res. 1993;35:567–576. doi: 10.1002/jnr.490350513. [DOI] [PubMed] [Google Scholar]
- Chen EH, Olson EN. Unveiling the mechanisms of cell-cell fusion. Science. 2005;308:369–373. doi: 10.1126/science.1104799. [DOI] [PubMed] [Google Scholar]
- Dayer AG, Cleaver KM, Abouantoun T, Cameron HA. New GABAergic interneurons in the adult neocortex and striatum are generated from different precursors. J Cell Biol. 2005;168:415–427. doi: 10.1083/jcb.200407053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doms RW, Keller DS, Helenius A, Balch WE. Role for adenosine triphosphate in regulating the assembly and transport of vesicular stomatitis virus G protein trimers. J Cell Biol. 1987;105:1957–1969. doi: 10.1083/jcb.105.5.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupressoir A, Marceau G, Vernochet C, Bénit L, Kanellopoulos C, Sapin V, Heidmann T. Syncytin-A and syncytin-B, two fusogenic placenta-specific murine envelope genes of retroviral origin conserved in Muridae. Proc Natl Acad Sci USA. 2005;102:725–730. doi: 10.1073/pnas.0406509102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eder C, DeCoursey TE. Voltage-gated proton channels in microglia. Prog Neurobiol. 2001;64:277–305. doi: 10.1016/s0301-0082(00)00062-9. [DOI] [PubMed] [Google Scholar]
- Ehninger D, Kempermann G. Regional effects of wheel running and environmental enrichment on cell genesis and microglia proliferation in the adult murine neocortex. Cereb Cortex. 2003;13:845–851. doi: 10.1093/cercor/13.8.845. [DOI] [PubMed] [Google Scholar]
- Florkiewicz RZ, Rose JK. A cell line expressing vesicular stomatitis virus glycoprotein fuses at low pH. Science. 1984;225:721–723. doi: 10.1126/science.6087454. [DOI] [PubMed] [Google Scholar]
- Gould E, Reeves AJ, Graziano MS, Gross CG. Neurogenesis in the neocortex of adult primates. Science. 1999;286:548–552. doi: 10.1126/science.286.5439.548. [DOI] [PubMed] [Google Scholar]
- Gould E, Vail N, Wagers M, Gross CG. Adult-generated hippocampal and neocortical neurons in macaques have a transient existence. Proc Natl Acad Sci USA. 2001;98:10910–10917. doi: 10.1073/pnas.181354698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graeber MB, Streit WJ, Kreutzberg GW. The microglial cytoskeleton: vimentin is localized within activated cells in situ. J Neurocytol. 1988;17:573–580. doi: 10.1007/BF01189811. [DOI] [PubMed] [Google Scholar]
- Hernandez LD, Hoffman LR, Wolfsberg TG, White JM. Virus-cell and cell-cell fusion. Annu Rev Cell Dev Biol. 1996;12:627–661. doi: 10.1146/annurev.cellbio.12.1.627. [DOI] [PubMed] [Google Scholar]
- Kaplan MS. Neurogenesis in the 3-month-old rat visual cortex. J Comp Neurol. 1981;195:323–338. doi: 10.1002/cne.901950211. [DOI] [PubMed] [Google Scholar]
- Katchanov J, Harms C, Gertz K, Hauck L, Waeber C, Hirt L, Priller J, von Harsdorf R, Bruck W, Hortnagl H, Dirnagl U, Bhide PG, Endres M. Mild cerebral ischemia induces loss of cyclin-dependent kinase inhibitors and activation of cell cycle machinery before delayed neuronal cell death. J Neurosci. 2001;21:5045–5053. doi: 10.1523/JNEUROSCI.21-14-05045.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koketsu D, Mikami A, Miyamoto Y, Hisatsune T. Nonrenewal of neurons in the cerebral neocortex of adult macaque monkeys. J Neurosci. 2003;23:937–942. doi: 10.1523/JNEUROSCI.23-03-00937.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornack DR, Rakic P. Cell proliferation without neurogenesis in adult primate neocortex. Science. 2001;294:2127–2130. doi: 10.1126/science.1065467. [DOI] [PubMed] [Google Scholar]
- Kuan CY, Schloemer AJ, Lu A, Burns KA, Weng WL, Williams MT, Strauss KI, Vorhees CV, Flavell RA, Davis RJ, Sharp FR, Rakic P. Hypoxia-ischemia induces DNA synthesis without cell proliferation in dying neurons in adult rodent brain. J Neurosci. 2004;24:10763–10772. doi: 10.1523/JNEUROSCI.3883-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis PF, Emerman M. Passage through mitosis is required for oncoretroviruses but not for the human immunodeficiency virus. J Virol. 1994;68:510–516. doi: 10.1128/jvi.68.1.510-516.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magavi SS, Leavitt BR, Macklis JD. Induction of neurogenesis in the neocortex of adult mice. Nature. 2000;405:951–955. doi: 10.1038/35016083. [DOI] [PubMed] [Google Scholar]
- Mi S, Lee X, Li X, Veldman GM, Finnerty H, Racie L, LaVallie E, Tang XY, Edouard P, Howes S, Keith JC, McCoy JM. Syncytin is a captive retroviral envelope protein involved in human placental morphogenesis. Nature. 2000;403:785–789. doi: 10.1038/35001608. [DOI] [PubMed] [Google Scholar]
- Miller MW, Nowakowski RS. Use of bromodeoxyuridine-immunohistochemistry to examine the proliferation, migration and time of origin of cells in the central nervous system. Brain Res. 1988;457:44–52. doi: 10.1016/0006-8993(88)90055-8. [DOI] [PubMed] [Google Scholar]
- Nakatomi H, Kuriu T, Okabe S, Yamamoto S, Hatano O, Kawahara N, Tamura A, Kirino T, Nakafuku M. Regeneration of hippocampal pyramidal neurons after ischemic brain injury by recruitment of endogenous neural progenitors. Cell. 2002;110:429–441. doi: 10.1016/s0092-8674(02)00862-0. [DOI] [PubMed] [Google Scholar]
- Okada A, Lansford R, Weimann JM, Fraser SE, McConnell SK. Imaging cells in the developing nervous system with retrovirus expressing modified green fluorescent protein. Exp Neurol. 1999;156:394–406. doi: 10.1006/exnr.1999.7033. [DOI] [PubMed] [Google Scholar]
- Ory DS, Neugeboren BA, Mulligan RC. A stable human-derived packaging cell line for production of high titer retrovirus/vesicular stomatitis virus G pseudotypes. Proc Natl Acad Sci USA. 1996;93:11400–11406. doi: 10.1073/pnas.93.21.11400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal R, Barenholz Y, Wagner RR. Pyrene phospholipid as a biological fluorescent probe for studying fusion of virus membrane with liposomes. Biochemistry. 1988;27:30–36. doi: 10.1021/bi00401a006. [DOI] [PubMed] [Google Scholar]
- Pawelek JM. Tumour-cell fusion as a source of myeloid traits in cancer. Lancet Oncol. 2005;6:988–993. doi: 10.1016/S1470-2045(05)70466-6. [DOI] [PubMed] [Google Scholar]
- Pomerantz J, Blau HM. Nuclear reprogramming: a key to stem cell function in regenerative medicine. Nat Cell Biol. 2004;6:810–816. doi: 10.1038/ncb0904-810. [DOI] [PubMed] [Google Scholar]
- Priller J, Persons DA, Klett FF, Kempermann G, Kreutzberg GW, Dirnagl U. Neogenesis of cerebellar Purkinje neurons from gene-marked bone marrow cells in vivo. J Cell Biol. 2001;155:733–738. doi: 10.1083/jcb.200105103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puri A, Winick J, Lowy RJ, Covell D, Eidelman O, Walter A, Blumenthal R. Activation of vesicular stomatitis virus fusion with cells by pretreatment at low pH. J Biol Chem. 1988;263:4749–4753. [PubMed] [Google Scholar]
- Rakic P. Adult neurogenesis in mammals: an identity crisis. J Neurosci. 2002;22:614–618. doi: 10.1523/JNEUROSCI.22-03-00614.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche S, Bressanelli S, Rey FA, Gaudin Y. Crystal structure of the low-pH form of the vesicular stomatitis virus glycoprotein G. Science. 2006;313:187–191. doi: 10.1126/science.1127683. [DOI] [PubMed] [Google Scholar]
- Roe T, Reynolds TC, Yu G, Brown PO. Integration of murine leukemia virus DNA depends on mitosis. EMBO J. 1993;12:2099–2108. doi: 10.1002/j.1460-2075.1993.tb05858.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkisian MR, Li W, Di Cunto F, D'Mello SR, LoTurco JJ. Citron-kinase, a protein essential to cytokinesis in neuronal progenitors, is deleted in the flathead mutant rat. J Neurosci. 2002;22(RC217):1–5. doi: 10.1523/JNEUROSCI.22-08-j0001.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spalding KL, Bhardwaj RD, Buchholz BA, Druid H, Frisén J. Retrospective birth dating of cells in humans. Cell. 2005;122:133–143. doi: 10.1016/j.cell.2005.04.028. [DOI] [PubMed] [Google Scholar]
- van Praag H, Schinder AF, Christie BR, Toni N, Palmer TD, Gage FH. Functional neurogenesis in the adult hippocampus. Nature. 2002;415:1030–1034. doi: 10.1038/4151030a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vignery A. Osteoclasts and giant cells: macrophage-macrophage fusion mechanism. Int J Exp Pathol. 2000;81:291–304. doi: 10.1111/j.1365-2613.2000.00164.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vignery A. Macrophage fusion: are somatic and cancer cells possible partners? Trends Cell Biol. 2005;15:188–193. doi: 10.1016/j.tcb.2005.02.008. [DOI] [PubMed] [Google Scholar]
- Walikonis RS, Jensen ON, Mann M, Provance DW, Mercer JA, Kennedy MB. Identification of proteins in the postsynaptic density fraction by mass spectrometry. J Neurosci. 2000;20:4069–4080. doi: 10.1523/JNEUROSCI.20-11-04069.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weimann JM, Charlton CA, Brazelton TR, Hackman RC, Blau HM. Contribution of transplanted bone marrow cells to Purkinje neurons in human adult brains. Proc Natl Acad Sci USA. 2003a;100:2088–2093. doi: 10.1073/pnas.0337659100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weimann JM, Johansson CB, Trejo A, Blau HM. Stable reprogrammed heterokaryons form spontaneously in Purkinje neurons after bone marrow transplant. Nat Cell Biol. 2003b;5:959–966. doi: 10.1038/ncb1053. [DOI] [PubMed] [Google Scholar]
- Yang Y, Geldmacher DS, Herrup K. DNA replication precedes neuronal cell death in Alzheimer's disease. J Neurosci. 2001;21:2661–2668. doi: 10.1523/JNEUROSCI.21-08-02661.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Ghosh HP. Characterization of the putative fusogenic domain in vesicular stomatitis virus glycoprotein G. J Virol. 1994;68:2186–2193. doi: 10.1128/jvi.68.4.2186-2193.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]