

Abstract
Although brain-derived neurotrophic factor (BDNF) potently regulates neuronal connectivity in the developing CNS, the mechanism by which BDNF influences the formation and/or maintenance of glutamatergic synapses remains unknown. Details about the subcellular localization of the BDNF receptor, TrkB, relative to synaptic and nonsynaptic proteins on excitatory neurons should provide insight into how BDNF might exert its effects during synapse formation. Here, we investigated the subcellular localization of tyrosine kinase receptor B (TrkB) relative to synaptic vesicle-associated proteins and NMDA receptors using immunocytochemistry, confocal microscopy, and time-lapse imaging in dissociated cultures of cortical neurons before, during, and after the peak of synapse formation. We find that TrkB is present in puncta on the surface and intracellularly in both dendrites and axons throughout development. Before synapse formation, some TrkB puncta in dendrites colocalize with NMDA receptors, and almost all TrkB puncta in axons colocalize with synaptic vesicle proteins. Clusters of TrkB fused to the enhanced green fluorescent protein (TrkB-EGFP) are highly mobile in both axons and dendrites. In axons, TrkB-EGFP dynamics are almost identical to vesicle-associated protein (VAMP2-EGFP), and these proteins are often transported together. Finally, surface TrkB is found in structures that actively participate in synapse formation: axonal growth cones and dendritic filopodia. Over time, surface TrkB becomes enriched at glutamatergic synapses, which contain both catalytic and truncated TrkB. These results suggest that TrkB is in the right place at the right time to play a direct role in the formation of glutamatergic synapses between cortical neurons.
Keywords: synaptic vesicle transport, visual cortex, synaptogenesis, neurotrophin, BDNF, TrkB-EGFP
Introduction
Neurotrophins and their tyrosine kinase receptors (Trks) play many roles during CNS development (for review, see McAllister et al., 1999; Cohen-Cory, 2002). Although there is ample evidence that brain-derived neurotrophic factor (BDNF) promotes the formation of inhibitory synapses (Rutherford et al., 1997; Marty et al., 2000; Seil and Drake-Baumann, 2000; Yamada et al., 2002), a role for BDNF in excitatory synapse formation is controversial. BDNF treatment increases numbers of excitatory synapses (Vicario-Abejon et al., 1998), TrkB knock-out mice have decreased numbers of excitatory CNS synapses (Martinez et al., 1998), and conditional TrkB knock-outs suggest that BDNF acts both presynaptically and postsynaptically to regulate glutamatergic synapse formation (Luikart et al., 2005). However, a previous report suggests that BDNF influences glutamatergic synapse formation only indirectly through homeostatic mechanisms (Elmariah et al., 2004). Thus, precisely where and how BDNF binds to TrkB to influence the formation and/or maintenance of CNS synapses remains unknown.
Localization of the BDNF receptor TrkB should provide insight into how BDNF might exert its effects during synapse formation. In general, BDNF and TrkB are highly expressed in the developing CNS during times of neuronal differentiation (for review, see McAllister et al., 1999). Yet the subcellular distribution of TrkB remains controversial, despite numerous papers on this issue. TrkB protein is often reported as very densely packed and diffuse throughout dendrites and somas (Fryer et al., 1996; Yan et al., 1997; Kryl et al., 1999), but recent reports suggest that TrkB is present in distinct puncta either throughout the dendrites (Swanwick et al., 2004) or specifically in the dendritic membrane (Elmariah et al., 2004). The presence of TrkB in axons is also debated, with some reporting TrkB in axons in both developing and adult neurons (Yan et al., 1997; Kryl et al., 1999; Swanwick et al., 2004), whereas others find TrkB present in axonal initial segments but mostly absent from axons and growth cones (Drake et al., 1999; Elmariah et al., 2004). TrkB is also found in dendritic spines and postsynaptic densities of excitatory synapses, but it is unclear whether TrkB is enriched there (Wu et al., 1996; Drake et al., 1999). Although TrkB can be found both presynaptically and postsynaptically in excitatory synapses in mature (Drake et al., 1999; Aoki et al., 2000) and developing (Swanwick et al., 2004; Petralia et al., 2005) neurons, the localization of TrkB in axons and dendrites relative to presynaptic and postsynaptic proteins before, during, and after synapse formation remains unknown. There is also almost nothing known about the distribution of TrkB receptors in the plasma membranes of axons and dendrites during development, where they must be present to be functional. Finally, there is little known about the dynamics of TrkB receptors before, during, and after synapse formation. Answers to these questions are essential to create unbiased models for how TrkB signaling might play a role in CNS synapse formation.
Here, we address these issues using immunocytochemistry, confocal microscopy, and time-lapse imaging in dissociated cultures of cortical neurons before, during, and after synapse formation. We find that TrkB is present in clusters at the surface and intracellularly in both dendrites and axons throughout development. Before synapse formation, some TrkB clusters colocalize with NMDA receptors (NMDARs) in dendrites and almost all TrkB in axons colocalizes with synaptic vesicle (SV) proteins. TrkB clusters are highly mobile in both axons and dendrites. In isolated axons, the dynamics of most TrkB-EGFP are almost identical to vesicle-associated membrane protein 2 (VAMP2)-EGFP dynamics, and these proteins are transported together. Moreover, surface TrkB is found in structures that actively participate in synapse formation: dendritic filopodia and axonal growth cones. Over time, surface TrkB becomes enriched at glutamatergic synapses, which contain both catalytic and truncated TrkB. These results suggest that TrkB is in the right place at the right time to play a direct role in the formation of glutamatergic synapses between cortical neurons.
Materials and Methods
All studies were conducted with an approved protocol from the University of California, Davis, Animal Care and Use Committee, in compliance with National Institutes of Health guidelines for the care and use of experimental animals.
Neuronal cultures.
Neuronal cultures were prepared from postnatal day 0–2 rat visual cortex essentially as described previously (Sabo and McAllister, 2003). Briefly, the visual cortex was excised, dissociated by papain treatment and plated on poly-l-lysine-coated coverslips in 12-well culture plates at a density of 105 cells/mm2. After plating, the coverslips were inverted over astrocyte monolayers and grown in MEM containing 10% horse serum and N2 supplement.
Immunocytochemistry.
TrkB antibodies were used only if their labeling pattern matched the distribution observed with another TrkB antibody raised against a different epitope in a different species. Primary antibodies used were as follows: mouse anti-NR1 (54.1 clone; 1:100; PharMingen, San Diego, CA), guinea-pig anti-vGlut (1:2000; Chemicon, Temecula, CA), rabbit anti-synapsin 1 (1:1000; Chemicon), mouse anti-microtubule-associated protein 2 (MAP2) (1:1000; Sternberger Monoclonals, Lutherville, MD), mouse anti-VAMP2 (1:300; Synaptic Systems, Goettingen, Germany), chicken anti-TrkB extracellular domain [TrkBec (LR); 1:250; a generous gift from Dr. Louis Reichardt, University of California (UCSF), San Francisco, CA], rabbit anti-TrkB extracellular domain [TrkBec (Up); 1:100; Upstate, Charlottesville, VA], and rabbit anti-TrkB intracellular domain (TrkBin; 1:100; a generous gift from Dr. David Kaplan, Hospital for Sick Children, Toronto, Ontario, Canada). Phalloidin-Alexa 548 (1:13; Invitrogen, Carlsbad, CA) was used to label actin filaments.
In most experiments, neurons were fixed with 4% paraformaldehyde and 4% sucrose in PBS at 4°C for 10 min, permeabilized with 0.25% Triton X-100 in PBS for 5 min, blocked with 10% BSA in PBS for 30 min, and incubated with primary antibodies diluted in 3% BSA in PBS for 1 h at room temperature or overnight at 4°C. After washing three times with 3% BSA in PBS for 5 min, coverslips were incubated in secondary antibody diluted in 3% BSA in PBS for 45 min. Coverslips were mounted in fluoromount (Fisher, Pittsburgh, PA) containing DABCO [1,4-diazabicyclo(2.2.2)octane] (Sigma, St. Louis, MO). Because the NR1 antibody does not recognize paraformaldehyde-fixed NR1, staining using the NR1 antibody was performed with fixation in methanol for 5 min at −20°C and incubation with primary antibodies for 5 h at room temperature or overnight at 4°C. TrkB staining after paraformaldehyde fixation and methanol fixation were identical.
For surface TrkB (sTrkB) staining, neurons were incubated in TrkBec (Up) antibody for 10 min at room temperature before fixation. After washing three times for 3 min with culture medium, coverslips were rinsed with PBS before proceeding with fixation and labeling with additional antibodies as described above. For combined sTrkB and TrkBec staining, sTrkB was labeled using TrkBec (Up) followed by fixation, permeabilization, and staining with TrkBec (LR) as described above. Surface staining for TrkB when followed by methanol fixation was not different from TrkB surface staining followed by 4% paraformaldehyde fixation (data not shown). To confirm that the labeling pattern seen with the above surface-labeling method was not caused by antibody-induced clustering, another set of neurons was fixed with 2% paraformaldehyde in PBS with 4% sucrose in nonpermeabilizing conditions then was stained for TrkBec (Up). Using this fixation protocol, no staining was obtained for the intracellular marker, MAP2, and surface staining for TrkB looked identical to surface staining before fixation (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Other than the indicated panels in supplementary Figure 1 (available at www.jneurosci.org as supplemental material), images shown of sTrkB labeling were all from neurons that were labeled before fixation. For all sTrkB labeling, the antibody used was TrkBec (Up) from Upstate. TrkBec (LR) was only used for staining the overall population of TrkB [TrkBec (LR)] in Figure 1.
Figure 1.
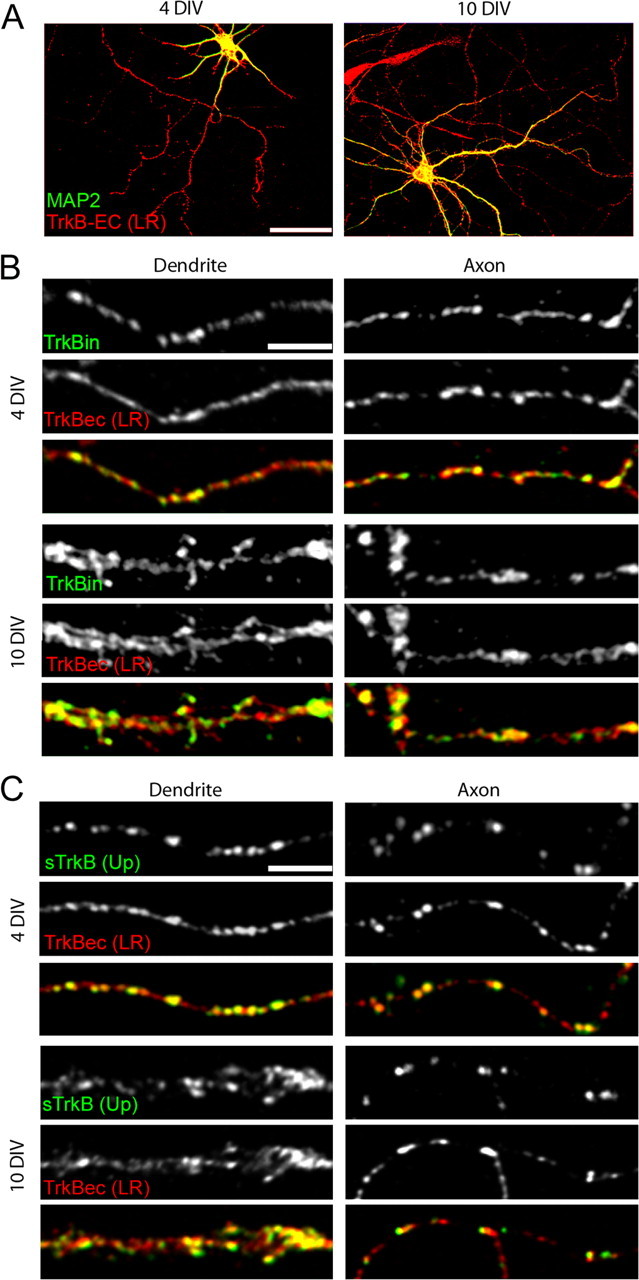
TrkB is found in both axons and dendrites of cortical neurons. A–C, Neurons were labeled with an antibody to the extracellular domain of TrkB [TrkBec (LR); A–C, red] and MAP2 (dendritic marker; A, green), full-length TrkB (TrkBin; B, green) or surface TrkB [sTrkB using TrkBec (Up); C, green]. Colocalization is indicated by yellow in the overlays. A, TrkB is present in axons, dendrites, and cell bodies before (4 DIV) and at the peak of synapse formation (10 DIV) in cortical cultures. B, Full-length TrkB (TrkBin) is expressed in both dendrites and axons and is highly colocalized with TrkBec (LR), suggesting that TrkBec is a reliable indicator of the presence of TrkBin in cortical neurons at the ages examined. In dendrites, TrkB shows a punctate pattern over a low diffuse background, and in axons, TrkB is mainly punctate at both 4 and 10 DIV. C, Surface TrkB [sTrkB; TrkBec (Up)] is widely distributed in puncta on both dendrites and axons and appears to be a subset of the total TrkB [labeled here with TrkBec (LR)]. Scale bars: A, 50 μm; B, C, 5 μm.
Microscopy.
Mounted coverslips were imaged using an Olympus (Tokyo, Japan) Fluoview 2.1 laser scanning confocal microscope with a 60× PlanApo oil immersion objective [1.4 numerical aperture (NA)] on an IX70 inverted microscope. Images for each fluorophore were acquired sequentially with 1–3× digital zoom and 2× Kalman averaging. Optimal parameters for imaging each fluorescence channel for each coverslip were chosen manually to minimize image saturation and maximize signal.
Image analysis.
Quantification of immunocytochemistry and colocalization was performed using Image Pro Plus software (Media Cybernetics, Silver Spring, MD). First, images were thresholded by subtracting an average background fluorescence measured as the mean plus two times the SD of the intensity of the total fluorescence in the dendritic shaft or axon. This choice of thresholding eliminates ∼95% of pixels and therefore makes it unlikely that puncta will be identified that are derived from background fluorescence while preserving detection of lightly labeled protein clusters characteristic of developing neurons. After thresholding, puncta were selected manually and the template of selected puncta was imposed on the other thresholded fluorescence channels. Puncta were considered colocalized when the fluorescence intensity of one channel was above background within a 0.5 μm radius from the center of a punctum in another channel [as in the study by Washbourne et al. (2002)]. Dendritic fields consisted of images with 2–3 dendrites, usually with axons crossing them. The density of axonal contacts increased with age. Axon fields consisted of images of 1–2 axons not in contact with dendrites. The probability of random colocalization of sTrkB with synapses was calculated by multiplying the density of synapses with the density of sTrkB puncta and the area of dendrite quantified. Additional data analyses were performed using Excel (Microsoft, Redmond, WA) and Statview (SAS, Institute, Cary, NC). Images were processed and prepared for print using Illustrator and Photoshop (Adobe, San Jose, CA). All data are presented as mean ± SEM and all p values represent significance tested by ANOVA factorial with Fisher's PLSD test.
Cross-covariation analysis was performed on the axonal distribution of TrkB-EGFP and VAMP2 in transfected cells and endogenous TrkB and VAMP2 in nontransfected cells to determine whether TrkB-EGFP has a similar distribution as endogenous TrkB and whether transfection alters the endogenous VAMP2 distribution. Twenty to 30 μm lines were drawn along the axons and fluorescence intensity profiles were generated for each label. These intensities were compared by cross-covariation analysis with Matlab software as described previously (Sabo et al., 2001). TrkB movements were tracked in Image Pro (Media Cybernetics). The fraction of mobile puncta was defined as the ratio of puncta that moved at any time during 5 min of imaging to the total number of puncta in the area of the axon analyzed at the beginning of imaging. For the movement behavior analysis, only the puncta that moved and could be tracked for >5 frames were included. To confirm comovement of mobile TrkB-EGFP and VAMP2-DsRed clusters, colocalization of peaks in each fluorescence channel that were at least 2× the local background intensity was detected using line-scan analysis. Line-scan analysis was then used in combination with frame-by-frame analysis of the time-lapse movies to determine mobility and directionality of comovement of puncta that could be tracked for at least four frames.
Transfection.
The TrkB-EGFP construct was a generous gift from Dr. Rosalind Segal (Harvard University, Cambridge, MA) (Watson et al., 1999). Neurons were transfected at 3–4 d in vitro (DIV) using a calcium phosphate transfection protocol (Xia et al., 1996). Culture medium was replaced with DMEM supplemented with 1 mm kynurenate, 1 mm of MgCl2, and 0.5 mm HEPES, pH 7.5. The DNA/calcium phosphate precipitate was prepared by mixing one volume of DNA in 250 mm CaCl2 with an equal volume of 2× HEPES-buffered sucrose. The precipitate was allowed to form at room temperature in the dark for 25–30 min. Transfection with 3 μg of DNA per 18 mm coverslip yielded healthy, lightly transfected neurons for imaging. After incubation with DNA complexes for 30 min, cells were washed three times with warmed DMEM. Conditioned growth medium was then added back to the cells and the plates returned to the incubator.
Live imaging.
Sixteen to 24 h after transfection, 18 mm coverslips were imaged in a custom-built imaging chamber (Warner, Hamden, CT) in artificial CSF (ACSF) (Washbourne et al., 2002). Neurons selected for imaging had healthy morphologies and low levels of EGFP-tagged protein expression. Imaging was conducted on an Eclipse TE300 Nikon (Tokyo, Japan) inverted fluorescence microscope using a 60× oil immersion objective (1.4 NA). Fluorophores were excited at their absorption maxima using a TILL Photonics (Martinsried, Germany) monochromator combined with a triple bandpass filter specific for FITC/Texas Red/Cy5 (Chroma, Battleboro, VT). Images were acquired every 15 s with a Coolsnap HQ CCD camera (Roper Scientific, Tucson, AZ) and Simple PCI software (C-Imaging; Compix, Cranberry Township, PA) for an average of 15 min. For dual-color imaging, channels were collected sequentially using a Zeiss (Oberkochen, Germany) Pascal confocal system and a 40× Plan Neofluor 0.75 NA objective. Images were collected every 10 s with scan times of 1.9–3.9 s. Lasers were Ar 488 and HeNe 543. Detection filters were bandpass 505–530 nm for EGFP and long-pass 560 nm for DsRed. Laser power was kept low (6% or less for 488 nm Ar and 18% or less for 543 nm HeNe) to avoid phototoxicity.
Subcellular fractionation.
Synaptic plasma membranes were obtained by subcellular fractionation according to the procedures published previously (Blackstone et al., 1992). All procedures were performed at 4°C in the presence of protease inhibitors (complete Mini/EDTA-free; Roche, Welwyn Garden City, UK). Briefly, 3-week-old male rat forebrains were homogenized in 10 volumes of HEPES-buffered sucrose (0.32 m sucrose and 4 mm HEPES/NaOH, pH 7.4) with a glass-Teflon homogenizer at 900 rpm. The homogenate was centrifuged at 900 × g for 10 min, and the nuclear fraction was discarded. The supernatant (S1) was then centrifuged at 10,000 × g for 15 min to yield the crude synaptosomal fraction (P2). P2′ (washed crude synaptosomal fraction) was obtained by resuspending the P2 pellet in 10 volumes of HEPES-buffered sucrose and then centrifuging at 10,000 × g for 15 min. P2′ was subsequently lysed by hypo-osmotic shock in water, immediately adjusted to 4 mm HEPES, and mixed constantly for 30 min. The lysate was spun at 25,000 × g for 20 min to yield a crude synaptic vesicle fraction (S3) and a synaptosomal membrane fraction (P3). The pellet was then resuspended in HEPES-buffered sucrose and layered on top of a discontinuous gradient containing 0.8, 1.0, and 1.2 m sucrose. The gradient was spun at 150,000 × g for 2 h in a swinging bucket rotor. Synaptic plasma membranes were collected in the interface between 1.0 and 1.2 m sucrose and resuspended in PBS, pH 7.4. The fractionation was confirmed by Western blotting using an antibody against synaptophysin (anti-mouse, 1:1000; Sigma, St. Louis, MO), a synaptic vesicle marker that becomes enriched throughout the fractionation (Blackstone et al., 1992). The presence of TrkB in each fraction was determined using an antibody against its extracellular domain (rabbit anti-TrkB, 1:100; Upstate).
Results
TrkB is present in axons and dendrites of cortical neurons before and during synapse formation
To propose unbiased models for the role for BDNF/TrkB signaling in excitatory synapse formation, we first determined the subcellular localization of TrkB in dissociated cortical neurons both before synapses had formed (4 DIV) and at the peak of synaptogenesis (10 DIV) (Washbourne et al., 2002). Cultures of dissociated neurons were used because they allowed for clear visualization and quantification of the subcellular distribution of immunostained proteins across dendrites and axons at multiple developmental stages. Neurons at each age were fixed and immunostained using antibodies to TrkB. Although there are numerous antibodies that we could have selected to label TrkB, antibodies were specifically chosen based on whether they matched the distribution of other TrkB antibodies raised against a different epitope in a different species. The two TrkBec antibodies that we used provided identical staining patterns: one antibody was purchased from Upstate [TrkBec (Up)], the other was a generous gift from Dr. Louis Reichardt's laboratory at UCSF [TrkBec (LR)]. In addition, the TrkBin antibody was used because, as expected, it specifically labeled a subset of TrkBec.
TrkB was found in all cortical neurons, including both excitatory and inhibitory neurons. Excitatory neurons were identified by their pyramidal-like morphologies and large size, which reliably correlated with a lack of GABA immunostaining (data not shown). The subcellular distribution of TrkB was then examined specifically in excitatory neurons. TrkB was found in all of the major compartments of each neuron (cell bodies, dendrites, and axons) both before (4 DIV) and during the peak of (10 DIV) synapse formation (Fig. 1A). Dendrites were defined by the presence of MAP2 staining and axons by its absence. The distribution of TrkB was highly punctate with a diffuse background in dendrites, whereas it was mainly punctate in axons (Fig. 1B,C). This distribution did not change between 4 and 10 DIV (Fig. 1 B,C).
The TrkB receptor has two main isoforms: the full-length catalytic receptor, which contains an intracellular tyrosine kinase domain, and the truncated receptor, in which the intracellular catalytic domain is absent. Because the functions of TrkB in synapse development are thought to be mediated mainly by its catalytic isoform (Dechant, 2001; Huang and Reichardt, 2003), it was important to determine whether and where full-length TrkB is present in excitatory cortical neurons. Using an antibody directed against the catalytic domain of the full-length TrkB (TrkBin), we found that full-length TrkB was also present in all excitatory cortical neurons. TrkBin localized to both axons and dendrites throughout development, showing a very similar distribution to the total TrkB population described above (TrkBin) (Fig. 1B, green panels). When neurons were double-labeled with antibodies that recognize the extracellular [TrkBec (LR)] and intracellular (TrkBin) domains of TrkB, 87.21 ± 3.96% (n = 9 dendritic fields) and 90.89 ± 1.81% (n = 10 dendritic fields) of TrkBec (LR) puncta colocalized with TrkBin puncta in dendrites at 4 and 10 DIV, respectively. In axons, TrkBin staining was punctate throughout the length of the axon and did not change between 4 and 10 DIV. At 4 and 10 DIV, 88.33 ± 3.06% (n = 10 axon fields) and 80.54 ± 2.56% (n = 10 axon fields) of TrkBec (LR) puncta colocalized with TrkBin in axons, respectively. The high colocalization of TrkBec puncta with TrkBin indicates that most clusters of TrkB contain the full-length isoform of TrkB. This localization of full-length TrkB within axons and dendrites before and during synapse formation suggests that TrkB signaling could play a role in the earliest stages of synaptogenesis.
BDNF activates TrkB signaling by binding to the extracellular domain of TrkB that is located at the cell surface. Therefore, only subcellular compartments that express TrkB at the cell surface are capable of mediating the effects of BDNF during synaptogenesis. To determine which subcellular compartments contain sTrkB, we incubated living neurons for 10 min at 25°C with TrkBec (Up). Neurons were then fixed and immunostained for MAP2 to label dendrites. Using this protocol, we found that TrkB was present at the surface of all neurons at 4 and 10 DIV. Similar to total TrkB, sTrkB (Up) was highly punctate in both axons and dendrites at both ages analyzed (Fig. 1C, green panels). To ensure that this punctate labeling pattern was not a result of antibody-induced clustering of TrkB, another set of neurons were labeled for TrkBec (Up) after a light (2%) paraformaldehyde fixation, which did not permeabilize the cells as confirmed by a lack of MAP2 staining (data not shown). Because the same distribution pattern of sTrkB was obtained with both protocols (supplemental Fig. 1, available at www.jneurosci.org as supplemental material), our data indicate that TrkB receptors are clustered at the surfaces of both axons and dendrites of developing excitatory neurons.
Throughout development, the majority of TrkB clusters were localized at the cell surface. In 4 DIV axons, 75.23 ± 4.78% (n = 7 axon fields) of TrkBec (LR) puncta contained sTrkB (Up). The fraction of TrkB puncta that localized to the cell surface was not significantly different in 10 DIV axons (60.38 ± 5.65%; n = 7 axon fields; p = 0.07). In dendrites, 74.49 ± 8.66% (n = 6 dendritic fields) and 80.12 ± 3.13% (n = 7 dendritic fields) of TrkBec (LR) colocalized with sTrkB (Up), at 4 DIV and 10 DIV, respectively (p = 0.53). This broad expression of TrkB at the neuronal surface suggests that both axons and dendrites are likely to be responsive to BDNF before and during the peak of synaptogenesis.
TrkB receptors are highly mobile in dendrites and colocalize with NMDA receptors before and during the peak of synapse formation
Because a number of synaptic proteins are found in highly mobile clusters during synaptogenesis (Ahmari et al., 2000; Washbourne et al., 2002; Sabo and McAllister, 2003; Shapira et al., 2003; Gerrow et al., 2006), we next determined whether TrkB is also trafficked in mobile transport packets before synapses are formed. To that end, 3–4 DIV cortical neurons were transfected with a plasmid encoding full-length TrkB-EGFP (Watson et al., 1999). To avoid possible effects of TrkB over-expression, we imaged neurons with normal morphologies and the lowest transfection levels that permitted image collection. To determine the level of overexpression of TrkB-EGFP in neurons, we immunostained coverslips containing transfected neurons with antibodies to GFP and to TrkBec (Up); the fluorescence intensity and density of TrkBec puncta was then compared in neighboring transfected and nontransfected neurons (Washbourne et al. 2002) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). On average, the fluorescence intensity of TrkB clusters was 1.9 times higher in transfected versus neighboring nontransfected neurons (transfected clusters, 22,962 ± 1739 a.u., n = 11; nontransfected clusters, 12,060 ± 990 a.u., n = 11; p < 0.0001) (supplemental Fig. 2A, available at www.jneurosci.org as supplemental material). Yet, the density of TrkB clusters was the same in transfected versus nontransfected neurons (transfected neurons, 5.6 ± 0.25 puncta/20 μm, n = 11; nontransfected neurons, 5.3 ± 0.24 puncta/20 μm, n = 10) (supplemental Fig. 2B, available at www.jneurosci.org as supplemental material). Therefore, although the amount of TrkB in each cluster was higher in transfected neurons, the distribution of TrkB clusters was not changed in TrkB-EGFP-transfected cells. Thus, the localization and behavior of TrkB-EGFP is likely to reflect that of endogenous TrkB.
Similar to endogenous TrkB, TrkB-EGFP was punctate throughout all dendrites (Fig. 2A). To determine whether these TrkB clusters were mobile in dendrites of young neurons, the dynamics of TrkB-EGFP were visualized using time-lapse imaging of 3–4 DIV neurons transfected with TrkB-EGFP. Movies were obtained by imaging neurons every 15 s for an average of 15 min. TrkB-EGP was highly dynamic in dendrites, with clusters moving in both the retrograde (toward the cell body) and anterograde (away from the cell body) directions (Fig. 2B, black arrowheads). Unfortunately, the high vesicle density and large volume of dendrites made it impossible to quantify movements of individual TrkB-EGFP puncta in dendrites because we could not follow puncta for more than a couple of frames. Nonetheless, a prominent streaming of small particles could be seen, showing that some clusters of TrkB-EGFP are highly mobile in dendrites during the earliest stages of synapse formation (supplemental movie 1, available at www.jneurosci.org as supplemental material). There is also a large proportion of TrkB that is stationary in dendrites even in these young neurons that have made few synapses (Fig. 2B, white arrowheads). Thus, like other synaptic proteins, there are at least two populations of nonsynaptic TrkB clusters in dendrites: those that are highly mobile and those that are stationary.
Figure 2.

TrkB-EGFP is highly mobile in dendrites and colocalizes with the NMDA receptor subunit NR1. A, A 3 DIV neuron transfected with TrkB-EGFP and imaged ∼24 h after transfection. Fluorescent images are inverted to better illustrate the TrkB-EGFP puncta. TrkB-EGFP is found in a highly punctate distribution along the dendrites. Scale bar, 20 μm. B, TrkB-EGFP is present in both stationary (white arrowhead) and mobile (black arrowheads) puncta that are transported in both retrograde (left) and anterograde (right) directions. Scale bar, 5 μm. C, Dendrites double-labeled for NR1 (top, green in overlay) and sTrkB (middle, red in overlay) at 4 and 10 DIV. Arrowheads indicate some of the NR1 puncta colocalized with sTrkB. Scale bar, 5 μm. D, The distribution of TrkB relative to nonsynaptic and synaptic NR1. A large fraction of NR1 puncta colocalize with sTrkB before, during, and after the peak of synaptogenesis. However, the proportion of NR1 with sTrkB that is at synapses dramatically increases with age.
We demonstrated previously that an early step in postsynaptic assembly is the recruitment of clusters of NMDARs to postsynaptic sites (Washbourne et al., 2002). Therefore, we next investigated whether surface TrkB colocalized with NR1, the obligate subunit of the NMDAR, before and during synapse formation. Triple-immunostaining of 4 and 10 DIV neurons with sTrkB (Up), NR1, and the vesicular glutamate transporter (vGlut1) allowed us to determine the percentage of NR1 found at synapses without sTrkB, at synapses with sTrkB, in nonsynaptic clusters with sTrkB, and in nonsynaptic clusters without TrkB. Colocalization was defined as puncta overlap or apposition and glutamatergic synapses were defined as colocalized clusters of NR1 and vGlut. In general, we found that NR1 colocalized with sTrkB both at 4 and 10 DIV (Fig. 2C, white arrowheads). The percentage of NR1 that colocalized with sTrkB did not significantly change between 4, 10, and even 16 DIV. A total of 61.18 ± 3.22% (n = 19 dendritic fields), 68.08 ± 3.47% (n = 20 dendritic fields) and 71.00 ± 3.47% of NR1 colocalized with sTrkB at 4, 10, and 16 DIV, respectively. However, the percentage of NR1 colocalized with sTrkB that is found at glutamatergic synapses increased significantly during synaptogenesis, from 27.16 ± 3.05% (n = 19 dendritic fields) at 4 DIV to 40.43 ± 2.74% (n = 20 dendritic fields) at 10 DIV (p = 0.0017) and 52.95 ± 2.82% (n = 18 dendritic fields) by 16 DIV (p = 0.0017 from 4 to 10 DIV; p < 0.0001 from 4 to 16 DIV; p = 0.0033 from 10 to 16 DIV) (Fig. 2D). Concomitantly, the population of nonsynaptic NR1 that was not associated with sTrkB decreased with development from 28.11 ± 2.80% (n = 19 dendritic fields) at 4 DIV to 19.06 ± 2.84% by 10 DIV (n = 20 dendritic fields; p = 0.0034) and 11.82 ± 1.92% (n = 18 dendritic fields) by 16 DIV (p < 0.0001 from 4 to 16 DIV, nonsignificant between 10 and 16 DIV) (Fig. 2D). This high association of NR1 with sTrkB at nonsynaptic sites at 4 DIV and its subsequent localization to synapses at 10 DIV is consistent with an instructive role for postsynaptic TrkB in trafficking of NMDARs to new synapses during early stages of synapse formation.
TrkB receptor clusters are highly mobile in axons before most synapses are formed
Although there is ample evidence that TrkB is retrogradely transported in dorsal root ganglia and sciatic nerve axons after exposure to BDNF (Bhattacharyya et al., 1997; Watson et al., 1999; Heerssen et al., 2004), it is unclear how TrkB is originally transported anterogradely from the cell body into axons and axonal growth cones in early development. To address this issue, we transfected young neurons (3–4 DIV) with TrkB-EGFP and monitored TrkB behavior in axons 24 h post-transfection using time-lapse imaging as described above. Axons are reliably identified using morphological criteria at this age (Sabo and McAllister, 2003). Similar to dendritic TrkB, axonal TrkB-EGFP was also found in highly mobile clusters (Fig. 3A–C, supplemental movie 2, available at www.jneurosci.org as supplemental material). On average, 57.5 ± 2.6% (n = 427 puncta, nine axon fields) of TrkB-EGFP puncta were mobile during 5 min imaging periods. TrkB-EGFP clusters had widely varying sizes and intensities (Fig. 3D,E), and the behavior of TrkB-EGFP puncta was complex and heterogeneous. Mobile TrkB-EGFP puncta often coalesced (Fig. 3F) and split (Fig. 3G), suggesting that the puncta might correspond to packets of vesicles traveling together.
Figure 3.

TrkB-EGFP is highly mobile in axons. A–I, Axons of 4–5 DIV pyramidal neurons transfected with TrkB-EGFP. The dynamics of TrkB-EGFP transport packets are complex. TrkB-EGFP puncta can be transported retrogradely (A), anterogradely (B) or bidirectionally (C). Black arrowheads indicate moving puncta. Stationary puncta were also observed (B, white arrowheads). TrkB-EGFP transport packets vary in intensity and apparent size (D, E) and can coalesce (F) or split (G) rapidly. H, Plot of the mean percent of time that the three TrkB-EGFP populations remained paused (*p < 0.001, between retrograde and bidirectional puncta). I, The mean instantaneous velocity for each population of TrkB-EGFP puncta was plotted against the average distance of axon traveled for each of the three TrkB-EGFP populations. It is important to note that the values for average distance of axon traveled for the retrograde and anterograde populations are limited by the image size as these populations of TrkB-EGFP puncta cannot be tracked for as many frames as bidirectional puncta (Table 1) because they often leave the image before the end of the imaging period. So, for anterograde and retrograde puncta the values of average distance of axon traveled should be read as “equal to or higher than” the indicated value. Scale bars, 5 μm.
To quantify the movements of individual TrkB-EGFP puncta, we analyzed the movements of puncta that could be tracked for at least five consecutive frames. Only puncta that were bright enough to be discernable from background were tracked although a streaming of dimmer puncta was sometimes perceptible (supplemental movie 2, available at www.jneurosci.org as supplemental material). To quantify the movements further, we subdivided the mobile puncta into three categories depending on their direction of movement: (1) the anterograde group (puncta that moved strictly away from the cell body), (2) the retrograde group (puncta that moved strictly toward the cell body), and (3) the bidirectional group (puncta that changed direction at any point during imaging) (Table 1, Fig. 3A). Retrograde TrkB-EGFP puncta were usually brighter than the puncta that moved anterogradely, suggesting that they contain more protein than either other class of puncta and making them easier to visualize and track (Table 1). Because this variance in intensity between the categories could result in variable detection of the separate populations, estimates of the percent of total puncta moving in each direction are inherently biased; however, we can report several reliable measures of the movements of those vesicles that we could track.
Table 1.
Characteristics of the different categories of TrkB-EGFP puncta
| Retrograde | Anterograde | Bidirectional | |
|---|---|---|---|
| Mean instantaneous velocity (μm/s) | 0.34 ± 0.06 | 0.32 ± 0.14 | 0.11 ± 0.02 |
| Average distance traveled in axon (μm) | ≥76.5 ± 11.3 | ≥52.5 ± 21.9 | 13.4 ± 2.2 |
| Percentage of time stopped | 34 ± 7 | 43 ± 12 | 64 ± 4 |
| Number of frames | 37.3 ± 5.8 | 29.1 ± 7.2 | 46.2 ± 3.0 |
| Puncta intensity (a.u.) | 31.58 ± 2.92 | 13.43 ± 9.9 | 19.72 ± 1.39 |
| n | 16 | 7 | 35 |
TrkB-EGFP puncta in axons from 4 DIV cortical neurons could be divided into three categories based on the characteristics of their movements during time-lapse imaging. The movement characteristics shown above were measured by tracking TrkB-EGFP puncta from frame-to-frame from time-lapse movies collected every 15 s for 15 min.
Of 58 mobile puncta that we could track for more than five consecutive frames to obtain velocity measurements, 28% moved in the retrograde direction, 12% moved anterogradely, and 60% were bidirectional. Retrograde and anterograde-moving TrkB-EGFP puncta had similar mean velocities (0.26 ± 0.06 μm/s, n = 16 puncta and 0.25 ± 0.15 μm/s, n = 7 puncta, respectively), whereas bidirectional puncta had a lower mean velocity (0.06 ± 0.02 μm/s; n = 35 puncta; p = 0.0002, retrograde and p = 0.008, anterograde). Because the mean velocity depends on both the instantaneous velocity and the number of frames during which a punctum actually moved, the lower mean velocity of bidirectional puncta could result from either or both of these factors. Quantification of mean instantaneous velocities revealed that bidirectional puncta moved at significantly lower instantaneous velocities (0.11 ± 0.02 μm/s; n = 35 puncta) than retrograde (0.34 ± 0.06 μm/s; n = 16 puncta; p = 0.0002) and anterograde puncta (0.32 ± 0.14 μm/s; n = 7 puncta; p = 0.048). Also contributing to their low mean velocities, bidirectional puncta spent more of their time stopped than anterograde and retrograde puncta; 64 ± 4% (n = 35 puncta) of the time for bidirectional as compared with 34 ± 7% (n = 16 puncta) and 43 ± 12% (n = 7 puncta) for anterograde and retrograde puncta, respectively (Fig. 3H).
Retrograde and anterograde puncta frequently traversed most of the length of the axon during the imaging period, whereas bidirectional puncta appeared to scan a smaller portion of the axon. When the distances traveled by each punctum were quantified, the average length of axon traveled by bidirectional puncta was significantly smaller (13.4 ± 2.2 μm; n = 35 puncta) than that traveled by both anterograde (52.5 ± 21.9 μm; n = 7 puncta; p = 0.0047) and retrograde (76.5 ± 11.3 μm; n = 16 puncta; p < 0.0001) puncta. The observed difference in the distance traveled is likely an underestimate of the actual differences between these populations because we were typically able to follow retrograde and anterograde puncta for fewer frames than bidirectional puncta because anterograde and retrograde puncta often left the field of view during imaging (37 ± 6 frames, n = 16 retrograde puncta; 29 ± 7 frames, n = 7 anterograde puncta; 46 ± 3 frames, n = 35 bidirectional puncta). Overall, bidirectional puncta have lower instantaneous velocities and scan a smaller area of the axon than anterograde or retrograde puncta (Fig. 3I). Together, these results suggest that axonal TrkB is present in mobile transport packets in axons that are in the right place at the right time to influence presynaptic terminal formation.
TrkB is transported with synaptic vesicle proteins before synaptogenesis
Because the qualitative mobility and average instantaneous velocities of TrkB-EGFP transport packets were similar to the mobility of SV proteins in axons at a similar age (Sabo et al., 2006), we next hypothesized that TrkB might be transported with SV proteins in axons. To determine whether TrkB colocalizes with SV proteins in axons of young cortical neurons before synapses are formed, 3–4 DIV neurons were double-immunostained for TrkBec (Up) and synapsin I, an SV-associated protein. The two proteins showed a remarkably high degree of colocalization in isolated axons (Fig. 4). At 4 DIV, 84.3 ± 4.4% (n = 6 axon fields) of synapsin I puncta colocalized with TrkBec puncta in axons before contact with dendrites (Fig. 4A–B). At 10 DIV, colocalization was even higher, with 94.8 ± 1.4% (n = 9 axon fields) of synapsin I puncta colocalizing with TrkBec puncta in sections of axons not in contact with dendrites and, therefore, not at synapses (Fig. 4 A–B). Moreover, most of the TrkB that colocalized with SV clusters was present at the axonal surface because 76.83 ± 1.90% of sTrkB colocalized with VAMP2, an integral membrane SV protein, and 83.02 ± 1.80% of VAMP colocalized with sTrkB (Fig. 4C).
Figure 4.
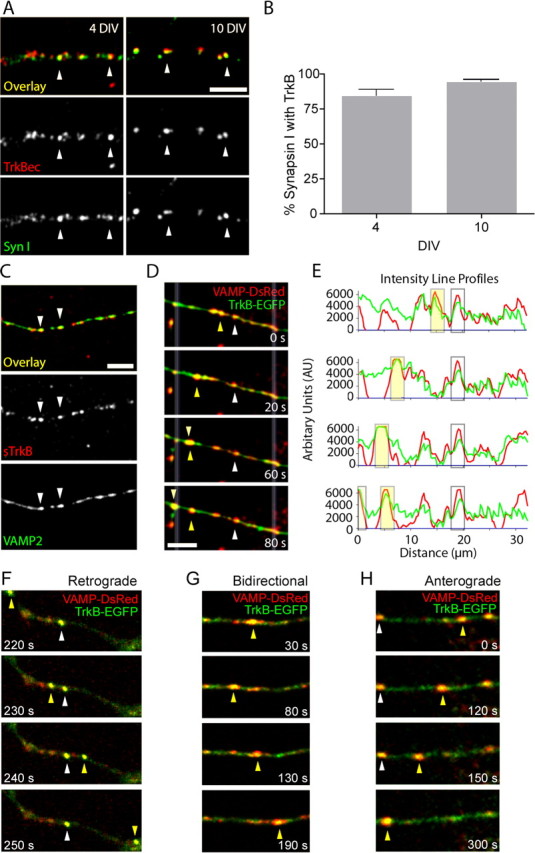
TrkB is almost completely colocalized with and trafficked with SV proteins in axons before most synapses have formed. A, Immunostaining for synapsin I (bottom, green in overlay) and TrkBec (Up) (middle, red in overlay) at 4 DIV (left) and 10 DIV (right). Arrowheads indicate some of the colocalized puncta. B, Synapsin I and TrkB are highly colocalized in isolated axons at both 4 and 10 DIV. C, Surface TrkB colocalizes with VAMP2 (arrowheads). D–H, Axons of neurons cotransfected with TrkB-EGFP (green) and VAMP2-DsRed (red) showed high colocalization of these proteins. D, Two moving puncta show clear simultaneous transport of both TrkB-EGFP and VAMP2-DsRed (arrowheads). E, Fluorescence intensity profiles along a line drawn through the axon between the white, faded lines in (D). Peaks of fluorescence intensity of TrkB-EGFP (green line) and VAMP2-DsRed (red line) show puncta moving together. The yellow box around intensity peaks corresponds to a moving punctum (yellow arrow) and the clear box corresponds to the static punctum (white arrow) in D. F–H, TrkB-EGFP (green) and VAMP2-DsRed (red) are transported together (yellow in overlays) retrogradely (F), bidirectionally (G), and anterogradely (H). The position of mobile puncta is indicated in each frame by a yellow arrowhead, stationary puncta are indicated by white arrowheads. The time that each image was collected is indicated in each panel in seconds. Scale bars, 5 μm.
Given that 57.5 ± 2.6% (n = 427 puncta; nine axon fields) of TrkB-EGFP puncta were mobile and their movement characteristics were similar to those of VAMP2-EGFP, the high degree of colocalization with SV proteins suggested that TrkB might be transported with SV precursor transport packets in axons. To address this issue, 3–4 DIV cortical neurons were cotransfected with TrkB-EGFP and VAMP2-DsRed. We have showed previously that the distribution of VAMP2-DsRed in axons is similar to the distributions of endogenous VAMP and VAMP2-EGFP (Sabo and McAllister, 2003). Furthermore, VAMP2-DsRed colocalized and moved together with synaptophysin-EGFP, another SV marker (supplemental Fig. 3, available at www.jneurosci.org as supplemental material), demonstrating the appropriateness of this approach. To ensure that TrkB-EGFP exhibited the same distribution relative to SV proteins as endogenous TrkB, we stained transfected and nontransfected cells with TrkBec (Up) and VAMP2 antibodies. To quantify colocalization in both sets of neurons, we then used cross-covariation analysis (Sabo et al., 2001). Cross-covariation is insensitive to absolute differences in fluorescence intensities and quantifies the correlations between distribution patterns overall rather than at the level of individual puncta, making the comparison between transfected and untransfected cells reliable even though the puncta in transfected cells were generally brighter. Fluorescence intensities for VAMP2 and TrkBec in nontransfected axons and for TrkB-EGFP and VAMP2 in transfected axons showed similar high degrees of cross-correlation, indicating that endogenous TrkB and TrkB-EGFP were equally highly colocalized with VAMP2 in nontransfected and transfected neurons, respectively (supplemental Fig. 2C–E, available at www.jneurosci.org as supplemental material).
When both TrkB-EGFP and VAMP2-DsRed were expressed at similar levels, they clearly moved together within axons, even before contact with neighboring neurons (n = 18 axon fields) (Fig. 4D–E). Of those puncta that we could identify unambiguously above background using line-scan analysis (n = 280 total puncta), 89.3% were colocalized with VAMP2-DsRed; only 2.9% of total contained VAMP alone and 7.9% of total contained TrkB alone (Table 2). On average, 71.8% of all identifiable TrkB-EGFP and VAMP2-DsRed puncta were mobile (n = 201 puncta) and 92.0% of mobile puncta moved together with VAMP2-DsRed. Furthermore, the kinds of movements of TrkB-EGFP puncta were similar in singly and doubly transfected neurons (Fig. 4F–H). For those mobile TrkB-EGFP and VAMP2-DsRed colocalized puncta that we could track, 24.3% moved retrogradely, 12.4% anterogradely, and 63.2% bidirectionally (compared with ∼27.6, 12.1, and 60.3% for the respective categories in TrkB-EGFP-transfected neurons). Mobile puncta containing VAMP alone comprised, on average, only 3.0% of the total number of mobile puncta. There were only subtle differences in direction of movement of VAMP2-DsRed exclusive puncta compared with comoving TrkB-EGFP and VAMP2-DsRed: 66.7% were bidirectional, 16.7% moved anterogradely, and 16.7% moved retrogradely. Similarly, mobile puncta containing TrkB alone comprised only 5.0% of total mobile puncta. Compared with comoving TrkB-EGFP and VAMP2-DsRed, a higher percentage of TrkB alone puncta were stationary (54.5% for TrkB-EGFP alone compared with 26.0% for colocalized puncta), but the overall directionality of movement was generally similar for these puncta as for colocalized puncta: 60.0% were bidirectional, 10.0% moved anterogradely, and 30.0% moved retrogradely. It is important to note that these numbers are likely an underestimate of the amount of TrkB-EGFP mobility. As mentioned above, there appears to be a significant amount of very fast movements (streaming) of very small TrkB-EGFP puncta that we cannot track; because we do not see a similar behavior for VAMP2-DsRed, it is possible that this fast-moving TrkB-EGFP population may not be transported together with VAMP2-DsRed. Nevertheless, a surprisingly high percent (92%) of TrkB-EGFP clusters that we could track were indeed transported together with VAMP2-DsRed.
Table 2.
Characteristics of the different categories of mobility of TrkB-EGFP and VAMP-DsRed colocalized puncta
| Anterograde | Retrograde | Bidirectional | Stationary | Total | Total mobile | |
|---|---|---|---|---|---|---|
| TrkB/VAMP puncta | 23 [12.4%] | 45 [24.3%] | 117 [63.2%] | 65 (26.0%) | 250 (89.3%) | 185 [92.0%] |
| VAMP alone puncta | 1 [16.7%] | 1 [16.7%] | 4 [66.7%] | 2 (25.0%) | 8 (2.9%) | 6 [3.0%] |
| TrkB alone puncta | 1 [10.0%] | 3 [30.0%] | 6 [60.0%] | 12 (54.5%) | 22 (7.9%) | 10 [5.0%] |
| Total puncta | 25 (8.9%) [12.4%] | 49 (17.5%) [24.4%] | 127 (45.4%) [63.2%] | 79 (28.2%) | 280 | 201 (71.8%) |
Three to four DIV cortical neurons were cotransfected with TrkB-EGFP and VAMP-DsRed. Twenty-four to 36 h after transfection, transfected neurons were imaged for mobility of colocalized puncta in axons; images were captured every 10 s for 10 min. All puncta containing either fluorophore that were 2× the local background intensity (defined using line-scan analysis) and could be tracked for at least four frames were included in the analysis. The numbers in the table correspond to the number of puncta with each behavior pooled from a total of 12 axons from 12 neurons. Numbers in parentheses correspond to the percent of total puncta within each category and numbers in brackets the percent of mobile puncta. Most TrkB-EGFP puncta were colocalized and moved together with VAMP-DsRed. The mobility of the small proportion of VAMP-DsRed alone and TrkB-EGFP alone puncta was generally similar to that of the colocalized puncta.
Although we cannot know whether TrkB-EGFP remains on the surface during cotransport with VAMP2-DsRed (for possible mechanisms, see Discussion), most of the endogenous TrkB that colocalized with SV clusters was present at the axonal surface in fixed neurons because surface labeling for TrkB followed by immunolabeling of endogenous VAMP2 indicated that 76.83 ± 1.90% of sTrkB colocalized with VAMP2 (Fig. 4C). In sum, these data indicate that TrkB on the surface of axons is highly colocalized with SV precursor transport packets, and TrkB-EGFP is transported with SV precursors before synapses are formed. Thus, BDNF could potentially influence the trafficking of presynaptic proteins by activating TrkB receptors that are trafficked with them.
Axonal growth cones and dendritic filopodia express surface TrkB before synapse formation
The high colocalization of TrkB with both presynaptic and postsynaptic proteins before synapses form suggests that TrkB could play an instructive role in the earliest stages of synapse formation. If so, then TrkB should be expressed in the axonal and dendritic structures that are the sites of synapse formation, namely dendritic filopodia and axonal growth cones. To address this issue, neurons were stained for sTrkB and MAP2, to visualize dendrites, or phalloidin, an actin marker to visualize axonal growth cones. In dendrites of 4 DIV cortical neurons, TrkB was found at the surface of dendritic filopodia and dendritic growth cones (Fig. 5A, arrows). To determine whether TrkB is in dynamic dendritic filopodia that could still be looking for a partner, we transfected 3–4 DIV neurons with TrkB-EGFP and imaged them 24 h later. TrkB-EGFP was present on dynamic dendritic filopodia (Fig. 5B) and on active dendritic growth cones (Fig. 5C). Similarly, all axonal growth cones selected by phalloidin staining expressed endogenous TrkB at their surface (n = 25 growth cones) (Fig. 5D), indicating that the presence of TrkB does not depend on the shape or size of the growth cone. Within axonal growth cones, endogenous TrkB localized both along the length of the growth cones and at the tips of their filopodia. In TrkB-EGFP-transfected neurons, TrkB was present in actively growing axonal growth cones (Fig. 5E) and axonal filopodia (Fig. 5F). These results show that TrkB clusters are present at the surface of axonal growth cones and dendritic filopodia while they are searching for their partners, possibly making them sensitive to BDNF secretion from nearby neurons.
Figure 5.

TrkB is located in dendritic filopodia and axonal growth cones. A, Section of a dendrite tipped with a dendritic growth cone from a neuron double immunostained for sTrkB (green) and phalloidin (actin marker; red). Arrows point to sTrkB in dendritic filopodia. B, C, Dendritic filopodia and growth cones of 4 DIV neurons transfected with TrkB-EGFP. TrkB-EGFP is at the tips (arrowheads) of active dendritic filopodia. D, An axonal growth cone from a neuron double immunostained for sTrkB (red) and phalloidin (green). Arrows point to sTrkB puncta found in filopodia of this axonal growth cone. E, F, TrkB-EGFP is present in active axonal growth cones (E) and filopodia (F) (arrowheads). Scale bars, 5 μm.
Surface TrkB becomes increasingly localized to synapses through development
If signaling through TrkB receptors is integrally involved in glutamatergic synapse formation, TrkB should be recruited to synapses before or in parallel with essential presynaptic proteins, postsynaptic proteins or both. To address this issue, we quantified the distribution of sTrkB (Up) in relation to presynaptic and postsynaptic proteins at nonsynaptic sites and at synapses (defined by colocalized vGlut1 and NR1 puncta) before, during, and after the peak of synapse formation. To that end, neurons were triple immunostained for sTrkB, NR1, and vGlut1 at 4, 10, and 16 DIV. As expected, the density of synapses increased over time (Fig. 6A). The number of synapses per 20 μm of dendrite was 4.45 ± 0.47 (n = 10 dendritic fields), 14.71 ± 1.69 (n = 10 dendritic fields), and 33.95 ± 2.86 (n = 8 dendritic fields) at 4, 10, and 16 DIV, respectively (p = 0.0003 from 4 to 10 DIV and p < 0.0001 from 10 to 16 DIV). Consistent with our hypothesis, the localization of sTrkB became more synaptic in parallel with the increased synapse density (Fig. 6B). At 4 DIV, 20.69 ± 2.33% (n = 19 dendritic fields) of sTrkB was associated with synapses, whereas 79.31 ± 2.33% (n = 19 dendritic fields) of sTrkB was nonsynaptic. By 10 DIV, the percentage of sTrkB at synapses was significantly higher, reaching 39.61 ± 1.71% (n = 20 dendritic fields; p < 0.0001). At 16 DIV, an even higher percentage of sTrkB colocalized with synapses (54.86 ± 2.36%; n = 19 dendritic fields; p < 0.0001 from 10 to 16 DIV) with only 42.98 ± 2.41% left in the nonsynaptic pool. The degree of colocalization of TrkB and synapses, at each stage of development, was significantly higher than that predicted by chance alone (chance predictions: 3.82 ± 0.4, 5.62 ± 0.61, and 22.4 ± 2.68% at 4, 10, and 16 DIV, respectively; p < 0.0001) (for calculation, see Materials and Methods). These data suggest that sTrkB is recruited from a primarily nonsynaptic pool in young neurons to a more synaptic localization in mature cortical neurons.
Figure 6.

Surface TrkB becomes increasingly localized to synapses through development. A, Sections of dendrites from neurons triple immunostained for sTrkB, vGlut1, and NR1. Glutamatergic synapses are defined as sites of colocalized vGlut1 and NR1 on dendrites. Qualitatively, there is a clear increase in sTrkB (blue) at synapses over time. B, The amount of sTrkB at synapses increases as neurons mature (orange bars). Significance as compared with the earlier age is indicated as *p < 0.0001. C, The distribution of synaptic and nonsynaptic sTrkB relative to vGlut1 and NR1 before (4 DIV), during (10 DIV), and after (16 DIV) the peak of synapse formation. Nonsynaptic sTrkB colocalized with vGlut1 in axons and NR1 in dendrites becomes recruited to synapses with time. Scale bars, 5 μm.
We also determined how the recruitment of TrkB to synapses correlated with the recruitment of either VGlut1 to presynaptic terminals or NR1 to postsynaptic densities. The percentage of sTrkB that colocalized with vGlut1 increased with age (Fig. 6C, yellow and orange bars), mainly because of increased enrichment of sTrkB at synapses. This was accompanied by a concomitant decrease in the percentage of nonsynaptic TrkB associated with vGlut1 over the same time frame (19.32 ± 2.35% at 4 DIV to 8.73 ± 1.17% at 16 DIV; n = 19 dendritic fields for both; p = 0.0002). The amount of sTrkB associated with NR1 also increased significantly over time (46.75 ± 2.73% at 4 DIV to 74.08 ± 2.11% at 16 DIV; n = 19 dendritic fields for both; p < 0.0001) mostly because of an increase in the fraction of sTrkB at synapses with age (Fig. 6C, red and orange bars). However, in contrast to the colocalization of nonsynaptic sTrkB with vGlut1, the percentage of sTrkB found with nonsynaptic NR1 was relatively constant: 26.17 ± 2.93% (n = 19 dendritic fields) of TrkB was found with nonsynaptic NR1 at 4 DIV, whereas 19.62 ± 1.79% (n = 19 dendritic fields) of sTrkB remained colocalized with nonsynaptic NR1 at 16 DIV. The early enrichment of sTrkB at synapses as neurons develop combined with the high degree of association of TrkB with presynaptic and postsynaptic proteins before synapse formation is consistent with TrkB being recruited to nascent synapses before or in parallel with synaptic proteins.
Surface TrkB is found at most glutamatergic synapses from the earliest stage of their formation
If signaling through TrkB receptors is an instructive signal for excitatory synapse formation, then the majority of glutamatergic synapses should contain TrkB even very early in development. To determine to what extent synapses contain TrkB as neurons develop, we quantified the fraction of glutamatergic synapses with sTrkB before (4 DIV), during (10 DIV), and after (16 DIV) the peak of synapse formation. Neurons were triple immunostained for sTrkB, NR1, and vGlut and the percentage of synapses that contained sTrkB at each of the three ages was quantified. Consistent with our hypothesis, the percentage of synapses that contain sTrkB was remarkably similar at all three ages. Approximately three quarters of glutamatergic synapses had TrkB at their plasma membranes throughout the stages of development examined [71.73 ± 3.11% (n = 19 dendritic fields), 74.47 ± 3.42% (n = 20 dendritic fields), and 73.62 ± 2.71% (n = 19 dendritic fields) at 4, 10, and 16 DIV, respectively] (Fig. 7A) even though the number of synapses dramatically increased during the same period (see above) (Fig. 6A). Although the resolution of the light microscope does not allow us to determine whether synaptic TrkB is localized to presynaptic or postsynaptic structures in mature synapses, our data suggests that it may be localized to both sides of the synapse during early stages of synapse formation. Moreover, the observation that a large majority of synapses contain sTrkB even at the earliest ages strongly suggests that TrkB accumulates at those synapses and is inserted into the synaptic membrane early in their formation.
Figure 7.

TrkB is found at synapses during development both in vitro and in vivo. A, The proportion of synapses that contain sTrkB is high and does not change over time in vitro. Thus, sTrkB is found at most glutamatergic synapses from the earliest stage of their formation. B, Full-length TrkB is found in SPMs in vivo. Synaptic plasma membranes were obtained by subcellular fractionation. The Western blot of the various fractions shows that full-length, catalytic TrkB (145 kDa) and truncated TrkB (95 kDa), as well as synaptophysin, are enriched in synaptic plasma membranes of juvenile rats. S1, Supernatant 1; S2, supernatant 2; P2, precipitate 2 (crude synaptosomes); S3, supernatant 3 (crude synaptic vesicles); P3, precipitate 3 (synaptosomes).
Both full-length and truncated TrkB receptors are found at synapses in vivo in juvenile rat cortex
In order for the TrkB receptors found at these young synapses to signal in response to BDNF, the full-length TrkB receptor must be present at these synapses. Because the antibodies used to label sTrkB are raised against the extracellular domain of the receptor, sTrkB staining cannot discern between full-length and truncated TrkB isoforms. To determine whether full-length TrkB is present at the surfaces of synapses in developing cortex, we biochemically isolated synaptic plasma membranes (SPMs) (Fig. 7B) from 3-week-old rat brains. The isolated synaptic fractions were immunoblotted for TrkB and synaptophysin, an SV protein that is enriched in SPMs (Fig. 7B, bottom lane). Both full-length TrkB and truncated TrkB (predicted molecular weights 145 and 95 kDa, respectively) were found in SPM preparations (n = 3 SPM preparations) (Fig. 7B). These results indicate that the catalytic form of the BDNF receptor is present at synapses in the developing cortex, supporting the hypothesis that BDNF-dependent TrkB signaling could directly and locally influence nascent synapses as they develop.
Discussion
To elucidate the molecular mechanisms by which TrkB, a plasma membrane receptor for BDNF, could affect glutamatergic synapse formation it is essential to know its subcellular location relative to synaptic and nonsynaptic proteins and to understand its dynamics at early stages of development. Here, we addressed these issues using immunocytochemistry, confocal microscopy, and time-lapse imaging in dissociated cultures of cortical neurons before, during, and after the peak of synapse formation. We found that TrkB is present in puncta on the surfaces of both dendrites and axons throughout development. Before synaptogenesis, some sTrkB clusters colocalize with NMDARs in dendrites and almost all axonal TrkB colocalizes with SV proteins. TrkB-EGFP puncta are highly dynamic, moving in both anterograde and retrograde directions in axons and dendrites. In axons, TrkB-EGFP dynamics are similar to VAMP2-EGFP dynamics, and these proteins are often transported together. Finally, dendritic filopodia and axonal growth cones, structures that actively participate in synapse formation, express TrkB at their plasma membrane. Over time, surface TrkB becomes enriched at glutamatergic synapses, which contain both catalytic and truncated TrkB. Overall, our results are consistent with a direct, site-specific role for TrkB in excitatory synaptogenesis.
TrkB localization during synaptogenesis
Although it is clear that neurotrophins and their receptors are highly expressed in the developing nervous system during times of active neuronal growth and differentiation (for review, see McAllister et al., 1999; Cohen-Cory, 2002), reports on the subcellular distribution of TrkB in developing neurons have been inconsistent and insufficient to determine whether TrkB is in the right place at the right time to directly regulate synaptogenesis between cortical neurons. Although the precise reasons for the discrepancies in TrkB distribution among published reports are unclear, it is possible that differences in the specific antibodies used could account for different patterns of staining. To ensure accuracy in our immunolabeling, we selected TrkBec antibodies based on finding two antibodies whose patterns of staining were identical. Many of the other TrkB antibodies we tried resulted in distinct and irreproducible staining patterns (data not shown); it is possible that these other antibodies recognize different glycosylated forms of TrkB. Alternatively, the imaging parameters used in other studies could mask the presence of TrkB in axons because optimizing parameters for imaging the much brighter TrkB in thick dendrites would make the axonal TrkB signal undetectable. Regardless of the reasons for previous discrepancies, our antibody selection criteria allows us to reliably define the distribution of the majority of TrkBec in neurons. Using this stringent approach, we found that TrkB was present in discrete puncta that often colocalized with synaptic markers in both dendrites and axons of dissociated cortical neurons before, during, and after synapse formation.
TrkB-EGFP trafficking before synaptogenesis
Activation of Trk receptors in axons generally leads to ligand-induced endocytosis and retrograde transport of the endocytosed receptors along the axon to the cell body (Ehlers et al., 1995; Tsui-Pierchala and Ginty, 1999; Watson et al., 1999; Bhattacharyya et al., 2002). This retrograde transport is critical for mediating the retrograde survival signal from axon terminals in peripheral neurons (Ye et al., 2003; Heerssen et al., 2004). As a result, retrograde transport of Trk receptors in axons has been the focus of most TrkB trafficking studies in developing and mature neurons (for review, see Yano and Chao, 2004); the transport of TrkB in dendrites has not been described, nor have other forms of TrkB transport in axons.
Here, we show that TrkB-EGFP is found in mobile puncta in both axons and dendrites of young cortical neurons before the bulk of synaptogenesis. Although the high density of TrkB-EGFP puncta in dendrites precluded detailed analysis of their behavior, a large number of mobile TrkB puncta were clearly observed in both dendrites and axons, moving anterogradely, retrogradely, and bidirectionally. These results confirm that TrkB is retrogradely transported in axons of young cortical neurons and that, in addition to retrograde transport, TrkB-EGFP is also anterogradely transported from the cell body to distal portions of the axon, consistent with TrkB being synthesized in the cell body and transported on intracellular vesicles to axons and dendrites (for review, see Yano and Chao, 2004). However, qualitative analysis in dendrites (supplemental movie 1, available at www.jneurosci.org as supplemental material) and quantitative analysis in axons (supplemental movie 2, available at www.jneurosci.org as supplemental material, Table 1, Fig. 3) indicate that the large majority of TrkB-EGFP puncta in both axons and dendrites undergo local, short-range movements in which they rapidly change direction during trafficking. This kind of short-range, bidirectional transport is characteristic of the transport of presynaptic and postsynaptic protein precursors during their trafficking in axons and dendrites before synapses are formed (Washbourne et al., 2002, 2004; Sabo and McAllister, 2003). Although the function of this bidirectional transport is unknown, it could provide a mechanism for TrkB receptor puncta to scan a specific, localized area of the axon and thereby sample potential BDNF secretion from a larger area than if they were stationary.
Colocalization and cotransport of TrkB with synaptic proteins before synaptogenesis
The site of TrkB localization is critical for proposing models for how BDNF signaling might alter synapse formation. There is strong evidence that BDNF can exert its effects through both the presynaptic axon terminal and the postsynaptic density in mature neurons. TrkB is found in both presynaptic terminals and postsynaptic densities of excitatory synapses in the developing and mature CNS (Drake et al., 1999; Petralia et al., 2005) and BDNF is transported to, and released from, both presynaptic and postsynaptic sites in an activity-dependent manner (Haubensak et al., 1998; Kohara et al., 2001). We show here that BDNF could also influence synapse formation on both sides of the synapse through TrkB because full-length, sTrkB colocalizes with both presynaptic and postsynaptic proteins before synapse formation.
TrkB signaling potently modulates postsynaptic neurotransmitter receptor function at excitatory synapses (for review, see Poo, 2001). BDNF treatment leads to phosphorylation of the NR1 NMDAR subunit within minutes of treatment (Jarvis et al., 1997; Suen et al., 1997; Levine et al., 1998; Slack and Thompson, 2002) and causes clustering of NMDARs after 48 h (Elmariah et al., 2004). The high colocalization of NMDAR and TrkB before and during synaptogenesis and the enrichment of TrkB at synapses of very young neurons suggest that TrkB could play a similar role at developing glutamatergic synapses. For example, the binding of BDNF to TrkB might influence synapse formation by altering the clustering of NMDARs at sites of axodendritic contact or change the function or stability of NMDARs at nascent synapses. Approximately 60–70% of NR1 puncta colocalized with sTrkB before, during, and after synapse formation. Importantly, both NR1 and sTrkB became increasingly localized to synapses with age. Although it has been demonstrated that acute activation of TrkB receptors does not direct the rapid clustering of NMDARs (Dalva et al., 2000), it remains possible that more focal application of BDNF to TrkB puncta in dendrites would alter NMDAR trafficking in young neurons during synaptogenesis. Nevertheless, the high association of NR1 with sTrkB at nonsynaptic sites at 4 DIV and their subsequent localization to synapses at 10 DIV implies that TrkB is in the right place at the right time to influence postsynaptic assembly or maturation.
In addition to its effects on NMDARs, BDNF also influences SV trafficking in mature synapses through increasing the expression of many SV proteins (Takei et al., 1998), enhancing synapsin 1 phosphorylation (Jovanovic et al., 1996), and altering the number of docked SVs at excitatory synapses (Martinez et al., 1998; Pozzo-Miller et al., 1999; Tyler and Pozzo-Miller, 2001; Carter et al., 2002). In addition, synaptic activity in CNS neurons is rapidly enhanced by BDNF through a presynaptic locus (for review, see Poo, 2001). Our data suggest that TrkB could play a similar role during synapse formation by regulating the trafficking of SV precursors. We show here that, early in development, SV proteins are highly colocalized (>80%) with sTrkB in axons before synaptogenesis and that the large majority (∼94%) of TrkB-EGFP is cotransported with VAMP2-DsRed along these isolated axons. Perhaps BDNF signaling through TrkB directly mobilizes SV precursors to accumulate at nascent synapses in young cortical neurons.
The high (>80%) colocalization of VAMP2 with sTrkB and the very high percentage of these proteins that are transported together (>93%) suggest that TrkB receptors that were in the plasma membrane at some point during surface-labeling are transported with intracellular SV precursor transport packets. The mechanism by which a surface transmembrane receptor is cotransported with SV proteins located on intracellular vesicles is unknown. There are numerous possibilities. First, sTrkB could be present in lipid rafts (Suzuki et al., 2001) and could move solely within the plasma membrane, tethered to the intracellular SV precursor organelle through an intracellular linker protein, such as spectrin. Although atypical, this kind of transport was recently demonstrated for the cotransport of NCAM and trans-Golgi-network organelles in young neurons (Sytnyk et al., 2002). Second, TrkB could be found on separate transport vesicles that rapidly cycle with the membrane to allow for surface immunolabeling and are transported together with the SV precursors within axons before synapse formation. Such rapid cycling of transmembrane proteins during their trafficking before synapse formation has been demonstrated previously for NMDA receptors in young cortical neurons (Washbourne et al., 2004). Finally, TrkB could be a component of the multiprotein complex found on SV precursor organelles (Ahmari et al., 2000; Zhai et al., 2001) and could be surface-labeled during cycling of those precursors, which is known to occur during their transport (Matteoli et al., 1992; Kraszewski et al., 1995). Future experiments using biochemical techniques and measures of cycling of sTrkB receptors should uncover the mechanism of sTrkB and SV precursor cotransport. Regardless of the mechanism, the observation that they are transported together suggests the possibility that BDNF signaling through TrkB could directly alter the transport of the colocalized SV precursors and thereby influence their accumulation at nascent synapses.
Implications of the distribution of TrkB for models of excitatory synapse formation
There is ample evidence that TrkB signaling is important for the formation of excitatory synapses. BDNF treatment causes the precocious formation of excitatory synapses in culture (Vicario-Abejon et al., 1998; Aguado et al., 2003) and BDNF injection enhances synapse formation in vivo (Alsina et al., 2001). In addition, TrkB knock-out animals show reduced numbers of hippocampal excitatory synapses (Martinez et al., 1998), and an elegant previous study using a series of conditional TrkB knock-out animals has demonstrated a direct role for TrkB signaling in excitatory synapse formation (Luikart et al., 2005). However, other groups report that BDNF influences excitatory synapse number only indirectly through homeostatic mechanisms resulting from a direct effect of BDNF in enhancing inhibitory synapse number (Elmariah et al., 2004).
In general, these previous results support a role for BDNF in synapse maturation, but the specific mechanisms by which TrkB signaling is translated into increases in glutamatergic synapse number is unknown. Our results show that TrkB is present at the right place at the right time on excitatory neurons to play an important, direct role in excitatory synapse formation. In addition, the presence of TrkB in the neuronal structures that actively participate in synapse formation (dendritic filopodia and axonal growth cones) further suggest that the role for TrkB in synaptogenesis might well be instructive. Moreover, our data suggest that TrkB must be inserted into new synapses rapidly because the same proportion of synapses (∼75%) contain sTrkB at all stages of development examined. Conversely, these results also suggest that there is a small population of excitatory synapses that do not contain TrkB, suggesting that all excitatory synapses may not require TrkB signaling for their formation. Nevertheless, the high degree of association of sTrkB with SVs and NMDARs before synapse formation and the enrichment of both sTrkB and these synaptic proteins at synapses during their formation, strongly suggest that TrkB may play a direct, local role in excitatory synapse formation.
How might TrkB signaling lead to recruitment of SV precursors and NMDARs to nascent synapses? Current models for synapse formation suggest that diffusible molecules such as BDNF act as “priming” molecules, providing a permissive environment for synapse formation (Waites et al., 2005). In this model, BDNF signaling would activate TrkB receptors, thereby leading to the clustering of synapse-inducing factors, such as trans-synaptic adhesion molecules. In turn, these inducing molecules initiate a signal transduction cascade that actively recruits presynaptic and postsynaptic proteins to the nascent synapse. Although we provide no data on the relative time course of recruitment of TrkB and synaptic proteins to new synapses, our results are inconsistent with this model. This current model would not predict that a synaptogenic molecule would be colocalized, much less cotransported, with synaptic proteins in neurons before synapses are formed, as we show here. Instead, our results suggest a new model in which synaptogenic molecules could influence the trafficking of synaptic protein transport packets through a direct association. In the future, it will be important to test this prediction not only for TrkB signaling, but also for the activity of other synaptogenic molecules.
Footnotes
This work was supported by the Pew Charitable Trusts and National Institutes of Health Grant RO1 EY13584 (A.K.M.). R.A.G. is a scholar from the Gulbenkian PhD Program in Biomedicine supported by Fundacao para a Ciencia e Tecnologia. We thank L. Reichardt (University of California, San Francisco, San Francisco, CA) for the chicken anti-TrkBec antibody, D. R. Kaplan (Hospital for Sick Children, Toronto, Ontario, Canada) for TrkBin antibody, M. Ehlers (Duke University, Durham, NC) for the SPM protocol, and R. Segal (Harvard University, Cambridge, MA) for the TrkB-EGFP construct. We thank Afonso Salcedo for help with movies. Special thanks to Carol Erickson for advice, support, and help with this manuscript.
References
- Aguado F, Carmona MA, Pozas E, Aguilo A, Martinez-Guijarro FJ, Alcantara S, Borrell V, Yuste R, Ibanez CF, Soriano E. BDNF regulates spontaneous correlated activity at early developmental stages by increasing synaptogenesis and expression of the K+/Cl− co-transporter KCC2. Development. 2003;130:1267–1280. doi: 10.1242/dev.00351. [DOI] [PubMed] [Google Scholar]
- Ahmari SE, Buchanan J, Smith SJ. Assembly of presynaptic active zones from cytoplasmic transport packets. Nat Neurosci. 2000;3:445–451. doi: 10.1038/74814. [DOI] [PubMed] [Google Scholar]
- Alsina B, Vu T, Cohen-Cory S. Visualizing synapse formation in arborizing optic axons in vivo: dynamics and modulation by BDNF. Nat Neurosci. 2001;4:1093–1101. doi: 10.1038/nn735. [DOI] [PubMed] [Google Scholar]
- Aoki C, Wu K, Elste A, Len G, Lin S, McAuliffe G, Black IB. Localization of brain-derived neurotrophic factor and TrkB receptors to postsynaptic densities of adult rat cerebral cortex. J Neurosci Res. 2000;59:454–463. doi: 10.1002/(SICI)1097-4547(20000201)59:3<454::AID-JNR21>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya A, Watson FL, Bradlee TA, Pomeroy SL, Stiles CD, Segal RA. Trk receptors function as rapid retrograde signal carriers in the adult nervous system. J Neurosci. 1997;17:7007–7016. doi: 10.1523/JNEUROSCI.17-18-07007.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya A, Watson FL, Pomeroy SL, Zhang YZ, Stiles CD, Segal RA. High-resolution imaging demonstrates dynein-based vesicular transport of activated Trk receptors. J Neurobiol. 2002;51:302–312. doi: 10.1002/neu.10062. [DOI] [PubMed] [Google Scholar]
- Blackstone CD, Moss SJ, Martin LJ, Levey AI, Price DL, Huganir RL. Biochemical characterization and localization of a non-N-methyl-d-aspartate glutamate receptor in rat brain. J Neurochem. 1992;58:1118–1126. doi: 10.1111/j.1471-4159.1992.tb09370.x. [DOI] [PubMed] [Google Scholar]
- Carter AR, Chen C, Schwartz PM, Segal RA. Brain-derived neurotrophic factor modulates cerebellar plasticity and synaptic ultrastructure. J Neurosci. 2002;22:1316–1327. doi: 10.1523/JNEUROSCI.22-04-01316.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen-Cory S. The developing synapse: construction and modulation of synaptic structures and circuits. Science. 2002;298:770–776. doi: 10.1126/science.1075510. [DOI] [PubMed] [Google Scholar]
- Dalva MB, Takasu MA, Lin MZ, Shamah SM, Hu L, Gale NW, Greenberg ME. EphB receptors interact with NMDA receptors and regulate excitatory synapse formation. Cell. 2000;103:945–956. doi: 10.1016/s0092-8674(00)00197-5. [DOI] [PubMed] [Google Scholar]
- Dechant G. Molecular interactions between neurotrophin receptors. Cell Tissue Res. 2001;305:229–238. doi: 10.1007/s004410100378. [DOI] [PubMed] [Google Scholar]
- Drake CT, Milner TA, Patterson SL. Ultrastructural localization of full-length TrkB immunoreactivity in rat hippocampus suggests multiple roles in modulating activity-dependent synaptic plasticity. J Neurosci. 1999;19:8009–8026. doi: 10.1523/JNEUROSCI.19-18-08009.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers MD, Kaplan DR, Price DL, Koliatsos VE. NGF-stimulated retrograde transport of trkA in the mammalian nervous system. J Cell Biol. 1995;130:149–156. doi: 10.1083/jcb.130.1.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmariah SB, Crumling MA, Parsons TD, Balice-Gordon RJ. Postsynaptic TrkB-mediated signaling modulates excitatory and inhibitory neurotransmitter receptor clustering at hippocampal synapses. J Neurosci. 2004;24:2380–2393. doi: 10.1523/JNEUROSCI.4112-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fryer RH, Kaplan DR, Feinstein SC, Radeke MJ, Grayson DR, Kromer LF. Developmental and mature expression of full-length and truncated TrkB receptors in the rat forebrain. J Comp Neurol. 1996;374:21–40. doi: 10.1002/(SICI)1096-9861(19961007)374:1<21::AID-CNE2>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Gerrow K, Romorini S, Nabi SM, Colicos MA, Sala C, El-Husseini A. A preformed complex of postsynaptic proteins is involved in excitatory synapse development. Neuron. 2006;49:547–562. doi: 10.1016/j.neuron.2006.01.015. [DOI] [PubMed] [Google Scholar]
- Haubensak W, Narz F, Heumann R, Lessmann V. BDNF-GFP containing secretory granules are localized in the vicinity of synaptic junctions of cultured cortical neurons. J Cell Sci. 1998;111:1483–1493. doi: 10.1242/jcs.111.11.1483. [DOI] [PubMed] [Google Scholar]
- Heerssen HM, Pazyra MF, Segal RA. Dynein motors transport activated Trks to promote survival of target-dependent neurons. Nat Neurosci. 2004;7:596–604. doi: 10.1038/nn1242. [DOI] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF. Trk receptors: roles in neuronal signal transduction. Annu Rev Biochem. 2003;72:609–642. doi: 10.1146/annurev.biochem.72.121801.161629. [DOI] [PubMed] [Google Scholar]
- Jarvis CR, Xiong ZG, Plant JR, Churchill D, Lu WY, MacVicar BA, MacDonald JF. Neurotrophin modulation of NMDA receptors in cultured murine and isolated rat neurons. J Neurophysiol. 1997;78:2363–2371. doi: 10.1152/jn.1997.78.5.2363. [DOI] [PubMed] [Google Scholar]
- Jovanovic JN, Benfenati F, Siow YL, Sihra TS, Sanghera JS, Pelech SL, Greengard P, Czernik AJ. Neurotrophins stimulate phosphorylation of synapsin I by MAP kinase and regulate synapsin I-actin interactions. Proc Natl Acad Sci USA. 1996;93:3679–3683. doi: 10.1073/pnas.93.8.3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohara K, Kitamura A, Morishima M, Tsumoto T. Activity-dependent transfer of brain-derived neurotrophic factor to postsynaptic neurons. Science. 2001;291:2419–2423. doi: 10.1126/science.1057415. [DOI] [PubMed] [Google Scholar]
- Kraszewski K, Mundigl O, Daniell L, Verderio C, Matteoli M, De Camilli P. Synaptic vesicle dynamics in living cultured hippocampal neurons visualized with CY3-conjugated antibodies directed against the lumenal domain of synaptotagmin. J Neurosci. 1995;15:4328–4342. doi: 10.1523/JNEUROSCI.15-06-04328.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kryl D, Yacoubian T, Haapasalo A, Castren E, Lo D, Barker PA. Subcellular localization of full-length and truncated Trk receptor isoforms in polarized neurons and epithelial cells. J Neurosci. 1999;19:5823–5833. doi: 10.1523/JNEUROSCI.19-14-05823.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine ES, Crozier RA, Black IB, Plummer MR. Brain-derived neurotrophic factor modulates hippocampal synaptic transmission by increasing N-methyl-d-aspartic acid receptor activity. Proc Natl Acad Sci USA. 1998;95:10235–10239. doi: 10.1073/pnas.95.17.10235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luikart BW, Nef S, Virmani T, Lush ME, Liu Y, Kavalali ET, Parada LF. TrkB has a cell-autonomous role in the establishment of hippocampal Schaffer collateral synapses. J Neurosci. 2005;25:3774–3786. doi: 10.1523/JNEUROSCI.0041-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez A, Alcantara S, Borrell V, Del Rio JA, Blasi J, Otal R, Campos N, Boronat A, Barbacid M, Silos-Santiago I, Soriano E. TrkB and TrkC signaling are required for maturation and synaptogenesis of hippocampal connections. J Neurosci. 1998;18:7336–7350. doi: 10.1523/JNEUROSCI.18-18-07336.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marty S, Wehrle R, Sotelo C. Neuronal activity and brain-derived neurotrophic factor regulate the density of inhibitory synapses in organotypic slice cultures of postnatal hippocampus. J Neurosci. 2000;20:8087–8095. doi: 10.1523/JNEUROSCI.20-21-08087.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matteoli M, Takei K, Perin MS, Sudhof TC, De Camilli P. Exo-endocytotic recycling of synaptic vesicles in developing processes of cultured hippocampal neurons. J Cell Biol. 1992;117:849–861. doi: 10.1083/jcb.117.4.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister AK, Katz LC, Lo DC. Neurotrophins and synaptic plasticity. Annu Rev Neurosci. 1999;22:295–318. doi: 10.1146/annurev.neuro.22.1.295. [DOI] [PubMed] [Google Scholar]
- Petralia RS, Sans N, Wang YX, Wenthold RJ. Ontogeny of postsynaptic density proteins at glutamatergic synapses. Mol Cell Neurosci. 2005;29:436–452. doi: 10.1016/j.mcn.2005.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poo MM. Neurotrophins as synaptic modulators. Nat Rev Neurosci. 2001;2:24–32. doi: 10.1038/35049004. [DOI] [PubMed] [Google Scholar]
- Pozzo-Miller LD, Gottschalk W, Zhang L, McDermott K, Du J, Gopalakrishnan R, Oho C, Sheng ZH, Lu B. Impairments in high-frequency transmission, synaptic vesicle docking, and synaptic protein distribution in the hippocampus of BDNF knock-out mice. J Neurosci. 1999;19:4972–4983. doi: 10.1523/JNEUROSCI.19-12-04972.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford LC, DeWan A, Lauer HM, Turrigiano GG. Brain-derived neurotrophic factor mediates the activity-dependent regulation of inhibition in neocortical cultures. J Neurosci. 1997;17:4527–4535. doi: 10.1523/JNEUROSCI.17-12-04527.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabo SL, McAllister AK. Mobility and cycling of synaptic protein-containing vesicles in axonal growth cone filopodia. Nat Neurosci. 2003;6:1264–1269. doi: 10.1038/nn1149. [DOI] [PubMed] [Google Scholar]
- Sabo SL, Ikin AF, Buxbaum JD, Greengard P. The Alzheimer amyloid precursor protein (APP) and FE65, an APP-binding protein, regulate cell movement. J Cell Biol. 2001;153:1403–1414. doi: 10.1083/jcb.153.7.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabo SL, Gomes RA, McAllister AK. Formation of presynaptic terminals at predefined sites along axons. J Neurosci. 2006;26:10813–10825. doi: 10.1523/JNEUROSCI.2052-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seil FJ, Drake-Baumann R. TrkB receptor ligands promote activity-dependent inhibitory synaptogenesis. J Neurosci. 2000;20:5367–5373. doi: 10.1523/JNEUROSCI.20-14-05367.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapira M, Zhai RG, Dresbach T, Bresler T, Torres VI, Gundelfinger ED, Ziv NE, Garner CC. Unitary assembly of presynaptic active zones from Piccolo-Bassoon transport vesicles. Neuron. 2003;38:237–252. doi: 10.1016/s0896-6273(03)00207-1. [DOI] [PubMed] [Google Scholar]
- Slack SE, Thompson SW. Brain-derived neurotrophic factor induces NMDA receptor 1 phosphorylation in rat spinal cord. NeuroReport. 2002;13:1967–1970. doi: 10.1097/00001756-200210280-00027. [DOI] [PubMed] [Google Scholar]
- Suen PC, Wu K, Levine ES, Mount HT, Xu JL, Lin SY, Black IB. Brain-derived neurotrophic factor rapidly enhances phosphorylation of the postsynaptic N-methyl-d-aspartate receptor subunit 1. Proc Natl Acad Sci USA. 1997;94:8191–8195. doi: 10.1073/pnas.94.15.8191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, Ito J, Takagi H, Saitoh F, Nawa H, Shimizu H. Biochemical evidence for localization of AMPA-type glutamate receptor subunits in the dendritic raft. Brain Res Mol Brain Res. 2001;89:20–28. doi: 10.1016/s0169-328x(01)00051-1. [DOI] [PubMed] [Google Scholar]
- Swanwick CC, Harrison MB, Kapur J. Synaptic and extrasynaptic localization of brain-derived neurotrophic factor and the tyrosine kinase B receptor in cultured hippocampal neurons. J Comp Neurol. 2004;478:405–417. doi: 10.1002/cne.20295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sytnyk V, Leshchyns'ka I, Delling M, Dityateva G, Dityatev A, Schachner M. Neural cell adhesion molecule promotes accumulation of TGN organelles at sites of neuron-to-neuron contacts. J Cell Biol. 2002;159:649–661. doi: 10.1083/jcb.200205098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takei N, Numakawa T, Kozaki S, Sakai N, Endo Y, Takahashi M, Hatanaka H. Brain-derived neurotrophic factor induces rapid and transient release of glutamate through the non-exocytotic pathway from cortical neurons. J Biol Chem. 1998;273:27620–27624. doi: 10.1074/jbc.273.42.27620. [DOI] [PubMed] [Google Scholar]
- Tsui-Pierchala BA, Ginty DD. Characterization of an NGF-P-TrkA retrograde-signaling complex and age-dependent regulation of TrkA phosphorylation in sympathetic neurons. J Neurosci. 1999;19:8207–8218. doi: 10.1523/JNEUROSCI.19-19-08207.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyler WJ, Pozzo-Miller LD. BDNF enhances quantal neurotransmitter release and increases the number of docked vesicles at the active zones of hippocampal excitatory synapses. J Neurosci. 2001;21:4249–4258. doi: 10.1523/JNEUROSCI.21-12-04249.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicario-Abejon C, Collin C, McKay RD, Segal M. Neurotrophins induce formation of functional excitatory and inhibitory synapses between cultured hippocampal neurons. J Neurosci. 1998;18:7256–7271. doi: 10.1523/JNEUROSCI.18-18-07256.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waites CL, Craig AM, Garner CC. Mechanisms of vertebrate synaptogenesis. Annu Rev Neurosci. 2005;28:251–274. doi: 10.1146/annurev.neuro.27.070203.144336. [DOI] [PubMed] [Google Scholar]
- Washbourne P, Bennett JE, McAllister AK. Rapid recruitment of NMDA receptor transport packets to nascent synapses. Nat Neurosci. 2002;5:751–759. doi: 10.1038/nn883. [DOI] [PubMed] [Google Scholar]
- Washbourne P, Liu XB, Jones EG, McAllister AK. Cycling of NMDA receptors during trafficking in neurons before synapse formation. J Neurosci. 2004;24:8253–8264. doi: 10.1523/JNEUROSCI.2555-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson FL, Heerssen HM, Moheban DB, Lin MZ, Sauvageot CM, Bhattacharyya A, Pomeroy SL, Segal RA. Rapid nuclear responses to target-derived neurotrophins require retrograde transport of ligand-receptor complex. J Neurosci. 1999;19:7889–7900. doi: 10.1523/JNEUROSCI.19-18-07889.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu K, Xu JL, Suen PC, Levine E, Huang YY, Mount HT, Lin SY, Black IB. Functional trkB neurotrophin receptors are intrinsic components of the adult brain postsynaptic density. Brain Res Mol Brain Res. 1996;43:286–290. doi: 10.1016/s0169-328x(96)00211-2. [DOI] [PubMed] [Google Scholar]
- Xia Z, Dudek H, Miranti CK, Greenberg ME. Calcium influx via the NMDA receptor induces immediate early gene transcription by a MAP kinase/ERK-dependent mechanism. J Neurosci. 1996;16:5425–5436. doi: 10.1523/JNEUROSCI.16-17-05425.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada MK, Nakanishi K, Ohba S, Nakamura T, Ikegaya Y, Nishiyama N, Matsuki N. Brain-derived neurotrophic factor promotes the maturation of GABAergic mechanisms in cultured hippocampal neurons. J Neurosci. 2002;22:7580–7585. doi: 10.1523/JNEUROSCI.22-17-07580.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Q, Radeke MJ, Matheson CR, Talvenheimo J, Welcher AA, Feinstein SC. Immunocytochemical localization of TrkB in the central nervous system of the adult rat. J Comp Neurol. 1997;378:135–157. [PubMed] [Google Scholar]
- Yano H, Chao MV. Mechanisms of neurotrophin receptor vesicular transport. J Neurobiol. 2004;58:244–257. doi: 10.1002/neu.10321. [DOI] [PubMed] [Google Scholar]
- Ye H, Kuruvilla R, Zweifel LS, Ginty DD. Evidence in support of signaling endosome-based retrograde survival of sympathetic neurons. Neuron. 2003;39:57–68. doi: 10.1016/s0896-6273(03)00266-6. [DOI] [PubMed] [Google Scholar]
- Zhai RG, Vardinon-Friedman H, Cases-Langhoff C, Becker B, Gundelfinger ED, Ziv NE, Garner CC. Assembling the presynaptic active zone: a characterization of an active one precursor vesicle. Neuron. 2001;29:131–143. doi: 10.1016/s0896-6273(01)00185-4. [DOI] [PubMed] [Google Scholar]