

Abstract
The ataxia mutation (axJ) is a recessive neurological mutation that results in reduced growth, ataxia, and hindlimb muscle wasting in mice. The axJ gene encodes ubiquitin-specific protease 14 (Usp14), a deubiquitinating enzyme (DUB) that associates with the proteasome via its ubiquitin-like (Ubl) domain and is involved in processing ubiquitin chains. Analysis of Usp14 gene products demonstrated that Usp14 undergoes alternative pre-mRNA splicing to produce a full-length form of Usp14 that is capable of binding proteasomes and a form that contains a deletion in the Ubl domain. The full-length form of Usp14 is the only form that appears to be reduced in the axJ mice. Transgenic rescue of the axJ mice with neuronal-specific expression of Usp14 demonstrated that the full-length form of Usp14 was sufficient to restore viability and motor system function to the axJ mice. Biochemical analysis showed that the ubiquitin hydrolyase activity of this form of Usp14 is dependent on the presence of proteasomes, and neuronal expression of full-length Usp14 was able to restore the levels of monomeric ubiquitin in the brains of axJ mice. However, the axJ-rescued mice still displayed the Purkinje cell axonal swellings that are seen in the axJ mice, indicating that this cerebellar alteration is not the primary cause of the axJ movement disorders. These results show that the motor defects observed in the axJ mice are attributable to a neuropathic disease rather than to a muscular disorder and suggest that changes in proteasomal function may contribute to neurological dysfunction in the axJ mice.
Keywords: ataxia, proteasome, ubiquitin, synapse, mutation, transgenic
Introduction
The ubiquitin proteasome system (UPS) controls numerous cellular processes such as cell cycle progression and development by regulating the degradation of intracellular proteins. This regulation occurs in a temporal or spatial manner via the covalent attachment of a polyubiquitin chain to a substrate, which targets it for degradation by the proteasome (Glickman and Ciechanover, 2002). Alterations in the UPS have been hypothesized to contribute to defects in immune regulation (Gregory et al., 2003), cancer (Kondo and Kaelin, 2001; Gregory et al., 2003; Kovalenko et al., 2003), and chronic neurological diseases such as Parkinson's disease, Huntington's disease, and spinocerebellar ataxias (Alves-Rodrigues et al., 1998; McNaught et al., 2001; Dawson and Dawson, 2003).
The ataxia (axJ) mutation is one of the few genetic lesions in the UPS that leads to neurological disease. This mutation results in reduced expression of ubiquitin-specific protease 14 (Usp14) and causes several defects in mice, including ataxia and muscle wasting (D'Amato and Hicks, 1965). Unlike many chronic neurological diseases (Alves-Rodrigues et al., 1998), the axJ mice do not have detectable ubiquitinated protein aggregates or display significant neuronal loss. Instead, these mice exhibit altered synaptic activity in both the CNS and PNS (Wilson et al., 2002). These findings support a direct role of the UPS in the regulation of synaptic function and demonstrate that synaptic defects may be a primary cause of neurological disease.
Usp14 is one of >70 deubiquitinating enzymes (DUBs) encoded in the mouse genome (Soboleva and Baker, 2004). DUBs are responsible for disassembling ubiquitin chains at the proteasome, cleaving ubiquitin precursors, and editing ubiquitin side chains (Guterman and Glickman, 2004) and are therefore essential for regulating protein stability and localization. Observations with Usp14 and its homolog ubiquitin-specific protease 6 (Ubp6) indicate that one of the functions of Usp14 is to recycle ubiquitin at the proteasome (Leggett et al., 2002; Chernova et al., 2003, Anderson et al., 2005). Although Usp14 is one of three DUBs that have been shown to associate with mammalian proteasomes (Guterman and Glickman, 2004), the finding that the loss of Usp14 results in neuronal dysfunction (Wilson et al., 2002) indicates that these proteasomal-bound DUBs act on specific substrates and are not redundant.
This study was initiated to better understand the in vivo function or functions of Usp14. We recently determined that Usp14 is alternatively spliced to produce two forms of Usp14, one that is capable of binding to proteasomes and one that has a deletion in the proteasome-binding domain. To examine the effect of the axJ mutation on the expression of these two spliced forms of Usp14, we characterized the expression of the Usp14 gene products in wild-type and axJ mice. Transgenic mice that specifically express Usp14 only in neurons also were constructed to determine the site of neuromuscular dysfunction in the axJ mice. The Usp14 transgene was evaluated for its ability to complement the biochemical and behavioral defects of the axJ mice in order to determine how Usp14 functions in the nervous system and to gain new insights into how alterations in the UPS lead to neurological disease.
Materials and Methods
Animals.
Wild-type C57BL/6J and Usp14axJ mice (The Jackson Laboratory, Bar Harbor, ME) have been maintained in our breeding colony at the University of Alabama at Birmingham, which is accredited fully by the Association for Assessment and Accreditation of Laboratory Animal Care International. Homozygous Usp14axJ mice (which we refer to as axJ mice) were generated by intercrossing axJ/+ siblings and could be identified phenotypically by 3 weeks of age. Transgenic animals were generated by pronuclear injection of the Thy1-Usp14LF transgene into C57BL/6J fertilized eggs. All research complied with the United States Animal Welfare Act and other federal statutes and regulations relating to animals and experiments involving animals and adhered to principles stated in the Guide for the Care and Use of Laboratory Animals, United States National Research Council.
Total RNA isolation.
RNA was isolated from mouse brain by using RNA-STAT (Tel-Test, Friendswood, TX).
Reverse transcription PCR.
Mouse embryonic day 7 (E7) and E15 cDNA libraries were purchased from Clontech (Mountain View, CA). The P28 library was constructed by using the first-strand synthesis kit from Invitrogen (Carlsbad, CA). Approximately 2 μl of reverse-transcribed cDNA was used for PCR amplification in a PerkinElmer 9700 thermocycler (Wellesley, MA) with Platinum Pfx DNA Polymerase (Invitrogen). Twenty PCR cycles were used to amplify the cDNAs.
Two-dimensional gel electrophoresis.
Total protein extracts were generated by using the ReadyPrep Protein Extraction kit (Bio-Rad, Hercules, CA). Proteasome extracts were precipitated before isoelectric focusing, using the two-dimensional (2D) gel cleanup kit (Amersham Biosciences, Piscataway, NJ). ReadyStrip IPG (immobilized pH gradient) strips (11 cm), pH 4–7, were hydrated overnight with protein extracts. Linear focusing conditions were as follows: 250 V for 15 min, 8000 V for 2.5 h, equaling 35,000 V h total. Strips were equilibrated in a solution containing 6 m urea, 0.375 m Tris-HCl, pH 8.8, 2% SDS, 20% glycerol, and 2% DTT for 10 min with shaking. Strips subsequently were incubated for 10 min in a solution containing 6 m urea, 0.375 m Tris-HCl, pH 8.8, 2% SDS, 20% glycerol, and 2% iodoacetamide. Strips then were rinsed in 1× SDS PAGE running buffer and placed into a 4–15% Criterion gel (Bio-Rad) and sealed with agarose.
Construction of Thy1-Usp14LF transgene.
The full-length Usp14 cDNA, including the Usp14 Kozak consensus sequence, was generated by using reverse transcription-PCR (RT-PCR) and was cloned into the XhoI site of the pThy1.2 expression cassette (gift from Dr. Pico Caroni at the Friedrich Institute, Basel, Switzerland). The transgene was excised from the vector by using EcoRI and NdeI and prepared for microinjection via standard procedures.
Antibodies.
Antibodies to Usp14 were generated by using recombinant Usp14 (amino acids 100–493) to immunize New Zealand White rabbits (Covance, Radnor, PA). Usp14 polyclonal antisera from rabbit R138 was affinity-purified and used for immunoblotting at a dilution of 1:2000. The JLA20 actin antibody and β-tubulin antibody were purchased from Developmental Hybridoma Core (Iowa City, IA) and were diluted 1:5000 and 1:2000, respectively. The control regulatory proteasome subunit 1 (Rpt1) and Rpt4 monoclonal antibodies (mAbs; Biomol, Plymouth Meeting, PA) and the calbindin antibody (Swant, Bellinzona, Switzerland) were used at a 1:1000 dilution.
Isolation of mouse proteins.
Mice 4–6 weeks of age and of appropriate genotype were killed by CO2 asphyxiation. Tissues were homogenized in 1–3 ml of homogenization buffer containing the following (in mm): 50 Tris, pH 7.5, 150 NaCl, 5 MgCl2, 2 N-ethylmaleimide, plus 0.5% SDS and Complete protease inhibitors from Roche (Indianapolis, IN). After homogenization the tissues were sonicated for 10 s and then centrifuged at 17,000 × g for 10 min at 4°C. Supernatants were removed and immediately frozen at –80°C. Protein concentrations were determined by using the bicinchoninic acid (BCA) protein assay kit from Pierce (Rockford, IL).
Isolation of proteasomes.
Proteasomes were isolated as described by Borodovsky et al. (2001).
Immunoblotting.
Proteins were resolved on either 8% Tris-glycine gels or 4–20% Tris-glycine NUPAGE gels (Invitrogen) and transferred onto either nitrocellulose or polyvinylidene difluoride (PVDF) membranes. The polyclonal Usp14 R138 antisera was diluted in PBS containing 3% BSA. Primary antibodies were detected by using an anti-mouse or anti-rabbit HRP-conjugated antibody (Southern Biotechnology Associates, Birmingham, AL) and Luminol reagents (Pierce).
Quantitation of immunoblots.
Blots were scanned by a Hewlett Packard Scanjet 3970 (Palo Alto, CA) and quantitated by using UN-SCAN-IT software (Silk Scientific, Orem, UT). Each value represents the average and SE from at least six blots, using at least three different animals of each genotype.
Labeling of proteasome-associated DUBs.
Approximately 5 μg of proteasomes was diluted into a solution containing the following (in mm): 50 Tris, pH 7.5, 250 sucrose, 1 DTT, and 2 ATP. Then 1 μl of a 0.2 μg/μl solution of hemagglutinin-tagged ubiquitin vinylmethyl ester (HA-Ub-VME) was added, and the reaction was incubated at room temperature for 30 min to label the active DUBs. SDS sample buffer was added, and the reaction was boiled for 5 min. Proteins were resolved on a 4–12% bis-Tris NUPAGE gel and then were transferred onto PVDF membranes. The labeled DUBs were detected by probing the blot with the 12CA5 anti-HA monoclonal antibody (Roche) and were visualized with Luminol reagents (Pierce). Approximately 50 ng of recombinant Usp14 (Biomol) was incubated with proteasomes at room temperature for 2 h before the addition of hemagglutinin-tagged ubiquitin vinyl sulfone (HA-Ub-VS).
Culture and staining of rat hippocampal neurons.
Neurons were cultured as described in Price and Brewer (2001). The E15–E18 rat hippocampal neurons were dissociated, plated, and allowed to grow in culture for 7–14 d before staining. Coverslips were washed three times with PBS containing the following (in mm): 2.67 KCl, 1.47 KH2PO4, 137.93 NaCl, and 8.10 Na2HPO4. Cells were fixed for 20 min with 4% paraformaldehyde (PFA) in sodium phosphate buffer containing 100 mm NaPO4. After fixation the coverslips were washed an additional two times with PBS. Cells were blocked in a solution containing 3% normal goat serum (NGS) and 0.3% Triton X-100 in PBS. Primary antibodies were diluted in blocking solution and applied to the coverslips overnight at 4°C. The next day the coverslips were washed three times in PBS containing 1% NGS. Alexa Fluor secondary antibodies from Invitrogen were diluted 1:500 in 1% NGS and applied to the coverslips for 1 h in the dark at room temperature. Coverslips then were washed in PBS containing 1% NGS, followed by PBS containing 1% NGS and 4′,6-diamidino-2-phenylindole (DAPI), and finally in PBS before being mounted and imaged.
Transfection of rat hippocampal neurons.
Neurons were transfected by using the Amaxa rat hippocampal neuron transfection kit (Gaithersburg, MD) according the manufacturer's protocol.
African green monkey kidney cell culture and transfection.
African green monkey kidney cells (COS-7 cells) were maintained in DMEM (Invitrogen, Gaithersburg, MD) containing 10% fetal bovine serum, 50 U/ml penicillin, and 50 μg/ml streptomycin (Invitrogen). Cells were maintained by passaging the cells every 3 d via trypsinization and subsequent replating of ∼1 × 106 cells. For transfection 9 × 105 COS-7 cells were plated on a 100 mm tissue culture dish the day before transfection. Each 100 mm dish was transfected with 3 μg of the appropriate DNA construct, using the Fu-Gene 6 transfection reagent (Roche) and following the manufacturer's protocol. Cells were allowed to express the construct for ∼24 h before collection in a solution containing the following (in mm): 200 Tris-HCl, pH 7.4, 750 NaCl, 5 EDTA, plus 5% NP-40 and Complete protease inhibitors (Roche). After collection the cells were sonicated for 10 s and then centrifuged at 17,000 × g for 10 min at 4°C. Supernatants were removed and frozen immediately at –80°C. Protein concentrations were determined by using the BCA protein assay kit from Pierce.
Immunofluorescence.
Mouse brains were submersion-fixed with either methanol–Carnoy (methacarn) or 4% PFA overnight at 4°C, followed by 70% EtOH for 24 h. Tissues fixed in 4% PFA were embedded in paraffin, and 10 μm sections were cut for staining. Methacarn-fixed brains were cut on a vibratome at 100 μm. Sections were blocked at room temperature for 1 h in PBS containing 10% normal goat sera, 1% BSA, and 0.1% Triton X-100. Primary and secondary antibodies were diluted in PBS containing 2% normal goat antisera, 0.1% BSA, and 0.1% Triton X-100. Images were acquired by using an Olympus (Tokyo, Japan) IX 70 inverted microscope equipped with epifluorescence optics. The camera used was a Retiga 1300 cooled CCD, firewire, high-resolution monochromatic camera purchased from QImaging (Burnaby, British Columbia, Canada). We used an 83000 Pinkel filter set from Chroma Technology (Rockingham, VT). A Mac 2000 electronic filter wheel with an automatic shutter separately housed the excitation filters. All image acquisition was performed with IPLab Spectrum imaging software from Scanalytics (Fairfax, VA). The University of Alabama at Birmingham High Resolution Imaging Facility assisted in sample preparation and imaging.
Elevated beam assay.
Assays were performed as described in Stanley et al. (2005). Mice were placed on the middle of a 60 cm rod that was 2 cm in diameter and was elevated 30 cm above the bench by metal supports. Mice were allowed to walk to a platform located at the end of the beam. Each trial consisted of five repetitions of this assay. Mice were videotaped to quantitate the number of falls and foot slips (one or both hindlimbs slipped from the beam) for each trial.
Results
Usp14 is alternatively spliced
Analysis of RT-PCR products generated by using Usp14-specific primers demonstrated the presence of two Usp14 transcripts that are developmentally regulated and are detected in both neuronal and non-neuronal tissues (Fig. 1 A). Cloning and sequencing of these Usp14 transcripts demonstrated that they are generated by alternative splicing of exon 4, which encodes ∼33 amino acids of the ubiquitin-like (Ubl) domain required for proteasome binding (Fig. 1 B). The cDNA of Usp14 that contains exon 4 has been designated Usp14LF, and the cDNA that lacks exon 4 is referred to as Usp14SF. No other alternatively spliced cDNAs were identified for Usp14.
Figure 1.

Usp14 is alternatively spliced. A, The left panel depicts Usp14 RT-PCR products from tissues of adult (P64) mice, and the right panel shows the Usp14 RT-PCR products from E7 and E15 embryos and whole brain from P28 wild-type mice demonstrating the two forms of Usp14 produced from the alternative splicing of exon 4. Primers flanking exon 4 were used for amplification of reverse-transcribed cDNAs. B, Schematic diagram of the cDNAs isolated from brain. The full-length cDNA is referred to as Usp14LF, and the splice variant lacking exon 4 (asterisk) is referred to as Usp14SF. C, Immunoblot analysis of 6-week-old wild-type (wt) and axJ brains demonstrating the two forms of Usp14. Blots were probed with polyclonal antisera to Usp14 and reprobed for β-actin as a loading control. D, Immunoblot analysis demonstrating the different migration patterns of Usp14LF and Usp14SF expressed from wild-type E15 embryos, COS-7 cells transfected with expression vectors for Usp14LF or Usp14SF, and neuronal proteasome fractions from wild-type (P28) mice. Blots were probed with polyclonal antisera to Usp14. E, Immunoblot of Usp14 showing the similar migration pattern between recombinant Usp14 and Usp14 found in neuronal proteasomes from wild-type mice. Blots were probed with polyclonal antisera to Usp14. F, Two-dimensional electrophoresis gel of proteasome fractions from wild-type mice immunoblotted with polyclonal antisera to Usp14.
Protein expression of Usp14LF and Usp14SF
The predicted molecular weights of Usp14LF and Usp14SF are 56 and 52 kDa, respectively. However, immunoblot analysis of whole brain extracts from wild-type mice demonstrated the presence of two immunoreactive bands, one that migrated at 52 kDa and a second band that migrated at 66 kDa (Fig. 1C). The 52 kDa immunoreactive band was not detected in proteasome fractions from the brains of wild-type mice (Fig. 1 D) and was greatly reduced in the immunoblots from wild-type E15 embryos (Fig. 1 D), which produce only a small amount of alternatively spliced Usp14SF mRNA (Fig. 1 A). Although the 66 kDa band could not be detected in the brain extracts from axJ mice, wild-type levels of the 52 kDa band were still observed in the axJ mice (Fig. 1C). To test whether these two bands correlated with the two forms of Usp14 produced from the alternative splicing of exon 4 of Usp14, we transfected COS-7 cells with mammalian expression vectors containing either Usp14LF or Usp14SF. Examination of protein extracts made from these transfected cells confirmed that Usp14LF migrates at 66 kDa, whereas Usp14SF migrates at 52 kDa (Fig. 1D). In addition, when we compared the migration pattern of Usp14 from the proteasome fractions with recombinant Usp14LF, which was produced in bacteria and therefore lacks any post-translational modification, both samples exhibited the same migration pattern during electrophoresis (Fig. 1 E).
To examine any potential post-translational modification to Usp14LF, such as phosphorylation, we separated proteasome fractions from the brains of wild-type mice by 2D gel electrophoresis and immunoblotted them with an antibody to Usp14. In these gels Usp14 migrated at a range of isoelectric focusing points (pIs) from ∼5.0 to 5.1 (Fig. 1 F). The predicted change in pI is ∼0.05 pI units per phosphate group added to Usp14, indicating that Usp14LF may contain at least three to four phosphorylated residues.
Usp14LF is activated in the presence of proteasomes
Previous work on the yeast ortholog of Usp14, Ubp6, demonstrated a 300-fold increase in activity when Ubp6 associated with the proteasome (Leggett et al., 2002). To determine whether Usp14 also requires proteasomes to activate its ubiquitin hydrolyase activity, we examined the activity of recombinant Usp14LF in the presence and absence of proteasome fractions prepared from the brains of axJ mice. In this assay the ubiquitin hydrolyase activity of Usp14 was examined by the ability of Usp14 to be labeled with HA-Ub-VS. Because HA-Ub-VS is an active-site probe, only catalytically active DUBs will be covalently modified by this probe (Borodovsky et al., 2001) and can be detected by an anti-HA antibody. As previously shown (Anderson et al., 2005), Usp14 was labeled with the HA-Ub-VS probe in the positive control lane of neuronal proteasomes isolated from wild-type mice (Fig. 2). No activity of Usp14 against the HA-Ub-VME substrate was observed from proteasomes isolated from axJ mice or from recombinant Usp14LF produced from bacterial cultures (Fig. 2). However, when recombinant Usp14LF was mixed with the proteasome fraction isolated from axJ mice, we detected significant labeling of Usp14 (Fig. 2). To determine whether the labeling was dependent on contact with proteasomes and not attributable to a soluble factor in the preparations, we also measured the ubiquitin-hydrolyzing activity from supernatant fractions from the proteasome preparations of wild-type mice both in the presence and absence of recombinant Usp14LF. Neither supernatant fraction was able to activate the catalytic activity of Usp14 (Fig. 2), indicating that the ubiquitin-hydrolyzing activity was dependent on the binding of Usp14 to the proteasomes.
Figure 2.

Usp14 is catalytically active only in the presence of proteasomes. The top panel indicates the catalytically active DUBs detected in wild-type (wt) and axJ proteasome fractions in the presence and absence of recombinant Usp14. The preparations were incubated with HA-Ub-VS and probed with the anti-HA antibody 12C5. The locations of the two proteasome-bound DUBs are indicated to the right. The middle panel depicts total Usp14 detected by polyclonal Usp14 antisera. The blots also were probed with an antibody to the 19S proteasome component Rpt4 as a control.
Subcellular localization of Usp14 in neurons
We have shown previously that Usp14 cofractionates with neuronal proteasomes (Anderson et al., 2005) and that the loss of Usp14 results in synaptic transmission defects (Wilson et al., 2002), indicating that Usp14 may be located with proteasomes near the synapse. To determine the subcellular distribution of Usp14 in neurons, we examined Usp14 expression from cultured primary rat hippocampal neurons by using indirect immunofluorescence. Similar to previous results (Mengual et al., 1996; Wojcik and DeMartino, 2003), a predominantly nuclear and perinuclear pattern was observed after neurons were stained with antibodies to the 26S proteasome (Fig. 3A). Staining for the proteasome also was detected throughout the cytoplasm of the hippocampal neurons. The distribution of endogenous Usp14 overlapped the staining of the cytoplasmic proteasomes, with the most predominant signal observed in the perinuclear region (Fig. 3A).
Figure 3.

Usp14 is a cytosolic protein in primary neuronal cultures. A, Rat primary hippocampal neurons were stained with DAPI (blue) and with antibodies to Usp14 (green) and 26S proteasomes (red). B, Primary rat hippocampal neurons were transfected with myc-tagged Usp14LF and stained with DAPI (blue) and a 9E10 anti-myc antibody to detect the myc-tagged Usp14LF (green). Red boxes depict the neurite processes of the transfected neurons. Images were collected at 40× magnification.
To verify the distribution of Usp14 with a different antibody, we next examined the expression pattern from primary hippocampal neurons transfected with myc-tagged Usp14LF. The myc-tagged Usp14LF showed a similar perinuclear localization pattern as the endogenous Usp14, with some nuclear staining (Fig. 3B). In addition, cytosolic Usp14 staining also was detected throughout the cell soma and in the neurite processes (Fig. 3B).
Generation of Usp14LF transgenic mice
The axJ mice suffer from a severe neuromuscular disorder characterized by tremor, muscle wasting, and hindlimb spasticity. We therefore were interested in determining whether the defects in the axJ mice were attributable solely to the loss of Usp14 expression in the nervous system or whether loss of Usp14 in muscle also contributed to disease in the axJ mice. To determine whether neuronal-specific expression of Usp14 could rescue the growth and motor defects of the axJ mice, we cloned Usp14LF behind the pan-neuronal Thy1.2 promoter (Caroni, 1997) to generate Thy1-Usp14LF transgenic mice (Fig. 4A). We chose to express Usp14LF in these studies because our previous research demonstrated a role for Usp14 in recycling ubiquitin at the proteasome (Anderson et al., 2005), because it is the predominant form of Usp14 detected in the brain (Fig. 1), and because it is the only form of Usp14 that shows reduced expression in the axJ mice (Fig. 1C).
Figure 4.

The Thy1-Usp14LF transgene is expressed only in the nervous system. A, A full-length Usp14 cDNA was cloned into the Thy1.2 expression cassette to allow for neuronal-specific expression of Usp14LF. B, Tissue extracts from wild-type (wt), axJ, and axJ Tg mice were prepared and immunoblotted with anti-Usp14 polyclonal antibodies to detect Usp14 levels. Blots were probed with an antibody to β-tubulin as a loading control. C, Quantitation of immunoblots from five sets of wild-type, axJ, and axJ Tg animals showing the fold increase of Usp14LF expression in the tissues. Error bars indicate SE.
Two independent founder lines, 9002 and 9045, were generated for the Thy1-Usp14LF transgene. Immunoblot analysis indicated that the Thy1-Usp14LF transgene was robustly expressed at postnatal day 7 (P7) and was maintained in each line until at least 20 weeks after birth (data not shown). Southern analysis of the founder lines demonstrated that both lines have a similar number of transgene insertions. No growth or behavioral defects were observed in either founder line, and both lines demonstrated similar expression patterns of the Thy1-Usp14LF transgene (data not shown). Although both lines yielded identical results throughout the study, only the results of the 9002 founder line are reported below.
To test the ability of the Thy1-Usp14LF transgene to rescue the defects observed in the axJ mice, we generated mice that were homozygous for the axJ mutation and contained the Thy1-Usp14LF transgene. These mice have been designated axJ Tg. To ensure that the transgene only expressed Usp14LF in neuronal tissues, we prepared protein extracts from the brain, spinal cord, muscle, liver, kidney, and heart of wild-type, axJ, and axJ Tg mice and immunoblotted them with an antibody to Usp14. When compared with the levels of Usp14LF expressed in the axJ mice, increased expression of Usp14LF from the Thy1-Usp14LF transgene was detected only in the brain and spinal cord (Fig. 4B). These levels corresponded to a 4.5-fold increase of Usp14LF in the brain and a sixfold increase in the spinal cord of the axJ Tg mice (Fig. 4C) as compared with the levels normally found in wild-type mice.
Thy1-Usp14LF rescues the axJ growth defects and lethality
To determine whether the growth defects and juvenile death of the axJ mice were rescued with the Thy1-Usp14LF transgene, we monitored body mass and survival rates for the axJ, axJ Tg, and wild-type mice. Similar to previous results (Anderson et al., 2005), all of the axJ mice died within 8 weeks of age (Fig. 5A). In contrast, all of the wild-type and axJ Tg mice survived to the end of the 20 week test period (Fig. 5A). A comparison of the body weights of the axJ Tg mice with those of the axJ and wild-type mice demonstrated that neuronal-specific expression of Usp14LF was able to restore body weights to wild-type levels (Fig. 5B), indicating that both the axJ postnatal lethality and growth defects are attributable to the loss of Usp14 in the nervous system. This improvement was evident by 6 weeks of age, the time at which the axJ mice normally show a remarkable decrease in body weights when compared with wild-type mice (Fig. 5B), and continued throughout the 20 week test period.
Figure 5.

Thy1-Usp14LF rescues the postnatal lethality and reduced growth in axJ mice. A, Survival curves for wild-type (wt), axJ, and axJ Tg mice (n = 5). B, Body weights were obtained over a 20 week period for wild-type, axJ, and axJ Tg mice (n = 5). Error bars indicate SE.
Usp14LF interacts with proteasomes and restores the cellular levels of ubiquitin
To examine whether the neuronal expression of Usp14LF rescued the biochemical defects observed in the axJ mice, we first examined the proteasomes from wild-type, axJ, and axJ Tg mice for the presence of Usp14. As we previously reported (Anderson et al., 2005), Usp14 was found in the proteasome fractions from wild-type animals and was greatly reduced in the fractions from axJ mice (Fig. 6A). Immunoblot analysis of axJ Tg proteasomes demonstrated that transgenic Usp14LF was able to stably associate with neuronal proteasomes (Fig. 6A).
Figure 6.

Usp14LF associates with proteasomes and restores the levels of ubiquitin in axJ mice. A, The left panel shows the immunoblot of proteasome fractions from the brains of wild-type (wt), axJ, and axJ Tg mice probed with polyclonal Usp14 antisera. The blots also were probed for the 19S proteasomal component Rpt4 as a positive control for proteasomes. The right panel shows catalytically active DUBs from neuronal proteasome fractions from wt, axJ, and axJ Tg mice that were labeled with HA-Ub-VME and immunoblotted with the anti-HA mAb 12CA5. The identities of the known DUBs are indicated at the right. B, Immunoblot analysis of hippocampal proteasome fractions from wild-type mice and wild-type mice containing the Usp14LF transgene (wt Tg) probed for Usp14 and the control 19S proteasomal component Rpt1 (left panel) and active DUBs labeled with HA-Ub-VME and immunoblotted with the anti-HA mAb 12CA5 (right panel). Graphs depict quantitation of the blots. C, Immunoblot analysis of hippocampal extracts measuring levels of conjugated and monomeric ubiquitin in wild-type, axJ, and axJ Tg mice. As loading controls the blots were reprobed with a monoclonal antibody to β-actin. Error bars indicate SE.
To determine whether the proteasome-bound Usp14 was catalytically active, we incubated proteasomes from wild-type, axJ, and axJ Tg mice with the HA-Ub-VME probe that is directed to the active-site of DUBs. Similar to previous results (Anderson et al., 2005), Usp14 and ubiquitin C-terminal hydrolyase 37 (UCH37) were the only DUBs that labeled with the HA-Ub-VME probe in wild-type mice (Fig. 6A). The axJ proteasomes lacked Usp14 but retained wild-type levels of UCH37 (Fig. 6A). In addition to stably associating with neuronal proteasomes, Usp14LF expressed from the Thy1-Usp14LF transgene was also catalytically active (Fig. 6A).
To elucidate whether we could increase the amount of Usp14 stably associated with the proteasome, we next measured the amount of Usp14 bound to proteasomes from wild-type mice containing the Usp14LF transgene. Because the Thy1.2 promoter previously has been shown to exhibit mosaic expression patterns (Caroni, 1997), proteasomes were prepared from the hippocampus, where expression of Thy1-Usp14LF is robust (see Fig. 8 K), and the level of proteasome-associated Usp14 could be evaluated in the wild-type mice containing the transgene. There was a 2.5-fold increase in total Usp14 and a threefold increase in labeling of catalytically active Usp14 in the proteasomes from wild-type mice that contained the Thy1-Usp14LF transgene (Fig. 6 B, wt Tg) as compared with proteasomes from wild-type mice, indicating that neuronal proteasomes are capable of binding more Usp14 than what normally is observed in the wild-type mice.
Figure 8.
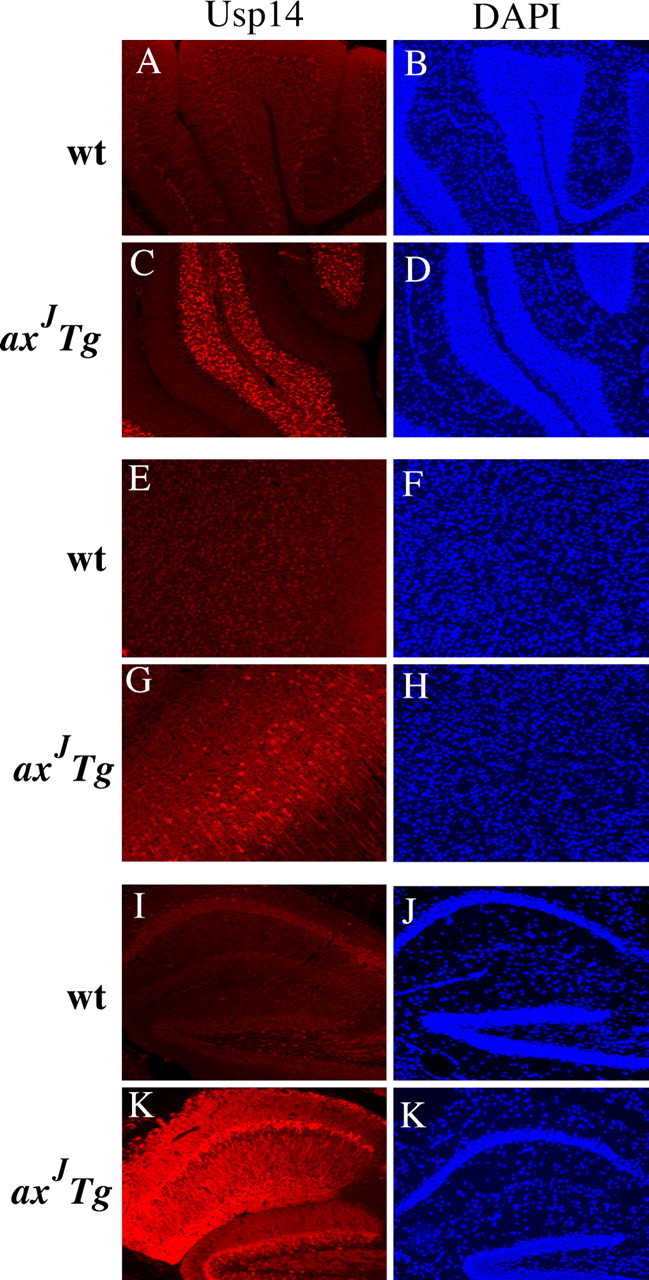
Expression patterns of Thy1-Usp14LF in the CNS. Shown is Usp14 (red) staining in the cerebellum (A, C), cortex (E, G), and hippocampus (I, K) of wild-type (wt) and axJ Tg mice. Endogenous Usp14 is not detected in wild-type tissues. DAPI staining in the cerebellum (B, D), cortex (F, H), and hippocampus (J, L) is shown in blue. Images were taken at 20× magnification.
To determine whether transgenic expression of Thy1-Usp14LF restored the levels of monomeric ubiquitin in the axJ mice, we examined the steady-state levels of ubiquitin in wild-type, axJ, and axJ Tg mice. Similar to previous results (Anderson et al., 2005), there was a 30% loss of monomeric ubiquitin in axJ brain extracts as compared with controls (Fig. 6C). In contrast, immunoblot analysis of hippocampal extracts showed that the levels of monomeric ubiquitin were restored to wild-type levels in the axJ Tg mice (Fig. 6C), indicating that the Ubl-containing form of Usp14 is sufficient for the proper maintenance of ubiquitin levels in the brain.
Thy1-Usp14LF rescues the motor defects of the axJ mice
The axJ mice display severe hindlimb paralysis and muscle wasting by 8 weeks of age, which is easily demonstrated by their inability to traverse an elevated beam. To determine whether the motor system defects observed in the axJ mice were rescued by the Thy1-Usp14LF transgene, we measured the performance of the wild-type, axJ, and axJ Tg mice on the elevated beam assay described by Stanley et al. (2005). In this assay all of the 8-week-old wild-type animals were capable of traversing the beam without any falls, and all of the axJ mice fell off the beam (Fig. 7A). Examination of the axJ Tg mice demonstrated that transgenic expression of Usp14LF specifically within neurons was able to restore the ability of the axJ mice to traverse the elevated beam (Fig. 7A). However, when we measured the number of times the hind feet of the animals slipped from the elevated beam during the trial, the axJ Tg mice demonstrated an increase in the number of foot slips as compared with wild-type controls (Fig. 7 B), possibly indicating the presence of cerebellar abnormalities in the axJ Tg mice that were not rescued by the Thy1-Usp14LF transgene.
Figure 7.

The Thy1-Usp14LF transgene rescues the axJ motor dysfunction. A, Elevated beam walking assay of wild-type (wt), axJ, and axJ Tg mice demonstrates that neuronal-specific expression of Usp14LF greatly reduces the number of falls per trial seen in the axJ mice (n = 4). B, The frequency of front or rear paw slips off the elevated beam is greater in axJ Tg mice than in wild-type mice. Error bars indicate SE.
Thy1-Usp14LF is not detected in Purkinje cells of axJ Tg mice
To determine whether the Thy1-Usp14LF transgene restored Usp14 expression in the various layers of the cerebellar cortex, we used immunostaining to examine the expression pattern of Usp14LF in the cerebellum of the axJ Tg mice. Our Usp14 antisera could not detect the endogenous Usp14 in the tissues from wild-type mice but could readily detect Usp14LF when it was overexpressed in the cerebellum of the axJ Tg mice (Fig. 8C). Although high levels of Usp14 expression were observed in the granule cell layer, expression of Usp14LF from the Thy1-Usp14LF transgene was not detected in the Purkinje cell layer of the axJ Tg mice (Fig. 8C). The only other neuronal population that lacked detectable expression of the Thy1-Usp14LF transgene was the CA3 pyramidal cells of the hippocampus (data not shown).
When we looked at the distribution of Usp14 in the brain, transgenic expression of Thy1-Usp14LF resulted in a mosaic pattern of expression of Usp14LF in the cortex (Fig. 8E–H) and fairly uniform expression in the hippocampus (Fig. 8I–L). In agreement with previous results (Fig. 3), Usp14 was detected primarily in the cytosol of the cortical and hippocampal pyramidal neurons (Fig. 8G,K).
axJ Tg mice display Purkinje cell axonal swellings
Previous studies on the axJ mice reported the presence of Purkinje cell axonal swellings that were hypothesized to be responsible for the motor defects of the axJ mice (D'Amato and Hicks, 1965). Because Usp14LF was not detected from the Thy1-Usp14LF transgene in the Purkinje cells of the axJ Tg mice, indirect immunofluorescence microscopy was used to examine the Purkinje cell axons of wild-type, axJ, and axJ Tg mice to determine whether the Purkinje cell pathology was still present in the cerebellum of the axJ Tg mice. In agreement with previous studies (D'Amato and Hicks, 1965), large varicosities could be detected along the Purkinje cell axons of the axJ mice as early as 3 weeks of age (Fig. 9A). Surprisingly, not only was the Purkinje cell axonal pathology still present in the axJ Tg mice at 8 weeks of age (Fig. 9B), but the number of the swellings increased between 8 and 12 weeks of age (data not shown), indicating that this pathology cannot be responsible for the overt motor defects of the axJ mice. However, the remaining Purkinje cell pathology may be related to the increased number of foot slips in the axJ Tg mice.
Figure 9.

Purkinje cell axonal pathology in axJ and axJ Tg mice. A, Indirect immunofluorescence microscopy was used to examine Purkinje cell axons from axJ and wild-type (wt) mice during development by using calbindin (red) antibodies and DAPI (blue). B, Indirect immunofluorescence microscopy demonstrated that axJ Tg mice have a similar Purkinje cell disease as the axJ mice. Arrowheads indicate the presence of normal Purkinje cell axons in wild-type mice, and arrows represent abnormal Purkinje cell axons in axJ and axJ Tg mice. Images were taken at 40× magnification.
Discussion
The results presented in this study have uncovered several new and important aspects of Usp14. Usp14 encodes a ubiquitin-specific protease that is expressed ubiquitously in mammalian tissues (Anderson et al., 2005) and is thought to regulate ubiquitin levels at the proteasome. By using transgenic animals that specifically express Usp14 within neurons, we have demonstrated that the postnatal lethality, stunted growth, and overt motor defects in the axJ mice are solely attributable to the loss of Usp14 expression in the nervous system. Although our previous studies identified an intracisternal A-particle (IAP) insertion into intron 5 of Usp14 in the axJ mice, which suggested that the axJ phenotype was attributable to the loss of Usp14 expression, the axJ mutation was the only known mutant allele of Usp14, and it is known that IAP insertions can have long-range effects on gene expression. The transgenic complementation of the axJ mice with a Usp14 cDNA therefore provides genetic proof that the axJ mutation resides in Usp14. This work has also demonstrated that, although Usp14 is expressed in a variety of tissues in mammals, Usp14 has an essential, non-redundant function in the nervous system and that the axJ neuromuscular defects are attributable to a neuropathy and not to a muscular disorder.
In addition to the full-length proteasome-bound form of Usp14LF, our studies have identified a novel splice form of Usp14, Usp14SF, that is expressed in both neuronal and non-neuronal tissues, is developmentally regulated, and contains a 33 aa deletion in the Ubl domain. Consistent with the observation that the Ubl is required for stable association with the proteasome (Leggett et al., 2002; Chernova et al., 2003), Usp14SF was not able to fractionate with proteasomes from the brains of wild-type mice (Fig. 1 D). Surprisingly, the insertion of the IAP into intron 5 of the axJ Usp14 (Wilson et al., 2002) does not appear to affect the expression of Usp14SF in the brains of the axJ mice. Because the IAP exerts its effect by altering Usp14 pre-mRNA splicing, it is possible that the IAP has a much greater effect on processing the Usp14LF pre-mRNA. The finding that the IAP insertion can exert variable effects on Usp14 protein levels is supported by our observation that the levels of Usp14LF are reduced only modestly in the testes of the axJ mice, whereas they are greatly reduced in brain and muscle (Anderson et al., 2005).
Immunoblot analysis of brain extracts from wild-type mice demonstrated the presence of two immunoreactive Usp14 bands that migrate to the same positions as the Usp14 proteins from extracts prepared from COS-7 cells transfected with the Ubl-containing Usp14LF construct and the alternatively spliced Usp14SF construct. We estimate that the steady-state level of Usp14LF is approximately five times greater than that of Usp14SF. Although Usp14SF migrates according to its predicted molecular weight, Usp14LF migrates significantly slower than its predicted molecular weight of 56 kDa. Comparison of the migration patterns of the two forms of Usp14 with recombinant Usp14LF clearly demonstrated that the altered migration of Usp14LF is attributable to the presence of the Ubl domain. Ubiquitin domains are extremely stable and therefore may be less susceptible to denaturation, resulting in reduced migration in our gels.
Although our studies with 2D gel electrophoresis suggest that Usp14LF is phosphorylated, phosphorylation does not appear to contribute to the reduced migration of Usp14LF in our gels. However, the phosphorylation of Usp14 could have important effects on the activity of Usp14. Recent studies have shown that the Usp14 homolog in Arabidopsis can interact with calmodulin and therefore may be phosphorylated by calcium/calmodulin-dependent protein kinase II (CaMKII) (Moon et al., 2005). Examination of the Usp14 protein sequence revealed a potential CaMKII phosphorylation position at amino acids 428–431. In addition, the crystal structure of Usp14 was solved recently and shown to contain two loops that block the catalytic domain of Usp14 (Hu et al., 2005). These loops were hypothesized to be displaced after proteasome binding to allow for the activation of Usp14. Because the CaMKII site is located in one of these blocking loops, phosphorylation of Usp14 by CaMKII may provide a mechanism to regulate the activity of Usp14 on the proteasome in a calcium-dependent manner.
Although a specific mechanism of proteasome malfunction has not been shown in the axJ mice, our data, along with Ubp6 studies in yeast and proteasome inhibition studies in EL4 cell lines (Borodovsky et al., 2001; Leggett et al., 2002; Hu et al., 2005), are consistent with the requirement of Usp14LF to bind to the proteasome to become catalytically active. In this study we have shown that recombinant Usp14LF becomes catalytically active only in the presence of proteasomes and that cell extracts devoid of proteasomes are unable to activate the ubiquitin hydrolyase activity of Usp14. Approximately 50% of Usp14LF was found within the neuronal proteasome fraction of wild-type mice (Fig. 2), and overexpression of Usp14LF resulted in an increase in the amount of Usp14LF that associates with neuronal proteasomes (Fig. 6A), suggesting that not all proteasomes contain Usp14LF. Therefore, by controlling the association of Usp14LF with the proteasome, it may be possible to modulate the deubiquitinating activity of the proteasome and thus control protein turnover in a temporal or spatial manner.
Our studies also have indicated that Usp14 is predominantly a cytoplasmic protein. This was unexpected, because previous reports demonstrated that the Usp14 homolog Ubp6 is predominantly a nuclear protein in yeast (Chernova et al., 2003). In addition, although some neuronal proteasomes are found in the cytoplasm, neuronal proteasomes have been shown to be localized primarily to the nucleus (Mengual et al., 1996; Wojcik and DeMartino, 2003). The cytoplasmic distribution of Usp14 therefore suggests that cytoplasmic proteasomes may have a different profile of associated DUBs from nuclear proteasomes and that they therefore may regulate different functions or activities at the proteasome. Cytoplasmic proteasomes are found in the cell soma, dendrites, and axons as well as synaptic terminals, where they have been suggested to regulate the stability of proteins involved in the function of synapses (Mengual et al., 1996; Aravamudan and Broadie, 2003; Speese et al., 2003; Bingol and Schuman, 2005). The demonstration that Usp14 is localized to the cell soma and neurites in primary hippocampal neurons and in brain sections is consistent with our previous findings that the loss of Usp14 alters synaptic activity (Wilson et al., 2002) and suggests that Usp14 may have a direct role in modulating synaptic function.
The rescue of the neuromuscular defect in the axJ mice by neuronal-specific expression of Usp14LF also supports our studies that indicate Usp14 functions at the presynapse (Wilson et al., 2002). Because our Thy1-Usp14LF construct expresses only the Ubl-containing form of Usp14, these experiments also suggest that the essential function of Usp14 is likely to be on the proteasome. Although it is formally possible that rescue of the axJ mice is attributable to a proteasome-independent function, we believe that our data are more consistent with a model whereby Usp14 functions on the proteasome. Our previous studies on the axJ mice and those on the Usp14 homolog in yeast demonstrated that Usp14/Ubp6 functions to recycle ubiquitin at the proteasome (Leggett et al., 2002; Chernova et al., 2003; Anderson et al., 2005). Examination of the axJ Tg mice in this study (Fig. 6C) demonstrated that the Ubl-containing form of Usp14 is essential for maintaining ubiquitin levels in the brain. Because Usp14 is predominantly a cytosolic protein, it suggests that the activities associated with the 19S subunit may vary with its subcellular location and that the 30% loss of monomeric ubiquitin levels is likely to be attributable to defects in cytosolic proteasomes. Whether there are potential focal sites of ubiquitin depletion in the axJ mice, such as at the synapse, is not known. The requirement of Usp14 to disassemble ubiquitin side chains at the proteasome therefore may be necessary for maintaining the local concentration of ubiquitin at sites of intense UPS activity or for rescuing proteins from degradation by removing their ubiquitin side chain. Given that Usp14 is expressed in a variety of tissues and that the loss of Usp14 results in decreased levels of ubiquitin in most tissues, either the nervous system is uniquely sensitive to decreased ubiquitin levels or Usp14 has another function or functions in the nervous system.
Our behavioral tests demonstrated that, although the Thy1-Usp14LF transgene restores most of the motor system defects of the axJ mice, mild coordination defects still exist in the axJ Tg mice. Examination of Usp14LF expression patterns in mice demonstrated that the only neuronal populations that did not express detectable levels of Usp14 were the CA3 hippocampal pyramidal cells and Purkinje cells of the cerebellum. The lack of expression of the Thy1-Usp14LF transgene in the Purkinje cells is consistent with the increase in the number of foot slips on the elevated beam test and with the presence of the Purkinje cell axonal swellings in the axJ Tg mice. Although the initial description of the axJ mice demonstrated the presence of Purkinje cell axonal swellings, which were suggested to be causal to the axJ motor defects, our data do not support a primary role of the Purkinje cell axonal pathology in the axJ muscle wasting, tremor, and ataxia. However, given the robust expression of Thy1-Usp14LF in the granule cell layer and the remaining Purkinje cell disease, the inability to correct the Purkinje axonal swellings indicates that Usp14 acts in a cell autonomous manner. Additional investigation into the expression patterns of the Thy1-Usp14LF transgene should provide important insights into the dysfunctional neuronal circuit or circuits in the axJ mice.
It is becoming increasingly clear that changes in the activity of the UPS play an important role in both synaptic plasticity and disease (Wilson et al., 2002; Speese et al., 2003; Yi and Ehlers, 2005; Karpova et al., 2006; Patrick, 2006). It is therefore possible that Usp14 has evolved a specialized function in neurons to regulate the ubiquitin side chain length of proteins bound to the proteasome. Because proteins with short ubiquitin chains are thought to be poor substrates for the proteasome (Guterman and Glickman, 2004), Usp14 may be involved in the release and rescue of proteins from proteasomal degradation. An essential question that remains is whether Usp14 exhibits substrate specificity or acts globally on proteins targeted to the proteasome. Given that there appears to be selective neuronal vulnerability to the axJ mutation, we believe that it is likely that Usp14 functions in the processing of specific substrates. In addition, because Usp14 resides on the regulatory particle of the proteasome, it is poised at a critical position to modulate protein degradation and to influence processes vital to maintaining neuronal viability.
Footnotes
This work was supported by the Evelyn F. McKnight Brain Institute, National Institutes of Health (NIH)–National Institute of Neurological Disorders and Stroke Grant NS047533, a March of Dimes Basil O'Conner Award, and NIH Grant P30NS47466 for tissue processing and staining. We thank Drs. Hidde Ploegh and Greg Korbel at the Whitehead Institute for generously supplying the HA-Ub-VME probe used in these studies and the Transgenic Animal/Embryonic Stem Cell Resource at University of Alabama at Birmingham for generating the Thy1-Usp14LF transgenic mice. We also thank Drs. Gail Johnson, Michael Brenner, Dan Finley, John Hanna, and Rohan Baker for their helpful review of this manuscript.
References
- Alves-Rodrigues A, Gregori L, Figueiredo-Pereira ME. Ubiquitin, cellular inclusions, and their role in neurodegeneration. Trends Neurosci. 1998;21:516–520. doi: 10.1016/s0166-2236(98)01276-4. [DOI] [PubMed] [Google Scholar]
- Anderson C, Crimmins S, Wilson JA, Korbel GA, Ploegh HL, Wilson SM. Loss of Usp14 results in reduced levels of ubiquitin in ataxia mice. J Neurochem. 2005;95:724–731. doi: 10.1111/j.1471-4159.2005.03409.x. [DOI] [PubMed] [Google Scholar]
- Aravamudan B, Broadie K. Synaptic Drosophila UNC-13 is regulated by antagonistic G-protein pathways via a proteasome-dependent degradation mechanism. J Neurobiol. 2003;54:417–438. doi: 10.1002/neu.10142. [DOI] [PubMed] [Google Scholar]
- Bingol B, Schuman EM. Synaptic protein degradation by the ubiquitin proteasome system. Curr Opin Neurobiol. 2005;15:536–541. doi: 10.1016/j.conb.2005.08.016. [DOI] [PubMed] [Google Scholar]
- Borodovsky A, Kessler BM, Casagrande R, Overkleeft HS, Wilkinson KD, Ploegh HL. A novel active site-directed probe specific for deubiquitylating enzymes reveals proteasome association of USP14. EMBO J. 2001;20:5187–5196. doi: 10.1093/emboj/20.18.5187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caroni P. Overexpression of growth-associated proteins in the neurons of adult transgenic mice. J Neurosci Methods. 1997;71:3–9. doi: 10.1016/s0165-0270(96)00121-5. [DOI] [PubMed] [Google Scholar]
- Chernova TA, Allen KD, Wesoloski LM, Shanks JR, Chernoff YO, Wilkinson KD. Pleiotropic effects of Ubp6 loss on drug sensitivities and yeast prion are due to depletion of the free ubiquitin pool. J Biol Chem. 2003;278:52102–52115. doi: 10.1074/jbc.M310283200. [DOI] [PubMed] [Google Scholar]
- D'Amato CJ, Hicks SP. Neuropathologic alterations in the ataxia (paralytic) mouse. Arch Pathol. 1965;80:604–612. [PubMed] [Google Scholar]
- Dawson TM, Dawson VL. Molecular pathways of neurodegeneration in Parkinson's disease. Science. 2003;302:819–822. doi: 10.1126/science.1087753. [DOI] [PubMed] [Google Scholar]
- Glickman MH, Ciechanover A. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev. 2002;82:373–428. doi: 10.1152/physrev.00027.2001. [DOI] [PubMed] [Google Scholar]
- Gregory RC, Taniguchi T, D'Andrea AD. Regulation of the Fanconi anemia pathway by monoubiquitination. Semin Cancer Biol. 2003;13:77–82. doi: 10.1016/s1044-579x(02)00102-5. [DOI] [PubMed] [Google Scholar]
- Guterman A, Glickman MH. Deubiquitinating enzymes are IN/(trinsic to proteasome function) Curr Protein Pept Sci. 2004;5:201–211. doi: 10.2174/1389203043379756. [DOI] [PubMed] [Google Scholar]
- Hu M, Li P, Song L, Jeffrey PD, Chenova TA, Wilkinson KD, Cohen RE, Shi Y. Structure and mechanisms of the proteasome-associated deubiquitinating enzyme USP14. EMBO J. 2005;24:3747–3756. doi: 10.1038/sj.emboj.7600832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpova A, Mikhaylova M, Thomas U, Knopfel T, Behnisch T. Involvement of protein synthesis and degradation in long-term potentiation of Schaffer collateral CA1 synapses. J Neurosci. 2006;26:4949–4955. doi: 10.1523/JNEUROSCI.4573-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo K, Kaelin WG., Jr The von Hippel–Lindau tumor suppressor gene. Exp Cell Res. 2001;264:117–125. doi: 10.1006/excr.2000.5139. [DOI] [PubMed] [Google Scholar]
- Kovalenko A, Chable-Bessia C, Cantarella G, Israel A, Wallach D, Courtois G. The tumour suppressor CYLD negatively regulates NF-κB signaling by deubiquitination. Nature. 2003;424:801–805. doi: 10.1038/nature01802. [DOI] [PubMed] [Google Scholar]
- Leggett DS, Hanna J, Borodovsky A, Crosas B, Schmidt M, Baker RT, Walz T, Ploegh H, Finley D. Multiple associated proteins regulate proteasome structure and function. Mol Cell. 2002;10:495–507. doi: 10.1016/s1097-2765(02)00638-x. [DOI] [PubMed] [Google Scholar]
- McNaught KS, Olanow CW, Halliwell B, Isacson O, Jenner P. Failure of the ubiquitin-proteasome system in Parkinson's disease. Nat Rev Neurosci. 2001;2:589–594. doi: 10.1038/35086067. [DOI] [PubMed] [Google Scholar]
- Mengual E, Arizti P, Rodrigo J, Gimenez-Amaya JM, Castano JG. Immunohistochemical distribution and electron microscopic subcellular localization of the proteasome in the rat CNS. J Neurosci. 1996;16:6331–6341. doi: 10.1523/JNEUROSCI.16-20-06331.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon BC, Choi MS, Kang YH, Kim MC, Cheong MS, Park CY, Yoo JH, Koo SC, Lee SM, Lim CO Cho MJ, Chung WS. Arabidopsis ubiquitin-specific protease 6 (AtUBP6) interacts with calmodulin. FEBS Lett. 2005;579:3885–3890. doi: 10.1016/j.febslet.2005.05.080. [DOI] [PubMed] [Google Scholar]
- Patrick GN. Synapse formation and plasticity: recent insights from the perspective of the ubiquitin proteasome system. Curr Opin Neurobiol. 2006;16:90–94. doi: 10.1016/j.conb.2006.01.007. [DOI] [PubMed] [Google Scholar]
- Price PJ, Brewer GJ. Serum-free media for neural cell cultures: adult and embryonic. In: Fedoroff S, Richardson A, editors. Protocols for neural cell culture. Ed 3. Totowa, NJ: Humana; 2001. pp. 255–264. [Google Scholar]
- Soboleva TA, Baker RT. Deubiquitinating enzymes: their functions and substrate specificity. Curr Protein Pept Sci. 2004;5:191–200. doi: 10.2174/1389203043379765. [DOI] [PubMed] [Google Scholar]
- Speese SD, Trotta N, Rodesch CK, Aravamudan B, Broadie K. The ubiquitin proteasome system acutely regulates presynaptic protein turnover and synaptic efficacy. Curr Biol. 2003;13:899–910. doi: 10.1016/s0960-9822(03)00338-5. [DOI] [PubMed] [Google Scholar]
- Stanley JL, Lincoln RJ, Brown TA, McDonald LM, Dawson GR, Reynolds DS. The mouse beam walking assay offers improved sensitivity over the mouse rotarod in determining motor coordination deficits induced by benzodiazepines. J Psychopharmacol. 2005;19:221–227. doi: 10.1177/0269881105051524. [DOI] [PubMed] [Google Scholar]
- Wilson SM, Bhattacharyya B, Rachel RA, Coppola V, Tessarollo L, Householder DB, Fletcher CF, Miller RJ, Copeland NG, Jenkins NA. Synaptic defects in ataxia mice result from a mutation in Usp14, encoding a ubiquitin-specific protease. Nat Genet. 2002;32:420–425. doi: 10.1038/ng1006. [DOI] [PubMed] [Google Scholar]
- Wojcik C, DeMartino GN. Intracellular localization of proteasomes. Int J Biochem Cell Biol. 2003;35:579–589. doi: 10.1016/s1357-2725(02)00380-1. [DOI] [PubMed] [Google Scholar]
- Yi JJ, Ehlers MD. Ubiquitin and protein turnover in synapse function. Neuron. 2005;47:629–632. doi: 10.1016/j.neuron.2005.07.008. [DOI] [PubMed] [Google Scholar]