

Abstract
An increase in the expression of the proinflammatory cytokine tumor necrosis factor α (TNF-α) has been observed in patients with amyotrophic lateral sclerosis (ALS) and in the mice models of the disease. TNF-α is a potent activator of macrophages and microglia and, under certain conditions, can induce or exacerbate neuronal cell death. Here, we assessed the contribution of TNF-α in motor neuron disease in mice overexpressing mutant superoxide dismutase 1 (SOD1) genes linked to familial ALS. This was accomplished by the generation of mice expressing SOD1G37R or SOD1G93A mutants in the context of TNF-α gene knock out. Surprisingly, the absence of TNF-α did not affect the lifespan or the extent of motor neuron loss in SOD1 transgenic mice. These results provide compelling evidence indicating that TNF-α does not directly contribute to motor neuron degeneration caused by SOD1 mutations.
Keywords: ALS, amyotrophic lateral sclerosis, degeneration, microglia, motoneuron, motor neuron, neuroinflammation, TNF-α
Introduction
Amyotrophic lateral sclerosis (ALS) is a progressive, adult-onset neurodegenerative disorder that affects primarily motor neurons in the cortex, brainstem, and spinal cord. The selective degeneration of these neurons leads to atrophy of skeletal muscle and, ultimately, to paralysis and death within 1–5 years. ALS occurs in both sporadic (90% of cases) and familial forms, which are clinically and pathologically similar. Missense mutations in the gene encoding the free radical-scavenging metalloenzyme, copper, zinc superoxide dismutase (SOD1) are responsible for 20% of familial ALS cases (Rosen et al., 1993). To date, more than 115 mutations have been found in the SOD1 gene. Transgenic mice overexpressing various SOD1 mutants develop an ALS-like phenotype through a gain of unknown toxic properties (Gurney et al., 1994). Several mechanisms have been proposed to explain motor neuron death in ALS, including glutamate-induced excitotoxicity (Rothstein, 1995), cytoskeletal abnormalities (Julien et al., 2005), protein aggregation (Julien, 2001), oxidative stress (Cleveland and Rothstein, 2001), and, more recently, toxicity via extracellular SOD1 (Urushitani et al., 2006).
Accumulating evidence indicates that non-neuronal cells may contribute to neurodegenerative processes in ALS (Clement et al., 2003; Boillee et al., 2006). Reactive astrocytes and activated microglia can be found in the spinal cord and motor cortex of patients with ALS and in SOD1 mice models of the disease (Engelhardt and Appel, 1990; Kawamata et al., 1992; Alexianu et al., 2001; Nguyen et al., 2002). Furthermore, gliosis is associated with an increase in the production of various potentially cytotoxic molecules, including reactive oxygen species, nitric oxide, proteases, and cytokines (Nguyen et al., 2002). Interestingly, an increased level of proinflammatory cytokines interleukin-1β (IL-1β), IL-6, and tumor necrosis factor-α (TNF-α) have been reported in SOD1 mice models and in ALS patients (Sekizawa et al., 1998; Poloni et al., 2000; Elliott, 2001; Nguyen et al., 2001; Hensley et al., 2003).
TNF-α is a potent proinflammatory cytokine and can elicit trophic or toxic biological responses depending on the pathways elicited by binding of its receptor subtypes, TNFR1 and TNFR2 (Viviani et al., 2004). TNF-α receptors are constitutively expressed on both neurons and glia in the CNS, and TNF-α can be synthesized and released by astrocytes, microglia, and some neurons (Zou and Crews, 2005). Production of TNF-α is closely associated with disease progression in SOD1 transgenic mice (Elliott, 2001; Hensley et al., 2003), and several studies have designated TNF-α as a pathogenic mediator in many CNS diseases with an inflammatory component (Ghezzi and Mennini, 2001; Viviani et al., 2004). Although TNF-α can mediate motor neuron death (Terrado et al., 2000; Robertson et al., 2001; He et al., 2002), its role in disease pathogenesis mediated by SOD1 mutants remains unclear. Here, to elucidate the contribution of TNF-α to the neurodegenerative processes in ALS caused by SOD1 mutations, we generated mice overexpressing SOD1G37R or SOD1G93A in the context of TNF-α gene knock-out. We then assessed whether the absence of TNF-α affected the development of motor neuron pathology.
Materials and Methods
Animals.
TNF-α knock-out (B6;129S6-Tnf tm1Gkl/j; 003008) and SOD1G93A [B6SJL-TgN(SOD1-G93A)1Gur/J; 002726] mice were acquired from The Jackson Laboratory (Bar Harbor, ME). SOD1G37R (line 29) transgenic mice were a gift from Drs. P. Wong and D. Price from John Hopkins University (Baltimore, MD). The SOD1G93A mice were crossed for at least six generations onto a C57BL/6 background before the start of the experimentation with the TNF-α knock-out mice. The SOD1G37R transgenic mice have been maintained as C57BL/6 for many years in our laboratory. The TNF-α knock-out mice were backcrossed for three generations onto C57BL/6 background before breeding with SOD1 transgenic mice. Our studies used a large number of mice derived by breeding TNF-α+/− mice with mutant TNF-α+/−; SOD1+/− to assess the effect of both TNF-α loss (−/−) and TNF-α reduction (+/−) on survival of mutant SOD1 transgenic mice. Hence, all analyses were performed with littermate controls. Mice were genotyped by Southern blot as described previously (Pasparakis et al., 1996; Couillard-Despres et al., 1998) or by PCR analysis in accordance with The Jackson Laboratory protocols. For mice in the SOD1G37R background, presymptomatic, early symptomatic, and end-stage mice refer to 7, 10, and 12 months, respectively. For mice in the SOD1G93A background, presymptomatic, early symptomatic, and end-stage mice refer to 2, 3, and 4 months, respectively. The use and maintenance of the mice described in this article were performed in accordance with the Guide of Care and Use of Experimental Animals of the Canadian Council on Animal Care.
Tissue collection, immunohistochemistry and in situ hybridization.
Mice were anesthetized and transcardially perfused with 0.9% NaCl and fixed with 4% paraformaldehyde. Tissue sample preparations, immunohistochemistry, and in situ hybridization procedures using S35 cRNA probes were performed as described previously (Laflamme et al., 1999; Nguyen et al., 2001). The primary antibodies used in this study were mouse monoclonal anti-glial fibrillary acidic protein (GFAP) (MAB360; Chemicon, Temecula, CA) and rat monoclonal anti-Mac-2 (galactose-specific lectin) (TIB-166; ATCC, Manassas, VA). The cDNA vectors for Toll-like receptor (TLR-2), inhibitor κBα (IκBα), IL-1β, and TNF-α were provided by Dr. S. Rivest (Laval University, Quebec, Quebec, Canada). Counts of L5 ventral root axons were done with Image-1 software (Universal Imaging Corporation, Downington, PA). The data were analyzed by one-way ANOVA, followed by Tukey's post hoc analysis.
ELISA assay.
TNF-α knock-out mice and control littermates were injected intraperitoneally with 1 mg/kg lipopolysaccharide (from Escherichia coli; serotype O55:B5; L2880; Sigma, St. Louis, MO), and sera were collected 4 h later. TNF-α and IL-1β were measured using mouse Quantikine immunoassays (MTA00, MLB00B; R & D Systems, Minneapolis, MN) according to the instructions of the manufacturer. ELISA plates were then read in a spectra max 340pc plate reader and analyzed using Soft max pro 3.11 software.
Quantification of proinflammatory transcripts.
The mRNA hybridization signals for IL-1β, TLR-2, and IκBα revealed on dipped NTB2 nuclear emulsion slides were analyzed and quantified under dark-field illumination at a magnification of 10× as described previously (Nguyen et al., 2001). Briefly, counts were made of clusters of silver grain hybridization signal present in every sixth section of the transverse lumbar spinal cord, each cluster corresponding to accumulation of silver grain labeling in individual cells. Colocalization of clusters with thionin-labeled cells was verified under bright-field illumination at higher magnification. The data were analyzed by unpaired t test.
Results
Absence of TNF-α does not influence disease progression or axonal degeneration in ALS mice
To assess the contribution of TNF-α in ALS pathogenesis, we generated SOD1G37R and SOD1G93A in the context of TNF-α gene knock-out. TNF-α-deficient mice are viable and fertile and show no apparent phenotypic abnormalities (Pasparakis et al., 1996). To confirm the absence of TNF-α in knock-out mice, TNF-α- and IL-1β-specific ELISA assays were performed. No TNF-α could be detected in knock-out mice (TNF-α, 1.8 ± 1.2 ng/ml; IL-1β, 118.4 ± 7.7 ng/ml) compared with TNF-α+/+ control littermates (TNF-α, 642.2 ± 43.2 ng/ml; IL-1β, 189.0 ± 19.9 ng/ml).
The effect of TNF-α deficiency on severity of motor neuron disease was examined using cohorts of TNF-α+/+; SOD1G93A and TNF-α−/−; SOD1G93A, TNF-α+/+; SOD1G37R, TNF-α−/−; SOD1G37R, as well as mutant SOD1 transgenic mice in TNF-α heterozygous background. As shown in Figure 1, the lack or reduction of TNF-α did not influence rate of mortality in mutant SOD1 transgenic mice. The TNF-α+/+; SOD1G93A and TNF-α−/−; SOD1G93A had a comparable median lifespan of 142 and 133 d, respectively. Similarly, the absence of TNF-α did not affect survival of mice expressing SOD1G37R with a median survival of 368 d for TNF-α+/+; SOD1G37R and 363 d for TNF-α−/−; SOD1G37R. Furthermore, motor axon loss in L5 ventral roots was quantified at presymptomatic, early symptomatic, and end stage of disease in mutant SOD1 transgenic mice with or without TNF-α. Absence of TNF-α did not affect the degree of axonal degeneration in SOD1G93A or SOD1G37R transgenic mice (Table 1).
Figure 1.
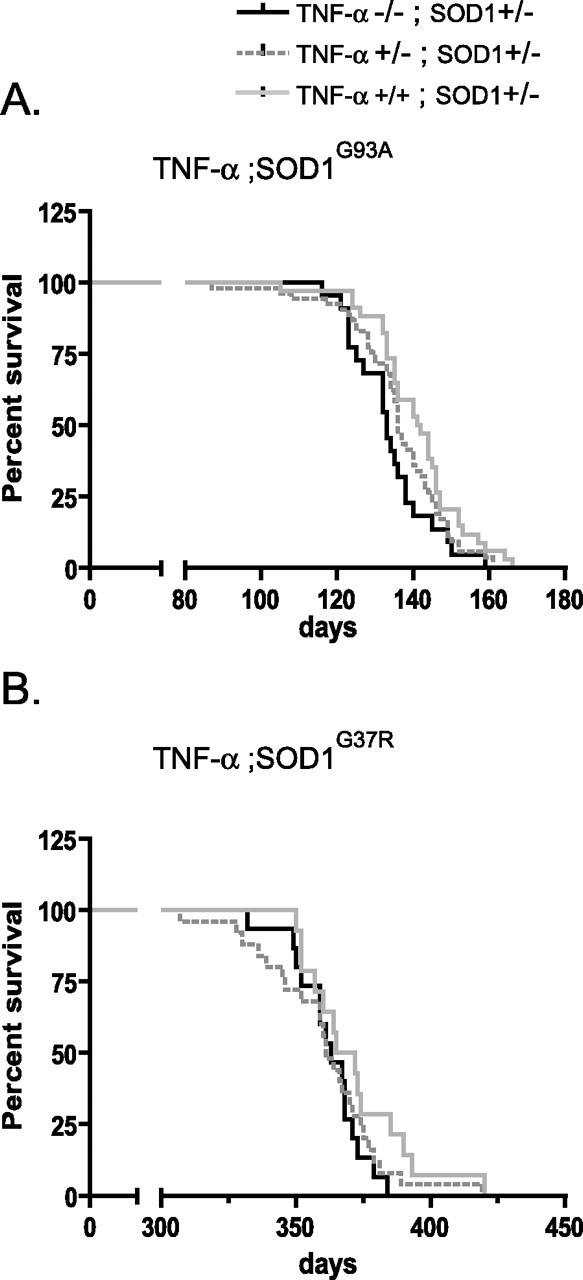
Absence of TNF-α does not affect survival of SOD1G93A or SOD1G37R mice. Kaplan–Meier survival curves: A, TNF-α−/−; SOD1G93A (n = 22), TNF-α+/−; SOD1G93A (n = 53), and TNF-α+/+; SOD1G93A (n = 34; log rank test for survival, p = 0.09); B, TNF-α−/−; SOD1G37R (n = 15), TNF-α+/−; SOD1G37R (n = 25), and TNF-α+/+; SOD1G37R (n = 14) (log rank test for survival, p = 0.26).
Table 1.
Axonal degeneration in SOD1 mice deficient in TNF-α
| Genotype | TNF-α KO; SOD1G93A |
TNF-α KO; SOD1G37R |
|---|---|---|
| Number of axons at 3 months of age (SEM) | Number of axons at 10 months of age (SEM) | |
| TNF-α+/+ | 1041 (43) | 1002 (59) |
| TNF-α+/+;SOD1 | 687 (93) | 563 (126) |
| TNF-α+/−;SOD1 | 769 (55) | 595 (72) |
| TNF-α−/−;SOD1 | 733 (31) | 450 (56) |
Motor axon counts in L5 ventral roots of TNF-α +/+; SOD1 and TNF-α −/−; SOD1 mice. No significant difference in the number of motor axon counts was found in TNF-α −/−; SOD1G93A compared with TNF-α +/+; SOD1G93A and TNF-α −/−; SOD1G37R compared with TNF-α +/+; SOD1G37R at early symptomatic and end stage of the disease. n = 3 mice per group.
No change in gliosis attributable to TNF-α deficiency in ALS mice
Astrocytosis and microgliosis are non-neuronal events that likely contribute to the neurodegenerative process in ALS. TNF-α is a potent microglial activator and is also involved in the induction of reactive astrogliosis. We therefore investigated whether the absence of TNF-α in mutant SOD1 mice had any effect on the expression of Mac-2, a marker of microglial activation, and GFAP, a marker of astrogliosis. Immunoreactivity was assessed in the spinal cord of normal mice, TNF-α−/−; SOD1, and TNF-α+/+; SOD1 mice at early symptomatic and end stages of disease. No obvious difference for these markers could be identified at any stage of the disease (Fig. 2). At both early symptomatic (Fig. 2B,C,G,H) and end stage (Fig. 2D,E,I,J) of disease, comparable numbers of Mac-2 (Fig. 2A–E) and GFAP (Fig. 2F–J) labeled cells were present in the lumbar ventral horns of TNF-α−/−; SOD1G93A and TNF-α+/+; SOD1G93A. Analogous results were obtained for mice expressing SOD1G37R (data not shown). The levels of Mac-2 and GFAP expression were also determined by Western blot analysis. No significant differences were detected between the ALS mice with or without TNF-α (data not shown).
Figure 2.

Absence of TNF-α does not influence gliosis. Immunohistochemistry on lumbar spinal cord for Mac-2 (A–E) and GFAP (F–J) did not reveal any apparent differences in gliosis in TNF-α−/−; SOD1G93A mice (C, E, H, J) compared with TNF-α+/+; SOD1G93A mice (B, D, G, I) at early symptomatic (B,C and G, H) or end-stage (D,E and I,J) of disease. Scale bars, 50 μm. n = 5–7 mice per group.
Detection of proinflammatory molecules in the spinal cord of ALS mice lacking TNF-α
Neurofilament-κB (NF-κB) is a key transcription factor involved in the induction of proinflammatory molecules, such as TNF-α, IL-1β, IL-6, and TLR-2 (O'Neill and Kaltschmidt, 1997). Activation of NF-κB also induces the transcription of its inhibitory factor IκBα to modulate inflammatory processes. To examine whether compensatory mechanisms occur in the spinal cord of SOD1 mice in the context of TNF-α deficiency, mRNA encoding IκBα, IL-1β, and TLR-2 were assayed by in situ hybridization. No change in IκBα mRNA expression could be detected between TNF-α+/+; SOD1 mice and TNF-α−/−; SOD1 mice (Fig. 3A,B,F,G,P,S). However, a significantly greater number of cells positive for IL-1β (Fig. 3A,B,H–J,Q) and TLR-2 (Fig. 3A,B,K–O,R) were counted in the lumbar spinal cord of TNF-α−/−; SOD1G93A compared with TNF-α+/+; SOD1G93A controls at the early symptomatic stage of disease but was similar at end stage of disease. A comparable tendency occurred in TNF-α−/−; SOD1G37R compared with TNF-α+/+; SOD1G37R, but it was not statistically significant (Fig. 3T,U). To further search for potential compensatory changes, a microarray analysis of spinal cord RNA expression was performed on early symptomatic TNF-α−/−; SOD1G93A mice and TNF-α+/+; SOD1G93A mice (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). No major difference was detected between the two groups, notably in TNFR1 and TNFR2 expression levels.
Figure 3.

mRNA expression and quantification of proinflammatory molecules in SOD1 mice with or without TNF-α. In situ hybridization for TNF-α, IκBα, IL-1β, and TLR-2 in TNF-α+/+; SOD1G93A (A) and TNF-α−/−; SOD1G93A (B); 40× magnification. Scale bar, 50 μm (dark-field photomicrographs). Bright-field photomicrographs of TNF-α (C–E), IκBα (F, G), IL-1β (H–J), and TLR-2 (K–O) for TNF-α+/+; SOD1G93A (C, D; F, H; K, L) and for TNF-α−/−; SOD1G93A (E, G, I, J, M–O); 100× magnification. Scale bar, 25 μm. No TNF-α could be detected in TNF-α knock-out mice (B) compared with control (A). Quantification of IκBα (P, S), IL-1β (Q, T), and TLR-2 (R, U) at early symptomatic and end stage of disease in TNF-α+/+; SOD1 transgenic mice and TNF-α−/−; SOD1 mice. No significant difference in the expression of IκBα was detected in SOD1 mice in the absence of TNF-α (P, S). A significant increase in the mRNA expression of IL-1β (Q) (p = 0.02) and TLR-2 (R) (p = 0.01) was observed in TNF-α−/−; SOD1G93A mice compared with TNF-α+/+; SOD1G93A mice at early symptomatic but not at end stage of disease. IL-1β (T) and TLR-2 (U) hybridization signals were not significantly different in mice of SOD1G37R background. Error bars correspond to SEM. * indicates a significant difference between groups. n = 5–7 mice per group. Arrows indicate IκBα-positive cells.
Discussion
Neuroinflammation has been detected in affected tissue of ALS cases and is a feature of mutant SOD1 transgenic mice models of the disease (Nguyen et al., 2002). Several studies have shown that expression of proinflammatory mediators such as TNF-α and IL-1β is an early event in mutant SOD1 transgenic mice (Alexianu et al., 2001; Elliott, 2001; Nguyen et al., 2001). In fact, expression of TNF-α correlates with the onset and progression of paralysis in mutant SOD1 mice (Elliott, 2001; Nguyen et al., 2001; Hensley et al., 2003). However, neither the presence of antigenic nor bioactive TNF-α correlated with disease severity, duration, or weight loss in ALS patients (Poloni et al., 2000). There is conflicting literature on the neuroprotective versus neurotoxic effect of TNF-α both in vivo and in vitro (Ghezzi and Mennini, 2001). TNF-α is one of the most potent proinflammatory cytokine and is capable of activating microglia and causing neurotoxicity in systems in which the neuronal population has been compromised (Robertson et al., 2001; Zou and Crews, 2005). Furthermore, neutralization of TNF-α can reduce neuronal damage (Terrado et al., 2000), and TNF-α is able to mediate motor neuron cell death in certain experimental paradigms (He et al., 2002). Interestingly, administration of thalidomide, a potent anti-inflammatory and immunomodulatory drug, whose effects include inhibition of TNF-α synthesis, delays death in SOD1G93A mice (Kiaei et al., 2006). Hence, the hypothesis of a central role for TNF-α in exacerbating disease pathogenesis in various context of neurodegeneration including ALS has emerged. However, it must be considered that, although TNF-α can induce apoptosis, activation of TNF receptors in neurons can also modulate the expression of proteins such as Bcl-2, manganese superoxide dismutase, and calcium-regulating proteins in ways that increase cellular resistance to apoptosis (Guo et al., 2004). Furthermore, TNF-α can induce IL-6 and leukemia inhibitory factor, two cytokines that have protective effects against motor neuron degeneration in wobbler mice (Ikeda et al., 1995, 1996). It is also noteworthy that motor neurons in mnd mice, a spontaneous mutation of mouse that causes a late-onset motor dysfunction and eventual paralysis, do not degenerate at presymptomatic stage despite the presence of high levels of TNF-α (Mennini et al., 2004).
Unexpectedly, we report here that disruption of the TNF-α gene in mice failed to influence onset, severity, or progression of disease caused by SOD1 mutations. Moreover, absence of TNF-α had no effect on axonal degeneration in mutant SOD1 mice. These results suggest that TNF-α is not a crucial contributor to motor neuron degeneration in this model. Surprisingly, the absence of TNF-α did not influence the appearance of gliosis or the expression levels for mRNA encoding IκBα in mutant SOD1 mice. However, an increase in transcripts encoding for IL-1β and TLR-2 at early symptomatic stage of the disease was observed in TNF-α−/−; SOD1G93A mice compared with TNF-α+/+; SOD1G93A controls but not in TNF-α−/−; SOD1G37R compared with TNF-α+/+; SOD1G37R. Hence, it is possible that, in TNF-α−/−; SOD1G93A mice, the upregulation of IL-1β and TLR-2 may be part of a compensatory process. Indeed, IL-1β has a leading role in the activation of the inflammatory response and shares the same collection of downstream effectors as TNF-α (Allan et al., 2005). The TLR family is a major class of pattern-recognition receptor that has emerged as a central player in the initiation and tailoring of both innate and subsequent adaptive immune responses (Beutler, 2004; Iwasaki and Medzhitov, 2004). Moreover, TLR-2 has been found to ligate a broad array of ligands, both pathogen associated and endogenously derived (Jack et al., 2005). Interestingly, another cytokine, lymphotoxin-α, has also been shown to bind TNF receptors (Ware, 2005). However, a study by Kuprash et al. (2002) concluded that TNF-α and lymphotoxin-α have primarily nonredundant functions in vivo (Kuprash et al., 2002).
Previous studies indicate that TNF-α can enhance or inhibit neuronal injury (Zou and Crews, 2005; Turrin and Rivest, 2006). The effects of TNF-α is determined in part by duration, extent of expression, and the state of the surrounding microenvironment (Allan and Rothwell, 2001). Indeed, TNF-α does not directly induce neuronal death in healthy brain tissue or on normal neurons (Viviani et al., 2004). However, TNF-α can enhance the effect of neuronal insults and act synergistically with other cytokines to promote neurodegeneration (Hemmer et al., 2001; He et al., 2002). The dual effects of TNF-α may also be explained by the different signaling pathways activated by TNFR1 and TNFR2. TNFR1 is thought to be responsible for the cytotoxic effect of TNF-α, whereas TNFR2 would mediate neurotrophic functions (Viviani et al., 2004). Interestingly, the expression of both receptors is increased in the spinal cord of mutant SOD1 transgenic mice (Veglianese et al., 2006).
Although an increase in the expression of the proinflammatory cytokine TNF-α has been observed in ALS patients and in the mice models of the disease, the results presented here suggest that TNF-α alone is not a key factor in motor neuron degeneration caused by SOD1 mutations.
Footnotes
This work was supported by the Canadian Institutes of Health Research (CIHR) and the Robert Packard Center for ALS Research at Johns Hopkins University. J.-P.J. holds a Canada Research Chair in Neurodegeneration. G.G. is the recipient of a CIHR Doctoral Research Award. F.D. is a recipient of a Le Fonds de la Recherche en Santé du Québec Studentship. We thank Jean-Nicolas Audet, Roxanne Larivière, and Mélanie Lalancette-Hébert for their technical assistance.
References
- Alexianu ME, Kozovska M, Appel SH. Immune reactivity in a mouse model of familial ALS correlates with disease progression. Neurology. 2001;57:1282–1289. doi: 10.1212/wnl.57.7.1282. [DOI] [PubMed] [Google Scholar]
- Allan SM, Rothwell NJ. Cytokines and acute neurodegeneration. Nat Rev Neurosci. 2001;2:734–744. doi: 10.1038/35094583. [DOI] [PubMed] [Google Scholar]
- Allan SM, Tyrrell PJ, Rothwell NJ. Interleukin-1 and neuronal injury. Nat Rev Immunol. 2005;5:629–640. doi: 10.1038/nri1664. [DOI] [PubMed] [Google Scholar]
- Beutler B. Inferences, questions and possibilities in Toll-like receptor signalling. Nature. 2004;430:257–263. doi: 10.1038/nature02761. [DOI] [PubMed] [Google Scholar]
- Boillee S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, Kassiotis G, Kollias G, Cleveland DW. Onset and progression in inherited ALS determined by motor neurons and microglia. Science. 2006;312:1389–1392. doi: 10.1126/science.1123511. [DOI] [PubMed] [Google Scholar]
- Clement AM, Nguyen MD, Roberts EA, Garcia ML, Boillee S, Rule M, McMahon AP, Doucette W, Siwek D, Ferrante RJ, Brown RH, Jr, Julien JP, Goldstein LS, Cleveland DW. Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science. 2003;302:113–117. doi: 10.1126/science.1086071. [DOI] [PubMed] [Google Scholar]
- Cleveland DW, Rothstein JD. From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Nat Rev Neurosci. 2001;2:806–819. doi: 10.1038/35097565. [DOI] [PubMed] [Google Scholar]
- Couillard-Despres S, Zhu Q, Wong PC, Price DL, Cleveland DW, Julien JP. Protective effect of neurofilament heavy gene overexpression in motor neuron disease induced by mutant superoxide dismutase. Proc Natl Acad Sci USA. 1998;95:9626–9630. doi: 10.1073/pnas.95.16.9626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott JL. Cytokine upregulation in a murine model of familial amyotrophic lateral sclerosis. Brain Res Mol Brain Res. 2001;95:172–178. doi: 10.1016/s0169-328x(01)00242-x. [DOI] [PubMed] [Google Scholar]
- Engelhardt JI, Appel SH. IgG reactivity in the spinal cord and motor cortex in amyotrophic lateral sclerosis. Arch Neurol. 1990;47:1210–1216. doi: 10.1001/archneur.1990.00530110068019. [DOI] [PubMed] [Google Scholar]
- Ghezzi P, Mennini T. Tumor necrosis factor and motoneuronal degeneration: an open problem. Neuroimmunomodulation. 2001;9:178–182. doi: 10.1159/000049024. [DOI] [PubMed] [Google Scholar]
- Guo Z, Iyun T, Fu W, Zhang P, Mattson MP. Bone marrow transplantation reveals roles for brain macrophage/microglia TNF signaling and nitric oxide production in excitotoxic neuronal death. Neuromol Med. 2004;5:219–234. doi: 10.1385/NMM:5:3:219. [DOI] [PubMed] [Google Scholar]
- Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX, Chen W, Zhai P, Sufit RL, Siddique T. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- He BP, Wen W, Strong MJ. Activated microglia (BV-2) facilitation of TNF-alpha-mediated motor neuron death in vitro. J Neuroimmunol. 2002;128:31–38. doi: 10.1016/s0165-5728(02)00141-8. [DOI] [PubMed] [Google Scholar]
- Hemmer K, Fransen L, Vanderstichele H, Vanmechelen E, Heuschling P. An in vitro model for the study of microglia-induced neurodegeneration: involvement of nitric oxide and tumor necrosis factor-alpha. Neurochem Int. 2001;38:557–565. doi: 10.1016/s0197-0186(00)00119-4. [DOI] [PubMed] [Google Scholar]
- Hensley K, Fedynyshyn J, Ferrell S, Floyd RA, Gordon B, Grammas P, Hamdheydari L, Mhatre M, Mou S, Pye QN, Stewart C, West M, West S, Williamson KS. Message and protein-level elevation of tumor necrosis factor alpha (TNF alpha) and TNF alpha-modulating cytokines in spinal cords of the G93A-SOD1 mouse model for amyotrophic lateral sclerosis. Neurobiol Dis. 2003;14:74–80. doi: 10.1016/s0969-9961(03)00087-1. [DOI] [PubMed] [Google Scholar]
- Ikeda K, Iwasaki Y, Tagaya N, Shiojima T, Kinoshita M. Neuroprotective effect of cholinergic differentiation factor/leukemia inhibitory factor on wobbler murine motor neuron disease. Muscle Nerve. 1995;18:1344–1347. doi: 10.1002/mus.880181122. [DOI] [PubMed] [Google Scholar]
- Ikeda K, Kinoshita M, Tagaya N, Shiojima T, Taga T, Yasukawa K, Suzuki H, Okano A. Coadministration of interleukin-6 (IL-6) and soluble IL-6 receptor delays progression of wobbler mouse motor neuron disease. Brain Res. 1996;726:91–97. [PubMed] [Google Scholar]
- Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5:987–995. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- Jack CS, Arbour N, Manusow J, Montgrain V, Blain M, McCrea E, Shapiro A, Antel JP. TLR signaling tailors innate immune responses in human microglia and astrocytes. J Immunol. 2005;175:4320–4330. doi: 10.4049/jimmunol.175.7.4320. [DOI] [PubMed] [Google Scholar]
- Julien JP. Amyotrophic lateral sclerosis unfolding the toxicity of the misfolded. Cell. 2001;104:581–591. doi: 10.1016/s0092-8674(01)00244-6. [DOI] [PubMed] [Google Scholar]
- Julien JP, Millecamps S, Kriz J. Cytoskeletal defects in amyotrophic lateral sclerosis (motor neuron disease) Novartis Found Symp. 2005;264:183–192. 192–196, 227–230. [PubMed] [Google Scholar]
- Kawamata T, Akiyama H, Yamada T, McGeer PL. Immunologic reactions in amyotrophic lateral sclerosis brain and spinal cord tissue. Am J Pathol. 1992;140:691–707. [PMC free article] [PubMed] [Google Scholar]
- Kiaei M, Petri S, Kipiani K, Gardian G, Choi DK, Chen J, Calingasan NY, Schafer P, Muller GW, Stewart C, Hensley K, Beal MF. Thalidomide and lenalidomide extend survival in a transgenic mouse model of amyotrophic lateral sclerosis. J Neurosci. 2006;26:2467–2473. doi: 10.1523/JNEUROSCI.5253-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuprash DV, Alimzhanov MB, Tumanov AV, Grivennikov SI, Shakhov AN, Drutskaya LN, Marino MW, Turetskaya RL, Anderson AO, Rajewsky K, Pfeffer K, Nedospasov SA. Redundancy in tumor necrosis factor (TNF) and lymphotoxin (LT) signaling in vivo: mice with inactivation of the entire TNF/LT locus versus single-knockout mice. Mol Cell Biol. 2002;22:8626–8634. doi: 10.1128/MCB.22.24.8626-8634.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laflamme N, Lacroix S, Rivest S. An essential role of interleukin-1β in mediating NF-κB activity and COX-2 transcription in cells of the blood–brain barrier in response to a systemic and localized inflammation but not during endotoxemia. J Neurosci. 1999;19:10923–10930. doi: 10.1523/JNEUROSCI.19-24-10923.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mennini T, Bigini P, Cagnotto A, Carvelli L, Di Nunno P, Fumagalli E, Tortarolo M, Buurman WA, Ghezzi P, Bendotti C. Glial activation and TNFR-I upregulation precedes motor dysfunction in the spinal cord of mnd mice. Cytokine. 2004;25:127–135. doi: 10.1016/j.cyto.2003.10.008. [DOI] [PubMed] [Google Scholar]
- Nguyen MD, Julien JP, Rivest S. Induction of proinflammatory molecules in mice with amyotrophic lateral sclerosis: no requirement for proapoptotic interleukin-1beta in neurodegeneration. Ann Neurol. 2001;50:630–639. doi: 10.1002/ana.1256. [DOI] [PubMed] [Google Scholar]
- Nguyen MD, Julien JP, Rivest S. Innate immunity: the missing link in neuroprotection and neurodegeneration? Nat Rev Neurosci. 2002;3:216–227. doi: 10.1038/nrn752. [DOI] [PubMed] [Google Scholar]
- O'Neill LA, Kaltschmidt C. NF-kappa B: a crucial transcription factor for glial and neuronal cell function. Trends Neurosci. 1997;20:252–258. doi: 10.1016/s0166-2236(96)01035-1. [DOI] [PubMed] [Google Scholar]
- Pasparakis M, Alexopoulou L, Episkopou V, Kollias G. Immune and inflammatory responses in TNF alpha-deficient mice: a critical requirement for TNF alpha in the formation of primary B cell follicles, follicular dendritic cell networks and germinal centers, and in the maturation of the humoral immune response. J Exp Med. 1996;184:1397–1411. doi: 10.1084/jem.184.4.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poloni M, Facchetti D, Mai R, Micheli A, Agnoletti L, Francolini G, Mora G, Camana C, Mazzini L, Bachetti T. Circulating levels of tumour necrosis factor-alpha and its soluble receptors are increased in the blood of patients with amyotrophic lateral sclerosis. Neurosci Lett. 2000;287:211–214. doi: 10.1016/s0304-3940(00)01177-0. [DOI] [PubMed] [Google Scholar]
- Robertson J, Beaulieu JM, Doroudchi MM, Durham HD, Julien JP, Mushynski WE. Apoptotic death of neurons exhibiting peripherin aggregates is mediated by the proinflammatory cytokine tumor necrosis factor-alpha. J Cell Biol. 2001;155:217–226. doi: 10.1083/jcb.200107058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O'Regan JP, Deng HX, Rahmani Z, Krizus A, McKenna-Yasek D, Cayabyab A, Gaston SM, Berger R, Tanzi RE, Halperin JJ, Herzfeldt B, Van den Bergh R. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- Rothstein JD. Excitotoxicity and neurodegeneration in amyotrophic lateral sclerosis. Clin Neurosci. 1995;3:348–359. [PubMed] [Google Scholar]
- Sekizawa T, Openshaw H, Ohbo K, Sugamura K, Itoyama Y, Niland JC. Cerebrospinal fluid interleukin 6 in amyotrophic lateral sclerosis: immunological parameter and comparison with inflammatory and non-inflammatory central nervous system diseases. J Neurol Sci. 1998;154:194–199. doi: 10.1016/s0022-510x(97)00228-1. [DOI] [PubMed] [Google Scholar]
- Terrado J, Monnier D, Perrelet D, Vesin D, Jemelin S, Buurman WA, Mattenberger L, King B, Kato AC, Garcia I. Soluble TNF receptors partially protect injured motoneurons in the postnatal CNS. Eur J Neurosci. 2000;12:3443–3447. doi: 10.1046/j.1460-9568.2000.00240.x. [DOI] [PubMed] [Google Scholar]
- Turrin NP, Rivest S. Tumor necrosis factor α but not interleukin 1β mediates neuroprotection in response to acute nitric oxide excitotoxicity. J Neurosci. 2006;26:143–151. doi: 10.1523/JNEUROSCI.4032-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urushitani M, Sik A, Sakurai T, Nukina N, Takahashi R, Julien JP. Chromogranin-mediated secretion of mutant superoxide dismutase proteins linked to amyotrophic lateral sclerosis. Nat Neurosci. 2006;9:108–118. doi: 10.1038/nn1603. [DOI] [PubMed] [Google Scholar]
- Veglianese P, Lo Coco D, Bao Cutrona M, Magnoni R, Pennacchini D, Pozzi B, Gowing G, Julien JP, Tortarolo M, Bendotti C. Activation of the p38MAPK cascade is associated with upregulation of TNF alpha receptors in the spinal motor neurons of mouse models of familial ALS. Mol Cell Neurosci. 2006;31:218–231. doi: 10.1016/j.mcn.2005.09.009. [DOI] [PubMed] [Google Scholar]
- Viviani B, Bartesaghi S, Corsini E, Galli CL, Marinovich M. Cytokines role in neurodegenerative events. Toxicol Lett. 2004;149:85–89. doi: 10.1016/j.toxlet.2003.12.022. [DOI] [PubMed] [Google Scholar]
- Ware CF. Network communications: lymphotoxins, LIGHT, and TNF. Annu Rev Immunol. 2005;23:787–819. doi: 10.1146/annurev.immunol.23.021704.115719. [DOI] [PubMed] [Google Scholar]
- Zou JY, Crews FT. TNF alpha potentiates glutamate neurotoxicity by inhibiting glutamate uptake in organotypic brain slice cultures: neuroprotection by NF kappa B inhibition. Brain Res. 2005;1034:11–24. doi: 10.1016/j.brainres.2004.11.014. [DOI] [PubMed] [Google Scholar]