

Abstract
Vanilloid receptor type 1 (TRPV1) is a ligand-gated nonselective cation channel that is considered to be an important integrator of various pain stimuli such as endogenous lipids, capsaicin, heat, and low pH. In addition to expression in primary afferents, TRPV1 is also expressed in the CNS. To test the hypothesis that the CNS plays a differential role in the effect of TRPV1 antagonists in various types of pain, the analgesic effects of two TRPV1 antagonists with similar in vitro potency but different CNS penetration were compared in vivo. Oral administration of either A-784168 (1-[3-(trifluoromethyl)pyridin-2-yl]-N-[4-(trifluoromethylsulfonyl)phenyl]-1,2,3,6-tetrahydropyridine-4-carboxamide) (good CNS penetration) or A-795614 (N-1H-indazol-4-yl-N′-[(1R)-5-piperidin-1-yl-2,3-dihydro-1H-inden-1-yl]urea) (poor CNS penetration) blocked capsaicin-induced acute pain with the same potency. In complete Freund's adjuvant (CFA)-induced chronic inflammatory pain, oral administration of either compound blocked thermal hyperalgesia with similar potency. Furthermore, intraplantar or intrathecal administration of A-784168 blocked CFA-induced thermal hyperalgesia, suggesting that both peripheral and CNS TRPV1 receptors may play a role in inflammatory thermal hyperalgesia. The effects of the two TRPV1 antagonists were further assessed in models presumably mediated by central sensitization, including CFA- and capsaicin-induced mechanical allodynia and osteoarthritic pain. In these models, the potency of the two compounds was similar after intrathecal administration. However, when administered orally, A-784168, with good CNS penetration, was much more potent than A-795614. Together, these results demonstrate that TRPV1 receptors in the CNS play an important role in pain mediated by central sensitization. In addition, these results demonstrate that significant CNS penetration is necessary for a TRPV1 antagonist to produce broad-spectrum analgesia.
Keywords: behavior, capsaicin, nociception, osteoarthritis, pharmacology, VR1
Introduction
Intradermal administration of capsaicin activates vanilloid receptors and induces nocifensive behaviors in rodents and pain in humans (Szolcsanyi, 1977; Carpenter and Lynn, 1981; Simone et al., 1987; LaMotte et al., 1991, 1992; Gilchrist et al., 1996). Vanilloid receptor type 1 (TRPV1) is a ligand-gated nonselective cation channel that can be activated not only by exogenous agents but also by many endogenous stimuli, including heat (>43°C), low pH, and anandamide (Caterina et al., 1997; Tominaga et al., 1998; Szallasi and Blumberg, 1999). In addition to direct activation, TRPV1 channel activity can also be modulated by inflammatory mediators, such as prostaglandins and bradykinin (Premkumar and Ahern, 2000; Chuang et al., 2001; Ferreira et al., 2004).
Activation of TRPV1 plays an important role in pain transmission and modulation. For example, TRPV1 knock-out mice exhibited reduced thermal nociception and a loss of inflammatory thermal hyperalgesia (Caterina et al., 2000; Davis et al., 2000). However, in these animals, mechanical allodynia associated with both inflammation and nerve injury remained unchanged. In contrast to these results, several novel TRPV1 antagonists, including N-(4-tert-butylphenyl)-4-(3-chloropyridin-2-yl)piperazine-1-carboxamide (BCTC) (Pomonis et al., 2003), AMG 9810 [(E)-3-(4-tert-butylphenyl)-N-(2,3-dihydrobenzo[b][1,4] dioxin-6-yl)acrylamide] (Gavva et al., 2005), and A-425619 [1-isoquinolin-5-yl-3-(4-trifluoromethyl-benzyl)-urea] (Gomtsyan et al., 2005; Honore et al., 2005), have demonstrated efficacy in reducing thermal hyperalgesia and mechanical allodynia associated with both chronic inflammation and neuropathy in rats. Together, these studies suggest that TRPV1 plays an important role in integrating multiple pain-producing stimuli.
Consistent with its key role in pain transmission, TRPV1 receptors have been found in both the peripheral nervous system and the CNS. TRPV1 receptors are expressed in small and medium size dorsal root ganglion (DRG) neurons and are localized on peripheral and central processes of the DRG cells (Guo et al., 1999; Carlton and Coggeshall, 2001; Valtschanoff et al., 2001). In the spinal cord, TRPV1 receptors are found both presynaptically and postsynaptically (Guo et al., 1999; Valtschanoff et al., 2001), and spinal administration of the TRPV1 receptor antagonist capsazepine has been shown to inhibit evoked activity of wide dynamic range neurons (Kelly and Chapman, 2002). In the brain, TRPV1 receptors have been identified in various regions known for their role in pain transmission or modulation (Mezey et al., 2000; Szabo et al., 2002; Roberts et al., 2004). For example, microinjection of capsaicin into the periaqueductal gray produces antinociceptive effects, suggesting a functional role of CNS TRPV1 (Palazzo et al., 2002; McGaraughty et al., 2003).
In this study, to enhance our understanding of the role of central and peripheral TRPV1 receptors in pain transmission, we compared the antinociceptive effects of two TRPV1 antagonists, A-784168 (1-[3-(trifluoromethyl)pyridin-2-yl]-N-[4-(trifluoromethylsulfonyl)phenyl]-1,2,3,6-tetrahydropyridine-4-carboxamide) and A-795614 (N-1H-indazol-4-yl-N′-[(1R)-5-piperidin-1-yl-2,3-dihydro-1H-inden-1-yl]urea), in several pain models following various routes of administration. These two compounds had comparable in vitro potency, plasma protein binding, and oral bioavailability but very different CNS penetration. Our results demonstrated that when administered directly into the CNS, both compounds effectively blocked nociception with comparable potency in a variety of pain models. In contrast, oral administration of the compound with greater CNS penetration was much more effective in blocking some pain states than the compound with poor CNS penetration.
Materials and Methods
In vitro studies
Materials.
Cell culture media, fetal bovine serum (FBS), and anandamide were obtained from Sigma-Aldrich (St. Louis, MO). Dulbecco's PBS, pH 7.4 (D-PBS) (with calcium, magnesium, and 1 mg/ml d-glucose), was obtained from Invitrogen (Carlsbad, CA). Fluo-4 AM was purchased from Tef Labs (Austin, TX). N-arachidonoyl-dopamine (NADA) was purchased from Tocris Cookson (Ellisville, MO). G-418 sulfate was obtained from Calbiochem (San Diego, CA).
Cloning and expression of the human TRPV1 receptor.
Cloning of the human TRPV1 (hTRPV1) receptor was described previously (Witte et al., 2002). Amino acid sequence was identical to a previously published sequence (Hayes et al., 2000), with the exception of one residue: an isoleucine-replaced valine at position 585. The hTRPV1 receptor was stably expressed in human embryonic kidney 293 (HEK293) and 1321N1 cells using standard lipid-mediated transfection methods. The cell lines were maintained in DMEM containing 10% (v/v) FBS and 300 μg/ml G-418 sulfate under a humidified 95% air and 5% CO2 atmosphere at 37°C.
Ca2+flux assay.
TRPV1-mediated elevation of intracellular calcium levels was measured using the fluorescent calcium chelating dye fluo-4, as described previously (Bianchi et al., 2006). Cells were incubated with the cell permanent acetoxymethyl (AM) ester form of Fluo-4 (2 μm) for 2 h at 25°C. Black-walled 96-well plates (BD Biosciences, San Jose, CA) with adherent cells were used to reduce light scattering. The unincorporated dye was removed from the cells by extensive washing with PBS. Aside from NADA, all test compounds were dissolved in DMSO (10 mm stocks). NADA was dissolved in ethanol (5 mg/ml). Test compounds were added to the cells at a delivery rate of 50 μl/s. In experiments using antagonists, the TRPV1 receptor antagonists were added to the cells 5 min before the addition of agonist, and the final assay volume was 200 μl. Acid activation was performed in a similar manner, except that ambient pH was lowered to pH 6.7. Acidic pH solutions were prepared by titration of D-PBS with 1 m HCl. Changes in fluorescence were recorded over time. The intensity of the fluorescence was captured by the CCD camera and digitally transferred to an interfaced personal computer. The peak increase in fluorescence over baseline (relative fluorescence units was calculated and expressed as the percentage of the maximal agonist response (in the absence of TRPV1 receptor antagonists). EC50 and IC50 values were calculated from curve fits of the concentration-effect data using a four-parameter logistic Hill equation (GraphPad Prism; GraphPad Software, San Diego, CA). Significant differences were calculated by unpaired, two-tailed Student's t tests.
Electrophysiology studies.
Lumbar (L4–L6) DRG were dissected from adult male Sprague Dawley rats (8 weeks of age; 250–300 g) and cultured as described previously (El Kouhen et al., 2005). For electrophysiological recordings, DRG neurons were maintained at room temperature in an extracellular solution (pH 7.4, 325 mOsm) consisting of the following (in mm): 155 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, 12 glucose. Patch pipettes composed of boroscilicate glass (1B150F-3; World Precision Instruments, Sarasota, FL) were pulled and fire-polished using a DMZ-Universal micropipette puller (Zeitz-Instruments, Martinsried, Germany). Pipettes (2–6 MΩ) were filled with an internal solution (pH 7.3, 295 mOsm) consisting of the following (in mm): 122.5 K-aspartate, 20 KCl, 1 MgCl2, 10 EGTA, 5 HEPES, 2 ATP·Mg. Standard whole-cell recording techniques were used for voltage-clamp studies using an Axopatch 200B amplifier (Molecular Devices, Foster City, CA).
Cells were continuously perfused with extracellular solution at a rate of 2 ml/min. Capsaicin was applied to individual neurons for 5 s using a piezoelectric-driven theta-tube application device (Burleigh Instruments, Fishers, NY) controlled by Molecular Devices pClamp 9 software (Molecular Devices). Control responses typically ran down for the first 5–10 min after whole-cell configuration. Therefore, capsaicin was applied alone at 2 min intervals until successive applications produced currents of similar amplitude. At this point, increasing concentrations of antagonist were preapplied to the neuron for ∼60 s, followed by coapplication of capsaicin and antagonist. Peak current amplitudes were measured and plotted as a function of antagonist concentration.
Data acquisition and analysis were performed using pClamp 9.0, and subsequent graphs were plotted and statistical analysis was done using GraphPad Prism (GraphPad Software). Antagonist concentration-response curves were fitted by a least-squares regression to the logistic equation provided in the GraphPad software: Y = min + [(max − min)/1 + 10(logEC50 − X)nH].
Pharmacokinetic and physicochemical properties.
In addition to their potency at TRPV1, A-784168 and A-795614 were also characterized for their pharmacokinetic and physicochemical properties. Rat plasma protein binding was determined by the following method: rat plasma (adjusted to pH 7.4) was spiked in duplicate with compound (5 μm). The spiked plasma samples were centrifuged at 86,000 × g for 18 h (Beckman XL-70, Type 25 rotor). The samples were then fractionated and extracted, and the free (unbound) drug was measured in the supernatant using liquid chromatography/mass spectrometry. Compounds were eluted from an octadecylsilyl (2 × 10 mm) Phenomenex column with a water (0.01% formic acid) and acetonitrile mobile phase, in which the percentage of acetonitrile represented 1, 100, 100, and 1% at 0.7, 1.2, 2.2, and 2.3 min, respectively. The column was equilibrated for an additional 0.7 min, and the flow rate was maintained at 0.5 ml/min. MS analysis was performed on a Sciex API2000 Biomolecular Mass Analyzer with a turbo ionspray interface. Analytes were ionized in the positive ion mode. Detection was in the single ion monitoring mode at the m/z of the respective compound.
From the pharmacokinetic profile of the compounds, it was determined that their volume of distribution (Vβ; low volume of distribution means that the compound remains mostly in the plasma; high volume of distribution means that the compound leaves the plasma and suggests good distribution to other tissues) were different, suggesting a possibility for different CNS penetration levels. To further characterize their CNS penetration, the plasma and brain levels of both compounds were quantified after oral administration at the same time point as the one used in the behavioral studies (1 h after oral dosing) corresponding to the peak exposure and peak effects of A-784168 and A-795614.
In vivo studies
Animals.
All animal protocols were approved by an Institutional Animal Care and Use Committee and are in accordance with the International Association for the Study of Pain. Male Sprague Dawley rats (Charles River Laboratories, Wilmington, MA), weighing 200–320 g for inflammatory pain studies or 170 g at the time of surgery for neuropathic pain studies, were used in the present study. Animals were group-housed and were given food and water ad libitum, except before oral administration of drugs when food was removed for 16 h before dosing.
Thermal test.
To assess sensitivity to thermal stimuli, radiant heat was applied to the plantar surface of each hindpaw, and withdrawal response latencies were determined (Hargreaves et al., 1988). Animals were placed under a clear plastic cage on a glass surface maintained at 30°C, which was elevated to allow maneuvering of a controlled, radiant heat source below (UARDG, Department of Anesthesiology, University of California, San Diego, La Jolla, CA). Each animal was adapted to the testing environment for 15–20 min before any stimulation. Heat stimuli were produced by a 50 Watt light bulb placed in a custom-built case, which enabled focusing of the light source. Each hindpaw received three stimuli, alternating between paws. The interstimulus interval for each paw was 5 min, and a 20 s cutoff was imposed on the stimulus duration to prevent tissue damage. Withdrawal latency for each paw was calculated as the mean of the two shorter latencies. The intensity of the lamp was adjusted and maintained to produce stable latencies of ∼9–10 s.
Mechanical test.
To assess sensitivity to mechanical stimuli, animals were placed in clear Plexiglas compartments with a mesh floor and were allowed to habituate for 20 min. A series of von Frey hairs (Stoetling, Kiel, WI) were then presented to the mid-plantar area of each paw, with a range of 2–14 g. The withdrawal thresholds were determined by the up-and-down method (Chaplan et al., 1994).
Capsaicin-induced acute pain.
Intraplantar (i.pl) injection of capsaicin (2.5 μg/10 μl) rapidly produced nocifensive behavior (licking, biting, flinching behaviors), presumably mediated by the direct activation of local TRPV1 receptors. Duration of nocifensive behaviors was assessed in the first 5 min after injection of capsaicin.
Complete Freund's adjuvant– induced thermal hyperalgesia and mechanical allodynia.
The animals received an intraplantar injection of 50% complete Freund's adjuvant (CFA) (150 μl) to the right hindpaw. Thermal hyperalgesia and mechanical allodynia were assessed 48 h after CFA injection as described above.
Osteoarthritic pain.
Sodium monoiodoacetate (MIA) (3 mg in 0.05 ml of sterile isotonic saline) was injected into the knee joint cavity with a 26 gauge needle under light 2% isoflurane anesthesia to induce unilateral knee joint osteoarthritis. As described previously, hindlimb weight bearing was reduced ipsilaterally after intraarticular injection of MIA into the knee. Pharmacological studies were performed 4 d after MIA injection (Bove et al., 2003).
Capsaicin-induced mechanical allodynia.
Intraplantar injection of capsaicin (10 μg/10 μl) produced acute nocifensive behaviors for the first 5 min, a primary thermal hyperalgesia in the immediate vicinity of the injection site and a secondary mechanical allodynia in a large region surrounding the injection site that persisted for several hours. The secondary mechanical allodynia was assessed 3 h after capsaicin injection.
Locomotor assay.
Spontaneous locomotor activity was recorded in an open field using photobeam activity monitors (AccuScan Instruments, Columbus, OH) for 30 min, starting 1 h after oral dosing of the compounds.
Intrathecal catheter implantation.
Rats (200–300 g) were anesthetized with isoflurane gas and mounted in a conventional stereotaxic instrument. As described previously (Yaksh and Rudy, 1976), a midline incision was made on the skull, extending from a line between the ears to a point ∼2 cm caudal. The fascia and superficial neck muscle were retracted to expose the occipital crest. A small incision was then made on the middle line of the atlanto-occipital membrane, using the tip of a fresh 18–20 gauge disposable needle as a cutting edge. The end of the catheter (PE-5 tubing ∼10 cm long) was inserted through the incision in the dura over cisterna and threaded caudally to the vicinity of the lumbar enlargement. The 8.00 cm length served to place the catheter tip at the rostral margin of the lumbar enlargement for ∼300 g rats. The distal end of the catheter was secured to the skin and neck muscle by suture. An injection of saline solution was made at this time to clear the catheter of any debris that may have accumulated during insertion.
Intracerebroventricular surgery.
Rats (250–350 g) were anesthetized with 2% isoflurane and stereotaxically implanted with three stainless mounting screws. A stainless steel guide cannula (22 gauge) was stereotaxically positioned 1.0 mm posterior to bregma, 1.6 mm lateral (left) to the sagittal suture, and 4.5 mm below surface of the skull. Screws were secured with cranioplastic mixture.
Drug.
A-784168 and A-795614 were synthesized at Abbott Laboratories. For oral administration, A-784168 in 10% dehydrated ethanol, 20% polysorbate 80, and 70% polyethylene glycol 400 (PEG) and A-795614 in 100% PEG were administered 1 h before testing using a volume of 2 ml/kg. For intrathecal administration, compounds were dissolved in 10% di-methyl-sulfoxide/90% hydroxyl-β-cyclodextrin, injected in a volume of 10 μl, and followed by a 10 μl saline flush. The pretreatment time for intrathecal administration was 10 min. The same vehicle and pretreatment time was used for intracerebroventricular administration. The same vehicle was also used for intraplantar injection; however, the injection volume was 50 μl, and pretreatment time was 20 min.
Statistics.
The significance of the data was determined by ANOVA followed by Fisher's post hoc analysis. The level of significance was set at p < 0.05. Data are presented as mean ± SEM. The ED50 value was determined as the concentration of a compound that produced 50% effect.
Results
In vitro pharmacological profiles of A-784168 and A-795614
A-784168 and A-795614 were identified as TRPV1 antagonists in a capsaicin-induced calcium flux assay using a fluorometric imaging plate reader. Both A-784168 and A-795614 potently and competitively inhibited capsaicin-induced calcium flux into 1321N1 astrocytoma cells expressing recombinant human TRPV1. Schild analysis yielded a single high-affinity site and slopes of 0.923 for both compounds with pA2 = 8.09 and 8.05 for A-784168 and A-794614, respectively (Fig. 1). The IC50 values for inhibiting TRPV1 activation by 50 nm capsaicin were 25.0 ± 2.0 and 14.0 ± 1.0 nm for A-784168 and A-795614, respectively (Table 1). Both compounds also inhibited pH 5.5-induced activation of recombinant TRPV1 expressed in HEK293 cells with similar potency (IC50 = 14.0 ± 2.3 and 8.6 ± 0.8 nm for A-784168 and A-795614, respectively) as well as activation by 3 μm NADA (IC50 = 33.7 ± 9.6 and 21.8 ± 1.7 nm for A-784168 and A-795614, respectively) and 10 μm anandamide (IC50 = 35.1 ± 10.6 and 23.4 ± 0.7 nm for A-784168 and A-795614, respectively). Both compounds also blocked 1 μm capsaicin-induced currents in rat dorsal root ganglion neurons using electrophysiologic methods (IC50 = 10 and 1.3 nm for A-784168 and A-795614, respectively).
Figure 1.
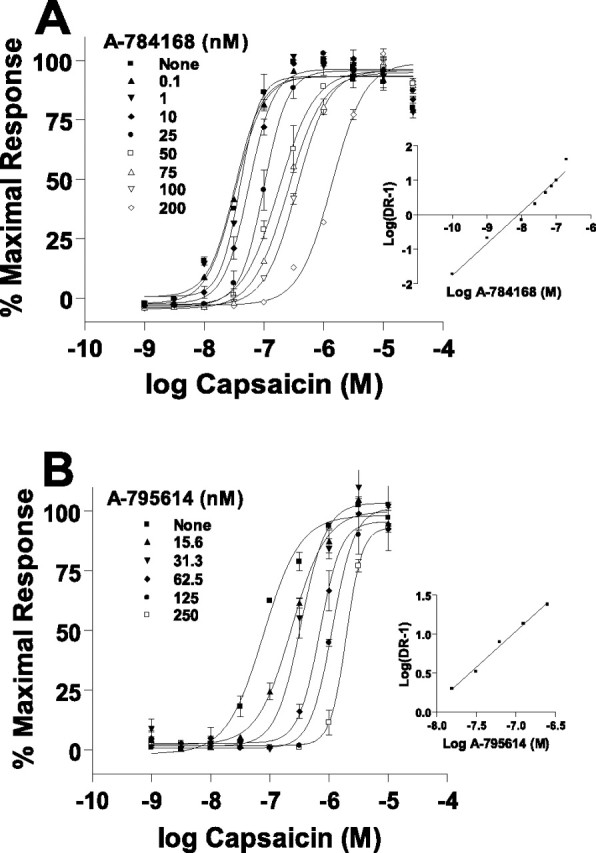
Both A-784168 and A-795614 are competitive antagonists with capsaicin at TRPV1. A, Concentration-effect curves for capsaicin-induced, increased intracellular Ca2+ in the presence of increasing concentrations of A-784168. Curves are plotted as a percentage of the maximal response obtained in the absence of an antagonist. Inset, Schild plot analysis of the antagonism produced by A-784168. B, Concentration-effect curves for capsaicin-induced, increased intracellular Ca2+ in the presence of increasing concentrations of A-795614. Curves are plotted as a percentage of the maximal response obtained in the absence of an antagonist. Inset, Schild plot analysis of the antagonism produced by A-795614.
Table 1.
Comparison of A-784168 and A-795614

Pharmacokinetic and in vitro properties of A-784168 and A-795614 are shown. A-784168 and A-785614 demonstrated similar in vitro potency, plasma protein binding, and oral bioavailability. However, the two TRPV1 antagonists have substantially different volumes of distribution (Vβ) and CNS penetration.
The selectivity of both compounds was evaluated against other TRP channels and 74 different neurotransmitter receptors and ion channels (Cerep, Paris, France). In addition to being potent TRPV1 antagonists, neither compound activated or inhibited TRPA1 (tested up to 50 μm). A-795614 had no activity against TRPM8. A-784168 was a very weak antagonist of TRPM8 (IC50 = 20.8 μm) compared with its activity against TRPV1. Neither compound had affinity for adenosine (A1, A2A, or A3), angiotensin (AT1, AT2), α1, α2, or β-adrenergic receptors, benzodiazepine, bombesin, CCR1, CXCR2, CGRP, cannabinoid (CB1 or CB2), cholecystikinin (CCK1 and CCK2), dopamine (D1, D2, D3, D4, or D5), endothelin (ETA and ETB), GABA, galanin (GAL-1), histamine (H2, H3), ML1, muscarinic acetylcholine (M1, M2, M3, M4, and M5), neurokinin (NK1, NK2, and NK3), neuropeptide Y (Y1 and Y2), neurotensin (NT1), opioid (δ, κ, and μ), ORL1, PACAP, PCP, prostanoid (thromboxane and prostacyclin), purinergic (P2X, P2Y), serotonin (5-HT3, 5-HT5A), somatostatin, TNF-α, VIP1, and vasopressin (V1A) receptors. A-784168 did demonstrate binding to human 5-HT2c, but no functional activation or inhibition (tested up to 10 μm) could be observed in calcium flux assays.
Pharmacokinetic profiles of A-784168 and A-795614 in rats
A-784168 and A-795614 had similar plasma binding (93.5 ± 0.9 vs 94.1 ± 0.5%) and oral bioavailability (33 vs 21%) (Table 1). However, these two compounds had very different volumes of distribution, 12.4 L/kg for A-784168 versus 0.64 L/kg for A-795614. As suggested by the volume of distribution data, the CNS penetration for these two compounds was also very different. After oral administration (30 μmol/kg), the plasma and brain levels for A-784168 were 745 ± 175 ng/ml and 1093 ± 203 ng/g, producing a plasma:brain ratio of 1:1.5. After oral administration (10 μmol/kg), the plasma and brain levels for A-795614 were 4520 ± 740 ng/ml and 38 ± 7 ng/g, producing a plasma:brain ratio of 1:0.008.
Capsaicin-induced acute pain
Intraplantar injection of capsaicin rapidly produced nocifensive behaviors, presumably mediated by direct activation of local TRPV1 receptors. Oral administration of A-784168 and A-795614 1 h before capsaicin injection reduced capsaicin-induced nocifensive behaviors with the same potency and maximum efficacy (ED50 values of 10 μmol/kg, p.o. for both compounds with 90 ± 5 and 86 ± 7% effect at 30 μmol/kg, per os (p.o.) for A-784168 and A-795614, respectively) (Fig. 2). These results were consistent with the fact that the two compounds had similar in vitro potency and oral bioavailability (Table 1).
Figure 2.

Oral administration of A-784168 (A) and A-795614 (B) blocked acute nocifensive behavior in the capsaicin model. Significance by ANOVA and Fisher's PLSD. *p < 0.05, **p < 0.01 versus vehicle. Error bars represent SEM (n = 12 per group).
CFA-induced chronic inflammatory thermal hyperalgesia
CFA injection into the paw produced thermal hyperalgesia. The paw withdrawal latency (PWL) to thermal stimulation was significantly decreased 48 h after CFA injection (PWL control, 10.29 ± 0.26 s vs PWL inflamed, 5.43 ± 0.11 s; p < 0.01) (Fig. 3). Oral administration of A-784168 or A-795614 decreased CFA-induced thermal hyperalgesia in a dose-dependent manner with ED50 values of 6 and 12 μmol/kg, p.o. (Fig. 3A,B; Table 2).
Figure 3.
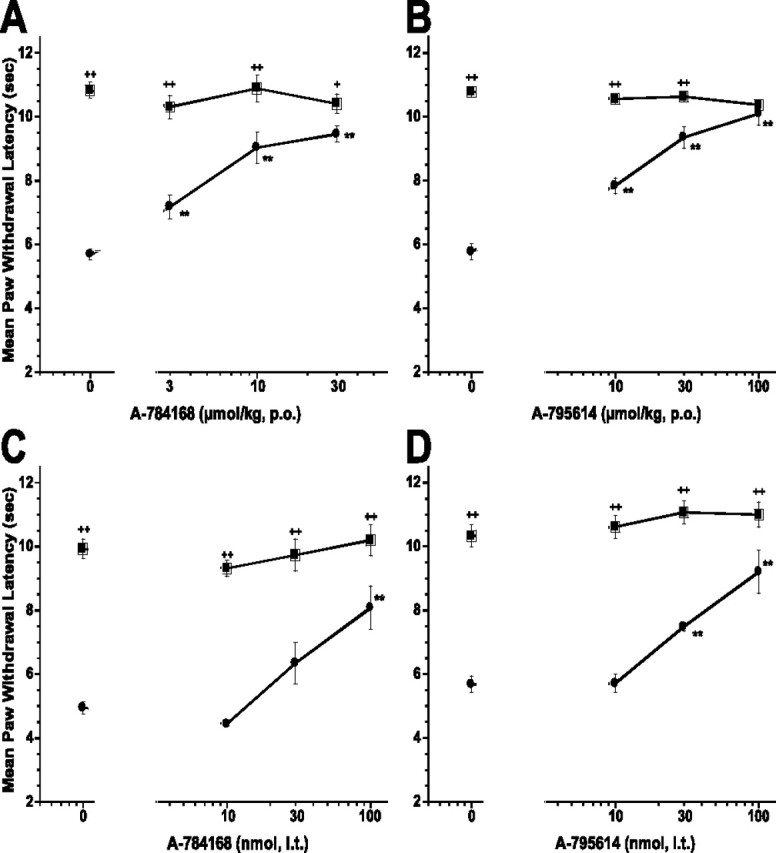
Oral administration of A-784168 (A) and A-795614 (B) and intrathecal administration of A-784168 (C) and A-795614 (D) blocked CFA-induced thermal hyperalgesia. Circles, Paw withdrawal latencies ipsilateral to the injury; squares, paw withdrawal latencies contralateral to the injury. Significance by ANOVA and Fisher's PLSD. **p < 0.01 versus vehicle; +p < 0.05, ++p < 0.01 ipsilateral versus contralateral paw. Error bars represent SEM (n = 6 per group).
Table 2.
Potency and efficacious plasma concentrations of A-784168 and A-795614 after oral or local administration in CFA-induced thermal hyperalgesia
| Oral administration | Intrathecal administration | Intraplantar administration in the ipsilateral paw | Intracerebreventricular administration | |
|---|---|---|---|---|
| A-784168 | ED50 = 6 μmol/kgPlasma conc = 0.2 μg/ml | ED50 = 62 nmolPlasma conc = 0.03 μg/ml | ED50 = 320 nmolPlasma conc = 0.015 μg/ml | ED50 = 54 nmolPlasma conc = 0.043 μg/ml |
| A-795614 | ED50 = 12 μmol/kgPlasma conc = 2 μg/ml | ED50 = 44 nmolPlasma conc = 0.12 μg/ml | Not tested | Not tested |
Plasma samples were collected at the time of testing for thermal hyperalgesia. Plasma concentrations (conc) of administered compounds which yield ED50 are shown.
To determine whether TRPV1 receptor antagonists were active in the CNS, various doses of each compound were delivered by intrathecal administration. A-784168 and A-795614 fully blocked thermal hyperalgesia in the CFA model of chronic inflammatory pain after intrathecal administration with ED50 values of 62 and 44 nmol, respectively (Fig. 3C,D). To confirm that these effects were attributable to activity of the TRPV1 antagonists in the CNS and not to leakage into the periphery, plasma samples from these animals were evaluated. The plasma concentrations (0.03 μg/ml for A-784168 and 0.12 μg/ml for A-795614) of the two compounds after intrathecal administration of their respective ED50 doses were substantially below the plasma levels (0.2 μg/ml for A-784168 and 2 μg/ml for A-795614) required for 50% effect after oral administration (Table 2). Similar to its intrathecal effect, intracerebroventricular administration of A-784168 also produced a significant reduction in CFA-induced thermal hyperalgesia with an ED50 value of 54 nmol (plasma at ED50 = 0.043 μg/ml). Because direct injection into the CNS yielded analgesic effects at plasma levels substantially below those required for analgesic effects after oral administration, these results suggest that the CNS is at least one site of action for TRPV1 antagonists in chronic inflammatory pain.
To determine whether peripheral TRPV1 receptors also played a role in blocking thermal hyperalgesia in the CFA model, A-784168 was administered intraplantarly. Either ipsilateral or contralateral injection of A-784168 produced a significant reduction in CFA-induced thermal hyperalgesia with ED50 values of 319 and 734 nmol, respectively (Fig. 4). Interestingly, ipsilateral administration of 300 nmol A-784168 produced 49 ± 8% effect (p < 0.01). However, the plasma concentration resulting from ipsilateral administration of this dose was much lower than the plasma level associated with 50% effect after oral administration (0.015 μg/ml vs 0.2 μg/ml) (Table 2). These results suggest that peripheral TRPV1 receptors do play a role in the CFA model of chronic inflammatory pain. In contrast, the plasma concentration of A-784168 associated with 50% effect after administration into the contralateral paw was 0.14 μg/ml, which is similar to the plasma level associated with 50% effect after oral administration, indicative of a systemic effect. In addition, after intraplantar administration into the noninflamed paw, antinociceptive effects on the noninflamed paw were observed at the 1000 nmol dose. Such antinociceptive effects were not observed after oral administration (data not shown), suggesting a local antinociceptive effect of A-784168 against acute thermal pain.
Figure 4.

Intraplantar administration of A-784168 reduced CFA-induced thermal hyperalgesia. A, Administration of A-784168 into ipsilateral paw blocked CFA-induced thermal hyperalgesia in a dose-dependent manner with an ED50 value of 319 nmol, i.pl. B, Administration of A-784168 into contralateral paw blocked CFA-induced thermal hyperalgesia in a dose-dependent manner with an ED50 value of 734 nmol, i.pl. Circles, Paw withdrawal latencies ipsilateral to the injury; squares, paw withdrawal latencies contralateral to the injury. Significance by ANOVA and Fisher's PLSD. *p < 0.05, **p < 0.01 versus vehicle; ++p < 0.01 ipsilateral versus contralateral paw. Error bars represent SEM (n = 6 per group).
CFA-induced chronic inflammatory mechanical allodynia
In addition to thermal hyperalgesia, CFA injection into the paw also produced mechanical allodynia. The paw withdrawal threshold to von Frey hairs was significantly reduced 48 h after CFA injection (control 13.1 ± 0.4 g vs inflamed 3.8 ± 0.3 g; p < 0.01). Oral administration of A-784168 significantly reduced CFA-induced mechanical allodynia with an ED50 value of 76 μmol/kg, p.o. (94 ± 12% effect at 300 μmol/kg, p.o.) (Fig. 5A). However, oral administration of A-795614 produced weaker effects (53 ± 12% at 300 μmol/kg, p.o.; p < 0.01) (Fig. 5B). To investigate the involvement of the CNS in CFA-induced mechanical allodynia, A-784168 and A-795614 were administered intrathecally. After intrathecal administration, the two compounds exhibited similar potency in blocking CFA-induced mechanical allodynia, with ED50 values of 40 and 82 nmol for A-784168 and A-795614, respectively (Fig. 5C,D).
Figure 5.
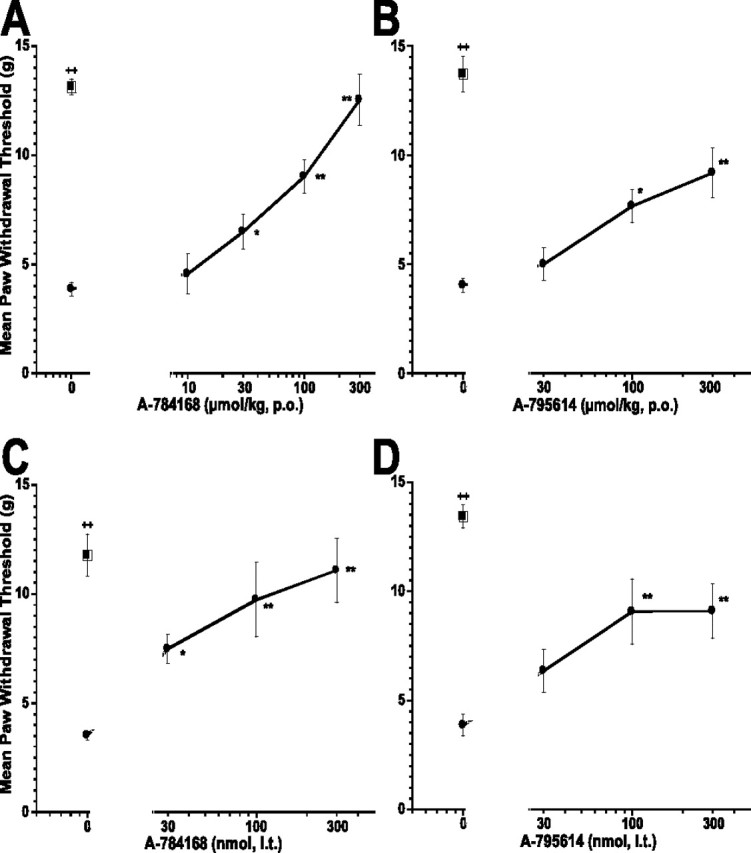
Oral administration of A-784168 (A) and A-795614 (B) and intrathecal administration of A-784168 (C) and A-795614 (D) reduced CFA-induced mechanical allodynia. Circles, Paw withdrawal threshold ipsilateral to the injury; squares, paw withdrawal threshold contralateral to the injury. Significance by ANOVA and Fisher's PLSD. *p < 0.05, **p < 0.01 versus vehicle; ++p < 0.01 ipsilateral versus contralateral paw. Error bars represent SEM (n = 6–12 per group).
MIA-induced osteoarthritic pain
MIA was injected into rats to induce osteoarthritis of the knee joint. The effect of A-784168 or A-795614 on weight-bearing differences (WBDs) between injured and noninjured hindlimbs was assessed 4 d after MIA injection. The WBD for the vehicle group was 51.9 ± 2.2 g. A-784168 p.o. dose-dependently reduced the WBD with an ED50 value of 8 μmol/kg, p.o. (Fig. 6A). The effect of A-784168 at 30 μmol/kg, p.o. was 85 ± 4.7% (p < 0.01). A-795614 also significantly reduced the WBD (p < 0.01) (Fig. 6B). However, the potency of A-795614 after oral administration was much lower than A-784168; A-795614 exhibited an ED50 value of 280 μmol/kg, p.o., and only 53 ± 10.2% effect (p < 0.01) at 300 μmol/kg, p.o. To determine whether TRPV1 receptors in the CNS play a role in blocking WBD in the OA model, A-784168 or A-795614 was administered intrathecally. In contrast to oral administration, intrathecal A-784168 and A-795614 showed similar potency in blocking MIA-induced WBD with ED50 values of 22 and 26 nmol, respectively (Fig. 6C,D). In addition, intracerebroventricular administration of A-784168 also significantly reduced the MIA-induced WBD with 65 ± 6% effect at 100 nmol with an ED50 value of 64 nmol (data not shown). Together, these results suggest that TRPV1 receptors in the CNS play a major role in osteoarthritic pain.
Figure 6.
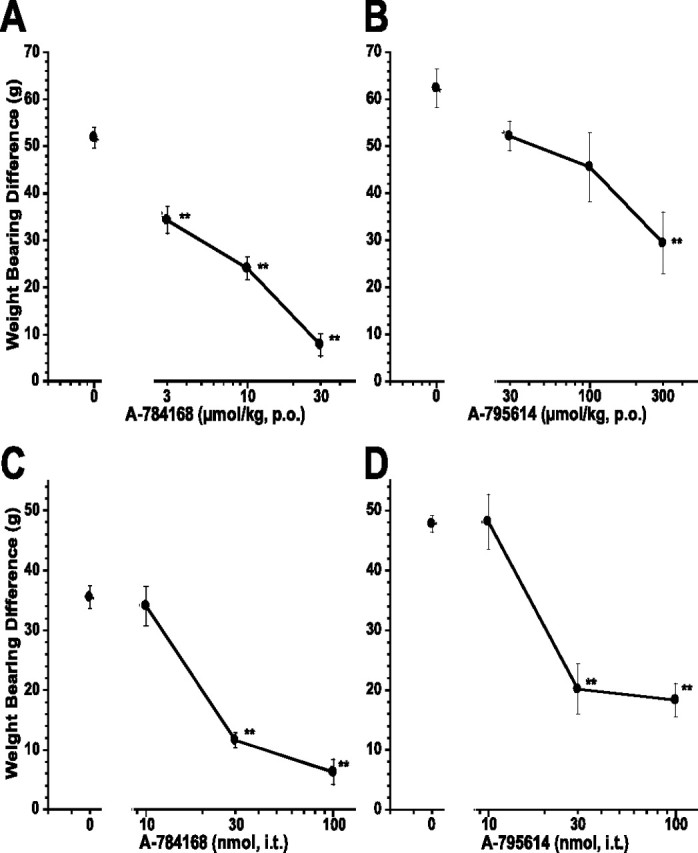
Oral administration of A-784168 (A) and A-795614 (B) and intrathecal administration of A-784168 (C) and A-795614 (D) blocked weight-bearing difference in the model of osteoarthritic pain. Circles, Weight-bearing difference between ipsilateral and contralateral paws. Significance by ANOVA and Fisher's PLSD. **p < 0.01 versus vehicle. Error bars represent SEM (n = 6 per group).
Capsaicin-induced secondary mechanical allodynia
Intraplantar injection of capsaicin produces initial acute nocifensive behaviors, a primary thermal hyperalgesia in the immediate vicinity of the injection site and a secondary mechanical allodynia in a large region surrounding the injection site that lasts for several hours. Central sensitization is considered to mediate this secondary allodynia. Oral administration of either A-784168 or A-795614 2 h after capsaicin injection significantly reduced capsaicin-induced secondary mechanical allodynia but with different potencies (Fig. 7A,B). Oral administration of A-784168 produced 87 ± 20% effect (p < 0.01) at 100 μmol/kg with an ED50 value of 40 μmol/kg. However, A-795614 only produced 60% effect (p < 0.01) at 300 μmol/kg, p.o. with ED50 value of 165 μmol/kg. In contrast, the potencies for the two compounds to inhibit secondary mechanical allodynia after intrathecal administration were similar (ED50 = 60 nmol for A-784168; ED50 = 77 nmol for A-795614) (Fig. 7C,D). These results confirmed that the CNS plays a significant role in relieving pain mediated by central sensitization.
Figure 7.
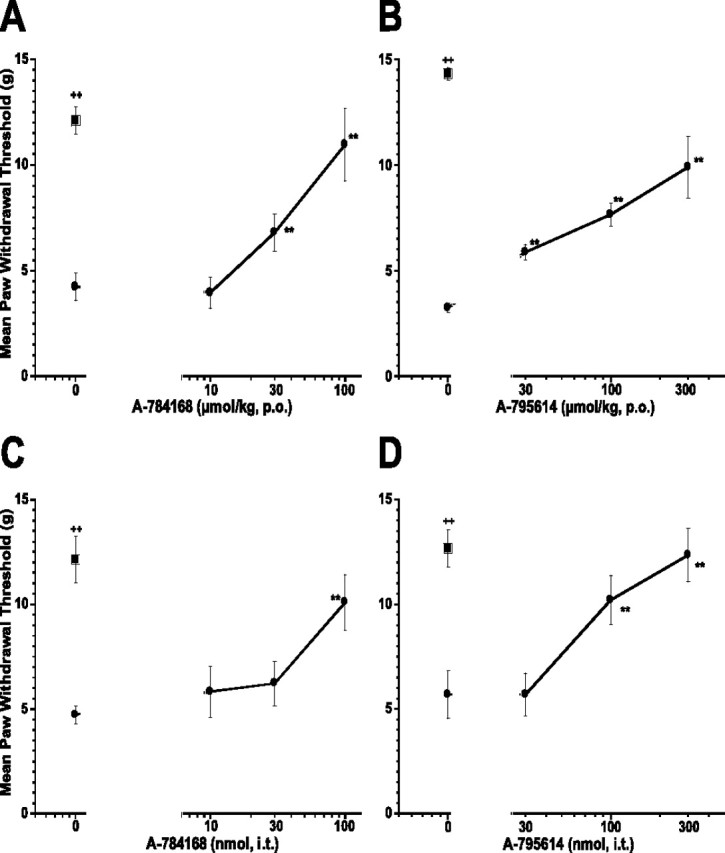
Oral administration of A-784168 (A) and A-795614 (B) and intrathecal administration of A-784168 (C) and A-795614 (D) reduced secondary mechanical allodynia in the capsaicin model. Circles, Paw withdrawal threshold ipsilateral to the injury; squares, paw withdrawal threshold contralateral to the injury. Significance by ANOVA and Fisher's PLSD. **p < 0.01 versus vehicle; ++p < 0.01 ipsilateral versus contralateral paw. Error bars represent SEM (n = 6 per group).
Spontaneous locomotor activity
Spontaneous locomotor activity in the vehicle groups were 5381 ± 724.9 U (A-784168 experiment) and 3938 ± 424 U (A-795614 experiment). A-784168 and A-795614 tested up to 300 μmol/kg after oral administration (n = 8 per group) did not show any significant effects on spontaneous locomotor activity (A-784168, F(3,28) = 0.42, p > 0.05; A-795614, F(3,28) = 2.59, p > 0.05). These results suggest that the effects on behavioral thresholds in the pain models are not attributable to any nonspecific effects on motor performance.
Discussion
In this report, we studied the role that the CNS plays in the analgesic effects of TRPV1 antagonists. A-784168 and A-795614, which have similar in vitro potency, plasma protein binding, and oral bioavailability but differ in CNS penetration, were evaluated in a variety of animal pain models. When administered orally, A-784168 and A-795614 blocked capsaicin-induced acute nocifensive behavior with the same potency and efficacy, indicating that both compounds behave as selective TRPV1 antagonists at peripheral sites in vivo. The present study also demonstrated that A-784168 (good CNS penetration) was more potent and efficacious than A-795614 (poor CNS penetration) in several pain models after oral administration. Interestingly, such differences in potency after oral administration across various pain models disappeared when compounds were administered directly into the CNS. These results demonstrate that the blockade of centrally located TRPV1 receptors is a major mechanism of action for TRPV1 antagonist-induced potent analgesia in some pain models.
In the CFA model of chronic inflammatory pain, orally administered A-795614 and A-784168 completely blocked CFA-induced thermal hyperalgesia with similar potency (ED50 values of 6 and 12 μmol/kg, p.o., respectively), consistent with their similar in vitro potency. Because the two compounds have very different degrees of CNS penetration, these results suggest that peripheral TRPV1 receptors appear to play a significant role in CFA-induced thermal hyperalgesia. Indeed, intraplantar administration of A-784168 dose-dependently blocked CFA-induced thermal hyperalgesia with an ED50 value of 319 nmol. At this dose, plasma concentrations were only 0.015 μg/ml, a plasma level much lower than the plasma level (0.2 μg/ml) required for 50% effect after oral administration, confirming the local effect of A-784168 after intraplantar administration. A-784168 also produced anti-hyperalgesic effects when dosed in the noninflamed paw (ED50 value of 734 nmol) with a plasma level of 0.14 μg/ml, suggesting that these effects are caused by systemic diffusion. These data also suggest that compound distribution is different after injection into an inflamed versus noninflamed tissue. Injection of a two-fold higher dose into noninflamed tissues versus inflamed tissues resulted in 10-fold higher plasma levels. Differences in tissue volume or tissue content could be partly responsible for these observations. After intrathecal application, both compounds effectively blocked thermal hyperalgesia in the CFA model, suggesting that the CNS also played a role in the effect of TRPV1 antagonists on thermal hyperalgesia. These results suggest that both the CNS and the periphery play important roles in the activity of TRPV1 antagonists on CFA-induced thermal hyperalgesia. The lower plasma level associated with the ED50 value for A-784168 (0.2 μg/ml) compared with A-795614 (2 μg/ml) in this model is consistent with TRPV1 receptors in the CNS contributing to the potency of A-784168 after oral administration. Interestingly, in agreement with the decrease in acute thermal nociception in TRPV1 knock-out mice, local administration of A-784168 in the noninflamed paw of CFA rats resulted in antinociceptive effect thermal stimuli. These results were confirmed in naive rats and were observed at plasma levels for which no effects on thermal pain were observed after A-784168 systemic administration, suggesting that local TRPV1 receptors play a role in acute thermal pain transmission.
Orally administered A-784168 and A-795614 also reduced CFA-induced mechanical allodynia. Several other novel selective TRPV1 antagonists (e.g., capsazepine, BCTC, and AMG 9810) have been reported to reduce mechanical allodynia associated with inflammation (Pomonis et al., 2003; Walker et al., 2003; Gavva et al., 2005). In contrast, mechanical allodynia associated with inflammation developed normally in TRPV1 knock-out mice (Caterina et al., 2000). It is possible that compensatory mechanisms resulted in the lack of an observed effect of TRPV1 gene ablation on mechanical allodynia. Another potential explanation for this discrepancy could be that the antagonists tested against inflammatory mechanical allodynia showed efficacy through another mechanism than TRPV1. Although one cannot exclude that A-784168 and A-795614 could cross-react with a target yet unidentified, these two TRPV1 antagonists have been shown to be inactive or weakly active at other TRP channels such as TRPA1 and TRPM8 in addition to being inactive at 74 different neurotransmitter receptors and ion channels (Cerep), suggesting that the behavioral effects produced by these two compounds are TRPV1 mediated. Interestingly, A-784168, which has better CNS penetration, was more potent than A-795614 in reducing mechanical allodynia when administered orally. In contrast, intrathecal administration of A-784168 and A-795614 resulted in similar potency in blocking CFA-induced mechanical allodynia. These results suggest that TRPV1 receptors in the CNS play a greater role in mechanical allodynia than thermal hyperalgesia in the CFA model. Although the present data clearly show a role for CNS TRPV1 receptors in pain transmission and modulation, they do not allow for a more detailed identification of the central site(s) of action. The fact that in vivo efficacy was observed after both intrathecal and intracerebroventricular administration strongly suggests that both spinal and supraspinal sites are playing a role in the analgesic activity of A-784168. However, diffusion between the two sites after central administration cannot be fully excluded. Additional studies using direct injection of TRPV1 antagonists in various brain regions could help answer this question.
Previous receptor expression studies have demonstrated that TRPV1 receptors are upregulated in both the periphery and the CNS during chronic inflammation (Carlton and Coggeshall, 2001; Ji et al., 2002; Luo et al., 2004). In addition, NGF, which has been shown to activate peripheral nociceptors, to affect gene expression at the cell body by retrograde signaling (Donnerer et al., 1992), and to be upregulated in peripheral tissue and DRG after inflammation and tissue injury (Woolf et al., 1994; Shu and Mendell, 1999; Amaya et al., 2004), can induce the upregulation of TRPV1 receptor expression and increase transport of TRPV1 to the peripheral nociceptor terminals (Ji et al., 2002). These studies support that the blockade of TRPV1 receptors in both the periphery and the CNS plays a role in chronic inflammatory pain.
The present study demonstrated that orally administered A-784168, which has good CNS penetration, was much more potent in reversing weight-bearing difference in the OA model than A-795614, which has poor CNS penetration. In contrast, the two compounds were equipotent when administered intrathecally. These results suggest that TRPV1 receptors in the CNS play a role in osteoathritic pain. Several lines of evidence support a role for TRPV1 in osteoarthritic pain: (1) selective TRPV1 antagonist, A-425619, previously described by our laboratory, reversed the weight-bearing difference observed in this pain model (Honore et al., 2005); (2) TRPV1 receptor expression is increased in the joint capsule of osteoarthritic rats (Cho and Valtschanoff, 2004); and (3) a decrease in pH at the site of bone resorption (Baron et al., 1985; Blair et al., 1989) may activate TRPV1 receptors in osteoarthritis. These data support a role for peripheral TRPV1 receptors in OA pain and other bone disease-related pains. Interestingly, in our series of experiments, the centrally located TRPV1 receptors were shown to play a major role in OA pain.
Intraplantar injection of either capsaicin (Simone et al., 1987; Gilchrist et al., 1996) or acid (Hamamoto et al., 1998) results in the development of both thermal hyperalgesia and secondary mechanical allodynia. The secondary mechanical allodynia observed after capsaicin injection is more robust than the thermal hyperalgesia, both in terms of duration of action and the size of the area of secondary allodynia (Gilchrist et al., 1996). It is believed that the mechanisms underlying thermal hyperalgesia and mechanical allodynia after capsaicin injection are different. Thermal hyperalgesia appears to be caused by peripheral sensitization of nociceptors, because it is restricted to the immediate area of injection. In contrast, secondary mechanical allodynia appears to be mediated by central sensitization, because the sensitivity is observed in a much larger area surrounding the site of injection (Simone et al., 1987, 1989; LaMotte et al., 1991). In the present study, oral administration of both A-784168 and A-795614 reversed capsaicin-induced secondary mechanical allodynia but A-784168, with good CNS penetration, was more potent than A-795614. When directly administered to the spinal cord, the two compounds exhibited similar potency in blocking capsaicin-induced secondary mechanical allodynia. These data further suggest that central TRPV1 receptors play a role in models of secondary mechanical allodynia.
In summary, the present study demonstrates that TRPV1 antagonists effectively reduce thermal hyperalgesia and mechanical allodynia associated with inflammatory and osteoarthritic pain. Systemic administration of two proprietary TRPV1 receptor antagonists, which differed substantially in their CNS penetration, revealed potency differences in some but not all pain models. Intrathecal administration of the TRPV1 receptor antagonists produced analgesic activity with similar potency in all models evaluated. These results demonstrated that TRPV1 receptors in the CNS play an important role in pain mediated by central sensitization. Significant CNS penetration appeared to be required for full efficacy of an orally administered TRPV1 antagonist across a wide array of pain states.
References
- Amaya et al., 2004.Amaya F, Shimosato G, Nagano M, Ueda M, Hashimoto S, Tanaka Y, Suzuki H, Tanaka M. NGF and GDNF differentially regulate TRPV1 expression that contributes to development of inflammatory thermal hyperalgesia. Eur J Neurosci. 2004;20:2303–2310. doi: 10.1111/j.1460-9568.2004.03701.x. [DOI] [PubMed] [Google Scholar]
- Baron et al., 1985.Baron R, Neff L, Louvard D, Courtoy PJ. Cell-mediated extracellular acidification and bone resorption: evidence for a low pH in resorbing lacunae and localization of a 100-kD lysosomal membrane protein at the osteoclast ruffled border. J Cell Biol. 1985;101:2210–2222. doi: 10.1083/jcb.101.6.2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi et al., 2006.Bianchi BR, Lee CH, Jarvis MF, El Kouhen R, Moreland RB, Faltynek CR, Puttfarken PS. Modulation of human TRPV1 receptor activity by extracellular proteins and host cell expression system. Eur J Pharmacol. 2006;537:20–30. doi: 10.1016/j.ejphar.2006.03.003. [DOI] [PubMed] [Google Scholar]
- Blair et al., 1989.Blair HC, Teitelbaum SL, Ghiselli R, Gluck S. Osteoclastic bone resorption by a polarized vacuolar proton pump. Science. 1989;245:855–857. doi: 10.1126/science.2528207. [DOI] [PubMed] [Google Scholar]
- Bove et al., 2003.Bove SE, Calcaterra SL, Brooker RM, Huber CM, Guzman RE, Juneau PL, Schrier DJ, Kilgore KS. Weight bearing as a measure of disease progression and efficacy of anti-inflammatory compounds in a model of monosodium iodoacetate-induced osteoarthritis. Osteoarth Cartil. 2003;11:821–830. doi: 10.1016/s1063-4584(03)00163-8. [DOI] [PubMed] [Google Scholar]
- Brown et al., 2005.Brown BS, Zheng GZ, Schmidt RG, Keddy RG, Koenig JR, Jinkerson TK, McDonald HA, Cui M, Honore P, Marsh KC, Darbyshire JF, Surowy CS, Moreland R, Jarvis MF, Faltynek CR, Lee C-H. A-784168, a novel TRPV1 receptor antagonist as analgesic agent; 230th ACS National Meeting; Washington, DC. 2005. [Google Scholar]
- Carlton and Coggeshall, 2001.Carlton SM, Coggeshall RE. Peripheral capsaicin receptors increase in the inflamed rat hindpaw: a possible mechanism for peripheral sensitization. Neurosci Lett. 2001;310:53–56. doi: 10.1016/s0304-3940(01)02093-6. [DOI] [PubMed] [Google Scholar]
- Carpenter and Lynn, 1981.Carpenter SE, Lynn B. Vascular and sensory responses of human skin to mild injury after topical treatment with capsaicin. Br J Pharmacol. 1981;73:755–758. doi: 10.1111/j.1476-5381.1981.tb16812.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caterina et al., 1997.Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature. 1997;389:816–824. doi: 10.1038/39807. [DOI] [PubMed] [Google Scholar]
- Caterina et al., 2000.Caterina MJ, Leffler A, Malmberg AB, Martin WJ, Trafton J, Petersen-Zeitz KR, Koltzenburg M, Basbaum AI, Julius D. Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science. 2000;288:306–313. doi: 10.1126/science.288.5464.306. [DOI] [PubMed] [Google Scholar]
- Chaplan et al., 1994.Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Cho and Valtschanoff, 2004.Cho W, Valtschanoff JG. Vanilloid receptor VR1-positive primary afferents in mouse model of single joint inflammatory osteoarthritis. Soc Neurosci Abstr. 2004;30:288.15. [Google Scholar]
- Chuang et al., 2001.Chuang HH, Prescott ED, Kong H, Shields S, Jordt SE, Basbaum AI, Chao MV, Julius D. Bradykinin and nerve growth factor release the capsaicin receptor from PtdIns(4,5)P2-mediated inhibition. Nature. 2001;411:957–962. doi: 10.1038/35082088. [DOI] [PubMed] [Google Scholar]
- Davis et al., 2000.Davis JB, Gray J, Gunthorpe MJ, Hatcher JP, Davey PT, Overend P, Harries MH, Latcham J, Clapham C, Atkinson K, Hughes SA, Rance K, Grau E, Harper AJ, Pugh PL, Rogers DC, Bingham S, Randall A, Sheardown SA. Vanilloid receptor-1 is essential for inflammatory thermal hyperalgesia. Nature. 2000;405:183–187. doi: 10.1038/35012076. [DOI] [PubMed] [Google Scholar]
- Donnerer et al., 1992.Donnerer J, Schuligoi R, Stein C. Increased content and transport of substance P and calcitonin gene-related peptide in sensory nerves innervating inflamed tissue: evidence for a regulatory function of nerve growth factor in vivo. Neuroscience. 1992;49:693–698. doi: 10.1016/0306-4522(92)90237-v. [DOI] [PubMed] [Google Scholar]
- El Kouhen et al., 2005.El Kouhen R, Surowy CS, Bianchi BR, Neelands TR, McDonald HA, Niforatos W, Gomtsyan A, Lee CH, Honore P, Sullivan JP, Jarvis MF, Faltynek CR. A-425619 [1-isoquinolin-5-yl-3-(4-trifluoromethyl-benzyl)-urea], a novel and selective transient receptor potential type V1 receptor antagonist, blocks channel activation by vanilloids, heat, and acid. J Pharmacol Exp Ther. 2005;314:400–409. doi: 10.1124/jpet.105.084103. [DOI] [PubMed] [Google Scholar]
- Ferreira et al., 2004.Ferreira J, da Silva GL, Calixto JB. Contribution of vanilloid receptors to the overt nociception induced by B2 kinin receptor activation in mice. Br J Pharmacol. 2004;141:787–794. doi: 10.1038/sj.bjp.0705546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavva et al., 2005.Gavva NR, Tamir R, Qu Y, Klionsky L, Zhang TJ, Immke D, Wang J, Zhu D, Vanderah TW, Porreca F, Doherty EM, Norman MH, Wild KD, Bannon AW, Louis JC, Treanor JJ. AMG 9810 [(E)-3-(4-t-butylphenyl)-N-(2,3-dihydrobenzo[b][1,4] dioxin-6-yl)acrylamide], a novel vanilloid receptor 1 (TRPV1) antagonist with antihyperalgesic properties. J Pharmacol Exp Ther. 2005;313:474–484. doi: 10.1124/jpet.104.079855. [DOI] [PubMed] [Google Scholar]
- Gilchrist et al., 1996.Gilchrist HD, Allard BL, Simone DA. Enhanced withdrawal responses to heat and mechanical stimuli following intraplantar injection of capsaicin in rats. Pain. 1996;67:179–188. doi: 10.1016/0304-3959(96)03104-1. [DOI] [PubMed] [Google Scholar]
- Gomtsyan et al., 2005.Gomtsyan A, Bayburt EK, Schmidt RG, Zheng GZ, Perner RJ, Didomenico S, Koenig JR, Turner S, Jinkerson T, Drizin I, Hannick SM, Macri BS, McDonald HA, Honore P, Wismer CT, Marsh KC, Wetter J, Stewart KD, Oie T, Jarvis MF, et al. Novel transient receptor potential vanilloid 1 receptor antagonists for the treatment of pain: structure-activity relationships for ureas with quinoline, isoquinoline, quinazoline, phthalazine, quinoxaline, and cinnoline moieties. J Med Chem. 2005;48:744–752. doi: 10.1021/jm0492958. [DOI] [PubMed] [Google Scholar]
- Guo et al., 1999.Guo A, Vulchanova L, Wang J, Li X, Elde R. Immunocytochemical localization of the vanilloid receptor 1 (VR1): relationship to neuropeptides, the P2X3 purinoceptor and IB4 binding sites. Eur J Neurosci. 1999;11:946–958. doi: 10.1046/j.1460-9568.1999.00503.x. [DOI] [PubMed] [Google Scholar]
- Hamamoto et al., 1998.Hamamoto DT, Ortiz-Gonzalez XR, Honda JM, Kajander KC. Intraplantar injection of hyaluronic acid at low pH into the rat hindpaw produces tissue acidosis and enhances withdrawal responses to mechanical stimuli. Pain. 1998;74:225–234. doi: 10.1016/s0304-3959(97)00185-1. [DOI] [PubMed] [Google Scholar]
- Hargreaves et al., 1988.Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- Hayes et al., 2000.Hayes P, Meadows HJ, Gunthorpe MJ, Harries MH, Duckworth DM, Cairns W, Harrison DC, Clarke CE, Ellington K, Prinjha RK, Barton AJL, Medhurst AD, Smith GD, Topp S, Murdock P, Sanger GJ, Terrett J, Jenkins O, Benham CD, Randall AD, et al. Cloning and functional expression of a human orthologue of rat vanilloid receptor-1. Pain. 2000;88:205–215. doi: 10.1016/S0304-3959(00)00353-5. [DOI] [PubMed] [Google Scholar]
- Honore et al., 2005.Honore P, Wismer CT, Mikusa J, Zhu CZ, Zhong C, Gauvin DM, Gomtsyan A, El Kouhen R, Lee CH, Marsh K, Sullivan JP, Faltynek CR, Jarvis MF. A-425619 [1-Isoquinolin-5-yl-3-(4-trifluoromethyl-benzyl)-urea], a novel transient receptor potential type V1 receptor antagonist, relieves pathophysiological pain associated with inflammation and tissue injury in rats. J Pharmacol Exp Ther. 2005;314:410–421. doi: 10.1124/jpet.105.083915. [DOI] [PubMed] [Google Scholar]
- Ji et al., 2002.Ji RR, Samad TA, Jin SX, Schmoll R, Woolf CJ. p38 MAPK activation by NGF in primary sensory neurons after inflammation increases TRPV1 levels and maintains heat hyperalgesia. Neuron. 2002;36:57–68. doi: 10.1016/s0896-6273(02)00908-x. [DOI] [PubMed] [Google Scholar]
- Kelly and Chapman, 2002.Kelly S, Chapman V. Spinal administration of capsazepine inhibits noxious evoked responses of dorsal horn neurons in non-inflamed and carrageenan inflamed rats. Brain Res. 2002;935:103–108. doi: 10.1016/s0006-8993(02)02552-0. [DOI] [PubMed] [Google Scholar]
- LaMotte et al., 1991.LaMotte RH, Shain CN, Simone DA, Tsai EF. Neurogenic hyperalgesia: psychophysical studies of underlying mechanisms. J Neurophysiol. 1991;66:190–211. doi: 10.1152/jn.1991.66.1.190. [DOI] [PubMed] [Google Scholar]
- LaMotte et al., 1992.LaMotte RH, Lundberg LE, Torebjork HE. Pain, hyperalgesia and activity in nociceptive C units in humans after intradermal injection of capsaicin. J Physiol (Lond) 1992;448:749–764. doi: 10.1113/jphysiol.1992.sp019068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo et al., 2004.Luo H, Cheng J, Han JS, Wan Y. Change of vanilloid receptor 1 expression in dorsal root ganglion and spinal dorsal horn during inflammatory nociception induced by complete Freund's adjuvant in rats. NeuroReport. 2004;15:655–658. doi: 10.1097/00001756-200403220-00016. [DOI] [PubMed] [Google Scholar]
- McGaraughty et al., 2003.McGaraughty S, Chu KL, Bitner RS, Martino B, El Kouhen R, Han P, Nikkel AL, Burgard EC, Faltynek CR, Jarvis MF. Capsaicin infused into the PAG affects rat tail flick responses to noxious heat and alters neuronal firing in the RVM. J Neurophysiol. 2003;90:2702–2710. doi: 10.1152/jn.00433.2003. [DOI] [PubMed] [Google Scholar]
- Mezey et al., 2000.Mezey E, Toth ZE, Cortright DN, Arzubi MK, Krause JE, Elde R, Guo A, Blumberg PM, Szallasi A. Distribution of mRNA for vanilloid receptor subtype 1 (VR1), and VR1-like immunoreactivity, in the central nervous system of the rat and human. Proc Natl Acad Sci USA. 2000;97:3655–3660. doi: 10.1073/pnas.060496197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palazzo et al., 2002.Palazzo E, de Novellis V, Marabese I, Cuomo D, Rossi F, Berrino L, Maione S. Interaction between vanilloid and glutamate receptors in the central modulation of nociception. Eur J Pharmacol. 2002;439:69–75. doi: 10.1016/s0014-2999(02)01367-5. [DOI] [PubMed] [Google Scholar]
- Pomonis et al., 2003.Pomonis JD, Harrison JE, Mark L, Bristol DR, Valenzano KJ, Walker K. N-(4-Tertiarybutylphenyl)-4-(3-cholorphyridin-2-yl)tetrahydropyrazine-1(2H)-carbox-amide (BCTC), a novel, orally effective vanilloid receptor 1 antagonist with analgesic properties: II. In vivo characterization in rat models of inflammatory and neuropathic pain. J Pharmacol Exp Ther. 2003;306:387–393. doi: 10.1124/jpet.102.046268. [DOI] [PubMed] [Google Scholar]
- Premkumar and Ahern, 2000.Premkumar LS, Ahern GP. Induction of vanilloid receptor channel activity by protein kinase C. Nature. 2000;408:985–990. doi: 10.1038/35050121. [DOI] [PubMed] [Google Scholar]
- Roberts et al., 2004.Roberts JC, Davis JB, Benham CD. [3H]Resiniferatoxin autoradiography in the CNS of wild-type and TRPV1 null mice defines TRPV1 (VR-1) protein distribution. Brain Res. 2004;995:176–183. doi: 10.1016/j.brainres.2003.10.001. [DOI] [PubMed] [Google Scholar]
- Shu and Mendell, 1999.Shu X, Mendell LM. Nerve growth factor acutely sensitizes the response of adult rat sensory neurons to capsaicin. Neurosci Lett. 1999;274:159–162. doi: 10.1016/s0304-3940(99)00701-6. [DOI] [PubMed] [Google Scholar]
- Simone et al., 1987.Simone DA, Ngeow JY, Putterman GJ, LaMotte RH. Hyperalgesia to heat after intradermal injection of capsaicin. Brain Res. 1987;418:201–203. doi: 10.1016/0006-8993(87)90982-6. [DOI] [PubMed] [Google Scholar]
- Simone et al., 1989.Simone DA, Baumann TK, Collins JG, LaMotte RH. Sensitization of cat dorsal horn neurons to innocuous mechanical stimulation after intradermal injection of capsaicin. Brain Res. 1989;486:185–189. doi: 10.1016/0006-8993(89)91293-6. [DOI] [PubMed] [Google Scholar]
- Szabo et al., 2002.Szabo T, Biro T, Gonzalez AF, Palkovits M, Blumberg PM. Pharmacological characterization of vanilloid receptor located in the brain. Brain Res Mol Brain Res. 2002;98:51–57. doi: 10.1016/s0169-328x(01)00313-8. [DOI] [PubMed] [Google Scholar]
- Szallasi and Blumberg, 1999.Szallasi A, Blumberg PM. Vanilloid (capsaicin) receptors and mechanisms. Pharmacol Rev. 1999;51:159–212. [PubMed] [Google Scholar]
- Szolcsanyi, 1977.Szolcsanyi J. A pharmacological approach to elucidation of the role of different nerve fibres and receptor endings in mediation of pain. J Physiol (Paris) 1977;73:251–259. [PubMed] [Google Scholar]
- Tominaga et al., 1998.Tominaga M, Caterina MJ, Malmberg AB, Rosen TA, Gilbert H, Skinner K, Raumann BE, Basbaum AI, Julius D. The cloned capsaicin receptor integrates multiple pain-producing stimuli. Neuron. 1998;21:531–543. doi: 10.1016/s0896-6273(00)80564-4. [DOI] [PubMed] [Google Scholar]
- Valtschanoff et al., 2001.Valtschanoff JG, Rustioni A, Guo A, Hwang SJ. Vanilloid receptor VR1 is both presynaptic and postsynaptic in the superficial laminae of the rat dorsal horn. J Comp Neurol. 2001;436:225–235. [PubMed] [Google Scholar]
- Walker et al., 2003.Walker KM, Urban L, Medhurst SJ, Patel S, Panesar M, Fox AJ, McIntyre P. The VR1 antagonist capsazepine reverses mechanical hyperalgesia in models of inflammatory and neuropathic pain. J Pharmacol Exp Ther. 2003;304:56–62. doi: 10.1124/jpet.102.042010. [DOI] [PubMed] [Google Scholar]
- Witte et al., 2002.Witte DG, Cassar SC, Masters JN, Esbenshade T, Hancock AA. Use of a fluorescent imaging plate reader-based calcium assay to assess pharmacological differences between the human and rat vanilloid receptor. J Biomol Screening. 2002;7:466–475. doi: 10.1177/108705702237679. [DOI] [PubMed] [Google Scholar]
- Woolf et al., 1994.Woolf CJ, Safieh-Garabedian B, Ma QP, Crilly P, Winter J. Nerve growth factor contributes to the generation of inflammatory sensory hypersensitivity. Neuroscience. 1994;62:327–331. doi: 10.1016/0306-4522(94)90366-2. [DOI] [PubMed] [Google Scholar]
- Yaksh and Rudy, 1976.Yaksh TL, Rudy TA. Chronic catheterization of the spinal subarachnoid space. Physiol Behav. 1976;17:1031–1036. doi: 10.1016/0031-9384(76)90029-9. [DOI] [PubMed] [Google Scholar]