

Abstract
Mutations in the genes for amyloid precursor protein (APP) and presenilins (PS1, PS2) increase production of β-amyloid 42 (Aβ42) and cause familial Alzheimer's disease (FAD). Transgenic mice that express FAD mutant APP and PS1 overproduce Aβ42 and exhibit amyloid plaque pathology similar to that found in AD, but most transgenic models develop plaques slowly. To accelerate plaque development and investigate the effects of very high cerebral Aβ42 levels, we generated APP/PS1 double transgenic mice that coexpress five FAD mutations (5XFAD mice) and additively increase Aβ42 production. 5XFAD mice generate Aβ42 almost exclusively and rapidly accumulate massive cerebral Aβ42 levels. Amyloid deposition (and gliosis) begins at 2 months and reaches a very large burden, especially in subiculum and deep cortical layers. Intraneuronal Aβ42 accumulates in 5XFAD brain starting at 1.5 months of age (before plaques form), is aggregated (as determined by thioflavin S staining), and occurs within neuron soma and neurites. Some amyloid deposits originate within morphologically abnormal neuron soma that contain intraneuronal Aβ. Synaptic markers synaptophysin, syntaxin, and postsynaptic density-95 decrease with age in 5XFAD brain, and large pyramidal neurons in cortical layer 5 and subiculum are lost. In addition, levels of the activation subunit of cyclin-dependent kinase 5, p25, are elevated significantly at 9 months in 5XFAD brain, although an upward trend is observed by 3 months of age, before significant neurodegeneration or neuron loss. Finally, 5XFAD mice have impaired memory in the Y-maze. Thus, 5XFAD mice rapidly recapitulate major features of AD amyloid pathology and may be useful models of intraneuronal Aβ42-induced neurodegeneration and amyloid plaque formation.
Keywords: Alzheimer's disease, transgenic mice, Aβ42, amyloid plaques, intraneuronal Aβ, neuron loss
Introduction
Amyloid plaques, deposits of the β-amyloid (Aβ) peptide (Glenner and Wong, 1984), are defining lesions in Alzheimer's disease (AD) brain. A strong genetic association exists between early-onset familial forms of AD (FAD) and the 42 amino acid species of the Aβ peptide (Aβ42) (for review, see Hutton et al., 1998; Younkin, 1998; Sisodia et al., 1999). Aβ is a proteolytic product of the amyloid precursor protein (APP), and autosomal dominant mutations in the genes for APP, presenilin1 (PS1), and presenilin2 (PS2) increase production of Aβ42 and cause FAD. Aβ42 is more fibrillogenic than shorter Aβ peptides, and elevated Aβ42 concentrations are thought to drive the formation of the insoluble fibrils that compose amyloid plaques. The robust association of Aβ42 with FAD argues strongly in favor of a causative role for Aβ42 in the etiology of AD.
Aβ is cleaved from APP by the sequential action of two proteases, the β- and γ-secretases (for review, see Sisodia et al., 1999; Selkoe, 2001). Initially, the β-secretase cuts APP at the N terminus of the Aβ domain to produce the membrane-bound fragment C99 and the secreted APP ectodomain APPsβ. Subsequently, C99 is cleaved by the γ-secretase to generate the C terminus of Aβ. γ-Secretase cleavage is not precise and produces a series of Aβ peptides of 38–43 amino acids in length. Under normal conditions, a higher proportion of Aβ is produced as the 40 amino acid Aβ40, and less is made as Aβ42. FAD mutations in APP that cluster near the β- and γ-secretase cleavage sites either elevate the production of total Aβ, as is the case with the K670N/M671L substitution [Swedish mutation (Mullan et al., 1992a)], or increase the generation of Aβ42 specifically, as occurs with the I716V [Florida mutation (Eckman et al., 1997)] and V717I [London mutation (Goate et al., 1991)] substitutions. PS1 and PS2 appear to be subunits of the γ-secretase complex (Esler and Wolfe, 2001), and presenilin FAD mutations also elevate Aβ42 generation specifically.
Previous studies have suggested that Aβ42-elevating FAD mutations act in an additive manner to increase Aβ42 generation when incorporated within the same molecule. In particular, the intramolecular combination of I716V and V717I mutations within APP doubled Aβ42 production compared with the individual mutations (Eckman et al., 1997), as did the two PS1 FAD mutations M146L and L286V when introduced together into PS1 (Citron et al., 1998). Moreover, the intermolecular combination of Aβ42-elevating mutations in APP and PS1 also additively increased Aβ42 production (Citron et al., 1998).
To additively drive Aβ42 generation and create a very rapid AD amyloid model, we made transgenic mice that coexpress a total of five FAD mutations [APP K670N/M671L (Swedish) + I716V (Florida) + V717I (London) and PS1 M146L+ L286V]. These “5XFAD” transgenic mice develop cerebral amyloid plaques and gliosis at 2 months of age, achieve massive Aβ42 burdens, and have reduced synaptic markers, increased p25 levels, neuron loss, and memory impairment in the Y-maze. Interestingly, we observe accumulation of intraneuronal Aβ42 before plaques form. Intraneuronal Aβ is aggregated, and amyloid plaques appear to originate from neuron cell bodies with intraneuronal Aβ. Accumulation of intraneuronal Aβ has been observed in the brains of AD (Gouras et al., 2000; Takahashi et al., 2002; Cataldo et al., 2004; Takahashi et al., 2004), Down's syndrome (DS) (Gouras et al., 2000; Gyure et al., 2001; Busciglio et al., 2002; Mori et al., 2002; Cataldo et al., 2004), and AD mouse models (Takahashi et al., 2002; Oddo et al., 2003; Schmitz et al., 2004), implying potential involvement of intraneuronal Aβ in AD pathology (for review, see Echeverria and Cuello, 2002; Tseng et al., 2004; Gouras et al., 2005). Our results with 5XFAD mice suggest that intraneuronal Aβ aggregates may have a role in neurodegeneration, neuron loss, and amyloid plaque formation.
Materials and Methods
Generation of transgenic mice.
The FAD mutations APP K670N/M671L (770 residue isoform numbering) + I716V + V717I and PS1 M146L + L286V were introduced into APP(695) and PS1 cDNAs by site-directed mutagenesis, then subcloned into exon 2 of the mouse Thy1 transgene cassette (Moechars et al., 1996), and sequenced following standard procedures. The APP transgene includes the 5′ untranslated region, which contains a putative interleukin-1β (IL-1β) translational enhancer element (Rogers et al., 1999). After purification of the transgenes from vector sequence, the transgenes were added together in equal proportions and microinjected into the pronuclei of single-cell C57/B6XSJL hybrid embryos by the staff at the Northwestern University Transgenic and Targeted Mutagenesis Laboratory (Hogan et al., 1994; Jankowsky et al., 2001; Oddo et al., 2003). Founder transgenic mice were identified by PCR and confirmed by Southern blot analysis. The three highest APP and PS1 transgene coexpressing lines (Tg6799, Tg7031, and Tg 7092) were bred with B6/SJL F1 hybrids (Taconic Farms, Germantown, NY) for additional analysis.
Immunoblot analysis.
Mice were anesthetized with pentobarbital (50 mg/kg) and transcardially perfused with PBS (Invitrogen, Carlsbad, CA) buffer containing protease inhibitor cocktail (Calbiochem, La Jolla, CA). Brains were removed and bisected down the midline; one hemibrain was used for biochemical analysis and the other for histological analysis. For immunoblots and ELISAs, hemibrains were homogenized in four volumes of PBS (Invitrogen) containing protease inhibitor cocktail (Calbiochem). Brain homogenates were then aliquoted into two equal portions, one for immunoblot analysis and the other for ELISA. Two times concentrated radioimmunoprecipitation assay buffer (100 mm NaCl, 1% NP-40, 0.25% sodium deoxycholate, 1% SDS, 1 mm EDTA, and 50 mm Tris HCl, pH 7.4) was added to the homogenate for immunoblotting and briefly sonicated. Protein concentrations were determined using a BCA kit (Pierce, Rockford, IL), and 10–20 μg of protein was run on 10 or 10–14.5% Tris-HCl SDS-PAGE gels (Bio-Rad, Hercules, CA). After transfer onto polyvinylidene difluoride membrane, blots were incubated in the following primary antibodies: anti-APP (22C11; Chemicon, Temecula, CA), 1:10,000; anti-PS1 N terminal (PS1NT) (gift from Gopal Thinakaran, University of Chicago, Chicago, IL) (Thinakaran et al., 1998), 1:5000; and anti-actin (Sigma, St. Louis, MO), 1:10,000. Peroxidase-conjugated secondary antibodies were applied, and immunoblot signals were detected by enhanced chemiluminescence [Amersham Biosciences (Arlington Heights, IL) or Pierce] and quantified using an Eastman Kodak (Rochester, NY) Image Analyzer.
For neuron marker and neurodegeneration pathway immunoblots, cortex homogenates were prepared in 1% Triton X-100, PBS, protease inhibitor cocktail (Calbiochem), and Halt Phosphatase Inhibitor Cocktail (Pierce). Protein concentrations were determined using a BCA kit (Pierce), and 5–15 μg of protein was run on 4–12% Bis–Tris gels (Invitrogen), immunoblotted as above, and incubated with the following antibodies: postsynaptic density-95 (PSD-95) (Chemicon), 1:1000; syntaxin (Chemicon), 1:1000; and p35 (Santa Cruz Biotechnology, Santa Cruz, CA), 1:1000. Detection was performed as described above.
Aβ40 and Aβ42 ELISAs.
For Aβ ELISAs, PBS/protease inhibitor brain homogenates were supplemented with guanidine HCl to a final concentration of 5 m, sonicated briefly, and serially diluted with ELISA sample buffer. Final guanidine HCl concentrations were below 0.1 m. Protein concentrations were determined using a BCA kit (Pierce). Sample duplicates were then run on Aβ40 and Aβ42 specific sandwich colorimetric ELISAs following the protocol of the manufacturer (BioSource, Camarillo, CA). Optical densities at 450 nm of each well were read on a Victor 1420 plate reader (Wallac, Gaithersburg, MD), and sample Aβ40 and Aβ42 concentrations were determined by comparison with the Aβ40 and Aβ42 standard curves. All readings were in the linear range of the assay. Finally, sample Aβ40 and Aβ42 concentration values were normalized to total brain protein concentrations and expressed in nanograms of Aβ per milligram total protein. The average of the duplicates was determined, and then the mean and SEM for each age of a given line was calculated.
Synaptophysin ELISA.
Synaptophysin levels in whole-brain homogenates were quantified with a capture ELISA as described previously (Schlaf et al., 1998), with the following modifications. The SY38 monoclonal synaptophysin antibody (Chemicon) was used to coat plates at the concentration of 1.75 μg/ml, whereas a rabbit polyclonal synaptophysin antibody (0.35 μg/ml; Calbiochem) and an HRP-conjugated goat anti-rabbit IgG antibody (1:10,000; Jackson ImmunoResearch, West Grove, PA) were used for detection with a tetramethylbenzidine development system (Sigma). Because of the lack of purified synaptophysin, a mouse corticohippocampal homogenate served as a standard, as described previously (Craft et al., 2004).
Histology and immunostaining.
After perfusion and dissection, one hemibrain of each mouse was fixed in paraformaldehyde/PBS and cryopreserved in 30% sucrose/PBS containing 0.01% sodium azide. Brains were sectioned sagittally on a freezing microtome at 30 μm. Serial sections were collected in 0.1 m phosphate buffer, pH 7.6. Free-floating sections (in most cases adjacent) were then incubated overnight at room temperature with the following primary antibodies: rabbit anti-Aβ42 (1:2500; BioSource), rabbit anti-Aβ40 (1:2500; BioSource), biotinylated mouse anti-Aβ (4G8, 1:10,000; Signet Laboratories, Dedham, MA), rabbit anti-Aβ (R1282, 1:500; gift from Dennis Selkoe, Harvard Medical School, Brigham & Women's Hospital, Boston, MA), rabbit anti-GFAP (G9269, 1:10,000; Sigma), rat anti-F4/80 (MCAP497, 1:10,000; Serotec, Oxford, UK), and anti-neuronal-specific nuclear protein (NeuN) (1:500; Sigma). After washes, the sections were incubated with secondary biotinylated goat anti-rabbit or goat anti-mouse at 1:2000 for 2 h at room temperature. The Vector Laboratories (Burlingame, CA) ABC kit was used with DAB as chromogen to visualize the reaction product. The sections were then mounted on charged slides, counterstained with hematoxylin, dehydrated in a series of alcohols, cleared in xylene, and coverslipped. In some cases, after primary antibody incubation, sections were prepared for immunofluorescence microscopy by incubating with FITC- or tetramethylrhodamine isothiocyanate-labeled goat anti-rabbit or goat anti-mouse secondary antibodies. Thioflavin S or thiazin red staining was also used to visualize amyloid. Cresyl violet was used to stain Nissl substance for visualizing neurons for estimating neuronal loss. Light microscopy was performed on a Nikon (Tokyo, Japan) E800 microscope with a Spot Advanced digital camera for capturing images, and confocal images were captured on a LSM510 META system (Zeiss, Oberkochen, Germany).
Spontaneous alternation Y-maze task.
Spontaneous alternation performance was tested as described previously (Ohno et al., 2004). Each mouse was placed in the center of the symmetrical Y-maze and was allowed to explore freely through the maze during an 8 min session. The sequence and total number of arms entered was recorded. Experiments were done blind with respect to the genotype of the mice. Percentage alternation is as follows: number of triads containing entries into all three arms/maximum possible alternations (the total number of arms entered − 2) × 100.
Results
Transgenic strategy and generation of 5XFAD mice
The majority of AD transgenic mouse models take ∼6–12 months, or longer, to form amyloid plaques (for review, see Spires and Hyman, 2005). Because many types of investigations would be facilitated by more rapid plaque development, we endeavored to accelerate amyloid deposition by combining multiple FAD mutations in transgenic mice. Previous studies have shown that mice coexpressing FAD mutant APP and PS1 transgenes exhibit additive increases in Aβ42 production (Borchelt et al., 1996, 1997; Citron et al., 1997; Holcomb et al., 1998), although rarely have more than two Aβ42-elevating FAD mutations been combined in transgenic animals. Intrigued by the reports that intramolecular combination of FAD mutations in either APP or PS1 act additively to increase Aβ42 generation (Eckman et al., 1997; Citron et al., 1998), we established transgenic mouse lines that co-overexpress human APP695 with the Swedish (K670N, M671L), Florida (I716V), and London (V717I) FAD mutations in the same APP molecule and human PS1 harboring two FAD mutations, M146L and L286V (Fig. 1 A). The Swedish mutation increases the production of total Aβ, whereas APP Florida and London, and PS1 M146L and L286V, specifically increase the production of Aβ42 (for review, see Sisodia et al., 1999; Selkoe, 2001). From the previous studies, it seemed likely that coexpression of these Aβ42-elevating mutations, in both intermolecular and intramolecular combination, would increase Aβ42 generation in an additive manner in transgenic mice. Because Aβ42 is thought to drive amyloid plaque formation and AD pathogenesis, we reasoned that these transgenics might exhibit more rapid amyloid deposition than previous models and allow us to investigate the overall effects of very high Aβ42 levels in the brain.
Figure 1.

5XFAD transgenes and expression levels. A, Schematic diagram of 5XFAD APP and PS1 transgenes. FAD mutations in Thy1–APP and Thy1–PS1 transgenes are indicated by arrowheads. Sw, Swedish mutation; Lon, London mutation; Fl, FL mutation. Small gray rectangles represent Thy1 exons. Coding regions of APP and PS1 are represented by larger rectangles and are shown inserted into Thy1 exon 2 of the mouse Thy1 transgene expression cassette (Moechars et al., 1996). The APP transgene has the 5′ untranslated region (5′UTR) of APP containing an IL-1β responsive translational element (Rogers et al., 1999). B, Immunoblot analysis of 5XFAD lines showing expression levels of transgene-derived APP and PS1. Whole-brain homogenate (15 μg) from 6-month-old 5XFAD lines (Tg6799, Tg7092, and Tg7031), Tg2576, and nontransgenic controls were run on SDS-PAGE and immunoblotted, followed by detection with APP (22C11; top row) and PS1 (PS1NT; middle row) antibodies. Blots were stripped and incubated with actin antibody (bottom row) as a loading control. A representative selection of male and female mice for each line is shown. Note that 5XFAD lines express less APP than Tg2576, yet all develop amyloid deposits much earlier than Tg2576 (see Figs. 3, 4).
Transgenic lines were produced by pronuclear coinjection of APP and PS1 transgenes, each using the neuron-specific mouse Thy1 promoter to drive overexpression in the brain (Moechars et al., 1996). Previous work has demonstrated that coinjection results in efficient transgene cointegration, often together at the same genomic locus, thus enabling stable germ-line transmission and transgene coinheritance (Jankowsky et al., 2001; Oddo et al., 2003). Of 17 total founders, four were positive for the APP transgene only, two were positive for the PS1 transgene only, and 11 were positive for both APP and PS1 transgenes (data not shown). Transgenic offspring of most of the bigenic founders were double positive for APP and PS1 transgenes, indicating stable genomic cointegration and germ-line transmission of both transgenes. Because the bigenics carry five FAD mutations, we collectively refer to them as 5XFAD mice.
To determine the levels of APP and PS1 transgene expression, hemizygous transgenic offspring of the 5XFAD founders were killed, and brain homogenates were prepared for immunoblot analysis with anti-APP (22C11) and anti-PS1NT (Thinakaran et al., 1998) antibodies (Fig. 1 B). After initial analyses, we chose for additional study three APP and PS1 coexpressing 5XFAD lines that overexpress APP at high (Tg6799), medium (Tg7092), and low (Tg7031) levels. The well established Tg2576 mouse model of AD, which overexpresses APP with the Swedish mutation (Hsiao et al., 1996), was used as a control in these studies and makes slightly more APP than our highest expressing line, Tg6799. The three 5XFAD lines have been maintained as hemizygotes on a B6/SJL hybrid background for more than 10 generations and show stable germ-line transmission and expression of both APP and PS1 transgenes, demonstrating that the 5XFAD mice breed as single transgenics.
5XFAD mice rapidly accumulate massive Aβ42 levels in brain
To determine whether 5XFAD mice exhibited additively increased Aβ42 production, we measured cerebral levels of Aβ40 and Aβ42 in whole-brain homogenates using specific sandwich ELISAs. As predicted, the three 5XFAD transgenic lines had dramatically higher levels of Aβ42 than those of Aβ40 (Fig. 2). Aβ42 levels increased in a nearly linear manner as 5XFAD mice aged. The Aβ42 load and the rate of the Aβ42 increase were proportional to the level of APP transgene expression for a given line. For example, Tg6799 had the highest APP expression level, the greatest Aβ42 burden (∼140 ng/mg total brain protein), and the fastest rate of Aβ42 accumulation (Fig. 2A). In contrast, Tg7031 had the lowest levels of both APP and Aβ42 (∼60 ng/mg total brain protein) and had the slowest rate of Aβ42 increase (Fig. 2C).
Figure 2.
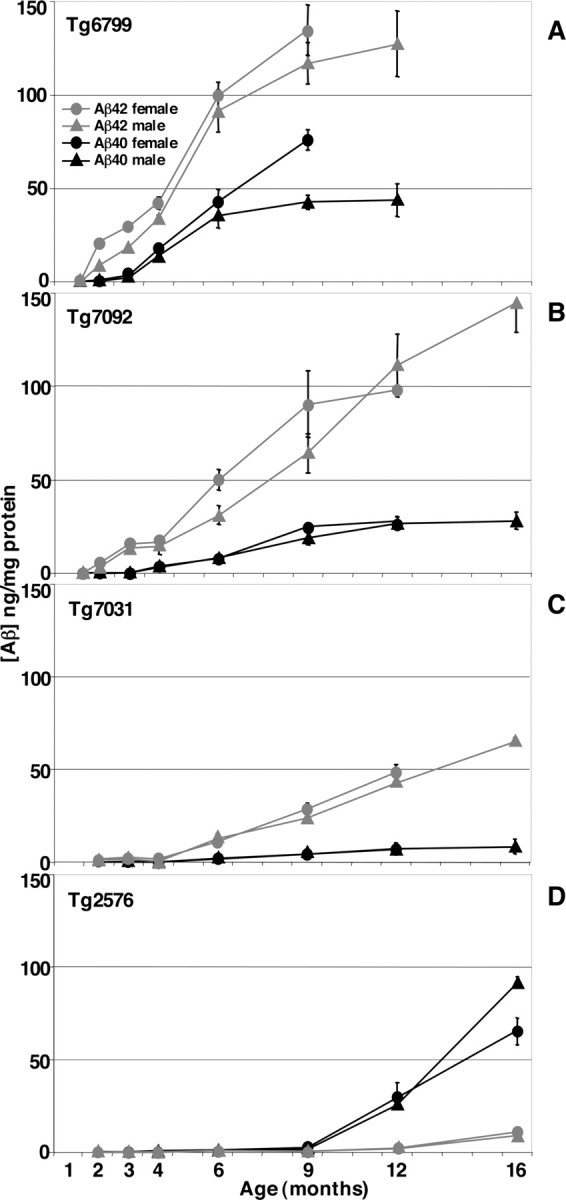
Rapid increase of cerebral Aβ42 levels in 5XFAD lines. Whole-brain homogenates from 1.5- to 16-month-old 5XFAD mice (A, Tg6799; B, Tg7092; C, Tg7031) and Tg2576 (D) were prepared in 5 m guanidine HCl to solubilize amyloid plaques, and 1 mg of total protein was analyzed by Aβ40 (black) and Aβ42 (gray) end-specific sandwich ELISAs. Aβ concentrations are expressed in nanograms per milligram of total brain protein. The means of male (triangles) and female (circles) samples are plotted separately. Four to five mice of each genotype per gender and age were analyzed. Error bars represent SEM. At young ages, 5XFAD mice generated Aβ42 almost exclusively. Aβ40 levels began to rise at older ages but were always much lower than Aβ42 levels. In contrast, for a given age, Tg2576 had very little Aβ42 compared with that of the 5XFAD lines, and Aβ40 levels were much greater than Aβ42 levels for Tg2576 at all ages. Note that the Aβ42 levels of the three 5XFAD lines are directly proportional to APP transgene expression levels (see Fig. 1B).
Cerebral Aβ42 levels began rising at very young ages in 5XFAD mice: 1.5, 2, and 4 months for Tg6799, Tg7092, and Tg7031, respectively (Fig. 2). For example, in the highest expressing line, Tg6799, at 1.5 months (the earliest age we tested), brain Aβ42 levels were ∼0.3–0.7 ng/mg protein (background is ∼0.1 ng/mg), but, by 2 months, Aβ42 levels rose to ∼9–21 ng/mg protein, a very steep increase in just ∼2 weeks. Interestingly, young 5XFAD mice produced Aβ42 almost exclusively. With age, Aβ40 levels also began to rise in transgenic brains, but the Aβ40 increase typically lagged ∼2–3 months behind the Aβ42 increase for a given line. In addition, the rate of the Aβ40 increase was less than that of Aβ42 for all lines. The Aβ42/Aβ40 ratios were very high in young mice, averaging ∼25, but decreased to ∼5 as a result of the increasing Aβ40 levels with age.
Levels of Aβ42 in the 5XFAD lines far exceeded those in Tg2576, which had ∼0.1–0.5 ng Aβ42/mg total brain protein up to 9 months, ∼2 ng Aβ42/mg total brain protein at 12 months, and ∼10 ng Aβ42/mg total brain protein at 16 months (Fig. 2D). Moreover, the Aβ42/Aβ40 ratio of Tg2576 was much lower than that of the 5XFAD lines, reaching a maximum of ∼0.1–0.2 at 9–16 months. Finally, we noted that young female Tg6799 and Tg7092 mice tended to have slightly higher Aβ42 levels than age-matched males, but this trend appeared to diminish at older ages. Together, our data demonstrate that 5XFAD mice accumulate massive amounts of cerebral Aβ42 at young ages and generate much more Aβ42 than Aβ40, suggesting cumulative effects of the five FAD mutations on Aβ42 production.
5XFAD mice display very early amyloid deposition and gliosis
Given the very high levels of Aβ42 that rapidly accumulated in 5XFAD brains, we anticipated that 5XFAD mice would show abundant amyloid deposition at young ages. Indeed, 5XFAD brain sections processed for histological detection of amyloid exhibited robust plaque deposition that was proportional to cerebral Aβ42 levels for a given line. Deposits were immunoreactive with 4G8 (anti-Aβtotal) (Fig. 3A–C), anti-Aβ42, and anti-Aβ40 end-specific antibodies (Fig. 4A–H) and were positive after staining with thioflavin S (Fig. 3E,F) and thiazin red (Fig. 3G), demonstrating that the deposits are composed of aggregated β-pleated sheet amyloid.
Figure 3.
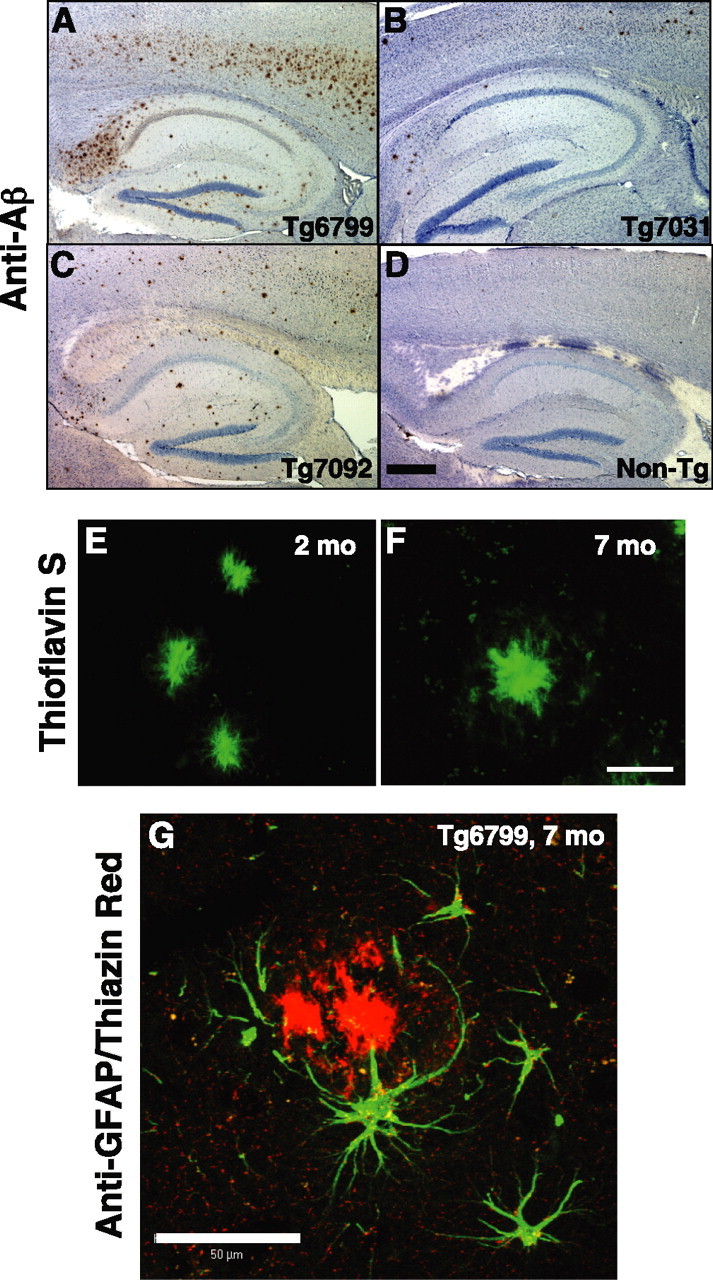
Amyloid deposition and gliosis in 5XFAD lines. Parasagittal serial sections of brains from Tg6799 (A, E–G), Tg7031 (B), Tg7092 (C), and nontransgenic control (D) mice were stained with anti-Aβ (4G8; A–D), thioflavin S (E, F), or anti-GFAP and thiazin red (G) and imaged by bright-field (A–D) or confocal (E–G) microscopy. Ages of mice were 2 (E), 6 (A–D), and 7 (F, G) months. A–D, Shown are sections including the hippocampus and cortex of representative 6-month-old 5XFAD mice from each line stained with anti-Aβ (brown) and hematoxylin (blue). Across the 5XFAD lines, the relative amounts of amyloid burden (and gliosis; data not shown) correlated with Aβ42 levels as measured by ELISA (Fig. 2). Scale bar (in D): A–D, 1 mm. E, F, Brain sections of representative 2-month-old (E) and 7-month-old (F) Tg6799 mice were stained with thioflavin S (green). Amyloid deposits labeled strongly with thioflavin S, indicating that Aβ was in an aggregated β-pleated sheet conformation. Note that plaque sizes (∼20 μm) are similar to those of large neuron cell bodies (data not shown). Scale bar (in F): E, F, 20 μm. G, Activated astrocytes around a Tg6799 plaque. Brain section of a representative 7-month-old Tg6799 mouse costained with thiazin red (to label β-pleated sheet amyloid) and anti-GFAP antibody (green). The astrocytes adjacent to the amyloid plaque display ramified processes and express GFAP, indicative of an activated phenotype associated with inflammation. Scale bar, 50 μm.
Figure 4.
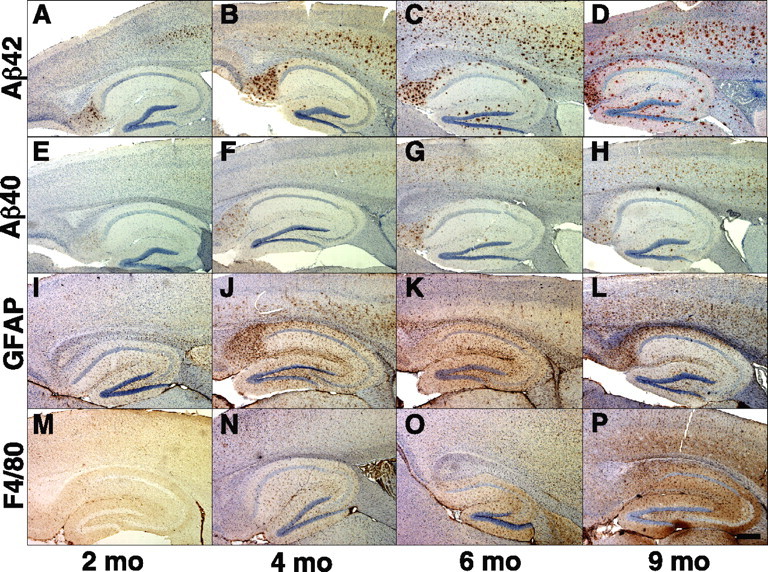
Rapid cerebral amyloid deposition and gliosis in 5XFAD line Tg6799. Parasagittal serial sections of brains from 2 (A, E, I, M), 4 (B, F, J, N), 6 (C, G, K, O), and 9 (D, H, L, P) month Tg6799 mice were stained with antibodies recognizing Aβ42 C terminus (A–D), Aβ40 C terminus (E–H), GFAP (I–L), and F4/80 (M–P) and counterstained with hematoxylin. Shown are sections including the hippocampus and cortex of representative Tg6799 mice from each age. Amyloid deposition began in the deep cortex and subiculum at 2 months in this line and then spread into other brain regions at older ages. Note that Aβ42 staining is much stronger than that of Aβ40 at every age and that the densities of astrocytes (GFAP) and microglia (F4/80) parallel that of the Aβ42 staining. Scale bar, 1 mm.
Tg6799, the line with the highest Aβ42 levels, showed the earliest amyloid deposition by anti-Aβ42 immunohistochemistry and thioflavin S staining, starting at 2 months (Figs. 3E, 4A) and following closely the initial rise in Aβ42 level by ELISA observed at 1.5 months (Fig. 2A). Deposition in Tg7092 and Tg7031 was delayed relative to that in Tg6799, beginning at 4 months (data not shown) and 6 months (Fig. 3B), respectively, reflecting the correspondingly lower Aβ42 levels for these lines. In contrast, Tg2576 mice did not begin to deposit amyloid until ∼9–12 months of age (data not shown) (Hsiao et al., 1996). As expected, the rate of deposition and the total amyloid burden in 5XFAD mice were directly proportional to cerebral Aβ42 levels for a given line; amyloid deposition increased rapidly in Tg6799 with age, reaching massive levels by 6 months (Fig. 4C), whereas the plaque loads in Tg7092 and Tg7031 increased more slowly and were quantitatively less than that of Tg6799 (data not shown). We also performed anti-Aβ40 immunohistochemistry on Tg6799 brain sections and observed substantially weaker Aβ40 immunoreactivity than that of Aβ42 in amyloid deposits (Fig. 4E–H), thus paralleling the lower Aβ40 levels as measured by ELISA (Fig. 2A).
All three 5XFAD lines had qualitatively similar spatial patterns of amyloid deposition in the brain. In young mice, plaques first appeared in deep layers of the cortex and in the subiculum (Fig. 4A,B). As mice aged, amyloid deposits spread to fill much of the cortex, subiculum, and hippocampus (Fig. 4C,D). Plaques were also observed in thalamus, brainstem, and olfactory bulb in older mice, but deposits were less numerous in these brain regions (data not shown). Amyloid deposition did not appear in the cerebellum of 5XFAD mice (data not shown). As expected, the spatial pattern of plaques in 5XFAD mice agreed well with those of previously reported Thy1 promoter-driven APP transgenic mice (Sturchler-Pierrat et al., 1997; Moechars et al., 1999; Oddo et al., 2003). Interestingly, amyloid deposits in 5XFAD mice were typically smaller (∼20 μm) but vastly more numerous than those observed in Tg2576 (data not shown), possibly because elevated Aβ42 levels may have resulted in greater plaque seed density in the 5XFAD lines. Together, our results demonstrate that the very high cerebral Aβ42 levels caused by the five FAD mutations drive the early formation of amyloid deposits that rapidly increase to large numbers, most notably in 5XFAD line Tg6799.
Neuroinflammation is a hallmark of AD brain and is characterized by the presence of activated astrocytes and microglia surrounding amyloid plaques (for review, see Akiyama et al., 2000). Similar to AD, APP transgenic mouse models have elevated inflammation markers and gliosis associated with amyloid deposits (Games et al., 1995; Hsiao et al., 1996; Sturchler-Pierrat et al., 1997). The 5XFAD lines also exhibited neuroinflammation, as demonstrated by astrogliosis and microgliosis (Figs. 3G, 4I–P). Immunostaining for glial activation markers GFAP (astrocytes) and F4/80 (microglia) revealed that gliosis was proportional to Aβ42 levels and amyloid deposition in 5XFAD brains and began at approximately the time when plaques initially appeared in each line. Tg6799 had GFAP-positive astrocytes and F4/80-positive microglia beginning at 2 months of age (Fig. 4I,M), whereas the appearance of activated glia in Tg7092 and Tg7031 started at 4 and 6 months, respectively (data not shown). The number of activated astrocytes and microglia increased with age for each line, paralleling the age-related rise in amyloid burden. By 9 months, Tg6799 had robust gliosis in cortex and hippocampus (Fig. 4L,P), whereas gliosis in Tg2576 did not become significant until after 9 months of age (data not shown). The spatial pattern of gliosis in 5XFAD brain closely followed the distribution of amyloid deposits (Fig. 4I–P). Similar to AD, GFAP-immunoreactive astrocytes were often observed surrounding amyloid deposits in 5XFAD brain (Fig. 3G). As a whole, our data indicate that 5XFAD mice display characteristics of neuroinflammation that appear similar to those observed in AD brain.
In addition to amyloid plaques, neurofibrillary tangles composed of hyperphosphorylated tau are characteristic lesions found in AD brain (Lee et al., 1991). To determine whether our 5XFAD mice exhibited abnormal tau phosphorylation similar to that observed in neurofibrillary tangles, we immunostained sections of Tg6799 brain from aged mice with antibody AT8 (Biernat et al., 1992). We did not observe significant staining of 5XFAD brain sections with this abnormal tau phosphorylation antibody (data not shown), but the presence of other phospho-tau epitopes has not yet been investigated. Similar to other APP single and APP/PS1 double-transgenic mice, we did not notice any neurofibrillary tangles in 5XFAD brain.
5XFAD mice have robust intraneuronal Aβ labeling
Recent studies have reported intraneuronal Aβ in AD, Down's syndrome, and APP transgenic brains (for review, see Echeverria and Cuello, 2002; Tseng et al., 2004; Gouras et al., 2005), suggesting that intraneuronal Aβ may play a role in AD pathology. In some cases, plaques in AD brain have appeared to originate from neuron cell bodies that contained intraneuronal Aβ (Gouras et al., 2000; D'Andrea et al., 2001). Interestingly, we also observed intraneuronal Aβ in the brains of 5XFAD mice, especially in the highest expressing line, Tg6799. Immunostaining of Tg6799 brain sections with anti-Aβ42 C terminus, 4G8, and R1282 (anti-Aβtotal) (Joachim et al., 1991) antibodies revealed strong subcellular labeling primarily within large pyramidal neurons of cortical layer 5 and the subiculum (Fig. 5A–C,G) (supplemental data, available at www.jneurosci.org as supplemental material). Anti-Aβ42 end-specific immunoreactivity was predominantly punctate, suggesting that Aβ42 accumulation occurred within vesicular compartments (Fig. 5A,C). The anti-Aβ42 C terminus antibody does not recognize full-length APP or APP C-terminal fragments (CTFs); therefore, the staining pattern accurately reflects Aβ42 intraneuronal localization. The intraneuronal Aβ immunostaining first appeared at 1.5 months of age in Tg6799 (Fig. 5A, inset), coinciding with the initial rise in cerebral Aβ42 levels and occurring immediately before the formation of amyloid deposits at 2 months in this line. Staining with 4G8 (Fig. 5B) and R1282 (Fig. 5G) (supplemental data, available at www.jneurosci.org as supplemental material) antibodies also showed punctate intraneuronal labeling, as well as diffuse lower-intensity staining in some neurons. In addition to total Aβ, 4G8 and R1282 also recognize full-length APP and APP CTFs, which may be responsible for this diffuse immunostaining. It is noteworthy that large 4G8-labeled intraneuronal accumulations were observed near the region of the axon hillock (Fig. 5B, black arrows).
Figure 5.

Intraneuronal Aβ accumulation in Tg6799: intracellular Aβ aggregates and plaque formation. Brain sections from representative 1.5- to 6-month-old Tg6799 mice were processed for immunocytochemistry with anti-Aβ antibodies (A–C, G) or stained with thioflavin S (D–F). Some sections were counterstained with hematoxylin (A, B) or with an antibody that recognizes the neuronal marker NeuN (green stain in C, G). Anti-Aβ antibodies used were specific for the C terminus of Aβ42 (A, brown; C, red) or recognized all forms of Aβ (B, 4G8, brown; G, R1282, red). Tg6799 ages were 1.5 (A, inset), 2 (A, C, D, F, G), 3 (E), and 6 (B) months. Intraneuronal Aβ accumulations were primarily observed as small puncta, although occasional large irregular accumulations were seen near the axon hillock (black arrows; B). Importantly, thioflavin S staining revealed intraneuronal Aβ aggregates consisting of β-pleated sheet amyloid accumulations within soma (punctate signals; D, E) and neurites (linear structures, white arrows; F). Note the thioflavin S-positive amyloid plaque (right; E). In some sections (G), an amyloid deposit (white arrowhead) was observed to originate from a neuron cell body with abnormal, disrupted-appearing morphology. This neuron had very little anti-NeuN antibody staining (green), indicating that it was likely in the process of degeneration. In addition, other neurons in the section appeared to be in various stages of degeneration (e.g., bottom right; G) and were also in close proximity to amyloid plaques (e.g., far left center; G). Note the presence of Aβ-filled neurites passing through the section (red linear structures, white arrows; G). Interestingly, plaque sizes were typically of the same magnitude as neuronal soma. Additional amyloid plaques can be seen at the bottom left in B and at the top right in G. For a movie of a three-dimensional reconstruction of the Z-stack sections in G, see supplemental material (available at www.jneurosci.org). Scale bars, 20 μm.
The observation of intense intraneuronal Aβ42 immunostaining in Tg6799 suggested that Aβ42 concentrations within intracellular puncta may have been high and implied the possibility that intraneuronal Aβ42 could have been aggregated. To investigate this hypothesis, we examined thioflavin S-stained Tg6799 brain sections for labeled intraneuronal puncta. Indeed, thioflavin S staining revealed a punctate, subcellular pattern within cell bodies of large pyramidal neurons of Tg6799 brain (Fig. 5D,E), demonstrating that intraneuronal Aβ42 had formed β-pleated sheet aggregates. These thioflavin S-positive puncta were of similar number and spatial pattern as the intraneuronal Aβ42-immunostained puncta and were not observed in negative control mice (data not shown). The approximate 1:1 ratio of thioflavin S-positive and Aβ42-positive puncta suggested that much of the intraneuronal Aβ42 was in an aggregated state. In addition to puncta within neuron cell bodies, thioflavin S-positive linear structures appearing to be neurites were observed in Tg6799 cortex (Fig. 5F). Together, these observations suggest that intraneuronal Aβ42 concentrations are high enough in intracellular compartments to form aggregates within the cell bodies and neurites of large pyramidal neurons in Tg6799 mice.
The presence of thioflavin S-positive intracellular Aβ42 aggregates within Tg6799 neurons suggested the possibility of an “inside-out” process in which intraneuronal Aβ42 aggregates lead to neurodegeneration and amyloid plaque formation. If so, we reasoned that we might observe neurons at a stage of degeneration with plaques appearing to originate from cell bodies. Indeed, Tg6799 brain sections stained with anti-Aβ antibody occasionally showed intraneuronal Aβ-containing soma in close physical association with amyloid deposits (Fig. 5G, arrowhead) (supplemental plaque seed movie, available at www.jneurosci.org as supplemental material). These neurons stained weakly with antibodies that recognize the neuronal marker NeuN and had abnormal morphologies, suggesting that the neurons were in the process of degeneration. Plaque-associated neurons were relatively rare within Tg6799 brain sections, implying that only a few neurons had made the transition to this stage at the time of fixation.
In support of the hypothesis that some plaques may form from intraneuronal Aβ aggregates within degenerating neurons, we observed that the highest density of amyloid deposits occurred in deep cortical layers and subiculum, the same regions that had the greatest number of intraneuronal Aβ-containing neurons in Tg6799 brain. Some neurites were also labeled with anti-Aβ antibody (Fig. 5G, arrows). As a whole, our results show that (1) 5XFAD mice accumulate intraneuronal Aβ before the start of amyloid deposition, (2) intraneuronal Aβ aggregates occur mainly in large pyramidal neurons of deep cortical layers and subiculum, and (3) some amyloid plaques appear to originate from intracellular Aβ-containing neurons that have abnormal, degenerated morphologies.
5XFAD mice exhibit neurodegeneration and neuron loss
Given our observations that some 5XFAD neurons contained intraneuronal Aβ aggregates, displayed disrupted-appearing morphologies, and were in close physical contact with amyloid deposits, we hypothesized that intraneuronal Aβ might be associated with neurodegeneration and neuron loss in 5XFAD brain. To initially investigate this possibility, we measured synaptophysin levels in whole-brain homogenates of Tg6799 mice by ELISA, which has greater accuracy and precision than immunoblot analysis. We observed that whole-brain synaptophysin levels decreased in Tg6799 brains beginning at 4 months of age and that, by 9 months, synaptophysin levels were ∼75% of those in age-matched nontransgenic controls (Fig. 6A), demonstrating significant degeneration of synapses in 5XFAD mice. To confirm these results, we performed immunoblot analysis of homogenates from cortexes of 3-, 9-, and 12-month-old Tg6799 mice with antibodies against syntaxin and PSD-95 (Fig. 6B,C) and found that the levels of these synaptic markers were also reduced at 9 and 12 months of age compared with control. Together, these results demonstrate synaptic degeneration in Tg6799 brain.
Figure 6.

Reduced synaptophysin, syntaxin, and PSD-95 levels in Tg6799: synaptic degeneration. A, Whole-brain homogenates from 2- to 12-month-old Tg6799 and 9-month-old nontransgenic control (9C) mice were analyzed for synaptophysin by ELISA. Synaptophysin levels are expressed as the percentage of 9-month-old control whole-brain synaptophysin levels (9C is 100%). No significant gender differences were observed in Tg6799 synaptophysin levels at any given age (data not shown), so values from age-matched males and females were pooled (3 mice for each gender and age). Note the steady decline in synaptophysin levels starting at 4 months in Tg6799, eventually falling to ∼75% of control levels at 9 months. B, C, Immunoblot analysis of synaptic markers syntaxin and PSD-95 in cortex homogenates (15 mg) from Tg6799 (Tg) and nontransgenic (wild type, WT) mice (3, 9, and 12 months old). Equal loading was determined by ponceau red staining of the blots (data not shown). Immunoblot signals were captured using a PhosphorImager (B), quantified, and expressed as percentage of age-matched control (C). Note that synaptic markers are significantly reduced by 9 months of age in Tg6799, indicating neurodegeneration. Error bars represent SEM.
Because of the spatial pattern of intraneuronal Aβ, we suspected that degeneration in Tg6799 may be restricted to specific neuronal populations. We reasoned that we might observe loss of certain neuron subpopulations in Tg6799 brain and that loss would most likely occur in the intracellular Aβ-containing neuron populations. To test this, we stained brain sections of 9-month-old Tg6799 and nontransgenic controls with cresyl violet and examined them for evidence of neuron loss. As predicted, we found that large pyramidal neurons in cortical layer 5 were visibly reduced in number in Tg6799 compared with control (Fig. 7A–D). Moreover, cortical layer 1 was significantly thinner in Tg6799 relative to control (Fig. 7A,B), most likely attributable to the loss of layer 5 neuron dendrites that project to and ramify in layer 1. In addition, neurons in Tg6799 subiculum appeared very pale or were missing entirely (Fig. 7F). Neurons in other cortical layers, hippocampus, and elsewhere in the brain appeared relatively intact by visual inspection, although we did not perform neuron counting and it is possible that additional neuron populations were also affected.
Figure 7.
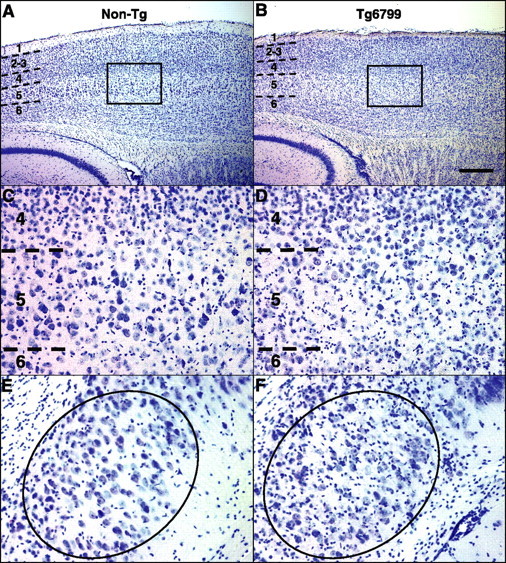
Neuron loss in cortical layer 5 and subiculum of Tg6799. Parasagittal sections of representative 9-month-old nontransgenic (Non-Tg) control (A, C, E) and Tg6799 (B, D, F) brains were stained with cresyl violet and micrographed to image layer 5 cortex (A–D) and subiculum (E, F). Numbers indicate cortical layers, and dashed lines identify boundaries between layers. Boxes in A and B delineate areas of cortex shown at higher magnification in C and D, respectively, to clearly visualize layer 5. B, D, Note that large pyramidal neurons in cortical layer 5 are visibly reduced in number in Tg6799. Moreover, layer 1 of Tg6799 cortex (B) is thinner than that of nontransgenics (A). Neuron loss is also visible in Tg6799 subiculum (oval, F) compared with that of nontransgenics (oval, E). In addition, note the large number of glia in Tg6799 subiculum (small darkly staining cells, F). Neurons are distinguishable from glial cells because of the larger size of neuronal cell body and the usual appearance of a prominent nuclear nucleolus. Layer 5 and subiculum exhibited the highest levels of both intraneuronal Aβ accumulation and amyloid deposition in Tg6799 (for Aβ42 immunostaining of a section adjacent to that in B, see Fig. 4D) Scale bar (in B): A, B, 1 mm; C–F, 100 μm.
It is important to note that the neuron loss in cortical layer 5 and subiculum occurred in the brain regions with the highest levels of both intraneuronal Aβ accumulation and amyloid plaque burden (Fig. 4D). We also observed that glial cells, identified by small nuclei that stained darkly with cresyl violet, were present in much larger numbers throughout the plaque-bearing regions of Tg6799 brain (Fig. 7F) compared with nonplaque regions and control. Neurons were easily distinguished from glial cells based on the larger neuron cell body and the prominent nuclear nucleolus. Together, our results indicate that 5XFAD mice exhibit significant neurodegeneration and loss of large pyramidal neurons, which appear to correlate with intraneuronal Aβ and amyloid plaques, respectively.
To gain insight into the molecular mechanism of synaptic degeneration and neuron loss in Tg6799, we performed immunoblot analysis of cortex homogenates from 3-, 9-, and 12-month-old mice using an antibody against the neurodegeneration pathway marker p25. We found that levels of p25 were increased to ∼150% of control in Tg6799 cortex at 9 and 12 months, with an upward trend at 3 months when intraneuronal Aβ aggregates were present (Fig. 8A,B). Thus, elevated p25 correlates with synaptic degeneration and neuron loss in 5XFAD brain and may be associated with intraneuronal Aβ aggregates.
Figure 8.

p25 levels are increased in Tg6799 mice. Immunoblot analysis of the cdk5 activating subunit p25 in cortex homogenates (15 mg) from Tg6799 (Tg) and nontransgenic (wild-type, WT) mice (3, 9, and 12 months old). Equal loading was determined by ponceau red staining of the blots (data not shown). Immunoblot signals were captured using a PhosphorImager (A), quantified, and expressed as percentage of age-matched control (B). Note that p25 levels are significantly elevated by 9 months of age in Tg6799 but that a trend toward higher values can be seen at 3 months. Error bars represent SEM.
5XFAD mice develop spatial memory deficits
AD is an amnestic disorder, and it has been shown that high cerebral Aβ levels correlate with memory impairments in APP transgenic mice (for review, see Ashe, 2001). To determine whether our 5XFAD mice also develop behavioral deficits, we tested 2-month-old and 4- to 5-month-old Tg6799 mice for spontaneous alternation performance in the Y-maze (Ohno et al., 2004). This learning task does not involve any training, reward, or punishment and allowed us to assess hippocampus-dependent spatial working memory in Tg6799. At 2 months of age, spontaneous alternation in the Y-maze for Tg6799 was similar to that for age-matched nontransgenic littermates. However, by 4–5 months of age, Tg6799 mice were significantly impaired in this task compared with nontransgenic control mice (Fig. 9A). The number of arms entered during the test was comparable for Tg6799 and control mice at both ages, indicating that Tg6799 had normal motor function and exploratory activity (Fig. 9B). Tg6799 mice have also been tested in trace and contextual fear conditioning paradigms and the Morris water maze, and they also exhibited deficits in these other hippocampus-dependent memory tasks (Ohno et al., 2006). These results demonstrate that, similar to other AD transgenic models, 5XFAD mice develop memory deficits with age.
Figure 9.

Memory deficits in Tg6799 mice with age. A, Spatial working memory of Tg6799 and nontransgenic littermate (Wild-type) mice was assessed by spontaneous alternation in the Y-maze (n = 12–20). Tg6799 mice showed normal alternation performance in the Y-maze at 2 months of age, but they performed poorly at 4–5 months of age (only slightly >50% chance levels) compared with nontransgenic littermate mice (F(3,62) = 13.96; p < 0.01). B, The total number of arm entries was not different between Tg6799 and nontransgenic (Wild-type) control mice, indicating similar levels of motor and exploratory activity in the Y-maze (n = 12–21). Each column represents the mean ± SEM. **p < 0.01 versus nontransgenic compared by ANOVA and post hoc Fisher's PLSD test.
Discussion
5XFAD mice: rapid amyloid pathology models
To substantially accelerate amyloid deposition in vivo, we made transgenic mouse lines that co-overexpress APP with three FAD mutations (K670N/M671L, I716V, and V717I) and PS1 with two FAD mutations (M146L and L286V) with the neuron-specific Thy1 promoter. 5XFAD mice breed as single transgenics and produce massive levels of cerebral Aβ42, far exceeding those of Aβ40. Other AD transgenic mice have demonstrated that Aβ42-elevating FAD mutations in APP and PS1 additively increase Aβ42 production and accelerate amyloid deposition when in intermolecular combination (for review, see Spires and Hyman, 2005). Here, we show that FAD mutations when combined together both intramolecularly and intermolecularly act in an additive manner to markedly increase the generation of Aβ42 in vivo. As a consequence, 5XFAD mice develop AD-like amyloid pathology starting at a very young age (2 months, in line Tg6799). The 5XFAD pathologic phenotype consists of amyloid plaques, gliosis, neurodegeneration, and memory deficits. In addition, 5XFAD mice exhibit intraneuronal Aβ and obvious neuron loss, a fundamental characteristic of AD lacking in most AD transgenic models.
Recent studies have described intraneuronal Aβ accumulation in AD and DS brain and have reported that amyloid plaques appear to originate from neuron cell bodies or processes (for review, see Echeverria and Cuello, 2002; Tseng et al., 2004; Gouras et al., 2005). Some AD transgenic mice also display intraneuronal Aβ (Takahashi et al., 2002; Oddo et al., 2003; Schmitz et al., 2004). Importantly, 5XFAD intraneuronal Aβ aggregates into β-pleated sheet amyloid, as determined by thioflavin S staining, and it labels with anti-Aβ42 end-specific antibody and can appear physically associated with amyloid plaques originating from neurons, similar to AD and DS.
We did not expect 5XFAD mice to develop neurofibrillary tangles, because tangle pathology has not been reported in APP single- or APP/PS1 double-transgenic mice. However, unlike other APP transgenic mice, 5XFAD did not display abnormally phosphorylated tau epitopes (Sturchler-Pierrat et al., 1997). The reason for the lack of phospho-tau immunoreactivity in 5XFAD brain is unclear, and additional analyses of pathologic tau epitopes in 5XFAD are required. Transgenic mice expressing human tau with the P301L frontotemporal dementia mutation develop neurofibrillary tangles when coexpressing an FAD mutant APP transgene (Lewis et al., 2001; Oddo et al., 2003) or after injection with Aβ42 (Gotz et al., 2001), indicating that Aβ42 promotes tangle formation. Crosses of 5XFAD with P301L tau mice will be valuable for investigating the effects of very high Aβ42 levels on the development of neurofibrillary tangles.
Although 5XFAD mice recapitulate major characteristics of AD-relevant amyloid pathology, caveats must be considered when interpreting the 5XFAD phenotype and its relationship to human AD. First, no cases of AD are caused by multiple FAD mutations, and unanticipated effects on APP processing could potentially occur as a consequence of the mutation combination in 5XFAD mice. Second, the Aβ42/Aβ40 ratio appears to be higher in 5XFAD than in human AD, thus raising the possibility that a greater level of Aβ42 toxicity exists in 5XFAD than is typical in AD brain. In general, all AD transgenic mouse models face similar criticisms because of the overexpression of one or more FAD mutations, yet they have provided invaluable insight into AD pathogenesis and therapeutic strategies. The fact that 5XFAD amyloid pathology is similar to, but more rapid than, that found in AD and other AD transgenics indicates that 5XFAD mice are potentially relevant and useful models of AD-like amyloidosis, although, like all animal models, caution should be exercised when relating the 5XFAD phenotype to human AD.
Intraneuronal Aβ, neurodegeneration, and amyloid plaque formation
In the highest expressing 5XFAD line, Tg6799, intraneuronal Aβ is evident at 1.5 months of age, just before the first appearance of amyloid deposits at 2 months. In DS and APP transgenics, intraneuronal Aβ accumulation has also been observed before the formation of plaques (Gyure et al., 2001; Busciglio et al., 2002; Oddo et al., 2003; Cataldo et al., 2004; Schmitz et al., 2004), suggesting the possibility that build-up of Aβ within neurons may play a role in plaque formation and neurodegeneration. Importantly, intraneuronal Aβ42 was observed in the brains of young (∼3–4 years old) DS patients (Gouras et al., 2000; Mori et al., 2002), years before plaques develop. In Tg6799, intraneuronal Aβ accumulation is prominent within large pyramidal neurons of layer 5 cortex and subiculum, possibly a result of the Thy1 transgene promoter expression pattern (Feng et al., 2000). Much of the intraneuronal Aβ is within soma, although intracellular Aβ is also observed in neuronal processes. Immunoreactivity with anti-Aβ42 end-specific antibodies demonstrates that intraneuronal staining identifies bona fide Aβ42 rather than APP or APP CTFs.
Intraneuronal Aβ labeling in 5XFAD brain is primarily punctate, suggesting vesicular compartmentalization. We have not yet determined the subcellular compartments in which intraneuronal Aβ inclusions occur. Based on extensive intracellular localization studies of APP and the secretases, we speculate that aggregated intraneuronal Aβ likely occurs within acidic compartments such as endosomes. Interestingly, Takahashi et al. (2002) have identified multi-vesicular bodies, or late endosomes, as compartments in which intracellular Aβ accumulates in AD and APP transgenic brains. We also observe large intraneuronal Aβ accumulations occurring near the axon hillock. This latter observation, together with anti-Aβ antibody and thioflavin S staining in neurites, suggests that aggregated intraneuronal Aβ may interfere with axonal and/or dendritic transport. Future studies will determine the subcellular compartments in which Aβ accumulates and the effects of intraneuronal Aβ aggregates on intracellular trafficking in 5XFAD mice.
β-Sheet-structured Aβ may be neurotoxic (Urbanc et al., 2002), and several observations suggest that the thioflavin S-positive intraneuronal Aβ aggregates in 5XFAD mice may cause neurodegeneration: (1) presynaptic markers synaptophysin and syntaxin are reduced; (2) postsynaptic marker PSD-95 is reduced; (3) intraneuronal Aβ-containing neurons frequently exhibit weak NeuN immunostaining and abnormal morphologies and sometimes are physically associated with amyloid deposits; (4) large pyramidal neurons of layer 5 cortex and subiculum are markedly lost, exhibit prominent intraneuronal Aβ staining, and are located in regions of high plaque burden; and (5) p25 levels are increased. Importantly, p25 elevation may be an early event in 5XFAD brain, trending upward by 3 months of age and temporally coinciding with the accumulation of intraneuronal Aβ. By 9 months of age, the p25 increase in 5XFAD brain is significant and correlates with marked synaptic degeneration and neuron loss. Calpain cleavage generates p25, which in turn activates cyclin-dependent kinase 5 (cdk5), a potential player in the connection between Aβ toxicity, neurodegeneration, and neurofibrillary tangle formation in AD (for review, see Cruz and Tsai, 2004). Although our work does not unequivocally prove that intraneuronal Aβ aggregates cause neuronal degeneration, it provides initial insights into a potential molecular mechanism that may link intraneuronal Aβ and neurodegeneration. Taken as a whole, our results suggest that intraneuronal Aβ42 aggregates may play a role in neurodegeneration, neuron loss, and the formation of amyloid plaques.
Robust neuronal loss has not been observed in most APP transgenic mouse models. However, a recent APP/PS1 line coexpressing APP K670N/M671L plus V717I and PS1 M233T plus L235P (APPSLPS1KI) exhibits significant neuron death in CA1/2 of the hippocampus (Casas et al., 2004; Schmitz et al., 2004). Like 5XFAD mice, the neuron loss in APPSLPS1KI mice is preceded by intraneuronal Aβ, although intraneuronal Aβ does not appear to cause amyloid plaque formation in this model. Other APP transgenics also display intraneuronal Aβ but do not have significant neuron death (Takahashi et al., 2002; Oddo et al., 2003). Why intraneuronal Aβ does not always correlate with neuron loss and plaque formation in APP transgenics is unclear, but the reason may be related to the different combinations of FAD mutants, expression levels, and promoters used in each line. Regardless, neuron loss is correlated with previous appearance of thioflavin S-positive intraneuronal Aβ aggregates in both 5XFAD and APPSLPS1KI mice.
Lee et al. (2005) have shown that β-site APP-cleaving enzyme 1 (BACE1) overexpression in Tg2576 mice causes Aβ production to shift from the axonal compartment to the somatodendritic compartment in which it is efficiently degraded, thus reducing steady-state Aβ levels and plaque deposition. In contrast, we observe soma-derived deposits in 5XFAD brain, which may be a result of high Aβ42 generation in 5XFAD relative to Tg2576. It is well established that Aβ42 becomes aggregated more readily than Aβ40, especially under acidic conditions (as occurs in endosomes). We speculate that high Aβ42 concentrations within acidic intracellular compartments could lead to Aβ42 aggregation, which in turn may impair Aβ-degrading mechanisms or inhibit Aβ secretion in the soma. As a result, Aβ42 aggregates may accumulate, damage membranes, cause neuron lysis, and form plaque seeds. It is conceivable that such conditions may occur in AD with Aβ42-elevating FAD mutations. Although it is unclear whether Aβ42 levels within sporadic AD neurons become high enough to allow intraneuronal Aβ42 aggregation and plaque formation, the observation that amyloid plaques appear to originate from degenerated neuron cell bodies in AD (Gouras et al., 2000; D'Andrea et al., 2001) is consistent with this hypothesis.
Recently, transgenic mice expressing a BRI protein–Aβ42 fusion construct that generates only Aβ42 in the brain have firmly established that Aβ42 is essential for parenchymal and vascular amyloid deposition (McGowan et al., 2005). In contrast to 5XFAD mice, little evidence of intraneuronal Aβ was found in BRI–Aβ42 transgenic brain. The absence of intraneuronal Aβ accumulation in BRI–Aβ42 mice may be related to the different processing and trafficking pathways that generate Aβ from BRI–Aβ42 fusion protein compared with full-length APP: BRI–Aβ42 is processed by furin in the trans-Golgi (potentially allowing efficient Aβ42 secretion), whereas APP is cleaved by secretases in endosomes (possibly favoring Aβ42 aggregation). Thus, intraneuronal Aβ42 accumulation may partly depend on the intracellular compartment in which Aβ42 is generated.
The apolipoprotein E4 (ApoE4) is a well established risk factor for AD that has been shown to enhance Aβ deposition in vivo (for review, see Bales et al., 2002), although the mechanisms involved are poorly understood and it is unknown whether ApoE isoform affects the aggregation of intraneuronal Aβ. ApoE is secreted by astrocytes and then is endocytosed by neurons in which it may encounter Aβ in endosomes. It is conceivable that, under conditions in which intraneuronal Aβ42 concentrations in endosomes are high, interaction between Aβ42 and ApoE4 may facilitate intracellular aggregation. Although the amount of extracellular ApoE in the brain is likely to far exceed that of intracellular ApoE, it is possible that local concentrations of endocytosed ApoE within intraneuronal compartments may reach high levels. Thus, ApoE isoform may affect both intracellular and extracellular Aβ aggregation. Crossing 5XFAD with different ApoE isoform transgenic mice will allow investigation of this question.
Plaques in 5XFAD mice are smaller, more numerous, and more densely packed than those in Tg2576. We propose that the very high Aβ42/Aβ40 ratio in 5XFAD may promote plaque nidus formation. Our results are consistent with those of Hyman et al. (1995) showing that higher Aβ production with normal Aβ42/Aβ40 ratio (e.g., DS) causes larger plaque size, whereas greater plaque initiation (e.g., AD with ApoE4 or Aβ42-elevation mutations) favors higher plaque number.
It should be stressed that multiple pathways of amyloid plaque formation are likely to exist, in addition to the one involving intraneuronal Aβ aggregates in soma. For example, we also observe thioflavin S-positive Aβ aggregates within neurites of 5XFAD brain (Fig. 5F). It is not yet known whether these structures are axons or dendrites. Plaques are frequently seen in soma-poor regions of neuropil and white matter (e.g., stratum oriens and stratum radiatum of the hippocampus, molecular layer of the dentate gyrus, and fimbria) (Fig. 4D), consistent with the hypothesis that Aβ accumulation within neurites may contribute to degeneration and deposit formation (Takahashi et al., 2002). In addition, plaques appear to form from Aβ released at the neuron terminal (Lazarov et al., 2002; Sheng et al., 2002), considered to be a major site of Aβ generation and secretion. Our results do not exclude the possibility that a portion of plaques are derived from Aβ secreted at the terminal, and it is probable that both intraneuronal Aβ aggregates and extracellular Aβ may contribute to deposit formation in 5XFAD brain. As noted above, high Aβ42 production in 5XFAD neurons may enhance intracellular Aβ accumulation relative to Aβ secretion. Interestingly, a recent study has indicated an inverse dynamic relationship between intracellular and extracellular pools of Aβ that appears to influence deposit formation in 3XTg-AD mouse brain (Oddo et al., 2006). Thus, pathways leading to amyloid plaque formation are likely to be diverse and complex, possibly involving several distinct processes that occur in parallel during AD.
Memory impairment and intraneuronal Aβ
In the 3XTg-AD mouse model (Oddo et al., 2003), long-term retention memory deficits in the Morris water maze correlate with intraneuronal Aβ at 4 months, an age when mice do not have amyloid deposits (Billings et al., 2005). In contrast, 5XFAD mice do not have a Y-maze deficit at 2 months of age, although intraneuronal Aβ starts accumulating at 1.5 months. Unlike the water maze, the Y-maze task is not capable of assaying long-term memory retention, which may be responsible for the different behavioral results of the two models. Testing 2- to 4-month-old 5XFAD mice in the Morris water maze may reveal memory retention impairments that correlate with intraneuronal Aβ. Conversely, we note that the 5XFAD Y-maze deficit appears at 4–5 months of age and correlates with the initial decline in whole-brain synaptophysin levels, raising the possibility that 5XFAD Y-maze memory dysfunction may be related to neurodegeneration.
In a previous study using Tg2576 mice, we showed that ablation of Aβ by targeted BACE1 gene deletion rescued the Y-maze deficit of young pre-plaque Tg2576, suggesting that soluble Aβ assemblies or oligomers, but not amyloid plaques, may be responsible for poor memory performance of Tg2576 (Ohno et al., 2004). However, we did not explore whether intraneuronal Aβ may have been involved in Tg2576 memory dysfunction. Multiple forms of Aβ are present in 4- to 5-month-old 5XFAD mice, including amyloid plaques, intraneuronal Aβ aggregates, and Aβ oligomers (Ohno et al., 2006). Therefore, the exact form(s) of Aβ responsible for the poor performance of 5XFAD in the Y-maze is currently unknown and requires additional analysis. Finally, we note that, in addition to the Y-maze, 5XFAD mice also exhibit memory impairments in trace and contextual fear conditioning tests and the Morris water maze at 4–6 months of age (younger mice were not tested) (Ohno et al., 2006). Thus, in a variety of behavioral paradigms, 5XFAD mice display hippocampus-dependent memory deficits similar to those of other AD transgenic models.
In conclusion, 5XFAD mice are very rapid-onset amyloid plaque models that recapitulate major pathologic and behavioral characteristics of AD. Importantly, 5XFAD mice provide evidence that intraneuronal Aβ aggregates may play a role in neurodegeneration, neuron loss, and amyloid plaque formation, suggesting that reduction of intraneuronal Aβ could be of potential therapeutic benefit for AD.
Footnotes
This work was supported by National Institute on Aging Grants P01 AG021184 (Project 1, L.V.E.; Project 4, R.V.; Histology Core, R.B.) and R01 AG022560 (R.V.) We are indebted to Lynn Doglio and her staff at the Northwestern Transgenic and Targeted Mutagenesis Laboratory for transgene microinjection and production of founder transgenic mice. We also thank the following individuals for their valuable input on this project: Fred Van Leuven for the Thy1 transgene cassette vector, Leong Chew for assistance with confocal imaging, Gopal Thinakaran for the PS1NT antibody, Dennis Selkoe for the R1282 antibody, and Marsel Mesulam, Richard Miller, Gunnar Gouras, Frank Laferla, and Sam Sisodia for thoughtful comments on this manuscript.
References
- Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, et al. Inflammation and Alzheimer's disease. Neurobiol Aging. 2000;21:383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashe KH. Learning and memory in transgenic mice modeling Alzheimer's disease. Learn Mem. 2001;8:301–308. doi: 10.1101/lm.43701. [DOI] [PubMed] [Google Scholar]
- Bales KR, Dodart JC, DeMattos RB, Holtzman DM, Paul SM. Apolipoprotein E, amyloid, and Alzheimer disease. Mol Interv. 2002;2:363–375. doi: 10.1124/mi.2.6.363. [DOI] [PubMed] [Google Scholar]
- Biernat J, Mandelkow E-M, Schroter C, Lichtenberg-Kraag B, Steiner B, Berling B, Meyer H, Mercken M, Vandermeeren M, Goedert M, et al. The switch of tau protein to an Alzheimer-like state includes the phosphorylation of two serine-proline motifs upstream of the microtubule binding region. EMBO J. 1992;11:1593–1597. doi: 10.1002/j.1460-2075.1992.tb05204.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billings LM, Oddo S, Green KN, McGaugh JL, Laferla FM. Intraneuronal Abeta causes the onset of early Alzheimer's disease-related cognitive deficits in transgenic mice. Neuron. 2005;45:675–688. doi: 10.1016/j.neuron.2005.01.040. [DOI] [PubMed] [Google Scholar]
- Borchelt DR, Thinakaran G, Eckman CB, Lee MK, Davenport F, Ratovitsky T, Prada CM, Kim G, Seekins S, Yager D, Slunt HH, Wang R, Seeger M, Levey AI, Gandy SE, Copeland NG, Jenkins NA, Price DL, Younkin SG, Sisodia SS. Familial Alzheimer's disease-linked presenilin 1 variants elevate Abeta1–42/1–40 ratio in vitro and in vivo. Neuron. 1996;17:1005–1013. doi: 10.1016/s0896-6273(00)80230-5. [DOI] [PubMed] [Google Scholar]
- Borchelt DR, Ratovitski T, van Lare J, Lee MK, Gonzales V, Jenkins NA, Copeland NG, Price DL, Sisodia SS. Accelerated amyloid deposition in the brains of transgenic mice coexpressing mutant presenilin 1 and amyloid precursor proteins. Neuron. 1997;19:939–945. doi: 10.1016/s0896-6273(00)80974-5. [DOI] [PubMed] [Google Scholar]
- Busciglio J, Pelsman A, Wong C, Pigino G, Yuan M, Mori H, Yankner BA. Altered metabolism of the amyloid beta precursor protein is associated with mitochondrial dysfunction in Down's syndrome. Neuron. 2002;33:677–688. doi: 10.1016/s0896-6273(02)00604-9. [DOI] [PubMed] [Google Scholar]
- Casas C, Sergeant N, Itier JM, Blanchard V, Wirths O, van der Kolk N, Vingtdeux V, van de Steeg E, Ret G, Canton T, Drobecq H, Clark A, Bonici B, Delacourte A, Benavides J, Schmitz C, Tremp G, Bayer TA, Benoit P, Pradier L. Massive CA1/2 neuronal loss with intraneuronal and N-terminal truncated Abeta42 accumulation in a novel Alzheimer transgenic model. Am J Pathol. 2004;165:1289–1300. doi: 10.1016/s0002-9440(10)63388-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cataldo AM, Petanceska S, Terio NB, Peterhoff CM, Durham R, Mercken M, Mehta PD, Buxbaum J, Haroutunian V, Nixon RA. Abeta localization in abnormal endosomes: association with earliest Abeta elevations in AD and Down syndrome. Neurobiol Aging. 2004;25:1263–1272. doi: 10.1016/j.neurobiolaging.2004.02.027. [DOI] [PubMed] [Google Scholar]
- Citron M, Westaway D, Xia W, Carlson G, Diehl T, Levesque G, Johnson-Wood K, Lee M, Seubert P, Davis A, Kholodenko D, Motter R, Sherrington R, Perry B, Yao H, Strome R, Lieberburg I, Rommens J, Kim S, Schenk D, Fraser P, St George Hyslop P, Selkoe D. Mutant presenilins of Alzheimer's disease increase production of 42-residue amyloid β-protein in both transfected cells and transgenic mice. Nat Med. 1997;3:67–72. doi: 10.1038/nm0197-67. [DOI] [PubMed] [Google Scholar]
- Citron M, Eckman CB, Diehl TS, Corcoran C, Ostaszewski BL, Xia W, Levesque G, St. George Hyslop P, S.G Y, Selkoe DJ. Additive effects of PS1 and APP mutations on secretion of the 42-residue amyloid beta-protein. Neurobiol Dis. 1998;5:107–116. doi: 10.1006/nbdi.1998.0183. [DOI] [PubMed] [Google Scholar]
- Craft JM, Watterson DM, Frautschy SA, Van Eldik LJ. Aminopyridazines inhibit beta-amyloid-induced glial activation and neuronal damage in vivo. Neurobiol Aging. 2004;25:1283–1292. doi: 10.1016/j.neurobiolaging.2004.01.006. [DOI] [PubMed] [Google Scholar]
- Cruz JC, Tsai LH. Cdk5 deregulation in the pathogenesis of Alzheimer's disease. Trends Mol Med. 2004;10:452–458. doi: 10.1016/j.molmed.2004.07.001. [DOI] [PubMed] [Google Scholar]
- D'Andrea MR, Nagele RG, Wang HY, Peterson PA, Lee DH. Evidence that neurones accumulating amyloid can undergo lysis to form amyloid plaques in Alzheimer's disease. Histopathology. 2001;38:120–134. doi: 10.1046/j.1365-2559.2001.01082.x. [DOI] [PubMed] [Google Scholar]
- Echeverria V, Cuello AC. Intracellular A-beta amyloid, a sign for worse things to come? Mol Neurobiol. 2002;26:299–316. doi: 10.1385/MN:26:2-3:299. [DOI] [PubMed] [Google Scholar]
- Eckman CB, Mehta ND, Crook R, Perez-tur J, Prihar G, Pfeiffer E, Graff-Radford N, Hinder P, Yager D, Zenk B, Refolo LM, Prada CM, Younkin SG, Hutton M, Hardy J. A new pathogenic mutation in the APP gene (I716V) increases the relative proportion of A beta 42(43) Hum Mol Genet. 1997;6:2087–2089. doi: 10.1093/hmg/6.12.2087. [DOI] [PubMed] [Google Scholar]
- Esler WP, Wolfe MS. A portrait of Alzheimer secretases-new features and familiar faces. Science. 2001;293:1449–1454. doi: 10.1126/science.1064638. [DOI] [PubMed] [Google Scholar]
- Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, Nerbonne JM, Lichtman JW, Sanes JR. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000;28:41–51. doi: 10.1016/s0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F, Guido T, Hagopian S, Johnson-Wood K, Khan K, Lee M, Leibowitz P, Lieberburg I, Little S, Masliah E, McConglue L, et al. Alzheimer-type neuroptahology in transgenic mice overexpressing V717F b-amyloid precursor protein. Nature. 1995;373:523–527. doi: 10.1038/373523a0. [DOI] [PubMed] [Google Scholar]
- Glenner GG, Wong CW. Alzheimer's disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun. 1984;120:885–890. doi: 10.1016/s0006-291x(84)80190-4. [DOI] [PubMed] [Google Scholar]
- Goate A, Chartier-Harlin MC, Mullan M, Brown J, Crawford F, Fidani L, Giuffra L, Haynes A, Irving N, James L, Mant R, Newton P, Rooke K, Roques P, Talbot C, Pericak-Vance M, Roses A, Williamson R, Rossor M, Owen M, Hardy J. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease. Nature. 1991;349:704–706. doi: 10.1038/349704a0. [DOI] [PubMed] [Google Scholar]
- Gotz J, Chen F, van Dorpe J, Nitsch RM. Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Aβ42 fibrils. Science. 2001;293:1491–1495. doi: 10.1126/science.1062097. [DOI] [PubMed] [Google Scholar]
- Gouras GK, Tsai J, Naslund J, Vincent B, Edgar M, Checler F, Greenfield JP, Haroutunian V, Buxbaum JD, Xu H, Greengard P, Relkin NR. Intraneuronal Abeta42 accumulation in human brain. Am J Pathol. 2000;156:15–20. doi: 10.1016/s0002-9440(10)64700-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouras GK, Almeida CG, Takahashi RH. Intraneuronal Abeta accumulation and origin of plaques in Alzheimer's disease. Neurobiol Aging. 2005;26:1235–1244. doi: 10.1016/j.neurobiolaging.2005.05.022. [DOI] [PubMed] [Google Scholar]
- Gyure KA, Durham R, Stewart WF, Smialek JE, Troncoso JC. Intraneuronal abeta-amyloid precedes development of amyloid plaques in Down syndrome. Arch Pathol Lab Med. 2001;125:489–492. doi: 10.5858/2001-125-0489-IAAPDO. [DOI] [PubMed] [Google Scholar]
- Hogan B, Beddington R, Costantini F, Lacy E. second Edition. Plainview, NY: Cold Spring Harbor Laboratory; 1994. Manipulating the mouse embryo: a laboratory manual. [Google Scholar]
- Holcomb L, Gordon MN, McGowan E, Yu X, Benkovic S, Jantzen P, Wright K, Saad I, Mueller R, Morgan D, Sanders S, Zehr C, O'Campo K, Hardy J, Prada CM, Eckman C, Younkin S, Hsiao K, Duff K. Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat Med. 1998;4:97–100. doi: 10.1038/nm0198-097. [DOI] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–103. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- Hutton M, Perez-Tur J, Hardy J. Genetics of Alzheimer's disease. Essays Biochem. 1998;33:117–131. doi: 10.1042/bse0330117. [DOI] [PubMed] [Google Scholar]
- Hyman BT, West HL, Rebeck GW, Buldyrev SV, Mantegna RN, Ukleja M, Havlin S, Stanley HE. Quantitative analysis of senile plaques in Alzheimer's disease: observation of log-normal size distribution and molecular epidemiology of differences associated with apolipoprotein E genotype and trisomy 21 (Down syndrome) Proc Natl Acad Sci USA. 1995;92:3586–3590. doi: 10.1073/pnas.92.8.3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowsky JL, Slunt HH, Ratovitski T, Jenkins NA, Copeland NG, Borchelt DR. Co-expression of multiple transgenes in mouse CNS: a comparison of strategies. Biomol Eng. 2001;17:157–165. doi: 10.1016/s1389-0344(01)00067-3. [DOI] [PubMed] [Google Scholar]
- Joachim CL, Games D, Morris J, Ward P, Frenkel D, Selkoe D. Antibodies to non-beta regions of the beta-amyloid precursor protein detect a subset of senile plaques. Am J Pathol. 1991;138:373–384. [PMC free article] [PubMed] [Google Scholar]
- Lazarov O, Lee M, Peterson DA, Sisodia SS. Evidence that synaptically released beta-amyloid accumulates as extracellular deposits in the hippocampus of transgenic mice. J Neurosci. 2002;22:9785–9793. doi: 10.1523/JNEUROSCI.22-22-09785.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EB, Zhang B, Liu K, Greenbaum EA, Doms RW, Trojanowski JQ, Lee VM. BACE overexpression alters the subcellular processing of APP and inhibits Abeta deposition in vivo. J Cell Biol. 2005;168:291–302. doi: 10.1083/jcb.200407070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee VM-Y, Balin BJ, Otvos L, Trojanowski JQ. A major subunit of paired helical filaments and derivatized forms of normal tau. Science. 1991;251:675–678. doi: 10.1126/science.1899488. [DOI] [PubMed] [Google Scholar]
- Lewis J, Dickson DW, Lin WL, Chisholm L, Corral A, Jones G, Yen SH, Sahara N, Skipper L, Yager D, Eckman C, Hardy J, Hutton M, McGowan E. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science. 2001;293:1487–1491. doi: 10.1126/science.1058189. [DOI] [PubMed] [Google Scholar]
- McGowan E, Pickford F, Kim J, Onstead L, Eriksen J, Yu C, Skipper L, Murphy MP, Beard J, Das P, Jansen K, Delucia M, Lin WL, Dolios G, Wang R, Eckman CB, Dickson DW, Hutton M, Hardy J, Golde T. Abeta42 is essential for parenchymal and vascular amyloid deposition in mice. Neuron. 2005;47:191–199. doi: 10.1016/j.neuron.2005.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moechars D, Lorent K, De Strooper B, Dewachter I, Van Leuven F. Expression in brain of amyloid precursor protein mutated in the alpha-secretase site causes disturbed behavior, neuronal degeneration and premature death in transgenic mice. EMBO J. 1996;15:1265–1274. [PMC free article] [PubMed] [Google Scholar]
- Moechars D, Dewachter I, Lorent K, Reverse D, Baekelandt V, Naidu A, Tesseur I, Spittaels K, Haute CV, Checler F, Godaux E, Cordell B, Van Leuven F. Early phenotypic changes in transgenic mice that overexpress different mutants of amyloid precursor protein in brain. J Biol Chem. 1999;274:6483–6492. doi: 10.1074/jbc.274.10.6483. [DOI] [PubMed] [Google Scholar]
- Mori C, Spooner ET, Wisniewsk KE, Wisniewski TM, Yamaguch H, Saido TC, Tolan DR, Selkoe DJ, Lemere CA. Intraneuronal Abeta42 accumulation in Down syndrome brain. Amyloid. 2002;9:88–102. [PubMed] [Google Scholar]
- Mullan M, Crawford F, Houlden H, Axelman K, Lilius L, Winblad B, Lannfelt L. A pathogenic mutation for probable Alzheimer's disease in the APP gene at the N-terminus of beta-amyloid. Nat Genet. 1992a;1:345–347. doi: 10.1038/ng0892-345. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Smith IF, Green KN, Laferla FM. A dynamic relationship between intracellular and extracellular pools of Abeta. Am J Pathol. 2006;168:184–194. doi: 10.2353/ajpath.2006.050593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno M, Sametsky EA, Younkin LH, Oakley H, Younkin SG, Citron M, Vassar R, Disterhoft JF. BACE1 deficiency rescues memory deficits and cholinergic dysfunction in a mouse model of Alzheimer's disease. Neuron. 2004;41:27–33. doi: 10.1016/s0896-6273(03)00810-9. [DOI] [PubMed] [Google Scholar]
- Ohno M, Chang L, Tseng W, Oakley H, Citron M, Klein WL, Vassar R, Disterhoft JF. Temporal memory deficits in Alzheimer's mouse models: rescue by genetic deletion of BACE1. Eur J Neurosci. 2006;23:251–260. doi: 10.1111/j.1460-9568.2005.04551.x. [DOI] [PubMed] [Google Scholar]
- Rogers JT, Leiter LM, McPhee J, Cahill CM, Zhan SS, Potter H, Nilsson LN. Translation of the Alzheimer amyloid precursor protein mRNA is up-regulated by interleukin-1 through 5′-untranslated region sequences. J Biol Chem. 1999;274:6421–6431. doi: 10.1074/jbc.274.10.6421. [DOI] [PubMed] [Google Scholar]
- Schlaf G, Salje C, Wetter A, Stuertz K, Felgenhauer K, Mader M. Determination of synapsin I and synaptophysin in body fluids by two-site enzyme-linked immunosorbent assays. J Immunol Methods. 1998;213:191–199. doi: 10.1016/s0022-1759(98)00027-1. [DOI] [PubMed] [Google Scholar]
- Schmitz C, Rutten BP, Pielen A, Schafer S, Wirths O, Tremp G, Czech C, Blanchard V, Multhaup G, Rezaie P, Korr H, Steinbusch HW, Pradier L, Bayer TA. Hippocampal neuron loss exceeds amyloid plaque load in a transgenic mouse model of Alzheimer's disease. Am J Pathol. 2004;164:1495–1502. doi: 10.1016/S0002-9440(10)63235-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- Sheng JG, Price DL, Koliatsos VE. Disruption of corticocortical connections ameliorates amyloid burden in terminal fields in a transgenic model of Aβ amyloidosis. J Neurosci. 2002;22:9794–9799. doi: 10.1523/JNEUROSCI.22-22-09794.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sisodia SS, Kim SH, Thinakaran G. Function and dysfunction of the presenilins. Am J Hum Genet. 1999;65:7–12. doi: 10.1086/302475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spires TL, Hyman BT. Transgenic models of Alzheimer's disease: learning from animals. NeuroRx. 2005;2:423–437. doi: 10.1602/neurorx.2.3.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturchler-Pierrat C, Abramowski D, Duke M, Wiederhold KH, Mistl C, Rothacher S, Ledermann B, Burki K, Frey P, Paganetti PA, Waridel C, Calhoun ME, Jucker M, Probst A, Staufenbiel M, Sommer B. Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc Natl Acad Sci USA. 1997;94:13287–13292. doi: 10.1073/pnas.94.24.13287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi RH, Milner TA, Li F, Nam EE, Edgar MA, Yamaguchi H, Beal MF, Xu H, Greengard P, Gouras GK. Intraneuronal Alzheimer Abeta42 accumulates in multivesicular bodies and is associated with synaptic pathology. Am J Pathol. 2002;161:1869–1879. doi: 10.1016/s0002-9440(10)64463-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi RH, Almeida CG, Kearney PF, Yu F, Lin MT, Milner TA, Gouras GK. Oligomerization of Alzheimer's β-amyloid within processes and synapses of cultured neurons and brain. J Neurosci. 2004;24:3592–3599. doi: 10.1523/JNEUROSCI.5167-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thinakaran G, Regard JB, Bouton CM, Harris CL, Price DL, Borchelt DR, Sisodia SS. Stable association of presenilin derivatives and absence of presenilin interactions with APP. Neurobiol Dis. 1998;4:438–453. doi: 10.1006/nbdi.1998.0171. [DOI] [PubMed] [Google Scholar]
- Tseng BP, Kitazawa M, LaFerla FM. Amyloid beta-peptide: the inside story. Curr Alzheimer Res. 2004;1:231–239. doi: 10.2174/1567205043332045. [DOI] [PubMed] [Google Scholar]
- Urbanc B, Cruz L, Le R, Sanders J, Ashe KH, Duff K, Stanley HE, Irizarry MC, Hyman BT. Neurotoxic effects of thioflavin S-positive amyloid deposits in transgenic mice and Alzheimer's disease. Proc Natl Acad Sci USA. 2002;99:13990–13995. doi: 10.1073/pnas.222433299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younkin SG. The role of Abeta 42 in Alzheimer's disease. J Physiol (Paris) 1998;92:289–292. doi: 10.1016/s0928-4257(98)80035-1. [DOI] [PubMed] [Google Scholar]