

Abstract
Dendritic morphology determines many aspects of neuronal function, including action potential propagation and information processing. However, the question remains as to how distinct neuronal dendrite branching patterns are established. Here, we report that postsynaptic density-95 (PSD-95), a protein involved in dendritic spine maturation and clustering of synaptic signaling proteins, plays a novel role in regulating dendrite outgrowth and branching, independent of its synaptic functions. In immature neurons, overexpression of PSD-95 decreases the proportion of primary dendrites that undergo additional branching, resulting in a marked reduction of secondary dendrite number. Conversely, knocking down PSD-95 protein in immature neurons increases secondary dendrite number. The effect of PSD-95 is activity-independent and is antagonized by cypin, a nonsynaptic protein that regulates PSD-95 localization. Binding of cypin to PSD-95 correlates with formation of stable dendrite branches. Finally, overexpression of PSD-95 in COS-7 cells disrupts microtubule organization, indicating that PSD-95 may modulate microtubules to regulate dendritic branching. Whereas many factors have been identified which regulate dendrite number, our findings provide direct evidence that proteins primarily involved in synaptic functions can also play developmental roles in shaping how a neuron patterns its dendrite branches.
Keywords: PSD-95, dendrite branching, hippocampal neurons, cypin, microtubule, activity-independent
Introduction
Dendrite branching plays an important role in normal brain function. Branching patterns, the relationship between the primary dendrites arising from the cell body and higher order dendrites arising from primary dendrites, appear to be cell-type specific and play a role in determining how information is received and processed by a neuron (Vetter et al., 2001; Schaefer et al., 2003). Dendritic arbors are shaped by intracellular factors, such as calcium/calmodulin-dependent protein kinase II (Fink et al., 2003), the small GTPases RhoA, Rac1, and Cdc42 (Threadgill et al., 1997; Ruchhoeft et al., 1999; Li et al., 2000), novel genes identified in Drosophila (Gao et al., 1999; Moore et al., 2002; Grueber et al., 2003; Ye et al., 2004), β-catenin (Yu and Malenka, 2003), glutamate receptor-interacting protein (GRIP) (Hoogenraad et al., 2005), and a calcium-responsive transactivator called CREST (Aizawa et al., 2004). Although these proteins regulate dendritic morphology, it has not been determined how a neuron decides in what manner its dendrites will branch (i.e., increasing proportion of primary dendrites that branch, and number of branches from a single branch point).
The establishment of mature dendrites and their branches is a multistep process and can be observed using dissociated rat hippocampal neurons in culture. Primary dendrites are extended from the cell body from 1 d in vitro (DIV) until at least 10 DIV (stages 2–4) (Dotti et al., 1988). Dendritic branches are then extended from the primary dendrites beginning at ∼6 DIV until ∼12 DIV (stage 4) (Dotti et al., 1988; Akum et al., 2004). After this burst of branching, some of the primary and secondary dendrites are pruned from 12 DIV until ∼3 weeks in culture until the process of spine formation and maturation occurs (stage 5) (Dotti et al., 1988).
Postsynaptic density-95 (PSD-95) is a postsynaptic scaffolding protein that acts to recruit signaling components at the synapse (Kim and Sheng, 2004) and to promote maturation of dendritic spines (El-Husseini et al., 2000). However, PSD-95 is expressed in immature neurons before synaptogenesis, and a large majority of PSD-95 clusters are not synaptic (Okabe et al., 2001; Prange and Murphy, 2001). Until now, no function has been attributed to this nonsynaptic pool of PSD-95. Our current studies demonstrate a role for PSD-95 in determining dendrite outgrowth and branching in immature neurons (12 DIV). We find that PSD-95 acts as a stop signal for branching and that overexpression of PSD-95 results in decreased secondary dendrite number because of a decreased proportion of primary dendrites that branch. Decreasing PSD-95 expression results in increased secondary dendrite number. Furthermore, we find that the effects of PSD-95 are independent of neuronal activity and can be regulated by cypin, a cytosolic PSD-95 interactor (Akum et al., 2004). Binding of cypin to PSD-95 decreases PSD-95 clustering and promotes the establishment of stable dendritic branches. Finally, overexpression of PSD-95 decreases the number of dynamic microtubules, which may underlie its effects on branching. Thus, nonsynaptic PSD-95 acts to regulate the branching pattern of hippocampal dendrites.
Materials and Methods
In accordance with the policy of Rutgers University governing the use of animals in research, all animal work described here was reviewed and approved by the Rutgers University Animal Care and Facilities Committee.
Antibodies.
Characterization of the rabbit polyclonal antibody raised against cypin has been described previously (Firestein et al., 1999). A rat anti-green fluorescent protein (GFP) was a kind gift from Dr. Shu-Chan Hsu of Rutgers University (Piscataway, NJ). Mouse monoclonals anti-PSD-95 (045, 046) were purchased from Affinity BioReagents (Golden, CO). Rabbit anti-PSD-95 was characterized previously (Firestein et al., 1999). Both monoclonal 046 and rabbit anti-PSD-95 are specific for PSD-95 (Firestein et al., 1999; Sans et al., 2000). Mouse anti-MAP2 and rabbit anti-DsRed were purchased from BD PharMingen (San Diego, CA) and rabbit anti-synaptophysin was purchased from Zymed (San Francisco, CA).
Antisense oligonucleotide knockdown of PSD-95 mRNA expression in cultured hippocampal neurons.
The sense and antisense oligonucleotides corresponded to nucleotides 435–449 (5′-GAATGGGTCACCTCC-3′) of rat PSD-95/synapse-associated protein 90 (SAP-90) mRNA (GenBank accession number M96853), identical to that used by Sattler et al. (1999). PSD-95 antisense, sense, and antisense for cortactin (5′-GTCCTCCTGCTCTCTCTCCT-3′) oligonucleotides were filter-sterilized in culture medium and added to a final concentration of 5 μm during media changes at 4, 6, 8, and 10 d after seeding. On DIV 10, cultures were simultaneously treated with oligonucleotides and transfected with cDNA encoding GFP to assess dendritic morphology of individual hippocampal neurons. For prolonged antisense oligodeoxynucleotide (ODN) knockdown of PSD-95 until 17 DIV, cultures were treated as above and the regimen was extended to include treatments on 12, 14, and 16 d after seeding. GFP expression was allowed to progress for 48 h, after which cells were fixed with methanol and double-labeled with a rat antibody to GFP and a mouse monoclonal antibody to PSD-95 (045). Alternatively, neurons were collected at 12 DIV and lysed in 25 mm Tris-HCl, pH 7.4, 1 mm EDTA, 1 mm EGTA, 1 mm PMSF. Lysates were analyzed by western blotting with a mouse mAb to PSD-95 (046) to assess the extent of PSD-95 knockdown. A basic local alignment search tool search revealed that the oligonucleotide sequences exhibited no similarity to any other known mammalian cDNA sequences.
U1 snRNA constructs.
The U1 snRNA constructs were designed as described previously (Akum et al., 2004). The anti-PSD-95 U1 snRNA plasmids were produced by PCR with an oligonucleotide containing the 5′ end mutation (5′-ACACTCTAGAAGGAGCAAGGG-CAGGGGAGATACCATGATCACGA-3′). The PCR fragment was inserted into unique XbaI and EcoRI sites of the vector SG702D.
Neuronal culture, antagonist treatment, immunohistochemistry, and transfection.
Hippocampal cultures were prepared from rat embryos at 18 d gestation as described previously (Firestein et al., 1999). For transfection, the neurons were grown for 10 d in culture and transfected with the appropriate constructs using Effectene (Qiagen, Hilden, Germany). For antagonist treatment, neurons were treated with 50 μm dl-2-amino-5-phosphonovalerate acid (APV), 10 μm 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX), and 1 μm tetrodotoxin (TTX) immediately after transfection (at 10 DIV) for 24 h. For immunocytochemistry, neurons were fixed at 12 or 17 DIV with ice-cold methanol for 15 min and stained with the following primary antibodies: mouse anti-PSD95 (1:200), rat anti-GFP (1:1000), rabbit anti-synaptophysin (1:500), or rabbit anti-DsRed (1:1000). Staining was visualized using secondary antibodies conjugated to Cy2 or Cy3 (Jackson ImmunoResearch, West Grove, PA).
Assessment of dendrite number.
Neurons were fixed and stained as described above. Labeled cells were visualized by immunofluorescence using an Olympus Optical (Tokyo, Japan) IX50 microscope with a Cooke Sensicam CCD cooled camera, fluorescence imaging system and ImagePro software (MediaCybernetics, Silver Spring, MD). Dendrite number was counted as described previously (Akum et al., 2004), using a semiautomated procedure implemented in the statistical software package R. All decisions that involved the identification of dendrites and branch points were made by the investigator and were not automated. Specifically, digital micrographs were contrast-enhanced and, using the aforementioned software package, the number of primary dendrites for each neuron was recorded by digitally marking the points from which dendritic segments arise at the soma boundary, followed by a second round of marking at the first branch point marking the end of the primary dendritic segment. If the primary dendrite did not branch, the mark was made at the terminus of the primary dendrite. The secondary dendrite number was determined after a third round of marking at the next branch point along the length of the secondary dendrite, marking the end of the secondary segment. If the secondary dendrite did not branch, the mark was made at the terminus of the segment. Processes arising from the soma or from higher-order dendritic segments that were shorter than 3 μm were rejected, because they could not be distinguished from filopodia. The software also recorded the relationships between primary and secondary dendrites, allowing for additional analysis such as the determination of the proportion of primary and secondary dendrites that branch, as well as the number of excess secondary dendrites formed at a branch point. We defined the latter as the number of secondary dendrites in excess of two that arise from a single branch point. Only secondary dendrites that clearly arose from a single branch point shared by at least two other secondary dendrites were classified as excess secondary dendrites. The formation of multiple dendrites from a single branch point is a normal branching pattern that has been shown to occur in vivo (Fan et al., 2005; Rai et al., 2005). To reduce the possibility of researcher bias, the investigator performing the above procedure was always blinded to the experimental conditions. For U1 snRNA knockdown experiments, all neurons expressing the knockdown constructs were counted so as not to bias the data. Data for Sholl analysis were collected using a semiautomated program in R, in which the soma boundary is approximated by an ellipsoid and dendrite intersections are assessed at radial distances from the soma.
Statistical analyses of branching patterns.
To analyze the dendrite patterning we used a three-stage model. The first stage models the number of primary dendrites via a Poisson model with parameter L1. L1 is allowed to be cell-type specific and, thus, denotes the expected number of primary dendrites forming in a given cell type. We assessed the goodness of fit of the Poisson model for all constructs. The second stage of the model addresses branching of primary dendrites. We model the proportion of primary dendrites that branch via a Binomial model with parameters N (the number of primaries) and p (the proportion of branching primaries), and let X denote the number of branching primaries. The parameter p can loosely be referred to as branching probability, but note that we are not modeling an evolutionary branching process. We allow the parameter p to depend on cell type and the number of primaries, N. We found that branching proportion is significantly related to the log of the number of primaries; that is, branching proportion decreases as the number of primaries increases for all constructs, an effect we can think of as “crowding.” We assessed the goodness of fit of the binomial model for all constructs. The third stage of the model addresses secondary dendrite numbers. We modeled each branch point as generating secondary branching, in excess of two, via a Poisson mixed-effects model with parameter L2 (the random effect is used to account for intraneuron dependencies). As an example, an L2 value of 1 would denote that we expect on average each branch point to be associated with three (two plus one) secondary dendrites. We allowed L2 to be cell-type specific and dependent on the number of branching primaries, X, or number of primaries, N. For our data, we saw no significant impact on L2 from N or X. We assessed the goodness of fit of the third-stage Poisson model for all constructs.
To determine which of the parameters (L1, p, L2) were cell-type specific, we used an exhaustive model search and analysis of deviance (ANODEV). The p values presented are based on ANODEV (χ2 tests). We selected the most parsimonious model that fits the data via AIC (Akaike, an information criterion). The p values reported were obtained by comparing the best subset models and adjusting for the number of comparisons (including the number of subset models of each “size” equals the number of constructs that differ) using Bonferroni test for multiple comparisons.
To analyze the proximal Sholl profiles (1–63 μm proximal to soma), we used a Poisson generalized linear mixed model (GLMM). The random effect takes the intraneuron dependencies into account (a repeated measures ANOVA is not appropriate here because dendrite counts are obtained at various radial distances for the same neuron). The fixed effects are distance and construct. We determined whether the Sholl profiles are significantly different between constructs by testing the main group and group-distance interaction effects. The p values were obtained using ANODEV (χ2 tests). The most parsimonious model that supports the data was selected using AIC. The p values reported were obtained by selecting the best subset model of construct differences (adjusting for multiple comparisons). As a post-processing step, the differential distance effects were adjusted for multiple comparisons across all distances <63 μm.
COS-7 cell culture, transfection, and quantification of microtubule disruption.
COS-7 cells were plated at 70–80% confluence and maintained in DMEM (Invitrogen, Carlsbad, CA), supplemented with 7.5% fetal bovine serum and 7.5% horse serum in a 7% CO2 atmosphere. For transfection, COS-7 cells were replated on poly-d-lysine-coated 22 mm glass-bottom dishes (World Precision Instruments, Sarasota, FL) at an approximate density of 500 cells/mm2. Cells were transfected with 1.0 μg of the indicated plasmid DNA encoding the indicated proteins using the LipofectAMINE 2000 (Invitrogen) method following the manufacturer's instructions. Cells were processed for immunofluorescence, as described above, using a mouse monoclonal antibody to acetylated tubulin (Sigma, St. Louis, MO) to visualize microtubules. Antibodies were visualized using secondary antibodies conjugated to Cy5 (Jackson ImmunoResearch). For live-cell imaging, culture medium was replaced with Ringer's solution (115 mm KCl, 2 mm CaCl2, 2.5 mm KCl, 10 mm HEPES, pH 7.4) 18–24 h after transfection, and images were acquired on a Nikon (Tokyo, Japan) inverted microscope (TE2000) equipped with a heated stage, a 60× Plan Fluor oil-immersion objective (numeric aperture, 1.3), a CCD camera (PXL1400, Roper Scientific, Tucson, AZ), and IPLab software (Scanalytics, Rockville, MD). For quantification of microtubule disruption, 20 random micrographs were taken for each replicate in the Cy5-tubulin channel. Transfected cells were marked before counting, so that the investigator could identify transfected cells but was blind to the transfection condition. The following criteria was used to define microtubule (MT) irregularity: (1) absence of a prominent microtubule organizing center (MTOC), (2) circuitous or winding MT bundles, and (3) dense MT “cages” surrounding an area devoid of tubulin fluorescence signal.
Results
Overexpression of PSD-95 inhibits dendritic branching
Sans et al. (2000) have shown previously that PSD-95 is not expressed in postnatal day 2 rat pups, but that expression does occur by postnatal day 10. In light of this work, we analyzed whether our cultures express PSD-95 at 10 and 12 DIV, times equivalent to postnatal day 8 and 10, when dendrite branching is occurring, and at 17 and 24 DIV, respectively, time points when dendritic spines are forming. We found that PSD-95 is expressed by 10 DIV, and expression increases through 24 DIV (Fig. 1 A,B). This pattern of expression was determined using two antibodies that we and others (Sans et al., 2000) have found to be specific for PSD-95. We then assessed whether clusters of PSD-95 are synaptic at 12 DIV by quantitating their apposition to synaptophysin staining. As seen in Figure 1 B, a majority of PSD-95 clusters are not synaptic at 12 DIV (synaptic clusters, 37.4 ± 1.6%; n = 36) and we observed little PSD-95 in the cell body (data not shown), whereas a majority of PSD-95 clusters become synaptic by 17 DIV (Fig. 1 C, arrowheads) and a prominent pool of nonsynaptic PSD-95 clusters is present in the soma (Fig. 1 C, inset, arrows).
Figure 1.
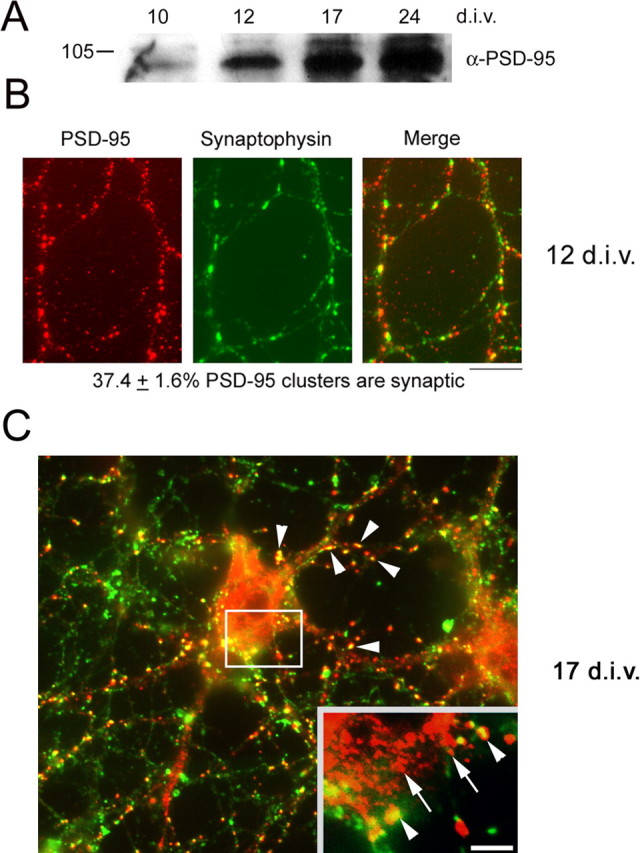
PSD-95 is expressed and is mostly nonsynaptic in hippocampal neurons at 10 DIV. A, Hippocampal neurons were grown for the indicated times and extracts were subjected to SDS-PAGE and Western blotting with an antibody to PSD-95. B, Neurons cultured for 12 DIV were double-labeled for PSD-95 and synaptophysin. Apposition of stained clusters was quantitated. Only 37.4 + 1.6% of PSD-95 clusters was synaptic (n = 36 neurons). C, Neurons cultured for 17 DIV were double-labeled for PSD-95 and synaptophysin. At this time point, a majority of dendritic PSD-95 clusters colocalize with synaptophysin (arrowheads), whereas most somatic PSD-95 clusters are smaller in size and do not colocalize with synaptophysin (arrows, inset). A minority of somatic PSD-95 clusters colocalize with synaptophysin (arrowheads, inset). Scale bars: B, C, 10 μm, inset, 3.5 μm.
Previous studies have shown that PSD-95 promotes spine stability (El-Husseini et al., 2000), one aspect of dendritic morphology in mature neurons with clusters of PSD-95 localized at synaptic sites. Because we found that PSD-95 is also present in young neurons in nonsynaptic clusters (Fig. 1), we postulated that PSD-95 could participate in the development of immature dendrites. To address this, we overexpressed PSD-95 in cultured hippocampal neurons from 10 to 12 DIV (stage 4) (Dotti et al., 1988; Akum et al., 2004) and counted primary and secondary dendrites. We found that overexpression of PSD-95 for 48 h results in a significant decrease in the number of secondary dendrites but no change in the number of primary dendrites (Fig. 2 A,B). Furthermore, both the average (Fig. 2 C) and the total (Fig. 2 C, right) dendrite length per neuron was decreased. Additional analysis indicated that this decrease in dendrite length results, at least in part, from a significant decrease in the average length per neuron of primary (Fig. 2 D, left), but not secondary (Fig. 2 D, right) dendritic segments. This suggests that the reduction in the number of secondary dendrites by PSD-95 might result from a premature termination of primary dendrite outgrowth, before these dendrites have a chance to form a branch. We also observed a marginally significant decrease in the total secondary dendrite length per neuron (Fig. 2 E, right), likely reflecting the loss of secondary dendrite number after overexpression of PSD-95 (Fig. 2 B). Thus, our data suggest that PSD-95 inhibits dendrite development in immature neurons.
Figure 2.

Overexpression of PSD-95 decreases secondary dendrite number. A, Hippocampal neurons were transfected with cDNAs encoding GFP + DsRed1 by itself (vector) or GFP + PSD-95-DsRed1 (PSD-95) at 10 DIV. Scale bar, 10 μm. B, Dendrite number was assessed at 12 DIV. Overexpression of PSD-95-DsRed1 decreased secondary dendrite number over control but not primary dendrite number. n values are as follows: vector, 28; PSD-95, 27. ***p < 0.001 by Welch t test. C, Average and total dendrite lengths per neuron were assessed for control (GFP) and PSD-95 overexpressing neurons. n = 15 for both groups. *p < 0.05 by Welch t test. D, The average length of primary, but not secondary dendrite segments per neuron was decreased when neurons overexpressed PSD-95-DsRed1 compared with control. *p < 0.05 by Student's t test. E, Neither the total primary nor total secondary dendrite length per neuron was significantly altered with overexpression of PSD-95-DsRed1 compared with control. Note that with expression of PSD-95-DsRed1, there was a marginal decrease in total secondary dendrite length per neuron, likely resulting from the decrease in the number of secondary dendrites. p = 0.0508 by Welch t test. F, Left, Proportion of primary dendrites that branch is lower in the neurons that overexpress PSD-95 (Binomial ANODEV, p = 0.00039). Middle, PSD-95 has no effect on the number of excess secondary dendrites (Poisson ANODEV, p = 0.39). Right, PSD-95 has no significant effect on the proportion of secondary dendrites that branch. G, Neither primary nor secondary dendrite number was altered when neurons overexpressed PSD-95 family members SAP-97 and SAP-102 from 10–12 DIV. H, Sholl analysis of neurons shows that overexpression of PSD-95 (asterisks) results in significantly fewer dendrites at 36–63 μm from the soma compared with GFP (open squares). For each distance (9–63 μm), the effect of PSD-95 was quantified using Poisson GLMM, and p values were adjusted using Bonferroni corrections for multiple comparisons across all distances. In the range 36–63 μm, all adjusted p values were <0.002 with effect sizes increasing from 36 to 63 μm (a 36% decrease and 48% decrease, respectively). Error bars indicate SE.
We further analyzed the decrease in secondary dendrite number elicited by PSD-95. Specifically, we asked whether PSD-95 decreases secondary dendrite numbers by decreasing the proportion of primary dendrites that branch or by decreasing the number of secondary dendrites generated at a branch point. Using Poisson and binomial generalized linear (mixed) models (for statistical model, see supplemental Fig. 1, available at www.jneurosci.org as supplemental material), we estimated the nonparametric density estimates of (1) the proportion of primary dendrites that branch and (2) the number of secondary dendrites in excess of two, that are generated at each branch point (Fig. 2 D). Interestingly, we found that overexpression of PSD-95 decreased the proportion of primary dendrites that branch (Fig. 2 D, left). However, when a primary dendrite of a PSD-95-expressing neuron does branch, it gives rise to the same number of secondary branches as that from a GFP-expressing neuron (Fig. 2 D, right). We further asked whether this effect was restricted to proximal dendrite branching, or whether PSD-95 can also inhibit the formation of higher order branches. As shown in Figure 2 F, PSD-95 expression did not change the proportion of secondary dendrites that give rise to tertiary branches. Together, our data suggest that PSD-95 decreases the number of secondary dendrites by decreasing the proportion of primary dendrites that branch. These data also suggest that PSD-95 acts to inhibit branching proximal to the cell body and that decreases in additional branching is caused by the decreased number of secondary dendrites that can give rise to tertiary dendrites.
To determine whether these observations reflect a specific role of PSD-95 in the regulation of dendritic branching or a general effect by proteins with similar domain structures, we overexpressed the PSD-95 family members SAP-97 and SAP-102, which bear a domain organization very similar to PSD-95. As shown in Figure 2 G, neither SAP-97 nor SAP-102 elicited a significant change in the number of primary and secondary dendrites. These results are consistent with a specific role for PSD-95 in the regulation of dendritic branching in immature neurons.
To elucidate the role of PSD-95 in higher-order dendritic branching, we performed Sholl analysis by measuring the number of dendrites that cross ellipsoids at different radial distances from the cell body (see Materials and Methods). As seen in Figure 2 H, overexpression of PSD-95 results in a decrease in dendrite number proximal to the soma and shorter overall dendritic length, as evidenced by the number of intersections falling to zero more rapidly than that of control (GFP-expressing) neurons. The results of our Sholl analysis agree with the dendrite length measurements presented in Figure 2 C–E. Thus, our data suggest that PSD-95 acts as a stop signal for secondary and higher-order dendrite formation by decreasing the proportion of primary dendrites that branch. In addition, our Sholl analysis suggests that PSD-95 also suppresses higher-order dendrite formation (Fig. 2 E).
Knockdown of PSD-95 protein results in increased dendrite branching
If PSD-95 acts as a stop signal for branching, then decreasing PSD-95 expression should result in an increase in secondary dendrites. To test this hypothesis, we knocked down PSD-95 using a previously described method: treatment with a 15-nucleotide oligomer phosphodiester antisense ODN (Sattler et al., 1999; Tao and Johns, 2001; Tao et al., 2001; Hou et al., 2003, 2005). As in these studies, treatment with sense ODN served as a negative control (Sattler et al., 1999; Tao and Johns, 2001; Tao et al., 2001; Hou et al., 2003, 2005). Furthermore, we used an ODN for cortactin as a second negative control because cortactin has been shown to play no role in regulating primary and secondary dendrite number (Hoogenraad et al., 2005). In our hands, the antisense ODN for PSD-95 decreased PSD-95 expression by 65% versus control (sense ODN) treatment (n = 3) (Fig. 3 B) and the ODN for cortactin had no effect on PSD-95 expression (data not shown). This was assessed by Western blotting, representing the neuronal population as a whole. Nonparametric analysis of these neurons results shows that decreased PSD-95 levels resulted in a modest increase in the number of primary dendrites, and a significant increase in the number of secondary dendrites (Fig. 3 A,C). ODN for cortactin showed no change in primary and secondary dendrite number compared with sense control (7.9 ± 0.52 primary dendrites and 12.9 ± 0.79 secondary dendrites for cortactin vs 7.7 ± 0.43 primary dendrites and 12.5 ± 0.74 secondary dendrites for sense; p > 0.05 by Tukey–Kramer multiple comparisons test). We then analyzed whether knocking down PSD-95 affects dendrite length. We found that the average dendrite length and total dendrite length decreased when neurons were treated with antisense ODN compared with sense ODN (Fig. 3 D). We further determined that this decrease in dendrite length results from a decrease in the average lengths per neuron of both the primary and secondary dendritic segments (Fig. 3 E). Thus, when PSD-95 was knocked down, there were more secondary dendrites, which were shorter in length. This decrease in length may be attributable to the fact that increased branching exhausts a limiting reagent required for dendrite outgrowth. This notion is supported by the finding that, although the average segment length is decreased (Fig. 3E), the total length per neuron of primary and secondary dendritic segments remains unchanged (Fig. 3 F) when PSD-95 is knocked down.
Figure 3.
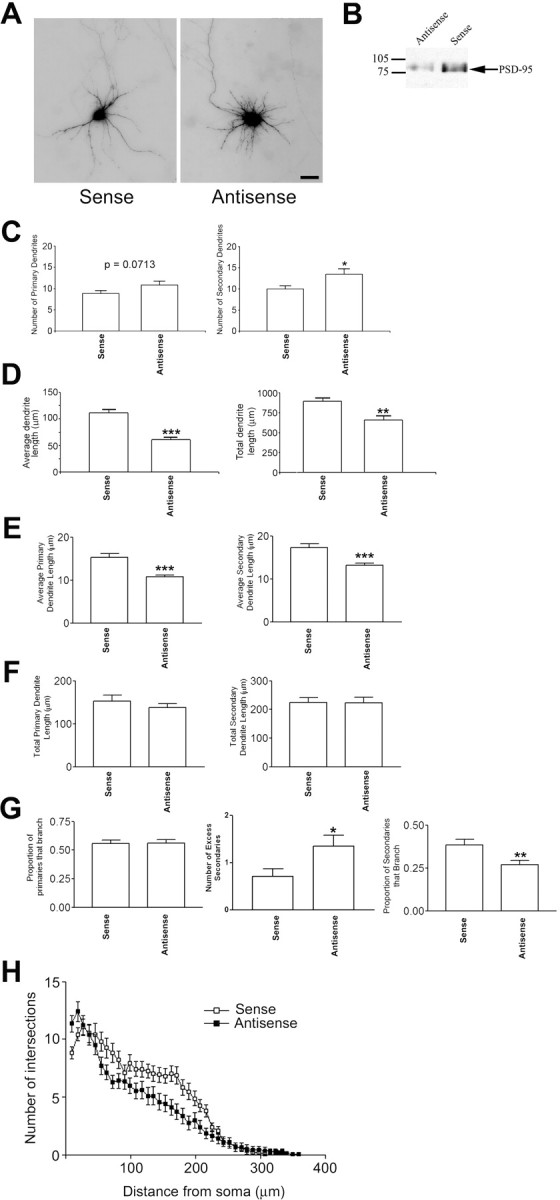
Antisense oligonucleotide knockdown of PSD-95 results in increased primary and secondary dendrite number. A, GFP fluorescence of representative hippocampal neurons that were treated with 5 μm PSD-95-specific sense (left) or antisense (right) oligonucleotides at 4, 6, 8, and 10 DIV. Cultures were transfected with cDNA encoding GFP at 10 DIV to visualize dendrites of individual neurons. B, Homogenates from sense- or antisense-treated hippocampal neuronal cultures were subjected to SDS-PAGE and Western blotting with a mouse monoclonal antibody to PSD-95 to confirm the knockdown of PSD-95 protein expression. A representative blot is shown. The average decrease in PSD-95 expression for treatment with antisense oligonucleotide was 65% (n = 3 experiments). C, Primary and secondary dendrite number was assessed at 12 DIV. Knockdown of PSD-95 results in a nonsignificant increase in primary dendrites (p = 0.0713 by Mann–Whitney test). The number of secondary dendrites was higher in antisense-treated cultures (*p = 0.0298 as determined by Mann–Whitney test; sense, n = 32; antisense, n = 33). D, Average and total dendrite lengths were assessed for control (sense) and sense oligonucleotide-treated neurons. n = 28 for both groups. **p < 0.01; ***p < 0.001 by Mann–Whitney test. E, The average length per neuron of primary (left) and secondary (right) dendrites were significantly decreased when cultures were treated with PSD-95 antisense ODNs compared with sense control. ***p < 0.001 by Mann–Whitney test. F, The total length per neuron of primary (left) and secondary (right) dendrites was unchanged when cultures were treated with PSD-95 antisense ODNs compared with sense control. G, Left, The proportion of primaries that branch is not significantly different between sense- and antisense-treated neurons. Middle, Treatment with antisense ODN increases the number of excess secondary dendrites compared with sense ODN (p = 0.039 after Bonferroni multiple comparisons test). Right, Treatment with antisense ODN significantly decreased the proportion of secondary dendrites that branch. **p < 0.01 by Mann–Whitney test. H, Sholl analysis shows that antisense significantly increases dendrite number compared in the region 9–18 μm from the soma boundary (p < 0.0001). Multiple comparison adjustments were made using Bonferroni multiple comparisons test for the differential effects of the constructs and across distances 9–63 μm. Scale bar, 10 μm. Error bars indicate SE.
Interestingly, our statistical model (supplemental Fig. 1, available at www.jneurosci.org as supplemental material) shows that, although the number of excess secondary dendrites was significantly higher when PSD-95 was knocked down (Fig. 3 G, middle), knock-down of PSD-95 had no effect on the proportion of primary dendrites that branch, even after taking into account the increase in the number of primary dendrites (Fig. 3 G, left). Thus, the increase in the number of secondary dendrites seen when PSD-95 is knocked down cannot be accounted for merely by the fact that there are more primary dendrites. Rather, these data support the idea that when PSD-95 is decreased or absent, primary dendrites give rise to more secondary branches originating from the branch point. In addition, consistent with the exhaustion of a limiting reagent (and therefore premature termination of higher-order dendritic arborization), we found that these secondary dendrites are less likely to form tertiary branches (Fig. 3 G, right). Together with the PSD-95 overexpression data, these data support the notion that PSD-95 regulates secondary dendrite number by controlling primary dendrite branching and tertiary dendrite number by controlling secondary branching. To further understand how decreasing PSD-95 expression affects the entire neuron, we performed Sholl analysis. As seen in Figure 3 H, antisense treatment results in an increase in the number of proximal dendrites, and a decrease in the overall length of dendrites, as evidenced by a more rapid decay of the curve for antisense ODN-treated neurons compared with the curve for sense ODN-treated neurons. Sholl analysis also shows that treatment with ODN for cortactin exhibits similar dendrite numbers to those of the sense control ODN (data not shown; p = 0.355), strongly suggesting that PSD-95 has a specific effect on dendrite number. These data are in agreement with our analysis of proximal dendrite number and dendrite lengths (Fig. 3 C–F).
Because ODN most likely knocks down PSD-95 protein in all neurons in the culture, we asked whether knocking down PSD-95 in individual neurons, acting in a cell autonomous manner, would result in increased dendrite branching. Thus, we transfected neurons with a cDNA encoding a 5′ end-mutated U1 snRNA corresponding to a region in the 3′ untranslated region (3′UTR) of PSD-95 (Fig. 4). When we expressed this construct from 10 to 17 DIV, both cluster size and intensity significantly decreased (60 and 25% decrease, respectively) (Fig. 4 A). We then assessed dendrite number in these neurons compared with that of neurons transfected with a control vector (nonmutated U1 snRNA) (Fig. 4 B,C). We found that PSD-95 knockdown had no significant effect on primary dendrite number (Fig. 4 B,C, left), but significantly increased secondary dendrite number (Fig. 4 B,C, right). In addition, to control for nonspecific effects of the U1 transgene, we coexpressed the transgene with a PSD-95 construct lacking the U1 target sequence (Fig. 4 B,C). We found that expression of exogenous PSD-95 when endogenous PSD-95 is knocked down brings the number of secondary dendrites back to control levels (Fig. 4 B,C, right). Together, our antisense and 5′ end-mutated U1 snRNA experiments suggest that knock down of PSD-95 results in increased dendrite branching.
Figure 4.
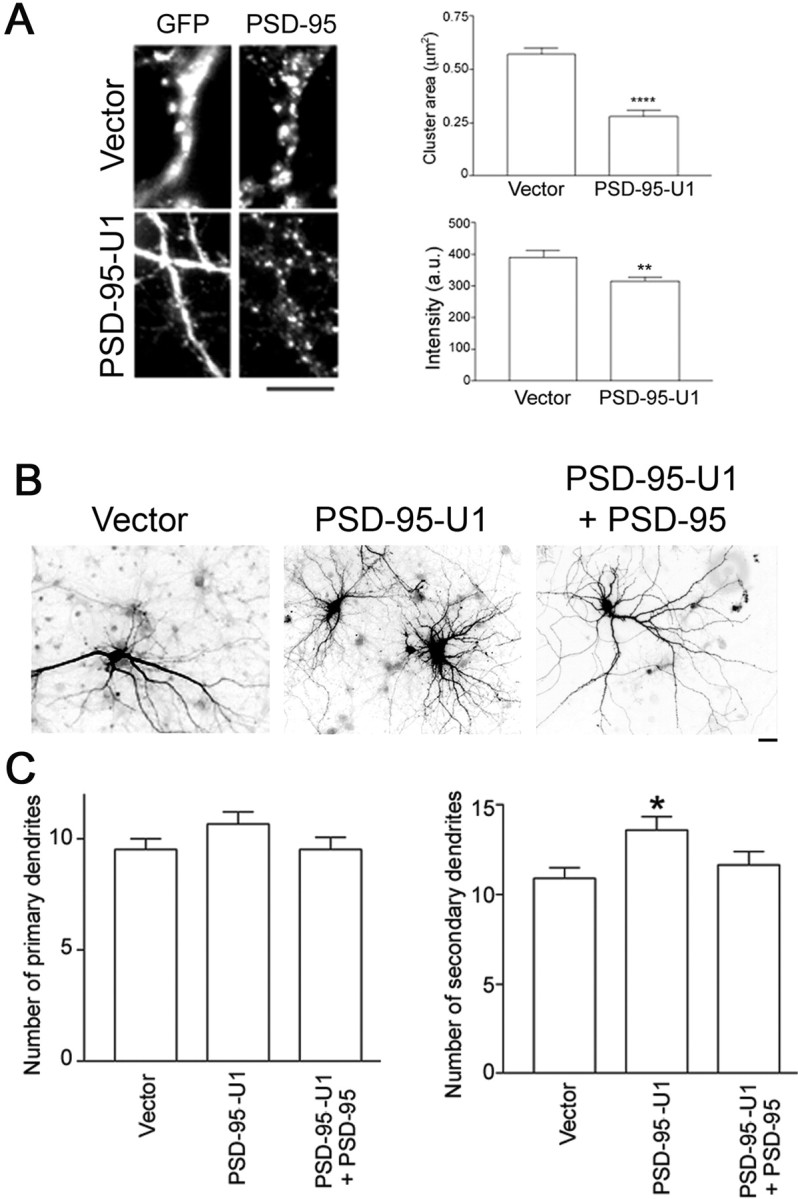
Knocking down PSD-95 protein levels by 5′ end-mutated U1 snRNA results in increased secondary dendrite number. A, Left, Hippocampal neurons were transfected with cDNAs encoding the indicated constructs on DIV 10. Endogenous PSD-95 protein was assessed by immunostaining at 17 DIV. Right, Transfected neurons were analyzed for size and intensity of PSD-95 clusters. n values are as follows: vector, 10; PSD-95–3′UTR-U1, 8. **p < 0.01 and ****p < 0.0001 by Student's t test (cluster area) or Mann–Whitney test (intensity). B, Hippocampal neurons were transfected with cDNAs encoding the indicated constructs at 10 DIV. C, Dendrite number was assessed at 17 DIV. *p < 0.05 as determined by ANOVA followed by Dunnett multiple-comparisons test compared with vector control. PSD-95-U1 + PSD-95 is constructed a plasmid with bicistronic coding for PSD-95-GFP lacking the 3′UTR (U1 recognition site) and PSD-95-U1. PSD-95-GFP restores dendrite numbers to control values (vector). Coexpression of PSD-95-U1 with PSD-95-DsRed1 also restores dendrite numbers to control values (data not shown). n values are as follows: vector, 26; PSD-95–3′UTR-U1, 20; PSD-95–3′UTR-U1 + PSD-95, 21. Scale bars: A, B, 10 μm. Error bars indicate SE.
PSD-95 inhibits dendrite branching in an activity-independent manner
One mechanism by which PSD-95 may affect dendrite branching is by modulating neuronal activity (see Fig. 9 A). For example, PSD-95 has been shown to increase surface expression of NMDA receptors in hippocampal neurons (Lin et al., 2004). In turn, neuronal activity has been shown to alter dendrite branching (McAllister et al., 1996; Jin et al., 2003; Yu and Malenka, 2003). To address whether decreased secondary branching is caused by increased signaling by NMDA receptors, we treated control (GFP-expressing) or PSD-95-overexpressing neurons with APV, an NMDA receptor antagonist. We then counted the number of primary and secondary dendrites. As shown in Figure 5, A and B, APV significantly decreased primary dendrite number and, as expected, overexpression of PSD-95 had no effect on primary dendrite number. Quite strikingly, both treatment with APV and overexpression of PSD-95 each resulted in a 33% decrease in secondary dendrite number, and their effects were greater when neurons were subjected to both conditions simultaneously (Fig. 5 B). Thus, treatment with APV does not occlude the effects of PSD-95 overexpression; rather, the effects of both are partially additive.
Figure 9.

Model for PSD-95-regulated dendrite branching. A, PSD-95 has been shown to increase the surface expression of NMDA (Lin et al., 2004) and AMPA receptors (Chen et al., 2000; El-Husseini et al., 2000; Schnell et al., 2002; Cuadra et al., 2004; Ehrlich and Malinow, 2004). *Because neuronal activity positively regulates dendritic branching (McAllister et al., 1996; Jin et al., 2003; Yu and Malenka, 2003), PSD-95 overexpression would be expected to increase dendritic branching. However, we found that PSD-95 overexpression has a negative impact on dendritic branching, suggesting that PSD-95 acts via a mechanism that is independent of and dominant over any concurrent PSD-95-induced increases in neuronal activity. B, In control neurons (GFP), PSD-95 (filled circles) acts to inhibit secondary dendrite growth, whereas PSD-95 overexpression results in decreased secondary dendrites. Conversely, when PSD-95 is knocked down, secondary dendrite number is transiently increased (dashed lines). Some PSD-95 clusters in the cell body may be decreased (smaller filled circles), resulting in an insignificant (p = 0.0713) increase in primary dendrite number at 12 DIV, but becoming significantly larger when PSD-95 knockdown treatment is maintained until 17 DIV. This suggests that a longer knockdown regimen is needed to relieve the inhibition on primary dendrite outgrowth by somatic PSD-95, which is localized at a higher density than dendritic PSD-95 (Fig. 1 C). Overexpression of cypin causes an increase in primary and secondary dendrites, some of which are pruned later in development (dashed lines). When cypin-PDZ is expressed, PSD-95 clusters are unchanged and secondary dendrite formation is inhibited. C, PSD-95 acts as a stop signal for secondary dendrite formation, possibly by disrupting MT growth and organization. Decreased PSD-95 clustering, regulated by the PDZ binding of cypin, results in increased secondary dendrite number. Alternatively, cypin-independent regulation of dendrite formation, such as inhibition of primary dendrite outgrowth by somatic PSD-95, may exist. In parallel, cypin increases both primary and secondary dendrite formation by promoting microtubule assembly.
Figure 5.
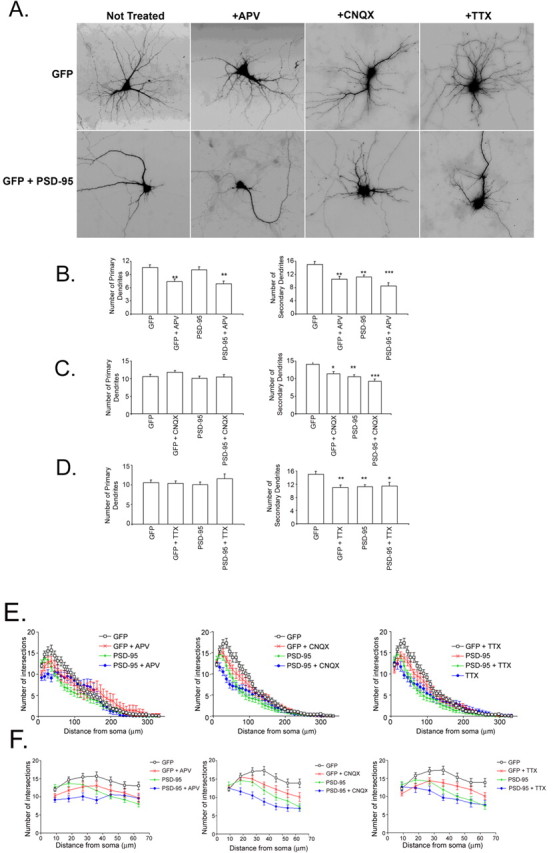
PSD-95 inhibition of branching is activity-independent. A, Hippocampal neurons were transfected with cDNAs encoding GFP by itself (vector) or with PSD-95-dsRed1 (PSD-95) at 10 DIV and were treated with either vehicle, 50 μm APV, 10 μm CNQX, or 1 μm TTX for 24 h. Dendrite number was assessed at 12 DIV. B, APV treatment but not PSD-95 overexpression decreases primary dendrite number. Both PSD-95 and APV decreased secondary dendrite number, and the effect is larger when both treatments are performed. n values are as follows: GFP, 10; GFP + APV, 10; PSD-95, 10; PSD-95 + APV, 14. **p < 0.01; ***p < 0.001 by ANOVA with Tukey–Kramer test for correction for multiple comparisons. C, Neither CNQX treatment nor PSD-95 overexpression changes the number of primary dendrites. Both conditions decrease the number of secondary dendrites. n values are as follows: GFP, 30; GFP + CNQX, 28; PSD-95, 27; PSD-95 + CNQX, 22. *p < 0.05; **p < 0.01; ***p < 0.001 by ANOVA with Tukey–Kramer test for correction for multiple comparisons. D, Neither TTX treatment nor PSD-95 overexpression changes the number of primary dendrites. n values are as follows: GFP, 30; GFP + TTX, 26; PSD-95, 27; PSD-95 + TTX, 17. *p < 0.05; **p < 0.01 by ANOVA with Tukey–Kramer test for correction for multiple comparisons. E, Sholl analysis shows that none of the treatments (APV, CNQX, TTX) block PSD-95 effects on dendrite branching. In fact, the effects are likely additive with PSD-95 overexpression proximal to the cell body. F, Proximal Sholl analysis (from E) within the first 63 μm of the soma. Left, APV does not block the effect of PSD-95 (p > 0.38 for all distances, testing for block or cancellation of the effect of PSD-95 by APV + 95). Middle, CNQX does not block the effect of PSD-95 (p > 0.99 for all distances, testing for block or cancellation of the effect of PSD-95 by CNQX + 95). Right, TTX does not block the effect of PSD-95 (p > 0.99 testing for block or cancellation of the effect of PSD-95 by TTX). All analyses were done using Poisson GLMM and ANODEV. Error bars indicate SE.
It has been reported that overexpression of PSD-95 can also affect the synaptic localization of AMPA receptors (Chen et al., 2000; El-Husseini et al., 2000; Schnell et al., 2002; Cuadra et al., 2004; Ehrlich and Malinow, 2004). To test whether PSD-95 affects dendrite branching by increasing the number of synaptic AMPA receptors, we treated control (GFP-expressing) or PSD-95-overexpressing neurons with CNQX, an AMPA receptor antagonist. As shown in Figure 5, A and C, neither CNQX nor PSD-95 overexpression affected primary dendrite number. However, both treatments resulted in a significant decrease in secondary dendrite number, and exposure to CNQX with PSD-95 overexpression resulted in a slightly larger decrease in secondary number (Fig. 5 C). Like APV treatment, CNQX treatment did not block the effects of PSD-95 overexpression on dendrite number. We then tested the effect of spontaneous electrical activity on PSD-95-mediated decreases in secondary dendrite number by treating the transfected neurons with TTX. As seen in Figure 5, A and D, TTX had no effect on primary dendrites. Furthermore, both PSD-95 overexpression and TTX treatment resulted in decreased secondary dendrite number (Fig. 5 D), and TTX did not block the effects of PSD-95 (Fig. 5 D). Finally, to further confirm that a block in activity does not affect PSD-95 action on dendrite number, we performed Sholl analysis (Fig. 5 E,F) on the entire neuron to assess overall effects of activity-blocking agents (Fig. 5 E) and then focused on the 63 μm closest to the cell body (Fig. 5 F) to assess proximal effects (Hoogenraad et al., 2005). As seen in Figure 5, E and F, neither APV, CNQX, nor TTX blocked the effects of PSD-95 on dendrite number. In fact, the effects of these drugs and PSD-95 were additive. Thus, because none of the inhibitors of activity blocked the effect of PSD-95 on secondary dendrite number, our data support the idea that PSD-95 acts intrinsically to regulate dendrite number in an activity-independent manner (see Fig. 9 A).
Interactions between PSD-95 and cypin in controlling dendrite number
Previous studies have shown that cypin, a PSD-95 interacting protein, enhances dendritic branching (Akum et al., 2004). Therefore, it is possible that PSD-95 reduces branching by inhibiting cypin function. Conversely, cypin could increase branching by negatively regulating PSD-95. Indeed, cypin negatively regulates PSD-95 localization (Firestein et al., 1999). To test these two hypotheses, we simultaneously overexpressed both cypin and PSD-95 for 48 h and counted primary and secondary dendrites at 12 DIV to determine whether the effect of one molecule or the other would predominate. We found that overexpressing PSD-95 attenuated the increase in primary and secondary dendrite number promoted by overexpressing cypin (Fig. 6 A,B). Furthermore, overexpression of PSD-95 significantly decreased secondary dendrite number below basal levels (GFP) even when cypin was overexpressed (Fig. 6 A,B). Our results suggest that elevated PSD-95 levels can suppress dendrite branching, even when cypin levels are elevated, suggesting that PSD-95 stops dendrite branching and that cypin regulates branching by antagonizing PSD-95.
Figure 6.

PSD-95 stops cypin-promoted dendritic branching. A, Hippocampal neurons were transfected with cDNAs encoding the indicated constructs on DIV 10. Scale bar, 10 μm. B, Dendrite number was assessed at 12 DIV. Overexpression of PSD-95-DsRed1 decreased cypin-promoted increases in primary and secondary dendrite number (cypin + PSD-95). PSD-95 decreased secondary dendrite number below basal (GFP) levels regardless of cypin overexpression. n values are as follows: GFP, 33; GFP + PSD-95-dsRed, 10; cypin, 30; cypin + PSD-95-dsRed, 20. *p < 0.05 and ***p < 0.001 by Kruskal–Wallis test followed by Dunn's multiple-comparisons test. C, left, Overexpression of PSD-95 decreases the proportion of primary dendrites that branch (ANODEV, p = 0.0003). PSD-95 blocks the effect of cypin (p = 0.81). Right, Cypin increases the number of excess secondary dendrites (ANODEV, p = 0.00002), and overexpression of PSD-95 blocks this effect. Error bars indicate SE.
In addition to regulating dendrite number, the clustered distribution of PSD-95 suggested that PSD-95 might regulate the pattern of dendritic branching. To understand how PSD-95 acts to regulate dendrite patterning, we analyzed the statistical distribution of the proportion of primary dendrites that branch and the formation of excess secondary dendrites. As seen in Figure 6 C (left), overexpression of PSD-95 with cypin decreased the proportion of primary dendrites that branch compared with overexpression of cypin alone. Cypin overexpression also increased the number of excess secondary dendrites, and coexpression of PSD-95 blocked this effect (Fig. 6 C, right). Thus, PSD-95 blocks all effects of cypin on dendrite formation and branching, suggesting that the two proteins may act in a sequential pathway to regulate dendrite morphology with PSD-95 acting downstream of cypin.
Because cypin increases dendrite number by antagonizing PSD-95, we were perplexed by our previous results that showed an increase in primary and secondary dendrite number by 12 DIV in neurons that overexpressed cypin-PDZ (PSD-95, discs large, zona occludens-1 domain), a cypin mutant that lacks the interaction motif for PSD-95 (Akum et al., 2004). We therefore analyzed neurons overexpressing wild-type cypin and cypin-PDZ counted the number of dendrites at 17 DIV, when neurons are mature and spines are beginning to form. Consistent with our previous results, cypin increased primary and secondary dendrite number at this time point (Fig. 7 A); however, the number of primary dendrites at 17 DIV was reduced when compared with that of 12 DIV (Fig. 7 A). Interestingly, there appeared to be excessive pruning of secondary dendrites in our cultures from 12 to 17 DIV for neurons that overexpressed cypin-PDZ (Fig. 7 A), resulting in no increase in primary or secondary dendrites over basal (GFP) conditions by 17 DIV (Fig. 7 A). Thus, elevating cypin levels may induce transient dendritic branching, but the resulting branches are only stable if cypin can bind to PSD-95.
Figure 7.
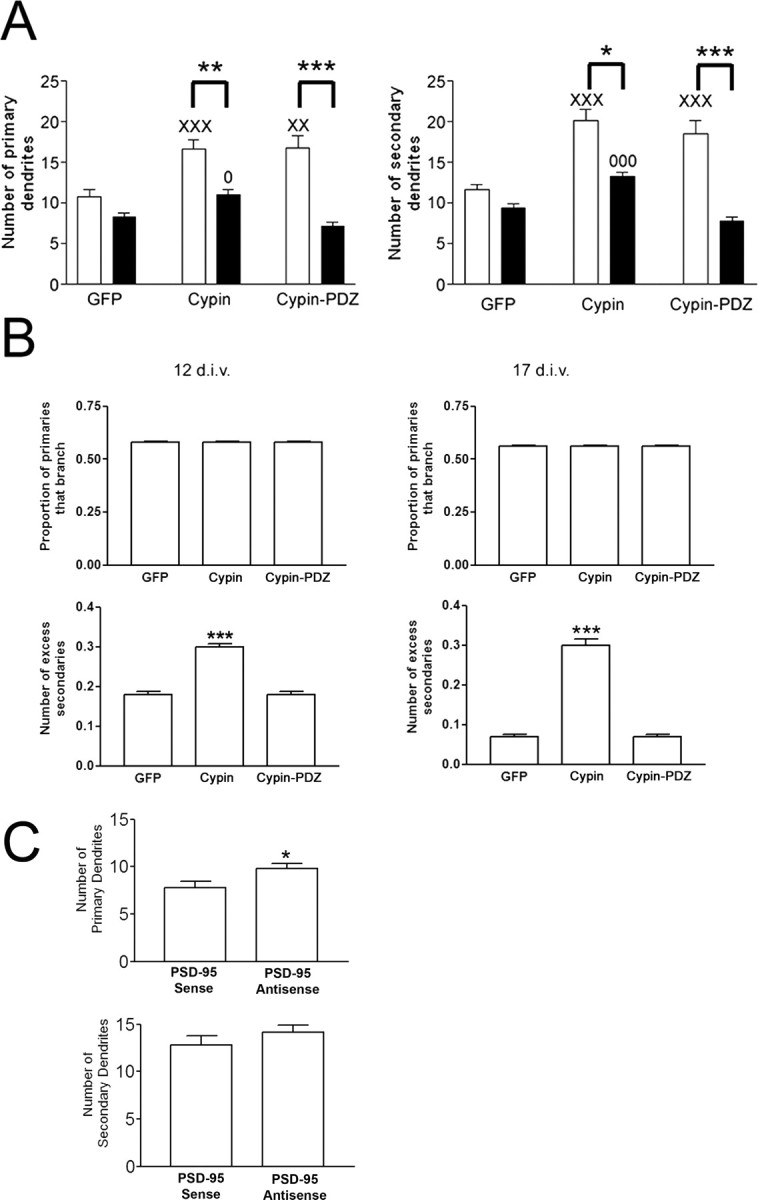
PSD-95 binding is essential for stabilizing cypin-promoted dendrites. A, Hippocampal neurons were transfected with cDNAs encoding the indicated constructs (N-terminally tagged with GFP) at 10 DIV and the number of primary and secondary dendrites were assessed at 12 (white bars) and 17 DIV (black bars). Overexpression of cypin resulted in increased primary and secondary dendrite number at both 12 and 17 DIV; however, overexpression with cypin-PDZ, a mutant that cannot interact with PSD-95, resulted in a transient increase in dendrite number at 12 but not 17 DIV. n values are as follows: 12 DIV, GFP, 33; cypin, 30; cypin-PDZ, 24; 17 DIV, GFP, 20; cypin, 29; cypin-PDZ, 12. *p < 0.05; ** p < 0.01; ***p < 0.001 by Kruskal–Wallis test followed by Dunn's multiple comparisons test as compared with GFP control. XX p < 0.01; XXX p < 0.001 versus GFP at 12 DIV. O p < 0.05; OOO p < 0.001 versus GFP at 17 DIV. B, Statistics collected for the samples of neurons in the different populations (GFP, cypin, cypin-PDZ). Left, 12 DIV; right, 17 DIV. The proportion of primary dendrites that branch is the same for all neurons (top row) for both time points (p = 0.17 and 0.49 respectively). The number of excess secondary branches is higher for neurons that overexpress cypin than GFP and cypin-PDZ. p < 1 × 10−16 for cypin and p = 0.8 for cypin-PDZ versus GFP control at 12 DIV; p = 2.8 × 10−7 for cypin and p = 0.37 for cypin-PDZ versus GFP control at 17 DIV. C, Hippocampal neuronal cultures were treated with PSD-95 antisense ODN or sense controls until 17 DIV. The increase in the number of secondary dendrites by treatment with PSD-95 antisense ODNs at 12 DIV was lost by 17 DIV. Note that there was a significant increase in the number of primary dendrites when cultures were treated with PSD-95 antisense compared with sense control at 17 DIV. *p < 0.05 by Mann–Whitney test. Error bars indicate SE.
Overexpression of cypin can increase dendrite number by both 12 and 17 DIV. We have shown previously that cypin binds to tubulin heterodimers and promotes microtubule assembly (Akum et al., 2004). Cypin-PDZ does promote microtubule assembly and, hence, increases dendrite number by 12 DIV. However, these new dendrites cannot be detected by 17 DIV, suggesting that the dendrites induced by this mutant protein are transient. To examine the pattern of these transient dendrites more closely, we analyzed the statistical distribution of neurons that overexpress GFP, cypin, or cypin-PDZ. We found that although the proportion of primary dendrites that branch is independent of whether any of the three proteins are expressed (Fig. 7 B, top two panels), the number of excess secondary branches is higher when cypin is overexpressed than when GFP and cypin-PDZ are overexpressed for both 12 and 17 DIV (Fig. 7 B, bottom two panels). Therefore, our data suggest that primary dendrites promoted by cypin and cypin-PDZ are pruned. They also support the idea that there are more secondary dendrite branches formed by cypin and that these branches remain stable. In contrast, cypin-PDZ cannot increase the number of secondary branches made at a branch point.
Consistent with the idea that cypin may be required to stabilize induced dendrite branches, we found that the increase in secondary dendrite branches could not be detected when PSD-95 antisense ODN treatment was maintained until 17 DIV (Fig. 7 C). However, we found that whereas the number of secondary dendrites was indistinguishable from sense control, there was a small yet significant increase in the number of primary dendrites after treatment with PSD-95 antisense ODN until 17 DIV. This may reflect the strong inhibition of primary dendrite outgrowth by PSD-95, which is heavily labeled in the soma (Fig. 1 C). Because treatment with PSD-95 ODN showed a marginally significant increase in the number of primary dendrites at 12 DIV (Fig. 3 C), a more prolonged period of antisense ODN treatment may be required to relieve the inhibition of primary dendrite outgrowth by PSD-95.
PSD-95 disrupts microtubule organization
We asked how PSD-95 might act independently of neuronal activity to regulate dendrite branching. We therefore tested the hypothesis that PSD-95 alters cytoskeletal organization. Indeed, it has been shown that PSD-95 is indirectly linked to tubulin and actin via intermediate proteins (Brenman et al., 1998; Naisbitt et al., 1999; Passafaro et al., 1999). To test whether expression of PSD-95 affects the organization of the cytoskeleton, we overexpressed it in COS-7 cells, a cell type in which changes in MT organization are more easily observable than in neurons (Fig. 8 A–C). In these cells, PSD-95-GFP or PSD-95-DsRed1 formed large aggregates in the perinuclear region, as well as smaller aggregates throughout the cell (Fig. 8 A, top row). Anti-acetylated tubulin immunofluorescence revealed that most cells transfected with PSD-95 exhibited a less prominent MTOC and irregular MT distribution pattern. In these cells, the MT bundles wound circuitously around a central axis or formed tight cages around larger PSD-95 aggregates. This is in contrast to control cells transfected with either GFP or DsRed1, localized mainly to the nucleus, that exhibited astral MT bundles originating from a single MTOC (Fig. 8 A, bottom row). These data suggest that PSD-95 disrupts the normal MT organization. In addition, quantitative analysis revealed that overexpression of PSD-95 significantly increased the proportion of cells that exhibited an irregular MT organization compared with neighboring untransfected cells or to cells transfected with GFP or DsRed1 alone (Fig. 8 C). For each of the indicated constructs, the percentage of transfected cells (Fig. 8 C, white bars) and neighboring untransfected cells (Fig. 8 C, black bars) exhibiting irregular MT arrangement was determined. We used the following criteria to define MT irregularity: (1) absence of a prominent MTOC, (2) circuitous or winding MT bundles, and (3) dense MT cages surrounding an area devoid of tubulin fluorescence signal. For quantification, the investigator was blind to the transfection conditions. As shown in Figure 8 C, the majority of cells transfected with PSD-95-GFP had irregular MTs compared with a small proportion of neighboring untransfected and GFP-transfected cells. Similarly, a majority of cells transfected with PSD-95-DsRed1 had irregular MTs compared with a small percentage of neighboring untransfected cells and cells transfected with DsRed1 alone. To exclude the possibility that this phenomenon was a byproduct of fixation, COS-7 cells were transfected with PSD-95-DsRed1 and tubulin-GFP to visualize MTs in living cells (Fig. 8 B). As above, when COS-7 cells were cotransfected with PSD-95-DsRed1 (Fig. 8 B, inset), the tubulin-GFP assembled into MT bundles that assumed a circuitous pattern (Fig. 8 B, bottom left), compared with neighboring cells transfected only with tubulin-GFP (Fig. 8 B, top right), which exhibited the normal MT arrangement. Thus, in combination with our data in neurons, these results suggest that PSD-95 may act to inhibit dendrite branching by disrupting microtubule organization.
Figure 8.

PSD-95 overexpression disrupts microtubule organization in COS-7 cells. A, COS-7 cells were transfected with cDNA encoding PSD-95 fused to either GFP or DsRed1 and labeled with a mouse monoclonal antibody to acetylated tubulin, to visualize MTs. Control cells were transfected with cDNA encoding GFP or DsRed1 alone. In these cells, PSD-95 fused to either GFP or DsRed1 formed large aggregates in the perinuclear region as well as smaller aggregates throughout the cell. Immunolabeling with anti-acetylated tubulin shows that, in contrast to control cells that exhibit astral MT bundles originating from a single MTOC, most cells transfected with PSD-95 exhibit an irregular MT organization such that the bundles wind circuitously around a central axis. Alternatively, MTs formed tight cages around large PSD-95 aggregates (data not shown). This suggests that PSD-95 alters the distribution of microtubules. To visualize the anti-acetylated tubulin antibody, a Cy5-conjugated goat anti-mouse secondary antibody was used. Scale bar, 40 μm. B, COS-7 cells were transfected with cDNA encoding PSD-95-DsRed1 and tubulin-GFP to visualize the above phenomenon in living cells. The tubulin-GFP was assembled into MT bundles that assumed a circuitous pattern in the cell cotransfected with cDNA encoding PSD-95-DsRed1 (bottom left), compared with a neighboring cell transfected only with cDNA encoding tubulin-GFP (top right), which exhibits a normal microtubule arrangement, as described above. The inset shows the dual-channel image, with PSD-95-DsRed1 fluorescence on the left and tubulin-GFP fluorescence on the right. Note that only the cell on the bottom-left is expressing both PSD-95-DsRed1 and tubulin-GFP. Scale bar: 30 μm; inset, 75 μm. C, Overexpression of PSD-95 fused to either GFP or DsRed1 significantly increased the proportion of cells exhibiting an irregular microtubule arrangement compared with neighboring untransfected cells, or to cells transfected with GFP or DsRed1 alone. For each of the indicated constructs, the percentage of transfected cells (white bars) and neighboring untransfected cells (black bars) exhibiting irregular MT arrangement was determined. As indicated, 72% (±10.6%) of cells transfected with PSD-95-GFP had irregular MTs compared with 16.9% (±2.7%) neighboring untransfected cells and 16.3% (±4.2%) of GFP-transfected cells. Similarly, 60.6% (±17.7%) of cells transfected with PSD-95-DsRed had irregular MTs compared with 15.9% (±0.12%) neighboring untransfected cells and 21.2% (±7.6%) of cells transfected with DsRed1 alone. n values are 2 for GFP, 3 for PSD-95-GFP, 2 for DsRed1, and 2 for PSD-95-DsRed1. *p < 0.05; **p < 0.01 by one-way ANOVA followed by Student–Newman–Keuls test for multiple comparisons. Error bars indicate SE.
Discussion
Regulation at the level of dendritic outgrowth, branching, and patterning is essential for the formation of distinct neuronal circuitry. Although many proteins have been identified that contribute to and influence these processes, little is known about the molecular and cellular mechanisms that regulate dendritic arbors. Our current data indicate that PSD-95, a postsynaptic scaffolding protein, also exhibits a nonsynaptic function in dendritic development. Specifically, our data suggest that PSD-95 acts as a stop signal for proximal dendrite outgrowth and branching. We also identified cypin, a PSD-95 binding protein, as a key regulator that counterbalances the PSD-95 inhibition of branching. Our data suggest that during dendrite maturation, the balance between PSD-95 and cypin influences the course of dendritic branching. A decrease in available PSD-95, either by knockdown strategy or by overexpressing cypin (Firestein et al., 1999; Akum et al., 2004), results in increased secondary dendrite number (Fig. 9 B). Overexpression of PSD-95 resulted in decreased secondary dendrite number; however, primary dendrites remained unchanged, possibly because of inhibition of primary dendrite formation by endogenous PSD-95. Furthermore, we found that the interaction between cypin and PSD-95 shapes dendrite patterning (Fig. 9 B,C), and overexpression of a cypin mutant protein that cannot interact with PSD-95 (cypin-PDZ) resulted in less excess secondary branches (i.e., less dendrites at the primary branch point) (Fig. 9 B). Finally, our data indicate that PSD-95 may inhibit secondary dendrite formation by disrupting the microtubule organization (Fig. 8 A–C). Thus, our data suggest that PSD-95 acts as a stop signal for proximal dendrite branching and that an underlying mechanism may involve binding by cypin to remove PSD-95 from branching sites to promote stable dendrite formation and branching.
The idea that PSD-95 acts as a brake for dendrite branching is quite interesting because the majority of studies have focused on the role of PSD-95 as a scaffolding protein at postsynaptic sites (Kim and Sheng, 2004). Our present study analyzes the role of nonsynaptic PSD-95 in immature neurons. Based on our data, we hypothesize that the clustering of PSD-95 serves to inhibit branching at precise locations. Furthermore, we hypothesize that a decrease in PSD-95 clustering at a potential branch point serves as a signal for dendrite branch formation. Indeed, we find that the majority of branch points (66.7 ± 4.7%) lack PSD-95 clusters. Thus, PSD-95 functions as a maturation protein because mature neurons branch less frequently while beginning to form dendritic spines (Dotti et al., 1988). This model is in agreement with previous data demonstrating that PSD-95 is involved in the morphological maturation of excitatory synapses (El-Husseini et al., 2000). Furthermore, the idea that PSD-95 mediates its effects by disrupting the microtubule cytoskeleton independent of neuronal activity gives insight into mechanisms that underlie how the branching pattern of a neuron is determined.
Why does PSD-95 overexpression affect the proportion of primary dendrites that branch and not the number of excess secondary dendrites at a branch point, whereas PSD-95 knockdown affects the number of excess secondary dendrites and not the proportion of primary dendrites that branch? One explanation is as follows (Fig. 9 B, top three panels). There are predetermined sites for dendrite branching in the neuron, and not all of these sites are saturated with PSD-95. With overexpression of PSD-95, more of these sites become saturated with PSD-95, thus decreasing the probability that a secondary dendrite will arise from a primary dendrite. In contrast, when PSD-95 is knocked down, PSD-95 levels will fall at a subset of the predetermined sites, resulting in the same probability that a particular dendrite will branch. Once PSD-95 drops below a certain level, the primary dendrite will continue to branch, yielding more secondary dendrites at that branch point.
Our data also indicate that PSD-95 may serve to strongly inhibit primary dendrite outgrowth. When hippocampal neuronal cultures were treated with PSD-95 antisense ODNs until 12 DIV, we found the number of secondary dendrites to be significantly increased with only a marginally significant increase in primary dendrites. However, when PSD-95 antisense ODN treatment was maintained until 17 DIV, we found that there was a significant increase in the number of primary dendrites in these neurons (Fig. 7 C). We hypothesize that this increase in primary dendrite formation after prolonged PSD-95 knockdown reveals the strong inhibition by nonsynaptic PSD-95 that is present in high abundance in the soma (Fig. 1 C). Accordingly, only prolonged treatment with PSD-95 antisense ODN can relieve the inhibition by PSD-95 on dendrite outgrowth (Fig. 9 B,C).
The role of PSD-95 in dendrite branching has not been addressed in studies using the PSD-95 knockout mouse (Migaud et al., 1998); however, this mouse expresses a truncated version of PSD-95 containing PDZ 1 and 2. Because this region is important for PSD-95 localization (Craven et al., 1999) and for cypin binding (Firestein et al., 1999), it is not the optimal mutant with which to study the role of PSD-95 in dendrite branching. Furthermore, compensatory mechanisms may occur. Our culture system allows temporal knockdown of PSD-95 protein for assessment of its role in dendrite branching and, thus, decreases the chance of compensatory mechanisms over a small developmental time period.
What is the physiological relevance of decreasing PSD-95 clusters during dendrite branching? PSD-95 has been shown to be involved in the targeting of AMPA receptors (Chen et al., 2000; Chetkovich et al., 2002; Schnell et al., 2002; Colledge et al., 2003) and Kv1.4 (Firestein et al., 1999), as well as signaling molecules such as neuronal nitric oxide synthase (Brenman et al., 1996) to the synapse. Localization of PSD-95 to postsynaptic sites occurs after axodendritic contact and neurotransmitter vesicle and protein complex recruitment to presynaptic sites and coincides with glutamate receptor recruitment (Friedman et al., 2000). Furthermore, NMDA receptor recruitment to synapses can be concurrent with PSD-95 recruitment (Washbourne et al., 2002). It is conceivable that the nonsynaptic PSD-95 clusters detected at early time points (Fig. 1 A) are in transit to developing synaptic sites. In order for a neuron to branch, it is plausible that it would have to deconstruct protein complexes that are destined for postsynaptic sites. Thus, decreased PSD-95 clusters would allow both this deconstruction and dendrite branching simultaneously. PSD-95 would then act as a link between these two processes.
The finding that PSD-95 acts via an activity-independent mechanism to regulate dendrite number provides an intriguing mechanism underlying the nonsynaptic effects of PSD-95. It is known that NMDA and AMPA receptors regulate dendrite branching (Cline and Constantine-Paton, 1990; Rajan and Cline, 1998; Heng et al., 1999; Rajan et al., 1999; Hirai and Launey, 2000; Li et al., 2000), and that PSD-95 is involved in targeting these receptors to the dendritic surface and synapse (Chen et al., 2000; El-Husseini et al., 2000; Schnell et al., 2002; Cuadra et al., 2004; Ehrlich and Malinow, 2004; Lin et al., 2004). However, we found that blocking the activity of these receptors has no effect on regulation of dendrite number by PSD-95. Instead, we found that PSD-95 disrupts the microtubule cytoskeleton, which may lead to decreased dendrite branching (Figs. 8, 9). This is quite different from recent reports of scaffolding proteins and their regulation of dendritic branching. For example, Hoogenraad et al. (2005) have found that the PDZ-containing protein GRIP exerts its effects on dendrite branching via the EphB2 receptor (Hoogenraad et al., 2005).
At the present, our data provide the groundwork for future studies into the cellular mechanisms underlying PSD-95 inhibition of dendritic branching. Previous studies have shown that PSD-95 can bind to adenomatous polyposis coli (APC) (Yanai et al., 2000; Takamori et al., 2006) and that proteins like the +TIPs (microtubule plus-end tracking proteins) interact with APC to regulate microtubule dynamics and targeting (Nakagawa et al., 2000). In addition, PSD-95 may associate with microtubules via CRIPT (cysteine-rich interactor of PDZ3) (Passafaro et al., 1999), and this interaction may be important in regulating dendrite branching. Together, our current data suggest a novel intrinsic regulation of dendrite outgrowth and branching by PSD-95, possibly by the alteration of microtubule dynamics.
Footnotes
This work was supported in part by a Busch Biomedical grant, New Jersey Governor's Council on Autism Pilot grant, National Science Foundation (NSF) Grant IBN-0234206, March of Dimes Foundation Grant 1-FY04-107, a Grant-in-Aid from the American Heart Association (B.L.F), National Institutes of Health (NIH) Grant R01 NS36241 (J.Q.Z.), NIH Grant R01 NS42023, a Grant-in-Aid from the American Heart Association (C.R.), and NSF Grant DMS-0306360 (R.J.J.). C.R. is a Pew Scholar in the Biomedical Sciences. B.F.A. is a Leathem Scholar of the Molecular Biosciences Graduate Program at Rutgers University. E.I.C. is supported by a Pharmaceutical Manufacturers of America Foundation Postdoctoral Fellowship. We thank members of the Firestein laboratory for comments on this manuscript.
References
- Aizawa H, Hu SC, Bobb K, Balakrishnan K, Ince G, Gurevich I, Cowan M, Ghosh A. Dendrite development regulated by CREST, a calcium-regulated transcriptional activator. Science. 2004;303:197–202. doi: 10.1126/science.1089845. [DOI] [PubMed] [Google Scholar]
- Akum BF, Chen M, Gunderson SI, Riefler GM, Scerri-Hansen MM, Firestein BL. Cypin regulates dendrite patterning in hippocampal neurons by promoting microtubule assembly. Nat Neurosci. 2004;7:145–152. doi: 10.1038/nn1179. [DOI] [PubMed] [Google Scholar]
- Brenman JE, Christopherson KS, Craven SE, McGee AW, Bredt DS. Cloning and characterization of postsynaptic density 93, a nitric oxide synthase interacting protein. J Neurosci. 1996;16:7407–7415. doi: 10.1523/JNEUROSCI.16-23-07407.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenman JE, Topinka JR, Cooper EC, McGee AW, Rosen J, Milroy T, Ralston HJ, Bredt DS. Localization of postsynaptic density-93 to dendritic microtubules and interaction with microtubule-associated protein 1A. J Neurosci. 1998;18:8805–8813. doi: 10.1523/JNEUROSCI.18-21-08805.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Chetkovich DM, Petralia RS, Sweeney NT, Kawasaki Y, Wenthold RJ, Bredt DS, Nicoll RA. Stargazin regulates synaptic targeting of AMPA receptors by two distinct mechanisms. Nature. 2000;408:936–943. doi: 10.1038/35050030. [DOI] [PubMed] [Google Scholar]
- Chetkovich DM, Chen L, Stocker TJ, Nicoll RA, Bredt DS. Phosphorylation of the postsynaptic density-95 (PSD-95)/discs large/zona occludens-1 binding site of stargazin regulates binding to PSD-95 and synaptic targeting of AMPA receptors. J Neurosci. 2002;22:5791–5796. doi: 10.1523/JNEUROSCI.22-14-05791.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cline HT, Constantine-Paton M. NMDA receptor agonist and antagonists alter retinal ganglion cell arbor structure in the developing frog retinotectal projection. J Neurosci. 1990;10:1197–1216. doi: 10.1523/JNEUROSCI.10-04-01197.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colledge M, Snyder EM, Crozier RA, Soderling JA, Jin Y, Langeberg LK, Lu H, Bear MF, Scott JD. Ubiquitination regulates PSD-95 degradation and AMPA receptor surface expression. Neuron. 2003;40:595–607. doi: 10.1016/s0896-6273(03)00687-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craven SE, El-Husseini AE, Bredt DS. Synaptic targeting of the postsynaptic density protein PSD-95 mediated by lipid and protein motifs. Neuron. 1999;22:497–509. doi: 10.1016/s0896-6273(00)80705-9. [DOI] [PubMed] [Google Scholar]
- Cuadra AE, Kuo SH, Kawasaki Y, Bredt DS, Chetkovich DM. AMPA receptor synaptic targeting regulated by stargazin interactions with the Golgi-resident PDZ protein nPIST. J Neurosci. 2004;24:7491–7502. doi: 10.1523/JNEUROSCI.1255-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dotti CG, Sullivan CA, Banker GA. The establishment of polarity by hippocampal neurons in culture. J Neurosci. 1988;8:1454–1468. doi: 10.1523/JNEUROSCI.08-04-01454.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich I, Malinow R. Postsynaptic density 95 controls AMPA receptor incorporation during long-term potentiation and experience-driven synaptic plasticity. J Neurosci. 2004;24:916–927. doi: 10.1523/JNEUROSCI.4733-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Husseini AE, Schnell E, Chetkovich DM, Nicoll RA, Bredt DS. PSD-95 involvement in maturation of excitatory synapses. Science. 2000;290:1364–1368. [PubMed] [Google Scholar]
- Fan Y, Zou B, Ruan Y, Pang Z, Xu ZC. In vivo demonstration of a late depolarizing postsynaptic potential in CA1 pyramidal neurons. J Neurophysiol. 2005;93:1326–1335. doi: 10.1152/jn.00734.2004. [DOI] [PubMed] [Google Scholar]
- Fink CC, Bayer KU, Myers JW, Ferrell JE, Jr, Schulman H, Meyer T. Selective regulation of neurite extension and synapse formation by the beta but not the alpha isoform of CaMKII. Neuron. 2003;39:283–297. doi: 10.1016/s0896-6273(03)00428-8. [DOI] [PubMed] [Google Scholar]
- Firestein BL, Brenman JE, Aoki C, Sanchez-Perez AM, El-Husseini AE, Bredt DS. Cypin: a cytosolic regulator of PSD-95 postsynaptic targeting. Neuron. 1999;24:659–672. doi: 10.1016/s0896-6273(00)81120-4. [DOI] [PubMed] [Google Scholar]
- Friedman HV, Bresler T, Garner CC, Ziv NE. Assembly of new individual excitatory synapses: time course and temporal order of synaptic molecule recruitment. Neuron. 2000;27:57–69. doi: 10.1016/s0896-6273(00)00009-x. [DOI] [PubMed] [Google Scholar]
- Gao FB, Brenman JE, Jan LY, Jan YN. Genes regulating dendritic outgrowth, branching, and routing in Drosophila . Genes Dev. 1999;13:2549–2561. doi: 10.1101/gad.13.19.2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grueber WB, Jan LY, Jan YN. Different levels of the homeodomain protein cut regulate distinct dendrite branching patterns of Drosophila multidendritic neurons. Cell. 2003;112:805–818. doi: 10.1016/s0092-8674(03)00160-0. [DOI] [PubMed] [Google Scholar]
- Heng JE, Zurakowski D, Vorwerk CK, Grosskreutz CL, Dreyer EB. Cation channel control of neurite morphology. Brain Res Dev Brain Res. 1999;113:67–73. doi: 10.1016/s0165-3806(98)00191-6. [DOI] [PubMed] [Google Scholar]
- Hirai H, Launey T. The regulatory connection between the activity of granule cell NMDA receptors and dendritic differentiation of cerebellar Purkinje cells. J Neurosci. 2000;20:5217–5224. doi: 10.1523/JNEUROSCI.20-14-05217.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoogenraad CC, Milstein AD, Ethell IM, Henkemeyer M, Sheng M. GRIP1 controls dendrite morphogenesis by regulating EphB receptor trafficking. Nat Neurosci. 2005;8:906–915. doi: 10.1038/nn1487. [DOI] [PubMed] [Google Scholar]
- Hou XY, Zhang GY, Zong YY. Suppression of postsynaptic density protein 95 expression attenuates increased tyrosine phosphorylation of NR2A subunits of N-methyl-d-aspartate receptors and interactions of Src and Fyn with NR2A after transient brain ischemia in rat hippocampus. Neurosci Lett. 2003;343:125–128. doi: 10.1016/s0304-3940(03)00365-3. [DOI] [PubMed] [Google Scholar]
- Hou XY, Zhang GY, Wang DG, Guan QH, Yan JZ. Suppression of postsynaptic density protein 95 by antisense oligonucleotides diminishes postischemic pyramidal cell death in rat hippocampal CA1 subfield. Neurosci Lett. 2005;385:230–233. doi: 10.1016/j.neulet.2005.05.054. [DOI] [PubMed] [Google Scholar]
- Jin X, Hu H, Mathers PH, Agmon A. Brain-derived neurotrophic factor mediates activity-dependent dendritic growth in nonpyramidal neocortical interneurons in developing organotypic cultures. J Neurosci. 2003;23:5662–5673. doi: 10.1523/JNEUROSCI.23-13-05662.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Sheng M. PDZ domain proteins of synapses. Nat Rev Neurosci. 2004;5:771–781. doi: 10.1038/nrn1517. [DOI] [PubMed] [Google Scholar]
- Li Z, Van Aelst L, Cline HT. Rho GTPases regulate distinct aspects of dendritic arbor growth in Xenopus central neurons in vivo. Nat Neurosci. 2000;3:217–225. doi: 10.1038/72920. [DOI] [PubMed] [Google Scholar]
- Lin Y, Skeberdis VA, Francesconi A, Bennett MV, Zukin RS. Postsynaptic density protein-95 regulates NMDA channel gating and surface expression. J Neurosci. 2004;24:10138–10148. doi: 10.1523/JNEUROSCI.3159-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister AK, Katz LC, Lo DC. Neurotrophin regulation of cortical dendritic growth requires activity. Neuron. 1996;17:1057–1064. doi: 10.1016/s0896-6273(00)80239-1. [DOI] [PubMed] [Google Scholar]
- Migaud M, Charlesworth P, Dempster M, Webster LC, Watabe AM, Makhinson M, He Y, Ramsay MF, Morris RG, Morrison JH, O'Dell TJ, Grant SG. Enhanced long-term potentiation and impaired learning in mice with mutant postsynaptic density-95 protein. Nature. 1998;396:433–439. doi: 10.1038/24790. [DOI] [PubMed] [Google Scholar]
- Moore AW, Jan LY, Jan YN. Hamlet, a binary genetic switch between single- and multiple-dendrite neuron morphology. Science. 2002;297:1355–1358. doi: 10.1126/science.1072387. [DOI] [PubMed] [Google Scholar]
- Naisbitt S, Kim E, Tu JC, Xiao B, Sala C, Valtschanoff J, Weinberg RJ, Worley PF, Sheng M. Shank, a novel family of postsynaptic density proteins that bind to the NMDA receptor/PSD-95/GKAP complex and cortactin. Neuron. 1999;23:569–582. doi: 10.1016/s0896-6273(00)80809-0. [DOI] [PubMed] [Google Scholar]
- Nakagawa H, Koyama K, Murata Y, Morito M, Akiyama T, Nakamura Y. EB3, a novel member of the EB1 family, preferentially expressed in the central nervous system, binds to a CNS-specific APC homologue. Oncogene. 2000;19:210–216. doi: 10.1038/sj.onc.1203308. [DOI] [PubMed] [Google Scholar]
- Okabe S, Miwa A, Okado H. Spine formation and correlated assembly of presynaptic and postsynaptic molecules. J Neurosci. 2001;21:6105–6114. doi: 10.1523/JNEUROSCI.21-16-06105.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passafaro M, Sala C, Niethammer M, Sheng M. Microtubule binding by CRIPT and its potential role in the synaptic clustering of PSD-95. Nat Neurosci. 1999;2:1063–1069. doi: 10.1038/15990. [DOI] [PubMed] [Google Scholar]
- Prange O, Murphy TH. Modular transport of postsynaptic density-95 clusters and association with stable spine precursors during early development of cortical neurons. J Neurosci. 2001;21:9325–9333. doi: 10.1523/JNEUROSCI.21-23-09325.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai KS, Murthy KD, Rao MS, Karanth KS. Altered dendritic arborization of amygdala neurons in young adult rats orally intubated with Clitorea ternatea aqueous root extract. Phytother Res. 2005;19:592–598. doi: 10.1002/ptr.1657. [DOI] [PubMed] [Google Scholar]
- Rajan I, Cline HT. Glutamate receptor activity is required for normal development of tectal cell dendrites in vivo. J Neurosci. 1998;18:7836–7846. doi: 10.1523/JNEUROSCI.18-19-07836.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajan I, Witte S, Cline HT. NMDA receptor activity stabilizes presynaptic retinotectal axons and postsynaptic optic tectal cell dendrites in vivo. J Neurobiol. 1999;38:357–368. doi: 10.1002/(sici)1097-4695(19990215)38:3<357::aid-neu5>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- Ruchhoeft ML, Ohnuma S, McNeill L, Holt CE, Harris WA. The neuronal architecture of Xenopus retinal ganglion cells is sculpted by rho-family GTPases in vivo. J Neurosci. 1999;19:8454–8463. doi: 10.1523/JNEUROSCI.19-19-08454.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sans N, Petralia RS, Wang YX, Blahos J, 2nd, Hell JW, Wenthold RJ. A developmental change in NMDA receptor-associated proteins at hippocampal synapses. J Neurosci. 2000;20:1260–1271. doi: 10.1523/JNEUROSCI.20-03-01260.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattler R, Xiong Z, Lu WY, Hafner M, MacDonald JF, Tymianski M. Specific coupling of NMDA receptor activation to nitric oxide neurotoxicity by PSD-95 protein. Science. 1999;284:1845–1848. doi: 10.1126/science.284.5421.1845. [DOI] [PubMed] [Google Scholar]
- Schaefer AT, Larkum ME, Sakmann B, Roth A. Coincidence detection in pyramidal neurons is tuned by their dendritic branching pattern. J Neurophysiol. 2003;89:3143–3154. doi: 10.1152/jn.00046.2003. [DOI] [PubMed] [Google Scholar]
- Schnell E, Sizemore M, Karimzadegan S, Chen L, Bredt DS, Nicoll RA. Direct interactions between PSD-95 and stargazin control synaptic AMPA receptor number. Proc Natl Acad Sci USA. 2002;99:13902–13907. doi: 10.1073/pnas.172511199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamori N, Shimomura A, Senda T. Microtubule-bundling activity of APC is stimulated by interaction with PSD-95. Neurosci Lett. 2006;403:68–72. doi: 10.1016/j.neulet.2006.04.045. [DOI] [PubMed] [Google Scholar]
- Tao F, Tao YX, Gonzalez JA, Fang M, Mao P, Johns RA. Knockdown of PSD-95/SAP90 delays the development of neuropathic pain in rats. NeuroReport. 2001;12:3251–3255. doi: 10.1097/00001756-200110290-00022. [DOI] [PubMed] [Google Scholar]
- Tao YX, Johns RA. Effect of the deficiency of spinal PSD-95/SAP90 on the minimum alveolar anesthetic concentration of isoflurane in rats. Anesthesiology. 2001;94:1010–1015. doi: 10.1097/00000542-200106000-00015. [DOI] [PubMed] [Google Scholar]
- Threadgill R, Bobb K, Ghosh A. Regulation of dendritic growth and remodeling by Rho, Rac, and cdc42. Neuron. 1997;19:625–634. doi: 10.1016/s0896-6273(00)80376-1. [DOI] [PubMed] [Google Scholar]
- Vetter P, Roth A, Hausser M. Propagation of action potentials in dendrites depends on dendritic morphology. J Neurophysiol. 2001;85:926–937. doi: 10.1152/jn.2001.85.2.926. [DOI] [PubMed] [Google Scholar]
- Washbourne P, Bennett JE, McAllister AK. Rapid recruitment of NMDA receptor transport packets to nascent synapses. Nat Neurosci. 2002;5:751–759. doi: 10.1038/nn883. [DOI] [PubMed] [Google Scholar]
- Yanai H, Satoh K, Matsumine A, Akiyama T. The colorectal tumour suppressor APC is present in the NMDA-receptor-PSD-95 complex in the brain. Genes Cells. 2000;5:815–822. doi: 10.1046/j.1365-2443.2000.00368.x. [DOI] [PubMed] [Google Scholar]
- Ye B, Petritsch C, Clark IE, Gavis ER, Jan LY, Jan YN. Nanos and Pumilio are essential for dendrite morphogenesis in Drosophila peripheral neurons. Curr Biol. 2004;14:314–321. doi: 10.1016/j.cub.2004.01.052. [DOI] [PubMed] [Google Scholar]
- Yu X, Malenka RC. Beta-catenin is critical for dendritic morphogenesis. Nat Neurosci. 2003;6:1169–1177. doi: 10.1038/nn1132. [DOI] [PubMed] [Google Scholar]