

Abstract
Spinal muscular atrophy (SMA) is a motor neuron degenerative disease caused by low levels of the survival motor neuron (SMN) protein and is linked to mutations or loss of SMN1 and retention of SMN2. How low levels of SMN cause SMA is unclear. SMN functions in small nuclear ribonucleoprotein (snRNP) biogenesis, but recent studies indicate that SMN may also function in axons. We showed previously that decreasing Smn levels in zebrafish using morpholinos (MO) results in motor axon defects. To determine how Smn functions in motor axon outgrowth, we coinjected smn MO with various human SMN RNAs and assayed the effect on motor axons. Wild-type SMN rescues motor axon defects caused by Smn reduction in zebrafish. Consistent with these defects playing a role in SMA, SMN lacking exon 7, the predominant form from the SMN2 gene, and human SMA mutations do not rescue defective motor axons. Moreover, the severity of the motor axon defects correlates with decreased longevity. We also show that a conserved region in SMN exon 7, QNQKE, is critical for motor axon outgrowth. To address the function of SMN important for motor axon outgrowth, we determined the ability of different SMN forms to oligomerization and bind Sm protein, functions required for snRNP biogenesis. We identified mutations that failed to rescue motor axon defects but retained snRNP function. Thus, we have dissociated the snRNP function of SMN from its function in motor axons. These data indicate that SMN has a novel function in motor axons that is relevant to SMA and is independent of snRNP biosynthesis.
Introduction
Spinal muscular atrophy (SMA) is the leading hereditary cause of infant mortality (Pearn, 1980; McAndrew et al., 1997). Pathologically, SMA is characterized by the loss of α motor neurons within the spinal cord, muscle denervation, and atrophy (Crawford and Pardo, 1996). Genetically, the disease is caused by loss of the survival motor neuron 1 (SMN1) and presence of the SMN2 gene, leading to low SMN protein levels (Lefebvre et al., 1995, 1997; Coovert et al., 1997). A base pair change within the splice enhancer of exon 7 of SMN2 results in a majority of SMN2 RNA lacking exon 7 (SMNΔ7) (Lorson et al., 1999; Monani et al., 1999; Lorson and Androphy, 2000). Exon 7 exclusion results in reduced oligomerization and decreased protein stability (Lorson et al., 1998; Lorson and Androphy, 2000; Young et al., 2000). Whereas the vast majority (94–96%) of SMA cases are caused by homozygous deletion of the SMN1 gene, point mutations in the patient population have also been identified (Wirth, 2000; Sun et al., 2005). SMN protein functions in spliceosomal small nuclear ribonucleoprotein (snRNP) biogenesis, a function that is required in all cell types and is dependent on Sm binding (Liu and Dreyfuss, 1996; Liu et al., 1997; Pellizzoni et al., 1999, 2002; Meister et al., 2000). Although there is precedent for tissue-specific RNA splicing in the nervous system (Jensen et al., 2000), SMN is present in all cell types, and no splicing defects have been reported in SMA patients.
SMN is also found in axons in vivo (Pagliardini et al., 2000) and at branch points and growth cones in cultured motoneurons (Jablonka et al., 2001). Sm proteins are not detected in PC12 axons (Sharma et al., 2005) and are at very low levels and do not colocalize with SMN in motoneuron axons in culture (Zhang et al., 2006). Thus, the question becomes, what function does SMN play in axons? Overexpression studies in chick forebrain neurons demonstrated that SMN is transported down axons; SMNΔ7 protein, in contrast, is not transported, and overexpression of this construct results in shorter axons (Zhang et al., 2003). SMN also binds the RNA binding protein heterogeneous nuclear ribonucleoprotein-R (hnRNP-R) (Rossoll et al., 2002), and SMN-deficient motoneurons have decreased growth cone β-actin (Rossoll et al., 2003). These data raise the possibility that SMN forms a complex in axons that may function to transport RNAs into growth cones, a process shown to be important for growth cone response to guidance cues (Piper and Holt, 2004).
We have shown that motor axon development in vivo is specifically perturbed when SMN levels are decreased (McWhorter et al., 2003). Here we show a strong correlation between motor axon defects and longevity, thus strengthening the link between these defects and SMA. In addition, we show that sequences in SMN exon 7 are critical for motor axon outgrowth, and, using both human and synthetic mutations, we show that SMN function in motor axons is independent of properties needed for snRNP assembly such as oligomerization and Sm binding. These data indicate that SMN has a critical function in motor axons that is independent of snRNP biogenesis.
Materials and Methods
Fish maintenance
Zebrafish and embryos were maintained at ∼28.5°C and staged by hours or days postfertilization (dpf) (Westerfield, 1995). Tg[gata2:gfp] fish, which express green fluorescent protein (gfp) in ventrally projecting motor axons from ∼2–4 dpf, were used for morpholino (MO) and RNA injections. This line is a hybrid between AB* and Tuebingen long fin backgrounds.
Antisense morpholino and synthetic RNA Injections
The antisense MO used to knockdown smn levels was described previously by McWhorter et al. (2003). Each embryo was injected with 9 ng of smn MO with or without 120 pg of synthetic RNA (RNA) using an MPPI-2 Pressure Injector (Applied Scientific Instrumentation, Eugene, OR). Dilutions were made of the MO or MO/RNA mixtures such that 1 nl would deliver the desired amount. To ensure this, the volume of the droplet from the injection needle was measured using a depression slide micrometer filled with mineral oil to measure the diameter of the droplet. The injection pressure and pulse duration was adjusted until the droplet diameter was 124 μm corresponding to 1 nl. The pressure did not exceed 10 psi, whereas the duration varied from 20 to 100 ms. This variability was dependent on the diameter of the glass injection pipette, which ranged between 8 and 10 μm.
RNA vectors
All untagged constructs were in the pCS2+ vector and were linearized with NotI. Following the protocol of the manufacturer, capped RNA was generated using the Sp6 mMESSAGE mMACHINE kit (Ambion, Austin, TX). hSMNΔ7 read-through (Wolstencroft et al., 2005) was linearized using SnaB1. hSMNΔ7-VDQNQKE was reported previously by Zhang et al. (2003), except here the red fluorescent protein tag was removed using BglII and BamHI, filling in the overhangs with Klenow and religating with T4 ligase. The hemagglutinin (HA)-tagged constructs were in pcDNA3 (except the read-through construct, which was in pCS2+ and processed as above) with the HA tag on the 5′ end of the RNA. Constructs were linearized with SmaI, and capped RNA was generated using T7 mMESSAGE mMACHINE kit (Ambion).
Visualization of GFP transgenic fish
Live Tg[gata2:gfp] fish were anesthetized with tricaine, mounted on glass coverslips for observation using a Zeiss (Oberkochen, Germany) Axioplan2 microscope and photographed using a Zeiss AxioCam MRc5 camera and AxioCam 4.2 software.
Longevity assay
Embryos injected with the smn MO or MO and RNA were scored as described, placed into individual wells of a 24-well culture plate, and observed for 7 d. At 7 dpf, the fish were transferred to housing tubes that were constructed from clear acrylic tubes and covered on the bottom end with nylon mesh; 24 tubes were housed within an 11 L tank, so that all 24 fish were exposed to the same water environment. The study was blinded so that the fish designation (severe, moderate, mild, no defects, and control) was unknown to those observing them daily. Each day the fish were observed twice and fed as normal. At the end of the assay, the results were unblinded, and results were analyzed as described below (see Statistical analysis).
Western blot analysis
MO-injected embryos were manually dechorionated and deyolked in a slurry of physiologic Ringer's solution (Westerfield, 1995), Ringer's ice chips, and protease inhibitors (Complete Mini, 1836170; Roche, Indianapolis, IN). Embryo samples were prepared and Western blots were performed as described previously (McWhorter et al., 2003; Monani et al., 2003) with the exception that the HA Western blots used TBS blotto instead of PBS/milk/BSA for blocking. Smn (1:1000; MANSMA7 or MANSMA21) (Young et al., 2000), β-actin (1:3000; catalog #6276-100; Abcam, Cambridge, MA), and HA (1:1000; HA.11 clone 16B2; Covance Research Products, Berkeley, CA) monoclonal antibodies were visualized using the ECL Detection kit (RPN 2109; Amersham Biosciences, Arlington Heights, IL). To quantitate the degree of Smn knockdown, films for three separate experiments were scanned into a Shimadzu (Tokyo, Japan) densitometer. Smn bands were standardized to β-actin and reported as average ± SD.
Chick forebrain culture and transfection
Chick forebrain neurons were cultured as described previously (Zhang et al., 2001, 2003). Briefly, chick forebrains (embryonic day 8) were dissected and trypsinized (0.15% in HBSS) at 37°C for 7 min. After washing in minimum essential medium eagle (MEM) twice, neurons were transfected with the constructs using DOTAP (N-[1-(2,3-dioleoyloxy)propyl]-N,N,N-trimethylammonium methylsulfate) liposomal reagent (Roche) and then plated on poly-l-lysine (0.4 mg/ml) and laminin (0.02 mg/ml) -coated coverslips. Cells were inverted onto a monolayer of astrocytes in N3-conditioned medium with 2% FBS and cultured for 4 d at 37°C in 5% CO2. N3-conditioned medium containing MEM supplemented with transferrin (0.2%), ovalbumin (0.1%), insulin (10 μg/ml), putrescine (32 μg/ml), sodium selenite (26 ng/ml), progesterone (12.5 ng/ml), hydrocortisone (9.1 ng/ml), T3 (3, 3′, 5′-triiodo-l-thyronine, sodium salt; 20 ng/ml), and BSA (10 μg/ml).
Neurons were fixed in paraformaldehyde (4% in PBS) and visualized using an Olympus Optical (Tokyo, Japan) BX61 microscope equipped with a 60× Plan-Neofluar objective, differential interference contrast optics, 100 W mercury arc lamp, and HiQ bandpass filters (Chroma Technology, Brattleboro, VT). Fluorescence images were acquired with narrow bandpass filter (cyanine 2) using a cooled CCD camera (SNHP HQ) that was run by IP Lab computer software (Scanalytics, Fairfax, VA). Exposure time was kept constant (1 s) and below grayscale saturation.
Binding curves
Glutathione S-transferase-fusion protein expression and purification.
The expression and purification were performed essentially as described by Lorson et al. (1998). A total of 100 ml of BL21 cells containing each SMN construct in the pGEX-3X or pGEX-5X-1 vector were cultured. The cells were induced with 1 mm isopropyl-β-d-thiogalactopyranoside for 3 h at 30°C. The cells were pelleted and resuspended in 10 ml of NETN buffer (20 mm Tris, pH 7.5, 500 mm NaCl, 1 mm EDTA, 4 mm NaF, 0.5% NP-40, and 1 mm DTT). The cells were chilled on ice, sonicated four times with a pulse of 30 s duration, and then centrifuged at 12,000 rpm for 20 min. The supernatant was removed and incubated with glutathione Sepharose beads (Amersham Biosciences) for 2 h at 4°C. The beads were washed four times with NETN and then resuspended in 500 μl of NETN. Protein concentration was determined by BCA assay, and then the proteins were visualized by 12% SDS-PAGE and staining with Coomassie blue.
In vitro transcription and translation.
The pcDNA3 vectors were used for in vitro transcription and translation of each insert to produce 35S-labeled protein. This was done as per Promega (Madison, WI) TNT in vitro Transcription and Translation kit specifications. Plasmid DNA was prepared using the Qiagen (Hilden, Germany) mini-prep kit. A reaction containing the master mix, radiolabeled methionine, and DNA template was set up and incubated for 90 min at 30°C.
Glutathione S-transferase-fusion protein binding assays.
The glutathione S-transferase-tagged proteins bound to glutathione beads (0.5, 1, 2, 4, and 6 μg) were incubated for 1 h at 4°C with 4 μl of 35S-labeled proteins in BB-100 buffer (25 mm Tris, pH 7.5, 10% glycerol, 0.1% NP-40, 100 mm NaCl, 5 mm MgCl2, 5 mm DTT, and 1 mm PMSF) . The beads were then washed three times with BB-400 buffer (25 mm Tris, pH 7.5, 10% glycerol, 0.1% NP-40, 400 mm NaCl, 5 mm MgCl2, 5 mm DTT, and 1 mm PMSF) . The bound protein was eluted with 6× SDS loading buffer, boiled, and then run on 12% SDS-PAGE gel. The gel was exposed to a phosphor screen and then quantitated with the Storm 860 PhosphoImager (GE Healthcare, Piscataway, NJ) and ImageQuant software (GE Healthcare).
Statistical analysis
Where stated, the quantitative data are presented as the mean ± SE. Distribution of larval classifications (severe, moderate, mild, and no defects) was analyzed using Mann–Whitney nonparametric rank test. Survival was evaluated using Kaplan–Meier survival curves, and statistical significance was verified via the log-rank test (SPSS version 13; SPSS, Chicago, IL).
Results
Classification of the motor axon defects observed in smn morphants
To address the function of SMN in motor axons, we asked what properties of SMN were necessary for motor axon outgrowth. We reduced the Smn level in zebrafish embryos using smn MO, added back mRNA (RNA) encoding different forms of human SMN (hSMN), and assayed the effect on motor axons. To standardize the assay, we developed a classification for the nerve defects observed in smn MO fish (smn morphants) at 2 dpf. Using a set of criteria, motor axonal phenotypes were classified as being severe, moderate, or mild (Table 1). For this and all characterization, we focused on nerves 7–16 on each side of Tg[gata2:gfp] embryos (Meng et al., 1997; McWhorter et al., 2003). Only fish with normal body morphology were scored; fish with delayed development or shortened, curved bodies (∼15% of smn morphants and ∼10% of control MO) were never examined because of possible secondary effects on motor axon outgrowth. After nerves were scored, individual fish could themselves be categorized according to the type and number of defects present (Table 2, Fig. 1). Using this classification system, we found smn morphants to be categorized as 15.5% severe, 22.3% moderate, 32.7% mild, and 29.1% unaffected. Western blot revealed that each class of embryos had an ∼64% decrease in Smn levels (Fig. 2) (severe, 66.8 ± 18.5%; moderate, 60.8 ± 21%; mild, 63.4 ± 18.5%; and no defects, 64.7 ± 14.2%). This is similar to the SMN levels found in human fetal SMA tissue (∼77–61% decrease in liver and spinal cord) (Lefebvre et al., 1997). Because of the mosaicism inherent in MO knockdown in zebrafish, this range of motor axon defects may be caused by different levels of Smn in individual motoneurons, which we cannot quantitate. Alternatively, this range of motor axon defects may be a natural consequence of low SMN levels.
Table 1.
Classifications for axon defects observed in smn MO-injected fish
| Motor axon defect | Classification |
|---|---|
| Truncated at the horizontal myoseptum | Severe |
| Truncated at the horizontal myoseptum and excessively branched | Severe |
| Excessively branched but not truncated | Moderate |
| Innervates neighboring myotome | Moderate |
| Ectopic branches or ventral roots but overall normal axon morphology | Mild |
| Defasciculated axons | Mild |
| Axons lacking stereotyped morphology but no excessive branches | Mild |
Motor axon defects caused by the reduction of Smn in zebrafish were observed at 2 dpf, and the severity was assigned to each type.
Table 2.
Fish classification determined by motor axon phenotypes
| Classification | Fish designation |
|---|---|
| Severe | Fish with at least 2 severe defects |
| Fish with >4 moderate defects | |
| Moderate | Fish with 1 severe defect |
| Fish with 2–4 moderate defects | |
| Fish with >4 mild defects | |
| Mild | Fish with 1 moderate defect |
| Fish with 2–4 mild defects |
The number and type of motor axon defects were determined (see Table 1), and each individual fish was assigned a classification.
Figure 1.

Classification of motor axon defects caused by Smn reduction. Motor axons were scored in Tg[gata2:gfp] embryos at 2 dpf. When compared with larvae in which motor axonal pathways follow the stereotypical pattern (A), severe fish display multiple truncations with (B) or without branching. Moderate fish show a more chimeric display of defects ranging from multiple nerves innervating the neighboring myotome (C) to a single truncation (D).
Figure 2.

Analysis of the Smn protein levels in the morphant classification groups. Smn Western blot analysis using protein from 2 dpf embryos scored as severe (lane 2), moderate (lane 3), mild (lane 4), and no defects (lane 5) compared with wild type (lane 1). β-Actin is used as a loading control.
As predicted based on our previous work (McWhorter et al., 2003), when we added back full-length hSMN, by coinjecting smn MO and hSMN RNA, we partially rescued the motor axon defects (Figs. 3, 4) (p < 0.001) (for all statistics, see supplemental Fig. 1, available at www.jneurosci.org as supplemental material). After coinjection with full-length hSMN, the severe phenotype was almost completely rescued with an average of 0.8% severe fish compared with 15.5% for smn morphant fish. We next examined SMNΔ7 RNA, the predominant RNA produced from the hSMN2 gene that generates a protein that cannot compensate for full-length SMN. After coinjection of this RNA with smn MO, there was no significant rescue of the motor axon phenotypes compared with smn MO alone (Fig. 4) (p = 0.97). The severe phenotype was observed in 13% of those fish that received both smn MO and SMNΔ7 RNA compared with 15.5% for smn MO alone. Control MO and hSMN RNA alone show very few motor axon defects. These results establish that we can use this assay to test which SMN protein forms can suppress the motor axon defects caused by low Smn levels.
Figure 3.

Diagrams of the human RNAs used to analyze SMN function in motor axons. SMN exons (ex) and the name of the constructs are denoted. Asterisks indicate the location of single nucleotide changes in the SMN sequence.
Figure 4.

Full-length hSMN, but not SMN lacking exon 7, rescues motor axon defects caused by reduction of Smn. Tg[gata2:gfp] zebrafish injected with smn MO and scored at 2 dpf (n = 407 fish, 8140 nerves) using criteria in Tables 1 and 2 resulted in a distribution of fish with motor nerve defects. Coinjection of smn MO with full-length hSMN RNA was able to partially rescue the nerve defects (p < 0.001; n = 110 fish, 2200 nerves), whereas coinjection with hSMNΔ7 did not (p = 0.97; n = 192 fish, 3840 nerves). Control (ct) MO (n = 86 fish, 1720 nerves) and hSMN RNA alone (n = 104 fish, 2080 nerves) are shown as controls. For details on the statistical analysis for this and similar figures, see Materials and Methods.
Human SMN mutations and the role of exon 7
Various missense mutations have been reported to give rise to SMA (Wirth, 2000). To test how mutant SMN proteins affect motor axon defects caused by low Smn levels, RNA encoding the human mutations G279V and Y272C were coinjected with smn MO. Motor axon defects in these fish were similar to smn morphants (Fig. 5) (p = 0.77 and 0.26, respectively) indicating that SMN forms that cause SMA are unable to rescue motor axon defects; this is consistent with these defects playing a role in this disease.
Figure 5.

RNAs encoding human SMA mutations fail to rescue motor axon defects caused by the reduction of Smn. Motor nerves in G279V RNA coinjected fish (p = 0.77; n = 117 fish, 2340 nerves) and Y272C RNA coinjected fish (p = 0.26; n = 89 fish, 1780 nerves) were not statistically different from smn morphant fish.
In SMN exon 7, there is a highly conserved motif, QNQKE (Fig. 6 A). It had been reported previously that the motif VDQNQKE, when added to SMN exons 1–6, resulted in a cellular distribution the same as full-length SMN when tested in cell culture (Zhang et al., 2003). To test whether this motif was capable of rescuing motor axon defects caused by low Smn levels, hSMNΔ7-VDQNQKE RNA (Fig. 3) was coinjected with smn MO into zebrafish embryos. These embryos displayed a rescue similar to that of full-length SMN (p = 0.54). The severe phenotype was not observed in any of the fish scored, whereas >60% showed no nerve defects (Fig. 6 B), demonstrating that the VDQNQKE motif has the ability to rescue the function that is disrupted by the loss of the entire exon 7. Interestingly, the G279V mutation has the sequence VFRQNQKE, and, as shown above, it fails to rescue the motor axons defects. Thus, although the 279V position is mutated the same in both of these proteins, they do not behave the same and VDQNQKE is capable of suppressing the motor axon phenotype.
Figure 6.

Addition of the VDQNQKE motif to the SMNΔ7 sequence rescues motor axon defects caused by the reduction of Smn. A, Alignment of exons 6 and 7 from different species. Arrow denotes the beginning of exon 7 (G279 in human). The QNQKE motif (red) has a high degree of evolutionary conservation most notably the first glutamine (Q282) and the glutamic acid (E286). B, Motor nerves in hSMNΔ7-VDQNQKE RNA (n = 117 fish, 2340 nerves) coinjected fish were significantly different when compared with motor nerves in smn morphant fish (p < 0.001) and equivalent to the rescue seen with the full-length hSMN RNA (p = 0.54).
To further test the importance of the QNQKE motif, we assayed a construct that restored SMN distribution in cells but was composed of random amino acids added to hSMNΔ7. In this construct, SMN exons 1–6 are followed by the first 30 nucleotides of exon 8 because of a mutated stop codon (Wolstencroft et al., 2005) (Fig. 3, hSMNΔ7 read-through). As a result, the hSMN exon 1–6 is followed by nine amino acids (EMLAASSTK). Using overexpression studies in chick forebrain neurons, we show that this construct, like full-length hSMN and hSMNΔ7-VDQNQKE, is transported down axons (Fig. 7 A–C). However, hSMNΔ7 read-through was not able to rescue the motor axon defects caused by low Smn levels (Fig. 7 D) (p = 0.08 compared with smn morphants). This result suggests that, whereas the addition of random amino acids to the end of the hSMNΔ7 protein allows normal distribution in the presence of wild-type protein, the function of SMN in motor axons is dependent on specific sequences in exon 7.
Figure 7.
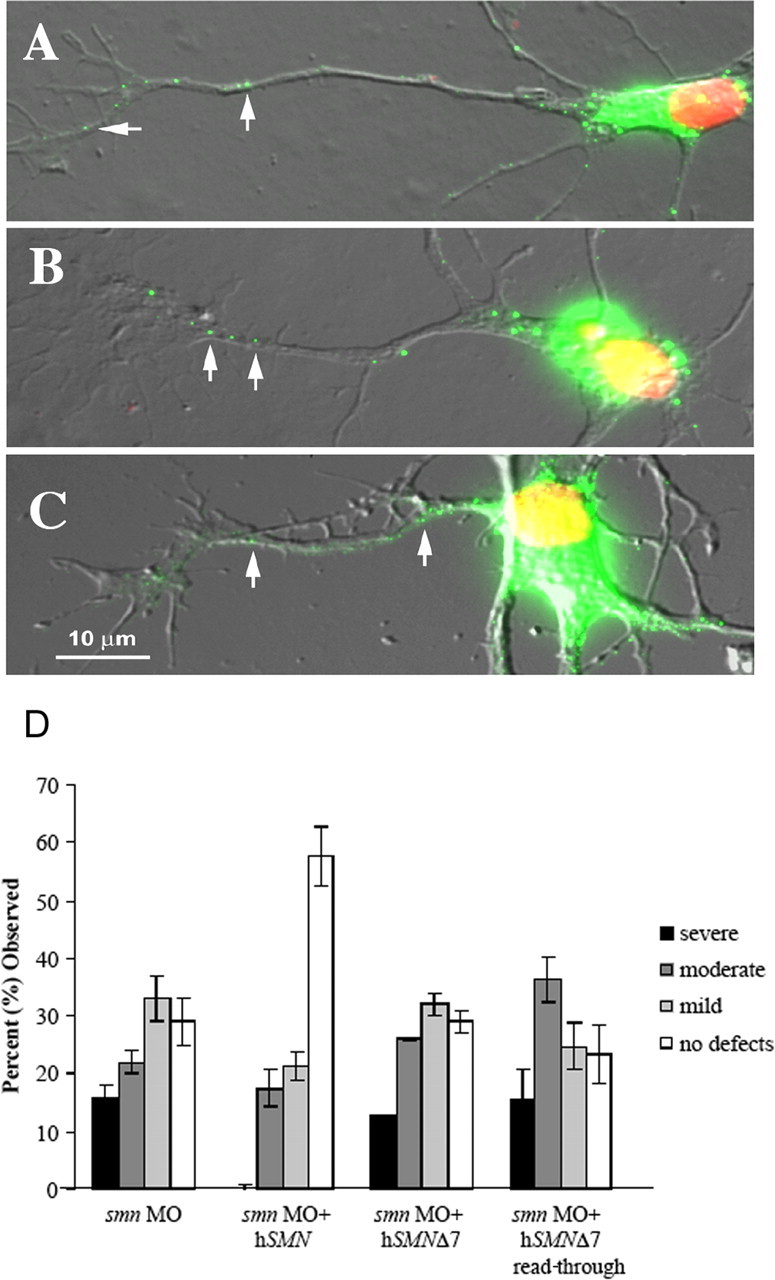
Amino acids added to SMNΔ7 enable transport into axons, but only exon 7-specific sequences rescue motor axon defects. Fluorescently tagged DNA constructs were transfected into cultured chick cortical neurons, and the distribution of the tagged protein was analyzed. Arrows indicate the presence of SMN protein in the axonal projections. Like wild-type SMN protein (A), SMNΔ7-VDQNQKE (B) and SMNΔ7 read-through (C) proteins are transported out of the nucleus and into the axons. D, SMNΔ7-VDQNQKE rescued motor axon defects in smn morphants (see Fig. 6). This was not the case with SMNΔ7 read-through (n = 85 fish, 1700 nerves), which was not statistically different from SMNΔ7 (p = 0.11) or smn MO alone (p = 0.08).
Specificity of the amino acids within the QNQKE motif
Evolutionary conservation of the first glutamine (Q) and the glutamic acid (E) within the QNQKE motif (Fig. 6 A) lead us to evaluate the functional importance of these two amino acids. By changing them individually to an alanine (Q282A and E286A, respectively) and coinjecting the resultant RNAs with the smn MO, we were able to test the functional significance of this exon 7 region (Fig. 8). E286A RNA rescued the axon defects caused by the reduction of Smn (Fig. 8) (p < 0.001 compared with smn morphants), demonstrating that, although it is conserved, the glutamic acid does not affect SMN function in motor axons. Conversely, Q282A RNA failed to rescue the motor axon defects caused by low levels of Smn (Fig. 8). In fact, adding this RNA caused an exacerbation of the severity of the motor axon defects observed with the smn MO alone (p = 0.02). To determine whether the Q282A RNA is acting as a dominant negative, it was injected alone into zebrafish. When compared with the injection of wild-type hSMN RNA, Q282A RNA led to motor axon defects, even in the absence of smn MO (p < 0.001) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). These data support the hypothesis that the exon 7 QNQKE motif, and in particular the highly conserved Q282, is critical for SMN function in motor axons.
Figure 8.

Evaluation of the QNQKE motif. Substitution of an alanine for the glutamic acid (E286A) followed by coinjection with smn MO rescues as well as hSMN RNA (p = 0.10; n = 152 fish, 3040 nerves). The same change at the first glutamine residue (Q282A; n = 106 fish, 2120 nerves) coinjected with smn MO leads to defects that are statistically worse than hSMN RNA coinjections (see Fig. 4) (p < 0.001).
SMN oligomerization and Sm binding does not correlate with the ability to rescue motor axon defects
SMN has a well characterized function in snRNP biogenesis (Liu et al., 1997) (for review, see Yong et al., 2004). However, it is unclear whether this is the functional deficit that causes SMA. Because we have identified SMN protein forms that alter the exon 7 QNQKE motif and either rescue (hSMNΔ7-VDQNQKE) or fail to rescue (hSMN Q282A) motor axon defects caused by low levels of Smn, we next asked whether these proteins could perform functions necessary for snRNP biogenesis such as self-association, Sm binding, and binding to wild-type SMN (Fig. 9). hSMNΔ7-VDQNQKE, which rescued motor axon defects in our assay, failed to efficiently self-associate, bind Sm protein, or bind SMN. Indeed, this protein was similar in its binding capacities to SMN lacking any exon 7 sequences such as hSMNΔ7 and hSMNΔ7 read-through. So although hSMNΔ7-VDQNQKE does not retain functions necessary for snRNP biogenesis, it rescues motor axon defects caused by low Smn levels. Conversely, Q282A bound both wild-type SMN and Sm protein with an affinity comparable with wild-type SMN; however, it completely failed to rescue motor axon defects. To confirm that we could dissociate motor axon function from snRNP functions, we tested the human mutation A111G (Sun et al., 2005). This SMN mutant protein undergoes normal Sm core assembly (Shpargel and Matera, 2005) and, in our binding assay, was able to bind SMN and had an intermediate level of self-association and Sm binding (Fig. 9). We found that this mutation could not rescue motor axon defects caused by low levels of Smn (Fig. 10). These results show that aspects of SMN function that are essential for snRNP biogenesis are not critical to the function of SMN in motor axons. Together, these data support the hypothesis that this exon 7 motif, and in particular glutamine 282, imparts an important function for SMN in axons that is independent of snRNP function (Table 3).
Figure 9.
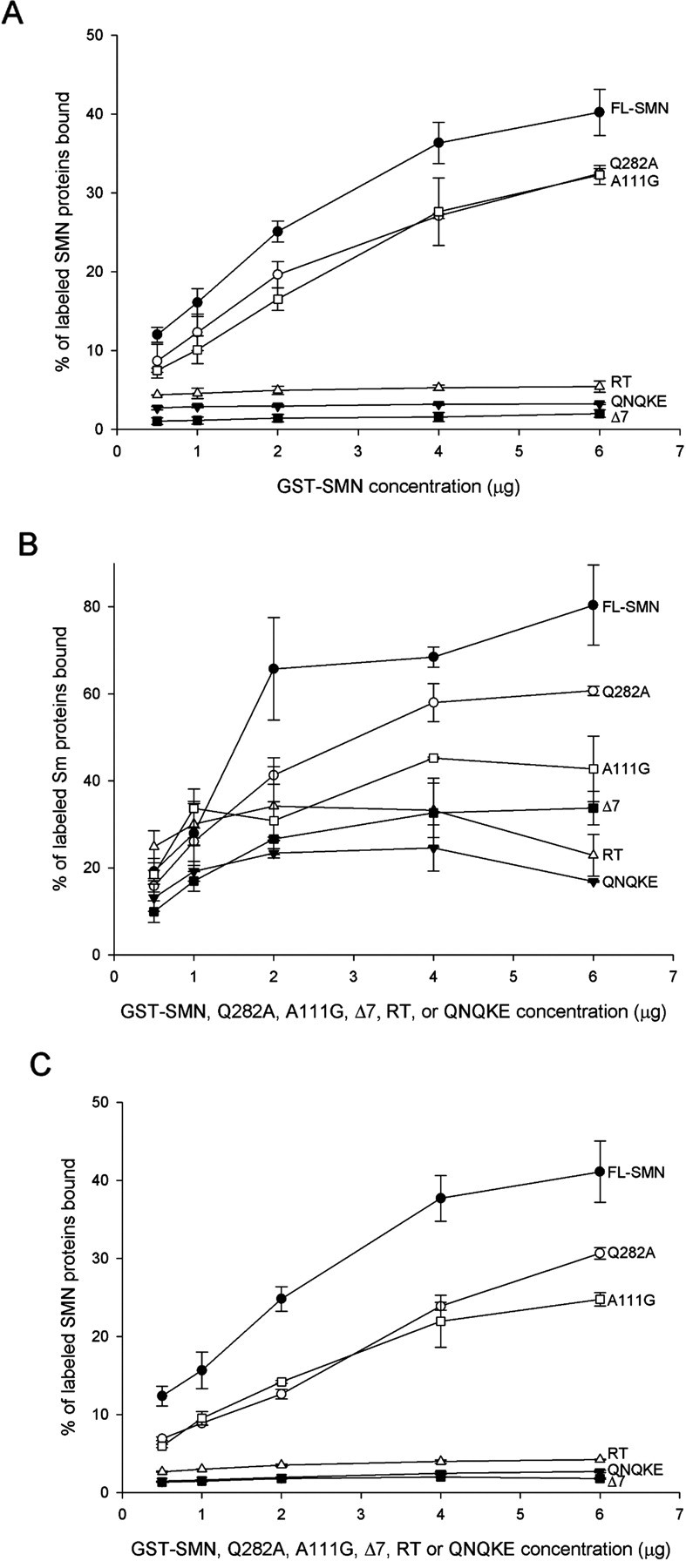
Analysis of SMN binding properties. The Q282A mutation maintains the ability to self-associate (A), bind to Sm proteins (B), and bind to wild-type SMN (C) and does so in a manner that is similar (within 2 SDs) to full-length SMN (FL-SMN). This is unlike the other three proteins tested: hSMNΔ7 (Δ7), hSMNΔ7-VDQNQKE (QNQKE), and hSMNΔ7 read-through (RT) that showed minimal ability to perform these functions. The human mutation A111G can self-associate and has moderate Sm and SMN binding. The Sm protein used in this assay is the neuronal form (SmN), which is most similar to SmB/B−.
Figure 10.

Sm core assembly does not lead to motor axon rescue. RNA encoding SMA mutation A111G, which retains the ability to undergo Sm core assembly, does not rescue motor axon defects caused by low Smn levels. n = 92 fish, 1840 nerves; p = 0.12 versus smn MO alone; p < 0.001 versus smn MO plus hSMN.
Table 3.
SMN rescues anomalies caused by low Smn levels without self-association or Sm binding
| Rescues motor axon defects | Self-associates | Binds Sm protein | |
|---|---|---|---|
| hSMN | + | + | + |
| hSMNΔ7 | − | − | − |
| hSMNΔ7-VDQNQKE | + | − | − |
| hSMN Q282A | − | + | + |
| hSMNΔ7 read-through | − | − | − |
| hSMN G279V | − | − | − |
| hSMN Y272C | − | − | − |
| hSMN A111G | − | + | + a |
The ability of the SMN forms to rescue the motor neuron defects caused by low levels of Smn, their capacity to self-associate, and their ability to bind Sm protein are listed.
a Shpargel and Matera (2005) showed no decreased in Sm core assembly.
To ensure that nonrescuing hSMN forms made stable protein, we added an HA epitope to the N terminus of the hSMN constructs, thus enabling us to specifically detect the human protein. Embryos were coinjected with the HA-tagged RNA, and smn MO and protein from these embryos was analyzed by Western blot (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). As expected, we detected full-length HA-tagged hSMN but could not detect HA-hSMNΔ7, consistent with previous experiments showing the instability of this protein form (Lorson and Androphy, 2000). hSMNΔ7 read-through, the human mutation A111G, and the synthetic mutation Q282A, which all failed to rescue motor axon defects, were all present at levels similar to full-length hSMN. Thus, the inability of these hSMN protein forms to rescue motor axon defects, with the exception of hSMNΔ7, was not attributable to insufficient protein levels.
Motor axon defects caused by low levels of Smn correlates with decreased longevity
SMA decreases longevity in both mouse models and human patients. If these motor axon defects are relevant to SMA, then we would expect them to correlate with a decreased lifespan. To test this, smn morphant fish were classified as severe, moderate, mild, or no defects, and along with control MO fish and wild types were monitored for 30 dpf. During this period, they were placed into individual housing tubes within one fish tank so they could be monitored separately but in a common environment. In blinded experiments, fish survival was monitored daily. Kaplan–Meier plots, in which 27 severe, 52 moderate, 42 mild, 20 no defects, 24 control injected, and 48 uninjected fish were evaluated (Fig. 11 A) (supplemental Fig. 4A, available at www.jneurosci.org as supplemental material). Death in the severe fish occurred rapidly and to a greater extent than in the other groups. This was found to be significantly different from all other groups analyzed (p < 0.006) (supplemental Fig. 4A, available at www.jneurosci.org as supplemental material). smn morphant fish with no motor axon defects had approximately the same amount of death as smn MO mild fish, but it occurred at a slower rate and was not significantly different from control MO or wild-type fish. Although there was more death observed in the control MO-injected fish compared with those that received no injection, this difference was not significant (p = 0.308). Western blot indicates that the overall Smn levels in these fish are similar (Fig. 2) and that Smn protein levels return to normal between 14 and 17 dpf (data not shown); thus, the severity of the motor axon defects observed in the fish that have reduced Smn protein levels correlates with survival of these fish.
Figure 11.
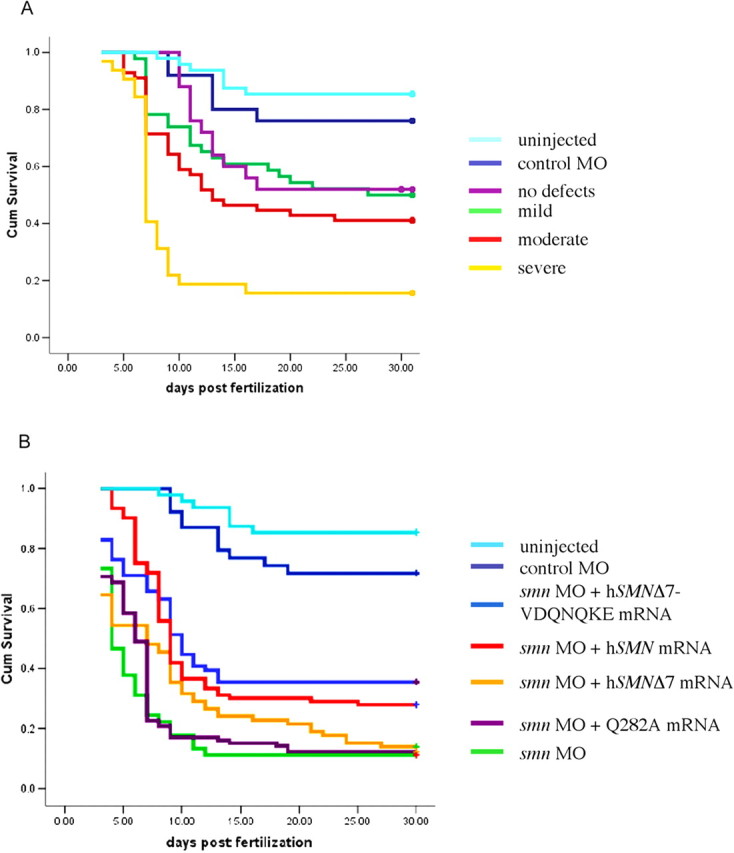
Severity of motor axon defects correlates with decreased survival. A, smn morphant fish were scored (severe, moderate, mild, and no defects), placed in individual housing tubes, and monitored up to 30 dpf. Severe fish (n = 27) died more abruptly, and to a greater extent, than the other groups (p = 0.006 vs moderates; p < 0.001 vs all other groups). Moderate (n = 52) and mild (n = 42) fish also showed a significant difference when compared with the control MO (n = 24) injected fish (p = 0.006 and 0.05, respectively). The fish with no observable differences (n = 20) were not significantly different from the mild category (p = 0.619) or the control MO group (p = 0.236). The control MO and uninjected (n = 48) fish had no difference in their survival (p = 0.308). B, To evaluate the effect of SMN forms on the survival of the fish, RNA was coinjected with smn MO and survival was monitored. Compared with the smn MO-injected fish, the p values are as follows for the coinjections: hSMN, p < 0.001, n = 93; hSMNΔ7, p = 0.152, n = 79; hSMNΔ7-VDQNQKE, p < 0.001, n = 76; Q282A, p = 0.224, n = 106; control MO, p < 0.001, n = 39; uninjected, p < 0.001, n = 48. The solid dots at the end of each plotted line are representative of fish that were still alive at the end of the survival assay. The difference in color of the last dot and the line is an artifact of the SPSS software.
Because motor axon defects correlated with survival, we would predict that adding back rescuing RNAs should increase survival. We first tested how long exogenous hSMN protein was present and found that HA-tagged hSMN was present at 3 dpf but gone by 6 dpf (supplemental Fig. 3B, available at www.jneurosci.org as supplemental material). We coinjected smn MO and RNAs that rescued motor axons and found that hSMN (n = 93) and hSMNΔ7-VDQNQKE (n = 76) could partially rescue longevity (p < 0.001 for both compared with smn MO morphants) (Fig. 11 B) (supplemental Fig. 4B, available at www.jneurosci.org as supplemental material). Thus, when motor axon defects are decreased, longevity is increased. Consistent with this, we found that hSMNΔ7 RNA (n = 79) was unable to rescue the death caused by the reduction of Smn (p = 0.152 compared with smn MO morphants). hSMN Q282A (n = 106), which like hSMNΔ7 was unable to rescue the motor neuron defects, also failed to improve survival (p = 0.224 compared with hSMNΔ7). These results suggest that the motor axon defects caused by low Smn levels have long-term consequences for survival and that SMN RNA forms that fail to rescue the motor axon defects, such as the Q282A, also failed to improve longevity.
Discussion
SMN has been shown to function in snRNP assembly; however, there is growing evidence that SMN may have other functions particularly in axons (Briese et al., 2005; Monani, 2005). We have shown that decreasing Smn levels during zebrafish development leads to motor axon defects. Although these defects can be partially rescued by the reintroduction of human SMN, SMN forms associated with SMA, such as hSMNΔ7 and the human mutations Y272C, G279V, and A111G, fail to do so supporting that these motor axon defects are an assay of SMN function and are relevant to SMA. Our data demonstrate that specific amino acids encoded by exon 7, QNQKE, are critical for the axonal function of SMN. It appears that the position or context of this domain is important because VDQNQKE corrects the motor axon defects, whereas the missense mutation G279V (VFRQNQKE), does not. SMN forms that do not efficiently oligomerize or bind Sm protein still correct motor axons defects, indicating that these properties, although essential for the function of SMN in snRNP assembly, are not critical for its function in motor axons.
SMN function
The predominant argument as to the role of mutant SMN in the genesis of SMA has centered on the known function of SMN in snRNP assembly. Although SMA patients do have a reduction in this capacity, this activity is still present in patient cells (40% in type I patient fibroblasts) (Wan et al., 2005). Because snRNP function is required in all cell types, the question remains, why SMA is a neuromuscular disease? Recent data have led to the hypothesis that, in addition to its role in snRNP biogenesis, SMN has an additional function in axons (for review, see Briese et al. 2005; also discussed below). Our findings support the hypothesis of an additional role for SMN in axons that is functionally independent of snRNP biogenesis. hSMNΔ7-VDQNQKE is able to rescue motor axon anomalies caused by the reduction of Smn but cannot self-associate or bind Sm protein, two properties essential for snRNP activity. Conversely, the Q282A mutation causes an aggravation of motor axon defects, above that of the smn MO alone, but retains functions required for snRNP assembly. Finally, the human mutation A111G oligomerizes and has Sm binding capacity and normal Sm core assembly (Shpargel and Matera, 2005) but fails to rescue motor axons in our assay. Together (Table 3), these data support the two-role hypothesis and implicate an snRNP-independent function of SMN in axons.
Comparison of G279V (VFRQNQKE) and hSMNΔ7-VDQNQKE, which rescues motor axons, shows that the VDQNQKE is an alteration that restores wild-type function (i.e., an intragenic suppressor). This restoration of function may be attributable to a change in the three-dimensional structure, amino acid spacing, or charge. These types of reversions are informative because they have the potential to identify novel functional domains. Indeed, identification of this intragenic suppressor followed by site-directed mutagenesis of the Q282 in the wild-type protein (GFRQNQKE) has identified this region as important for SMN function in motor axons. Identification of intragenic suppressors has been used to study protein function especially in yeast, worms, and flies. Our data show that this approach can be applied to neurological disorders and sets a precedent for this approach as a means toward understanding aberrant protein function in other nervous system diseases.
Because the A111G and the Y272C mutations have an intact GFRQNQKE motif, it suggests that these mutations effect other domains relevant to SMN function in axons. Indeed, the A111G mutation falls within the tudor domain, which has been shown to bind methylarginine proteins (Cote and Richard, 2005), such as Sm proteins (Buhler et al., 1999; Pellizzoni et al., 1999). However, it is highly likely that other methylarginine proteins bind this region as well (Cote and Richard, 2005). Although A111G has decreased affinity for Sm protein (Fig. 9), it can still assemble the Sm core (Shpargel and Matera, 2005). It is possible that the SMN A111G tudor domain has a more substantial decrease in affinity for a protein important for axonal function, thus leading to motor axon defects.
Zebrafish motor axons as an assay for SMN function
Tissue culture data revealed that motor axons from SMA mice have impaired outgrowth (Rossoll et al., 2003). Although a Drosophila model of SMA has shown defects at the neuromuscular junction (Chan et al., 2003), no other in vivo model of SMA except zebrafish (McWhorter et al., 2003; Winkler et al., 2005) has been reported as having motor axon outgrowth defects. It is possible that zebrafish motor axons are less refractory to low levels of Smn protein or that there is no compensatory mechanisms in this animal. Regardless, the forms of SMN that cause disease in humans do not rescue motor axon defects in zebrafish, indicating that motor axon defects can be used to assay SMN function. The longevity data also show a strong link between the severity of the motor axon defects and death. Protein from injected RNA is gone by 6 dpf, and, despite a decrease in motor axon defects, the fish were not rescued to control levels in our longevity study (Fig. 11). This suggests that SMN is not only needed for motor axon outgrowth but also in other events, perhaps maintenance of motor nerves or muscle–nerve interactions. Another study knocked down Gemin2 in zebrafish and found motor axon defects that could be rescued by introduction of snRNPs (Winkler et al., 2005). This study, however, examined motor axon defects in fish with abnormal morphology (developmentally delayed, shortened body axis, kinked tail), thus raising the possibility that the motor axon defects were secondary to the abnormal body morphology. It is possible that reintroduction of snRNPs rescued the developmental defects and secondarily the motor axon defects. Determining the cell autonomy of Gemin2 will help resolve this issue because we have shown previously that SMN functions cell autonomously in motoneurons (McWhorter et al., 2003).
Indications that SMN might function outside of snRNP assembly first came from analysis of mouse spinal cord sections that revealed that SMN was present in dendrites and axons (Pagliardini et al., 2000). In addition, SMN was present at axon branch points and growth cones in cultured motoneurons and neuronal-like cells (Jablonka et al., 2001; Fan and Simard, 2002). This was supported by work from Zhang et al. (2003) that showed that SMN–GFP in neuronal cultures was transported down axons in granules in a microtubule-dependent manner. It has also been shown that Sm proteins are not present or only present at very low levels in axons, implying that SMN has different binding partners in axons (Sharma et al., 2005; Zhang et al., 2006); such axon binding partners may include the RNA binding protein hnRNP-R (Rossoll et al., 2002, 2003). Work from Rossoll et al. (2003) showed that motoneurons from SMA mice are short and have less β-actin in their growth cones, and this effect is mediated by both SMN and hnRNP-R (Rossoll et al., 2003). Along with our in vivo data showing that motor axons extend abnormally when Smn levels are decreased (McWhorter et al., 2003) and the work presented here, a clearer picture is beginning to emerge. In addition to a complex involved in snRNP assembly, SMN may also form a complex with proteins such as hnRNP-R to bind and transport RNAs down axons to growth cones. This complex would not depend on SMN oligomerization but would be mediated by the C-terminal QNQKE motif and perhaps the tudor domain. Extending axons need localized protein translation to respond to guidance cues and for stabilizing synapses (Piper and Holt, 2004). Decreased SMN levels may impair this process, leading to developmental and functional deficits that could ultimately cause axon retraction, muscle denervation, and motoneuron death. We can test these ideas in zebrafish by analyzing the long-term effects of Smn knockdown and whether RNA transport and localized protein translation in the growth cone in vivo is compromised in Smn-deficient animals.
Footnotes
This work was supported by National Institutes of Health Grants RO1NS41649 and RO1NS050414 (C.E.B., A.H.M.B.), RO1NS038650 (A.H.M.B.), RO1NS41584 (C.L.), and P30-NS045758. We thank the Spinal Muscular Atrophy Foundation for supporting both C.E.B. and G.J.B., FightSMA for supporting C.L., the Families of SMA for supporting M.L.M., and The Miracle for Madison Fund at The Ohio State University. We thank the fish facility staff for fish care, Erin Horan for help monitoring the fish in housing tubes, and Lei Xing for assistance with cDNA constructs.
References
- Briese M, Esmaeili B, Sattelle DB. Is spinal muscular atrophy the result of defects in motor neuron processes? BioEssays. 2005;27:946–957. doi: 10.1002/bies.20283. [DOI] [PubMed] [Google Scholar]
- Buhler D, Raker V, Luhrmann R, Fischer U. Essential role for the tudor domain of SMN in spliceosomal U snRNP assembly: implications for spinal muscular atrophy. Hum Mol Genet. 1999;8:2351–2357. doi: 10.1093/hmg/8.13.2351. [DOI] [PubMed] [Google Scholar]
- Chan YB, Miguel-Aliaga I, Franks C, Thomas N, Trulzsch B, Sattelle DB, Davies KE, van den Heuvel M. Neuromuscular defects in a Drosophila survival motor neuron gene mutant. Hum Mol Genet. 2003;12:1367–1376. doi: 10.1093/hmg/ddg157. [DOI] [PubMed] [Google Scholar]
- Coovert DD, Le TT, McAndrew PE, Strasswimmer J, Crawford TO, Mendell JR, Coulson SE, Androphy EJ, Prior TW, Burghes AH. The survival motor neuron protein in spinal muscular atrophy. Hum Mol Genet. 1997;6:1205–1214. doi: 10.1093/hmg/6.8.1205. [DOI] [PubMed] [Google Scholar]
- Cote J, Richard S. Tudor domains bind symmetrical dimethylated arginines. J Biol Chem. 2005;280:28476–28483. doi: 10.1074/jbc.M414328200. [DOI] [PubMed] [Google Scholar]
- Crawford TO, Pardo CA. The neurobiology of childhood spinal muscular atrophy. Neurobiol Dis. 1996;3:97–110. doi: 10.1006/nbdi.1996.0010. [DOI] [PubMed] [Google Scholar]
- Fan L, Simard LR. Survival motor neuron (SMN) protein: role in neurite outgrowth and neuromuscular maturation during neuronal differentiation and development. Hum Mol Genet. 2002;11:1605–1614. doi: 10.1093/hmg/11.14.1605. [DOI] [PubMed] [Google Scholar]
- Jablonka S, Bandilla M, Wiese S, Buhler D, Wirth B, Sendtner M, Fischer U. Co-regulation of survival of motor neuron (SMN) protein and its interactor SIP1 during development and in spinal muscular atrophy. Hum Mol Genet. 2001;10:497–505. doi: 10.1093/hmg/10.5.497. [DOI] [PubMed] [Google Scholar]
- Jensen KB, Dredge BK, Stefani G, Zhong R, Buckanovich RJ, Okano HJ, Yang YY, Darnell RB. Nova-1 regulates neuron-specific alternative splicing and is essential for neuronal viability. Neuron. 2000;25:359–371. doi: 10.1016/s0896-6273(00)80900-9. [DOI] [PubMed] [Google Scholar]
- Lefebvre S, Burglen L, Reboullet S, Clermont O, Burlet P, Viollet L, Benichou B, Cruaud C, Millasseau P, Zeviani M, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80:155–165. doi: 10.1016/0092-8674(95)90460-3. [DOI] [PubMed] [Google Scholar]
- Lefebvre S, Burlet P, Liu Q, Bertrandy S, Clermont O, Munnich A, Dreyfuss G, Melki J. Correlation between severity and SMN protein level in spinal muscular atrophy. Nat Genet. 1997;16:265–269. doi: 10.1038/ng0797-265. [DOI] [PubMed] [Google Scholar]
- Liu Q, Dreyfuss G. A novel nuclear structure containing the survival of motor neurons protein. EMBO J. 1996;15:3555–3565. [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Fischer U, Wang F, Dreyfuss G. The spinal muscular atrophy disease gene product, SMN, and its associated protein SIP1 are in a complex with spliceosomal snRNP proteins. Cell. 1997;90:1013–1021. doi: 10.1016/s0092-8674(00)80367-0. [DOI] [PubMed] [Google Scholar]
- Lorson CL, Androphy EJ. An exonic enhancer is required for inclusion of an essential exon in the SMA-determining gene SMN. Hum Mol Genet. 2000;9:259–265. doi: 10.1093/hmg/9.2.259. [DOI] [PubMed] [Google Scholar]
- Lorson CL, Strasswimmer J, Yao JM, Baleja JD, Hahnen E, Wirth B, Le T, Burghes AH, Androphy EJ. SMN oligomerization defect correlates with spinal muscular atrophy severity. Nat Genet. 1998;19:63–66. doi: 10.1038/ng0598-63. [DOI] [PubMed] [Google Scholar]
- Lorson CL, Hahnen E, Androphy EJ, Wirth B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc Natl Acad Sci USA. 1999;96:6307–6311. doi: 10.1073/pnas.96.11.6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAndrew PE, Parsons DW, Simard LR, Rochette C, Ray PN, Mendell JR, Prior TW, Burghes AH. Identification of proximal spinal muscular atrophy carriers and patients by analysis of SMNT and SMNC gene copy number. Am J Hum Genet. 1997;60:1411–1422. doi: 10.1086/515465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McWhorter ML, Monani UR, Burghes AH, Beattie CE. Knockdown of the survival motor neuron (Smn) protein in zebrafish causes defects in motor axon outgrowth and pathfinding. J Cell Biol. 2003;162:919–931. doi: 10.1083/jcb.200303168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meister G, Buhler D, Laggerbauer B, Zobawa M, Lottspeich F, Fischer U. Characterization of a nuclear 20S complex containing the survival of motor neurons (SMN) protein and a specific subset of spliceosomal Sm proteins. Hum Mol Genet. 2000;9:1977–1986. doi: 10.1093/hmg/9.13.1977. [DOI] [PubMed] [Google Scholar]
- Meng A, Tang H, Ong BA, Farrell MJ, Lin S. Promoter analysis in living zebrafish embryos identifies a cis-acting motif required for neuronal expression of GATA-2. Proc Natl Acad Sci USA. 1997;94:6267–6272. doi: 10.1073/pnas.94.12.6267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monani UR. Spinal muscular atrophy: a deficiency in a ubiquitous protein; a motor neuron-specific disease. Neuron. 2005;48:885–895. doi: 10.1016/j.neuron.2005.12.001. [DOI] [PubMed] [Google Scholar]
- Monani UR, Lorson CL, Parsons DW, Prior TW, Androphy EJ, Burghes AH, McPherson JD. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum Mol Genet. 1999;8:1177–1183. doi: 10.1093/hmg/8.7.1177. [DOI] [PubMed] [Google Scholar]
- Monani UR, Pastore MT, Gavrilina TO, Jablonka S, Le TT, Andreassi C, DiCocco JM, Lorson C, Androphy EJ, Sendtner M, Podell M, Burghes AH. A transgene carrying an A2G missense mutation in the SMN gene modulates phenotypic severity in mice with severe (type I) spinal muscular atrophy. J Cell Biol. 2003;160:41–52. doi: 10.1083/jcb.200208079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagliardini S, Giavazzi A, Setola V, Lizier C, Di Luca M, DeBiasi S, Battaglia G. Subcellular localization and axonal transport of the survival motor neuron (SMN) protein in the developing rat spinal cord. Hum Mol Genet. 2000;9:47–56. doi: 10.1093/hmg/9.1.47. [DOI] [PubMed] [Google Scholar]
- Pearn J. Classification of spinal muscular atrophies. Lancet. 1980;1:919–922. doi: 10.1016/s0140-6736(80)90847-8. [DOI] [PubMed] [Google Scholar]
- Pellizzoni L, Charroux B, Dreyfuss G. SMN mutants of spinal muscular atrophy patients are defective in binding to snRNP proteins. Proc Natl Acad Sci USA. 1999;96:11167–11172. doi: 10.1073/pnas.96.20.11167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellizzoni L, Yong J, Dreyfuss G. Essential role for the SMN complex in the specificity of snRNP assembly. Science. 2002;298:1775–1779. doi: 10.1126/science.1074962. [DOI] [PubMed] [Google Scholar]
- Piper M, Holt C. RNA translation in axons. Annu Rev Cell Dev Biol. 2004;20:505–523. doi: 10.1146/annurev.cellbio.20.010403.111746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossoll W, Kroning AK, Ohndorf UM, Steegborn C, Jablonka S, Sendtner M. Specific interaction of Smn, the spinal muscular atrophy determining gene product, with hnRNP-R and gry-rbp/hnRNP-Q: a role for Smn in RNA processing in motor axons? Hum Mol Genet. 2002;11:93–105. doi: 10.1093/hmg/11.1.93. [DOI] [PubMed] [Google Scholar]
- Rossoll W, Jablonka S, Andreassi C, Kroning AK, Karle K, Monani UR, Sendtner M. Smn, the spinal muscular atrophy-determining gene product, modulates axon growth and localization of beta-actin mRNA in growth cones of motoneurons. J Cell Biol. 2003;163:801–812. doi: 10.1083/jcb.200304128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A, Lambrechts A, Hao le T, Le TT, Sewry CA, Ampe C, Burghes AH, Morris GE. A role for complexes of survival of motor neurons (SMN) protein with gemins and profilin in neurite-like cytoplasmic extensions of cultured nerve cells. Exp Cell Res. 2005;309:185–197. doi: 10.1016/j.yexcr.2005.05.014. [DOI] [PubMed] [Google Scholar]
- Shpargel KB, Matera AG. Gemin proteins are required for efficient assembly of Sm-class ribonucleoproteins. Proc Natl Acad Sci USA. 2005;102:17372–17377. doi: 10.1073/pnas.0508947102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Grimmler M, Schwarzer V, Schoenen F, Fischer U, Wirth B. Molecular and functional analysis of intragenic SMN1 mutations in patients with spinal muscular atrophy. Hum Mutat. 2005;25:64–71. doi: 10.1002/humu.20111. [DOI] [PubMed] [Google Scholar]
- Wan L, Battle DJ, Yong J, Gubitz AK, Kolb SJ, Wang J, Dreyfuss G. The survival of motor neurons protein determines the capacity for snRNP assembly: biochemical deficiency in spinal muscular atrophy. Mol Cell Biol. 2005;25:5543–5551. doi: 10.1128/MCB.25.13.5543-5551.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerfield M. The zebrafish book. Eugene, OR: University of Oregon; 1995. [Google Scholar]
- Winkler C, Eggert C, Gradl D, Meister G, Giegerich M, Wedlich D, Laggerbauer B, Fischer U. Reduced U snRNP assembly causes motor axon degeneration in an animal model for spinal muscular atrophy. Genes Dev. 2005;19:2320–2330. doi: 10.1101/gad.342005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirth B. An update of the mutation spectrum of the survival motor neuron gene (SMN1) in autosomal recessive spinal muscular atrophy (SMA) Hum Mutat. 2000;15:228–237. doi: 10.1002/(SICI)1098-1004(200003)15:3<228::AID-HUMU3>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Wolstencroft EC, Mattis V, Bajer AA, Young PJ, Lorson CL. A non-sequence-specific requirement for SMN protein activity: the role of aminoglycosides in inducing elevated SMN protein levels. Hum Mol Genet. 2005;14:1199–1210. doi: 10.1093/hmg/ddi131. [DOI] [PubMed] [Google Scholar]
- Yong J, Wan L, Dreyfuss G. Why do cells need an assembly machine for RNA-protein complexes? Trends Cell Biol. 2004;14:226–232. doi: 10.1016/j.tcb.2004.03.010. [DOI] [PubMed] [Google Scholar]
- Young PJ, Man NT, Lorson CL, Le TT, Androphy EJ, Burghes AH, Morris GE. The exon 2b region of the spinal muscular atrophy protein, SMN, is involved in self-association and SIP1 binding. Hum Mol Genet. 2000;9:2869–2877. doi: 10.1093/hmg/9.19.2869. [DOI] [PubMed] [Google Scholar]
- Zhang H, Xing L, Rossoll W, Wichterle H, Singer RH, Bassell GJ. Multiprotein complexes of the survival of motor neuron protein SMN with Gemins traffic to neuronal processes and growth cones of motor neurons. J Neurosci. 2006;26:8622–8632. doi: 10.1523/JNEUROSCI.3967-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang HL, Eom T, Oleynikov Y, Shenoy SM, Liebelt DA, Dictenberg JB, Singer RH, Bassell GJ. Neurotrophin-induced transport of a beta-actin mRNP complex increases beta-actin levels and stimulates growth cone motility. Neuron. 2001;31:261–275. doi: 10.1016/s0896-6273(01)00357-9. [DOI] [PubMed] [Google Scholar]
- Zhang HL, Pan F, Hong D, Shenoy SM, Singer RH, Bassell GJ. Active transport of the survival motor neuron protein and the role of exon-7 in cytoplasmic localization. J Neurosci. 2003;23:6627–6637. doi: 10.1523/JNEUROSCI.23-16-06627.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]