

Abstract
To get insight into the mechanisms that may lead to progression of temporal lobe epilepsy, we investigated gene expression during epileptogenesis in the rat. RNA was obtained from three different brain regions [CA3, entorhinal cortex (EC), and cerebellum (CB)] at three different time points after electrically induced status epilepticus (SE): acute phase [group D (1 d)], latent period [group W (1 week)], and chronic epileptic period [group M (3–4 months)]. A group that was stimulated but that had not experienced SE and later epilepsy was also included (group nS). Gene expression analysis was performed using the Affymetrix Gene Chip System (RAE230A). We used GENMAPP and Gene Ontology to identify global biological trends in gene expression data. The immune response was the most prominent process changed during all three phases of epileptogenesis. Synaptic transmission was a downregulated process during the acute and latent phases. GABA receptor subunits involved in tonic inhibition were persistently downregulated. These changes were observed mostly in both CA3 and EC but not in CB. Rats that were stimulated but that did not develop spontaneous seizures later on had also some changes in gene expression, but this was not reflected in a significant change of a biological process. These data suggest that the targeting of specific genes that are involved in these biological processes may be a promising strategy to slow down or prevent the progression of epilepsy. Especially genes related to the immune response, such as complement factors, interleukins, and genes related to prostaglandin synthesis and coagulation pathway may be interesting targets.
Keywords: epileptogenesis, gene ontology, immune response, CA3, entorhinal cortex, synaptic transmission, PCR, immunostaining, status epilepticus, seizure
Introduction
Mesial temporal lobe epilepsy (MTLE) is a severe epilepsy syndrome that may evolve after an initial insult such as complex febrile seizures, stroke, brain infections, head trauma, or status epilepticus (SE) (Mathern et al., 1996; Hauser, 1997). The initial insult is usually followed by a latent period, after which spontaneous epileptic seizures occur that can become more and more frequent over time (Engel, 1996; Mathern et al., 1996). Epileptic patients are treated commonly with antiepileptic drugs that may suppress seizures. Antiepileptogenic drugs that retard or prevent epileptogenesis after an initial insult are not yet available (Temkin, 2001; Schmidt and Rogawski, 2002; Loscher and Schmidt, 2004).
The MTLE syndrome is mimicked in the so-called post-SE rodent models. The induced SE is followed, after a latent period, by the occurrence of spontaneous epileptic seizures. It is hypothesized that the initial insult triggers a range of molecular, structural, and functional changes leading to the occurrence of spontaneous seizures. Microarray and SAGE gene expression profiling have been used as tools to identify molecular processes that may be epileptogenic. In recent years, several experimental large-scale genomic studies have shown changes of hundreds of genes shortly after SE. Most of these studies identified genes that are commonly associated with neuronal plasticity, gliosis, neuronal death, neurogenesis, and structural reorganization (Tang et al., 2002; Becker et al., 2003; Elliott et al., 2003; Lukasiuk et al., 2003). Notwithstanding the valuable lists of “epilepsy candidate genes” yielded by these studies, little insight was obtained regarding specific biological pathways with potential epileptogenic properties. Moreover, the reproducibility of the results obtained in different studies was somewhat disappointing, which might be attributable to the different microarray platforms, the relatively small numbers of animals used, the pooling of data from individual animals, and the fact that different brain areas were selected for analysis (Lukasiuk and Pitkanen, 2004). With the rapid advance of the microarray technique, the expression of many more genes can be studied and the reproducibility improved, so that biological pathways can be identified in more detail (Bammler et al., 2005; Irizarry et al., 2005). Insight in the epileptogenic process can be greatly enhanced by (1) taking samples at sequential time points, particularly during the early stages of epileptogenesis, (2) choosing brain areas with different sensitivities to epileptogenesis, and (3) including a group of animals that is treated similarly but that has not developed epilepsy. The present study was designed to include all three features; in addition, we analyzed each animal separately instead of pooling the results from several animals. The construction of biological pathway maps at a sequence of epileptic stages can provide insight in the dynamics of the biological processes relevant to epileptogenesis (Doniger et al., 2003). Therefore, we performed an analysis according to well defined biological processes based on Gene Ontology (GO) categories (Bammler et al., 2005). Identification of specific biological processes and biochemical pathways during critical phases of epileptogenesis is of crucial importance to construct strategies that may prevent the development of epilepsy.
Materials and Methods
Experimental animals.
Adult male Sprague Dawley rats (Harlan CPB, Zeist, The Netherlands) weighing 300–500 g were used in this study, which was approved by the University Animal Welfare committee. The rats were housed individually in a controlled environment (21 ± 1°C; humidity, 60%; lights on from 8:00 A.M. to 8:00 P.M.; food and water available ad libitum).
Electrode implantation and seizure induction.
At 3–4 months of age, rats were anesthetized with an intramuscular injection of ketamine (57 mg/kg; Alfasan, Woerden, The Netherlands) and xylazine (9 mg/kg; Bayer, Leverkusen, Germany), and placed in a stereotactic apparatus. To record hippocampal EEG, a pair of insulated stainless-steel electrodes (70 μm wire diameter; tips were 80 μm apart) were implanted into the left dentate gyrus under electrophysiological control as described previously (Gorter et al., 2001). A pair of stimulation electrodes was implanted in the angular bundle. Several weeks after electrode implantation, rats underwent tetanic stimulation (50 Hz) of the hippocampus in the form of a succession of trains of pulses every 13 s. Each train had a duration of 10 s and consisted of biphasic pulses (pulse duration, 0.5 ms; maximal intensity, 500 μA). Stimulation was stopped when the rats displayed sustained forelimb clonus and salivation for minutes, which usually occurred within 1 h. However, stimulation never lasted longer than 90 min. EEG signals were amplified (10×) via a field effect transistor on the headstage and then led to a differential amplifier (CyberAmp; Molecular Devices, Burlingame, CA), amplified (20×), filtered (1–60 Hz), and sampled by a seizure detection program at a frequency of 200 Hz per channel (Harmonie; Stellate Systems, Montreal, Quebec, Canada). EEG recordings were visually monitored and screened for seizure activity. Behavior was observed during electrical stimulation and several hours thereafter. Immediately after termination of the stimulation, periodic epileptiform discharges occurred at a frequency of 1–2 Hz and were accompanied by behavioral and EEG seizures (SE). Rats were killed at three successive time points: (1) at 1 d after SE (group D; acute phase; n = 3); (2) at 1 week after SE (group W; n = 6) (the rats in this group did not exhibit spontaneous seizures during the first week) (i.e., they were in the latent period); (3) at 3–4 months after SE (group M; n = 5); for this latter group, we selected rats that had developed a progressive form of epilepsy and that exhibited daily seizures; and (4) also at 3–4 months after stimulation but including only rats that did not display the initial SE and later chronic seizures (group nS; n = 5). Age-matched controls (4–7 months of age) that were implanted but not stimulated except for field potential recordings, were also included (group C; n = 6). The EEG of all chronic epileptic rats was monitored for at least several weeks, to ensure that seizure progression had occurred. When they exhibited an increasing number of seizures (progressive form of epilepsy), they were disconnected (1–2 months after SE) and reconnected 1 week before killing, for quantification of their daily seizures. All non-SE rats were monitored until killing. The number of rats per group was chosen as a compromise between the requirements of sufficient statistical power and the wish to keep costs of the analysis as low as possible.
Tissue collection.
After decapitation, the temporal lobe [which includes mainly the entorhinal cortex (EC) and parts of the perirhinal and posterior piriform cortex] was removed under RNase-free conditions by incision at the ventrocaudal part underneath the rhinal fissure until ∼5 mm posterior to bregma, as well as the hippocampus. Both hippocampi were sliced into smaller parts (200–300 μm) and the CA3 region was cut out of the slices in 4°C saline solution under a dissection microscope. The whole cerebellum (CB) was also removed. All material was frozen on dry ice and stored at −80°C until use. Microarray analyses were performed on ipsilateral hippocampal CA3 and EC from each rat in each group (two chips per animal; total n = 50) and on cerebellum of a subset of these rats [control (C), n = 5; 1 week (W), n = 5; chronic (M), n = 5], yielding a data set of 65 microarrays.
RNA isolation and Affymetrix Genechip processing.
After pottering the selected brain material in glass tubes, total RNA was isolated using TRIzol LS Reagent (Invitrogen, Groningen, The Netherlands) with Phase Lock Gel-Heavy tubes (Eppendorf, Hamburg, Germany), following the manufacturer's instructions. RNA was quantified using a nanodrop spectrophotometer (Ocean Optics, Dunedin, FL). Using the Superscript II cDNA kit (Invitrogen), total RNA was amplified and labeled according to the Affymetrix (Santa Clara, CA) Small Sample Labeling Protocol (vII). In short, in the first cycle, 100 ng of total RNA was reverse transcribed using an oligo-dT primer containing the T7 promotor (70°C; 6 min). The resultant double-strand cDNA was subjected to a first round of in vitro transcription (MEGAscript T7 kit; Ambion, Austin, TX). The cRNA product was purified using RNeasy Mini kit (Qiagen, Hilden, Germany). In the second cycle of amplification, cRNA was transcribed using an oligo-dT primer containing the T7 promotor (70°C; 6 min). Double-strand cDNA was subjected to in vitro transcription (EnzoBioarray High Yield RNA Transcript Labeling kit; Affymetrix) in the presence of biotinylated UTP and CTP. The cRNA product was purified using the sample cleanup module (RNeasy; Qiagen), quantified using a nanodrop spectrophotometer (Ocean Optics) and checked for the correct size distribution, using the Agilent 2100 bioanalyzer (Agilent Technologies, Palo Alto, CA). Samples (15 μg) were hybridized to the Rat RAE 230A GeneChip. After 16 h of hybridization, the GeneChips were washed and stained on a fluidics station (Affymetrix) and scanned in a confocal scanner (Agilent Affymetrix GeneArray Scanner) according to the Affymetrix GeneChip Expression Analysis Manual. The RAE230A GeneChip oligonucleotide microarray (Affymetrix) comprises 681,012 distinct oligonucleotide features, which are combined into probe sets. In the original Affymetrix configuration, 15,866 probe sets were generated, which represent 4699 well annotated full-length genes, 10,467 expressed sequence tags (ESTs), and 700 non-ESTs (excluding full-lengths). The expression levels and present/absent calls were calculated for all 15,866 probe sets with the affy package from Bioconductor in the R program for statistical computing, using the MAS5 algorithm (Gautier et al., 2004). To incorporate newly described gene sequences and other knowledge described since the original Affymetrix annotation, we also incorporated the updated probe set definitions according to Dai et al. (2005). To compare with previous studies, genes that were eliminated by the Dai annotation are still presented within each process but they are marked as (Dai). All probes set scaling was used to normalize overall intensities of the different arrays (MAS5.0).
Microarray data analysis.
Data transformations (log2 conversion), selection, and statistical analyses were performed with either Excel (version 9.0; Microsoft, Redmond, WA) or custom-written software. All statistical tests were performed on the log transformed intensities, using a combination of Excel and SigmaStat (SPSS, version 2). The first step in the analysis for the study reported here was to determine which genes to consider “present” or “absent.” The Affymetrix analysis provides a p value for the presence of each gene on each chip. A gene is considered present in an experimental group when p < 0.05 holds for all animals, or no more than one animal had a p > 0.07 or no more than two animals had 0.05 < p < 0.07. When a gene was “present” in at least one region at one time point, we included the gene in the comparisons. In this way, we do not ignore “inducible” or strongly repressed genes. We also excluded outliers based on the expression intensity within a group (defined as a data point more than twice the SDs away from the mean); such values were extremely rare and did not affect the outcome of the analysis reported here. Comparisons were made between control and experimental samples, taken at the same time points, requiring at least p < 0.05 for a significant difference (ANOVA). Because array analysis deals with large numbers of multiple comparisons, it is necessary to take into consideration the false discovery rate (FDR); for an in-depth discussion, see Benjamini et al. (2001). We calculate FDR as the ratio “expected false positives” to “observed positives.” There is no absolute level that FDR should obey; it depends on the subjective balance between missing genes that changed versus accepting genes that did not really change. In this study, we will consider changes in gene expression when p < 0.05 (but only if that gene belongs to a process that is significantly changed as a whole). For our data, this implies a FDR of <1% for genes that change similarly (p < 0.05) in the CA3 and EC region and a FDR of <15% for genes that change (p < 0.05) in only one of the two regions. If in a single region, the change of gene expression is significant at the p < 0.01 level, the FDR drops to a value <5% for our data. Fold changes in gene expression were calculated by dividing the mean intensity signal from a specific time or region by the mean intensity signal from the corresponding control samples. With the aim of obtaining a global impression of the “present” genes changed in this process, an unsupervised hierarchical clustering analysis was performed and scatter plots were constructed, using Spotfire Decision Site for Functional Genomics program. The dataset consisting of the significantly altered genes was entered into GenMAPP (Gene Map Annotator and Pathway Profiler), a computer program that is designed for viewing and analyzing genome-scale data on MAPPs representing biological pathways and any other grouping of genes. MAPPFinder is an accessory program that works with GenMAPP and uses the annotations from the GO Consortium to identify global biological trends in gene expression data. MAPPFinder relates microarray data meeting a user-defined criterion for a “significant” gene expression change to each term in the Gene Ontology hierarchy, and calculates the percentage of genes changed within each GO biological process, cellular component, and molecular function term. MAPPFinder then calculates the total number of genes changed within a “parent GO term” and all of its “children” (local MAPPs), and a statistical score (z score), giving a comprehensive picture of the gene expression changes associated with a particular GO term (Doniger et al., 2003).
Real-time quantitative PCR analysis.
To validate some of the changes that were detected on the microarrays, real-time quantitative PCR analysis was performed using RNA obtained from the same tissue samples as used in the array experiment. Samples were run in triplicate. Because the amount of tissue was limited, RNA of the rats belonging to the same experimental group was pooled. The concentration and purity of RNA (isolated using the TRIzol LS Reagent) were determined spectrophotometrically at 260/280 nm. Five micrograms of total RNA were reverse-transcribed into cDNA using 125 pmol two-base anchored oligo-dT primers [5′-(dT)14-d(A/G/C)-d(A/G/C/T); Amersham Biosciences, Roosendaal, The Netherlands]. The reverse transcription was performed in 50 μl reactions. Five nanomole oligo-dT primers were annealed to 5 μg of total RNA in a total volume of 20 μl by incubation at 72°C for 10 min and cooled to 4°C. Reverse transcription was performed by the addition of 25 μl of RT-mix, containing the following: 50 mm Tris-HCl, 75 mm KCl, 3 mm MgCl2, 20 mm DTT, 0.1 mm dNTPs (Amersham Biosciences), 30 U of RNase inhibitor (Roche Applied Science, Indianapolis, IN), and 400 U of Moloney murine leukemia virus reverse transcriptase (Invitrogen). This mixture was incubated at 37°C for 60 min, heated to 95°C for 10 min, and cooled to 4°C. Real-time monitoring of PCRs was performed using the LightCycler system (Roche Applied Science). PCR primers (Sigma-Genosys, Zwijndrecht, The Netherlands) were designed on the basis of the reported cDNA sequences and are listed in Table 1. For each PCR, a master mixture was prepared on ice, containing the following per sample: 1 μl of cDNA, 1 μl of FastStart Reaction Mix SYBR Green I (Roche Applied Science, Indianapolis, IN), 0.5 μl of 10 μm primers, and 1.6 μl of 25 mm MgCl2. The final volume was adjusted with H2O to 10 μl. After the reaction mixture was loaded into a glass capillary tube, the cycling conditions were performed as follows: initial denaturation at 95°C for 6 min, followed by 45 cycles of denaturation at 95°C for 15 s, annealing between 56 and 60°C for 5 s and extension at 72°C for 10–15 s. The temperature transition rate was set at 20°C/s. Fluorescent product was measured by a single acquisition mode at 72°C after each cycle. Separate calibration (standard) curves for the different primers and TBP1 (TATA box binding protein-like protein-1) (as reference) were constructed using serial dilutions of cDNA from rat hippocampus. The standard curve samples were included in each PCR. Standards were defined to contain an arbitrary starting concentration, because no primary calibrators exist. Hence, all calculated concentrations are relative to the concentration of the standard. For distinguishing specific from nonspecific products and primer dimers, a melting curve was obtained after amplification by holding the temperature at 65°C for 15 s followed by a gradual increase in temperature to 95°C at a rate of 0.1°C/s, with the signal acquisition mode set continuous. Quantification of data was performed using the LightCycler analysis software. Background fluorescence was removed by setting a noise band. The log-linear portion of the standard's amplification curve was selected. The crossing points were identified at the intersection of the best fit line through the log-linear region and the noise band. Using calibration curves, the concentration of a given product was calculated. The amount of each specific product was divided by the amount of a household protein (β-actin, cyclophilin, and TATA-Box-Protein-Like) for each sample and normalized to control values.
Table 1.
List of primers used for quantitative RT-PCR
| Gene | Forward primer | Reverse primer |
|---|---|---|
| Ftl | 5′-TCTCCTCAAGTTGCAGAAC-3′ | 5′-AGTGGCTTTCCAAGAAGT-3′ |
| Kncd2 | 5′-AGACACTCTGCTGGGCAGTTC-3′ | 5′-TTGAGGATGTGGCGGAAGAT-3′ |
| Gal | 5′-CTGCTAGCCTGGCTCCTGTT-3′ | 5′-TGTTCAGGGTCCAGCCTCTCT-3′ |
| Hmox1 | 5′-CAACCCCACCAAGTTCAAACA-3′ | 5′-AGGCGGTCTTAGCCTCCTCTG-3′ |
| Viaat | 5′-CCATCGGCATCATCGTGTT-3′ | 5′-CCAGTTCATCATGCAGTGGAA-3′ |
| Gria2 (Glur2) | 5′-AATCCTGGACTGTGAAAGGGATAA-3′ | 5′-CAATGATATAATGGTACCCTTTAACATGTT-3′ |
| β-actin | 5′-TGAAGATCAAGATCATTGCTCC-3′ | 5′-ACTCATCGTACTCCTGCTTGC-3′ |
| CycA | 5′-CCCACCGTGTTCTTCGACAT-3′ | 5′-AAACAGCTCGAAGCAGACGC-3′ |
| Tbp1 | 5′-CACTGTTGGTCCAGTTTTCCTT-3′ | 5′-CACAGCCGTGGAGCAGAT-3′ |
Tissue preparation and histology.
To confirm whether changes in gene expression indicated changes in protein expression, we performed a number of immunocytochemical analyses. For this procedure, a separate group of 15 rats was used with n = 3 in each group. Rats were deeply anesthetized with pentobarbital (Nembutal, 60 mg/kg, i.p.). The animals were perfused through the ascending aorta with 300 ml of 0.37% Na2S solution and 300 ml of 4% paraformaldehyde plus 0.2% glutaraldehyde in 0.1 m phosphate buffer, pH 7.4. The brains were postfixed in situ overnight at 4°C, dissected, and cryoprotected in 30% phosphate-buffered sucrose solution, pH 7.4. After overnight incubation at 4°C, the brain was frozen in isopentane (−25°C) and stored at −80°C until sectioning. The brain was cut on a sliding microtome, and 40 μm horizontal sections were collected in 0.1 m phosphate buffer for immunocytochemistry or histology. Horizontal sections of control and post-SE rats were washed in 0.05 m PBS, pH 7.4, and incubated for 30 min in 0.3% hydrogen peroxide in PBS to inactivate endogenous peroxidase. Sections were then washed (10 min, two times) in 0.05 m PBS, followed by washing (60 min, one time) in PBS plus 0.5% Triton X-100 plus 0.4% bovine serum albumin (BSA). Sections were incubated with CD11b/c (Ox-42; monoclonal mouse; BD Pharmingen, San Diego, CA; 1:100 as marker for microglia), Spp1 (Osteopontin; monoclonal mouse; 1:100; ITK Diagnostics, Uithoorn, The Netherlands), and Npy (polyclonal anti-rabbit; 1:3000; no. 7698 (7699/4); Swant, Bellinzona, Switzerland). After incubation in primary antibody for 24 h, sections were washed in PBS (10 min, three times) and incubated for 1.5 h in biotinylated sheep anti-rabbit or anti-mouse Ig (Amersham Biosciences; diluted 1:200 in PBS plus 0.1% Triton X-100 plus 0.4% BSA). Sections were washed in PBS (10 min, three times) and incubated for 1.5 h in streptavidin-horseradish peroxidase (Zymed Laboratories, San Francisco, CA), diluted 1:200 in PBS plus 0.1% Triton X-100 plus 0.4% BSA. After washing in 0.05 m Tris-HCl, pH 7.9, the sections were stained with 3,3′-diaminobenzidine tetrahydrochloride (30 mg; Sigma-Aldrich, Zwijndrecht, The Netherlands) and 750 μl of 1% hydrogen peroxide in a 100 ml solution of Tris-HCl. The staining reaction was monitored under the microscope and stopped by washing the sections in Tris-HCl. After mounting on gelatin-coated slides, the sections were air dried, dehydrated in alcohol and xylene, and coverslipped with Entellan (Merck, Darmstadt, Germany).
Western blot.
The CA3 and EC (C, 5; D, 3; W, 3; M, 6) were homogenized in lysis buffer containing the following (per 20 ml): 200 μl of 1 m Tris, pH 8.0; 1 ml of 3 m NaCl; 2 ml of 10% NP-40; 4 ml of 50% glycerol; 800 μl of Na-orthovanadate (10 mg/ml); 200 μl of 0.5 m EDTA, pH 8.0; 400 μl of protease inhibitors; 200 μl of 0.5 m NaF; and 11.2 ml of H2O. Fifty micrograms of total protein per lane, as determined using bicinchoninic acid method (Smith et al., 1985), were separated by SDS-PAGE, and transferred to nitrocellulose by electroblotting (Transblot SD; Bio-Rad, Hercules, CA). Blots were incubated with primary antibodies [goat anti-cox2 polyclonal antibody; catalog #100034; Cayman Chemical, Ann Arbor, MI; 1:500; mouse anti-Glur2 monoclonal antibody 6C4; 1:5000; gift from Dr. J. H. Morrison (Mount Sinai, New York, NY)] and the secondary antibody, anti-goat/mouse labeled with horseradish peroxidase (1:2500; DakoCytomation, High Wycombe, UK). Immunoreactivity was visualized with Lumi-Light Plus Western blotting substrate (Roche Diagnostics, Mannheim, Germany), and the blots were digitized using a Luminescent Image Analyzer (LAS-3000; Fuji Film, Tokyo, Japan). The optical density of each sample was measured using Scion Image (release β 3b; Scion Corporation, Frederick, MD) software. For each sample, the background was subtracted and optical density values were normalized with the amount of β-actin in each sample. Statistical analysis was performed using the Student's t test. Differences with p < 0.05 were considered significant.
Results
Severity of the SE and subsequent epilepsy
We collected material of rats that were killed at three time points after SE. All groups (D, W, M) experienced an SE of ∼10 h. The 1 d group (D) was killed 24 h after the electrical stimulation was stopped. The 1 week group (W) was killed at 1 week after SE in the latent period. The chronic epileptic rats (M) were killed 3–4 months after SE when they exhibited on average 8.3 ± 1.2 seizures (range, 5–12) per day. In addition, we analyzed the non-SE group (nS) of rats that experienced some seizures during the initial electrical stimulation but that did not develop SE or chronic seizures. During continuous EEG monitoring, we never detected a spontaneous seizure in the latter group.
Exploratory data analysis: general features
We organized our data according to the criteria proposed by Dai et al. (2005), using the most recent annotation of Unigene (January 2006; RN230A_RN_UG_6). After recalculation of the original Affymetrix probe set definitions, 10,179 Unigene-based probe sets were left (of the original 15,866 Affymetrix probe sets). Based on the criteria mentioned in Materials and Methods for “present” and “absent” calls, 6987 probe sets were finally accepted “present” in our experiment and used for additional analysis (69%).
Gene expression in CA3, temporal lobe, and cerebellum
In our experimental model of temporal lobe epilepsy (TLE), the epileptogenic area involves the hippocampus and the EC (Spencer and Spencer, 1994; Avoli et al., 2002; de Guzman et al., 2004), but most likely not in the CB (Bohnen et al., 1998), although some cerebellar abnormalities can be observed in human TLE patients (Niedermeyer, 2004; Hermann et al., 2005). We analyzed gene expression of the CB and not only of CA3 and EC, to incorporate in this study a region in the same animals that was most likely not involved in epileptogenesis. First, we performed a hierarchical clustering on the full probe sets of the three different brain areas of the control groups (C). This hierarchical clustering analysis shows that CB, CA3, and EC samples of the different rats form distinct clusters, which indicates that these regions have their own characteristic genetic profile. The analysis also suggests that the CA3 and EC region are more similar to each other than to the cerebellum (Fig. 1a).
Figure 1.

a, Hierarchical clustering showed that the profiles of genetic expression of control rats cluster according to brain area. b, Hierarchical clustering performed on the mean expression values of experimental animals showed that they cluster according to the stage of epileptogenesis, with the exception of the chronic phase. The analysis also showed that changes in gene expression were more similar in CA3 and EC in the early phases than in the chronic phase. The CB group completely separates from the other groups. Red depicts higher expression levels, and blue depicts lower; the deeper shade indicates a larger difference.
Time course of epileptogenesis
The fact that we analyzed rats that were killed at three different time points after SE, enabled us to identify changes related to critical epileptic stages in the course of epileptogenesis: the acute phase (D), the latent phase (W), and the chronic epileptic phase (M). A chronic group that had received stimulation but that had not developed SE was also included (nS). We performed hierarchical clustering on the mean expression values (log-transformed) of the different groups: C, nS, D, W, and M rats of all three regions: CB, CA, EC (Fig. 1b). First of all, it can be seen that CB clusters separately for all available three time points (C, W, M). Figure 1b also illustrates the different behavior of the D and W groups on the one hand, and the C and M groups on the other. Whereas the former form clusters according to time sample (D: CA-D and EC-D; W: CA-W and EC-W), the latter (M and C) cluster according to brain area (CA: CA-M and CA-C; EC: EC-M and EC-C). This indicates that the epileptogenic process causes changes of gene expression that are common to the two areas directly involved, CA3 and EC, but in the chronic phase the specificities of the brain area prevail over the epileptogenic changes. Thus, the changes in gene expression were much more similar in CA3 and EC in the early phases than in the chronic phase (see also Fig. 3). In Figure 2, we show the “fold change” versus p value (up to 0.15) at the three specific time points after SE in the CA3, EC, and CB region. From these plots, it is easy to see that at 1 d many more genes had a more than twofold change than at later time points. We also see that most genes with on average more than twofold change were significantly different from controls at p < 0.05. In the cerebellum, the scatter was much smaller, indicating that no large changes in gene expression were evident in this region, although a considerable number of genes could still be significantly changed at 1 week or during the chronic phase.
Figure 3.

Number of genes changed (upregulated or downregulated) at p < 0.01 of rats killed at three post-SE stages, D (1 d), W (1 week), and M (3–4 months post-SE), for the CA3 and EC region and the CB. The panels show the average signal expression for the total set of changed genes and accompanying Venn diagrams in which the numbers of downregulated (blue) and upregulated (red) genes exclusively in each area and simultaneously in two or three areas are indicated. For example, in b, 100 were upregulated only in CA3, and 419 only in EC; additionally, 356 were upregulated in both; thus, the fraction of genes that changed in both areas was 77% for CA3 (356 of 464), and for EC (356 of 786), the fraction was 45%. The FDR is indicated in blue (next to each circle).
Figure 2.

Scatter diagrams at the three time points after SE (acute, D; latent, W; chronic, M) in CA3, EC, and CB, showing fold change (with respect to control = 1) as a function of the p value of that change (up to 0.15). Significance (p < 0.05) is indicated with shaded area. The scatter is much larger during the acute and latent phases in both CA3 and EC than during chronic phase. Scatter is small in CB during both latent and chronic phases.
The total numbers of upregulated and downregulated genes in the different areas are indicated in Table 2 and were obtained using three different p values (0.05, 0.01, and 0.001). They relate to the Dai defined probe set. A considerable fraction of the genes that showed changed expression was similarly regulated in CA3 and EC. The overlap was stage dependent and appeared to be primarily related to the biological processes that were affected in both regions (see below). In Figure 3, the average gene profile patterns are shown for the genes that had significantly (p < 0.01) changed expression in CA3, EC, and CB region at D (Fig. 3a), W (Fig. 3b), and M (Fig. 3c) in comparison with controls. A similar pattern was observed in CA3 and EC region but not in the CB region. The number of genes that changed expression after SE (both upregulated and downregulated) was initially larger in the EC (1501) than in the CA3 (1225). Remarkably, in the chronicphase, however, the number of genes with altered expression was larger in the CA3 (505) than in the EC (209). The Venn diagrams (Fig. 3) represent, in addition to the number of genes that was significantly downregulated or upregulated in each region, also the number that changes similarly in any two or three regions. The largest overlap between CA3 and EC was found at 1 d after SE where 58% of the CA3 downregulated genes were also downregulated in EC and where 64% of the CA3 upregulated genes were also upregulated in EC. A similar calculation showed that, in the chronic phase (M), only 5% of the CA3 downregulated genes were also downregulated in EC, whereas ∼10% of the CA3 upregulated genes were also upregulated in EC (the percentages with respect to the EC are indicated in Fig. 3 and Table 2). This difference in overlap percentages between the early phases of the epileptogenic process (D, W) and the chronic phase (M) is remarkable. The changes in gene expression in CB were much smaller. Genes that changed expression in the CB also hardly overlapped with the other two regions. At 1 week, there were 24 of 103 cerebellar genes changed (both upregulated and downregulated, 23%), that changed similarly with either CA3 and/or EC. During the chronic phase, 5 of 182 cerebellar genes had similarly changed as in either CA3 or EC. From this exploratory analysis (summarized in Figs. 1 and 3 and Table 2), we conclude that changes in gene expression were similar in regions CA3 and EC in the early phases, whereas in the chronic phase (M) gene expression changes appeared to be rather specific for each brain area.
Table 2.
The total numbers of upregulated and downregulated genes in EC, CA3, both in EC and CA3 and CB, at 1 d (D), 1 week (W), 3–4 months after SE (M), and in the non-SE group (nS), obtained using three different p values (0.05, 0.01, and 0.001)
| D | W | M | nS | |
|---|---|---|---|---|
| EC | ||||
| p < 0.05 | 1241 up (13%) | 1107 up (15%) | 369 up (45%) | 285 up (68%) |
| 1307 down (13%) | 1133 down (15%) | 313 down (53%) | 326 down (64%) | |
| p < 0.01 | 761 up (4%) | 786 up (4%) | 114 up (29%) | 63 up (52%) |
| 740 down (4%) | 564 down (6%) | 95 down (35%) | 85 down (39%) | |
| p < 0.001 | 330 up (1%) | 449 up (1%) | 24 up (14%) | 6 up (55%) |
| 289 down (1%) | 194 down (2%) | 18 down (18%) | 13 down (25%) | |
| CA3 | ||||
| p < 0.05 | 1050 up (15%) | 775 up (20%) | 609 up (26%) | 230 up (58%) |
| 1128 down (15%) | 625 down (25%) | 627 down (25%) | 246 down (64%) | |
| p < 0.01 | 610 up (5%) | 464 up (7%) | 262 up (12%) | 53 up (59%) |
| 615 down (5%) | 221 down (14%) | 243 down (13%) | 46 down (68%) | |
| p < 0.001 | 285 up (1%) | 244 up (1%) | 77 up (4%) | 6 up (52%) |
| 192 down (2%) | 40 down (8%) | 54 down (6%) | 7 down (45%) | |
| EC and CA3 | ||||
| p < 0.05 | 726 up (1%) | 559 up (2%) | 87 up (10%) | 16 up (54%) |
| 763 down (1%) | 325 down (3%) | 53 down (16%) | 10 down (87%) | |
| p < 0.01 | 391 up (0%) | 356 up (0%) | 25 up (1%) | 0 up |
| 356 down (0%) | 100 down (0%) | 12 down (3%) | 0 down | |
| p < 0.001 | 162 up (0%) | 186 up (0%) | 15 up (1%) | 0 up |
| 82 down (0%) | 13 down (0%) | 4 down (2%) | 0 down | |
| CB | ||||
| p < 0.05 | 254 up (61%) | 334 up (46%) | ||
| 232 down (67%) | 359 down (43%) | |||
| p < 0.01 | 53 up (55%) | 99 up (31%) | ||
| 50 down (62%) | 83 down (37%) | |||
| p < 0.001 | 8 up (39%) | 16 up (19%) | ||
| 5 down (62%) | 10 down (31%) |
The FDR is indicated in parentheses.
Gene expression profiles
To go beyond the identification of series of individual genes that showed a changed expression associated with epileptogenesis, we determined groups of functionally related genes, based on the Gene Ontology system (GO). Three main classes of processes were distinguished according to the ordering of this ontology system: (1) biological processes, (2) molecular functions, and (3) cellular components (Doniger et al., 2003). In the following text, classified GO terms will be indicated in italics. In this analysis, we used GenMAPP and the associated MAPPFinder software. Because the hierarchical clustering shows that CA3 and EC cluster together at D and W, indicating a large similarity of the gene profile at these time points, we made profiles for the two regions combined. Because the hierarchical clustering separates CA3 from EC in the chronic phase (M), we also made profiles for CA3 and EC separately at this latter stage. Considering that we deal here with processes or functions that consist of combined sets of genes, we did not want to use a strict gene selection criterion and admitted any individual gene as long as its expression was changed at p < 0.05. The Z scores of the corresponding set of genes were calculated.
Biological processes during epileptogenesis
Acute phase (D)
We related the microarray data using D > C (p < 0.05) as criterion for a “significant” upregulation (or D < C for downregulation) for each term in the Gene Ontology hierarchy, and calculated the percentage of genes changed for each GO biological process, cellular component, and molecular function term. Table 3 shows the significant GO terms according to a Z score >1.96 (which corresponds to a p value of 0.05) according to the lists of ontology classes (processes, functions, and components); to limit the number of classes, we added the criterion that a class to be considered for additional analysis should comprise more than two genes. In Table 3, we present those that were upregulated at 1 d in both CA3 and EC in comparison with controls. A total of 726 probes met the criteria of upregulated in both regions. Of this collection of probes, 406 genes met the criterion and were linked to a GO term. A total of 763 probes met the criteria for downregulation in both regions. Of this collection of probes, 317 genes met the criterion and were linked to a GO term. In the second column, we show whether the process was also marked at one of the other time points (D, W, or M). The number of genes with a changed expression is indicated in the fourth column. The percentage of changed genes over the total number of genes that belong to a given GO term, is presented in the fifth column. We should remember that the same gene may belong to different GO processes. The main characteristic of the acute phase was that a large series of identified biological processes appeared to be related to transcriptional and translational processes and were involved in the short-term stress response to the SE. These changes were associated with the considerable amount of cell death and local tissue inflammatory reaction occurring immediately after the SE. At this stage, genes related to apoptosis and immune response were also enhanced. In contrast, processes associated with synaptic transmission, synaptic plasticity, calcium ion homeostasis, ion transport and cell adhesion were repressed at the same acute phase, which likely results in a disruption of normal neuronal and glial activity. Many upregulated processes are localized to “cellular component—nucleus” (Z score, 3.88), whereas downregulated processes belong mainly to the “cellular components—(plasma) membrane” or synaptic vesicle.
Table 3.
Significant GO terms according to a Z score >1.96 (which corresponds to a p value of 0.05) according to the lists of ontology classes (processes, functions, and components) at 1 d (D), the latent period (W), or in the chronic phase (M) in CA3 and EC
| GOID | No. genes up | % changed | Z score | ||
|---|---|---|---|---|---|
| Group D | 726 probes upregulated in both CA3 and EC (406 GO) | ||||
| Process | |||||
| 6357 | Regulation of transcription from RNA polymerase II promoter | 3 | 60 | 4.73 | |
| 6986 | Response to unfolded protein | 6 | 60 | 4.47 | |
| 45944 | Positive regulation of transcription from RNA polymerase II promoter | 3 | 38 | 4.15 | |
| 6950 | M | Response to stress | 3 | 50 | 4.08 |
| 6412 | Protein biosynthesis | 17 | 29 | 3.79 | |
| 6413 | Translational initiation | 5 | 42 | 3.51 | |
| 6355 | Regulation of transcription, DNA-dependent | 32 | 19 | 3.31 | |
| 6953 | M | Acute-phase response | 4 | 50 | 3.15 |
| 6915 | W | Apoptosis | 8 | 24 | 3.02 |
| 6886 | M | Intracellular protein transport | 15 | 19 | 2.82 |
| 6260 | DNA replication | 6 | 33 | 2.75 | |
| 6955 | WM | Immune response | 8 | 28 | 2.67 |
| 6350 | Transcription | 20 | 21 | 2.59 | |
| 6457 | M | Protein folding | 15 | 26 | 2.57 |
| 6888 | M | ER to Golgi transport | 6 | 32 | 2.45 |
| 42981 | W | Regulation of apoptosis | 7 | 37 | 2.31 |
| 8152 | Metabolism | 5 | 5 | 2.20 | |
| 30163 | Protein catabolism | 3 | 33 | 2.10 | |
| 7517 | Muscle development | 3 | 21 | 2.05 | |
| Function | |||||
| 8243 | Plasminogen activator activity | 3 | 100 | 4.52 | |
| 16563 | Transcriptional activator activity | 9 | 45 | 4.51 | |
| 3743 | Translation initiation factor activity | 11 | 42 | 4.51 | |
| 3676 | Nucleic acid binding | 18 | 14 | 3.93 | |
| 17017 | MAP kinase phosphatase activity | 3 | 75 | 3.72 | |
| 5544 | Calcium-dependent phospholipid binding | 4 | 50 | 3.15 | |
| 3690 | W | Double-stranded DNA binding | 4 | 50 | 3.15 |
| 51082 | Unfolded protein binding | 11 | 28 | 2.89 | |
| 4298 | Threonine endopeptidase activity | 4 | 40 | 2.57 | |
| 3723 | RNA binding | 14 | 25 | 2.54 | |
| 3677 | DNA binding | 20 | 16 | 2.48 | |
| 31072 | M | Heat shock protein binding | 5 | 33 | 2.38 |
| 3700 | Transcription factor activity | 17 | 21 | 2.28 | |
| Component | |||||
| 5634 | Nucleus | 82 | 19 | 3.88 | |
| 5622 | Intracellular | 12 | 15 | 2.78 | |
| 5839 | Proteasome core complex (sensu Eukaryota) | 4 | 40 | 2.57 | |
| 9986 | M | Cell surface | 3 | 33 | 2.34 |
| 5615 | W | Extracellular space | 10 | 23 | 2.19 |
| Group W | 559 probes upregulated in both CA3 and EC (298 GO) | ||||
|---|---|---|---|---|---|
| Process | |||||
| 6955 | DM | Immune response | 13 | 45 | 8.65 |
| 19886 | Antigen processing, exogenous antigen via MHC class II | 6 | 100 | 7.61 | |
| 19884 | Antigen presentation, exogenous antigen | 5 | 100 | 6.94 | |
| 9611 | Response to wounding | 4 | 67 | 4.82 | |
| 6954 | Inflammatory response | 5 | 42 | 4.50 | |
| 8283 | Cell proliferation | 4 | 33 | 4.25 | |
| 7242 | M | Intracellular signaling cascade | 18 | 20 | 3.74 |
| 6508 | Proteolysis | 19 | 19 | 3.43 | |
| 6935 | Chemotaxis | 3 | 43 | 3.32 | |
| 7165 | Signal transduction | 15 | 12 | 3.04 | |
| 6915 | D | Apoptosis | 4 | 12 | 2.56 |
| 6631 | Fatty acid metabolism | 5 | 22 | 2.53 | |
| 42981 | D | Regulation of apoptosis | 4 | 21 | 2.51 |
| 6817 | M | Phosphate transport | 4 | 27 | 2.29 |
| 30154 | Cell differentiation | 4 | 9 | 2.07 | |
| Function | |||||
| 3690 | MHC class II receptor activity | 5 | 100 | 6.94 | |
| 30106 | DM | Double-stranded DNA binding | 4 | 50 | 3.94 |
| 8233 | MHC class I receptor activity | 3 | 20 | 3.88 | |
| 4871 | Peptidase activity | 6 | 14 | 3.57 | |
| 16798 | Signal transducer activity | 5 | 31 | 3.15 | |
| 4872 | M | Hydrolase activity, acting on glycosyl bonds | 4 | 13 | 2.95 |
| 16787 | Receptor activity | 23 | 10 | 2.92 | |
| 16798 | Signal transducer activity | 5 | 31 | 3.15 | |
| 4872 | M | Hydrolase activity, acting on glycosyl bonds | 4 | 13 | 2.95 |
| 16787 | Receptor activity | 23 | 10 | 2.92 | |
| 5096 | Hydrolase activity | 17 | 25 | 2.54 | |
| 4896 | GTPase activator activity | 3 | 100 | 2.29 | |
| Component | |||||
| 5764 | M | Lysosome | 16 | 67 | 10.23 |
| 16020 | Membrane | 55 | 12 | 3.02 | |
| 5737 | M | Cytoplasm | 15 | 13 | 2.50 |
| 5615 | D | Extracellular space | 8 | 18 | 2.30 |
| 5886 | M | Plasma membrane | 4 | 11 | 2.02 |
| M | 609 probes upregulated in CA3 (307 GO) | ||||
| Process | |||||
| 6817 | W | Phosphate transport | 6 | 40 | 3.98 |
| 6953 | D | Acute-phase response | 3 | 38 | 2.66 |
| 7242 | W | Intracellular signaling cascade | 11 | 15 | 2.55 |
| 6888 | D | ER to Golgi transport | 5 | 26 | 2.46 |
| 7264 | Small GTPase mediated signal transduction | 13 | 18 | 2.42 | |
| 6886 | D | Intracellular protein transport | 13 | 16 | 2.36 |
| 6950 | D | Response to stress | 3 | 15 | 2.31 |
| 7155 | Cell adhesion | 7 | 18 | 2.24 | |
| 6955 | DW | Immune response | 3 | 18 | 2.24 |
| 16567 | Protein ubiquitination | 11 | 17 | 2.10 | |
| Function | |||||
| 3690 | D | Double-stranded DNA binding | 3 | 38 | 2.66 |
| 31072 | D | Heat shock protein binding | 4 | 27 | 2.23 |
| 16874 | Ligase activity | 5 | 17 | 2.12 | |
| 4842 | Ubiquitin-protein ligase activity | 12 | 17 | 2.08 |
| Group M | 369 probes upregulated in EC (200 GO) | ||||
|---|---|---|---|---|---|
| Process | |||||
| 15986 | ATP synthesis coupled proton transport | 4 | 24 | 2.93 | |
| 7218 | Neuropeptide signaling pathway | 3 | 21 | 2.33 | |
| 9058 | Biosynthesis | 4 | 33 | 2.32 | |
| 5975 | Carbohydrate metabolism | 5 | 12 | 2.22 | |
| 6865 | Amino acid transport | 3 | 23 | 2.18 | |
| 6457 | D | Protein folding | 8 | 14 | 2.05 |
| Function | |||||
| 5544 | D | Calcium-dependent phospholipid binding | 4 | 50 | 5.08 |
| 46933 | Hydrogen-transporting ATP synthase activity, rotational mechanism | 4 | 25 | 3.08 | |
| 46961 | Hydrogen-transporting ATPase activity, rotational mechanism | 4 | 25 | 3.08 | |
| 16798 | Hydrolase activity, acting on glycosyl bonds | 3 | 23 | 2.62 | |
| 31072 | D | Heat shock protein binding | 3 | 20 | 2.18 |
| Component | |||||
| 5764 | W | Lysosome | 6 | 25 | 4.21 |
| 9986 | D | Cell surface | 3 | 33 | 4.10 |
| 16469 | Proton-transporting two-sector ATPase complex | 4 | 25 | 2.93 | |
| 5737 | W | Cytoplasm | 13 | 11 | 2.69 |
| 5886 | W | Plasma membrane | 3 | 9 | 2.19 |
| Process | |||||
| 15986 | ATP synthesis coupled proton transport | 4 | 24 | 2.93 | |
| 7218 | Neuropeptide signaling pathway | 3 | 21 | 2.33 | |
| 9058 | Biosynthesis | 4 | 33 | 2.32 | |
| 5975 | Carbohydrate metabolism | 5 | 12 | 2.22 | |
| 6865 | Amino acid transport | 3 | 23 | 2.18 | |
| 6457 | D | Protein folding | 8 | 14 | 2.05 |
| Function | |||||
| 5544 | D | Calcium-dependent phospholipid binding | 4 | 50 | 5.08 |
| 46933 | Hydrogen-transporting ATP synthase activity, rotational mechanism | 4 | 25 | 3.08 | |
| 46961 | Hydrogen-transporting ATPase activity, rotational mechanism | 4 | 25 | 3.08 | |
| 16798 | Hydrolase activity, acting on glycosyl bonds | 3 | 23 | 2.62 | |
| 31072 | D | Heat shock protein binding | 3 | 20 | 2.18 |
| Component | |||||
| 5764 | W | Lysosome | 6 | 25 | 4.21 |
| 9986 | D | Cell surface | 3 | 33 | 4.10 |
| 16469 | Proton-transporting two-sector ATPase complex | 4 | 25 | 2.93 | |
| 5737 | W | Cytoplasm | 13 | 11 | 2.69 |
| 5886 | W | Plasma membrane | 3 | 9 | 2.19 |
| Group D | 763 probes downregulated in both CA3 and EC (317 GO) | ||||
|---|---|---|---|---|---|
| Process | |||||
| 7268 | Synaptic transmission | 4 | 24 | 4.65 | |
| 48167 | Regulation of synaptic plasticity | 3 | 50 | 3.92 | |
| 7417 | CNS development | 3 | 60 | 2.92 | |
| 6874 | Calcium ion homeostasis | 3 | 75 | 2.90 | |
| 6816 | W | Calcium ion transport | 6 | 29 | 2.84 |
| 7399 | Nervous system development | 5 | 22 | 2.77 | |
| 8654 | Phospholipid biosynthesis | 3 | 38 | 2.50 | |
| 6811 | Ion transport | 15 | 18 | 2.20 | |
| 7155 | Cell adhesion | 8 | 19 | 2.12 | |
| 6468 | W | Protein amino acid phosphorylation | 21 | 15 | 1.98 |
| Function | |||||
| 4685 | WM | Calcium- and calmodulin-dependent protein kinase activity | 5 | 100 | 6.71 |
| 4364 | Glutathione transferase activity | 3 | 60 | 3.73 | |
| 30170 | Pyridoxal phosphate binding | 3 | 60 | 3.73 | |
| 5516 | W | Calmodulin binding | 9 | 29 | 3.55 |
| 5262 | Calcium channel activity | 3 | 33 | 2.84 | |
| 16740 | Transferase activity | 35 | 16 | 2.83 | |
| 3779 | W | Actin binding | 10 | 23 | 2.75 |
| 5230 | Extracellular ligand-gated ion channel activity | 3 | 30 | 2.71 | |
| 5216 | W | Ion channel activity | 8 | 22 | 2.70 |
| 3774 | Motor activity | 5 | 31 | 2.55 | |
| 16301 | Kinase activity | 9 | 17 | 2.37 | |
| 4890 | GABAA receptor activity | 3 | 33 | 2.33 | |
| 19992 | Diacylglycerol binding | 4 | 29 | 2.32 | |
| 5509 | Calcium ion binding | 26 | 15 | 2.23 | |
| 4674 | W | Protein serine/threonine kinase activity | 16 | 16 | 2.22 |
| 5215 | Transporter activity | 14 | 17 | 2.19 | |
| 5515 | W | Protein binding | 30 | 12 | 2.17 |
| 3924 | W | GTPase activity | 4 | 27 | 2.15 |
| 5234 | Glutamate-gated ion channel activity | 3 | 30 | 2.11 | |
| 15297 | Antiporter activity | 3 | 43 | 2.11 | |
| 16757 | Transferase activity, transferring glycosyl groups | 5 | 23 | 1.96 | |
| Component | |||||
| 5954 | Calcium- and calmodulin-dependent protein kinase complex | 3 | 100 | 5.19 | |
| 5887 | Integral to plasma membrane | 4 | 21 | 2.80 | |
| 5886 | W | Plasma membrane | 6 | 17 | 2.59 |
| 8021 | Synaptic vesicle | 3 | 23 | 2.52 | |
| 16020 | W | Membrane | 61 | 14 | 2.48 |
| 45211 | M | Postsynaptic membrane | 6 | 24 | 2.34 |
| 16021 | M | Integral to membrane | 71 | 12 | 2.19 |
| Group W | 325 probes downregulated in both CA3 and EC (147 GO) | ||||
|---|---|---|---|---|---|
| Process | |||||
| 46928 | Regulation of neurotransmitter secretion | 3 | 75 | 6.69 | |
| 6695 | Cholesterol biosynthesis | 4 | 40 | 6.44 | |
| 6836 | Neurotransmitter transport | 4 | 22 | 3.41 | |
| 6629 | Lipid metabolism | 3 | 7 | 2.66 | |
| 6468 | D | Protein amino acid phosphorylation | 13 | 9 | 2.64 |
| 6816 | D | Calcium ion transport | 3 | 14 | 2.11 |
| Function | |||||
| 4685 | D | Calcium- and calmodulin-dependent protein kinase activity | 3 | 60 | 5.89 |
| 5516 | D | Calmodulin binding | 6 | 19 | 3.91 |
| 4674 | D | Protein serine/threonine kinase activity | 11 | 11 | 3.14 |
| 19992 | D | Diacylglycerol binding | 3 | 21 | 2.99 |
| 3924 | D | GTPase activity | 3 | 20 | 2.83 |
| 5216 | D | Ion channel activity | 3 | 8 | 2.34 |
| 20037 | Heme binding | 3 | 14 | 2.11 | |
| 5515 | D | Protein binding | 13 | 5 | 2.10 |
| 3779 | D | Actin binding | 5 | 11 | 2.08 |
| Component | |||||
| 8076 | Voltage-gated potassium channel complex | 4 | 20 | 3.27 | |
| 5886 | D | Plasma membrane | 4 | 11 | 2.67 |
| 16020 | D | Membrane | 33 | 7 | 2.67 |
| 5856 | Cytoskeleton | 6 | 13 | 2.38 |
| Group M | 627 probes downregulated in CA3 (279 GO) | ||||
|---|---|---|---|---|---|
| Function | |||||
| 4685 | Calcium- and calmodulin-dependent protein kinase activity | 3 | 60 | 4.04 | |
| 3899 | DNA-directed RNA polymerase activity | 4 | 44 | 3.78 | |
| 3735 | Structural constituent of ribosome | 9 | 24 | 3.35 | |
| 5198 | Structural molecule activity | 4 | 11 | 2.44 | |
| Component | |||||
| 5840 | Ribosome | 7 | 23 | 3.04 | |
| 45211 | W | Postsynaptic membrane | 5 | 20 | 1.98 |
| Group M | 313 probes downregulated in EC (110 GO) | ||||
|---|---|---|---|---|---|
| Process | |||||
| 6814 | Sodium ion transport | 3 | 15 | 2.82 | |
| Function | |||||
| 5516 | DW | Calmodulin binding | 5 | 16 | 3.87 |
| Component | |||||
| 45211 | W | Postsynaptic membrane | 3 | 12 | 2.34 |
The total number of genes upregulated or downregulated at a specific stage is indicated at the top; the number of GO-related terms is in parentheses. In the first column, the GO identity is shown; the second column indicates whether a process also occurred during another epileptogenic stage. The fourth column shows the number of genes that are upregulated or downregulated (“local term”); the fifth column shows the percentage of the genes in the specific GO term that is upregulated or downregulated; the sixth column indicates the Z score.
Latent period (W)
Several of the processes that were activated at 1 d were also activated at 1 week after SE. GO terms that were commonly upregulated at both time points included the following: immune response, apoptosis and double strand DNA binding. In the list of GO classes of upregulated genes ordered according to Z scores (Table 3), it is worth noting that at day 1 post-SE (D) the most prominent places are occupied by processes involved in transcription and translation, and response to unfolded protein, whereas at 1 week (W) the immune and inflammatory response and antigen processing, the response to wounding, and processes associated with the cellular component—“lysosomes” become prominent, and occupy the top positions of the list of Z scores. A total of 559 probes met the criteria of upregulated in both regions. Of this collection of probes, 298 genes met the criterion and were linked to a GO term. A total of 325 probes met the criteria of downregulated in both regions. Of this collection of probes, 147 genes met the criterion and were linked to a GO term. Some downregulated processes at 1 week (W) overlapped with those similarly changed at 1 d (D), namely protein phosphorylation, calcium ion transport, calmodulin binding, protein and actin binding, protein serine/threonine kinase activity, GTPase activity, ion channel activity and plasma membrane belong to this category. However, at this stage, processes involved in regulation of neurotransmitter secretion and cholesterol biosynthesis appeared at top places of downregulated processes according to Z scores. With respect to cellular components, there was a shift from the nucleus (at 1 d) to the lysosome, cytoplasm (plasma) membrane, and extracellular space. In particular, the cytoskeleton, membrane and voltage gated potassium channel complex were downregulated cellular components.
Chronic phase (M)
We found a much smaller number of GO processes that changed significantly in both regions in the chronic epileptic phase when rats have ∼5–10 seizures per day (3–4 months after SE). The overlap between genes that change in CA3 and EC was remarkably large especially in the acute and latent phases and was much higher than previously reported when hippocampus and temporal lobe were compared (Lukasiuk et al., 2003). Differences between regions, however, became evident in the chronic phase when especially immune and acute phase responses were significantly activated in CA3 but not in EC, whereas in the latter, and not in the former, neuropeptide signaling pathway and amino-acid transport were upregulated processes. Because the hierarchical clustering showed that CA3 and EC did not cluster together in this phase (Fig. 1b), we analyzed CA3 and EC separately. There were several region-specific changes at M (Table 3). In the CA3, the main changed processes were related to stress and immune response, whereas in the EC these were related to proton and amino acid transport, biosynthesis and neuropeptide signaling. Heat shock protein binding was the only GO term that was significantly upregulated in both CA3 and EC. Several GO terms that appeared significantly upregulated in the chronic phase were also apparent in the acute but not in the latent phase of epileptogenesis; in CA3 these include acute phase response, intracellular protein transport, response to stress and heat shock protein binding; in EC: protein folding, calcium dependent phospholipids binding, double stranded DNA binding, heat shock protein binding. These processes belong mainly to the cellular components: lysosome, cell surface, cytoplasm and plasma membrane. The postsynaptic membrane was a GO term that belonged to the downregulated structural components in acute and chronic phases (Table 3). GO terms that appeared in the latent as well as in the chronic phase included in CA3: immune response, intracellular signaling cascade and phosphate transport; in EC several cellular components appeared significantly affected: lysosome, cytoplasm, and plasma membrane.
Waves of gene induction
Genes that belong to a specific process were often induced or repressed at a specific time point during epileptogenesis. Examples of these different waves of induction or repression of genes are presented in Figure 4a–f. Here, we display the average response of the selected genes (p < 0.05) that were transiently (T) and significantly regulated in CA3 only at 1 d (TD) or at 1 week (TW) or both (TDW), genes that were only regulated during the chronic phase and not in the acute and latent phases (M#DW), genes that were induced or repressed during all phases (DWM), and genes that had a biphasic response with regulation in acute and chronic phases (“seizure related”). The figure indicates that regulation of expression of these genes was mostly similar in the EC except for the M#DW and DWM patterns where EC responded somewhat differently. We also analyzed the processes that were prominent in each pattern. These are indicated next to each specific pattern. Not surprisingly, many of the processes during the early period (TD, TW, and TDW) were mostly similar to what was observed in the CD and CW comparisons (Table 3). These show a prominent position for the immune response including antigen presentation, prostaglandin biosynthesis, and complement activation. Neurotransmitter secretion and transport were downregulated processes during these specific phases.
Figure 4.
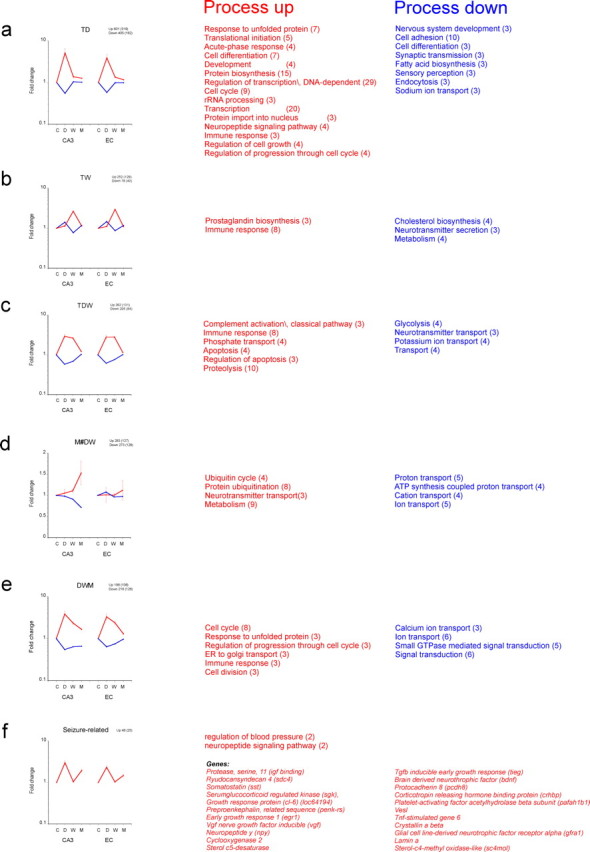
Different waves of gene expression in CA3 and EC. Average fold change (±SEM) of those genes that have significantly changed in CA3 (p < 0.01, with respect to control expression) transiently at 1 d only (TD) (a); at 1 week only (TW) (b); at both 1 d and 1 week (TDW) (c); at the chronic phase only (M#DW) (d); during all three time points (DWM) (e); and “seizure-related” (not changed during the latent period) (f). Except for the M#DW pattern, the average expression pattern is similar in EC. The number of genes involved in each pattern is indicated above each graph (GO-related genes in between brackets). Because there are only 25 genes identified with a seizure-related pattern, a list of these genes is presented. The processes that are significant with each specific pattern are mentioned next to each graph (number of genes involved between brackets).
Dynamic changes in GO processes at the gene level
To identify how genes belonging to a given GO process changed in the course of epileptogenesis, we determined the corresponding signal intensities for the three phases of the process as well as for the controls and the nS animals. Furthermore, we modified GENMAPP maps to be able to examine more precisely whether the changes in signal intensity, either in the “up” or the “down” sense, were statistically different from controls. Each gene is indicated in abbreviation and the full name plus associated GO term can be found in Table 4. We identified the most conspicuous genes that were involved in the stress response and that appeared not only prominently in the acute phase but also in the chronic phase. Some of the genes involved were heat shock proteins. Several of these genes are displayed on the left side of Figure 5a. For clarity, only a limited number of genes (usually six to eight) that changed most dramatically after SE are shown in each graph. The statistics of the genes within the GO process are displayed in color-coded maps next to the graph. Genes with a significant change of <0.01 are coded in red (up) or dark blue (down), and genes that changed with p < 0.05 are coded in pink (up) and light blue (down). In the lists, only the genes are shown that were present in the process and that were not ESTs. Most of the stress related genes displayed were acutely and manifold activated and restored in the latent phase. The genes encoding the heat shock proteins Hspb1 (Hsp27), Dnajb9 and Dnajc3, however, were still activated in the chronic phase; Crystalline a β (Cryab) showed a biphasic response with an upregulation during the acute phase and at the chronic phase in both EC and CA (and not in CB). The expression level of these genes can determine the fate of a cell in response to an injury; particularly the apoptosis-inhibitory Hspb1 and Hsp1a1 (Hsp70) genes (Didelot et al., 2006). Corticotropin-releasing hormone (Crh) was also significantly upregulated in the acute phase and tended to increase again in the chronic phase. Activation of this hormone, which by itself has some depolarizing effects (Hollrigel et al., 1998), leads to the systemic secretion of glucocorticoids, which in turn have immunosuppressant effects. Genes involved in oxidative stress are shown in Figure 5b. The nuclear factor kB1 (Nfkb1), which regulates genes encoding cytokines, cytokine receptors, cell adhesion molecules, proteins involved in coagulation, and genes involved in cell growth control, was acutely upregulated and reached an almost twofold increase at 1 week after SE (Fig. 5c). Among the stress-activated genes, fos oncogene (Fos) was among the most activated genes during the acute phase (∼30-fold in CA3) and was still significantly activated in CA3 in the chronic phase. This gene showed also activation in the cerebellum in the latent phase (p < 0.05). Apart from activation of transcription factors (Nfkb1 and Atf3) and immediate early genes (Fos and Jun), genes that were activated to prevent further production of reactive oxygen species (ROS) included the following: glutathione peroxidase (Gpx), thioredoxin reductase (Txnrd), superoxide dismutase (Sod), heme oxygenase 1 (Hmox1), and ferritin (Ftl). Most of these genes were activated in both acute and latent phases. Hmox1 was upregulated ∼10-fold in both CA3 and EC and was still activated in the chronic phase. Sod2 was acutely and chronically upregulated. Nuclear factor X (NFI), a gene that can contribute to ROS, was repressed in the latent and chronic phase (in EC). This gene has been previously shown to be repressed by oxidative stress (Morel et al., 1999). Oxidative stress changes the immune response, which is another major process that was activated not only shortly after SE but also during the latent and chronic phases (Table 3 and below). Apoptotic mechanisms via activation of caspases and proteases further contribute to cell death during the acute and latent phases (for more detailed description of gene regulation, see Gorter et al., 2006).
Table 4.
List of gene names and accompanying GO term (biological process)
| Gene symbol | Gene name | Biological process |
|---|---|---|
| A2m | α-2-Macroglobulin | Intracellular protein transport; acute-phase response; inflammatory response; response to wounding; negative regulation of calcium-mediated signaling |
| Abat | 4-Aminobutyrate aminotransferase | Synaptic transmission; GABA metabolism; neurotransmitter catabolism |
| Ada | Adenosine deaminase | Purine nucleotide metabolism; immune response; nucleotide metabolism; purine ribonucleoside monophosphate biosynthesis; antimicrobial humoral response (sensu Vertebrata); regulation of circadian sleep/wake cycle, sleep; adenosine metabolism |
| Anxa1 | Annexin A1 | Lipid metabolism; cell motility; inflammatory response; cell cycle; signal transduction; cell surface receptor linked signal transduction; insulin secretion; regulation of cell proliferation; arachidonic acid secretion |
| Ap3m2 | Adaptor-related protein complex 3, μ2 subunit | Transport; intracellular protein transport; neurotransmitter secretion |
| Apba2 | Amyloid β (A4) precursor protein-binding, family A, member 2 | Transport; intracellular protein transport; intracellular signaling cascade; neurogenesis; protein transport; synaptic vesicle exocytosis |
| Apoe | Apolipoprotein E | Response to reactive oxygen species; lipid transport; induction of apoptosis; cytoskeleton organization and biogenesis; synaptic transmission, cholinergic; synaptogenesis; learning and/or memory; circulation; regulation of axon extension; neurite regeneration; lipoprotein metabolism; cholesterol homeostasis; intracellular transport; regulation of neuronal synaptic plasticity |
| Atf3 | Activating transcription factor 3 | Gluconeogenesis; regulation of transcription, DNA-dependent |
| Atp1a2 | ATPase, Na+/K+ transporting, α2 polypeptide | Neurotransmitter uptake; cation transport; potassium ion transport; sodium ion transport; metabolism; monovalent inorganic cation transport; ATP hydrolysis coupled proton transport; sperm motility; hydrogen ion homeostasis |
| B2m | β-2 Microglobulin | Immune response; cellular defense response; antigen presentation, endogenous antigen; antigen processing, endogenous antigen via MHC class I |
| Bdnf | Brain-derived neurotrophic factor | Antiapoptosis; neurogenesis; feeding behavior; dendrite morphogenesis; regulation of metabolism; regulation of long-term neuronal synaptic plasticity; regulation of short-term neuronal synaptic plasticity |
| Bhlhb2 | Basic helix-loop-helix domain containing, class B2 | Regulation of transcription, DNA-dependent; neurogenesis; entrainment of circadian clock; negative regulation of neuronal synaptic plasticity |
| C1qa_predicted | Complement component 1, q subcomponent, α polypeptide (predicted) | Phosphate transport; complement activation, classical pathway; cell–cell signaling |
| C1qb | Complement component 1, q subcomponent, β polypeptide | Phosphate transport; immune response; complement activation, classical pathway |
| C1qg_predicted | Complement component 1, q subcomponent, γ polypeptide (predicted) | Phosphate transport; immune response; complement activation, classical pathway |
| C3 | Complement component 3 | Positive regulation of type IIa hypersensitivity; inflammatory response; complement activation; complement activation, alternative pathway; complement activation, classical pathway; signal transduction; G-protein-coupled receptor protein signaling pathway; positive regulation of phagocytosis |
| C3ar1 | Complement component 3a receptor 1 | Chemotaxis; complement activation; G-protein-coupled receptor protein signaling pathway |
| C4a | Complement component 4a | Inflammatory response; complement activation; complement activation, classical pathway; humoral defense mechanism (sensu Vertebrata); positive regulation of smooth muscle contraction |
| C8b | Complement component 8, β polypeptide | Complement activation, alternative pathway; complement activation, classical pathway; cytolysis |
| Cacna1a | Calcium channel, voltage-dependent, P/Q type, α1A subunit | Calcium ion transport; elevation of cytoplasmic calcium ion concentration; synaptic transmission; neurogenesis |
| Cacna1c | Calcium channel, voltage-dependent, L type, α1C subunit | Calcium ion transport; regulation of heart contraction rate |
| Cacna2d1 | Calcium channel, voltage-dependent, α2/δ subunit 1 | Ion transport |
| Cacnb3 | Calcium channel, voltage-dependent, β3 subunit | Calcium ion transport |
| Cacng3 | Calcium channel, voltage-dependent, γ subunit 3 | Calcium ion transport |
| Cacng4 | Calcium channel, voltage-dependent, γ subunit 4 | Calcium ion transport |
| Cacng7 | Calcium channel, voltage-dependent, γ subunit 7 | Calcium ion transport |
| Calb1 | Calbindin 1 | Learning and/or memory; locomotory behavior; vitamin D metabolism; regulation of synaptic plasticity |
| Calca | Calcitonin/calcitonin-related polypeptide, α | Skeletal development; inflammatory response; G-protein signaling, coupled to cAMP nucleotide second messenger; adenylate cyclase activation; phospholipase C activation; elevation of cytoplasmic calcium ion concentration; neuropeptide signaling pathway; cell–cell signaling; feeding behavior; regulation of blood pressure; negative regulation of smooth muscle contraction |
| Camk2a | Calcium/calmodulin-dependent protein kinase IIα subunit | G1/S transition of mitotic cell cycle; protein amino acid phosphorylation; protein amino acid phosphorylation; calcium ion transport; autophosphorylation; regulation of neurotransmitter secretion; plasticity |
| Camk2b | Calcium/calmodulin-dependent protein kinase IIβ subunit | G1/S transition of mitotic cell cycle; protein amino acid phosphorylation; calcium ion transport; signal transduction; autophosphorylation; plasticity |
| Camk2g | Calcium/calmodulin-dependent protein kinase IIγ | G1/S transition of mitotic cell cycle; response to hypoxia; calcium ion transport response to oxidative stress; autophosphorylation; regulation of long-term neuronal synaptic plasticity |
| Casp1 | Caspase 1 | Response to hypoxia; proteolysis and peptidolysis; apoptosis; induction of apoptosis; signal transduction; protein processing; regulation of apoptosis; positive regulation of I-κB kinase/NF-κB cascade |
| Cck | Cholecystokinin | Apoptosis; signal transduction |
| Ccl2 | Chemokine (C-C motif) ligand 2 | Protein amino acid phosphorylation; calcium ion homeostasis; antiapoptosis; chemotaxis; inflammatory response; immune response; humoral immune response; cell adhesion; signal transduction; G-protein signaling, coupled to cyclic nucleotide second messenger; JAK-STAT cascade; cell–cell signaling; response to pathogenic bacteria; organogenesis; viral genome replication |
| Ccl3 | Chemokine (C-C motif) ligand 3 | Calcium ion homeostasis; exocytosis; cell motility; chemotaxis; inflammatory response; immune response; cytoskeleton organization and biogenesis; signal transduction; G-protein-coupled receptor protein signaling pathway; cell–cell signaling; antimicrobial humoral response (sensu Vertebrata); neutrophil chemotaxis; immune cell chemotaxis; regulation of viral genome replication |
| Cd14 | CD14 antigen | Inflammatory response; immune response |
| Cd59 | CD59 antigen | Immune response; cell surface receptor linked signal transduction; blood coagulation; regulation of complement activation |
| Cd74 | CD74 antigen (invariant polpypeptide of major histocompatibility class II antigen-associated) | Protein folding; immune response |
| Cd81 | CD81 antigen | Activation of MAPK activity; phosphatidylinositol biosynthesis; protein localization; positive regulation of cell proliferation; response to wounding; phosphoinositide metabolism; positive regulation of B cell proliferation; regulation of growth; regulation of cell proliferation; positive regulation of 1-phosphatidylinositol 4-kinase activity; viral entry into host cell; virion attachment, binding of host cell surface receptor; positive regulation of peptidyl-tyrosine phosphorylation |
| Cdk5 | Cyclin-dependent kinase 5 | Cytokinesis; protein amino acid phosphorylation; cell cycle; cell-matrix adhesion; axonogenesis; central nervous system development; cell migration; regulation of cell migration; regulation of synaptic plasticity; synaptic vesicle endocytosis |
| Cfh | Complement component factor H | Complement activation, alternative pathway; regulation of complement activation |
| Cltb | Clathrin, light polypeptide (Lcb) | Neurotransmitter transport; intracellular protein transport; neurotransmitter secretion; synaptic vesicle coating |
| Crcp | Calcitonin gene-related peptide-receptor component protein | Response to stress; inflammatory response |
| Crh | Corticotropin releasing hormone | Response to stress; inflammatory response; immune response; signal transduction; synaptic transmission; pregnancy; parturition; learning and/or memory; long-term memory; induction of apoptosis by hormones |
| Crry | Complement receptor related protein | Negative regulation of complement activation |
| Cryab | Crystallin, αB | Eye development (sensu Mammalia); protein folding; muscle contraction; response to stress; sensory organ development; muscle development; visual perception; response to heat |
| Cx3cl1 | Chemokine (C-X3-C motif) ligand 1 | Chemotaxis; immune response; cell adhesion; signal transduction; cytokine and chemokine mediated signaling pathway; immune cell chemotaxis; positive regulation of inflammatory response; leukocyte adhesive activation; positive regulation of calcium-independent cell–cell adhesion |
| Cxcl1 | Chemokine (C-X-C motif) ligand 1 | Regulation of progression through cell cycle; inflammatory response; immune response; neutrophil chemotaxis; cellular physiological process |
| Cxcl10 | Chemokine (C-X-C motif) ligand 10 | Cell motility; chemotaxis; inflammatory response; immune response; signal transduction; muscle development; positive regulation of cell proliferation; protein secretion; positive regulation of cell migration |
| Dbi | Diazepam binding inhibitor | Acyl-CoA metabolism; steroid biosynthesis; transport |
| Dnaja1 | DnaJ-like protein | Protein folding; response to unfolded protein; DNA damage response, perception of DNA damage |
| Dnajb9 | DnaJ (Hsp40) homolog, subfamily B, member 9 | Protein folding; antiapoptosis; response to stress |
| Dnajc3 | Protein kinase inhibitor p58 | Protein folding; negative regulation of protein kinase activity |
| Dnajc5 | Cysteine string protein | Protein folding; synaptic transmission |
| Ece1 | Endothelin converting enzyme 1 | Proteolysis and peptidolysis; cell–cell signaling; protein processing |
| Egr1 | Early growth response 1 | Regulation of transcription, DNA-dependent; learning and/or memory; T cell differentiation; positive regulation of transcription from RNA polymerase II promoter |
| F10 | Coagulation factor X | Proteolysis and peptidolysis; blood coagulation |
| F2 | Coagulation factor 2 | Proteolysis and peptidolysis; acute-phase response; blood coagulation; wound healing |
| F2r | Coagulation factor II (thrombin) receptor | Regulation of progression through cell cycle; apoptosis; caspase activation; cell motility; signal transduction; G-protein-coupled receptor protein signaling pathway; tyrosine phosphorylation of STAT protein; STAT protein nuclear translocation; blood coagulation; response to wounding; morphogenesis; positive regulation of I-κB kinase/NF-κB cascade |
| F3 | Coagulation factor 3 | Immune response; blood coagulation |
| F5 | Coagulation factor 5 | Cell adhesion; blood coagulation |
| Faf1 | Fas-associated factor 1 | Apoptosis; induction of apoptosis; negative regulation of NF-κB-nucleus import |
| Fcgrt | Fc receptor, IgG, α chain transporter | Immune response; humoral immune response; pregnancy; antigen presentation |
| Fos | FBJ murine osteosarcoma viral oncogene homolog | NA |
| Ftl1 | Ferritin light chain 1 | Iron ion transport; iron ion homeostasis |
| Gabarap | GABA receptor associated protein | Microtubule cytoskeleton organization and biogenesis; protein targeting; transport; synaptic transmission; protein transport |
| Gabbr1 | GABAB receptor 1 | Osteoblast differentiation; G-protein-coupled receptor protein signaling pathway; negative regulation of adenylate cyclase activity; GABA signaling pathway; synaptic transmission |
| Gabra4 | GABAA receptor, subunit α4 | Transport; ion transport; chloride transport; GABA signaling pathway; synaptic transmission |
| Gabra5 | GABAA receptor, α5 | Behavioral fear response; transport; ion transport; chloride transport; signal transduction; GABA signaling pathway; synaptic transmission; brain development; associative learning |
| Gabra6 | GABAA receptor, α6 | Ion transport; chloride transport; signal transduction; GABA signaling pathway; synaptic transmission |
| Gabrb1 | GABA receptor, subunit β1 | Ion transport; chloride transport; chloride transport; signal transduction; GABA signaling pathway; synaptic transmission |
| Gabrd | GABAA receptor, δ | Ion transport; chloride transport; signal transduction; GABA signaling pathway; synaptic transmission |
| Gabrg1 | GABAA receptor, γ1 | Ion transport; chloride transport; GABA signaling pathway; synaptic transmission |
| Gabrg3 | GABAA receptor, subunit γ3 | Ion transport; chloride transport; GABA signaling pathway; synaptic transmission; response to drug |
| Gad1 | Glutamate decarboxylase 1 | Amino acid metabolism; glutamate decarboxylation to succinate; synaptic transmission; protein-pyridoxal-5-phosphate linkage; neurotransmitter biosynthesis |
| Gak | Cyclin G associated kinase | Regulation of progression through cell cycle; protein folding; protein amino acid phosphorylation; cell cycle |
| Gal | Galanin | Smooth muscle contraction; inflammatory response; neuropeptide signaling pathway; neurogenesis; negative regulation of cell proliferation; insulin secretion; growth hormone secretion; positive regulation of apoptosis; negative regulation of lymphocyte proliferation; regulation of immune response |
| Gclc | Glutamate-cysteine ligase, catalytic subunit | Cysteine metabolism; glutamate metabolism; glutathione biosynthesis; circulation |
| Gfra1 | Glial cell line-derived neurotrophic factor family receptor α1 | Cell surface receptor linked signal transduction; transmembrane receptor protein tyrosine kinase signaling pathway; neurogenesis; morphogenesis |
| Gpx1 | Glutathione peroxidase 1 | Response to reactive oxygen species; response to oxidative stress; induction of apoptosis by oxidative stress |
| Gria2 | Glutamate receptor, ionotropic, 2 | Transport; ion transport; potassium ion transport; signal transduction; synaptic transmission |
| Gria3 | Glutamate receptor, ionotropic, AMPA3 (α3) | Lipid metabolism; transport; ion transport; potassium ion transport; glutamate signaling pathway; synaptic transmission |
| Gria4 | Glutamate receptor, ionotropic, 4 | Ion transport; potassium ion transport; glutamate signaling pathway; synaptic transmission |
| Grik2 | Glutamate receptor, ionotropic, kainate 2 | Ion transport; potassium ion transport; glutamate signaling pathway; synaptic transmission |
| Grik4 | Glutamate receptor, ionotropic, kainate 4 | Ion transport; potassium ion transport; glutamate signaling pathway; synaptic transmission |
| Grin1 | Glutamate receptor, ionotropic, NMDA 1 | Ion transport; cation transport; potassium ion transport; calcium ion homeostasis; glutamate signaling pathway; synaptic transmission; learning and/or memory; response to ethanol; regulation of synaptic plasticity |
| Grin2a | Glutamate receptor, ionotropic, NMDA 2A | Ion transport; glutamate signaling pathway; synaptic transmission; learning and/or memory |
| Grin2c | Glutamate receptor, ionotropic, NMDA 2C | Ion transport; glutamate signaling pathway; synaptic transmission |
| Grina | Glutamate receptor, ionotropic, NMDA-associated protein 1 (glutamate binding) | NA |
| Grm1 | Glutamate receptor, metabotropic 1 | G-protein-coupled receptor protein signaling pathway; synaptic transmission; perception of pain; calcium-mediated signaling |
| Grm3 | Glutamate receptor, metabotropic 3 | G-protein-coupled receptor protein signaling pathway; negative regulation of adenylate cyclase activity; synaptic transmission; regulation of synaptic transmission |
| Grm4 | Glutamate receptor, metabotropic 4 | G-protein-coupled receptor protein signaling pathway; negative regulation of adenylate cyclase activity; metabotropic glutamate receptor signaling pathway; synaptic transmission |
| Grm8 | Glutamate receptor, metabotropic 8 | G-protein-coupled receptor protein signaling pathway; synaptic transmission; perception of smell |
| Hcn1 | Hyperpolarization-activated cyclic nucleotide-gated potassium channel 1 | Potassium ion transport; sodium ion transport |
| Hcn2 | Hyperpolarization activated cyclic nucleotide-gated potassium channel 2 | Potassium ion transport; sodium ion transport |
| Hmox1 | Heme oxygenase (decycling) 1 | Phospholipid metabolism; heme oxidation; intracellular signaling cascade; small GTPase mediated signal transduction; DNA damage response, signal transduction resulting in induction of apoptosis; positive regulation of I-κB kinase/NF-κB cascade; response to stimulus |
| Homer1 | Homer homolog 1 (Drosophila) | Metabotropic glutamate receptor signaling pathway |
| Hspa1a | Heat shock 70 kDa protein 1A | Telomere maintenance; inhibition of caspase activation; DNA repair; mRNA catabolism; protein folding; antiapoptosis; defense response; response to unfolded protein; response to heat |
| Hspb1 | Heat shock 27 kDa protein 1 | Regulation of translational initiation; protein folding; response to unfolded protein |
| Il18 | Interleukin 18 | Angiogenesis; immune response; cell–cell signaling; induction of apoptosis via death domain receptors; regulation of cell adhesion; sleep; chemokine biosynthesis; T helper 1 type immune response; T helper 2 type immune response; interleukin-2 biosynthesis; interferon-γ biosynthesis; positive regulation of activated T cell proliferation; interleukin-13 biosynthesis; granulocyte macrophage colony-stimulating factor biosynthesis; regulation of circadian sleep/wake cycle, non-REM sleep |
| Il1a | Interleukin 1α | Regulation of progression through cell cycle; apoptosis; antiapoptosis; chemotaxis; inflammatory response; immune response; cell–cell signaling; cell proliferation; cytokine and chemokine mediated signaling pathway |
| Il1b | Interleukin 1β | Regulation of progression through cell cycle; angiogenesis; apoptosis; inflammatory response; immune response; signal transduction; cell–cell signaling; cell proliferation; cytokine and chemokine mediated signaling pathway; antimicrobial humoral response (sensu Vertebrata); neutrophil chemotaxis; positive regulation of chemokine biosynthesis; positive regulation of interleukin-6 biosynthesis |
| Il2rg | Interleukin 2 receptor, γ (severe combined immunodeficiency) | Protein complex assembly; immune response; signal transduction; cell surface receptor linked signal transduction; cell proliferation |
| Il6 | Interleukin 6 | Programmed cell death, neutrophils; acute-phase response; immune response; negative regulation of chemokine biosynthesis; regulation of circadian sleep/wake cycle, non-REM sleep |
| Il6r | Interleukin 6 receptor | Immune response; cell surface receptor linked signal transduction; development; cell proliferation |
| Junb | Jun-B oncogene | Regulation of progression through cell cycle; regulation of transcription, DNA-dependent; positive regulation of transcription from RNA polymerase II promoter, mitotic; positive cellular physiological process |
| Kcna4 | Potassium voltage-gated channel, shaker-related subfamily, member 4 | Potassium ion transport |
| Kcna5 | Potassium voltage-gated channel, shaker-related subfamily, member 5 | Potassium ion transport |
| Kcnb1 | Potassium voltage-gated channel, Shab-related subfamily, member 1 | Potassium ion transport |
| Kcnc1 | Potassium voltage-gated channel, Shaw-related subfamily, member 1 | Potassium ion transport |
| Kcnc2 | Potassium voltage-gated channel, Shaw-related subfamily, member 2 | Potassium ion transport |
| Kcnc3 | Potassium voltage-gated channel, Shaw-related subfamily, member 3 | Potassium ion transport |
| Kcnd2 | Potassium voltage-gated channel, Shal-related family, member 2 | Regulation of action potential; transport; ion transport; cation transport; potassium ion transport; synaptic transmission |
| Kcnd3 | Potassium voltage-gated channel, Shal-related family, member 3 | Potassium ion transport |
| Kcnj11 | Potassium inwardly rectifying channel, subfamily J, member 11 | Potassium ion transport |
| Kcnj12 | Potassium inwardly rectifying channel, subfamily J, member 12 | Potassium ion transport; muscle contraction; regulation of heart contraction rate |
| Kcnj3 | Potassium inwardly rectifying channel, subfamily J, member 3 | Potassium ion transport |
| Kcnj8 | Potassium inwardly rectifying channel, subfamily J, member 8 | Potassium ion transport |
| Kcnk15 | Potassium channel, subfamily K, member 15 | Potassium ion transport |
| Lmna | Lamin A | Spermatogenesis; muscle development |
| Mgst1 | Microsomal glutathione S-transferase 1 | Glutathione metabolism |
| Nfix | Nuclear factor I/X | DNA replication; transcription; regulation of transcription, DNA-dependent; transcription from RNA polymerase II promoter; regulation of transcription |
| Nfkb1 | Nuclear factor of κ light chain gene enhancer in B-cells 1, p105 | Regulation of transcription, DNA-dependent; transcription from RNA polymerase II promoter; apoptosis; antiapoptosis; inflammatory response; response to oxidative stress; signal transduction; response to pathogenic bacteria; antibacterial humoral response (sensu Vertebrata); negative regulation of interleukin-12 biosynthesis |
| Nfkbia | Nuclear factor of κ light chain gene enhancer in B-cells inhibitor, α | Protein-nucleus import, translocation; apoptosis; cytoplasmic sequestering of NF-κB; response to pathogenic bacteria; regulation of cell proliferation |
| Nlgn1 | Neuroligin 1 | Protein targeting; cell adhesion; neurogenesis; synaptogenesis; synaptic vesicle targeting; calcium-dependent cell–cell adhesion; ion channel clustering; regulation of neuron differentiation |
| Npy | Neuropeptide Y | Calcium ion transport; cell motility; G-protein signaling, coupled to cyclic nucleotide second messenger; neuropeptide signaling pathway; synaptic transmission; digestion; feeding behavior; feeding behavior; circulation; regulation of blood pressure; cell proliferation |
| Npy5r | Neuropeptide Y receptor Y5 | G-protein-coupled receptor protein signaling pathway; synaptic transmission |
| Nqo1 | NAD(P)H dehydrogenase, quinone 1 | Electron transport; xenobiotic metabolism; nitric oxide biosynthesis; synaptic transmission, cholinergic; response to toxin |
| Opcml | Opioid-binding protein/cell adhesion molecule-like | Cell adhesion; neuron recognition |
| Pcdh8 | Protocadherin 8 | Somitogenesis; cell adhesion; homophilic cell adhesion; cell–cell signaling; synaptic transmission; long-term memory; morphogenesis of embryonic epithelium |
| Penk-rs | Preproenkephalin, related sequence | Immune response; neuropeptide signaling pathway |
| Pla2g4a | Phospholipase A2, group IVA (cytosolic, calcium-dependent) | Proteolysis and peptidolysis; platelet activating factor biosynthesis; icosanoid metabolism; phospholipid catabolism; lipid catabolism |
| Plat | Plasminogen activator, tissue | Protein modification; proteolysis and peptidolysis; blood coagulation; platelet-derived growth factor receptor signaling pathway |
| Plau | Plasminogen activator, urokinase | Proteolysis and peptidolysis; chemotaxis; signal transduction; blood coagulation; negative regulation of blood coagulation |
| Ppt | Palmitoyl-protein thioesterase | Protein modification; neurogenesis; visual perception; grooming behavior; adult locomotory behavior; regulation of synaptic plasticity |
| Prkcd | Protein kinase C, δ | Protein amino acid phosphorylation; induction of apoptosis; intracellular signaling cascade; humoral defense mechanism (sensu Vertebrata); B cell proliferation |
| Psmb9 | Proteosome (prosome, macropain) subunit, β type 9 | Proteolysis and peptidolysis; ubiquitin-dependent protein catabolism; immune response |
| Ptgds | Prostaglandin D2 synthase | Prostaglandin biosynthesis; prostaglandin metabolism; transport; regulation of circadian sleep/wake cycle, sleep |
| Ptgds2 | Prostaglandin D2 synthase 2 | Prostaglandin biosynthesis; prostaglandin metabolism; signal transduction; cell–cell signaling; locomotory behavior |
| Ptgs1 | Prostaglandin-endoperoxide synthase 1 | Prostaglandin biosynthesis; lipid metabolism; response to oxidative stress; physiological process; regulation of blood pressure; keratinocyte differentiation |
| Ptgs2 | Prostaglandin-endoperoxide synthase 2 | Regulation of progression through cell cycle; prostaglandin biosynthesis; cell motility; response to oxidative stress; physiological process; regulation of blood pressure; positive regulation of cell proliferation; negative regulation of cell proliferation; cyclooxygenase pathway; keratinocyte differentiation; regulation of inflammatory response |
| Scn2a1 | Sodium channel, voltage-gated, type 2, α1 polypeptide | Sodium ion transport; apoptosis; neurogenesis; generation of action potential |
| Scn6a | Sodium channel, voltage-gated, type 6, α polypeptide | Sodium ion transport; muscle contraction |
| S100b | S100 protein, β polypeptide | Energy reserve metabolism; regulation of protein biosynthesis; calcium ion homeostasis; induction of apoptosis; axonogenesis; central nervous system development; learning and/or memory; memory; cell proliferation; regulation of cytokine biosynthesis; positive regulation of complement activation; astrocyte activation; hyperphosphorylation; regulation of neuronal synaptic plasticity |
| Sc4 mol | Sterol-C4-methyl oxidase-like | Fatty acid metabolism; metabolism; steroid metabolism; sterol biosynthesis |
| Scn1a | Sodium channel, voltage-gated, type 1, α polypeptide | Sodium ion transport; generation of action potential |
| Scn1b | Sodium channel, voltage-gated, type I, β polypeptide | Sodium ion transport |
| Scn3a | Sodium channel, voltage-gated, type III, α polypeptide | Sodium ion transport; response to wounding; perception of pain |
| Scn3b | Sodium channel, voltage-gated, type III, β | Sodium ion transport |
| Sdc4 | Syndecan 4 | Cell adhesion; cell–cell signaling |
| Sgk | Serum/glucocorticoid regulated kinase | Protein amino acid phosphorylation; sodium ion transport; sodium ion homeostasis; apoptosis; response to stress |
| Slc6a1 | GABA transporter protein | Transport; neurotransmitter transport; synaptic transmission |
| Snap25 | Synaptosomal-associated protein 25 | Neurotransmitter uptake; exocytosis; synaptic transmission; neurotransmitter secretion; synaptic vesicle docking during exocytosis |
| Sod1 | Superoxide dismutase 1 | Activation of MAPK activity; DNA fragmentation during apoptosis; superoxide metabolism; response to oxidative stress; neurogenesis; removal of superoxide radicals |
| Spnb3 | β-Spectrin 3 | Vesicle-mediated transport |
| Spp1 | Secreted phosphoprotein 1 | Ossification; antiapoptosis; inflammatory response; cell adhesion; cell-matrix adhesion; cell–cell signaling; negative regulation of bone mineralization; immune cell chemotaxis; T helper 1 type immune response; positive regulation of T cell proliferation; regulation of myeloid cell differentiation |
| Sst | Somatostatin | Regulation of cell migration |
| Sstr1 | Somatostatin receptor 1 | G-protein-coupled receptor protein signaling pathway; G-protein signaling, coupled to cyclic nucleotide second messenger; glutamate signaling pathway; neuropeptide signaling pathway; cell–cell signaling; response to nutrients; digestion; negative regulation of cell proliferation; T cell differentiation |
| Stat3 | Signal transducer and activator of transcription 3 | Negative regulation of transcription from RNA polymerase II promoter; eye photoreceptor cell differentiation; regulation of transcription, DNA-dependent; regulation of transcription from RNA polymerase II promoter; cell motility; acute-phase response; signal transduction; intracellular signaling cascade; JAK-STAT cascade; neurogenesis |
| Stx4a | Syntaxin 4A (placental) | Neurotransmitter transport; intracellular protein transport; synaptic vesicle docking during exocytosis |
| Sv2a | Synaptic vesicle glycoprotein 2a | Transport; neurotransmitter transport; calcium ion homeostasis; synaptic transmission |
| Syn2 | Synapsin II | Synaptic transmission; regulation of neurotransmitter secretion |
| Syp | Synaptophysin | Transport; endocytosis; synaptic transmission; synaptic vesicle maturation; regulation of neuronal synaptic plasticity; synaptic vesicle membrane organization and biogenesis |
| Syt12 | Synaptotagmin 12 | Transport; regulation of neurotransmitter secretion |
| Syt4 | Synaptotagmin 4 | Transport; neurotransmitter secretion; regulation of calcium ion-dependent exocytosis; synaptic vesicle transport |
| Tbxas1 | Thromboxane A synthase 1 | Prostaglandin biosynthesis; electron transport; blood coagulation |
| Thbd | Thrombomodulin | Blood coagulation |
| Tieg | TGFB inducible early growth response | Induction of apoptosis; regulation of cell proliferation |
| Timp1 | Tissue inhibitor of metalloproteinase 1 | C21-steroid hormone biosynthesis |
| Tlr4 | Toll-like receptor 4 | Inflammatory response; immune response; signal transduction; I-κB kinase/NF-κB cascade; activation of NF-κB-inducing kinase; Toll signaling pathway; metabolism; detection of pathogenic bacteria; detection of fungi; T helper 1 type immune response; macrophage activation; positive regulation of interleukin-12 biosynthesis; innate immune response; positive regulation of interleukin-1 biosynthesis; positive regulation of interleukin-13 biosynthesis; positive regulation of interleukin-6 biosynthesis; mast cell activation; negative regulation of osteoclast differentiation |
| Tnfsf4 | Tumor necrosis factor (ligand) superfamily, member 4 | Immune response; T cell proliferation |
| Trh | Thyrotropin releasing hormone | Signal transduction; cell–cell signaling; hormone-mediated signaling |
| Trim9 | Tripartite motif-containing 9 | Synaptic vesicle exocytosis; protein ubiquitination; negative regulation of calcium ion-dependent exocytosis |
| Txnrd1 | Thioredoxin reductase 1 | Electron transport; thioredoxin pathway; response to oxidative stress; signal transduction |
| Ugt1a6 | UDP glycosyltransferase 1 family, polypeptide A6 | Glucuronate metabolism |
| Unc13c | unc-13 homolog C (C. elegans) | Synaptic transmission; regulation of neurotransmitter secretion |
| Vgf | VGF nerve growth factor inducible | Ovarian follicle development; generation of precursor metabolites and energy; sexual reproduction; neuron differentiation |
| Viaat | Vesicular inhibitory amino acid transporter | Transport; neurotransmitter transport; amino acid transport; glycine transport |
| Vip | Vasoactive intestinal polypeptide | Vasodilation |
| Znf179 | Zinc finger protein 179 | Immune response; protein ubiquitination |
Abbreviations are listed in alphabetical order.
Figure 5.
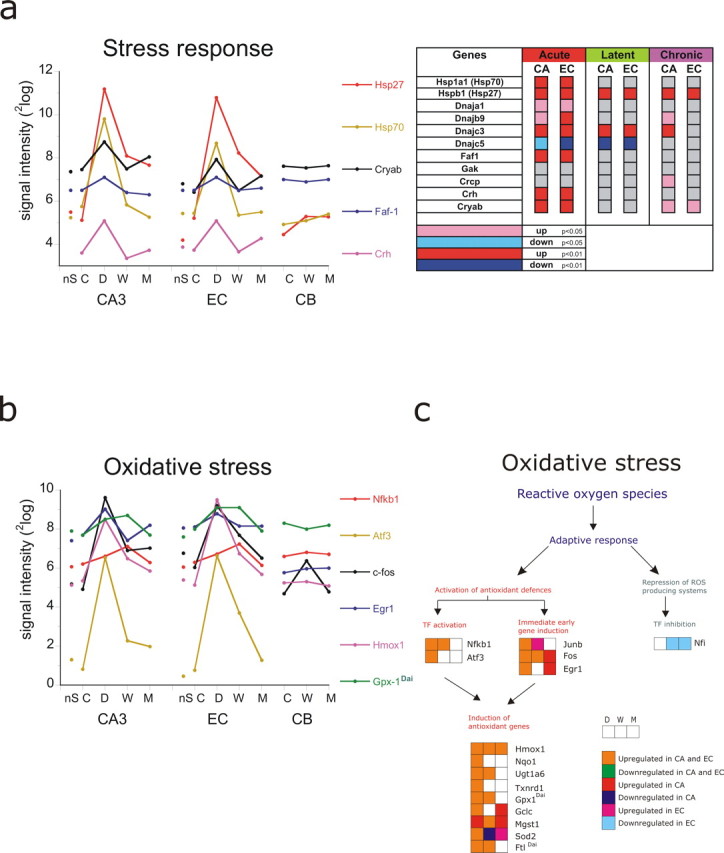
Changes in gene expression after SE related to stress response (a) and oxidative stress (b). Graphs show the signal intensity of six to eight genes in CA3, EC, and CB in the non-SE group (nS), the control group (C), 1 d after SE (D), 1 week after SE (W), and during the chronic phase (M). The statistics of the genes within the GO process are displayed in a color-coded map next to the graph. Genes with a significant change in CA3 or EC of <0.01 are coded in red (up) or dark blue (down), and genes that changed with p < 0.05 are coded in pink (up) and light blue (down). c, Scheme adapted from Morel et al. (1999) that shows the adaptive response during each phase (DWM) on reactive oxygen species. Orange indicates upregulated in CA3 and EC, and green is downregulated in CA3 and EC. Red is upregulated in CA3 only. Pink is upregulated in EC only. Blue is downregulated in CA3 only. Light blue is downregulated in EC only. Gene abbreviations are presented in Table 4.
Genes associated with immune response
The immune response was not only induced during both acute and latent phases of epileptogenesis but also during the chronic phase (in CA3) (Table 3). The immune response was most prominent at 1 week after SE (Z score, 8.65), but some genes peaked at 1 d after SE as shown in Figure 6a [e.g., Stat3, CD14 antigen, and secreted phosphoprotein 1 (Spp1 or osteopontin), a glycoprotein that promotes macrophage migration]. Hg2a (or CD74; H-2 class II histocompatibility antigen), proteosome (Psmb9, macropain), toll-like receptor 4 (Tlr4), and tumor necrosis factor superfamily 4 (Tnsf4) peaked at 1 week. Proenkephalin related sequence (Penk-rs) and crystalline a β (Cryab) showed a biphasic response with an upregulation during the acute phase and chronic phase. Zinc finger protein 179 and S100β were downregulated during the acute and latent phases. Regarding the interleukin and chemokines (Fig. 6b), we also observed waves of expression. IL6 and IL1b and the receptors IL1rII and Il2rg clearly peaked at 1 d after SE, whereas IL1a, IL18, and IL6 receptor peaked at 1 week after SE. Caspase 1, which cleaves the inactive pro-IL1b and inactive IL18 in the biologically active IL1b and active IL18 peaked at 1 week after SE and was still upregulated in the chronic phase (CA3). Tumor necrosis factor α (Tnfa) showed upregulation both during 1 d and 1 week after SE. Similarly to Tnfa, IL1 acts as an amplifier of the inflammatory and wound healing response. It induces expression of multiple inflammatory genes and neurotrophic factors in glial cells (Benveniste, 1998). IL6, which was also dramatically induced at 1 d after SE, has also antiinflammatory properties via inhibition of proinflammatory cytokines and induction of IL1ra. The small inducible chemokine Ccl2 (also Scya2 or MCP1, monocyte secretory protein JE), a chemokine that is involved in macrophage recruitment (Mahad et al., 2006), was the most activated gene of all genes present on the array and peaked at 1 d after SE.
Figure 6.

Changes in gene expression after SE related to immune response (a) and cytokine production (b). Graphs show the signal intensity of eight genes in CA3, EC, and CB in the non-SE group (nS), the control group (C), 1 d after SE (D), 1 week after SE (W), and during the chronic phase (M). The statistics of the genes within the GO process are displayed in a color-coded map next to each graph. Genes with a significant change in CA3 or EC of <0.01 are coded in red (up) or dark blue (down), and genes that changed with p < 0.05 are coded in pink (up) and light blue (down). White indicates “not present.” Gene abbreviations are presented in Table 4.
Inflammation, complement pathway, and coagulation pathway
Release and subsequent receptor binding of proinflammatory cytokines initiates the prostaglandin pathway and activates both the complement and the coagulation pathways, which are involved in the acute phase response. The acute phase response was not only activated immediately after SE but also during the chronic phase (CA3) (Table 3). In Figure 7, we illustrate the most relevant genes in these processes.
Figure 7.

Changes in gene expression after SE related to prostaglandin regulation (a), complement activation (b), and coagulation factors (c). Graphs show the signal intensity of six to eight genes in CA3, EC, and CB in the non-SE group (nS), the control group (C), 1 d after SE (D), 1 week after SE (W), and during the chronic phase (M). The statistics of the genes within the GO process are displayed in a color-coded map next to each graph. Notice the seizure-related regulation of Cox-2, the strong activation of coagulation factors (F10), and the strong activation of complement factors in the latent period, which can persist into the chronic phase [complement component 3 (C3)]. Gene abbreviations are presented in Table 4.
Prostaglandin pathway.
(Fig. 7a). Phospholipase A2 (Pla2g4a) activation and subsequent prostaglandin synthesis is considered to be a pivotal event in inflammation. Phospholipase A2, which hydrolyzes phospholipids, was strongly upregulated in the acute and latent phases. This protein has been previously shown to be upregulated during kindling epileptogenesis (Tu and Bazan, 2003). Cox-2 [prostaglandin–endoperoxide synthase 2 (Ptgs2)] showed a biphasic responses with upregulation in the acute and chronic phases, but not significant in the latent phase. Thromboxane A synthase 1 (Tbxas1) was upregulated in the acute and latent phases in both EC and CA3. Annexin I (Lipocortin; Anxa1) that regulates prostaglandin metabolism via inhibition of Pla2, was also acutely upregulated after SE.
The complement pathway
(Fig. 7b). Genes involved in the complement pathway (e.g., C1qa, C1qc, C3, C4a, Cfh) were also induced, and many of them remained elevated in the chronic phase. C1qa and C1qc were already induced in the acute phase. C3 and C4a were induced in the latent phase and showed the highest expression at the end of the latent period. Crry, an inhibitor of complement activation was also upregulated at 1 week, whereas CD59, another inhibitor of complement activation, was upregulated in CA3 at 1 week and during the chronic phase (CA3). α-2 macroglobulin (A2m), a member of the same protein family as the complement protein C3 and an inhibitor of metalloproteinases (Mmps), remained upregulated at the same level as during the latent period in both CA3 and EC region, indicating a continued challenge to counteract the proteolytic activities of Mmps (Cucullo et al., 2003).
The coagulation cascade
(Fig. 7c). Several coagulation factors (F2, F3, and F5) were acutely upregulated. Coagulation factor X (F10) was strongly induced in the latent phase. Tissue and urokinase plasminogen activators (Plat and Plau), which facilitate anticoagulant activity, were induced in the acute and latent phases, whereas Plat was still increased compared with control in the chronic phase (CA3). Inhibition of tissue plasminogen activity with neuroserpin has been shown to have antiseizure activity (Yepes et al., 2002).
Genes associated with synaptic transmission
The process of synaptic transmission was most significantly downregulated in the acute phase (Z score, 4.65). Figure 8a displays the list related to genes that code for proteins related to the synaptic release machinery. Although a recovery of expression to control levels was apparent along the epileptogenic process, most genes were still downregulated in the latent phase and some were even downregulated during the chronic phase such as in CA3: an amyloid β precursor protein-binding (Apba2), a putative vesicular trafficking protein, and β-spectrin (Spnb3), a gene involved in secretion; in EC: Unc13h3 (Munc13–3), neuroligin 1 (Nlgn1), which is a postsynaptic cell adhesion molecule, and Atp12, a Na+/K+ transporting ATPase. In contrast, Syntaxin 4 (Stx4) and synaptotagmin 4 (Syt4) were genes that were transiently upregulated during the acute phase. Syt4, an immediate early gene present in presynaptic terminals, is also upregulated after kainate seizures (Tocco et al., 1996). Synaptic vesicle protein Sv2a, the binding site for levetiracetam (Lynch et al., 2004), was only transiently downregulated in EC. Other genes involved in synaptic transmission were related to neurotransmitter receptors, the expression of which is shown below.
Figure 8.

Expression of genes related to synaptic transmission and plasticity. Changes in gene expression in the three regions after SE related to synaptic transmission (a) and plasticity (b). Graphs show the signal intensity of six to eight genes in CA3, EC, and CB in the non-SE group (nS), the control group (C), 1 d after SE (D), 1 week after SE (W) and during the chronic phase (M). The statistics of the genes within the GO process are displayed in a color-coded map next to each graph. Notice that most but not all of the synaptic proteins are downregulated after SE and recover to a large extent during epileptogenesis. b, Plasticity-related genes such as the CaMKIIa are downregulated after SE and remain so during all three phases. Protein kinase C δ (Prkcd) and brain-derived neurotrophic factor (Bdnf) are still upregulated during the chronic epileptic phase in CA3 and EC. Gene abbreviations are presented in Table 4.
Genes associated with synaptic plasticity
Epileptogenesis reflects a strong form of plasticity, and there are indications that it is related to memory impairments (Stefan and Pauli, 2002). Therefore, we also looked to plasticity-related genes and in particular to genes linked to learning and memory. The profiles are shown in Figure 8b. This process emerged as significantly downregulated during the acute phase (Table 3). CaMK-associated genes were downregulated in the early phase, and CaMkIIα and CaMkIIβ (CA3) were still downregulated in the chronic phase. The calcium- and calmodulin-dependent protein kinase activity emerged as a downregulated function during all three phases. CaMKII is one of the best candidates for being a molecular component of the learning and memory machinery in the mammalian brain (Silva et al., 1992; Mayford et al., 1996). Absence or a downregulation of CaMKIIα has previously been reported to be related to hyperexcitability and epilepsy (Butler et al., 1995; Churn et al., 2000; Simonato et al., 2002). Protein kinase δ (Prkcd) and brain-derived neurotrophic factor (Bdnf) were upregulated genes. Prkcd was upregulated during all three phases, whereas Bdnf displayed a biphasic pattern with upregulation in the acute and chronic phases but not in the latent phase. Prkcd can be an activator of the Ras/Raf/MEK/MAP kinase signaling pathway (Jackson and Foster, 2004). Interestingly the Ras signaling pathway has been shown to be involved in different kinds of learning and memory formation in the hippocampus (Giese et al., 2001) and amygdala (Merino and Maren, 2006).
Genes associated with ionic channels and receptor processes
As indicated above, synaptic transmission was a downregulated process during the early phase. This process consists of genes that encode presynaptic and postsynaptic proteins. Because there is ample evidence that epileptogenesis is associated with changes in voltage-gated ion channels and GABA and glutamate receptor expression, we focus here especially on these channel and receptor functions. Moreover ion channel activity and GABA and glutamate-gated receptor activities emerged as significant repressed functions during the acute phase (Table 3).
Ion channels
Figure 9 show the profiles of the changes in signal intensity of the voltage-gated channels during epileptogenesis and in the nS group in CA3, EC, and CB region, next to the gene maps. The maps representing sodium channel subunits shows that Scn1a, Scn2a, and Scn3b were downregulated during the early phase but had recovered in the chronic phase (except Scnb3). Scn6a was upregulated in the acute phase (CA3) and latent period and even more increased in the chronic phase in both regions (Fig. 9a).
Figure 9.
Expression of genes related to ion channels. Changes in gene expression in the three regions after SE related to sodium channels (a), potassium channels (b), and calcium channels (c). Graphs show the signal intensity of six to eight genes in CA3, EC, and CB in the non-SE group (nS), the control group (C), 1 d after SE (D), 1 week after SE (W), and during the chronic phase (M). The statistics of the genes within the GO process are displayed in a color-coded map next to each graph. Notice that different subunits of the ion channels are downregulated after SE but recover to a large extent. The exception is the Scn6a channel, which is persistently upregulated in CA3 and EC but not in CB. Gene abbreviations are presented in Table 4.
The voltage-gated potassium channel complex emerged as repressed component during the latent phase (Table 3). Several potassium channel genes that were present on the array were already downregulated during the acute phase, except for the inward rectifier gene [Kcnj8 (=Kir6.1)]. The expression recovered for most downregulated genes in the chronic epileptic phase, although some potassium channel genes in CA3 were still downregulated compared with control levels [e.g., Kcnc1 (=Kv3.1) and Kcnd2 (=Kv4.2, which represents an A-type potassium channel)] (Fig. 9b). Kcnmb4, a large conductance, calcium-activated potassium (BK) channel was notably downregulated in CA3 and EC in the acute and latent periods.
Calcium channel activity was repressed during the acute phase (Table 3). Several voltage-gated calcium channels were responsible for this downregulated activity. However, they appeared to be mostly recovered in the chronic phase, except for Cacnb3 in the EC (Fig. 9c).
Glutamate signaling
As shown in Figure 10a, most glutamate signaling-related genes were acutely downregulated (approximately twofold) but recovered to control levels after the latent period [except Gria1 (Glur1) and Gria2 (Glur2) and Grm1 (metabotropic GluR1) in EC, which remained below control levels]. NMDA receptor glutamate-binding chain (Grina) was upregulated, but only in the EC during the chronic phase. The only glutamate signaling molecule that was acutely upregulated was Vesl/homer, which appears to play a role in synaptic potentiation (Hennou et al., 2003). Increased expression of homer1a, however, has some antiepileptogenic effects as shown in kindled mice with overexpression of homer1a (Potschka et al., 2002).
Figure 10.
Expression of genes related to neurotransmitter receptors. Changes in gene expression in the three regions after SE related to glutamate signaling (a), GABA signaling (b), and neuropeptides (c). Graphs show the signal intensity of six to eight genes in CA3, EC, and CB in the non-SE group (nS), the control group (C), 1 d after SE (D), 1 week after SE (W), and during the chronic phase (M). The statistics of the genes within the GO process are displayed in a color-coded map next to each graph. Notice that several subunits of the glutamate receptor channels were significantly downregulated after SE and have recovered to a large extent during the chronic phase. a, Homer/Vesl was the exception with a biphasic, seizure-related upregulation. b, GABA receptor subunit α5 (CA3) and δ (EC), two subunits involved in tonic inhibition, were permanently downregulated. c, Several neuropeptides had a biphasic upregulation. Gene abbreviations are presented in Table 4.
GABA signaling
Figure 10b shows various genes that are related to GABA signaling. Most GABA receptor subunits were downregulated during the acute phase including α4 and α5, β1 subunit, and δ subunit. The genes encoding the GABA-B1 (Gabbr1; CA3) and GABA-B2 (Gabbr2; EC) receptor subunit were also downregulated. The genes that encode α5 and the δ GABA receptor subunit were most dramatically downregulated (around twofold). Still a significant downregulation of the α5 and δ was observed in the chronic phase in CA3 or EC, respectively. A GABA transporter (Slc6a1 or Gabt1) was also transiently downregulated in the acute phase. In contrast, a significant upregulation of GABA receptor-associated protein (GABArap) that is involved in postsynaptic clustering of synaptic GABAa receptor subunits (Coyle and Nikolov, 2003) was present during the acute and latent phases in EC. The vesicular inhibitory amino acid transporter Viaat was initially downregulated, but significantly upregulated in the chronic epileptic phase in EC (see below).
Neuropeptide signaling
Because several neuropeptides have strong antiseizure actions, we also looked in more detail to changes in gene expression of several neuropeptides. Moreover, the neuropeptide signaling pathway became an activated process in the chronic phase (EC). As shown in Figure 10c, these genes were almost all acutely and many fold upregulated. Various neuropeptides showed a “biphasic” response with an upregulation at 1 d after SE and in the chronic epileptic phase but not in the latent period in both CA3 and EC, although this only reached statistical significance for Npy, Penk-rs, and Trh and somatostatin (Sst). Npy5 receptor was transiently downregulated and somatostatin receptor 1 (Sstr1) was downregulated in EC during the latent phase. Diazepam binding inhibitor (Dbi), an endogenous neuropeptide for benzodiazepine receptors, is upregulated during the latent period in the EC. Interestingly, Dbi-derived peptide fragments induce a typical pattern of limbic seizures in rats (Vezzani et al., 1991).
Gene expression in the chronic epileptic phase
The animals in the chronic group (M group) exhibited a relatively high seizure incidence, on average 8.3 ± 1.2 seizures (range, 5–12) per day. This study was designed in such a way that we tried to compose groups of animals that could function as replicates. Because the M group has the longest experimental history, they carry the highest risk of being nonuniform. This could be related to their seizure history, seizure severity, recency of last seizure before decapitation, or eventually other environmental factors. A cluster analysis based on the 280 (GO-related) upregulated genes in the CA3 region of the 11 animals (M group and controls), however, gave a clear separation into two highly consistent groups (M and C; Cophenet correlation coefficient, 0.94) (data not shown). We also found that the variance in gene expression of the M group was not different from that of the C group (the F test comparing the variances in the two groups for each gene was not significant for 95% of the 280 genes). Because the experiments exhibited little variation within groups, it is not surprising that a correlation between gene expression and, for example, seizure recency before decapitation (range, 1–5 h) could not be detected in the M group. In addition, we found that none of the possible rankings of the recency values (or any other seizure-related parameter associated with an animal) showed a significant correlation with the expression of this set of 280 genes.
Replication of gene pattern profiles by PCR
To validate the microarray results, we investigated gene expression profiles of six different genes and three household genes (β-actin, TBP like protein, and cyclophilin-A) using quantitative PCR. Material from the same rats that was used for the array was used in the PCR experiment, but the material was pooled per time point. We chose genes that, according to the microarray data, were expressed with a characteristic time pattern. To avoid the possible influence of a single household gene that may also change expression because of the SE, we normalized over the average of all three household genes obtained at each time point. In most cases, gene profile patterns were nicely reproduced (Fig. 11), although the fold change could differ. Heme oxygenase (Hmox1) (see above) was significantly upregulated during all three phases, peaking at 1 d after SE. Ferritin-L (Ftl), a protein involved in iron ion homeostasis and previously shown to be upregulated during epileptogenesis (Gorter et al., 2005), was significantly upregulated at 1 d and at 1 week after SE. Viaat (Slc32a1), a vesicular GABA transporter, was significantly downregulated in the latent phase (CA3) but upregulated in the chronic phase (in EC). An upregulation of Viaat has been previously shown in rats with spontaneous seizures (Lamas et al., 2001). Kncd2, a potassium channel, and Gria2 (the Glur2 glutamate receptor) were mainly downregulated during the acute and latent phases. Gria2 or Glur2 also has been previously shown to be downregulated after kainate seizures and ischemic insult (Friedman et al., 1994; Gorter et al., 1997). Galanin (Gal), a neuropeptide with antiseizure properties (Mazarati et al., 1998; Haberman et al., 2003), was mostly activated at 1 d after SE and appeared to be slightly upregulated again in the chronic phase in EC.
Figure 11.

PCR comparison with array expression. Graphs showing the fold change (with respect to control) during the post-SE time points in CA3 and EC. PCR analysis shows a large correspondence with the array expression patterns of the different genes. Hmox1, Heme oxygenase 1; Ftl, Ferritin light chain 1; Kncd2, potassium voltage-gated channel, Shal-related family 2; Gria2, ionotropic glutamate receptor subunit 2; Gal, galanin; Viaat, vesicular inhibitory amino acid transporter.
Replication of gene profiles by immunostaining and Western blot
To determine whether the transcript regulation corresponded with protein expression, we performed immunohistochemistry (IHC) and Western blotting (WB) on tissue samples of rats that were killed at the same time point. The selected proteins included the following: CD11b/c (OX42), osteopontin (Spp1) and Npy (IHC) and Cox-2 and Glur2 (WB). CD11b/c, a marker for microglial activation (Fig. 12a), was clearly induced at 1 d after SE, peaked at 1 week after SE, and was mostly recovered in the chronic phase. Osteopontin protein expression was induced in glial cells with astrocyte and microglial morphology with highest expression at 1 d after SE (Fig. 12b). Npy expression was clearly induced in mossy fiber endings with a similar seizure-related time course as gene expression (Fig. 12c). Figure 12d shows the optical densities for Glur2 and Cox-2 protein expression in CA3 and EC, respectively. Glur2 was mainly downregulated at 1 d after SE and recovered during epileptogenesis to some extent. The biphasic pattern of Cox-2 observed in the array was reproduced by the Western blot.
Figure 12.

Immunohistochemistry and Western blot. Immunocytochemistry of CD11b/c (Ox42) (a), Spp1 (Osteopontin) (b), and Neuropeptide Y (Npy) (c). a, Microglial activation that was induced at 1 d, peaked at 1 week after SE, and recovers to large extent in the chronic phase. b, Spp1 expression in control (C) is present in neurons and glial cells with microglial morphology; at 1 d (D) is strongly induced in microglial cells with ameboid morphology and still increased in glial cells with microglial and astroglial morphology at 1 week (W). In the chronic phase, some microglial cell still show increased Spp1 expression. c, In control (C), the strongest Npy expression was found in individual neurons scattered throughout hippocampus; Npy expression was strongly induced at 1 d (D) in the mossy fiber endings in hilus and CA3, but also in CA1 neurons and granule cells and in cortical regions. In the latent period (W), Npy expression was much lower than at 1 d after SE. In the chronic epileptic phase (M), Npy expression increased again in mossy fibers in CA3 (arrows) but also in the mossy fibers that had sprouted to the inner molecular layer of the dentate gyrus (arrowheads). Scale bar: a, c, 400 μm; b, 80 μm. d, Two Western blots of ionotropic AMPA2 receptor Gria2 (also Glur2) taken from CA3 material and Cox-2 taken from the EC. Both patterns were similar to the pattern observed with the microarray gene expression.
Discussion
This microarray analysis of the process of epileptogenesis in this post-SE model of temporal lobe epilepsy in the rat, revealed dynamic changes in gene expression. Microarray results for selected genes were consistent with PCR results and immunostaining. Similarly to previous studies that looked at the acute effects in SE rat models (Becker et al., 2003; Lukasiuk et al., 2003; Hunsberger et al., 2005), we also found a dramatic upregulation of death and inflammation-related genes soon after electrically induced SE. However, our study yields novel information by including measurements at later time points and by identifying many more genes and biological processes involved at each phase.
The immune response is the most prominent process changed during all three phases of epileptogenesis
Within the immune response, prostaglandin synthesis and complement activation were prominent processes. Prostaglandin synthesis, illustrated by Cox-2 induction, was activated in the acute and chronic phases but not in the latent period, indicating that this process is related to the occurrence of seizure activity. Activation of prostaglandin receptors could increase intracellular calcium and subsequent glutamate release, which would increase excitability in the surrounding networks (Bezzi et al., 1998). An important component of the immune response is the activation of the complement pathway. Although complement factors might invade the brain via a leaky blood–brain barrier (BBB), part of the increased expression is likely to originate from activated glial cells (Gasque et al., 1997; Hosokawa et al., 2003). The complement system may be useful in eliminating aggregated and toxic proteins. However, an overactivation of the complement system can also have damaging effects through the activation of microglia and proinflammatory cytokines. Interestingly, sequential infusion of individual proteins of the membrane attack pathway into the hippocampus of freely moving rats induces seizures as well as cytotoxicity (Xiong et al., 2003). A recent microarray study on entorhinal cortex material of MTLE patients also has pointed to the involvement of complement activation (Jamali et al., 2006). The changes in the immune response, the activation of proinflammatory cytokines and associated processes found in our model may be related to the breakdown of the BBB that occurs directly after SE (van Vliet et al., 2006). This leads to intrusion of monocytes, neutrophils, and blood components into the neuropil, which might also destabilize the surrounding networks (Newman et al., 2005). The activation of coagulation factors genes, upregulation of plasminogen activators, and the induction of matrix metalloproteinases (Mmps) (Gorter et al., 2006) are likely associated with BBB damage. Many of these acute changes are also found after other excitotoxic insults such as ischemia (Tang et al., 2002; Lu et al., 2004), hemorrhage and hypoglycemia (Tang et al., 2002), and after brain trauma (Raghavendra Rao et al., 2003). Although seizures are observed within the first day of the postischemic period (Hartings et al., 2003), it is not known whether some of these animals later become epileptic. Thus, we assume that the dynamics of the changes and the localization of the changed gene expression changes are a crucial factor that determines whether epilepsy develops or not. This issue needs a more detailed analysis.
Can we observe waves of gene expression in the course of post-SE epileptogenesis?
Insight in the timing of the molecular changes is of crucial importance when we want to develop drugs that can be applied to prevent epilepsy or to stop the progression of epilepsy. We can identify a series of transient changes of gene expression peaking at different times. A typical time pattern is the seizure-related pattern, which consists of genes that are upregulated during the acute and the chronic phases, when seizures occur, but have a relatively normal expression during the latent period. This was the case of genes related to neuropeptide signaling and also with growth-related factors (Bdnf, Tieg, Vgf, Gfra1) and Cox-2. Activation of these genes could contribute to aggravate the epileptic condition, because increased excitability has been observed when the expression of some of these proteins is increased (Kokaia et al., 1995; Bezzi et al., 1998; Scharfman et al., 1999).
Similarly to what has been observed after brain ischemia (Raghavendra Rao et al., 2002; Lu et al., 2004), synaptic transmission was one of the processes that were repressed after SE. The most extensive downregulation was observed during the acute phase and might account for the temporary absence of seizures after the acute phase (i.e., in the latent period). This downregulation could reflect a homeostatic protective, rather than an epileptogenic response. It does not simply reflect neuronal loss, but it consists of a transient repression of gene expression because channel and receptor gene expression mostly recovers. The downregulation of glutamate receptor activity can help to limit additional excitotoxic damage. The seizure-related increase of neuropeptides can be seen as a compensatory response because several neuropeptides have antiepileptic properties (Baraban et al., 1997; Tallent and Siggins, 1999; Mazarati et al., 2001; Vezzani et al., 2002). Nevertheless, this increased gene expression is not very effective because rats continue to have frequent daily seizures, but we should note that Npy5 receptors are downregulated, which may be proepileptogenic (Nanobashvili et al., 2004).
Glia and ion homeostasis
The role of astrocytes in epileptogenesis is being recognized as contributing greatly to the origin of epileptic seizures (Tian et al., 2005). In our animal model, gliosis peaks at the end of the latent period (Aronica et al., 2000). A large number of the glial genes that have maximum induced expression at 1 week are genes linked to the immune and inflammatory response (Aloisi, 1999; Dong and Benveniste, 2001; Bonifati and Kishore, 2007). Other genes that are expressed in glial cells are related to water and ion homeostasis (Simard and Nedergaard, 2004). A gene that was persistently upregulated was the Scn6a, an atypical sodium channel gene that serves as a sodium-level sensor of the body fluid (Hiyama et al., 2002; Watanabe et al., 2006). This gene was originally considered to be a glial sodium channel (Gautron et al., 1992), although neuronal localization has also been observed (Grob et al., 2004). The persistent increased expression was the more remarkable because expression of other sodium channel subunits was reduced. Together with a repression of ion transport via the downregulation of several sodium, potassium, and calcium channels, transporters and glial redistribution of water channels (Aqp4) involved in water homeostasis (de Lanerolle and Lee, 2005), these changes point to a persistent and disturbed ion homeostasis and transport that could lead to increased excitability (Schwartzkroin et al., 1998; Bonan et al., 2000).
Genes and neuronal network dynamics
The most salient gene changes that can contribute to a change of the dynamics of the local networks are those involving GABA signaling. The most consistent downregulated genes were those encoding for the subunits α5 (CA3) and δ (EC). Both are extrasynaptic receptors that control tonic inhibition (Houser and Esclapez, 2003; Dibbens et al., 2004; Peng et al., 2004; Glykys and Mody, 2006). This leads us to assume that in our epileptic rats GABAergic tonic inhibition is likely impaired, which can lead to a disturbance of the dynamics of neuronal networks leading to epileptiform oscillations. GABA-α5 also emerged as downregulated gene in a number of clinical studies (de Lanerolle and Lee, 2005; Arion et al., 2006; Ozbas-Gerceker et al., 2006).
Suggested candidate targets
We may suggest a number of targets that could have antiepileptogenic or antiepileptic actions. In addition to possible targets involved in apoptosis and cell death including the cysteine and serine proteases and Mmps (discussed in Gorter et al., 2006), other targets are related to the immune response, in particular aiming at inhibition of microglial activation. However, because many proteins involved in the immune response do not only have proinflammatory and harmful effects but also antiinflammatory and beneficial effects, more knowledge concerning cytokine (inter)actions and the timing of such a treatment is crucial. Most studies show effects of different antiinflammatory treatments on acute seizure activity in experimental models (Vezzani and Granata, 2005), but antiepileptogenic effects of these treatments have not been reported. Similarly, although inhibition of complement activation has successfully reduced ischemic damage (De Simoni et al., 2003), no studies have been performed concerning epileptogenesis. Other strategies could consist in inhibiting proteins that are induced by seizures and that have proepileptogenic actions by amplifying the actions of glutamate, as Bdnf (Kokaia et al., 1995; Scharfman et al., 1999, 2002; He et al., 2004) and Cox-2 (Bezzi et al., 1998). GABA receptor subunits related to tonic inhibition are among the most persistent downregulated receptors and could be a target in any phase during epileptogenesis. Similarly, an intervention to counterbalance the atypical ion sodium channel Scn6a could be an interesting target worth exploring.
Conclusions
This microarray investigation yields a large number of genes related to important biological processes and functions, the expression of which changes significantly in the course of epileptogenesis. Some suggestions for the possible functional consequences of these changes are advanced. Furthermore, a few suggestions regarding possible targets that may be explored to counteract the epileptogenic process, or may be novel targets for antiepileptic drugs, are put forward.
Footnotes
This work was supported by the Nationaal Epilepsie Fonds (01-09, 03-03, and 05-11) and the Hersenstichting (“Brain Foundation”) in The Netherlands. We thank Erno Vreugdenhil, John Meerman, Wim Ghijsen, and Robert Stad for their helpful advice at an early stage of the experiments and Jurgo Verkooijen, Sandra Redeker, and Linda van Schaik for technical assistance.
References
- Aloisi F. The role of microglia and astrocytes in CNS immune surveillance and immunopathology. Adv Exp Med Biol. 1999;468:123–133. doi: 10.1007/978-1-4615-4685-6_10. [DOI] [PubMed] [Google Scholar]
- Arion D, Sabatini M, Unger T, Pastor J, Alonso-Nanclares L, Ballesteros-Yanez I, Garcia Sola R, Munoz A, Mirnics K, Defelipe J. Correlation of transcriptome profile with electrical activity in temporal lobe epilepsy. Neurobiol Dis. 2006;22:374–387. doi: 10.1016/j.nbd.2005.12.012. [DOI] [PubMed] [Google Scholar]
- Aronica E, Vliet van EA, Mayboroda O, Troost D, Lopes da Silva FH, Gorter JA. Upregulation of metabotropic glutamate receptor subtype mGluR3 and mGluR5 in reactive astrocytes in a rat model of mesial temporal lobe epilepsy. Eur J Neurosci. 2000;12:2333–2345. doi: 10.1046/j.1460-9568.2000.00131.x. [DOI] [PubMed] [Google Scholar]
- Avoli M, D'Antuono M, Louvel J, Kohling R, Biagini G, Pumain R, D'Arcangelo G, Tancredi V. Network and pharmacological mechanisms leading to epileptiform synchronization in the limbic system in vitro. Prog Neurobiol. 2002;68:167–207. doi: 10.1016/s0301-0082(02)00077-1. [DOI] [PubMed] [Google Scholar]
- Bammler T, Beyer RP, Bhattacharya S, Boorman GA, Boyles A, Bradford BU, Bumgarner RE, Bushel PR, Chaturvedi K, Choi D, Cunningham ML, Deng S, Dressman HK, Fannin RD, Farin FM, Freedman JH, Fry RC, Harper A, Humble MC, Hurban P, et al. Standardizing global gene expression analysis between laboratories and across platforms. Nat Methods. 2005;2:351–356. doi: 10.1038/nmeth754. [DOI] [PubMed] [Google Scholar]
- Baraban SC, Hollopeter G, Erickson JC, Schwartzkroin PA, Palmiter RD. Knock-out mice reveal a critical antiepileptic role for neuropeptide Y. J Neurosci. 1997;17:8927–8936. doi: 10.1523/JNEUROSCI.17-23-08927.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker AJ, Chen J, Zien A, Sochivko D, Normann S, Schramm J, Elger CE, Wiestler OD, Blumcke I. Correlated stage- and subfield-associated hippocampal gene expression patterns in experimental and human temporal lobe epilepsy. Eur J Neurosci. 2003;18:2792–2802. doi: 10.1111/j.1460-9568.2003.02993.x. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Drai D, Elmer G, Kafkafi N, Golani I. Controlling the false discovery rate in behavior genetics research. Behav Brain Res. 2001;125:279–284. doi: 10.1016/s0166-4328(01)00297-2. [DOI] [PubMed] [Google Scholar]
- Benveniste EN. Cytokine actions in the central nervous system. Cytokine Growth Factor Rev. 1998;9:259–275. doi: 10.1016/s1359-6101(98)00015-x. [DOI] [PubMed] [Google Scholar]
- Bezzi P, Carmignoto G, Pasti L, Vesce S, Rossi D, Rizzini BL, Pozzan T, Volterra A. Prostaglandins stimulate calcium-dependent glutamate release in astrocytes. Nature. 1998;391:281–285. doi: 10.1038/34651. [DOI] [PubMed] [Google Scholar]
- Bohnen NI, O'Brien TJ, Mullan BP, So EL. Cerebellar changes in partial seizures: clinical correlations of quantitative SPECT and MRI analysis. Epilepsia. 1998;39:640–650. doi: 10.1111/j.1528-1157.1998.tb01433.x. [DOI] [PubMed] [Google Scholar]
- Bonan CD, Walz R, Pereira GS, Worm PV, Battastini AM, Cavalheiro EA, Izquierdo I, Sarkis JJ. Changes in synaptosomal ectonucleotidase activities in two rat models of temporal lobe epilepsy. Epilepsy Res. 2000;39:229–238. doi: 10.1016/s0920-1211(00)00095-4. [DOI] [PubMed] [Google Scholar]
- Bonifati DM, Kishore U. Role of complement in neurodegeneration and neuroinflammation. Mol Immunol. 2007;44:999–1010. doi: 10.1016/j.molimm.2006.03.007. [DOI] [PubMed] [Google Scholar]
- Butler LS, Silva AJ, Abeliovich A, Watanabe Y, Tonegawa S, McNamara JO. Limbic epilepsy in transgenic mice carrying a Ca2+/calmodulin-dependent kinase II alpha-subunit mutation. Proc Natl Acad Sci USA. 1995;92:6852–6855. doi: 10.1073/pnas.92.15.6852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churn SB, Kochan LD, DeLorenzo RJ. Chronic inhibition of Ca2+/calmodulin kinase II activity in the pilocarpine model of epilepsy. Brain Res. 2000;875:66–77. doi: 10.1016/s0006-8993(00)02623-8. [DOI] [PubMed] [Google Scholar]
- Coyle JE, Nikolov DB. GABARAP: lessons for synaptogenesis. Neuroscientist. 2003;9:205–216. doi: 10.1177/1073858403009003013. [DOI] [PubMed] [Google Scholar]
- Cucullo L, Marchi N, Marroni M, Fazio V, Namura S, Janigro D. Blood-brain barrier damage induces release of alpha2-macroglobulin. Mol Cell Proteomics. 2003;2:234–241. doi: 10.1074/mcp.M200077-MCP200. [DOI] [PubMed] [Google Scholar]
- Dai M, Wang P, Boyd AD, Kostov G, Athey B, Jones EG, Bunney WE, Myers RM, Speed TP, Akil H, Watson SJ, Meng F. Evolving gene/transcript definitions significantly alter the interpretation of GeneChip data. Nucleic Acids Res. 2005;33:e175. doi: 10.1093/nar/gni179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Guzman P, D'Antuono M, Avoli M. Initiation of electrographic seizures by neuronal networks in entorhinal and perirhinal cortices in vitro. Neuroscience. 2004;123:875–886. doi: 10.1016/j.neuroscience.2003.11.013. [DOI] [PubMed] [Google Scholar]
- de Lanerolle NC, Lee TS. New facets of the neuropathology and molecular profile of human temporal lobe epilepsy. Epilepsy Behav. 2005;7:190–203. doi: 10.1016/j.yebeh.2005.06.003. [DOI] [PubMed] [Google Scholar]
- De Simoni MG, Storini C, Barba M, Catapano L, Arabia AM, Rossi E, Bergamaschini L. Neuroprotection by complement (C1) inhibitor in mouse transient brain ischemia. J Cereb Blood Flow Metab. 2003;23:232–239. doi: 10.1097/01.WCB.0000046146.31247.A1. [DOI] [PubMed] [Google Scholar]
- Dibbens LM, Feng HJ, Richards MC, Harkin LA, Hodgson BL, Scott D, Jenkins M, Petrou S, Sutherland GR, Scheffer IE, Berkovic SF, Macdonald RL, Mulley JC. GABRD encoding a protein for extra- or peri-synaptic GABAA receptors is a susceptibility locus for generalized epilepsies. Hum Mol Genet. 2004;13:1315–1319. doi: 10.1093/hmg/ddh146. [DOI] [PubMed] [Google Scholar]
- Didelot C, Schmitt E, Brunet M, Maingret L, Parcellier A, Garrido C. Heat shock proteins: endogenous modulators of apoptotic cell death. Handb Exp Pharmacol. 2006;172:171–198. doi: 10.1007/3-540-29717-0_8. [DOI] [PubMed] [Google Scholar]
- Dong Y, Benveniste EN. Immune function of astrocytes. Glia. 2001;36:180–190. doi: 10.1002/glia.1107. [DOI] [PubMed] [Google Scholar]
- Doniger SW, Salomonis N, Dahlquist KD, Vranizan K, Lawlor SC, Conklin BR. MAPPFinder: using Gene Ontology and GenMAPP to create a global gene-expression profile from microarray data. Genome Biol. 2003;4:R7. doi: 10.1186/gb-2003-4-1-r7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott RC, Miles MF, Lowenstein DH. Overlapping microarray profiles of dentate gyrus gene expression during development- and epilepsy-associated neurogenesis and axon outgrowth. J Neurosci. 2003;23:2218–2227. doi: 10.1523/JNEUROSCI.23-06-02218.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel J., Jr Clinical evidence for the progressive nature of epilepsy. Epilepsy Res Suppl. 1996;12:9–20. [PubMed] [Google Scholar]
- Friedman LK, Pellegrini-Giampietro DE, Sperber EF, Bennett MV, Moshe SL, Zukin RS. Kainate-induced status epilepticus alters glutamate and GABAA receptor gene expression in adult rat hippocampus: an in situ hybridization study. J Neurosci. 1994;14:2697–2707. doi: 10.1523/JNEUROSCI.14-05-02697.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasque P, Singhrao SK, Neal JW, Gotze O, Morgan BP. Expression of the receptor for complement C5a (CD88) is up-regulated on reactive astrocytes, microglia, and endothelial cells in the inflamed human central nervous system. Am J Pathol. 1997;150:31–41. [PMC free article] [PubMed] [Google Scholar]
- Gautier L, Cope L, Bolstad BM, Irizarry RA. affy–analysis of Affymetrix GeneChip data at the probe level. Bioinformatics. 2004;20:307–315. doi: 10.1093/bioinformatics/btg405. [DOI] [PubMed] [Google Scholar]
- Gautron S, Dos Santos G, Pinto-Henrique D, Koulakoff A, Gros F, Berwald-Netter Y. The glial voltage-gated sodium channel: cell- and tissue-specific mRNA expression. Proc Natl Acad Sci USA. 1992;89:7272–7276. doi: 10.1073/pnas.89.15.7272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giese KP, Friedman E, Telliez JB, Fedorov NB, Wines M, Feig LA, Silva AJ. Hippocampus-dependent learning and memory is impaired in mice lacking the Ras-guanine-nucleotide releasing factor 1 (Ras-GRF1) Neuropharmacology. 2001;41:791–800. doi: 10.1016/s0028-3908(01)00096-x. [DOI] [PubMed] [Google Scholar]
- Glykys J, Mody I. Hippocampal network hyperactivity after selective reduction of tonic inhibition in GABA A receptor alpha5 subunit-deficient mice. J Neurophysiol. 2006;95:2796–2807. doi: 10.1152/jn.01122.2005. [DOI] [PubMed] [Google Scholar]
- Gorter JA, Petrozzino JJ, Aronica EM, Rosenbaum DM, Opitz T, Bennett MV, Connor JA, Zukin RS. Global ischemia induces downregulation of Glur2 mRNA and increases AMPA receptor-mediated Ca2+ influx in hippocampal CA1 neurons of gerbil. J Neurosci. 1997;17:6179–6188. doi: 10.1523/JNEUROSCI.17-16-06179.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorter JA, van Vliet EA, Aronica E, Lopes da Silva FH. Progression of spontaneous seizures after status epilepticus is associated with mossy fibre sprouting and extensive bilateral loss of hilar parvalbumin and somatostatin-immunoreactive neurons. Eur J Neurosci. 2001;13:657–669. doi: 10.1046/j.1460-9568.2001.01428.x. [DOI] [PubMed] [Google Scholar]
- Gorter JA, Mesquita A, Vliet van EA, Lopes da Silva FH, Aronica E. Increased expression of ferritin, an iron storage protein, in specific regions of the parahippocampal cortex of epileptic rats. Epilepsia. 2005;46:1371–1379. doi: 10.1111/j.1528-1167.2005.11505.x. [DOI] [PubMed] [Google Scholar]
- Gorter JA, van Vliet EA, Rauwerda H, Breit T, van Schaik R, Vreugdenhil E, Redeker S, Aronica E, Lopes da Silva FH, Wadman WJ. Dynamic changes of proteases and protease inhibitors revealed by microarray analysis in CA3 and entorhinal cortex during epileptogenesis in the rat. Epilepsia. doi: 10.1111/j.1528-1167.2007.01290.x. in press. [DOI] [PubMed] [Google Scholar]
- Grob M, Drolet G, Mouginot D. Specific Na+ sensors are functionally expressed in a neuronal population of the median preoptic nucleus of the rat. J Neurosci. 2004;24:3974–3984. doi: 10.1523/JNEUROSCI.3720-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberman RP, Samulski RJ, McCown TJ. Attenuation of seizures and neuronal death by adeno-associated virus vector galanin expression and secretion. Nat Med. 2003;9:1076–1080. doi: 10.1038/nm901. [DOI] [PubMed] [Google Scholar]
- Hartings JA, Williams AJ, Tortella FC. Occurrence of nonconvulsive seizures, periodic epileptiform discharges, and intermittent rhythmic delta activity in rat focal ischemia. Exp Neurol. 2003;179:139–149. doi: 10.1016/s0014-4886(02)00013-4. [DOI] [PubMed] [Google Scholar]
- Hauser WA. Incidence and prevalence. In: Engel J Jr, Pedley TA, editors. Epilepsy: a comprehensive textbook. Philadelphia: Liipincott-Raven; 1997. [Google Scholar]
- He XP, Kotloski R, Nef S, Luikart BW, Parada LF, McNamara JO. Conditional deletion of TrkB but not BDNF prevents epileptogenesis in the kindling model. Neuron. 2004;43:31–42. doi: 10.1016/j.neuron.2004.06.019. [DOI] [PubMed] [Google Scholar]
- Hennou S, Kato A, Schneider EM, Lundstrom K, Gahwiler BH, Inokuchi K, Gerber U, Ehrengruber MU. Homer-1a/Vesl-1S enhances hippocampal synaptic transmission. Eur J Neurosci. 2003;18:811–819. doi: 10.1046/j.1460-9568.2003.02812.x. [DOI] [PubMed] [Google Scholar]
- Hermann BP, Bayless K, Hansen R, Parrish J, Seidenberg M. Cerebellar atrophy in temporal lobe epilepsy. Epilepsy Behav. 2005;7:279–287. doi: 10.1016/j.yebeh.2005.05.022. [DOI] [PubMed] [Google Scholar]
- Hiyama TY, Watanabe E, Ono K, Inenaga K, Tamkun MM, Yoshida S, Noda M. Na(x) channel involved in CNS sodium-level sensing. Nat Neurosci. 2002;5:511–512. doi: 10.1038/nn0602-856. [DOI] [PubMed] [Google Scholar]
- Hollrigel GS, Chen K, Baram TZ, Soltesz I. The pro-convulsant actions of corticotropin-releasing hormone in the hippocampus of infant rats. Neuroscience. 1998;84:71–79. doi: 10.1016/s0306-4522(97)00499-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosokawa M, Klegeris A, Maguire J, McGeer PL. Expression of complement messenger RNAs and proteins by human oligodendroglial cells. Glia. 2003;42:417–423. doi: 10.1002/glia.10234. [DOI] [PubMed] [Google Scholar]
- Houser CR, Esclapez M. Downregulation of the alpha5 subunit of the GABA(A) receptor in the pilocarpine model of temporal lobe epilepsy. Hippocampus. 2003;13:633–645. doi: 10.1002/hipo.10108. [DOI] [PubMed] [Google Scholar]
- Hunsberger JG, Bennett AH, Selvanayagam E, Duman RS, Newton SS. Gene profiling the response to kainic acid induced seizures. Brain Res Mol Brain Res. 2005;141:95–112. doi: 10.1016/j.molbrainres.2005.08.005. [DOI] [PubMed] [Google Scholar]
- Irizarry RA, Warren D, Spencer F, Kim IF, Biswal S, Frank BC, Gabrielson E, Garcia JG, Geoghegan J, Germino G, Griffin C, Hilmer SC, Hoffman E, Jedlicka AE, Kawasaki E, Martinez-Murillo F, Morsberger L, Lee H, Petersen D, Quackenbush J, et al. Multiple-laboratory comparison of microarray platforms. Nat Methods. 2005;2:345–350. doi: 10.1038/nmeth756. [DOI] [PubMed] [Google Scholar]
- Jackson DN, Foster DA. The enigmatic protein kinase Cdelta: complex roles in cell proliferation and survival. FASEB J. 2004;18:627–636. doi: 10.1096/fj.03-0979rev. [DOI] [PubMed] [Google Scholar]
- Jamali S, Bartolomei F, Robaglia-Schlupp A, Massacrier A, Peragut JC, Regis J, Dufour H, Ravid R, Roll P, Pereira S, Royer B, Roeckel-Trevisiol N, Fontaine M, Guye M, Boucraut J, Chauvel P, Cau P, Szepetowski P. Large-scale expression study of human mesial temporal lobe epilepsy: evidence for dysregulation of the neurotransmission and complement systems in the entorhinal cortex. Brain. 2006;129:625–641. doi: 10.1093/brain/awl001. [DOI] [PubMed] [Google Scholar]
- Kokaia M, Ernfors P, Kokaia Z, Elmer E, Jaenisch R, Lindvall O. Suppressed epileptogenesis in BDNF mutant mice. Exp Neurol. 1995;133:215–224. doi: 10.1006/exnr.1995.1024. [DOI] [PubMed] [Google Scholar]
- Lamas M, Gomez-Lira G, Gutierrez R. Vesicular GABA transporter mRNA expression in the dentate gyrus and in mossy fiber synaptosomes. Brain Res Mol Brain Res. 2001;93:209–214. doi: 10.1016/s0169-328x(01)00202-9. [DOI] [PubMed] [Google Scholar]
- Loscher W, Schmidt D. New horizons in the development of antiepileptic drugs: the search for new targets. Epilepsy Res. 2004;60:77–159. doi: 10.1016/j.eplepsyres.2004.06.004. [DOI] [PubMed] [Google Scholar]
- Lu XC, Williams AJ, Yao C, Berti R, Hartings JA, Whipple R, Vahey MT, Polavarapu RG, Woller KL, Tortella FC, Dave JR. Microarray analysis of acute and delayed gene expression profile in rats after focal ischemic brain injury and reperfusion. J Neurosci Res. 2004;77:843–857. doi: 10.1002/jnr.20218. [DOI] [PubMed] [Google Scholar]
- Lukasiuk K, Pitkanen A. Large-scale analysis of gene expression in epilepsy research: is synthesis already possible? Neurochem Res. 2004;29:1169–1178. doi: 10.1023/b:nere.0000023604.91584.6c. [DOI] [PubMed] [Google Scholar]
- Lukasiuk K, Kontula L, Pitkanen A. cDNA profiling of epileptogenesis in the rat brain. Eur J Neurosci. 2003;17:271–279. doi: 10.1046/j.1460-9568.2003.02461.x. [DOI] [PubMed] [Google Scholar]
- Lynch BA, Lambeng N, Nocka K, Kensel-Hammes P, Bajjalieh SM, Matagne A, Fuks B. The synaptic vesicle protein SV2A is the binding site for the antiepileptic drug levetiracetam. Proc Natl Acad Sci USA. 2004;101:9861–9866. doi: 10.1073/pnas.0308208101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahad D, Callahan MK, Williams KA, Ubogu EE, Kivisakk P, Tucky B, Kidd G, Kingsbury GA, Chang A, Fox RJ, Mack M, Sniderman MB, Ravid R, Staugaitis SM, Stins MF, Ransohoff RM. Modulating CCR2 and CCL2 at the blood-brain barrier: relevance for multiple sclerosis pathogenesis. Brain. 2006;129:212–223. doi: 10.1093/brain/awh655. [DOI] [PubMed] [Google Scholar]
- Mathern GW, Babb TL, Leite JP, Pretorius K, Yeoman KM, Kuhlman PA. The pathogenic and progressive features of chronic human hippocampal epilepsy. Epilepsy Res. 1996;26:151–161. doi: 10.1016/s0920-1211(96)00052-6. [DOI] [PubMed] [Google Scholar]
- Mayford M, Bach ME, Huang YY, Wang L, Hawkins RD, Kandel ER. Control of memory formation through regulated expression of a CaMKII transgene. Science. 1996;274:1678–1683. doi: 10.1126/science.274.5293.1678. [DOI] [PubMed] [Google Scholar]
- Mazarati A, Langel U, Bartfai T. Galanin: an endogenous anticonvulsant? Neuroscientist. 2001;7:506–517. doi: 10.1177/107385840100700607. [DOI] [PubMed] [Google Scholar]
- Mazarati AM, Liu H, Soomets U, Sankar R, Shin D, Katsumori H, Langel U, Wasterlain CG. Galanin modulation of seizures and seizure modulation of hippocampal galanin in animal models of status epilepticus. J Neurosci. 1998;18:10070–10077. doi: 10.1523/JNEUROSCI.18-23-10070.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merino SM, Maren S. Hitting Ras where it counts: Ras antagonism in the basolateral amygdala inhibits long-term fear memory. Eur J Neurosci. 2006;23:196–204. doi: 10.1111/j.1460-9568.2005.04546.x. [DOI] [PubMed] [Google Scholar]
- Morel Y, Mermod N, Barouki R. An autoregulatory loop controlling CYP1A1 gene expression: role of H2O2 and NFI. Mol Cell Biol. 1999;19:6825–6832. doi: 10.1128/mcb.19.10.6825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanobashvili A, Woldbye DP, Husum H, Bolwig TG, Kokaia M. Neuropeptide Y Y5 receptors suppress in vitro spontaneous epileptiform bursting in the rat hippocampus. NeuroReport. 2004;15:339–343. doi: 10.1097/00001756-200402090-00026. [DOI] [PubMed] [Google Scholar]
- Newman TA, Galea I, van Rooijen N, Perry VH. Blood-derived dendritic cells in an acute brain injury. J Neuroimmunol. 2005;166:167–172. doi: 10.1016/j.jneuroim.2005.04.026. [DOI] [PubMed] [Google Scholar]
- Niedermeyer E. The electrocerebellogram. Clin EEG Neurosci. 2004;35:112–115. doi: 10.1177/155005940403500213. [DOI] [PubMed] [Google Scholar]
- Ozbas-Gerceker F, Redeker S, Boer K, Ozguc M, Saygi S, Dalkara T, Soylemezoglu F, Akalan N, Baayen JC, Gorter JA, Aronica E. Serial analysis of gene expression in the hippocampus of patients with mesial temporal lobe epilepsy. Neuroscience. 2006;138:457–474. doi: 10.1016/j.neuroscience.2005.11.043. [DOI] [PubMed] [Google Scholar]
- Peng Z, Huang CS, Stell BM, Mody I, Houser CR. Altered expression of the δ subunit of the GABAA receptor in a mouse model of temporal lobe epilepsy. J Neurosci. 2004;24:8629–8639. doi: 10.1523/JNEUROSCI.2877-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potschka H, Krupp E, Ebert U, Gumbel C, Leichtlein C, Lorch B, Pickert A, Kramps S, Young K, Grune U, Keller A, Welschof M, Vogt G, Xiao B, Worley PF, Loscher W, Hiemisch H. Kindling-induced overexpression of Homer 1A and its functional implications for epileptogenesis. Eur J Neurosci. 2002;16:2157–2165. doi: 10.1046/j.1460-9568.2002.02265.x. [DOI] [PubMed] [Google Scholar]
- Raghavendra Rao VL, Bowen KK, Dhodda VK, Song G, Franklin JL, Gavva NR, Dempsey RJ. Gene expression analysis of spontaneously hypertensive rat cerebral cortex following transient focal cerebral ischemia. J Neurochem. 2002;83:1072–1086. doi: 10.1046/j.1471-4159.2002.01208.x. [DOI] [PubMed] [Google Scholar]
- Raghavendra Rao VL, Dhodda VK, Song G, Bowen KK, Dempsey RJ. Traumatic brain injury-induced acute gene expression changes in rat cerebral cortex identified by GeneChip analysis. J Neurosci Res. 2003;71:208–219. doi: 10.1002/jnr.10486. [DOI] [PubMed] [Google Scholar]
- Scharfman HE, Goodman JH, Sollas AL. Actions of brain-derived neurotrophic factor in slices from rats with spontaneous seizures and mossy fiber sprouting in the dentate gyrus. J Neurosci. 1999;19:5619–5631. doi: 10.1523/JNEUROSCI.19-13-05619.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfman HE, Goodman JH, Sollas AL, Croll SD. Spontaneous limbic seizures after intrahippocampal infusion of brain-derived neurotrophic factor. Exp Neurol. 2002;174:201–214. doi: 10.1006/exnr.2002.7869. [DOI] [PubMed] [Google Scholar]
- Schmidt D, Rogawski MA. New strategies for the identification of drugs to prevent the development or progression of epilepsy. Epilepsy Res. 2002;50:71–78. doi: 10.1016/s0920-1211(02)00070-0. [DOI] [PubMed] [Google Scholar]
- Schwartzkroin PA, Baraban SC, Hochman DW. Osmolarity, ionic flux, and changes in brain excitability. Epilepsy Res. 1998;32:275–285. doi: 10.1016/s0920-1211(98)00058-8. [DOI] [PubMed] [Google Scholar]
- Silva AJ, Paylor R, Wehner JM, Tonegawa S. Impaired spatial learning in alpha-calcium-calmodulin kinase II mutant mice. Science. 1992;257:206–211. doi: 10.1126/science.1321493. [DOI] [PubMed] [Google Scholar]
- Simard M, Nedergaard M. The neurobiology of glia in the context of water and ion homeostasis. Neuroscience. 2004;129:877–896. doi: 10.1016/j.neuroscience.2004.09.053. [DOI] [PubMed] [Google Scholar]
- Simonato M, Bregola G, Armellin M, Del Piccolo P, Rodi D, Zucchini S, Tongiorgi E. Dendritic targeting of mRNAs for plasticity genes in experimental models of temporal lobe epilepsy. Epilepsia. 2002;43:153–158. doi: 10.1046/j.1528-1157.43.s.5.32.x. [DOI] [PubMed] [Google Scholar]
- Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC. Measurement of protein using bicinchoninic acid. Anal Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [Erratum (1987) 163:279] [DOI] [PubMed] [Google Scholar]
- Spencer SS, Spencer DD. Entorhinal-hippocampal interactions in medial temporal lobe epilepsy. Epilepsia. 1994;35:721–727. doi: 10.1111/j.1528-1157.1994.tb02502.x. [DOI] [PubMed] [Google Scholar]
- Stefan H, Pauli E. Progressive cognitive decline in epilepsy: an indication of ongoing plasticity. Prog Brain Res. 2002;135:409–417. doi: 10.1016/S0079-6123(02)35038-6. [DOI] [PubMed] [Google Scholar]
- Tallent MK, Siggins GR. Somatostatin acts in CA1 and CA3 to reduce hippocampal epileptiform activity. J Neurophysiol. 1999;81:1626–1635. doi: 10.1152/jn.1999.81.4.1626. [DOI] [PubMed] [Google Scholar]
- Tang Y, Lu A, Aronow BJ, Wagner KR, Sharp FR. Genomic responses of the brain to ischemic stroke, intracerebral haemorrhage, kainate seizures, hypoglycemia, and hypoxia. Eur J Neurosci. 2002;15:1937–1952. doi: 10.1046/j.1460-9568.2002.02030.x. [DOI] [PubMed] [Google Scholar]
- Temkin NR. Antiepileptogenesis and seizure prevention trials with antiepileptic drugs: meta-analysis of controlled trials. Epilepsia. 2001;42:515–524. doi: 10.1046/j.1528-1157.2001.28900.x. [DOI] [PubMed] [Google Scholar]
- Tian GF, Azmi H, Takano T, Xu Q, Peng W, Lin J, Oberheim N, Lou N, Wang X, Zielke HR, Kang J, Nedergaard M. An astrocytic basis of epilepsy. Nat Med. 2005;11:973–981. doi: 10.1038/nm1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tocco G, Bi X, Vician L, Lim IK, Herschman H, Baudry M. Two synaptotagmin genes, Syt1 and Syt4, are differentially regulated in adult brain and during postnatal development following kainic acid-induced seizures. Brain Res Mol Brain Res. 1996;40:229–239. doi: 10.1016/0169-328x(96)00055-1. [DOI] [PubMed] [Google Scholar]
- Tu B, Bazan NG. Hippocampal kindling epileptogenesis upregulates neuronal cyclooxygenase-2 expression in neocortex. Exp Neurol. 2003;179:167–175. doi: 10.1016/s0014-4886(02)00019-5. [DOI] [PubMed] [Google Scholar]
- van Vliet EA, da Costa Araújo S, Redeker S, van Schaik R, Aronica E, Gorter JA. Blood-brain barrier leakage may lead to progression of temporal lobe epilepsy. Brain. 2006 doi: 10.1093/brain/awl318. in press. [DOI] [PubMed] [Google Scholar]
- Vezzani A, Granata T. Brain inflammation in epilepsy: experimental and clinical evidence. Epilepsia. 2005;46:1724–1743. doi: 10.1111/j.1528-1167.2005.00298.x. [DOI] [PubMed] [Google Scholar]
- Vezzani A, Serafini R, Stasi MA, Samanin R, Ferrarese C. Epileptogenic activity of two peptides derived from diazepam binding inhibitor after intrahippocampal injection in rats. Epilepsia. 1991;32:597–603. doi: 10.1111/j.1528-1157.1991.tb04698.x. [DOI] [PubMed] [Google Scholar]
- Vezzani A, Michalkiewicz M, Michalkiewicz T, Moneta D, Ravizza T, Richichi C, Aliprandi M, Mule F, Pirona L, Gobbi M, Schwarzer C, Sperk G. Seizure susceptibility and epileptogenesis are decreased in transgenic rats overexpressing neuropeptide Y. Neuroscience. 2002;110:237–243. doi: 10.1016/s0306-4522(01)00581-4. [DOI] [PubMed] [Google Scholar]
- Watanabe E, Hiyama TY, Shimizu H, Kodama R, Hayashi N, Miyata S, Yanagawa Y, Obata K, Noda M. Sodium-level-sensitive sodium channel Na(x) is expressed in glial laminate processes in the sensory circumventricular organs. Am J Physiol. 2006;290:R568–R576. doi: 10.1152/ajpregu.00618.2005. [DOI] [PubMed] [Google Scholar]
- Xiong ZQ, Qian W, Suzuki K, McNamara JO. Formation of complement membrane attack complex in mammalian cerebral cortex evokes seizures and neurodegeneration. J Neurosci. 2003;23:955–960. doi: 10.1523/JNEUROSCI.23-03-00955.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yepes M, Sandkvist M, Coleman TA, Moore E, Wu JY, Mitola D, Bugge TH, Lawrence DA. Regulation of seizure spreading by neuroserpin and tissue-type plasminogen activator is plasminogen-independent. J Clin Invest. 2002;109:1571–1578. doi: 10.1172/JCI14308. [DOI] [PMC free article] [PubMed] [Google Scholar]