

Abstract
Mutations in the gene for the astrocyte specific intermediate filament, glial fibrillary acidic protein (GFAP), cause the rare leukodystrophy Alexander disease (AxD). To study the pathology of this primary astrocyte defect, we have generated knock-in mice with missense mutations homologous to those found in humans. In this report, we show that mice with GFAP-R76H and -R236H mutations develop Rosenthal fibers, the hallmark protein aggregates observed in astrocytes in AxD, in the hippocampus, corpus callosum, olfactory bulbs, subpial, and periventricular regions. Astrocytes in these areas appear reactive and total GFAP expression is elevated. Although general white matter architecture and myelination appear normal, when crossed with an antioxidant response element reporter line, the mutant mice show a distinct pattern of reporter-gene induction that is especially prominent in the corpus callosum, and histochemical staining reveals accumulation of iron in the same region. The mutant mice have a normal lifespan and show no overt behavioral defects, but are more susceptible to kainate-induced seizures. Although these mice demonstrate increased GFAP expression by themselves, further elevation of GFAP via crosses to GFAP transgenic animals leads to a shift in GFAP solubility, an increased stress response, and ultimately death. The mice do not display the full spectrum of pathology observed in human infantile AxD, but may more closely resemble the adult form of the disease. These studies provide formal proof linking GFAP mutations with Rosenthal fibers and oxidative stress, and correlate gliosis and GFAP protein levels to the severity of the disease.
Keywords: Alexander disease, GFAP, astrocyte, Rosenthal fiber, oxidative stress, leukodystrophy
Introduction
Alexander disease (AxD) is an autosomal dominant disorder caused by mutations in the astrocyte specific type III intermediate filament glial fibrillary acidic protein (GFAP) (Brenner et al., 2001). The hallmark pathology of this disease is the presence of Rosenthal fibers, ubiquitinated protein aggregates composed of GFAP, vimentin, and small heat shock proteins in the cytoplasm of astrocytes (Alexander, 1949; Iwaki et al., 1989). In the infantile form of AxD, which is the most common, symptoms appear within the first 2 years of life and typically include seizures, macrocephaly, bulbar dysfunction, and psychomotor retardation. Infantile onset is also associated with an aggressive course (mean survival <4 years after onset) and widespread abnormalities of frontal white matter, the latter feature leading to the usual classification of Alexander disease among the leukodystrophies. Later-onset juvenile and adult forms of AxD have also been recognized, originally based solely on the presence of Rosenthal fibers but more recently with genetic confirmation. These individuals have longer life expectancies and frequently demonstrate motor but not cognitive deficits, with pathology predominantly affecting the cerebellum and brainstem. Leukodystrophy is less severe in the juvenile and adult groups, and sometimes completely absent (Messing and Goldman, 2004; Salvi et al., 2005; Barkovich and Messing, 2006; van der Knaap et al., 2006).
Nearly all of the disease-causing mutations in GFAP occur as heterozygous single base-pair changes within the coding region, mostly in the conserved α-helices of the central rod domain, but with a few occurring in the C-terminal tail domain and in the precoil domain near the N terminus (Brenner et al., 2001; Li et al., 2005). Two mutation hot spots are apparent at R79 and R239. Approximately half of all published patients have mutations at these two codons, mostly predicting changes to either cysteine or histidine. For most of the mutations, genotype-phenotype correlations are not clear (for instance, R239C, R79C, and R79H mutations occur in both infantile and juvenile forms of AxD), although all of the patients with R239H mutations are infantile with particularly severe courses of disease. The mechanism by which GFAP mutations lead to Rosenthal fiber accumulation and the pathogenicity of the Rosenthal fibers themselves are not known.
We reported previously that transgenic mice carrying additional copies of the wild-type human GFAP gene and expressing elevated levels of protein replicate some features of human AxD (Messing et al., 1998; Hagemann et al., 2005). Nevertheless, the GFAP transgenics are not true models of human AxD because they express wild-type rather than mutant forms of the protein. To more accurately model the underlying molecular defect in AxD, we have generated knock-in mice with GFAP missense mutations homologous to those found in the human disorder. Here, we show a causal link between GFAP missense mutations identified in AxD patients and the formation of Rosenthal fibers and white matter stress. In addition, we find that elevated levels of GFAP, in concert with mutant forms of the protein, dramatically accelerates the course of disease.
Materials and Methods
Generation of GFAP mutant knock-in mice.
To generate targeting vectors for replicating Alexander disease (AxD) mutations in mice, we used a 6.5 kb BamHI genomic fragment from the GFAP gene (murine strain 129S6; Stratagene, Cedar Creek, TX; library no. 946305), previously isolated to construct a GFAP knock-out vector (McCall et al., 1996). This fragment, encompassing the first six exons of the GFAP gene and 2 kb of the 5′ promoter region, was used as a template for PCR-based site-directed mutagenesis (Weiner et al., 1994). Separate plasmids were generated containing specific mutations equivalent to R79H and R239H found in AxD patients. Primers flanking the six exons were used to sequence the resulting clones to confirm the site-directed mutation, and to screen against potential secondary mutations created in the coding sequence, intron splice sites, or promoter region through polymerase error.
A unique BstBI restriction site within intron 3 was used to insert a loxP-flanked phosphoglycerate kinase promoter-neomycin (neo) cassette in the targeting sequence for positive selection. After insertion of the neo cassette, the BamHI fragment was ligated into the vector pMC1-TKpA, containing the herpes thymidine kinase (TK) gene and polyadenylation signal (pA) for negative selection. A NotI site from the pMC1-TK vector linearizes the constructed plasmid with the pBluescript vector remaining attached to the TK gene.
AB-1 embryonic stem (ES) cells were electroporated with 10 μg of linearized targeting vector and treated with 350 μg/ml G418 and 200 nm FIAU (1-(2-deoxy-2-fluoro-β-d-arabinofuranosyl)-5-iodouracil) for positive and negative selection, respectively. The resulting clones were isolated and screened by Southern blot analysis of EcoRI digested genomic DNA with a 600bp BamHI/EcoRI fragment from intron 6 of the GFAP gene, as described previously (McCall et al., 1996). An EcoRI site within the neomycin cassette of the targeting vector allowed the detection of a truncated restriction product from the targeted allele. Positive ES cell clones with normal karyotypes were subsequently injected into C57BL/6J blastocysts. Chimeric male founders were bred with 129S6 females. To excise the loxP flanked neomycin cassette from intron 3, first filial generation (F1) mice carrying the targeted mutations were crossed with the Rosa26-Cre transgenic line in the FVB/N strain (Grippo et al., 2002), which maintains ubiquitous expression of Cre recombinase and allows germline deletions of the loxP flanked sequences. GFAP mutants in the F2 generation were then bred back to 129S6 or C57BL/6J strains. The experiments described here used early generation (F3–F5) mixed-strain mice (129S6 × FVB/N) with littermate controls, unless stated otherwise.
Transgenic and knock-out mice crossed with the GFAP mutant mice.
GFAP Tg73.7 transgenic (Messing et al., 1998), GFAP knock-out (McCall et al., 1996), Rosa26-Cre (Grippo et al., 2002), and antioxidant response element-human placental alkaline phosphatase (ARE-hPAP) reporter mice (Johnson et al., 2002) were generated as described previously. GFAP transgenic, ARE-hPAP, and Rosa26-Cre mice are maintained in the FVB/N background as hemizygotes, and GFAP knock-out mice are maintained as heterozygotes in the 129S6 strain.
Neurobehavioral testing.
To examine the mice for gross neurological deficits, mutants and littermate controls (n ≥ 8, males, 3 months old) were put through a battery of tests from the SHIRPA primary screen (Rogers et al., 1997). Briefly, this testing includes observation and scoring of body weight and position, spontaneous activity, respiration rate, presence of tremor, transfer arousal and locomotor activity in an open arena, palpebral closure, piloerection, startle response, gait, pelvic and tail elevation, touch response, positional passivity, trunk curl, visual placing, limb grasping, grip strength, body tone, pinnea reflex, corneal reflex, toe-pinch response, skin color, heart rate, limb tone, abdominal tone, lacrimation, salivation, provoked biting, righting reflex, contact righting reflex, negative geotaxis, fear (freezing during transfer arousal), irritability (struggle during supine restraint), aggression (provoked biting or attack), and vocalization (provoked during handling). A more detailed description of the protocol can be found on the Mouse Mutagenesis Program web site at http://www.mgu.har.mrc.ac.uk/facilities/mutagenesis/mutabase/shirpa_summary.html (MRC Mammalian Genetics Unit and UK Mouse Genome Centre, Harwell, Oxfordshire, UK).
Sequence analysis of genomic DNA and cDNA.
To confirm the sequence of targeted alleles, PCR products amplified from the mutant exons were sequenced. Amplification for the R76H mutation in exon 1 was performed with the primer pair 5′-GGC AGG ATG GAG CGG AGA CG-3′ and 5′-TCA GTT CAG CTG CCA GCG CCT-3′, and for R236H in exon 4 primers 5′-CGA GAA CCG AGC TGG AGT CT-3′ and 5′ TGG GCA AGA CTG GTC ATC TA-3′ were used. Sequence reactions were performed with Applied Biosytems (Foster City, CA) BigDye terminator cycle sequencing kit with purified PCR products and the same primers listed above, and analyzed with an Applied Biosytems DNA analyzer at the University of Wisconsin Biotechnology Center. Sequences were confirmed from multiple amplification reactions to screen against Taq polymerase errors.
To confirm the expression of full-length transcripts from the mutant alleles, brain RNA isolated by homogenization in Trizol (Invitrogen, Carlsbad, CA), was converted to cDNA in a reverse transcription reaction with SuperScript II (1 μg RNA in a 20 μl reaction) according to the manufacturer's protocol. GFAP cDNA was PCR amplified with the primers 5′-GGC AGG ATG GAG CGG AGA CG-3′ at the translation initiation site and 5′-AGG GAA GGA CAA CTG AGC GGA CAC-3′ 91 bp upstream of the poly-adenylation signal in the 3′-untranslated region. PCR products were cloned into the vector pCR2.1 (Invitrogen) and multiple products sequenced to identify mutant and wild-type transcripts. Mutant cDNA clones were sequenced in their entirety to show that transcripts were correctly spliced and that no additional mutations had been incorporated.
Western analysis of GFAP and MBP expression.
GFAP mutant mice were crossed with the GFAP-knock-out line (McCall et al., 1996) to verify expression of protein from the mutated allele. Protein was extracted from the brain by homogenization in 1% deoxycholate/1% Triton X-100/0.1% SDS in PBS with 100 μg/ml PMSF and Complete Protease Inhibitor Cocktail (Roche Applied Sciences, Indianapolis, IN). After centrifugation at 17,000 × g for 20 min at 4°C, supernatants were isolated and protein concentrations determined using the Bio-Rad (Hercules, CA) protein assay. Western analysis for GFAP was performed with 60 μg protein per lane on a 10% Tris-HCl gel, transferred onto polyvinylidene difluoride membrane (Bio-Rad). After blocking in 3% milk/3% normal goat serum in TBS, membranes were probed with monoclonal anti-GFAP (MAB3402; Millipore, Temecula, CA) and anti-β-actin (A-5441; Sigma-Aldrich, St. Louis, MO) primary antibodies diluted 1:1000 and 1:2000, respectively, in TBS with 0.05% Tween 20 for 16 h at 4°C. After washing with TBS/Tween 20, blots were incubated with AlexaFluor 488-tagged secondary antibodies (Invitrogen) diluted 1:500 for 2 h at room temperature. After additional washing, membranes were scanned with a Molecular Dynamics Storm PhosphorImager (Amersham Biosciences, Buckinghamshire, UK).
The same procedure was followed for detection of myelin basic protein (MBP) with the following exceptions: 50 μg protein was loaded along with standard curves of 5–90 μg protein on 15% Tris-HCl gels. MBP blots were blocked in 3% milk in TBS and probed with monoclonal anti-β-actin (A-5441; Sigma-Aldrich) and polyclonal anti-MBP (SC-13914; Santa Cruz Biotechnology, Santa Cruz, CA) at 1:2000 and 1:1000, respectively.
Quantitation of GFAP transcripts and protein.
Real-time PCR and ELISA analysis for GFAP expression was performed with samples from three female mice at 8 weeks of age for each genotype in the 129S6 background (generation 7 or higher). Brains were sagittally bisected for RNA and protein preparation. Quantitative PCR was performed as described previously (Hagemann et al., 2005). GFAP-specific sequences were amplified from brain transcript cDNA with PCR primers corresponding to exons 6–8 (5′-CAA CGT TAA GCT AGC CCT GGA CAT-3′ and 5′-CTC ACC ATC CCG CAT CTC CAC AGT-3′) in a reaction with SYBR green PCR master mix (Applied Biosystems, Foster City, CA), and quantitated with an Applied Biosystems 7500 Real-Time PCR System.
For GFAP ELISA, samples were homogenized in 0.5% Triton X-100, 2 mm Tris-HCl, pH 7.4, 2 mm EDTA, 1 mm PMSF supplemented with Complete Protease Inhibitor Cocktail (Roche Applied Sciences), and centrifuged at 17,000 × g for 20 min at 4°C. Supernatants were collected as the soluble fraction and the remaining pellet representing the insoluble fraction was disrupted in 2% SDS and boiled for 15 min. Protein concentrations of both fractions were quantitated separately with the Bio-Rad protein assay. ELISA was performed in microtiter plates coated with polyclonal anti-GFAP (1 μg/ml PBS; #Z334; Dako, Carpinteria, CA) and blocked with Blotto (5% milk in PBS). Protein samples were diluted in PBS with 0.5% Triton X-100/1% BSA and 1.6 μg soluble or 100 ng insoluble protein analyzed per well. Serial dilutions of purified GFAP (Research Diagnostics, Flanders, NJ) were used as standards. Monoclonal anti-GFAP was used for the detection antibody (MAB3402; Millipore) at 1 μg/ml in Blotto-TX (0.5% Triton X-100). Peroxidase-conjugated anti-mouse IgG antibody (A2304; Sigma) was used for the secondary antibody (1:1000 in Blotto-TX). Wells were rinsed with PBS (before addition of protein samples) or PBS/0.5% Triton X-100 (after incubation with protein samples) between all incubations. The presence of peroxidase activity was detected with the addition of SuperSignal ELISA Femto maximum sensitivity substrate (Pierce Biotechnology, Rockford, IL) and quantitated with a GloRunner microplate luminometer (Turner Biosystems, Sunnyvale, CA).
Electron microscopy of Rosenthal fibers.
Animals were anesthetized and perfused with 10 ml of 0.1 m sodium phosphate buffer, pH7.4, followed by 50 ml 2.5% glutaraldehyde/2% paraformaldehyde. Brains were removed and postfixed for 2 h before embedding in Polybed 812, and sections were viewed in a Philips CM120 scanning transmission electron microscope at the University of Wisconsin Medical School Electron Microscopy Facility (Madison, WI).
Immunohistochemistry and immunofluorescence.
For GFAP immunohistochemistry, animals were anesthetized and perfused with 20 ml of PBS, the brains removed and immersion fixed in methacarn overnight, and then paraffin embedded and sectioned for slides (6 μm). Tissue sections were deparaffinized, rehydrated and pretreated with 0.15% H2O2 for 10 min, followed by 2 h of blocking in PBS with 10% normal goat serum, 1% BSA, and 0.25% Triton X-100. Sections were incubated with monoclonal anti-GFAP (MAB3402; Millipore) diluted 1:500 in block overnight at 4°C, followed by biotinylated goat anti-mouse IgG (1:200) for 2 h, avidin/biotin-peroxidase complex solution for 30 min (Elite ABC kit; Vector Laboratories, Burlingame, CA), followed by Vector SG substrate for 10 min. For costaining with 5-bromo-4-chloro-3-indolyl phosphate (BCIP)/nitroblue tetrazoleum (NBT), 0.2 mg/ml 3,3′-diaminobenzidine (DAB; D5905; Sigma) with 0.006% H2O2 was used for the peroxidase substrate instead of Vector SG. Tissues were rinsed thoroughly with PBS between all incubations.
For GFAP and endothelin B receptor double immunofluorescence, animals were perfused with 4% paraformaldehyde, the brains removed and postfixed 4–5 h, and then cryoprotected in 30% sucrose before cutting 40 μm sections with a sliding microtome. Floating sections were permeabilized with 0.25% Triton X-100 in PBS with 10% normal goat serum and 1% BSA for 2 h. Primary antibodies were diluted in 2% NGS; 0.2% BSA; 0.25% Triton X-100 [1:500 mouse anti-GFAP, MAB3402 (Millipore); 1:100 rabbit anti-endothelin B receptor (Alomone Labs, Jerusalem, Israel)] and sections incubated overnight at 4°C, followed by incubation with secondary antibodies Alexafluor-594-conjugated goat anti-mouse IgG1 (1:200; Invitrogen) and FITC-conjugated goat anti-rabbit IgG (1:100, F0382; Sigma). Images were taken with a Nikon (Tokyo, Japan) C1 laser scanning confocal microscope.
Myelin histochemistry.
Animals were killed by CO2 asphyxiation at 3 months, brains immersion fixed in 10% formalin and cryoprotected in 30% sucrose, and 30 μm sections were prepared with a sliding microtome. Sections were mounted on slides and incubated in a solution of 0.2% gold (III) chloride trihydrate (G4022; Sigma) in 0.02 m sodium PBS, pH 7.4, for 3 h (Schmued, 1990). After rinsing twice in H2O for 5 min, tissues were fixed with 2.5% sodium thiosulfate for 5 min. Slides were subsequently rinsed in running tap water for 30 min, dehydrated, and coverslips were applied with Histochoice mounting media (Amresco, Solon, OH).
Myelin quantitation.
To look for myelin alterations, two methods were used: comparing anterior commissure size and semiquantitative Western blotting of MBP. The cross-sectional area of the anterior commissure was measured in 15 wild-type and 11 R236H/+ mice at 3 months of age using StereoInvestigator (MicroBrightField, Williston, VT). To adjust the raw measurements in consideration of brain size, the following formula was used: Y′ = Y − b (X − M), where Y′ is the adjusted value, Y is the raw measurement, b is the slope of the brain weight-commissure area regression line, X is the brain weight, and M is the mean brain size (Bishop and Wahlsten, 1999). The adjusted commissure areas were then compared by t test.
For the semiquantitative Westerns, standard curves from age- and strain-matched wild-type mice were used to determine the linear relationship between actin and MBP levels. The actin and MBP levels from eight R236H/+ mutants and eight wild-type female littermates at P21 were then used to calculate relative MBP levels for each sample. These relative values were compared using the t test. Each sample was analyzed on multiple blots, with 2–8 replicates per sample.
ARE-hPAP histochemistry and quantitative assay.
For alkaline phosphatase histochemistry, animals at 3 months of age were perfused with 10 ml cold PBS followed by 50 ml 4% paraformaldehyde. Brains were immersion fixed for 1 h, alcohol dehydrated, paraffin embedded, and sectioned for slides (6 μm). After deparaffinizing and rehydration, sections were heated at 65°C for 30 min in alkaline phosphatase (AP) buffer (0.1 m Tris, pH 9.5, 0.1 m NaCl, 5 mm MgCl2) to inactivate endogenous alkaline phosphatases. Sections were then incubated in AP buffer with 0.17 mg/ml BCIP (B6149; Sigma) for 48 h at 37°C, or with 0.17 mg/ml BCIP and 0.34 mg/ml NBT (N6639; Sigma) for 1 h at room temperature. Sections were rinsed, counterstained with eosin, dehydrated, and coverslips were applied.
To quantitate and compare the level of reporter-gene activity in the mutant lines at different ages, brains were collected from GFAP-mutant and wild-type mice, all also carrying the ARE-hPAP transgene, at 1, 2, 3, and 6 weeks, and 3 and 6 months of age (three samples per group). Half-brain samples (bisected at midline) were homogenized in 1 ml of lysate buffer [50 mm Tris-HCl, pH 7.5, 5 mm MgCl2, 100 mm NaCl/4% CHAPS (3-[(3-cholamidopropyl)dimethyl-ammonio]-1-propanesulfonate)] and centrifuged at 17,000 × g for 20 min at 4°C. Supernatants were collected and protein quantitated with the Bio-Rad protein assay. Samples were diluted to 100 ng/μl, 25 μl added to 75 μl 200 mm diethanolamine (DEA) in a microtiter plate, and heated for 20 min at 65°C to inactivate endogenous alkaline phosphatase activity. At room temperature, 100 μl chemiluminescent substrate CSPD (disodium-3-[4-methoxyspiro{1,2-dioxetane-3,2′-(5′-chloro)tricyclo[3.3.1.13,7]decan}-4-yl]phenyl phosphate) with Emerald enhancer (2×; Tropix; Applied Biosystems) in 5 mm MgCl2/150 mm DEA was added to each well, incubated at room temperature for 20 min, and enzyme activity quantified with a GloRunner microplate luminometer (Turner Biosystems).
Iron histochemistry.
To assess the distribution and concentration of iron in the brain, the histochemical method described by Hill and Switzer (1984) was used to stain frozen sections from both wild-type and R236H/+ mice at 6 weeks of age. Mice were perfused with 10% formalin in PBS before dissection. Brains were postfixed by immersion overnight, cryoprotected in 30% sucrose, frozen in Tissue-Tek Optimal Cutting Temperature Compound (Sakura Finetek, Torrance, CA), and 40 μm sections were cut with a sliding microtome and mounted on slides. Sections were treated with a modified Perl's solution (1% potassium ferrocyanide, 1% HCl) for 30 min, rinsed in H2O for 30 min, and stained with 5 mg/ml DAB chromogen in sodium phosphate buffer, pH 7.4, with 0.005% H2O2 for 15 min. Slides were rinsed again in H2O for 30 min, dehydrated, and coverslips were applied. Control slides were treated with DAB solution only and showed minimal background peroxidase activity.
Kainic acid induced seizures.
Wild-type and R236H/+ mice (n = 20 including 10 males and 10 females each, C57BL/6J generation 11, 3 months of age) were injected intraperitoneally with 20 mg/kg kainic acid (A.G. Scientific, San Diego, CA) and observed for 3 h after injection. Seizures were evaluated at 5 min intervals over a 3 h period based on a modified scoring system described previously (Racine, 1972): 0, normal; 1, immobile; 2, rigid; 3, repetitive movement (forelimb tremor, head bobbing); 4, rearing and falling, loss of posture; 5, repeated episodes of category 4 within 5 min period; 6, tonic seizure; 7, death. Animals that died were scored as 7 for the remaining time intervals. Although the only wild-type mouse that died was female, there were no differences observed between male and female mutant mice, and sexes for the two genotypes were combined for statistical analysis.
At 24 h postinjection, surviving animals were killed by CO2 asphyxiation, the brains removed, and immersion fixed in 10% formalin before embedding in paraffin for haematoxylin and eosin (H&E) staining. Adjacent sections were stained with FluoroJade B per the manufacturer's protocol (Millipore). Briefly, sections were incubated in 80% ethanol with 1% NaOH for 5 min, 70% ethanol 2 min, H2O 2 min, 0.06% KMnO4 20 min, H2O 2 min, 4 μg/ml FluoroJade B in 0.1% acetic acid 20 min, rinsed in H2O, dried, dehydrated in xylene, and coverslipped with DPX Mountant for histology (Sigma-Aldrich).
Results
Generating knock-in mice to replicate AxD GFAP mutations
To generate a mouse model of Alexander disease, we designed targeting vectors to replicate mutations found in human GFAP. Arg79 and Arg239 are mutation hotspots in AxD through transitions to cysteine or histidine codons. We have replicated changes at both positions with targeting constructs carrying the equivalent mutations R76H and R236H in the mouse gene (the mouse sequence is offset from the human sequence by the absence of three residues in the head domain). A genomic fragment of the mouse GFAP gene including the first six exons and promoter region was used as the base for the targeting vector (Fig. 1A). Sequences were modified by site-directed mutagenesis within exons 1 and 4 for the R76 and R236 residues, respectively, and a neomycin resistance gene was inserted within intron 3 for positive selection.
Figure 1.

Generation of GFAP knock-in mice. Gene targeting vectors were designed with mutations homologous to two mutations found in humans: R79H and R239H. A, The genomic organization of the murine GFAP gene is indicated above with the exons shown as solid numbered boxes. Targeting vectors were generated from a 6.5 kb BamHI fragment encompassing the promoter and exons 1 through 6. The equivalent positions for the human mutations are shown as R76 and R236 in exons 1 and 4, respectively. Relevant restriction sites including the BamHI fragment and the BstBI site used to insert the loxP-flanked neo cassette (dark gray box) are also indicated. The herpes TK gene is shown adjacent to the genomic BamHI fragment (light gray box). Homologous recombination with the targeting vector will introduce a single base mutation within the designated codon. The BamHI/EcoRI probe for screening targeted ES cells for recombination is indicated below intron 6 with the corresponding EcoRI fragments from genomic (6.5 kb) versus targeted (4 kb) alleles. The neo cassette was removed by crossing GFAP mutant mice with transgenic mice expressing Cre recombinase. The final gene structure retains the introduced mutation and the remaining loxP site (arrowhead) in intron 3. B, Southern blot analysis of the targeted ES cells for the different constructs shows the expected EcoRI fragments after homologous recombination: the wild-type alleles give a 6.5 kb band (lanes 1, 3), whereas the R76H line shows a 4 kb allele (lane 2), and the R236H line shows a 2 kb allele (lane 4, the neo cassette was cloned into this vector in the reverse orientation). C, Western blot analysis of heterozygous GFAP mutants and mutant mice crossed with GFAP knock-out mice shows expression of the mutant alleles. A + indicates a wild-type allele; − indicates the null allele. D, R236H/+ mice show a significant decrease in body weight compared with wild-type littermates among both male and female mice at 3 months. Decreases are also observed in R76H/+ male mice compared with wild-type. n ≥10 for each group. Error bars indicate SD.
Targeted ES cells were identified by Southern analysis (Fig. 1B) followed by sequencing to verify that the designed mutations had been retained. Positive ES cells were injected into blastocysts from C57BL/6J mice to generate chimeric founders, which were then bred with 129S6 mice to establish germline transmission of the mutant alleles.
To remove the loxP-flanked neomycin cassette from intron 3 of the targeted GFAP allele, F1 generation mice inheriting the mutations from founders were crossed with the Rosa26-Cre transgenic line, which maintains ubiquitous expression of Cre-recombinase (Fig. 1A). Transcription of the mutant allele was verified after excision of the neo cassette, by cloning and sequencing cDNAs of the GFAP coding sequence prepared from brain mRNA (data not shown).
GFAP protein expression was demonstrated by Western blot analysis (Fig. 1C). Heterozygous mutants R76H/+ and R236H/+ showed similar or increased GFAP band intensity compared with wild-type mice (+/+). To confirm translation of GFAP protein from the targeted alleles, mutant mice were crossed with GFAP knock-out (−/−) mice (McCall et al., 1996) to create heterozygotes with one mutant and one null allele. Western analysis for R76H/− and R236H/− animals demonstrated GFAP bands that were comparable in intensity to that seen in +/− heterozygotes (Fig. 1C).
As a preliminary screen for functional or neurobehavioral phenotypes, observations were made according to the SHIRPA protocol at 3 months of age (Rogers et al., 1997). This series of tests includes assessment of body weight, posture, activity, and any unusual behavior (excessive scratching or biting, tremors or convulsions, rearing, stereotyped or compulsive activity), followed by subjective scoring of transfer arousal, locomotor activity, startle response, and gait in an open arena. Muscle tone and strength are evaluated on a grid above the arena, and autonomic behavior is examined while the animal is restrained in the supine position. The R76H/+ and R236H/+ mice are smaller than control littermates, with the R236H/+ mice showing a more significant difference in body weight (Fig. 1D), but none of the mutant mice show an overt neurological phenotype, or exhibit unusual mortality.
Essentially the same phenotype is observed in mice that have been made congenic on either the 129S6 or C57BL/6J backgrounds. A limited number of mice were generated that are homozygous for the point mutations in an attempt to establish a breeding colony; these animals appeared to have more Rosenthal fibers and weigh less than heterozygotes, but were not otherwise characterized and remained viable up to 11 months of age.
Rosenthal fibers in GFAP mutant mice
Given that the hallmark feature of Alexander disease is the formation of Rosenthal fibers in astrocytes, we examined the brains of the GFAP mutant mice for these characteristic protein aggregates. Histological analysis of brain revealed Rosenthal fibers, especially within the hippocampus, olfactory bulb, corpus callosum and rostral extension, as well as in subpial, perivascular and periventricular regions, in both the R76H/+ and R236H/+ mice (Fig. 2). In general, the R236H/+ mice show more Rosenthal fibers than R76H/+ mice (supplemental Table 1, available at www.jneurosci.org as supplemental material).
Figure 2.
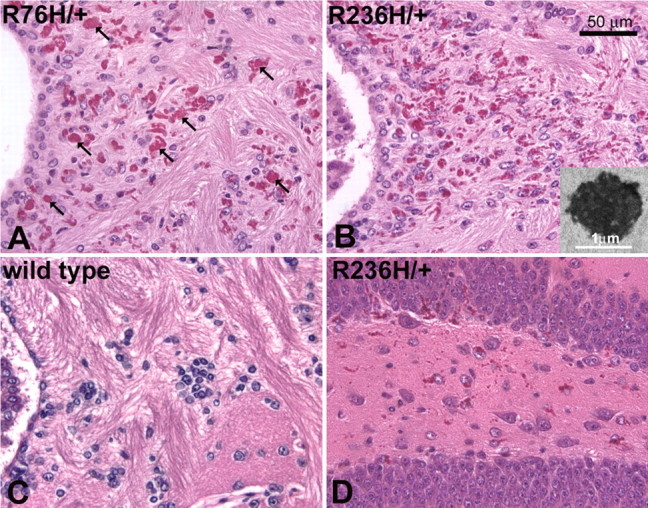
H&E staining shows Rosenthal fibers in GFAP mutant mice. Rosenthal fibers are especially prevalent in the hippocampus, corpus callosum, rostral migratory stream, olfactory bulb, periventricular, perivascular, and subpial regions. A, B, Periventricular white matter, as shown in both R76H/+ (A) and R236H/+ (B) mice at 3 months of age, demonstrates significant Rosenthal fiber accumulation (arrows indicate examples of Rosenthal fibers in A). B, Inset, Electron microscopy reveals Rosenthal fibers as dense osmiophilic aggregates with adjacent swirls of intermediate filaments. C, Rosenthal fibers are never seen in wild-type mice. D, Astrocytes in the hippocampus (dentate gyrus shown) are also laden with Rosenthal fibers as shown in R236H/+ mice (3 months).
In R236H/+ mice, Rosenthal fibers are apparent at the pial surfaces of the brainstem and ventral midbrain and forebrain by routine H&E staining as early as postnatal day 7 (P7). Rosenthal fibers are consistently found in the ventral subpial regions, but are more sparse in the subpial region of the cortex. In the olfactory bulb, Rosenthal fibers are found in the subpial region of the olfactory nerve layer, and by 6 weeks are also apparent in the glomerular layer. At 3 months, small aggregates are evident in the granular layer also. In the hippocampus, Rosenthal fibers can be found near the fissure at 7 d, and by 6 weeks are prominent throughout the hippocampus with a mixture of individual punctate aggregates and larger inclusions aligned within astrocytic processes. At 3 months, the Rosenthal fibers appear to localize more specifically to the dentate gyrus as well as the fissure. Periventricular Rosenthal fibers are generally prevalent near the third and fourth ventricles, again beginning to appear at P7. However, the lateral walls of the lateral ventricles show virtually no Rosenthal fibers at 6 weeks but intense Rosenthal fiber accumulation at 3 months. In the rostral migratory stream, Rosenthal fibers are apparent at 6 weeks, and are more prevalent at 3 months. Dense Rosenthal fiber formation is often evident in the corpus callosum, particularly near the lateral ventricle, and by 6weeks in the adjacent cingulum. In the R76H/+ mice, Rosenthal fibers are less prevalent, but the distribution is similar to that of R236H/+ mice. These data are summarized in supplemental Table 1 (available at www.jneurosci.org as supplemental material).
Rosenthal fibers appear in regions of the brain in which GFAP positive astrocytes are normally observed through immunohistochemical staining. Astrocytes appear hypertrophic in these regions, particularly in the R236H/+ mice at 3 months of age (Fig. 3A–F), with increased immunoreactivity for endothelin B receptor (Fig. 3G–L), suggesting a reactive state (Ishikawa et al., 1997; Baba, 1998; Wilhelmsson et al., 2004). Ubiquitination of Rosenthal fibers is variable, but apparent in both the R76H/+ and R236H/+ mice (Fig. 3M–O).
Figure 3.
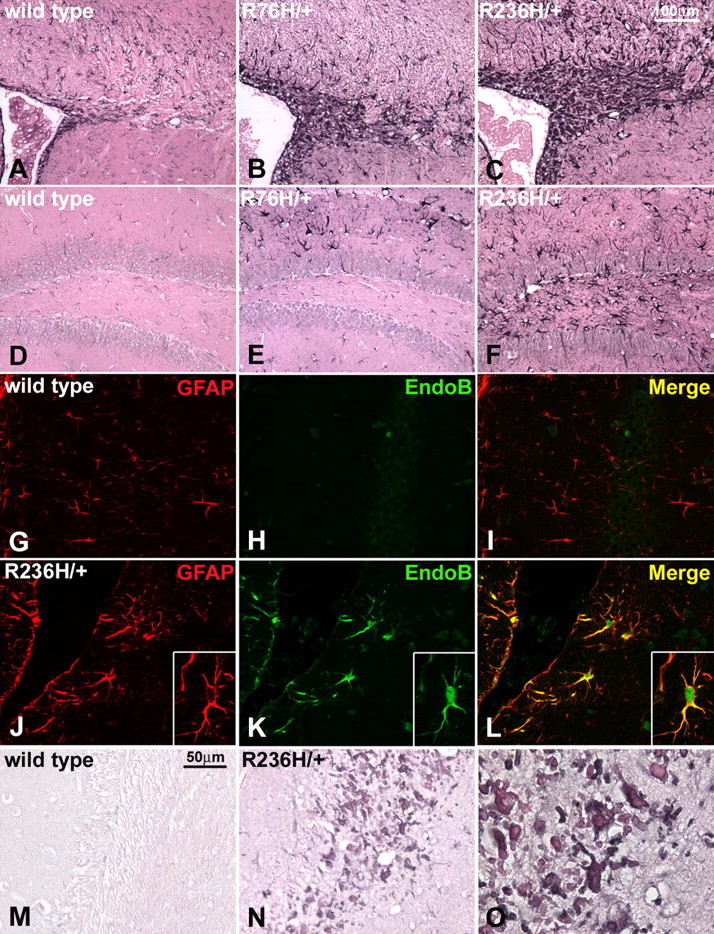
Increased expression of GFAP and reactive astrocytes with ubiquitinated Rosenthal fibers in GFAP mutant mice. A–F, GFAP immunohistochemistry in the periventricular white matter (A–C) and hippocampus (D–F) of R76H/+ (B, E) and R236H/+ (C, F) mice (3 months) shows an increase in GFAP expression in what appear to be reactive astrocytes, when compared with wild-type mice (A, D). Rosenthal fibers are apparent as eosinophilic aggregates demarcated by GFAP immunoreactivity, especially in the periventricular white matter (C). G–L, Immunofluorescent labeling for the endothelin B receptor shows increased expression in astrocytes from R236H/+ (J–L), compared with wild-type mice (G–I), indicating a reactive state. GFAP immunostaining is in red (G, J), the endothelin B receptor is labeled with green (H, K), and the images are merged to show colocalization (I, L) (40× objective). A reactive astrocyte is shown in the inset (J–L) with increased magnification (60× objective). M–O, Rosenthal fibers near the lateral ventricle adjacent to the hippocampus are heavily ubiquitinated as shown by immunohistochemical staining (dark blue-gray) in R236H/+ mice (N, O) compared with wild-type (M) at 3 months of age. N and O are images of the same section with increasing magnification. (M, N, 40× objective; O, 100× objective).
GFAP mutant mice with Rosenthal fibers show increased expression of GFAP
Both Western analysis and immunohistochemistry suggest that GFAP expression is increased in the R236H/+ mice (Figs. 1C, 3C,F). To quantitate GFAP expression, heterozygous mutants were analyzed at 8 weeks of age for total GFAP transcript and protein levels using quantitative real-time RT-PCR and ELISA, respectively. GFAP mRNA was elevated in both R76H/+ and R236H/+ mice (Fig. 4A), with R236H/+ expression being the highest.
Figure 4.

GFAP expression in mutant mice. A, Quantitative real-time PCR analysis of brain transcripts shows increased GFAP expression levels in both R76H/+ and R236H/+ heterozygous mice. B, C, For protein quantitation, brain lysates were separated into soluble (B) and insoluble (C) fractions for ELISA. D, GFAP quantified from mice with the indicated genotypes is expressed as an average nanogram of GFAP per milligram of protein, +/− SD (fold change, the ratio of mutant expression over that of wild-type). R76H/+ mice show a 1.5-fold increase in GFAP for both fractions (D), whereas R236H/+ mutants show a greater increase of 3.7-fold for insoluble GFAP compared with a twofold increase in soluble GFAP (D). All experiments were performed in triplicate with female mice at 8 weeks. For comparison to wild-type control animals: *p < 0.05; **p < 0.01; ***p < 0.001. Error bars indicate SD.
As an intermediate filament protein, GFAP normally exists in dynamic equilibrium between soluble and insoluble pools. To analyze expression and test whether the point mutations alter this equilibrium, the two fractions were separated for quantitation (see Materials and Methods). In R76H/+ and R236H/+ heterozygous mice, both soluble and insoluble GFAP were elevated (Fig. 4B,C). Although GFAP is elevated in both the R76H/+ and R236H/+ animals, the R236H/+ mutant demonstrated a greater increase in the insoluble fraction (Fig. 4D), suggesting that for R236H/+ mice the greater increase in total GFAP in part reflects a shift in equilibrium toward the insoluble pool.
Myelination is normal in GFAP mutant mice
The infantile form of AxD typically involves significant lesions of frontal lobe white matter. To assess myelination in the GFAP mutant mice, we performed histochemical staining for myelin using the gold chloride method. No qualitative differences were observed between mutants and age-matched controls (Fig. 5A,B). Immunohistochemical detection of MBP showed similar results (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). We also measured the area of the anterior commissure in sagittal midline sections from R236H/+ and age-matched controls, and no statistically significant differences were observed (Fig. 5C). As another measure of myelination, we evaluated expression of MBP using semiquantitative Western analysis (Fig. 5D). R236H/+ mice at P21 were compared with age- and sex-matched controls, and showed normal amounts of MBP. These data suggest that brain myelination is normal at an early age, and that loss of myelin is not apparent in the adult mouse.
Figure 5.

Normal white matter architecture and MBP expression in the GFAP-mutant mice. A, B, Histochemical staining for myelin in the R236H/+ mice (B) shows normal anatomical structures when compared with wild-type mice (A) at 3 months of age. C, Area measures of the anterior commissure (a.c.) show no difference between wild-type (WT) and R236H/+ mice (n = 15 wild-type and 11 R236H/+ mice). D, Semiquantitative Western blotting was performed to compare MBP levels between R236H mutants and wild-type littermates at an earlier time point of P21. The first four lanes show examples of wild-type and R236H/+ protein (50 μg/lane); the last four lanes are representative of samples used to generate standard curves for actin versus MBP with the protein quantity indicated above the lane. No significant differences were found for 18.8 or 21.5 kDa isoforms (t test, n = 8 females at 21 d of age). Error bars indicate SD.
Stress response and iron accumulation in the corpus allosum
Previously, we found that overexpression of wild-type GFAP with Rosenthal fiber formation is associated with an antioxidant stress response including increased expression of several genes regulated by the transcription factor Nrf2 (Nfe2l2, nuclear factor, erythroid derived 2, like 2) (Hagemann et al., 2005). To evaluate the CNS stress response in the GFAP point mutants, the mice were crossed with a transgenic reporter line carrying the gene for human placental alkaline phosphatase regulated by the antioxidant response element. R236H/+ mice that also carry the ARE-hPAP transgene show a distinct pattern of alkaline phosphatase activity. By P21, staining is present in the subpial regions of the frontal cortex, the ventral midbrain, and the hippocampal fissure. Faint staining is also present in the corpus callosum. At 6 weeks, staining in these areas increases, particularly within the corpus callosum and adjacent cingulum. By 3 months of age, reporter gene expression is more intense, again highlighting the corpus callosum, cingulum, and the hippocampus, as well as the rostral migratory stream, lateral olfactory tract, and glomerular layer of the olfactory bulb (Fig. 6A,B). The R76H/+ mice show a similar but lower intensity staining pattern (data not shown). To determine the identity of the cell type in which ARE activation occurs, we combined histochemical detection of hPAP activity with immunodetection of GFAP in R236H/+ mice, which revealed colocalization of staining in some (but not all) astrocytes (Fig. 6C). Quantitative analysis of hPAP activity from brain lysates reflects the same increase in reporter gene activity with age in the R236H/+ and R76H/+ mice, with R236H/+ > R76H/+ at 6 weeks and later (Fig. 6D).
Figure 6.

Stress response and iron accumulation in white matter of R236H/+ mice. A, B, GFAP mutant mice were crossed with the ARE-hPAP transgenic reporter line. Induction of hPAP expression through the ARE, as detected by histochemical staining (see Materials and Methods), indicates a cellular stress response. A, Wild-type GFAP;ARE-hPAP transgenic mice (wt) show very little or no alkaline phosphatase activity as indicated by the lack of blue staining. B, R236H/+ mice positive for the ARE-hPAP transgene show reporter expression in the corpus callosum, hippocampus, olfactory bulb, rostral migratory stream, and subpial regions (3 months of age shown). C, Immunohistochemical staining for GFAP (brown) shows colocalization with hPAP activity (purple) within an astrocyte near the hippocampal fissure (R236H/+; 100× objective). D, The stress response in R76H/+ mice and R236H/+ mice increases with age. Brain homogenates from R76H/+;ARE-hPAP and R236H/+;ARE-hPAP mice were analyzed for alkaline phosphatase activity with a chemiluminescent assay from 1 week to 6 months of age. Both the R76H/+ and R236H/+ lines show a significant increase in reporter activity compared with wild-type mice starting at 2 weeks (p < 0.005), that continues to increase with age. By 6 weeks, the R236H/+ mice show an increase in reporter activity over R76H/+ (p < 0.01) as well as wild-type mice. Error bars represent SD. E, F, Iron accumulation in white matter astrocytes is also apparent in GFAP mutant mice. E, In wild-type mice, iron staining mostly highlights oligodendrocytes, however in the R236H/+ mice (F), abnormal iron accumulation can be observed in the corpus callosum and other areas that coincide with the formation of Rosenthal fibers and a stress response (6 weeks of age; sagittal section; CA3, hippocampus; fi, fimbria; cc, corpus callosum).
Because several genes involved in iron homeostasis are regulated by ARE elements, including the ferritin heavy and light chain genes (Wasserman and Fahl, 1997; Tsuji et al., 2000), and because iron accumulation has been shown to cause or exacerbate other neurodegenerative disorders (Curtis et al., 2001; LaVaute et al., 2001; Qian and Shen, 2001), we analyzed the GFAP mutant mice for aberrant iron distribution. Histochemical analysis of iron deposition in wild-type mice typically shows staining of oligodendrocytes (Connor and Menzies, 1996). In the R236H/+ mice, cells with the morphologic features of hypertrophic astrocytes are stained in the corpus callosum, hippocampus and olfactory bulb, whereas staining of oligodendrocytes is weak and difficult to distinguish (Fig. 6E,F).
GFAP mutant mice show increased sensitivity to kainate induced-seizures
Seizures are a common feature of the AxD phenotype in human patients. Although the GFAP mutant mice do not show overt seizures, we surmised that exposure to a convulsant such as kainic acid might reveal an enhanced predisposition to seizure activity, especially considering the abundance of Rosenthal fibers in the hippocampus. As an initial test of this hypothesis, R236H/+ and wild-type mice were injected with kainic acid to induce status epilepticus and evaluate seizure severity. Seizures scored at 5 min intervals over a 3 h period showed a significant increase in severity in the mutants over time (Fig. 7A). Histological analysis of the hippocampus after 24 h showed damage in the regions of CA1 and/or CA3 in all surviving animals with severe seizures scoring ≥5 (repeated loss of postural control, tonic extension, or death), whereas animals scoring <5 exhibited no neuronal death, regardless of genotype (Fig. 7C–H). Of the R236H/+ mice, 50% had severe seizures (score ≥5), with five animals dying, compared with only 10% of wild-type with one lethal outcome (Fig. 7B).
Figure 7.

Increased sensitivity to kainate-induced seizures in R236H/+ mice. A, Seizure severity was scored at 5 min intervals in animals injected with 20 mg/kg kainic acid. R236H/+ animals showed a significant increase in the severity of seizures compared with wild-type mice over the 3 h postinjection period (p < 0.0001, two-way ANOVA; *p < 0.05, Bonferroni posttests; error bars indicate SEM). B, Maximal seizure scores during the observation period were recorded for R236H/+ mice and controls. A higher proportion of mutant mice achieve seizure scores of 5 or higher (*p = 0.0138, Fisher's exact test). C–H, Histochemical analysis of the hippocampus with standard H&E staining (C–E) or FluoroJade B staining (F–H) reveals cell death in CA1 and CA3 (CA1 shown) in animals with severe seizures scoring ≥5 (E, H) (R236H/+ shown), compared with animals with maximal seizure scores <5, which show no cell death regardless of genotype (C, F, wild-type; D, G, R236H/+). n = 20 for each genotype (10 males, 10 females, 3 months old).
Mutant mouse GFAP in combination with elevated wild-type human GFAP is lethal
Given that human Alexander disease typically reflects a combination of GFAP point mutations and upregulation of GFAP, perhaps because of associated gliosis, we hypothesized that significant morbidity might only occur when GFAP levels exceed a critical threshold. To test this hypothesis, we crossed the GFAP point mutant mice with a previously reported transgenic line that overexpresses wild-type human GFAP [Tg73.7 line, referred to here as GFAPTG (Messing et al., 1998)]. In a pure FVB/N genetic background, this GFAPTG line exhibits 7% mortality by 1 month, but the other 93% have apparently normal lifespans (T. L. Hagemann, unpublished observations). As noted above, there is no obvious mortality in the GFAP point mutant lines. In contrast, virtually all mice carrying either of the GFAP point mutations and the wild-type human transgene died at ∼30 d of age (Fig. 8). In this cohort, single mutant or GFAPTG littermates were fully viable through at least 3 months
Figure 8.

GFAP transgene (elevated levels of wild-type protein) combined with GFAP point mutations cause lethality. When GFAP mutant mice are crossed with the GFAP-transgenic line Tg73.7 (GFAPTG) which overexpresses wild-type human GFAP, all progeny carrying the transgene and either the R76H/+ or R236H/+ mutation die at ∼30 d of age. Single transgenic and single mutant littermates showed 100% survival (at least through 90 d). Numbers in parenthesis indicate the number of mice evaluated for each genotype.
At 24 d of age, the R236H/+;GFAPTG mice show increased Rosenthal fiber formation (data not shown) and higher levels of insoluble GFAP than that observed in the single mutant or transgenic mice (Fig. 9A,B,D). Introducing the ARE-hPAP reporter into the R236H/+;GFAPTG mouse reveals increased hPAP staining in the subpial regions, corpus callosum, and hippocampus (data not shown), reflected at the whole-brain level by an increase in ARE-reporter activity (Fig. 9C), suggesting further elevation of the stress response. However, histochemical analysis still shows normal white matter, and there is no evidence of cell death (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Full autopsy revealed no additional pathology, and the exact cause of death is not yet clear.
Figure 9.

Elevated levels of insoluble GFAP and an increased stress response in R236H/+;GFAPTG mice. A, B, Brains from wild-type, R236H/+,GFAPTG, and R236H/+;GFAPTG animals were analyzed at P24 by ELISA to quantitate GFAP protein levels in the soluble (A) and insoluble (B) pools. D, GFAPTG mice show an increase in both soluble and insoluble GFAP compared with mutant mice, and R236H/+;GFAPTG show an additional increase of insoluble GFAP compared with GFAPTG animals. C, R236H/+;GFAPTG mice have an increased CNS stress response. Brain homogenates were taken from the progeny of R236H/+ mice crossed with GFAPTG mice with the ARE-hPAP reporter transgene to quantify the stress response at P25. Reporter activity in R236H/+;GFAPTG mice was increased over the response observed in the R236H/+ mice (*p = 0.02). Differences in reporter activity between R236H/+ and GFAPTG mice, and GFAPTG mice and R236H/+; GFAPTG mice were not significant. n = 4 for each group; *p < 0.05; ***p < 0.001. Error bars indicate SD.
Discussion
Alexander disease is associated with de novo missense mutations in the gene for GFAP that are hypothesized to lead to a dominant gain of function in the encoded protein (Brenner et al., 2001; Li et al., 2002; Messing and Brenner, 2004). In this report, we have replicated two of the common human GFAP mutations in mice: R76H and R236H. During the early postnatal period, these GFAP mutant mice begin to form Rosenthal fibers, the same hallmark protein aggregates found in patients with AxD. The Rosenthal fibers are especially prominent in subpial, periventricular, and perivascular locations, which is similar to the distribution in the human disease. Analogous with the relative severity of disease observed in humans with these two mutations, the R236H/+ mice (equivalent to human R239H) exhibit a more dramatic phenotype than the R76H/+ mice with respect to astrocyte hypertrophy, stress response, and body weight. Both R76H/+ and R236H/+ mice show elevated levels of total GFAP expression. Separation of GFAP into soluble and insoluble compartments demonstrates that increases in GFAP were distributed proportionally in both pools for R76H/+ mice, but skewed toward the insoluble fraction in R236H/+ mutants. This data corresponds with recent transfection studies in cultured cells with human R239C GFAP that also suggested decreased solubility of mutant filaments (Hsiao et al., 2005).
Because the infantile form of Alexander disease is a white matter disorder, we examined the mutant mice for evidence of altered myelination. At the molecular level, early myelination in the R236H/+ mice appears to proceed normally, because MBP levels are the same as that of wild-type mice at P21. In the adult (3 months), histochemical staining for myelin shows normal white matter architecture, and area measures of the anterior commissure in the R236H/+ mutants are not statistically different from wild-type mice. Although these data show no major changes in myelination, the distribution of Rosenthal fibers in the corpus callosum of the GFAP mutants suggests that the pathology is concentrated in white matter regions. The appearance of Rosenthal fibers and white matter loss in humans with AxD are not always coincident (Crome, 1953; Friede, 1964), and the mechanism underlying myelin deficits is not known (Mignot et al., 2004). The GFAP mutant mice may more closely resemble juvenile or adult-onset AxD where white matter lesions are more sporadic than in infantile cases, and in one recent patient not present at all (Barkovich and Messing, 2006; van der Knaap et al., 2006). Additional examination of the mice at a later age may yet reveal subtle late onset degeneration of myelin, although we have not observed obvious changes in a limited number of animals aged to 2 years (Hagemann, unpublished observation).
The ARE-hPAP reporter mouse line further demonstrates a specific white matter stress response in the corpus callosum, as well as in the hippocampus and olfactory bulbs. Activation of the reporter gene correlates with the appearance of Rosenthal fibers, and at least some hPAP-expressing cells are clearly astrocytes based on double-labeling for GFAP. However, it is unclear whether Rosenthal fibers are the cause of, or the response to, stress in the cell. Given the membrane localization of hPAP, it is difficult to determine the percentage of astrocytes that express the reporter gene. We cannot exclude the possibility that some of the ARE activation takes place in microglia (Min et al., 2006).
The ARE sequence is found in the promoters of endogenous stress response genes, and is recognized by the transcription factor Nrf2 (Nfe2l2). Nrf2 transactivates a cadre of antioxidant and detoxifying genes including catalase, oxidoreductases such as Nqo1, and several genes involved with glutathione metabolism (Venugopal and Jaiswal, 1996; Wild et al., 1999; Ishii et al., 2000; Nguyen et al., 2000; Chanas et al., 2002). In addition, Nrf2 regulates both the ferritin heavy and light chain genes (Wasserman and Fahl, 1997; Tsuji et al., 2000). The aberrant distribution of iron observed in the GFAP mutants also corresponds anatomically with the presence of Rosenthal fibers and ARE-hPAP reporter induction, and may result from increased ferritin expression and iron accumulation within astrocytes.
Recent findings in two genetically unrelated leukodystrophies, vanishing white matter leukodystrophy (VWM) and megalencephalic leukoencephalopathy with subcortical cysts (MLC), support the conclusion that astrocytes play a vital role in the development and maintenance of myelin (Leegwater et al., 2002). VWM results from recessive mutations in the subunit genes for the ubiquitously expressed translation initiation factor 2B (eIF2B) complex (Dietrich et al., 2005). The number of GFAP-positive astrocytes is sometimes decreased in VWM white matter lesions (Francalanci et al., 2001; Dietrich et al., 2005), and primary cell cultures derived from the corpus callosum of one VWM patient showed normal development of oligodendrocytes but deficiency of astrocytes, suggesting an astrocyte-mediated deficit (Dietrich et al., 2005). In MLC, patients suffer from recessive mutations in the MLC1 gene, which encodes a putative membrane channel that localizes to the distal processes of astrocytes in the perivascular, subependymal, and subpial regions (Boor et al., 2005). MLC1 is also expressed in Bergmann glia and olfactory bulb astrocytes, but not in oligodendrocytes (Schmitt et al., 2003).
GFAP is elevated in humans with AxD (Hagemann et al., 2005; Tian et al., 2006), and increased expression of wild-type human GFAP by itself results in Rosenthal fibers in transgenic mice (Messing et al., 1998), suggesting that gliosis and elevated GFAP are consistent components of Alexander disease pathology. In the point mutant mice, hypertrophied astrocytes are apparent in regions of the brain that normally show high levels of GFAP expression, but are even more prevalent in the Tg73.7 line of GFAPTG mice. Compared with the knock-in mutants, these GFAPTG animals have more Rosenthal fibers, an increased CNS stress response, and a lower body weight; in the pure FVB/N background ∼7% die at ∼1 month of age (Hagemann, unpublished observations). To intensify GFAP expression in the point mutant mice, we crossed them with this line of GFAPTG mice. Double positive animals showed a lethal phenotype with an average age of death similar to that observed in the few GFAPTG animals that die prematurely. The R236H/+;GFAPTG mice show a synergistic increase in GFAP expression, almost doubling that found in the GFAPTG alone (Fig. 9B), demonstrating a clear correlation between phenotype severity and total GFAP levels. In GFAPTG mice, endogenous mouse GFAP transcripts account for ∼25–35% of the total elevated GFAP (depending on age) (Hagemann, unpublished observation). Although the specific ratio between mutant and wild-type protein in R236H/+;GFAPTG mice has not been determined, these data support the notion of a critical threshold for GFAP levels in the pathological processes leading to astrocyte dysfunction and death.
Humans with AxD frequently suffer from seizures. Although the GFAP mutant mice do not display an overt epileptic phenotype, treatment with kainic acid revealed an increased vulnerability to the convulsant. We were also able to observe two mice of the combination R236H/+;GFAPTG genotype during their terminal events, and both appeared to experience seizure-like episodes. It will be of interest to perform EEG monitoring on the point mutant animals as well as transgenic combinations to further characterize the nature of their CNS hyperexcitability. Recent evidence has shown that Ca2+-regulated glutamate release from astrocytes can lead to paroxysmal depolarization shifts, suggesting that astrocytes may be responsible for the synchronized neuronal firing leading to seizure activity (Tian et al., 2005).
Rosenthal fibers in the GFAP mutant mice are most prominent in corpus callosum, hippocampus, and olfactory bulb, and also coincide with regions involved in neural progenitor production such as the walls of the lateral ventricle, the rostral migratory stream, and the dentate gyrus of the hippocampus (Doetsch et al., 1999; Seri et al., 2001; Gritti et al., 2002). The list of functions currently attributed to astrocytes has grown beyond the classical housekeeping role to include modulating synaptic formation and activity (Allen and Barres, 2005; Volterra and Meldolesi, 2005), regulating cerebral blood flow (Takano et al., 2006), fulfilling a role as a stem cell and providing a niche for other progenitors (Doetsch, 2003). A cytoskeletal defect in astrocytes affecting cell proliferation, migration or interactions with other cells could have widespread effects on the CNS (Mignot et al., 2004). Whether these or other critical aspects of astrocyte function are compromised, such as those involving glutamate metabolism or transport, remains to be determined.
The GFAP point mutant mice provide formal proof that the mutations found in humans with AxD are responsible for Rosenthal fiber formation, gliosis with increased GFAP expression, and white matter stress. As with other neurodegenerative diseases (Watase and Zoghbi, 2003), a combination of transgenic and mutant mice will provide valuable tools for understanding how a primary astrocyte defect can lead to such a devastating disorder.
Footnotes
This work was supported by National Institutes of Health Grants NS42803 and HD03352. We sincerely thank Heide Peickert, Charanjeet Kaur, and Denice Springman for technical support; Ruth Sullivan for providing pathology services; Benjamin August for electron microscopy; and Michael Brenner, James E. Goldman, and Roy Quinlan for critical review and suggestions on this manuscript.
References
- Alexander WS. Progressive fibrinoid degeneration of fibrillary astrocytes associated with mental retardation in a hydrocephalic infant. Brain. 1949;72:373–381. doi: 10.1093/brain/72.3.373. [DOI] [PubMed] [Google Scholar]
- Allen NJ, Barres BA. Signaling between glia and neurons: focus on synaptic plasticity. Curr Opin Neurobiol. 2005;15:542–548. doi: 10.1016/j.conb.2005.08.006. [DOI] [PubMed] [Google Scholar]
- Baba A. Role of endothelin B receptor signals in reactive astrocytes. Life Sci. 1998;62:1711–1715. doi: 10.1016/s0024-3205(98)00133-7. [DOI] [PubMed] [Google Scholar]
- Barkovich AJ, Messing A. Alexander disease: not just a leukodystrophy anymore. Neurology. 2006;66:468. doi: 10.1212/01.wnl.0000200905.43191.4d. [DOI] [PubMed] [Google Scholar]
- Bishop KM, Wahlsten D. Sex and species differences in mouse and rat forebrain commissures depend on the method of adjusting for brain size. Brain Res. 1999;815:358–366. doi: 10.1016/s0006-8993(98)01088-9. [DOI] [PubMed] [Google Scholar]
- Boor PK, de Groot K, Waisfisz Q, Kamphorst W, Oudejans CB, Powers JM, Pronk JC, Scheper GC, van der Knaap MS. MLC1: a novel protein in distal astroglial processes. J Neuropathol Exp Neurol. 2005;64:412–419. doi: 10.1093/jnen/64.5.412. [DOI] [PubMed] [Google Scholar]
- Brenner M, Johnson AB, Boespflug-Tanguy O, Rodriguez D, Goldman JE, Messing A. Mutations in GFAP, encoding glial fibrillary acidic protein, are associated with Alexander disease. Nat Genet. 2001;27:117–120. doi: 10.1038/83679. [DOI] [PubMed] [Google Scholar]
- Chanas SA, Jiang Q, McMahon M, McWalter GK, McLellan LI, Elcombe CR, Henderson CJ, Wolf CR, Moffat GJ, Itoh K, Yamamoto M, Hayes JD. Loss of the Nrf2 transcription factor causes a marked reduction in constitutive and inducible expression of the glutathione S-transferase Gsta1, Gsta2, Gstm1, Gstm2, Gstm3 and Gstm4 genes in the livers of male and female mice. Biochem J. 2002;365:405–416. doi: 10.1042/BJ20020320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor JR, Menzies SL. Relationship of iron to oligodendrocytes and myelination. Glia. 1996;17:83–93. doi: 10.1002/(SICI)1098-1136(199606)17:2<83::AID-GLIA1>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Crome L. Megalencephaly associated with hyaline pan-neuropathy. Brain. 1953;76:215–228. doi: 10.1093/brain/76.2.215. [DOI] [PubMed] [Google Scholar]
- Curtis AR, Fey C, Morris CM, Bindoff LA, Ince PG, Chinnery PF, Coulthard A, Jackson MJ, Jackson AP, McHale DP, Hay D, Barker WA, Markham AF, Bates D, Curtis A, Burn J. Mutation in the gene encoding ferritin light polypeptide causes dominant adult-onset basal ganglia disease. Nat Genet. 2001;28:350–354. doi: 10.1038/ng571. [DOI] [PubMed] [Google Scholar]
- Dietrich J, Lacagnina M, Gass D, Richfield E, Mayer-Proschel M, Noble M, Torres C, Proschel C. EIF2B5 mutations compromise GFAP+ astrocyte generation in vanishing white matter leukodystrophy. Nat Med. 2005;11:277–283. doi: 10.1038/nm1195. [DOI] [PubMed] [Google Scholar]
- Doetsch F. The glial identity of neural stem cells. Nat Neurosci. 2003;6:1127–1134. doi: 10.1038/nn1144. [DOI] [PubMed] [Google Scholar]
- Doetsch F, Caille I, Lim DA, Garcia-Verdugo JM, Alvarez-Buylla A. Subventricular zone astrocytes are neural stem cells in the adult mammalian brain. Cell. 1999;97:703–716. doi: 10.1016/s0092-8674(00)80783-7. [DOI] [PubMed] [Google Scholar]
- Francalanci P, Eymard-Pierre E, Dionisi-Vici C, Boldrini R, Piemonte F, Virgili R, Fariello G, Bosman C, Santorelli FM, Boespflug-Tanguy O, Bertini E. Fatal infantile leukodystrophy: a severe variant of CACH/VWM syndrome, allelic to chromosome 3q27. Neurology. 2001;57:265–270. doi: 10.1212/wnl.57.2.265. [DOI] [PubMed] [Google Scholar]
- Friede RL. Alexander's disease. Arch Neurol. 1964;11:414–422. doi: 10.1001/archneur.1964.00460220076010. [DOI] [PubMed] [Google Scholar]
- Grippo PJ, Nowlin PS, Cassaday RD, Sandgren EP. Cell-specific transgene expression from a widely transcribed promoter using Cre/lox in mice. Genesis. 2002;32:277–286. doi: 10.1002/gene.10080. [DOI] [PubMed] [Google Scholar]
- Gritti A, Bonfanti L, Doetsch F, Caille I, Alvarez-Buylla A, Lim DA, Galli R, Verdugo JM, Herrera DG, Vescovi AL. Multipotent neural stem cells reside into the rostral extension and olfactory bulb of adult rodents. J Neurosci. 2002;22:437–445. doi: 10.1523/JNEUROSCI.22-02-00437.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagemann TL, Gaeta SA, Smith MA, Johnson DA, Johnson JA, Messing A. Gene expression analysis in mice with elevated glial fibrillary acidic protein and Rosenthal fibers reveals a stress response followed by glial activation and neuronal dysfunction. Hum Mol Genet. 2005;14:2443–2458. doi: 10.1093/hmg/ddi248. [DOI] [PubMed] [Google Scholar]
- Hill JM, Switzer RC., III The regional distribution and cellular localization of iron in the rat brain. Neuroscience. 1984;11:595–603. doi: 10.1016/0306-4522(84)90046-0. [DOI] [PubMed] [Google Scholar]
- Hsiao VC, Tian R, Long H, Der PM, Brenner M, Quinlan RA, Goldman JE. Alexander-disease mutation of GFAP causes filament disorganization and decreased solubility of GFAP. J Cell Sci. 2005;118:2057–2065. doi: 10.1242/jcs.02339. [DOI] [PubMed] [Google Scholar]
- Ishii T, Itoh K, Takahashi S, Sato H, Yanagawa T, Katoh Y, Bannai S, Yamamoto M. Transcription factor Nrf2 coordinately regulates a group of oxidative stress-inducible genes in macrophages. J Biol Chem. 2000;275:16023–16029. doi: 10.1074/jbc.275.21.16023. [DOI] [PubMed] [Google Scholar]
- Ishikawa N, Takemura M, Koyama Y, Shigenaga Y, Okada T, Baba A. Endothelins promote the activation of astrocytes in rat neostriatum through ET(B) receptors. Eur J Neurosci. 1997;9:895–901. doi: 10.1111/j.1460-9568.1997.tb01440.x. [DOI] [PubMed] [Google Scholar]
- Iwaki T, Kume-Iwaki A, Liem RK, Goldman JE. Alpha B-crystallin is expressed in non-lenticular tissues and accumulates in Alexander's disease brain. Cell. 1989;57:71–78. doi: 10.1016/0092-8674(89)90173-6. [DOI] [PubMed] [Google Scholar]
- Johnson DA, Andrews GK, Xu W, Johnson JA. Activation of the antioxidant response element in primary cortical neuronal cultures derived from transgenic reporter mice. J Neurochem. 2002;81:1233–1241. doi: 10.1046/j.1471-4159.2002.00913.x. [DOI] [PubMed] [Google Scholar]
- LaVaute T, Smith S, Cooperman S, Iwai K, Land W, Meyron-Holtz E, Drake SK, Miller G, Abu-Asab M, Tsokos M, Switzer R, III, Grinberg A, Love P, Tresser N, Rouault TA. Targeted deletion of the gene encoding iron regulatory protein-2 causes misregulation of iron metabolism and neurodegenerative disease in mice. Nat Genet. 2001;27:209–214. doi: 10.1038/84859. [DOI] [PubMed] [Google Scholar]
- Leegwater PA, Boor PK, Yuan BQ, van der SJ, Visser A, Konst AA, Oudejans CB, Schutgens RB, Pronk JC, van der Knaap MS. Identification of novel mutations in MLC1 responsible for megalencephalic leukoencephalopathy with subcortical cysts. Hum Genet. 2002;110:279–283. doi: 10.1007/s00439-002-0682-x. [DOI] [PubMed] [Google Scholar]
- Li R, Messing A, Goldman JE, Brenner M. GFAP mutations in Alexander disease. Int J Dev Neurosci. 2002;20:259–268. doi: 10.1016/s0736-5748(02)00019-9. [DOI] [PubMed] [Google Scholar]
- Li R, Johnson AB, Salomons G, Goldman JE, Naidu S, Quinlan R, Cree B, Ruyle SZ, Banwell B, D'Hooghe M, Siebert JR, Rolf CM, Cox H, Reddy A, Gutierrez-Solana LG, Collins A, Weller RO, Messing A, van der Knaap MS, Brenner M. Glial fibrillary acidic protein mutations in infantile, juvenile, and adult forms of Alexander disease. Ann Neurol. 2005;57:310–326. doi: 10.1002/ana.20406. [DOI] [PubMed] [Google Scholar]
- McCall MA, Gregg RG, Behringer RR, Brenner M, Delaney CL, Galbreath EJ, Zhang CL, Pearce RA, Chiu SY, Messing A. Targeted deletion in astrocyte intermediate filament (Gfap) alters neuronal physiology. Proc Natl Acad Sci USA. 1996;93:6361–6366. doi: 10.1073/pnas.93.13.6361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messing A, Brenner M. Models of Alexander disease. In: Lazzarini RA, editor. Myelin biology and disorders. Amsterdam: Elsevier; 2004. pp. 1115–1124. [Google Scholar]
- Messing A, Goldman JE. Alexander disease. In: Lazzarini RA, editor. Myelin biology and disorders. Amsterdam: Elsevier; 2004. pp. 851–866. [Google Scholar]
- Messing A, Head MW, Galles K, Galbreath EJ, Goldman JE, Brenner M. Fatal encephalopathy with astrocyte inclusions in GFAP transgenic mice. Am J Pathol. 1998;152:391–398. [PMC free article] [PubMed] [Google Scholar]
- Mignot C, Boespflug-Tanguy O, Gelot A, Dautigny A, Pham-Dinh D, Rodriguez D. Alexander disease: putative mechanisms of an astrocytic encephalopathy. Cell Mol Life Sci. 2004;61:369–385. doi: 10.1007/s00018-003-3143-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min KJ, Yang MS, Kim SU, Jou I, Joe EH. Astrocytes induce hemeoxygenase-1 expression in microglia: a feasible mechanism for preventing excessive brain inflammation. J Neurosci. 2006;26:1880–1887. doi: 10.1523/JNEUROSCI.3696-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen T, Huang HC, Pickett CB. Transcriptional regulation of the antioxidant response element Activation by Nrf2 and repression by MafK. J Biol Chem. 2000;275:15466–15473. doi: 10.1074/jbc.M000361200. [DOI] [PubMed] [Google Scholar]
- Qian ZM, Shen X. Brain iron transport and neurodegeneration. Trends Mol Med. 2001;7:103–108. doi: 10.1016/s1471-4914(00)01910-9. [DOI] [PubMed] [Google Scholar]
- Racine RJ. Modification of seizure activity by electrical stimulation II. Motor seizure. Electroencephalogr Clin Neurophysiol. 1972;32:281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- Rogers DC, Fisher EM, Brown SD, Peters J, Hunter AJ, Martin JE. Behavioral and functional analysis of mouse phenotype: SHIRPA, a proposed protocol for comprehensive phenotype assessment. Mamm Genome. 1997;8:711–713. doi: 10.1007/s003359900551. [DOI] [PubMed] [Google Scholar]
- Salvi F, Aoki Y, Della NR, Vella A, Pastorelli F, Scaglione C, Matsubara Y, Mascalchi M. Adult Alexander's disease without leukoencephalopathy. Ann Neurol. 2005;58:813–814. doi: 10.1002/ana.20634. [DOI] [PubMed] [Google Scholar]
- Schmitt A, Gofferje V, Weber M, Meyer J, Mossner R, Lesch KP. The brain-specific protein MLC1 implicated in megalencephalic leukoencephalopathy with subcortical cysts is expressed in glial cells in the murine brain. Glia. 2003;44:283–295. doi: 10.1002/glia.10304. [DOI] [PubMed] [Google Scholar]
- Schmued LC. A rapid, sensitive histochemical stain for myelin in frozen brain sections. J Histochem Cytochem. 1990;38:717–720. doi: 10.1177/38.5.1692056. [DOI] [PubMed] [Google Scholar]
- Seri B, Garcia-Verdugo JM, McEwen BS, Alvarez-Buylla A. Astrocytes give rise to new neurons in the adult mammalian hippocampus. J Neurosci. 2001;21:7153–7160. doi: 10.1523/JNEUROSCI.21-18-07153.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano T, Tian GF, Peng W, Lou N, Libionka W, Han X, Nedergaard M. Astrocyte-mediated control of cerebral blood flow. Nat Neurosci. 2006;9:260–267. doi: 10.1038/nn1623. [DOI] [PubMed] [Google Scholar]
- Tian GF, Azmi H, Takano T, Xu Q, Peng W, Lin J, Oberheim N, Lou N, Wang X, Zielke HR, Kang J, Nedergaard M. An astrocytic basis of epilepsy. Nat Med. 2005;11:973–981. doi: 10.1038/nm1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian R, Gregor M, Wiche G, Goldman JE. Plectin regulates the organization of glial fibrillary acidic protein in Alexander disease. Am J Pathol. 2006;168:888–897. doi: 10.2353/ajpath.2006.051028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuji Y, Ayaki H, Whitman SP, Morrow CS, Torti SV, Torti FM. Coordinate transcriptional and translational regulation of ferritin in response to oxidative stress. Mol Cell Biol. 2000;20:5818–5827. doi: 10.1128/mcb.20.16.5818-5827.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Knaap MS, Ramesh V, Schiffmann R, Blaser S, Kyllerman M, Gholkar A, Ellison DW, van der Voorn JP, van Dooren SJ, Jakobs C, Barkhof F, Salomons GS. Alexander disease: ventricular garlands and abnormalities of the medulla and spinal cord. Neurology. 2006;66:494–498. doi: 10.1212/01.wnl.0000198770.80743.37. [DOI] [PubMed] [Google Scholar]
- Venugopal R, Jaiswal AK. Nrf1 and Nrf2 positively and c-Fos and Fra1 negatively regulate the human antioxidant response element-mediated expression of NAD(P)H:quinone oxidoreductase1 gene. Proc Natl Acad Sci USA. 1996;93:14960–14965. doi: 10.1073/pnas.93.25.14960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volterra A, Meldolesi J. Astrocytes, from brain glue to communication elements: the revolution continues. Nat Rev Neurosci. 2005;6:626–640. doi: 10.1038/nrn1722. [DOI] [PubMed] [Google Scholar]
- Wasserman WW, Fahl WE. Functional antioxidant responsive elements. Proc Natl Acad Sci USA. 1997;94:5361–5366. doi: 10.1073/pnas.94.10.5361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watase K, Zoghbi HY. Modelling brain diseases in mice: the challenges of design and analysis. Nat Rev Genet. 2003;4:296–307. doi: 10.1038/nrg1045. [DOI] [PubMed] [Google Scholar]
- Weiner MP, Costa GL, Schoettlin W, Cline J, Mathur E, Bauer JC. Site-directed mutagenesis of double-stranded DNA by the polymerase chain reaction. Gene. 1994;151:119–123. doi: 10.1016/0378-1119(94)90641-6. [DOI] [PubMed] [Google Scholar]
- Wild AC, Moinova HR, Mulcahy RT. Regulation of gamma-glutamylcysteine synthetase subunit gene expression by the transcription factor Nrf2. J Biol Chem. 1999;274:33627–33636. doi: 10.1074/jbc.274.47.33627. [DOI] [PubMed] [Google Scholar]
- Wilhelmsson U, Li L, Pekna M, Berthold C-H, Blom S, Eliasson C, Renner O, Bushong E, Ellisman M, Morgan TE, Pekny M. Absence of glial fibrillary acidic protein and vimentin prevents hypertrophy of astrocytic processes and improves post-traumatic regeneration. J Neurosci. 2004;24:5016–5021. doi: 10.1523/JNEUROSCI.0820-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]