

The intrinsic properties of a neuron determine the translation of synaptic input to axonal output. It is this input–output relationship that is the heart of all nervous system activity. As such, the overall regulation of the intrinsic excitability of a neuron directly determines the output of that neuron at a given point in time, giving the cell a unique “functional identity.” To maintain this distinct functional output, neurons must adapt to changing patterns of synaptic excitation. These adaptations are essential to prevent neurons from either falling silent as synaptic excitation falls or becoming saturated as excitation increases. In the absence of stabilizing mechanisms, activity-dependent plasticity could drive neural activity to saturation or quiescence. Furthermore, as cells adapt to changing patterns of synaptic input, presumably the overall balance of intrinsic conductances of the cell must be maintained so that reliable output is achieved (Daoudal and Debanne, 2003; Turrigiano and Nelson, 2004; Frick and Johnston, 2005). Although these regulatory phenomena have been well documented, the molecular and physiological mechanisms involved are poorly understood.
This mini-symposium provides an opportunity to review recent work on the relationship between neuronal excitability and how this influences the unique output and function of a neuron. We take the perspective of an increasing scope of analysis, starting from the expression of single genes and ending with broader intercellular effects of modulators on cellular excitability. We first focus on how transcriptional events link the genome to neuronal excitability by examining quantitative levels of gene expression as they relate to functional identity of individual neurons. This includes focal sampling of ion channel expression by measuring the levels of mRNA for small numbers of channel genes involved in intrinsic excitability. This is then expanded to consider genome-wide screens of differences in gene expression correlated to neuronal cell identity and how this expression profiling can be applied to understand typical as well as pathological states of neuronal excitability. Finally, we explore concepts beyond gene expression related to cellular excitability and its role in determining cell identity, including intracellular posttranslational influences and intercellular neuromodulatory effects on intrinsic excitability.
Ion channel expression, excitability, and functional neuronal identity
One possible mechanism for stabilizing neuronal excitability is an activity-dependent regulation of ion channel expression that counters changes in synaptic input. For example, in Drosophila motor neurons, voltage-gated sodium current (INa) changes to compensate for altered levels of synaptic excitation (Mee et al., 2004). In the absence of synaptic excitation, INa is significantly elevated, whereas increased excitation results in a reduction in INa (Fig. 1A). Measurement of levels of paralytic (para) mRNA, which encodes for the sole voltage-gated Na+ channel in these neurons, indicates that regulation of INa is achieved through control of para mRNA level (Fig. 1B). The changes in mRNA are likely attributable, at least in part, to activity-dependent translational repression by the known translational repressor pumilio (pum). pum is both necessary and sufficient for the activity-dependent changes observed in para mRNA in differing synaptic activity mutants (Fig. 1C). Sequence analysis of the para transcript shows it to contain specific motifs, termed nanos response elements, in its multiple 5′ untranslated regions. These motifs have been shown previously necessary for Pum protein binding to hunchback (hb) RNA. Binding of Pum to hb is the initial step that results in translational repression of this transcript (Baines, 2005). The presence of these motifs in para indicates a possible direct interaction between Pum and para mRNA, resulting in suppression of translation, and possibly degradation, of the para transcript. Thus, this work provides detailed insight into the regulation of the expression of a single channel protein, para, and its ultimate influence on cellular excitability.
Figure 1.
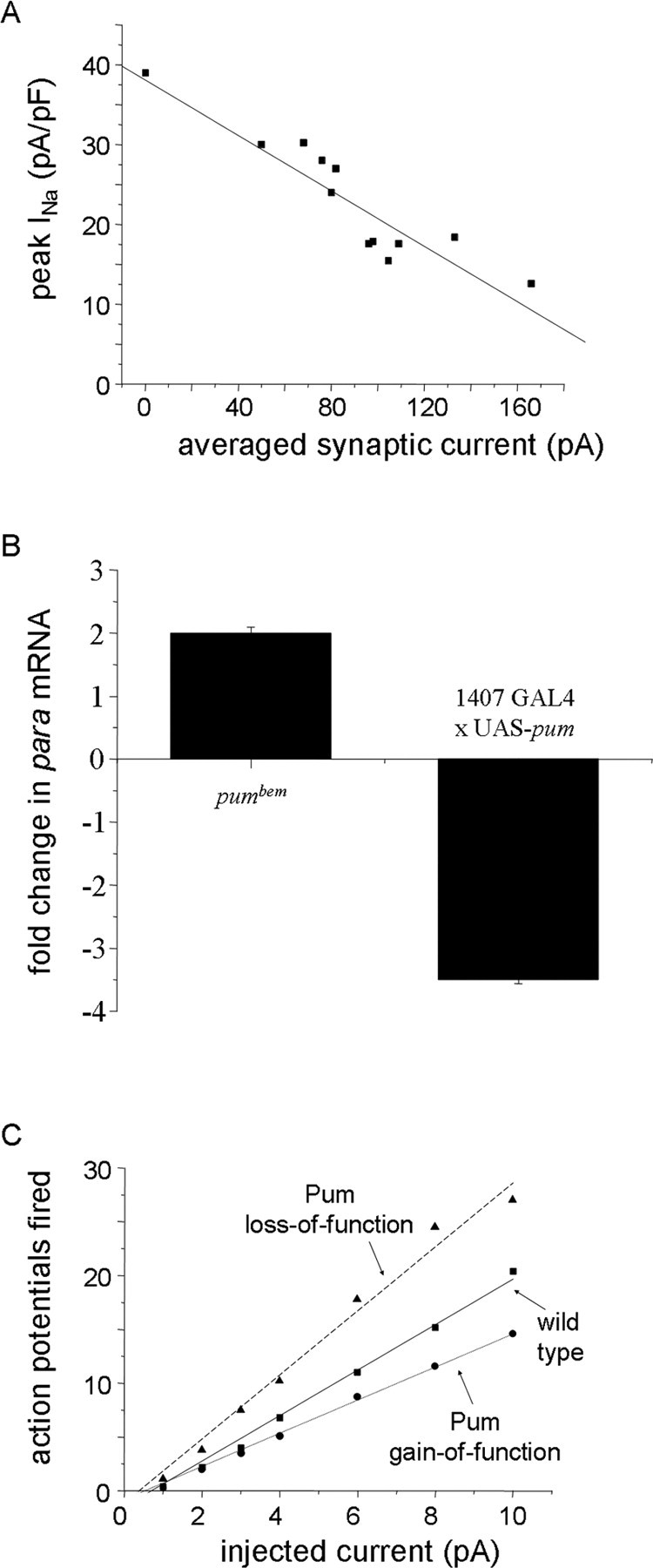
Exposure to synaptic excitation regulates INa and membrane excitability in Drosophila motoneurons. A, The amplitude of the voltage-gated INa recorded in Drosophila motoneurons is inversely related to the synaptic excitation that these neurons receive. B, The abundance of para mRNA in isolated CNS is significantly greater in the loss-of-function pumbem allele. By comparison, overexpression of UAS (upstream activating sequence)–pum in all of the neurons of the CNS (1407 GAL4) is sufficient to significantly reduce para mRNA. C, Membrane excitability, measured as action potential firing, shows a significant increase in Pumiliobem (loss-of-function) and a significant decrease after overexpression of UAS-pumilio (gain-of-function), respectively. The action potential firing rate was determined by injection of constant current (1–10 pA/500 ms) from a holding potential of −60 mV. A is reprinted from Baines and Pym (2006). B and C are reprinted from Mee et al. (2004).
Although altering intrinsic excitability may counter changes in synaptic input, changes in the contribution of only one ionic conductance may throw off the delicate balance of numerous membrane conductances responsible for the firing properties of a cell. Recent work indicates that such regulation of ion channel expression (and ultimately cellular excitability) does not occur independently for single ion channels. Although expression levels for a given ion channel can vary twofold to fourfold in a particular class of neuron (Schulz et al., 2006), some fixed relationships may exist among ion channels that may play a role in stabilizing functional neuronal output (MacLean et al., 2003; Schulz et al., 2006). By measuring the levels of mRNA for six different ion channels from the same single identified motor neurons of the crustacean stomatogastric ganglion, recent studies demonstrate that multiple ion channels in a single cell show correlated levels of expression (D. J. Schulz, J.-M. Goaillard, and E. Marder, unpublished observations). For example, in pyloric dilator neurons, six different significant pairwise correlations among these channel types were detected, which revealed an apparent four-way coregulation among shal (IA), para (INa), IH (IH), and BKKCa (IK[ Ca] ) mRNA. Furthermore, in six different individually identifiable cell types examined, no two cell types showed the same set of correlated channel expression levels. These results suggest that, not only do cells compensate for altered excitability, but each different cell type has a unique mechanism at least in part dependent on relative abundance of ion channel expression by which its distinct output is generated and maintained.
Identifying and understanding the properties of unique classes of neurons using genome-wide expression profiles
The fact that no two uniquely identified cell types show the same pattern of expression among six different ion channels points out that functional differences between a neuron and its neighbors result at least in part from differences in gene expression. An ambitious project has been launched to use these differences in gene expression to bring a systematic approach to the identification and classification of cell types in the mammalian brain (Nelson et al., 2006; Sugino et al., 2006). The strategy used brings together two powerful and relatively new technologies: transgenic mice and microarrays (Fig. 2). Genetically encoded markers such as green fluorescent protein can now be selectively expressed in highly restricted populations of neurons with remarkable precision. This allows them to be targeted for electrophysiological recordings to assess their intrinsic excitability properties. The fluorescent marker also allows the neurons to be purified so that mRNA can be extracted, amplified, and probed with microarrays. The result is a comprehensive genome-wide expression profile that can be used as an index of similarity between neuronal cell types and thus, one hopes, the basis for a systematic “neuronal taxonomy” (Sugino et al., 2006). These expression profiles also provide the identity of large numbers of cell type-specific genes that likely contribute to the distinct functional identities of the different neuronal cell types. Among these, of course, are genes coding for ion channels. These provide a starting point for investigations into the role of individual ion channels in determining electrophysiological phenotypes.
Figure 2.
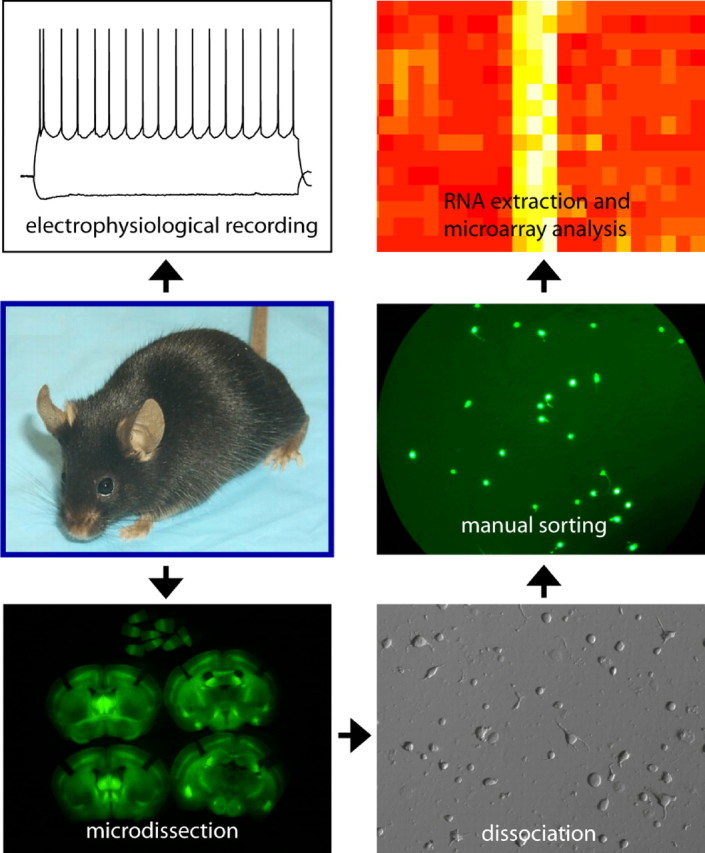
Mice in which subpopulations of neurons are genetically labeled are used for electrophysiological recordings and cell type-specific microarray analysis to obtain a correlated profile of firing properties and gene expression.
These concepts of expression profiling are already being used to understand normal neuronal function and susceptibility to pathology in neurons with important implications for human health, the midbrain dopaminergic (DA) neurons. In the midbrain, DA neurons are arranged in two overlapping nuclei, the substantia nigra (SN) and the ventral tegmental area (VTA). Selective degeneration or functional dysregulation of DA neurons are causally involved in important human disorders, such as Parkinson disease (PD), drug addiction, and schizophrenia (Seamans and Yang, 2004; Moore et al., 2005; Hyman et al., 2006). By combining single-cell electrophysiology and gene expression profiling (Fig. 3A), recent work highlighted the emerging role of ion channels in the pathophysiology of the DA midbrain system (Liss et al., 2005). Already under physiological control conditions, DA neurons in SN and VTA display a variety of distinct electrophysiological properties that are functionally correlated with the differential expression of related ion channel subunits (for review, see Korotkova et al., 2004; Liss and Roeper 2004). Ion channel gating orchestrates the electrical activity of DA neurons and in turn their dopamine release (Cragg et al., 1997). Furthermore, it has been demonstrated that neuronal activity is crucial for the survival of SN DA neurons in chronic neurodegenerative disease: parkinsonism-inducing toxins (the mitochondrial complex I blockers rotenone and 1-methyl-4-phenylpyridinium) selectively activate ATP-sensitive potassium (K-ATP) channels (Nicols, 2006) in highly vulnerable SN DA neurons but not in more resistant VTA DA neurons. Consequently, normal spontaneous activity of SN DA neurons is inhibited (Liss et al., 2005). Higher mRNA expression levels of K-ATP channels and lower mRNA levels of the mitochondrial uncoupling protein UCP-2 (Andrews et al., 2005) in SN DA neurons are correlated with the K-ATP channel-dependent selective silencing of these neurons (Fig. 3B). Genetic inactivation of the pore-forming K-ATP channel subunit Kir6.2 completely rescues the highly vulnerable SN DA population in the chronic in vivo 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) mouse model of PD (Fig. 3C) (Liss et al., 2005). Ongoing work aims to identify mechanisms that control activity and differential vulnerability of DA midbrain neurons, upstream and downstream from K-ATP channels, by analyzing gene expression of individual electrophysiologically characterized DA neurons from SN and VTA via microarray strategies. This single-cell approach complements and improves the specificity of recent elegant microarray studies using pools of microdissected SN and VTA DA neurons for global transcriptome comparison (Grimm et al., 2004; Chung et al., 2005; Greene et al., 2005).
Figure 3.
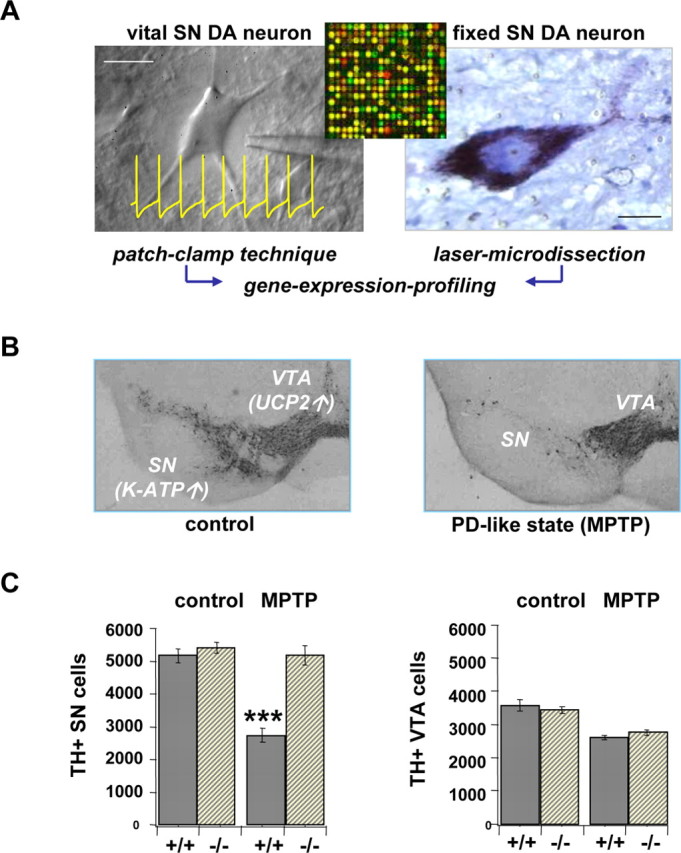
K-ATP channel activity triggers selective degeneration of SN DA neurons. A, A schematic overview of analyzing electrophysiological function and gene expression of individual dopaminergic neurons (mouse/human) combining brain slice patch clamping (inset, typical spontaneous activity of DA neurons) or laser microdissection with gene expression profiling (reverse transcription-PCR/microarrays). Scale bars, 15 μm. B, Dopaminergic neurons in SN and VTA [ marked by tyrosine hydroxylase (TH) immunostain] display differential vulnerability in the chronic mouse MPTP-model of PD. Differential mRNA levels of K-ATP channel subunits and UCP2 are indicated. C, Results of quantitative stereology indicated complete rescue of highly vulnerable SN DA neurons in the chronic MPTP PD model in a K-ATP channel knock-out mouse (Kir6.2 −/−; left). In contrast, MPTP-induced moderate loss of VTA DA neurons was independent from the presence (+ /+) or absence (−/−) of K-ATP channels (right).
Posttranslational events and the regulation of neuronal identity by cellular excitability
Although understanding the expression of genes involved in cellular excitability and functional identity continues to shed a great deal of light on fundamental questions of neuronal function, expression-level analyses reveal only a small portion of the mechanisms of the functional output of a cell. Posttranslational modification, particularly phosphorylation, can rapidly alter the properties of a given ion channel and therefore drastically alter the electrophysiological characteristics of the cell. A prime example of this type of regulation is a voltage-gated K+ channel Kv2.1, which mediates somatodendritic IK currents in central neurons (Murakoshi and Trimmer, 1999). Kv2.1 is expressed in large (∼2 μm diameter) clusters on the somata and proximal dendrites of principal pyramidal neurons (Lim et al., 2000) and also is highly and constitutively phosphorylated (Fig. 4A,B) (Park et al., 2006). Because of its high threshold for voltage-dependent activation (Fig. 4C), Kv2.1 does not appear to play a prominent role in repolarization of single action potentials (Du et al., 2000; Malin and Nerbonne, 2002). Furthermore, this unique characteristic of Kv2.1 undergoes dramatic changes under various pathological conditions. In seizure models, it has been demonstrated that resultant Ca2+ influx leads to dramatic dephosphorylation of Kv2.1, dispersion of Kv2.1 clusters, and a large leftward shift in the voltage activation curve of Kv2.1 channel through calcineurin-dependent mechanisms (Fig. 4C) (Misonou et al., 2004). Kv2.1 then is able to contribute to repolarizing membrane potentials, leading to suppression of overall excitability of neurons. Moreover, brief ischemia causes similar modulation of Kv2.1 (Fig. 4C) induced not by Ca2+ influx but by intracellular Ca2+ release from mitochondria (Misonou et al., 2005). These findings clearly show that, in response to hyperexcitable conditions, neurons use specific mechanisms to modulate Kv2.1 molecules by phosphorylation events, such that Kv2.1 channels become more active and homeostatically suppress neuronal excitability. This may represent a key but previously unrecognized contributor to a protection against hyperexcitability induced by epileptic seizures and brain ischemia.
Figure 4.

A, Dephosphorylation of Kv2.1 induced by seizures and ischemia. Rats were subjected to either kainate-induced continuous seizures (Kainate) or CO2-induced brain ischemia (Ischemia). Total brain membrane was prepared, and a fraction was treated in vitro with alkaline phosphatase (AP) to completely dephosphorylate Kv2.1. The arrow indicates the highly phosphorylated Kv2.1, and arrowhead indicates fully dephosphorylated Kv2.1. Numbers on the left denote the molecular weight standards. B, Dispersion of Kv2.1 clusters in brain by ischemia. Brain sections from control and ischemic rats were stained with a specific anti-Kv2.1 antibody. Images show pyramidal neurons in the cerebral cortex. It should be noted that kainate-induced seizures also resulted in dispersion of Kv2.1 clusters. Scale bar, 10 μm. C, Changes in the biophysical properties of IK/Kv2.1 by glutamate stimulation and ischemia in cultured neurons. IK currents were recorded in a cultured hippocampal neuron under whole-cell voltage clamp. The half-maximal activation membrane potential (G1/2) and the slope of the activation curve are shown (means ± SEM). The values are from Misonou et al. (2004, 2005).
Beyond intrinsic properties and cellular excitability: neuropeptidomics and the influence of modulatory substances on neuronal identity
An understanding of the intrinsic properties of an individual cell is instrumental to uncovering the mechanisms by which it generates a unique output, but in vivo the excitability of a cell is also shaped by its extracellular environment. In particular, the excitability of cells is often subject to extensive neuromodulation. Therefore, a fundamental understanding of how an individual cell produces a unique functional output requires not only an understanding its intrinsic channel density and subsequent intracellular phosphorylation events but also the identification of the full complement of neuromodulators acting on it. Neuropeptides are modulatory substances that can act via divergent and convergent pathways to provide a balance between the stability and flexibility of an individual neuron and the network(s) to which it belongs (Brezina and Weiss, 1997; Nusbaum et al., 2001; Swensen and Marder, 2001). A key to understanding this balance is uncovering the mechanisms of how a rich repertoire of neuropeptides can fine tune a well defined neural circuitry to produce multiple outputs at the cellular and network levels. Networks found in the stomatogastric nervous system of crustaceans are extensively modulated by neuromodulators released from projection neurons as well as peptide hormones secreted from neurohemal organs. Using a highly sensitive mass spectrometry-based peptide profiling and de novo sequencing strategy (Fig. 5A), a large number of novel peptides have been discovered, revealing that even a relatively simple neural network contains an unexpectedly rich diversity of neuropeptides (Fu and Li, 2005; Fu et al., 2005a,b; DeKeyser and Li, 2006). Using a combined mass spectrometric and electrophysiological approach, this work provides the first evidence that different members of the well known FMRFamide (Phe-Met-Arg-Phe-amide)-like peptide family exhibit differential sensitivity to extracellular peptidase activity (Fig. 5B), which might provide another mechanism for the modulation of excitability of specific neurons in a neural network (Cruz-Bermudez et al., 2006). Such combined peptidomic and physiological studies will help to elucidate the functional roles of neuropeptides in neuron and neural network plasticity and advance our understanding of the full complement of mechanisms contributing to the unique identity and output of any given neuron in the nervous system.
Figure 5.
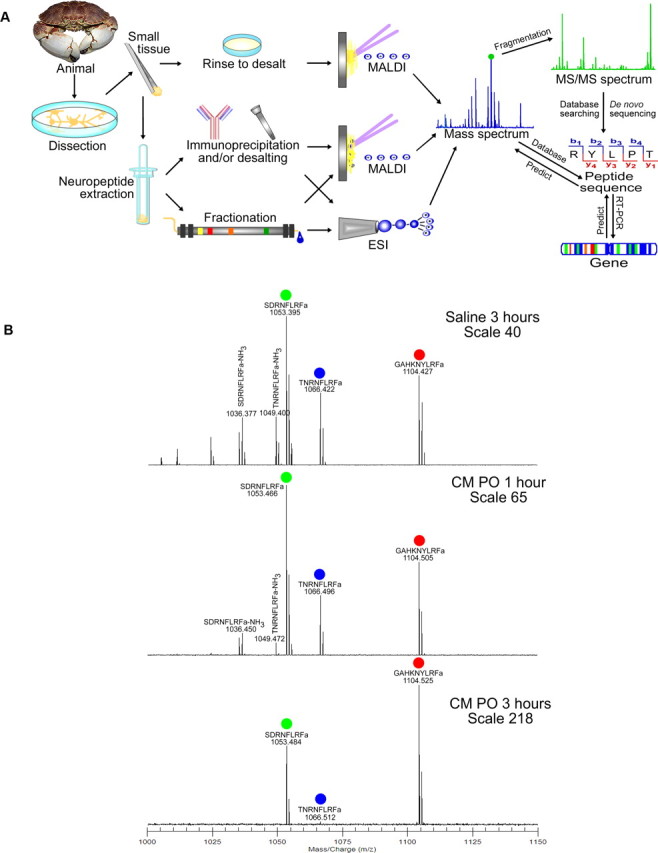
Development of a mass spectrometry-based peptidomic approach for understanding functional consequence of neuropeptide multiplicity. A, An overview of mass spectrometry-based strategy for neuropeptide discovery and characterization. B, Differential degradation of three FLRFamide (Phe-Leu-Arg-Phe-NH2) peptides by peptidase located in the pericardial organ monitored by matrix-assisted laser desorption/ionization Fourier transform mass spectrometry. A is reprinted from DeKeyser and Li (2006). B is adapted from Cruz-Bermudez et al. (2006). MS, Mass spectrometry; MALDI, matrix-assisted laser desorption/ionization; ESI, electrospray ionization; RT, reverse transcription; CM PO, Cancer magister pericardial organ.
Conclusion
The functional specialization of the many distinct classes of neurons that make up the nervous system is determined in large part by the specific sets and relative abundance of ion channels that neurons express. It is these ion channels that dictate the membrane characteristics through which each type of neuron translates its synaptic input into axonal output and ultimately determine the role an individual cell plays in a neural network. The goal of this mini-symposium has been to highlight research that is illuminating the relationships between neuronal excitability and neuronal identity, with the hopes of understanding not only what confers on a cell its unique identity but the underlying cellular mechanisms that allow a cell to maintain this identity in the face of changing synaptic input.
Footnotes
B.L. was supported by grants from the Royal Society, National Institutes of Health, Bundesministerium für Bildung und Forschung (National Genome Research Network), and the Hertie Foundation. L.L. was supported in part by the School of Pharmacy and Wisconsin Alumni Research Foundation at the University of Wisconsin-Madison, a National Science Foundation Career Award, the National Institutes of Health, and an Alfred P. Sloan Research Fellowship.
References
- Andrews ZB, Diano S, Horvath TL. Mitochondrial uncoupling proteins in the CNS: in support of function and survival. Nat Rev Neurosci. 2005;6:829–840. doi: 10.1038/nrn1767. [DOI] [PubMed] [Google Scholar]
- Baines RA. Neuronal homeostasis through translational control. Mol Neurobiol. 2005;32:113–121. doi: 10.1385/MN:32:2:113. [DOI] [PubMed] [Google Scholar]
- Baines RA, Pym EC. Determinants of electrical properties in developing neurons. Semin Cell Dev Biol. 2006;17:12–19. doi: 10.1016/j.semcdb.2005.11.003. [DOI] [PubMed] [Google Scholar]
- Brezina V, Weiss KR. Functional consequences of divergence and convergence in physiological signaling pathways. Mol Psychiatry. 1997;2:9–11. doi: 10.1038/sj.mp.4000191. [DOI] [PubMed] [Google Scholar]
- Chung CY, Seo H, Sonntag KC, Brooks A, Lin L, Isacson O. Cell type-specific gene expression of midbrain dopaminergic neurons reveals molecules involved in their vulnerability and protection. Hum Mol Genet. 2005;14:1709–1725. doi: 10.1093/hmg/ddi178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cragg S, Rice ME, Greenfield SA. Heterogeneity of electrically evoked dopamine release and reuptake in substantia nigra, ventral tegmental area, and striatum. J Neurophysiol. 1997;77:863–873. doi: 10.1152/jn.1997.77.2.863. [DOI] [PubMed] [Google Scholar]
- Cruz-Bermudez ND, Fu Q, Kutz-Naber KK, Christie AE, Li L, Marder E. Mass spectrometric characterization and physiological actions of GAHKNYLRFamide, a novel FMRFamide-like peptide from crabs of the genus Cancer. J Neurochem. 2006;97:784–799. doi: 10.1111/j.1471-4159.2006.03747.x. [DOI] [PubMed] [Google Scholar]
- Daoudal G, Debanne D. Long-term plasticity of intrinsic excitability: learning rules and mechanisms. Learn Mem. 2003;10:456–465. doi: 10.1101/lm.64103. [DOI] [PubMed] [Google Scholar]
- DeKeyser SS, Li L. Mass spectrometric charting of neuropeptides in arthropod neurons. Anal Bioanal Chem. 2006 doi: 10.1007/s00216-006-0596-x. in press. [DOI] [PubMed] [Google Scholar]
- Du J, Haak LL, Phillips-Tansey E, Russell JT, McBain CJ. Frequency-dependent regulation of rat hippocampal somato-dendritic excitability by the K+ channel subunit Kv2.1. J Physiol (Lond) 2000;522:19–31. doi: 10.1111/j.1469-7793.2000.t01-2-00019.xm. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frick A, Johnston D. Plasticity of dendritic excitability. J Neurobiol. 2005;64:100–115. doi: 10.1002/neu.20148. [DOI] [PubMed] [Google Scholar]
- Fu Q, Li L. De novo sequencing of neuropeptides using reductive isotopic methylation and investigation of ESI QTOF MS/MS fragmentation pattern of neuropeptides with N-terminal dimethylation. Anal Chem. 2005;77:7783–7795. doi: 10.1021/ac051324e. [DOI] [PubMed] [Google Scholar]
- Fu Q, Goy MF, Li L. Identification of neuropeptides from the decapod crustacean sinus glands using nanoscale liquid chromatography tandem mass spectrometry. Biochem Biophys Res Commun. 2005a;337:765–778. doi: 10.1016/j.bbrc.2005.09.111. [DOI] [PubMed] [Google Scholar]
- Fu Q, Kutz KK, Schmidt JJ, Hsu YW, Messinger DI, Cain SD, de la Iglesia HO, Christie AE, Li L. Hormone complement of the Cancer productus sinus gland and pericardial organ: An anatomical and mass spectrometric investigation. J Comp Neurol. 2005b;493:607–626. doi: 10.1002/cne.20773. [DOI] [PubMed] [Google Scholar]
- Greene JG, Dingledine R, Greenamyre JT. Gene expression profiling of rat midbrain dopamine neurons: implications for selective vulnerability in parkinsonism. Neurobiol Dis. 2005;18:19–31. doi: 10.1016/j.nbd.2004.10.003. [DOI] [PubMed] [Google Scholar]
- Grimm J, Mueller A, Hefti F, Rosenthal A. Molecular basis for catecholaminergic neuron diversity. Proc Natl Acad Sci USA. 2004;101:13891–13896. doi: 10.1073/pnas.0405340101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman SE, Malenka RC, Nestler EJ. Neural mechanisms of addiction: the role of reward-related learning and memory. Annu Rev Neurosci. 2006;29:565–598. doi: 10.1146/annurev.neuro.29.051605.113009. [DOI] [PubMed] [Google Scholar]
- Korotkova TM, Ponomarenko AA, Brown RE, Haas HL. Functional diversity of ventral midbrain dopamine and GABAergic neurons. Mol Neurobiol. 2004;29:243–259. doi: 10.1385/MN:29:3:243. [DOI] [PubMed] [Google Scholar]
- Lim ST, Antonucci DE, Scannevin RH, Trimmer JS. A novel targeting signal for proximal clustering of the Kv2.1 K+ channel in hippocampal neurons. Neuron. 2000;25:385–397. doi: 10.1016/s0896-6273(00)80902-2. [DOI] [PubMed] [Google Scholar]
- Liss B, Roeper J. Correlating function and gene expression of individual basal ganglia neurons. Trends Neurosci. 2004;27:475–481. doi: 10.1016/j.tins.2004.06.007. [DOI] [PubMed] [Google Scholar]
- Liss B, Haeckel O, Wildmann J, Miki T, Seino S, Roeper J. K-ATP channels promote the differential degeneration of dopaminergic midbrain neurons. Nat Neurosci. 2005;8:1742–1751. doi: 10.1038/nn1570. [DOI] [PubMed] [Google Scholar]
- MacLean JN, Zhang Y, Johnson BR, Harris-Warrick RM. Activity-independent homeostasis in rhythmically active neurons. Neuron. 2003;37:109–120. doi: 10.1016/s0896-6273(02)01104-2. [DOI] [PubMed] [Google Scholar]
- Malin SA, Nerbonne JM. Delayed rectifier K+ currents, IK, are encoded by Kv2 α-subunits and regulate tonic firing in mammalian sympathetic neurons. J Neurosci. 2002;22:10094–10105. doi: 10.1523/JNEUROSCI.22-23-10094.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mee CJ, Pym EC, Moffat KG, Baines RA. Regulation of neuronal excitability through pumilio-dependent control of a sodium channel gene. J Neurosci. 2004;24:8695–8703. doi: 10.1523/JNEUROSCI.2282-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misonou H, Mohapatra DP, Park EW, Leung V, Zhen D, Misonou K, Anderson AE, Trimmer JS. Regulation of ion channel localization and phosphorylation by neuronal activity. Nat Neurosci. 2004;7:711–718. doi: 10.1038/nn1260. [DOI] [PubMed] [Google Scholar]
- Misonou H, Mohapatra DP, Menegola M, Trimmer JS. Calcium- and metabolic state-dependent modulation of the voltage-dependent Kv2.1 channel regulates neuronal excitability in response to ischemia. J Neurosci. 2005;25:11184–11193. doi: 10.1523/JNEUROSCI.3370-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore DJ, West AB, Dawson VL, Dawson TM. Molecular pathophysiology of Parkinson's disease. Annu Rev Neurosci. 2005;28:57–87. doi: 10.1146/annurev.neuro.28.061604.135718. [DOI] [PubMed] [Google Scholar]
- Murakoshi H, Trimmer JS. Identification of the Kv2.1 K+ channel as a major component of the delayed rectifier K+ current in rat hippocampal neurons. J Neurosci. 1999;19:1728–1735. doi: 10.1523/JNEUROSCI.19-05-01728.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson SB, Sugino K, Hempel CM. The problem of neuronal cell types: a physiological genomics approach. Trends Neurosci. 2006;29:339–345. doi: 10.1016/j.tins.2006.05.004. [DOI] [PubMed] [Google Scholar]
- Nicols CG. KATP channels as molecular sensors of cellular metabolism. Nature. 2006;440:470–476. doi: 10.1038/nature04711. [DOI] [PubMed] [Google Scholar]
- Nusbaum MP, Blitz DM, Swensen AM, Wood D, Marder E. The roles of co-transmission in neural network modulation. Trends Neurosci. 2001;24:146–154. doi: 10.1016/s0166-2236(00)01723-9. [DOI] [PubMed] [Google Scholar]
- Park K-S, Mohapatra DP, Misonou H, Trimmer JS. Graded regulation of the Kv2.1 potassium channel by variable phosphorylation. Science. 2006;313:976–979. doi: 10.1126/science.1124254. [DOI] [PubMed] [Google Scholar]
- Schulz DJ, Goaillard J-M, Marder E. Variable channel expression in identified single and electrically coupled neurons in different animals. Nat Neurosci. 2006;9:356–362. doi: 10.1038/nn1639. [DOI] [PubMed] [Google Scholar]
- Seamans JK, Yang CR. The principal features and mechanisms of dopamine modulation in the prefrontal cortex. Prog Neurobiol. 2004;74:1–58. doi: 10.1016/j.pneurobio.2004.05.006. [DOI] [PubMed] [Google Scholar]
- Sugino K, Hempel CM, Miller MN, Hattox AM, Shapiro P, Wu C, Huang ZJ, Nelson SB. Molecular taxonomy of major neuronal classes in the adult mouse forebrain. Nat Neurosci. 2006;9:99–107. doi: 10.1038/nn1618. [DOI] [PubMed] [Google Scholar]
- Swensen AM, Marder E. Modulators with convergent cellular actions elicit distinct circuit outputs. J Neurosci. 2001;21:4050–4058. doi: 10.1523/JNEUROSCI.21-11-04050.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano GG, Nelson SB. Homeostatic plasticity in the developing nervous system. Nat Rev Neurosci. 2004;5:97–107. doi: 10.1038/nrn1327. [DOI] [PubMed] [Google Scholar]