

Abstract
Dopamine depletion is involved in the pathophysiology of Parkinson's disease, whereas hyperdopaminergia may play a fundamental role in generating endophenotypes associated with schizophrenia. Sleep disturbances are known to occur in both schizophrenia and Parkinson's disease, suggesting that dopamine plays a role in regulating the sleep–wake cycle. Here, we show that novelty-exposed hyperdopaminergic mice enter a novel awake state characterized by spectral patterns of hippocampal local field potentials that resemble electrophysiological activity observed during rapid-eye-movement (REM) sleep. Treatment with haloperidol, a D2 dopamine receptor antagonist, reduces this abnormal intrusion of REM-like activity during wakefulness. Conversely, mice acutely depleted of dopamine enter a different novel awake state characterized by spectral patterns of hippocampal local field potentials that resemble electrophysiological activity observed during slow-wave sleep (SWS). This dopamine-depleted state is marked by an apparent suppression of SWS and a complete suppression of REM sleep. Treatment with D2 (but not D1) dopamine receptor agonists recovers REM sleep in these mice. Altogether, these results indicate that dopamine regulates the generation of sleep–wake states. We propose that psychosis and the sleep disturbances experienced by Parkinsonian patients result from dopamine-mediated disturbances of REM sleep.
Keywords: dopamine, sleep, REM, psychosis, schizophrenia, Parkinson's
Introduction
Dopamine is critically involved in regulating processes responsible for the generation of complex movement and emotions (Carlsson, 1987). Altered central dopaminergic synaptic transmission has been implicated in several neurological and psychiatric disorders, such as Parkinson's disease, schizophrenia, and attention deficit hyperactivity disorder (Carlsson, 1987; Mazei-Robison et al., 2005; Greenwood et al., 2006). Individuals with these diseases demonstrate dramatic sleep disturbances, such as excessive daytime sleepiness (Adler, 2005), rapid-eye-movement (REM) sleep behavior disorder (Gagnon et al., 2002; Abbott, 2005), decreased REM sleep latency, and disturbed sleep architecture (Maggini et al., 1986; O'Brien et al., 2003). Overall, these observations suggest that dopamine may play a role in regulating the sleep–wake cycle as well.
Dramatic changes in neurotransmitter levels are known to occur as the brain progresses through the sleep–wake cycle. These changes are coordinated by complex interactions that systematically modulate the firing rate of cholinergic (Hobson et al., 1993), orexinergic (Lin et al., 1999), noradrenergic (Aston-Jones and Bloom, 1981), histaminergic (John et al., 2004), and serotonergic (Espana and Scammell, 2004) neurons. These neurons, in turn, send efferent projections to cortical and subcortical structures, generating patterns of brain and muscle activity characteristic of the three major brain states: waking, slow-wave sleep (SWS), and REM sleep. During waking, the cortex displays low-amplitude fast oscillations in the gamma (33–55 Hz) frequency range, and muscle activity is highest (Steriade et al., 1993). As the brain transitions to SWS, fast cortical oscillations are replaced by high-amplitude, low-frequency oscillations, and muscle activity decreases (Steriade et al., 1993; Hobson and Pace-Schott, 2002). During REM sleep, the cortex and subcortical structures display fast gamma oscillations similar to those observed during waking, and the hippocampus displays characteristic local field potential (LFP) oscillations in the 4–9 Hz range called theta rhythm (Vanderwolf, 1969; Timo-Iaria et al., 1970; Cantero et al., 2003). Additionally, muscle activity is primarily inhibited during REM sleep, with the exception of eye movements in humans and whisker movements in rodents (Aserinsky and Kleitman, 1953; Dement and Kleitman, 1957a,b; Jouvet et al., 1959). Although the mean firing rate of dopaminergic neurons does not change significantly throughout the sleep–wake cycle (Miller et al., 1983), REM sleep is also characterized by an increase in dopamine release (Maloney et al., 2002; Lena et al., 2005). Together with the sleep disturbances displayed by individuals with altered dopaminergic transmission, this evidence further supports the hypothesis that dopamine plays an important role in regulating the sleep–wake cycle.
To test this hypothesis, we investigated patterns of neural and electromyographic (EMG) activity throughout the sleep–wake cycle in mice with genetically and pharmacologically manipulated levels of dopamine. Dopamine transporter–knock-out (DAT-KO) mice lack the gene encoding the dopamine transporter (DAT), a transmembrane protein that is responsible for regulating the reuptake of synaptic dopamine and replenishing dopamine stores in the presynaptic terminal (Gainetdinov and Caron, 2003). As such, the DAT plays the key role in controlling dopamine homeostasis. Because of the loss of the DAT, DAT-KO mice exhibit profound depletion of intraneural dopamine stores as well as a fivefold increase in extracellular dopamine levels (Gainetdinov and Caron, 2003). Thus, when exposed to novelty, DAT-KO mice experience a marked period of behavioral hyperactivity (Gainetdinov et al., 1999). A similar phenotype is displayed by normal mice treated with amphetamine, which causes effective reversal of DAT-mediated dopamine transport and a subsequent 5- to 10-fold increase in extracellular dopamine (Jones et al., 1998).
Because intracellular dopamine stores originate from both de novo synthesis and DAT-mediated recycling of released dopamine, normal mice treated with the tyrosine hydroxylase inhibitor α-methyl-p-tyrosine (αMT) demonstrate only a partial (60%) reduction in striatal dopamine levels. Because DAT-KO mice are unable to recycle dopamine, dopamine concentrations are solely dependent on de novo synthesis. Thus, treatment with αMT produces a state in DAT-KO mice in which the striatal dopamine concentration is reduced to 0.2% of the level in control animals [dopamine-depleted DAT-KO (DDD) mice] (Sotnikova et al., 2005). By treating normal and DAT-KO mice with amphetamine and αMT, respectively, one can acutely manipulate dopaminergic tone and subsequently assess the role of dopamine in regulating the sleep–wake cycle.
Materials and Methods
Animal care and use.
All experiments were conducted in accordance with National Institutes of Health guidelines for the care and use of animals and with an approved animal protocol from the Duke University Institutional Animal Care and Use Committee. Experiments were performed with DAT-KO mice and their wild-type (WT) littermates that had been backcrossed into a C57B/6J background. Mice were housed four or five to a cage and maintained under standard laboratory conditions (12 h light/dark cycle) with food and water provided ad libitum. Adult mice were separated into individual cages and surgically implanted with electrodes and EMG wires. Experiments were conducted following a 1 week recovery.
Surgery.
Mice were anesthetized and placed in a stereotaxic device, and ground screws were secured to the cranium. Tungsten microwire array electrodes (diameter, 30 μm; impedance, 2 MΩ at 1 kHz, 5 nA) were implanted through a small cranial window into the dorsal hippocampus (stereotaxic coordinates: −2.3 mm anteroposterior, 1.6 mm mediolateral, and 1.8 mm dorsoventral from bregma) and anchored to ground screws using dental acrylic. Tungsten EMG wires were placed in the trapezius muscle, and skin was closed using surgical sutures.
Experimental setup.
All experiments were conducted in a novel environment and during the animals' normal light cycle to ensure a significant amount of sleep. The novel environment consisted of an empty cage bottom (11.5 × 7 × 4.5 inches) that was marked into six equal sections, and gross motor activity was determined from the number of section crosses during the waking period (see Fig. 1). Two pieces of rodent feed and a small piece of paper were placed in the cage bottom, and a water bottle was suspended from the side wall.
Figure 1.

Behavioral dynamics of DAT-KO and WT control mice. a, Locomotion of DAT-KO mice and WT control mice in a novel environment. b, DAT-KO mice display locomotor hyperactivity during the WK-N period compared with WT control mice (n = 7). There was no significant difference between DAT-KO and WT mice during the habituated period (Kruskal–Wallis test: df = 3, p < 0.0001; followed by Mann–Whitney test; *p < 0.01 for comparisons of DAT-KO and WT control mice during WK-N, p > 0.05 for comparisons during WK-H; n = 8 for DAT-KO; n = 7 for WT control mice). Error bars indicate SEM.
Data acquisition.
LFPs were preamplified (500×), filtered (0.3–400 Hz), and digitized at 500 Hz using a Digital Acquisition card (National Instruments, Austin, TX) and a Multi-Neuron Acquisition Processor (Plexon, Dallas, TX). Behaviors were recorded with a CCD video camera and a video cassette recorder. Video and neural recordings were synchronized with a millisecond precision timer (model VTG-55; For-A, Tokyo, Japan).
Construction of the two-dimensional state space map.
The two-dimensional state space was defined by two spectral amplitude ratios calculated by dividing integrated spectral amplitudes at selected frequency bands from LFPs recorded from the dorsal hippocampus. First, all data segments with amplitude saturation were discarded from the working data set (2.31 ± 0.75%; mean ± SEM of the total data per mouse). With Matlab (MathWorks, Natick, MA), a sliding window Fourier transform was applied to the LFP signal using a 2 s window with a 1 s step. The Fourier transform parameters were chosen to allow for a frequency resolution of 0.5 Hz. Then two spectral amplitude ratios were calculated by integrating the spectral amplitude (absolute value) over selected frequency bands for each data window. The ratios were heuristic, resulting from a thorough search for parameters aimed at the best separation of states. A low-cut frequency of 0.5 Hz was used to eliminate the DC component. For each animal, spectral amplitude ratios were further smoothed with a Hanning window of 20 s to reduce within-state variability. These two ratios were used to construct the two-dimensional state space in which each point represents 1 s of ongoing brain activity. The overlap of behavioral clusters was determined by applying the Matlab “inpolygon” function to the two-dimensional state map.
EMG activity.
Fourier transform was applied to the LFP signal using a 2 s window with a 1 s step. The Fourier transform parameters were chosen to allow for a frequency resolution of 0.5 Hz. EMG activity was then calculated by taking the root mean square of the spectral amplitude over selected frequency bands: 30–56 and 64–250 Hz. All EMG traces were high-pass filtered at 30 Hz.
Behavioral state identification using two-dimensional state map and EMG.
Parameters of the state map were chosen to generate cluster separation based on the high-amplitude theta (4–9 Hz) and gamma (33–55 Hz) oscillations characteristic of REM sleep, the absence of gamma oscillations and the high-amplitude delta (1–4 Hz) characteristic of SWS, and the high-amplitude gamma and theta oscillations characteristic of waking. Similar parameters have been shown to produce 95% cluster separation in state maps generated from LFP oscillations recorded from rat dorsal hippocampus. The minor scoring errors in these maps typically occurred in differentiating REM sleep from waking. Thus, EMG analysis was used to identify periods of atonia consistent with REM sleep and combined with the two-dimensional state map cluster scoring method for all sleep experiments conducted in this study. Behavioral state selections were confirmed using behavioral observations and direct LFP and EMG analysis in two WT and two DAT-KO mice. Spectral trajectories with high speeds, which represented transitions between behavioral states, were excluded from behavioral state cluster selections.
Selecting a 2 h habituated period.
To create representative REM and habituated waking (WK-H) spectrogram patterns, we empirically selected a two-hour period during the last 4 h of the recording in which the mouse experienced at least 10 min of waking and 2 min of REM sleep.
Peak theta powers distribution.
Behavioral state clusters were selected using Statemap, EMG analysis, and Matlab, and mean power spectrums were calculated for waking after novelty exposure (WK-N), WK-H, and REM. The f max (frequency, in the 4.5–11 Hz range, at which the max power occurs) was determined for each mean power spectrum, and the maximum power in a 1 Hz window surrounding f max was calculated for each second during the behavioral period. These peak power values were normalized to the maximum peak power observed during REM and placed into nine equally spaced bins. Importantly, the mean power spectrum observed during REM remained unchanged when DAT-KO mice were pretreated with haloperidol and when 12 h experimental recordings were conducted on DAT-KO mice in their home cage. This suggests that the peak theta power observed during REM can be used effectively as the baseline variable wherewith to normalize the peak theta power distribution.
Wake–REM similarity index.
Values were calculated by taking the sum of the absolute value of the difference between waking and REM peak theta power distributions and bounded between 1, for identical waking and REM distributions, and 0, as follows:
 |
Dopamine depletion and REM recovery.
All dopamine depletion studies were started 30 min after treatment with 250 mg/kg αMT. In REM recovery experiments, l-DOPA, Quinpirole, or SKF-81297 was administered after an additional 30 min baseline dopamine depletion recording period. To ensure that the recovery of REM sleep was attributable to the administration of exogenous dopaminergic agonists, and not enhanced tyrosine hydroxylase synthesis resulting from repeated dopamine depletion, the REM recovery recording period was limited to 6.5 h from the initial treatment with αMT. REM sleep was scored by the presence of 10 continuous seconds of LFP spectral ratios and EMG activity within the normal REM boundaries established during the initial baseline sleep recordings conducted in untreated mice. Periods of REM sleep were always preceded and followed by high-speed inter-cluster data points representing interstate transitions.
Confirmation of REM suppression using the sleep scoring method based on standard LFP and EMG analysis.
To confirm the absence of bouts of REM sleep in DDD mice, we used an additional sleep scoring method based on standard LFP and EMG analysis. First, we analyzed the baseline sleep recordings conducted in untreated DAT-KO mice and determined the maximum EMG power observed in REM sleep for each mouse. Next, we identified all 10 s epochs in DDD animals where EMG activity remained below the maximum value observed during normal REM sleep. This was done to maintain high sensitivity for bouts of REM sleep. This method was also highly selective for epochs of low muscle activity occurring during SWS and quiet waking, and 10 s epochs identified by this method often overlapped with each other. Next, we excluded all epochs in which mean fast (gamma) activity was lower than the mean fast activity observed across SWS during the baseline sleep recordings. Finally, we used video data to perform behavioral observations and determine state-specific postures of the mice during each epoch (e.g., curling quietly, moving, standing, rearing, etc.). This process excluded all 10 s epochs in dopamine-depleted mice and identified bouts of REM sleep in dopamine-depleted mice treated with l-DOPA and Quinpirole. In several DDD mice, there were no 10 s epochs in which EMG activity was lower than that observed during REM sleep. In other animals, there were no 10 s epochs of low EMG activity with high-frequency brain activity.
Theta power control in hyperactive animals.
Wake–REM Similarity Index (WRSI) values were determined for each 1 min period in which animals displayed 10–15 section crosses/min. These values were normalized over the number of observations for each animal (14 ± 2 1 min periods per mouse). All animals with five or less 1 min time periods of 10–15 sections crosses were excluded from the analysis.
Statistics.
Statistical significance of data from this study was analyzed by the Mann–Whitney test for single comparisons and the Kruskal–Wallis test, followed by the Mann–Whitney test for multiple comparisons. A p value <0.05 was considered significant.
Results
Mice with genetically induced hyperdopaminergia display REM-like neural oscillations after exposure to a novel environment
Twenty-one adult DAT-KO and 13 WT littermate control mice were surgically implanted with tungsten multielectrode arrays in the hippocampus and tungsten electrodes in the trapezius muscle. After a 1 week recovery period, animals were subjected to continuous hippocampal LFP and EMG recordings for 12 h, while their behaviors were recorded with a video camera. Eight DAT-KO mice were recorded in a novel cage, to induce behavioral hyperactivity (Fig. 1). All of the WT mice were recorded in the novel cage as controls, and 11 DAT-KO mice were subjected to continuous recordings for 8 h in their home cage, to determine their baseline sleep patterns. To quantitatively distinguish WK, SWS, and REM sleep, we used a method recently developed in our laboratory that separates wake and sleep states as distinct clusters in a two-dimensional state map derived from two LFP spectral ratios (Gervasoni et al., 2004). The first ratio (ratio 1) produces cluster separation based on high-frequency gamma (33–55 Hz) spectral oscillations (Steriade et al., 1993), whereas the second (ratio 2) produces cluster separation based primarily on theta (4–9 Hz) spectral oscillations (Vanderwolf, 1969; Timo-Iaria et al., 1970; Cantero et al., 2003) (Fig. 2). The real-time version of the two-dimensional state map predicted behavioral states consistent with standard methods for scoring sleep in mice based on direct observations of LFP activity, EMG activity, behavioral activity, and state-specific postures (Fig. 3). WK periods were characterized by high brain activity and high muscle activity, SWS was characterized by low brain activity and low muscle activity, and REM was characterized by high brain activity and negligible muscle activity (atonia).
Figure 2.

Determination of state map ratios from power spectrum analysis. Behavioral state maps were generated by plotting the following spectral ratios: x-axis, 2–4.5 Hz/2–9 Hz (Ratio 2); y-axis, 2–20 Hz/2–55 Hz (Ratio 1).
Figure 3.

LFP and EMG activity during state map predicted behavioral states. Mice were introduced into a novel cage and subjected to 12 h continuous LFP (hippocampus) and EMG (trapezius) recordings. Real-time two-dimensional behavioral state maps were generated by plotting the following spectral ratios: x-axis, 0.5–4.5 Hz/0.5–9 Hz; y-axis, 0.5–20 Hz/0.5–55 Hz. Raw LFP and EMG activity was analyzed during periods of WK, SWS, and REM sleep predicted by the two-dimensional state map. As demonstrated previously, WK was characterized by high brain activity and high muscle activity, SWS was characterized by low brain activity and low muscle activity, and REM was characterized by high brain activity and negligible muscle activity (atonia).
To ensure accurate identification of sleep–wake states, we combined analysis of EMG activity patterns with an off-line version of our two-dimensional state map for all experiments presented in this report. As observed previously in rats (Gervasoni et al., 2004), three spectral clusters were clearly visible in two-dimensional state maps obtained from WT control mice, corresponding to WK, SWS, and REM sleep (Fig. 4 a). Surprisingly, in the hyperdopaminergic DAT-KO mice recorded in the novel cage, a substantial overlap between WK and REM sleep clusters was observed in two-dimensional state maps generated from 12 h electrophysiological recordings. No alteration in the SWS cluster was observed in these animals (Fig. 4 a,b) (percentage of overlap values: DAT-KO vehicle, 48 ± 6%; WT vehicle, 10 ± 4%; mean ± SEM; n = 8 for both groups; Mann–Whitney test, p < 0.01). The increased percentage of WK-REM overlap values in DAT-KO mice suggests that a greater percentage of the LFP spectral ratios typically observed during REM sleep appear during periods when the animals are awake. Importantly, during REM sleep, the relative power spectrum of hippocampal LFP oscillations at different frequencies was equivalent between WT and DAT-KO animals (Fig. 4 c,d; Table 1).
Figure 4.
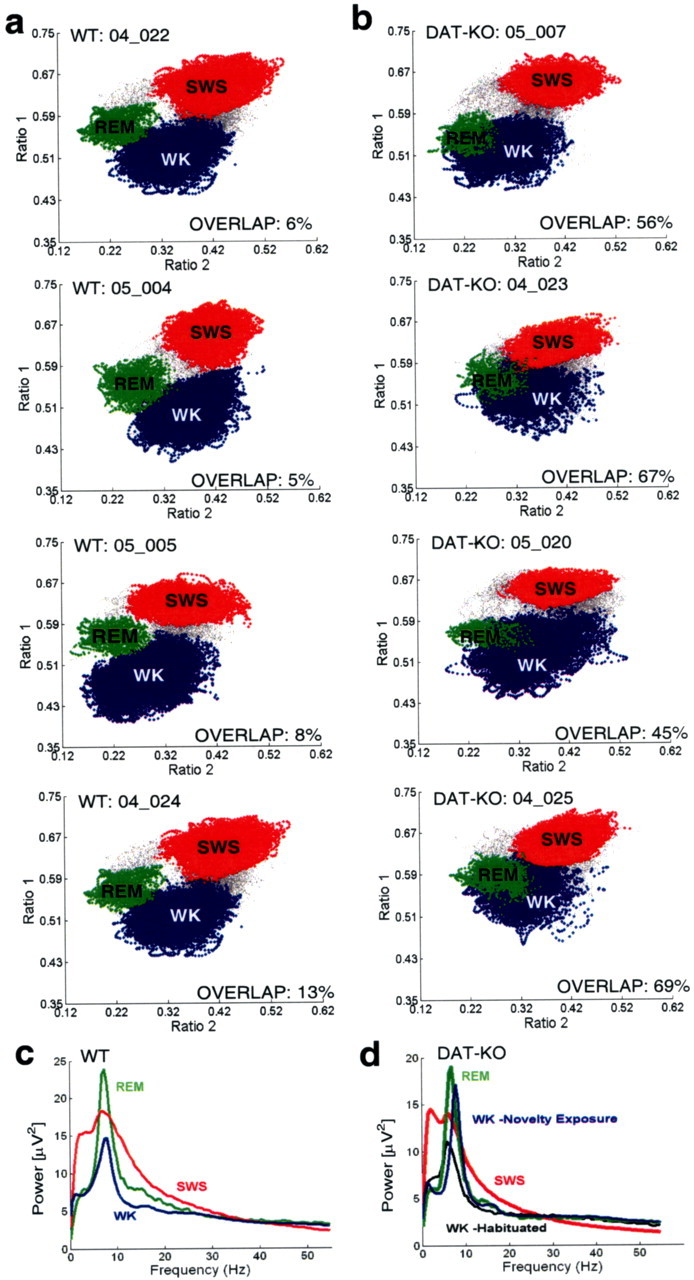
DAT-KO mice display novel REM-like awake state. Mice were introduced into a novel cage and subjected to 12 h continuous LFP (hippocampus) and EMG (trapezius) recordings. Two-dimensional behavioral state maps were generated by plotting the following spectral ratios: x-axis, 2–4.5 Hz/2–9 Hz; y-axis, 2–20 Hz/2–55 Hz. EMG data were used to disambiguate WK and REM clusters. All unassigned time points, typically corresponding to interstate transitions, were coded gray. a, WT mice displayed clear separation of the WK (blue), SWS (red), and REM (green) clusters. b, DAT-KO mice displayed distinct SWS clusters (red) but showed fused WK (blue) and REM (green) clusters. c, WT mice displayed state-dependant power spectral patterns characteristic of REM (green), SWS, and WK (blue) in rodents. REM was characterized by high-amplitude theta (4–9 Hz) and gamma (33–55 Hz) oscillations, SWS was characterized by high-amplitude delta (1–4 Hz) and low-amplitude gamma oscillations, and WK was characterized by high-amplitude gamma oscillations. d, DAT-KO mice displayed state-dependant power spectral oscillations characteristic of REM and SWS. WK spectrogram patterns displayed an REM-like distribution after exposure to novelty (blue) and normalized once the animals habituated to the novel environment (black).
Table 1.
Relative spectral power of WT and DAT-KO mice during REM sleep
| Delta | Theta | Beta | Gamma | |
|---|---|---|---|---|
| WT | 26 ± 2% | 48 ± 2% | 15 ± 1% | 10 ± 1% |
| DAT-KO | 29 ± 1% | 46 ± 1% | 15 ± 1% | 10 ± 0% |
There was no statistical difference between WT and DAT-KO relative spectral power at any frequency. p > 0.1 for comparisons of relative spectral power distributed within the delta (0.5–4 Hz), theta, beta (11–30 Hz), and gamma frequency ranges (Mann–Whitney test); n = 8 for WT and DAT-KO mice.
The increased overlap of WK and REM sleep clusters in DAT-KO mice was not present in state maps generated from LFP oscillations recorded during the last 4 h of the experimental period (Fig. 5), suggesting that the waking period in which the LFP spectral ratios assumed an REM-like character typically occurred during the initial period of exposure to novelty (Fig. 4 d), and subsequently subsided as the animals habituated to the new environment. Thus, we set out to compare spectrogram patterns of the hippocampal LFPs observed during the first 2 h of the recording (WK-N) with those observed after habituation to the novel environment, during both waking (WK-H) and REM sleep (note that there was no REM sleep in DAT-KO mice during the 2 h immediately after WK-N). The habituated period corresponded to 8–12 h after the beginning of the recording session. To further investigate the temporal nature of the striking WK-REM overlap in DAT-KO mice, we also calculated the peak spectral power, in the theta frequency range, over time.
Figure 5.

Habituated DAT-KO display normal behavioral state maps. Mice were introduced into a novel cage and subjected to 12 h continuous LFP (hippocampus) and EMG (trapezius) recordings. State maps were generated from LFPs, recorded 8–12 h after the animal was introduced into the novel cage, by plotting the following spectral ratios: x-axis, 2–4.5 Hz/2–9 Hz; y-axis, 2–20 Hz/2–55 Hz. EMG data were used to disambiguate WK and REM clusters. DAT-KO mice displayed clear separation of the WK (blue), SWS (red), and REM (green) clusters during the behaviorally habituated period.
WT mice displayed peak theta power distributions that were similar during WK-N and WK-H. Furthermore, peak theta power was distributed over a wider range during REM sleep than any of the waking periods in WT mice (Fig. 6 a). In contrast, DAT-KO mice displayed peak theta power distributions during the first 2 h in the novel environment that were virtually identical to those observed during REM sleep (Fig. 6 b). To quantify these differences, we defined a WRSI that measures the correlation between the peak theta power distribution of a given waking period and REM sleep. DAT-KO mice displayed significant WK/REM similarity during the WK-N period (i.e., animal awake after initial exposure to novelty). During the WK-H period (i.e., after habituation), however, the WK/REM similarity was much lower and statistically indistinguishable from that observed in WT mice (Fig. 6 c). These results demonstrate that after exposure to novelty, hyperdopaminergic DAT-KO mice enter a novel awake state characterized by patterns of hippocampal neural activation similar to those observed during REM sleep. Interestingly, this state is also characterized by significantly increased hippocampal oscillations in the gamma frequency range (Fig. 7).
Figure 6.
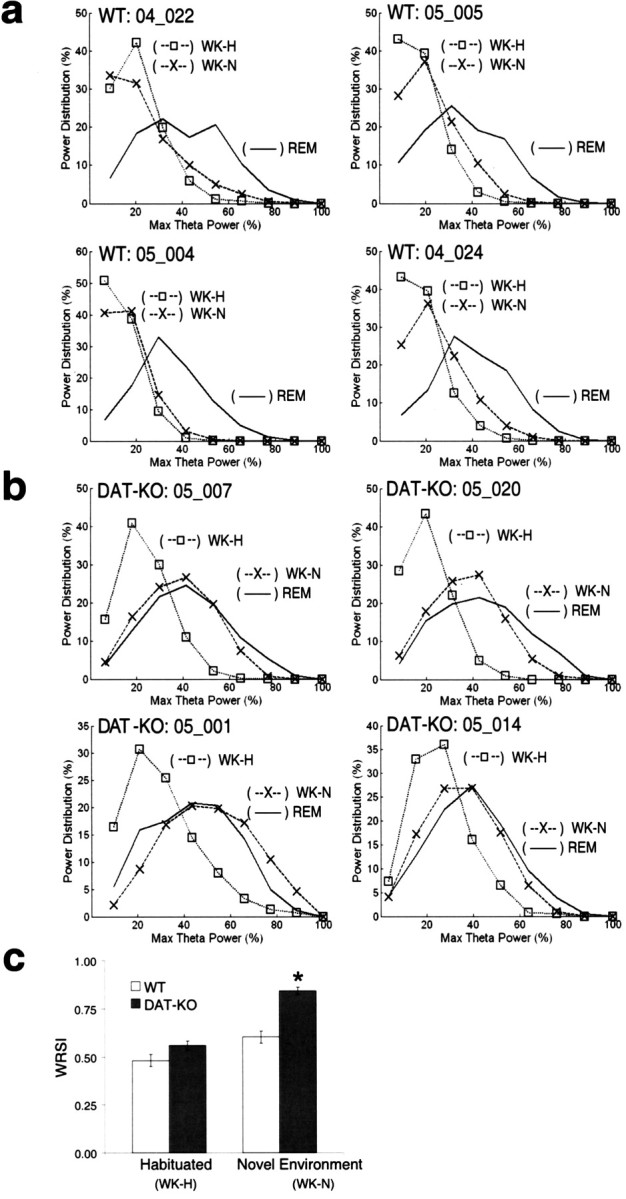
Hyperdopaminergia and WK-N are necessary for generation of the REM-like awake state. a, b, Peak theta power distributions were determined for WT (a) and DAT-KO (b) mice during awake periods in a novel (WK-N) environment (×), habituated (WK-H) environment (□), and REM sleep (solid line) behavioral periods and normalized to the maximum peak power observed during REM. c, The WRSI shows that the peak theta power distribution during WK-N in DAT-KO mice (n = 8) is significantly more similar to that of REM than the WK-H/DAT-KO, WK-H/WT, and WK-N/WT (n = 8) distributions (Kruskal–Wallis test: df = 3, p < 0.0001; followed by Mann–Whitney test; *p < 0.001). There was no statistical difference between WT and DAT-KO animals during the habituated period (p > 0.1).
Figure 7.

Novelty-induced hippocampal gamma oscillations in hyperdopaminergic mice. WT and DAT-KO mice were introduced into a novel cage and subjected to 12 h continuous LFP (hippocampus) and EMG (trapezius) recordings. Mean hippocampal gamma power was determined during the waking period immediately after WK-N and after habituation (WK-H). These values were then normalized to the mean gamma power observed during REM sleep for each animal. Novelty exposure significantly increased hippocampal gamma oscillations in DAT-KO mice (Kruskal–Wallis test: df = 3, p < = 0.01; followed by Mann–Whitney test; *p < 0.01) but not in WT mice (Mann–Whitney test, p > 0.05; n = 8 for DAT-KO and WT control mice). There was no statistical difference in hippocampal gamma power observed in DAT-KO and WT mice after habituation (Mann–Whitney test, p > 0.05). Treatment with 3.0 mg/kg amphetamine (Amp) significantly increased hippocampal gamma oscillations in WT control mice (Mann–Whitney test; # p < 0.05 compared with WT control mice/WK-N). Treatment with 0.3 mg/kg haloperidol (Hal) significantly reduced gamma oscillations in novelty-exposed DAT-KO mice (Mann–Whitney test; ## p < 0.05 compared with DAT-KO mice/WK-N).
Although WT and DAT-KO mice display similar abundances of WK, SWS, and REM during the dark cycle (Wisor et al., 2001), the argument could be raised that the REM-like neural oscillations observed in awake DAT-KO mice during the WK-N period may be attributable to subtle differences in the preceding dark-cycle sleep–wake patterns, and not exposure to novelty. To determine whether the REM-like neural oscillations observed in awake DAT-KO mice were indeed as a result of novelty exposure, we conducted 12 h electrophysiological recordings and behavioral observations across the light cycle of five DAT-KO mice in their home cage. The DAT-KO mice recorded in their home cage displayed WRSI values during the initial recording period that were statistically similar to those observed in novelty-exposed DAT-KO mice during the habituated period [WRSI values: DAT-KO home cage, 0.55 ± 0.08 (n = 5); DAT-KO habituated, 0.56 ± 0.02 (n = 8); mean ± SEM; p > 0.1, Mann–Whitney test]. These results demonstrate that the REM-like neural oscillations observed in DAT-KO mice during awake states are indeed attributable to exposure to the novel environment. Importantly, exposure to novelty does not further increase dopaminergic tone in DAT-KO mice (Gainetdinov et al., 1999). Therefore, because the “REM-like” patterns of neural activation cease as the animal habituates to the environment, whereas dopaminergic tone remains constant (Gainetdinov et al., 1999), our results suggest that hyperdopaminergia is not sufficient to generate the REM-like oscillations in awake DAT-KO mice.
Haloperidol reduces REM-like neural activity during wakefulness in DAT-KO mice
Next, we set out to determine whether hyperdopaminergia is necessary to generate the REM-like oscillations in awake novelty-exposed DAT-KO mice. Four DAT-KO mice were treated with 0.3 mg/kg haloperidol, which has been shown to attenuate novelty-induced behavioral hyperactivity (Spielewoy et al., 2000), and 12 h electrophysiological recordings and behavioral observations were repeated. Treatment with the D2 dopamine receptor antagonist haloperidol caused significant suppression of the REM-like neural alterations observed in novelty-exposed DAT-KO mice, resulting in reduced overlap of WK and REM clusters (Fig. 8). Furthermore, this was concomitant to a significant decrease in WK/REM similarity observed during the WK-N period of treated animals [WRSI values: DAT-KO vehicle, 0.84 ± 0.02 (n = 8); DAT-KO treated with haloperidol, 0.67 ± 0.02 (n = 4); mean ± SEM; p < 0.05, Mann–Whitney test]. These results demonstrate that the WK/REM similarity observed in novelty-exposed DAT-KO mice can be attenuated by blockade of the D2 dopamine receptor, suggesting that hyperdopamergia is indeed necessary to generate the REM-like neural oscillations in novelty-exposed mice. Moreover, these results demonstrate that the WK-REM similarity is mediated via the interaction of novelty exposure and activation of the D2 dopamine receptor pathway. Importantly, treatment with haloperidol also reduced hippocampal oscillations in the gamma frequency range [normalized gamma power: DAT-KO vehicle, 1.18 ± 0.05 (n = 8); DAT-KO treated with haloperidol, 1.02 ± 0.06 (n = 4); Mann–Whitney test, p < 0.05] (Fig. 7).
Figure 8.
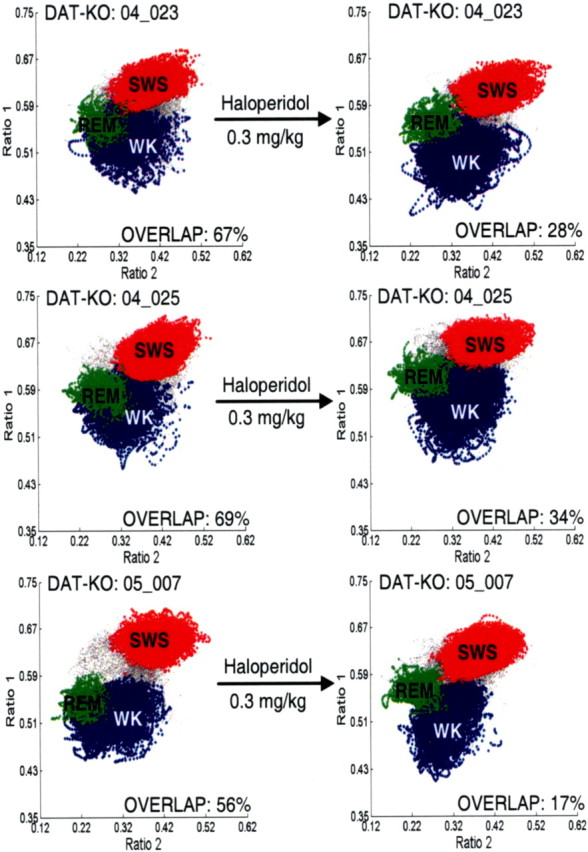
D2 antagonist attenuates REM-like awake state in novelty-exposed DAT-KO mice. After initial recordings (left column), DAT-KO animals were given intraperitoneal injections of a single dose of 0.3 mg/kg haloperidol (right column), placed in a novel environment, and subjected to additional 12 h recordings. The haloperidol-treated group displayed significantly less overlap of WK (blue) and REM (green) clusters compared with the untreated group (p < 0.05, Mann–Whitney test). All unassigned time points, typically corresponding to interstate transitions, are coded gray.
Mice with pharmacologically induced hyperdopaminergia display REM-like neural oscillations while awake
To investigate whether hyperdopaminergia would generate REM-like neural oscillations in the hippocampus of awake normal mice, and thus increase the WK/REM similarity, we treated five novelty-exposed WT control mice with amphetamine (Guix et al., 1992; Jones et al., 1998) and repeated our recording protocol. As predicted, WT mice treated with 3.0 mg/kg d-amphetamine displayed peak theta power distributions that were wider during WK-N than those observed during the WK-N period of untreated control mice and power spectral distributions that were similar to those observed during their periods of REM sleep (Fig. 9 a,b). These electrophysiological alterations were parallel to a significant increase in WK-N WRSI values in the amphetamine-treated group [WRSI values: WT vehicle, 0.59 ± 0.03 (n = 8); WT d-amphetamine, 0.81 ± 0.02 (n = 5); mean ± SEM; p < 0.01, Mann–Whitney test]. Moreover, the WK/REM similarity observed in the amphetamine-treated control mice was statistically indistinguishable from that observed in the hyperdopaminergic DAT-KO mice after exposure to novelty (p > 0.1, Mann–Whitney test). Interestingly, WT mice treated with amphetamine also displayed a significant increase in hippocampal oscillations in the gamma frequency range [normalized gamma power: WT vehicle, 1.02 ± 0.04 (n = 8); WT d-amphetamine, 1.19 ± 0.03 (n = 5); p < 0.01] (Fig. 7) that was statistically indistinguishable from that observed in novelty-exposed DAT-KO mice (p > 0.1, Mann–Whitney test) (Fig. 7).
Figure 9.
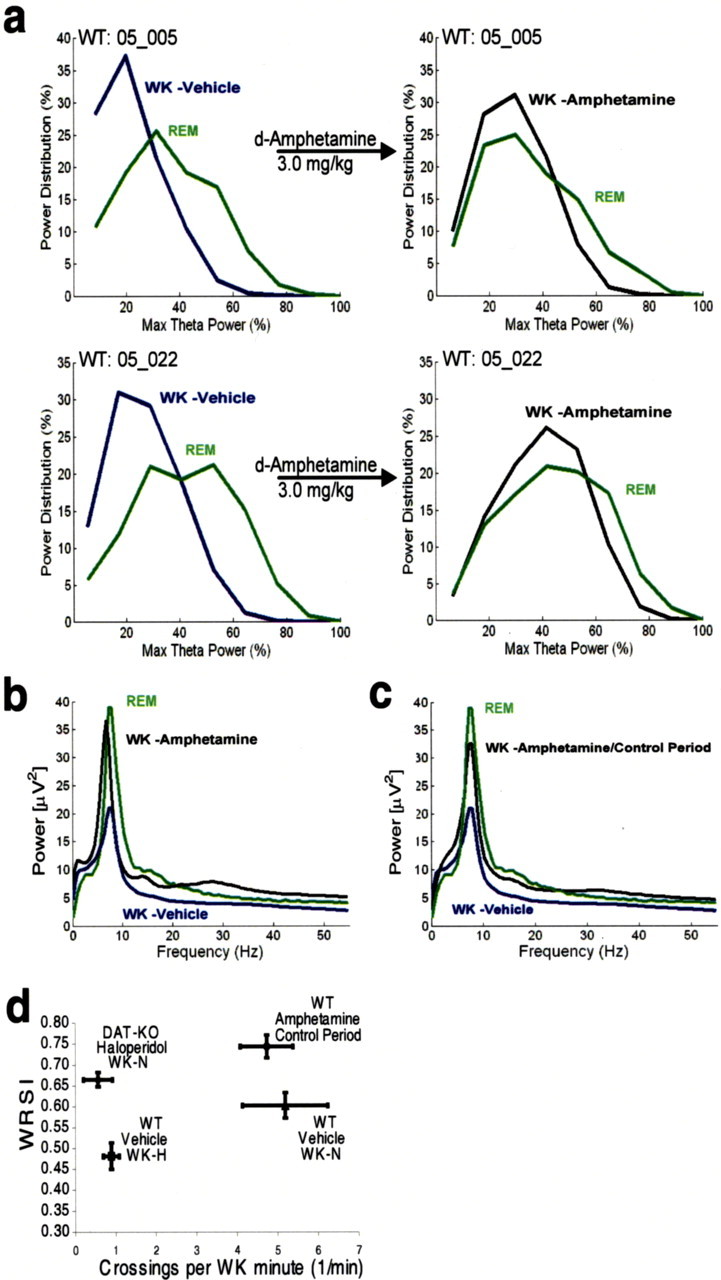
The role of hyperdopaminergia and locomotor hyperactivity in generating WK/REM similarity. After initial recordings (left column), WT animals were given intraperitoneal injections of a single dose of 3.0 mg/kg amphetamine (right column), placed in a novel environment, and subjected to 12 h recordings. a, b, WT mice treated with amphetamine displayed WK-N peak theta power distributions (a) and LFP power spectrum oscillations (b) that were similar to those observed during REM. c, Hippocampal LFP oscillations displayed REM-like distribution in amphetamine-treated WT mice even after the cessation of behavioral hyperactivity. d, WT mice treated with amphetamine (n = 5) displayed significantly elevated WRSI values after cessation of behavioral hyperactivity compared with untreated WT control mice (n = 5) during WK-N. DAT-KO mice treated with 0.3 mg/kg haloperidol (n = 4) displayed significantly elevated WRSI values during WK-N compared with WT control mice during WK-H (p < 0.05, Mann–Whitney test).
REM-like oscillations can be caused by hyperdopaminergia and novelty even in the absence of hyperlocomotive behavior
Although these results strongly suggest that the WK/REM similarity is mediated via the interaction of novelty and dopamine, and that hyperdopaminergia alone is sufficient to generate REM-like oscillations in awake WT control mice exposed to novelty, they also raise the possibility that the increased WRSI values observed during the WK-N period of the hyperdopaminergic mice could emerge because of changes in hippocampal theta oscillations resulting from enhanced exploratory behavior observed in these animals (Vanderwolf, 1969; Winson, 1974). To test whether the increased WK/REM similarity was simply the result of enhanced exploratory behavior rather than hyperdopaminergia, we determined WRSI values for hyperdopaminergic mice in three behaviorally controlled paradigms. First, we calculated WRSI values for all 1 min time periods during the WK-N period in which WT mice, DAT-KO mice, and WT mice treated with amphetamine displayed 10–15 section crosses. This level of behavioral locomotion was considered elevated for WT mice (5.2 ± 1.1 section crosses per WK minute), low for DAT-KO mice (22.4 ± 4.6 section crosses per WK minute), and average for WT mice treated with amphetamine (18.8 ± 2.3 section crosses per WK minute). Although the DAT-KO mice and amphetamine-treated WT mice displayed similar degrees of behavioral hyperactivity as the untreated WT mice [section crosses: WT, 12.1 ± 0.3 (n = 7); DAT-KO, 12.4 ± 0.3 (n = 7); WT-amphetamine, 12.5 ± 0.2 (n = 5); mean ± SEM; p > 0.1 for both comparisons, Mann–Whitney test], WK/REM similarity values were significantly elevated in the DAT-KO mice and amphetamine-treated WT mice when compared with the untreated WT mice [WRSI values: WT vehicle, 0.65 ± 0.03 (n = 7); DAT-KO vehicle, 0.77 ± 0.02 (n = 7); WT-amphetamine, 0.77 ± 0.00 (n = 5); mean ± SEM; Mann–Whitney test, p < 0.05 for both comparisons].
Next, we determined WRSI values for the five amphetamine-treated WT mice during the 2 h control period immediately after the cessation of behavioral hyperactivity. This period typically occurred 2 h after administration of amphetamine. Although the amphetamine-treated WT mice no longer displayed behavioral hyperactivity during the control period, they continued to display REM-like neural oscillations during wakefulness (Fig. 9 c). Importantly, WRSI values remained significantly elevated in the amphetamine-treated mice during the control period compared with WK-N WRSI values observed in untreated animals (Fig. 9 d, right side of the graph) [WRSI values: WT amphetamine/control period, 0.74 ± 0.03 (n = 5); WT vehicle/WK-N, 0.60 ± 0.03 (n = 8); mean ± SEM; p < 0.01, Mann–Whitney test; square crossings/WK minute values: WT amphetamine/control period, 4.7 ± 0.7; WT vehicle/WK-N, 5.2 ± 1.1; mean ± SEM; p > 0.1, Mann–Whitney test]. These results suggest that the WK/REM similarity observed in normal mice treated with amphetamine does not result from enhanced exploratory behavior, because it was observed even after the cessation of behavioral hyperactivity.
Next, we set out to compare WK/REM similarity values observed in DAT-KO mice with reduced behavioral hyperactivity profiles to those observed in normal mice. Thus, we compared WK-N WRSI values observed in haloperidol-treated DAT-KO mice with those observed in WT mice during the habituated period. Importantly, the WK/REM similarity observed in the haloperidol-treated DAT-KO mice was significantly greater than that observed in WT mice with similar behavioral profiles (Fig. 9 d, left side of the graph) [WRSI values: DAT-KO haloperidol, 0.67 ± 0.02 (n = 4); WT vehicle, 0.48 ± 0.03 (n = 8); mean ± SEM; p < 0.05, Mann–Whitney test; square crossings/WK minute values: DAT-KO haloperidol, 0.6 ± 0.4; WT vehicle, 0.9 ± 0.2; mean ± SEM; p > 0.1, Mann–Whitney test]. This result strongly suggests that both the hyperdopaminergic state and exposure to novelty, but not hyperlocomotion, are necessary and sufficient for the appearance of REM-like neural oscillations in awake WT and DAT-KO mice, because it was observed during periods of elevated, normal, and decreased locomotor activity compared with control mice with equivalent behavioral profiles.
Hippocampal theta oscillations are highly correlated with behaviors such as changes in posture or limb position, walking, head movements, and rearing and have been shown to vary on the order of hundreds of milliseconds (Vanderwolf and Baker, 1986). Thus, the argument can be raised that behavioral experiments that control locomotion across 1 min or 2 h time periods are not sufficient to account for behavior-associated increases in theta oscillations. To address this issue, we analyzed LFP and EMG recordings in DAT-KO and WT mice and found that DAT-KO mice appeared to display elevated theta oscillations during the WK-N period even at low levels of muscle activity (Fig. 10). Thus, we used video recordings to identify all 1 s intervals during the WK-N period in which animals were completely immobile (resting all four paws on the floor) and calculated the mean theta power relative to that observed during subsequent episodes of REM sleep. Even during periods of immobility, hyperdopaminergic DAT-KO mice displayed significantly elevated power in the theta frequency range compared with WT control mice [percentage of REM theta power: DAT-KO immobile, 87 ± 4% (n = 4); WT immobile, 66 ± 3% (n = 4); mean ± SEM; p < 0.05, Mann–Whitney test]. Importantly, the WT and DAT-KO animals displayed similar levels of theta power during REM sleep [REM theta power: DAT-KO, 9 ± 3 μV2 (n = 4); WT, 12 ± 4 μV2 (n = 4); mean ± SEM; p > 0.1, Mann–Whitney test]. These results demonstrate that DAT-KO mice display a significant increase in hippocampal theta oscillations, providing strong evidence that the WK/REM similarity is mediated via the interaction between enhanced dopaminergic transmission and novelty, rather than simply resulting from enhanced locomotor behavior.
Figure 10.

Activity independent theta oscillations during WK-N in DAT-KO mice. WT and DAT-KO mice were introduced into a novel cage and subjected to 12 h continuous LFP (hippocampus) and EMG (trapezius) recordings. Peak theta power and EMG activity was determined for each second period. WT mice displayed similar levels of theta power immediately after exposure to novelty (blue) and after habituation (red) at low levels of EMG activity (arrows). DAT-KO mice displayed higher theta power after exposure to novelty (blue) than after habituation (red) at low levels of EMG activity as seen by the increase in visible blue area.
Dopamine depletion alters sleep–wake states in WT and DAT-KO mice
After verifying that excess dopamine generates a different awake state in novelty-exposed mice, we set out to examine whether dopamine depletion would also alter sleep–wake states. After the initial baseline recording period, five WT and five DAT-KO mice were treated with αMT, and the recording protocol was repeated. WT mice treated with αMT demonstrated a partial (60%) reduction in striatal dopamine levels (Sotnikova et al., 2005). Although the WT animals treated with αMT neither experience changes in their motor behavioral profile (Sotnikova et al., 2005) nor significant shifts in their LFP spectral ratios during the 6 h recording period, they all demonstrate a reduction in the total REM sleep time [62 ± 18% reduction in total REM time (n = 5); mean ± SEM; p < 0.05, Mann–Whitney test] (Fig. 11 a). Although these results suggest that dopaminergic transmission mediates REM sleep in normal mice, they also raise the possibility that the reduction in total REM time observed in the αMT-treated WT mice may be attributable to secondary depletion of norepinephrine or epinephrine (Sotnikova et al., 2005). However, this is highly unlikely because manipulations that decrease norepinephrine have been shown to increase the abundance of REM sleep (Kaur et al., 2004) and because the main source of norepinephrine in the brain, the locus ceruleus, ceases firing during REM sleep (Aston-Jones and Bloom, 1981).
Figure 11.

Dopamine depletion suppresses REM sleep and generates novel awake state in DAT-KO mice. a, After baseline behavioral state recordings (left), WT mice were treated with a single dose of 250 mg/kg αMT intraperitoneally, placed in a novel environment, and subjected to 6 h LFP (hippocampus) and EMG (trapezius) recordings. Two-dimensional behavioral state maps were generated by plotting the following spectral ratios: x-axis, 0.5–4.5 Hz/0.5–9 Hz; y-axis, 0.5–20 Hz/0.5–55 Hz. EMG data were used to disambiguate WK and REM clusters. All unassigned time points, typically corresponding to interstate transitions, are coded gray. WT mice treated with αMT (right) continued to display REM sleep clusters during the 6 h recording period, although total REM time was dramatically reduced. b, c, After baseline behavioral state recordings in their home cage (left), DAT-KO mice were treated with a single dose of 250 mg/kg αMT intraperitoneally, placed in a novel environment, and subjected to 8 h LFP (hippocampus) and EMG (trapezius) recordings. b, DAT-KO mice treated with αMT displayed LFP spectral ratios that were indistinguishable from those observed during SWS sleep in untreated animals. Because our state map method did not produce cluster separation between WK and SWS in DDD mice, we termed this state “αMT.” This phenomenon lasted the entire 8 h period, and no REM clusters were observed. c, Dopamine-depleted DAT-KO mice also displayed significant increases in EMG activity during this period, corresponding to increased muscle rigidity. d, DAT-KO mice treated with αMT displayed high-amplitude, low-frequency LFP oscillations by during awake periods marked by high muscle tone. e, State-dependent hippocampal LFP power spectral data observed in DAT-KO mice before and after being treated with αMT. The αMT (black) state was characterized by a reduction in mean hippocampal theta spectral power compared with WK (blue) and REM (green) in untreated animals and a reduction in mean hippocampal gamma spectral power compared with WK, SWS (red), and REM.
DAT-KO mice lack the key controller of dopamine homeostasis (Gainetdinov and Caron, 2003). These mice display remarkable dysregulation of intracellular and extracellular dopamine compartmentalization, and dopamine concentrations are solely dependent on ongoing dopamine synthesis. Treatment with 250 mg/kg αMT in DAT-KO mice reduces striatal dopamine concentration to <0.2% of the level observed in control animals (Sotnikova et al., 2005). As a consequence, DAT-KO mice treated with αMT (DDD mice) display severe akinesia and rigidity, which resembles late stages of Parkinson's disease in humans (Sotnikova et al., 2005). Importantly, DDD mice demonstrate a similar reduction in norepinephrine levels as WT mice treated with αMT (Sotnikova et al., 2005). Thus, we predicted that if the reduction in REM time observed in WT mice treated with αMT was indeed attributable to dopamine depletion, DDD mice would demonstrate a much more profound suppression of REM sleep. Interestingly, treatment with αMT generated a novel awake state in DAT-KO mice characterized by LFP spectral ratios that were similar to those observed during periods of SWS in untreated animals (Figs. 3, 11 b) and increased EMG activity corresponding to muscle rigidity (Fig. 11 c). After analysis of hippocampal LFP activity, we found that awake DDD mice (high EMG activity, eyes open, and standing on all four paws) displayed high-amplitude, low-frequency LFP oscillations similar to those observed during SWS (Fig. 11 d). These high-amplitude, low-frequency LFP oscillations were observed in striatum and cortex as well (Costa et al., 2006). Moreover, this state was marked by a dramatic reduction in LFP oscillations in the gamma and theta frequency range (Fig. 11 e). From direct visual observations, these animals appeared not to enter into sleep of any kind. However, given that our LFP recordings could not be used to distinguish brain activity in the dopamine-depleted state from that observed during SWS, we focused our studies on investigating the absence of REM sleep in the dopamine-depleted DAT-KO mice.
Treatment with αMT led to complete REM suppression in DAT-KO mice, which lasted for the entire 8 h of our original recording sessions. To confirm that the DDD mice truly experience a complete suppression of REM sleep, we used a second method of scoring sleep–wake states based on standard analysis of 10 s epochs of LFP and EMG activity and behavioral observations. Using this method, we never observed epochs of muscular atonia accompanied by desynchronized brain activity. Thus, we were unable to identify any periods of REM sleep in DDD mice using this standard method. Later we discovered that complete REM suppression could be produced for up to 16 h by treating these mice with an additional dose of 250 mg/kg αMT immediately after the initial 8 h period of dopamine depletion (n = 2; data not shown). These results strongly suggest that dopamine is also involved in the induction of normal physiological REM sleep, because complete depletion of this neurotransmitter causes complete abolishment of REM sleep periods.
Activation of the D2 pathway recovers REM sleep in dopamine-depleted mice
Next, we set out to test whether administration of exogenous dopaminergic agonists would induce recovery of REM sleep in DDD mice. It has been shown that high doses of the dopamine precursor l-DOPA and combined D1/D2 receptor stimulation restore locomotion in DDD mice (Sotnikova et al., 2005). Here, five DDD mice were treated with a dose (50 mg/kg) of l-DOPA that is considered insufficient for restoring mobility (Sotnikova et al., 2005). Strikingly, DDD mice treated with these lower doses of l-DOPA displayed recovery of REM sleep during the ensuing 6 h recording period, although they did not recover mobility (Sotnikova et al., 2005) (Fig. 12 a) [REM onset time: 1.4 ± 0.3 h in DAT-KO mice treated with l-DOPA (n = 5) compared with 1.7 ± 0.3 h in DAT-KO mice treated with vehicle (n = 6); mean ± SEM; p > 0.1, Mann–Whitney test]. We were able to identify periods of REM sleep using both our state-space method and the standard method of scoring sleep–wake states. Moreover, LFP activity traces during these time periods displayed trains of theta oscillations characteristic of REM sleep and EMG traces showed atonia (Fig. 13). Interestingly, DDD mice treated with l-DOPA displayed significantly less bouts of REM sleep compared with untreated DAT-KO mice [REM bouts per mouse: 8 ± 2 in DDD mice treated with l-DOPA (n = 5) compared with 21 ± 3 in DAT-KO mice treated with vehicle (n = 6); mean ± SEM; p < 0.05, Mann–Whitney test], although mean time spent in each bout of REM sleep was the same [REM time per bout: 59 ± 7 s in DDD mice treated with l-DOPA (n = 39 total bouts) compared with 42 ± 3 in DAT-KO mice treated with vehicle (n = 123 total bouts); mean ± SEM; p > 0.1, Mann–Whitney test]. These results demonstrate that REM sleep can be recovered in DDD mice by administering an endogenous dopamine precursor, even at doses insufficient to restore motor behaviors. Importantly, treatment with l-DOPA recovers both norepinephrine and epinephrine to some extent in DDD mice. Thus, although these results strongly suggest that REM sleep is mediated via dopaminergic transmission, they do not eliminate the possibility that the suppression and recovery of REM sleep in DDD mice is attributable to depletion and recovery of other catecholamines.
Figure 12.
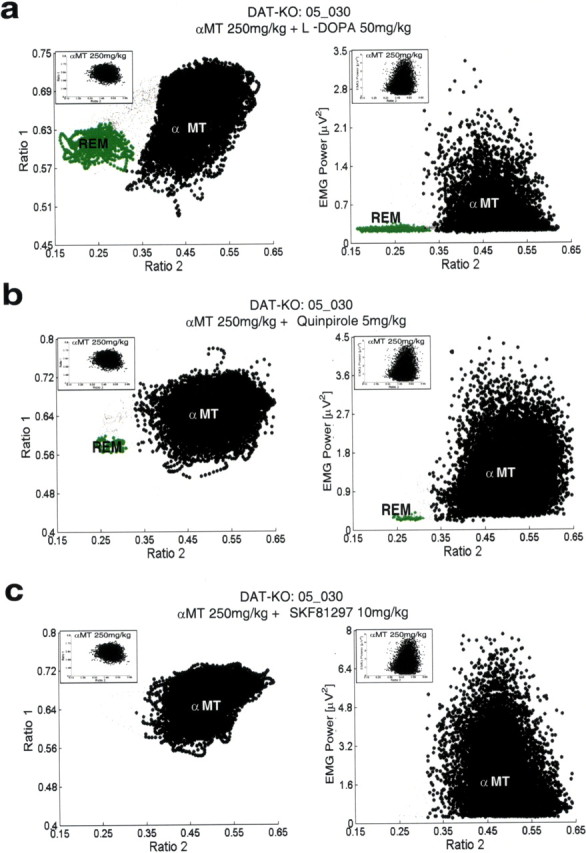
Selective recovery of REM sleep in DDD mice. DDD mice were treated intraperitoneally with a single dose of 50 mg/kg l-DOPA, 5 mg/kg Quinpirole, or 10 mg/kg SKF 81297, placed in a novel environment, and subjected to 6 h LFP (hippocampus) and EMG (trapezius) recordings. Two-dimensional behavioral state maps were generated by plotting the following spectral ratios: x-axis, 0.5–4.5 Hz/0.5–9 Hz; y-axis, 0.5–20 Hz/0.5–55 Hz. EMG data were used to disambiguate WK and REM clusters. All unassigned time points, typically corresponding to interstate transitions, are coded gray. a, Treatment with 50 mg/kg l-DOPA recovered a clear state map REM sleep cluster (left) that was marked by low EMG activity (right), although it did not completely reverse the αMT state observed in DDD mice. State maps and EMG plots observed in untreated DDD mice are displayed at the top left of each plot. b, DDD mice treated with the D2 dopamine receptor agonist Quinpirole (5 mg/kg) displayed a clear REM sleep cluster (left) that was marked by low EMG activity (right). Treatment with Quinpirole did not reverse the αMT observed in DDD mice. c, DDD mice treated with the D1 dopamine receptor agonist SKF 81297 (10 mg/kg) displayed neither recovery of an REM sleep cluster nor reversal of the αMT state.
Figure 13.

Raw LFP and EMG activity during selective recovery of REM sleep in DDD mice. DDD mice were treated intraperitoneally with a single dose of 50 mg/kg l-DOPA or 5 mg/kg Quinpirole, placed in a novel environment, and subjected to 6 h LFP (hippocampus) and EMG (trapezius) recordings. DDD mice displayed trains of theta oscillations and atonia during periods of REM sleep recovered by treatment with l-DOPA or Quinpirole, similar to that observed during REM sleep in untreated DAT-KO mice.
To determine whether REM suppression in DDD mice was directly attributable to depletion of dopamine, we treated DDD mice with direct D1 and D2 dopamine receptor agonists. When administered together (but not separately), D1 and D2 dopamine receptor agonists have been shown to recover movement in DDD mice (Sotnikova et al., 2005). Five DDD mice were treated with 5 mg/kg of the selective D2 dopamine receptor agonist Quinpirole, and the recording protocol was repeated. This dose was chosen because it has been shown to cause activation of D2 dopamine receptors sufficient to recovery of forward locomotion in DDD mice treated with a D1 dopamine receptor agonist. Again, although this treatment alone was not sufficient to induce recovery of mobility in DDD mice, Quinpirole recovered REM sleep during the ensuing 6 h recording period (Fig. 12 b) [REM onset time: 1.8 ± 0.5 h in DDD mice treated with Quinpirole (n = 5) compared with 1.7 ± 0.3 h in DAT-KO mice treated with vehicle (n = 6); mean ± SEM; p > 0.1, Mann–Whitney test]. Periods of REM sleep were identified by both our state-space method and the standard method of scoring sleep–wake states. Again, LFP activity traces during these time periods display trains of theta oscillations characteristic of REM sleep, and EMG traces demonstrate atonia (Fig. 13). Thus, activation of the D2 receptor was sufficient to recover REM sleep in DDD mice. Interestingly, DDD mice treated with Quinpirole demonstrate a similar number of bouts of REM sleep compared with DDD mice treated with l-DOPA [REM bouts: 10 ± 3 in DDD mice treated with Quinpirole (n = 5); mean ± SEM; p > 0.1, Mann–Whitney test], though the mean time spent in each of these bouts of REM is significantly reduced (REM time/bout: 37 ± 7 s in DDD mice treated with Quinpirole (n = 48 total bouts), mean ± SEM; p < 0.01, Mann–Whitney test]. Importantly, Quinpirole activates D2 dopamine receptors in DDD mice without recovering norepinephrine and epinephrine. Thus, these results demonstrate that dopamine does indeed mediate the generation of REM sleep. Interestingly, because DDD mice treated with Quinpirole demonstrate shorter bouts of REM sleep than DDD mice treated with l-DOPA, our findings also suggest that other catecholamines may play a role in regulating the maintenance of REM sleep (Ouyang et al., 2004). Curiously, selective activation of the D1 dopamine receptor pathway, via injection of the selective D1 dopamine receptor agonist SKF-81297 (10 mg/kg), did not induce any recovery of REM sleep (Fig. 12 c) (n = 5). This dose of SKF-81297 was chosen because it was sufficient to recover forward locomotion in DDD mice treated with a D2 dopamine receptor agonist (Sotnikova et al., 2005). Importantly, we never observed epochs of atonia that were accompanied by desynchronized brain activity. Thus, we were unable to identify any episodes of REM sleep using our state-space method or standard sleep-scoring method.
Altogether, these results indicate that selective activation of the D2, but not the D1, dopamine receptor pathway is sufficient to recover REM sleep in DDD mice. This is of particular importance given that the novelty-induced, REM-like neural oscillations observed in awake hyperdopaminergic mice could be attenuated by blocking the D2 pathway. Thus, our results demonstrate that both physiological REM sleep and the novelty-induced, REM-like neural oscillations are mediated by the D2 receptor pathway in DAT-KO mice, suggesting that the REM-like neural oscillations observed in awake hyperdopaminergic mice may result from aberrant activation of a physiological REM sleep pathway during awake behaving periods.
Discussion
Our findings demonstrate that novelty-exposed mice with genetically (DAT-KO mice) or pharmacologically (WT animals treated with amphetamine) induced hyperdopaminergia display a novel awake state characterized by hippocampal neural oscillations similar to those observed during REM sleep. Generally stated, REM-like electrophysiological activity appears during wakefulness in hyperdopaminergic mice. This activity is marked by a significant increase in hippocampal theta and gamma oscillations that is not trivially explained by enhanced exploratory behavior displayed by these mice. Additionally, treatment with the D2 dopamine receptor antagonist and antipsychotic agent haloperidol attenuates these REM-like hippocampal theta and gamma oscillations. Conversely, our findings also demonstrate that dopamine depletion diminishes REM sleep in normal mice and causes DAT-KO mice to enter a different novel awake state characterized by neural oscillations similar to those observed during SWS (increased cortical oscillations in the delta frequency range and a virtual absence of gamma oscillations), an apparent suppression of normal SWS, and a complete suppression of REM sleep. Finally, we showed that REM sleep can be recovered in these DDD mice by selective activation of the D2, but not the D1, dopamine receptor pathway. Altogether, these results demonstrate for the first time that dopamine plays a central role in regulating sleep–wake states and that this action is mediated via the D2 dopamine receptor pathway.
REM sleep is a brain state during unconsciousness in which the brain displays increased activation of cortical networks similar to that observed during waking and the body displays atonia (Steriade et al., 1993). The high-frequency brain activity observed during waking and REM sleep is modulated via the ascending reticular activating system (RAS), which arises from the brain stem and projects to the cortex. The RAS has two main branches. The first branch consists of cholinergic projections from the pedunculopontine and laterodorsal tegmental nuclei to the thalamus. The second branch projects to the basal forebrain and cortex and consists of serotonergic efferents from the dorsal and medial raphe nuclei, noradrenergic efferents from the locus ceruleus, histaminergic efferents from the tuberomammillary nucleus, and dopamine efferents from the ventral periaqeuductal gray matter. These efferents activate neurons in the basal forebrain, which in turn modulate the projections to the cortex (Saper et al., 2005). Both branches of the RAS converge on a population of cells in the sublateral dorsal nucleus [SLD; anatomically corresponding to the peri-locus ceruleus-α (peri-LCα) in cats] thought to be responsible for the generation of REM sleep. Indeed, activation of the SLD/peri-LCα via microinjection of glutamatergic agonists has been shown to generate a transient REM-like brain state accompanied by atonia (Boissard et al., 2002). The SLD/peri-LCα nuclei receive monoaminergic afferents, including dopamine afferents from the posterior hypothalamus. Interestingly, direct application of dopaminergic agonists to the caudal SLD/peri-LCα has been shown to generate an REM-like brain state that is not accompanied by atonia (Crochet and Sakai, 1999). Here, we show that mice with genetically and pharmacologically induced hyperdopaminergia demonstrate REM-like neural oscillations during awake behaving periods accompanied by an increase in hippocampal gamma oscillations. Moreover, we demonstrate that mice profoundly depleted of dopamine display an awake state characterized by the virtual suppression of hippocampal gamma oscillations and the absence of REM sleep. Together, this suggests that dopamine may modulate efferents from the SLD/peri-LCα responsible for coordinating the activation of thalamocortical networks during REM sleep. Thus, in the absence of dopamine, the cortex displays widely synchronized oscillations characteristic of thalamocortical network inhibition, whereas in the presence of excess dopamine, the cortex displays increased high-frequency oscillations. Indeed, the REM-like brain state observed in animals with dopamine agonists administered to the SLD/peri-LCα may result from activation of REM sleep relay tracks without the accompanying activation of upstream neurons responsible for the generation of muscle atonia.
Dopamine is critically involved in regulating neural processes responsible for complex movements and emotions (Carlsson, 1987). Consequently, altered central dopaminergic transmission has been implicated in several neurological and psychiatric disorders such as schizophrenia and Parkinson's disease. Dopaminergic dysfunction was first implicated in mediating psychosis when the therapeutic efficacy of classical antipsychotic agents (e.g., haloperidol) was found to be directly correlated with their affinity for the dopamine D2 receptor (Creese et al., 1976; Seeman et al., 1976). Furthermore, drugs that increase endogenous dopamine release (e.g., amphetamines) were found to induce psychosis in healthy individuals and exacerbate psychotic symptoms in schizophrenic patients (Snyder, 1972). Recent evidence also suggests that other pharmacological agents that induce psychosis in healthy individuals (e.g., PCP) demonstrate significant potency for the D2 dopamine receptor (Kapur and Seeman, 2002; Seeman et al., 2005).
Freud and Kraepelin, the founding fathers of modern psychiatry, proposed that psychosis resulted from the intrusion of the sleeping mind on the conscious mind (Freud, 1900; Heynick, 1993). Here, we demonstrate that mice with genetically or pharmacologically induced hyperdopaminergia display spectral changes in hippocampal theta oscillations after exposure to novel environments. Notably, these spectral changes result in increased neural similarity between waking and REM sleep. We also demonstrate that normal REM sleep can be suppressed in both normal and DAT-KO mice by diminishing dopaminergic tone. These results establish an important and previously unreported role of dopamine in regulating physiological REM sleep. Finally, we showed that REM sleep requires activation of the D2 dopamine receptor pathway, the very pathway implicated in mediating psychosis, and that the D2 dopamine receptor antagonist and antipsychotic agent haloperidol suppresses the novelty-induced, REM-like hippocampal neural oscillations observed in DAT-KO mice.
Hippocampal theta oscillations are driven by cholinergic input from the medial septum, which receives afferent projections from A10 dopaminergic neurons in the ventral tegmental area (Bjorklund and Lindvall, 1984). When injected in the medial septum or administered systemically, dopamine agonists increase theta oscillations (Miura et al., 1987; Kichigina, 2004). Moreover, systemic administration of dopamine antagonists decreases theta oscillations (Miura et al., 1987; Kichigina, 2004). Our results demonstrate that normal mice treated with amphetamine demonstrate a significant increase in hippocampal theta oscillations. Amphetamine induces a dopamine-dependent release of acetylcholine in the hippocampus (Nilsson et al., 1992). Thus, although amphetamine demonstrates efficacy for several monoamine transporters other than the DAT (Gainetdinov and Caron, 2003), the increased theta oscillations observed in the hippocampus of normal mice treated with amphetamine are likely attributable to the action of amphetamine at the DAT. This suggests that hippocampal theta oscillations may be directly correlated with dopaminergic tone under normal physiological conditions as well. REM is characterized by prominent hippocampal theta oscillations (Vanderwolf, 1969; Timo-Iaria et al., 1970; Cantero et al., 2003) that are highly correlated with dreaming (Aserinsky and Kleitman, 1953; Dement and Kleitman, 1957a, b; Jouvet et al., 1959). Furthermore, recent evidence suggests that REM is also characterized by an increase in mesolimbic dopamine release (Lena et al., 2005). This is consistent with our results, which demonstrate that the pathological REM-like hippocampal theta oscillations are modulated by antipsychotic and psychotomimetic agents that alter dopaminergic transmission. Moreover, mesolimbic forebrain regions that receive dopaminergic projections are not only selectively activated during periods of psychosis (Epstein et al., 1999), but they are also active during periods of REM sleep in healthy individuals as well (Hobson et al., 1998). Although evidence suggests that REM and dreaming are dissociable to some extent (Foulkes, 1962), dopamine also plays a role in dreaming (Hartmann, 1976; Solms, 2000; Gottesmann, 2005). Forebrain lesions involving dopaminergic ventromedial frontal fibers abolish dreaming (Solms, 2000). Indeed, transection of these fibers was the mainstay of treatment for patients with intractable psychosis before psychopharmacological therapy became available. In the majority of cases, patients subjected to this procedure reported cessation of dreaming (Solms, 2000). Additionally, along with its psychotomimetic properties, l-DOPA, a dopamine precursor, enhances dreaming in Parkinsonian patients (Jenkins and Groh, 1970; Sharf et al., 1978). Thus, we propose that dopamine plays a key role in the genesis of REM sleep, dreaming, and psychosis.
Our findings demonstrate that mice profoundly depleted of dopamine display a complete suppression of REM sleep and that treatment with the D2 dopamine receptor agonist Quinpirole recovers REM sleep in these animals. Importantly, the REM onset time in these animals is well beyond the time necessary for Quinpirole to activate D2 dopamine receptors (Cai et al., 2000). This demonstrates that activation of the D2 receptor pathway is necessary but not sufficient to generate REM sleep. Similarly, we demonstrate that hyperdopaminergia is necessary but not sufficient to generate REM-like neural oscillations in awake DAT-KO mice. Thus, we propose that REM sleep neural pathways are only activated when D2 dopamine receptors are activated on a background of reduced mesolimbic glutamatergic and serotonergic tone. Importantly, this is consistent with neurotransmitters levels observed during REM sleep (Trulson and Jacobs, 1979; Park et al., 1999; Lena et al., 2005), and during the waking period of DAT-KO mice immediately after exposure to novelty (Gainetdinov et al., 1999, 2001). This also predicts that psychosis may be generated by pharmacological agents that activate the D2 dopamine receptor while altering specific aspects of glutamatergic or serotonergic transmission (Kapur and Seeman, 2002; Seeman et al., 2005).
Parkinson's disease results from progressive destruction of dopaminergic neurons in the substantia nigra pars compacta. The disease typically becomes clinically apparent after destruction of 60–70% of dopaminergic neurons and is characterized by akinesia, rigidity, resting tremors, and gait disturbances (Fahn, 2003). Parkinsonian patients also endure severe sleep disturbances, such as excessive daytime sleepiness (Adler, 2005). Additionally, REM sleep behavior disorder occurs in 15–47% of individuals with Parkinson's disease (Gagnon et al., 2002) and, in many cases, predates motor symptoms classically associated with Parkinson's disease (Abbott, 2005). Here, we show that WT mice treated with αMT experience disturbance of REM sleep without changes in gross behavior (Sotnikova et al., 2005). Thus, our findings directly demonstrate that partial dopamine depletion causes disturbances of REM sleep without affecting motor functions. This provides a possible explanation as to why sleep disturbances often occur in the early stages of Parkinson's before motor symptoms become apparent.
Conclusion
Overall, the present observations demonstrate the central role of dopamine in regulating sleep–wake states. Moreover, they provide new hope for the discovery of novel antipsychotic agents and the development of objective diagnostic technologies for the early detection of Parkinson's disease based on electrophysiological analysis of brain activity obtained during waking, SWS, and REM sleep.
Footnotes
This work was supported by the Duke Medical Scientist Training Program, the Wakeman Foundation, the Ruth K. Broad Foundation, and the United Negro College Fund/Merck to K.D.; by a Pew Latin American Fellowship to S.R.; by the National Alliance for Research on Schizophrenia and Depression (NARSAD) and the National Institutes of Health (NIH) to M.G.C.; by the Michael J. Fox Foundation for Parkinson's Research to T.D.S., R.R.G. and M.G.C.; and by NIH, NARSAD, and the Hereditary Disease Foundation to M.A.L.N. We especially thank Freeman Hrabowski, Robert and Jane Meyerhoff, and the Meyerhoff Scholarship Program. We thank G. Lehew and Jim Meloy for technical assistance; L. Oliveira, T. Jones, and G. Wood for miscellaneous support; and S. Halkiotis for proofreading this manuscript.
References
- Abbott A. Neuroscience: while you were sleeping. Nature. 2005;437:1220–1222. doi: 10.1038/4371220a. [DOI] [PubMed] [Google Scholar]
- Adler CH. Nonmotor complications in Parkinson's disease. Mov Disord. 2005;20:S23–S29. doi: 10.1002/mds.20460. [DOI] [PubMed] [Google Scholar]
- Aserinsky E, Kleitman N. Regularly occurring periods of eye motility, and concomitant phenomena, during sleep. Science. 1953;118:273–274. doi: 10.1126/science.118.3062.273. [DOI] [PubMed] [Google Scholar]
- Aston-Jones G, Bloom FE. Activity of norepinephrine-containing locus coeruleus neurons in behaving rats anticipates fluctuations in the sleep-waking cycle. J Neurosci. 1981;1:876–886. doi: 10.1523/JNEUROSCI.01-08-00876.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorklund A, Lindvall O. Dopamine-containing systems in the CNS. In: Hokfelt T, editor. Handbook of chemical neuroanatomy. Amsterdam: Elsevier; 1984. pp. 52–122. [Google Scholar]
- Boissard R, Gervasoni D, Schmidt MH, Barbagli B, Fort P, Luppi PH. The rat ponto-medullary network responsible for paradoxical sleep onset and maintenance: a combined microinjection and functional neuroanatomical study. Eur J Neurosci. 2002;16:1959–1973. doi: 10.1046/j.1460-9568.2002.02257.x. [DOI] [PubMed] [Google Scholar]
- Cai G, Zhen X, Uryu K, Friedman E. Activation of extracellular signal-regulated protein kinases is associated with a sensitized locomotor response to D2 dopamine receptor stimulation in unilateral 6-hydroxydopamine-lesioned rats. J Neurosci. 2000;20:1849–1857. doi: 10.1523/JNEUROSCI.20-05-01849.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantero JL, Atienza M, Stickgold R, Kahana MJ, Madsen JR, Kocsis B. Sleep-dependent theta oscillations in the human hippocampus and neocortex. J Neurosci. 2003;23:10897–10903. doi: 10.1523/JNEUROSCI.23-34-10897.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsson A. Perspectives on the discovery of central monoaminergic neurotransmission. Annu Rev Neurosci. 1987;10:19–40. doi: 10.1146/annurev.ne.10.030187.000315. [DOI] [PubMed] [Google Scholar]
- Costa RM, Lin SC, Sotnikova TD, Gainetdinov RR, Cyr M, Caron MG, Nicolelis MAL. Rapid alterations in corticostriatal ensemble coordination during acute dopamine-dependent motor dysfunction. Neuron. 2006 doi: 10.1016/j.neuron.2006.07.030. in press. [DOI] [PubMed] [Google Scholar]
- Creese I, Burt DR, Snyder SH. Dopamine receptor binding predicts clinical and pharmacological potencies of antischizophrenic drugs. Science. 1976;192:481–483. doi: 10.1126/science.3854. [DOI] [PubMed] [Google Scholar]
- Crochet S, Sakai K. Effects of microdialysis application of monoamines on the EEG and behavioural states in the cat mesopontine tegmentum. Eur J Neurosci. 1999;10:3738–3752. doi: 10.1046/j.1460-9568.1999.00760.x. [DOI] [PubMed] [Google Scholar]
- Dement WC, Kleitman N. The relation of eye movements during sleep to dream activity: an objective method for the study of dreaming. J Exp Psychol. 1957a;53:339–346. doi: 10.1037/h0048189. [DOI] [PubMed] [Google Scholar]
- Dement W, Kleitman N. Cyclic variations in EEG during sleep and their relation to eye movements, body motility, and dreaming. Electroencephalogr Clin Neurophysiol Suppl. 1957b;9:673–690. doi: 10.1016/0013-4694(57)90088-3. [DOI] [PubMed] [Google Scholar]
- Epstein J, Stern E, Silbersweig D. Mesolimbic activity associated with psychosis in schizophrenia. Symptom-specific PET studies. Ann NY Acad Sci. 1999;877:562–574. doi: 10.1111/j.1749-6632.1999.tb09289.x. [DOI] [PubMed] [Google Scholar]
- Espana RA, Scammell TE. Sleep neurobiology for the clinician. Sleep. 2004;27:811–820. [PubMed] [Google Scholar]
- Fahn S. Description of Parkinson's disease as a clinical syndrome. Ann NY Acad Sci. 2003;991:1–14. doi: 10.1111/j.1749-6632.2003.tb07458.x. [DOI] [PubMed] [Google Scholar]
- Foulkes WD. Dream reports from different stages of sleep. J Abnorm Soc Psychol. 1962;65:14–25. doi: 10.1037/h0040431. [DOI] [PubMed] [Google Scholar]
- Freud S. London: Encyclopaedia Britannica; 1900. The interpretation of dreams, 1952 edition. [Google Scholar]
- Gagnon JF, Bedard MA, Fantini ML, Petit D, Panisset M, Rompre S, Carrier J, Montplaisir J. REM sleep behavior disorder and REM sleep without atonia in Parkinson's disease. Neurology. 2002;59:585–589. doi: 10.1212/wnl.59.4.585. [DOI] [PubMed] [Google Scholar]
- Gainetdinov RR, Caron MG. Monoamine transporters: from genes to behavior. Annu Rev Pharmacol Toxicol. 2003;43:261–284. doi: 10.1146/annurev.pharmtox.43.050802.112309. [DOI] [PubMed] [Google Scholar]
- Gainetdinov RR, Wetsel WC, Jones SR, Levin ED, Jaber M, Caron MG. Role of serotonin in the paradoxical calming effect of psychostimulants on hyperactivity. Science. 1999;283:397–401. doi: 10.1126/science.283.5400.397. [DOI] [PubMed] [Google Scholar]
- Gainetdinov RR, Mohn AR, Bohn LM, Caron MG. Glutamatergic modulation of hyperactivity in mice lacking the dopamine transporter. Proc Natl Acad Sci USA. 2001;98:11047–11054. doi: 10.1073/pnas.191353298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gervasoni D, Lin SC, Ribeiro S, Soares ES, Pantoja P, Nicolelis M. Global forebrain dynamics predict rat behavioral states and their transitions. J Neurosci. 2004;24:11137–11147. doi: 10.1523/JNEUROSCI.3524-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottesmann C. Dreaming and schizophrenia: a common neurobiological background. Sleep Biol Rhythms. 2005;3:64–74. [Google Scholar]
- Greenwood TA, Schork NJ, Eskin E, Kelsoe JR. Identification of additional variants within the human dopamine transporter gene provides further evidence for an association with bipolar disorder in two independent samples. Mol Psychiatry. 2006;11:125–133. doi: 10.1038/sj.mp.4001764. 115. [DOI] [PubMed] [Google Scholar]
- Guix T, Hurd YL, Ungerstedt U. Amphetamine enhances extracellular concentrations of dopamine and acetylcholine in dorsolateral striatum and nucleus accumbens of freely moving rats. Neurosci Lett. 1992;138:137–140. doi: 10.1016/0304-3940(92)90490-x. [DOI] [PubMed] [Google Scholar]
- Hartmann E. Schizophrenia: a theory. Psychopharmacol (Berl) 1976;49:1–15. doi: 10.1007/BF00427464. [DOI] [PubMed] [Google Scholar]
- Heynick F. Toronto: Wiley; 1993. Language and its dream disturbance. [Google Scholar]
- Hobson JA, Pace-Schott EF. The cognitive neuroscience of sleep: neuronal systems, consciousness and learning. Nat Rev Neurosci. 2002;3:679–693. doi: 10.1038/nrn915. [DOI] [PubMed] [Google Scholar]
- Hobson JA, Datta S, Calvo JM, Quattrochi J. Acetylcholine as a brain state modulator—triggering and long-term regulation of REM sleep. In: Cuello AC, editor. Cholinergic function and dysfunction. Amsterdam: Elsevier; 1993. pp. 389–404. [DOI] [PubMed] [Google Scholar]
- Hobson JA, Stickgold R, Pace-Schott EF. The neuropsychology of REM sleep dreaming. NeuroReport. 1998;9:R1–R14. doi: 10.1097/00001756-199802160-00033. [DOI] [PubMed] [Google Scholar]
- Jenkins RB, Groh RH. Mental symptoms in Parkinsonian patients treated with l-dopa. Lancet. 1970;2:177–179. doi: 10.1016/s0140-6736(70)92535-3. [DOI] [PubMed] [Google Scholar]
- John J, Wu MF, Boehmer LN, Siegel JM. Cataplexy-active neurons in the hypothalamus: implications for the role of histamine in sleep and waking behavior. Neuron. 2004;42:619–634. doi: 10.1016/s0896-6273(04)00247-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SR, Gainetdinov RR, Wightman RM, Caron MG. Mechanisms of amphetamine action revealed in mice lacking the dopamine transporter. J Neurosci. 1998;18:1979–1986. doi: 10.1523/JNEUROSCI.18-06-01979.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jouvet M, Michel F, Courjon J. On a stage of rapid cerebral electrical activity in the course of physiological sleep. C R Seances Soc Biol Fil. 1959;153:1024–1028. [PubMed] [Google Scholar]
- Kapur S, Seeman P. NMDA receptor antagonists ketamine and PCP have direct effects on the dopamine D(2) and serotonin 5-HT(2)receptors-implications for models of schizophrenia. Mol Psychiatry. 2002;7:837–844. doi: 10.1038/sj.mp.4001093. [DOI] [PubMed] [Google Scholar]
- Kaur S, Panchal M, Faisal M, Madan V, Nangia P, et al. Long term blocking of GABA-A receptor in locus coeruleus by bilateral microinfusion of picrotoxin reduced rapid eye movement sleep and increased brain Na-K ATPase activity in freely moving normally behaving rats. Behav Brain Res. 151:185–190. doi: 10.1016/j.bbr.2003.08.011. [DOI] [PubMed] [Google Scholar]
- Kichigina VF. Dopaminergic regulation of theta activity of septohippocampal neuron in the awake rabbit. Zh Vyssh Nerv Deiat Im I P Pavlova. 2004;54:210–215. [PubMed] [Google Scholar]
- Lena I, Parrot S, Deschaux O, Muffat-Joly S, Sauvinet V, Renaud B, Suaud-Chagny M, Gottesmann C. Variations in extracellular levels of dopamine, noradrenaline, glutamate, and aspartate across the sleep-wake cycle in the medial prefrontal cortex and nucleus accumbens of freely moving rats. J Neurosci Res. 2005;81:891–899. doi: 10.1002/jnr.20602. [DOI] [PubMed] [Google Scholar]
- Lin L, Faraco J, Li R, Kadotani H, Rogers W, Lin X, Qiu X, de Jong PJ, Nishino S, Mignot E. The sleep disorder canine narcolepsy is caused by a mutation in the hypocretin (orexin) receptor 2 gene. Cell. 1999;98:365–376. doi: 10.1016/s0092-8674(00)81965-0. [DOI] [PubMed] [Google Scholar]
- Maggini C, Guazzelli M, Pieri M, Lattanzi L, Ciapparelli A, Massimetti G, Rossi G. REM latency in psychiatric disorders: polygraphic study on major depression, bipolar disorder-manic, and schizophrenic disorder. New Trends Exp Clin Psychiatry. 1986;2:93–101. [Google Scholar]
- Maloney KJ, Mainville L, Jones BE. c-Fos expression in dopaminergic and GABAergic neurons of the ventral mesencephalic tegmentum after paradoxical sleep deprivation and recovery. Eur J Neurosci. 2002;15:774–778. doi: 10.1046/j.1460-9568.2002.01907.x. [DOI] [PubMed] [Google Scholar]
- Mazei-Robison MS, Couch RS, Shelton RC, Stein MA, Blakely RD. Sequence variation in the human dopamine transporter gene in children with attention deficit hyperactivity disorder. Neuropharmacology. 2005;49:724–736. doi: 10.1016/j.neuropharm.2005.08.003. [DOI] [PubMed] [Google Scholar]
- Miller JD, Farber J, Gatz P, Roffwarg H, German DC. Activity of mesencephalic dopamine and non-dopamine neurons across stages of sleep and walking in the rat. Brain Res. 1983;273:133–141. doi: 10.1016/0006-8993(83)91101-0. [DOI] [PubMed] [Google Scholar]
- Miura Y, Ito T, Kadokawa T. Effects of intraseptally injected dopamine and noradrenaline on hippocampal synchronized theta wave activity in rats. Jpn J Pharmacol. 1987;44:471–479. doi: 10.1254/jjp.44.471. [DOI] [PubMed] [Google Scholar]
- Nilsson OG, Leanza G, Bjorklund A. Acetylcholine release in the hippocampus: regulation by monoaminergic afferents as assessed by in vivo microdialysis. Brain Res. 1992;584:132–140. doi: 10.1016/0006-8993(92)90886-e. [DOI] [PubMed] [Google Scholar]
- O'Brien LM, Ivanenko A, Crabtree VM, Holbrook CR, Bruner JL, Klaus CJ, Gozal D. Sleep disturbances in children with attention deficit hyperactivity disorder. Pediatr Res. 2003;54:237–243. doi: 10.1203/01.PDR.0000072333.11711.9A. [DOI] [PubMed] [Google Scholar]
- Ouyang M, Hellman K, Abel T, Thomas SA. Adrenergic signaling plays a critical role in the maintenance of waking and in the regulation of REM sleep. J Neurophysiol. 2004;92:2071–2082. doi: 10.1152/jn.00226.2004. [DOI] [PubMed] [Google Scholar]
- Park SP, Lopez-Rodriguez F, Wilson CL, Maidment N, Matsumoto Y, Engel JJ. In vivo microdialysis measures of extracellular serotonin in the rat hippocampus during sleep-wakefulness. Brain Res. 1999;833:291–296. doi: 10.1016/s0006-8993(99)01511-5. [DOI] [PubMed] [Google Scholar]
- Saper CB, Scammell TE, Lu J. Hypothalamic regulation of sleep and circadian rhythms. Nature. 2005;437:1257–1263. doi: 10.1038/nature04284. [DOI] [PubMed] [Google Scholar]
- Seeman P, Lee T, Chau-Wong M, Wong K. Antipsychotic drug doses and neuroleptic/dopamine receptors. Nature. 1976;261:717–719. doi: 10.1038/261717a0. [DOI] [PubMed] [Google Scholar]
- Seeman P, Ko F, Tallerico T. Dopamine receptor contribution to the action of PCP, LSD and ketamine psychotomimetics. Mol Psychiatry. 2005;10:877–883. doi: 10.1038/sj.mp.4001682. [DOI] [PubMed] [Google Scholar]
- Sharf B, Moskovitz C, Lupton MD, Klawans HL. Dream phenomena induced by chronic levodopa therapy. J Neural Transm. 1978;43:143–151. doi: 10.1007/BF01579073. [DOI] [PubMed] [Google Scholar]
- Snyder SH. Catecholamines in the brain as mediators of amphetamine psychosis. Arch Gen Psychiatry. 1972;27:169–179. doi: 10.1001/archpsyc.1972.01750260021004. [DOI] [PubMed] [Google Scholar]
- Solms M. Dreaming and REM sleep are controlled by different brain mechanisms. Behav Brain Sci. 2000;23:843–850. doi: 10.1017/s0140525x00003988. [DOI] [PubMed] [Google Scholar]
- Sotnikova TD, Beaulieu JM, Barak LS, Wetsel WC, Caron MG, Gainetdinov RR. Dopamine-independent locomotor actions of amphetamines in a novel acute mouse model of Parkinson disease. PLoS Biol. 2005;3:e271. doi: 10.1371/journal.pbio.0030271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spielewoy C, Roubert C, Hamon M, Nosten-Bertrand M, Betancur C, Giros B. Behavioural disturbances associated with hyperdopaminergia in dopamine-transporter knockout mice. Behav Pharmacol. 2000;11:279–290. doi: 10.1097/00008877-200006000-00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steriade M, McCormick DA, Sejnowski TJ. Thalamocortical oscillations in the sleeping and aroused brain. Science. 1993;262:679–685. doi: 10.1126/science.8235588. [DOI] [PubMed] [Google Scholar]
- Timo-Iaria C, Negrao N, Schmidek WR, Hoshino K, Lobato de Menezes CE, Leme da Rocha T. Phases and states of sleep in the rat. Physiol Behav. 1970;5:1057–1062. doi: 10.1016/0031-9384(70)90162-9. [DOI] [PubMed] [Google Scholar]
- Trulson ME, Jacobs BL. Raphe unit activity in freely moving cats: correlation with level of behavioral arousal. Brain Res. 1979;163:135–150. doi: 10.1016/0006-8993(79)90157-4. [DOI] [PubMed] [Google Scholar]
- Vanderwolf CH. Hippocampal electrical activity and voluntary movement in the rat. Electroencephalogr Clin Neurophysiol. 1969;26:407–418. doi: 10.1016/0013-4694(69)90092-3. [DOI] [PubMed] [Google Scholar]
- Vanderwolf CH, Baker GB. Evidence that serotonin mediates non-cholinergic neocortical low voltage fast activity, non-cholinergic hippocampal rhythmical slow activity and contributes to intelligent behavior. Brain Res. 1986;374:342–356. doi: 10.1016/0006-8993(86)90428-2. [DOI] [PubMed] [Google Scholar]
- Winson J. Patterns of hippocampal theta rhythm in the freely moving rat. Electroencephalogr Clin Neurophysiol. 1974;36:291–301. doi: 10.1016/0013-4694(74)90171-0. [DOI] [PubMed] [Google Scholar]
- Wisor JP, Nishino S, Sora I, Uhl GH, Mignot E, Edgar DM. Dopaminergic role in stimulant-induced wakefulness. J Neurosci. 2001;21:1787–1794. doi: 10.1523/JNEUROSCI.21-05-01787.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]