

Abstract
We examined the effects of the chemokine fractalkine (CX3CL1) on EPSCs evoked by electrical stimulation of Schaffer collaterals in patch-clamped CA1 pyramidal neurons from rat hippocampal slices. Acute application of CX3CL1 caused a sustained reduction of EPSC amplitude, with partial recovery after washout. CX3CL1-induced EPSC depression is postsynaptic in nature, because paired-pulse ratio was maintained, amplitude distribution of spontaneous excitatory postsynaptic currents shifted to lower values, and whole-cell current responses to AMPA were reversibly inhibited. EPSC depression by CX3CL1 is mediated by CX3CL1 receptor (CX3CR1), because CX3CL1 was unable to influence EPSC amplitude in CA1 pyramidal neurons from CX3CR1 knock-out mice. CX3CL1-induced depression of both EPSC and AMPA current was not observed in the absence of afferent fiber stimulation or AMPA receptor activation, respectively, indicating the requirement of sustained receptor activity for its development. Findings obtained from hippocampal slices, cultured hippocampal neurons, and transfected human embryonic kidney cells indicate that a Ca2+-, cAMP-, and phosphatase-dependent process is likely to modulate CX3CL1 effects because of the following: (1) CX3CL1-induced depression was antagonized by intracellular BAPTA, 8Br-cAMP, phosphatase inhibitors, and pertussis toxin (PTX); (2) CX3CL1 inhibited forskolin-induced cAMP formation sensitive to PTX; and (3) CX3CL1 inhibited forskolin-induced Ser845 GluR1 phosphorylation, which was sensitive to PTX and dependent on Ca2+ and phosphatase activity. Together, these findings indicate that CX3CL1 negatively modulates AMPA receptor function at active glutamatergic synapses through cell-signaling pathways by influencing the balance between kinase and phosphatase activity.
Keywords: AMPA receptors, hippocampal slices, GluR1, phosphorylation, chemokines, EPSC depression, adenylate cyclase
Introduction
The modulation of AMPA-type glutamate receptors (GluRs) is a key event involved in many complex forms of synaptic plasticity, including long-term depression (LTD) and potentiation (Kameyama et al., 1998; Lee et al., 1998; Kullmann et al., 2000; Song and Huganir, 2002). In the hippocampus, several endogenous factors have been demonstrated to modify synaptic plasticity by modulating the level of AMPA-type glutamate receptor expression or phosphorylation at postsynaptic sites (Carroll et al., 1999; Lin et al., 2003; Wu et al., 2004). Many of these forms of plasticity are activity dependent, indicating that specific conditions or previous synaptic history may alter their modulation of synaptic function. Nevertheless, it is generally assumed that LTD can be achieved by the activation of intracellular pathways that leads to AMPA receptor dephosphorylation (Mulkey et al., 1993, 1994; Lee et al., 1998), whereas long-term synaptic potentiation (LTP) is driven by pathways that induce AMPA receptor phosphorylation (Malenka et al., 1989; Banke et al., 2000). It is believed that second messengers may regulate synaptic plasticity acting on the balance between kinase and phosphatase activity, and that the phosphorylation of GluR1 subunit plays a pivotal role on the mechanisms influencing this balance (Lee et al., 1998, 2000; Tavalin et al., 2002).
Chemokines are small chemotactic molecules widely expressed throughout the CNS, where they contribute to the regulation of cellular communication in the adult brain (Tran and Miller, 2003) and of cell migration and differentiation during development (Lu et al., 2002) and may exert neuroprotective effects in different neurotoxic experimental conditions (Araujo and Cotman, 1993; Meucci et al., 1998; Robinson et al., 1998; Bruno et al., 2000; Limatola et al., 2000; Hatori et al., 2002; Deiva et al., 2004; Krathwohl and Kaiser, 2004; Limatola et al., 2005). Several examples illustrating the neuromodulatory potential of chemokines and chemokine receptors in the CNS have been reported. Specifically, chemokines may alter synaptic transmitter release (Giovannelli et al., 1998; Meucci et al., 1998; Limatola et al., 2000; Ragozzino et al., 2002), modulate the functional properties of ionic channels (Meucci et al., 1998; Lax et al., 2002; Oh et al., 2002) and promote the release of glutamate from astrocytes (Bezzi et al., 2001). Among the chemokines, fractalkine (CX3CL1) and its receptor CX3CR1 are highly expressed in hippocampal neurons (Meucci et al., 1998, 2000; Hughes et al., 2002; Tarozzo et al., 2003; Limatola et al., 2005), where they induce a pertussis toxin (PTX)-sensitive increase of intracellular Ca2+ (Meucci et al., 2000) and reduce both spontaneous glutamate release (Meucci et al., 1998) and postsynaptic glutamatergic currents (Limatola et al., 2005). We wondered whether, in hippocampal tissue, CX3CL1 could function as an endogenous modulator of synaptic plasticity. Here, we show that in the rat hippocampus, CX3CL1 negatively modulates EPSCs at Schaffer collateral-CA1 synapses acting at postsynaptic sites of active synapses through mechanisms involving CX3CR1, reduces postsynaptic AMPA-evoked currents, and triggers signaling pathways that favor the reduction of Ser845 GluR1 phosphorylation.
Materials and Methods
Slice preparation.
Hippocampal slices were prepared from 14- to 22-d-old Sprague Dawley rats or homozygous wild-type (wt), knock-out (KO), and heterozygous littermates derived from breeding between heterozygous CX3CR1-KO mice on the C57BL/6 background (Haskell et al., 2001). All experiments followed international guidelines on the ethical use of animals from the European Communities Council Directive of 24 November 1986 (86/609/EEC). Rats were decapitated under halothane anesthesia, and whole brains were rapidly removed and incubated in chilled artificial CSF (ACSF) for 15 min. Transverse hippocampal slices (250 μm) were cut at 4°C, using a Vibratome (DSK, Dosaka EM, Kyoto, Japan). Before use, slices were maintained for at least 1 h at room temperature (22–25°C) in oxygenated (95% O2-5% CO2) ACSF, containing the following (in mm): 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 26 NaHCO3, 2 CaCl2, 1 MgCl2, and 10 glucose, pH 7.35. All recordings were performed at room temperature on slices submerged in ACSF in the recording chamber. The ACSF was perfused at a rate of ∼1 ml/min.
Patch-clamp recordings.
Neurons were visualized at 640× with Nomarski optics with an upright Zeiss (Thornwood, NY) Axioscope microscope. Patch-clamp recordings were obtained using glass electrodes (3–4 MΩ) filled with the following (in mm): 140 CsCl, 2 MgCl2, 10 HEPES, 2 MgATP, 0.2–20 BAPTA (5 mm where not otherwise indicated; pH 7.3, with CsOH). Bicuculline methyl-chloride (10 μm), unless otherwise indicated, was routinely added to the bath solution to block GABAA receptors, and recordings started at least 10 min after bicuculline addition to allow complete GABAergic block. Neurons were clamped at −70 mV, unless otherwise indicated. Membrane currents, recorded with a patch-clamp amplifier (Axopatch 200A; Molecular Devices, Foster city, CA), were filtered at 2 kHz, digitized (10 kHz), and acquired with LTP114 software package (courtesy of W. R. W. Anderson, Bristol University, UK) or Clampex8 software (Molecula Devices). Data were analyzed off-line either with LTP114 and/or Clampfit 9 (Molecular Devices).
Evoked synaptic currents.
EPSCs were evoked in CA1 pyramidal neurons by electrical stimulation with theta glass tubes pulled to a final tip diameter of 10–20 μm and filled with external solution. Stimulating electrodes were placed in the stratum radiatum to activate the Schaffer collateral pathway projecting to CA1. EPSCs were evoked by stimulating at 5–50 V for 150–300 μs every 4 s in the presence of bicuculline. The stimulus was adjusted to evoke EPSCs in the range 15–50 pA. Recorded EPSCs were routinely averaged over 1 min (n = 15) to analyze the time course of EPSC amplitude. Stable EPSCs were monitored for at least 10 min before applying CX3CL1 and successively monitored during slice exposure to CX3CL1. Closely spaced consecutive stimuli (50 ms interval) were used, and paired-pulse ratio was calculated as the ratio between the amplitude of the responses evoked by the second stimulus (P2) over the first (P1; P2/P1). The stability of the patch was checked by repetitively monitoring the input and series resistance during the experiment, and recordings were discarded when any of these parameters changed by >10%.
Drugs and application procedures.
CX3CL1 (rat, Calbiochem, La Jolla, CA; human, Peprotech, Rocky Hill, NJ), bicuculline (Tocris Cookson, Avonmouth, UK), tetrodotoxin (TTX), d-2-amino-5-phosphonopentanoic (d-APV), forskolin (FSK; stock solution 12 mm in DMSO), and 8Br-cAMP (stock solution 100 mm in H2O) were usually applied to the slice by bath perfusion. Because no substantial differences between rat and human CX3CL1 were detected, all experiments here reported were performed with rat CX3CL1, except for mouse preparations, where human CX3CL1 was used. Unless otherwise indicated, the CX3CL1 concentration routinely used was 2 nm. To activate postsynaptic glutamate receptors, AMPA (Tocris Cookson) or glutamate were delivered together with cyclothiazide (50 μm; Tocris Cookson) by pressure application (5–15 psi, 10–100 ms; Picospritzer II, General Valve, Fairfield, NJ) from glass micropipettes positioned above the slice over the soma of the recorded neuron. In some experiments, AMPA applications were delivered in a Ca2+-free solution (0 Ca2+/1 mm EGTA) while superfusing slices with standard medium. When necessary, calyculin A (Calbiochem), cyclosporin A (Alexis Biochemicals, San Diego, CA), okadaic acid, thapsigargin, 8Br-cAMP, ATPγS, and PTX were added to the patch pipette solution. PTX, stocked at 200 μg/ml, was activated in 10 mm dithiothreitol at 37°C for 15 min, diluted at 10 μg/ml in the pipette solution containing 20 mm nicotinic acid adenine dinucleotide, and further diluted 1:10 to a final concentration of 1 μg/ml after 10 min (Xie and Lewis, 1997). In all experiments based on intracellular dialysis, we waited for 20–30 min to attain intracellular drug diffusion. Control experiments showed that EtOH and DMSO, at their final concentrations in the intracellular solution (never higher than 1:1000 and 1:10000, respectively), or dithiothreitol plus nicotinic acid adenine dinucleotide did not alter the pattern of glutamatergic currents. Unless otherwise indicated, all drugs were purchased from Sigma Italia (Milan, Italy).
Electrophysiological data analysis.
Data are presented as mean ± SEM; statistical significance was assessed with paired t test, unless otherwise specified. Dose–response curves were fitted with a Hill equation (Origin software; Microcal Software, Northampton, MA). Spontaneous postsynaptic currents were acquired by pClamp software into separate files (5 min duration) and pooled during analysis by Clampfit 9.2 software. Amplitude distribution of EPSCs was fitted by a Gaussian (Origin software).
Slice preparation for biochemical experiments.
Hippocampal slices, prepared as described above, were collected in ACSF, transferred to a submersion-type holder chamber, and incubated at room temperature in ACSF bubbled with 95% O2 and 5% CO2 for 1 h. The slices were then transferred to a 24-well plate with mesh inserts (Corning, Corning, NY); each well was filled with ACSF. The multiwell plates were supplied with humidified oxygenated atmosphere (95% O2, 5% CO2) and placed in a 30°C water bath. The slices were left to equilibrate for 1 h before the experiment. The slices were gently transferred by using the mesh inserts to a well containing ACSF (control), FSK (1 μm), or 12-O-tetradecanoylphorbol-13-acetate (TPA; 1 μm) in the presence or absence of CX3CL1 (100 nm) for 15 min. Reactions were blocked in ice-cold ACSF, and slices were quickly frozen on dry ice and stored at −80°C. Homogenates of hippocampal slices were prepared by sonicating the slices in resuspension buffer (RB) consisting of the following (in mm): 10 Na phosphate, pH 7.0, 100 NaCl, 10 Na pyrophosphate, 50 NaF, 1 Na orthovanadate, 5 EDTA, 5 EGTA, 1 μm okadaic acid, and 10 U/ml aprotinin for 30 s. The homogenates were centrifuged at 12,000 × g for 5 min, and the crude membrane pellets were resuspended in RB buffer. Protein concentration was determined by BCA protein assay (Pierce, Rockford, IL); ∼20 μg of proteins per lane were loaded onto a SDS-polyacrylamide gel and analyzed by Western blotting with antibodies specific for phospho-GluR1 Ser845 or Ser831 (Upstate Biotechnology, Lake Placid, NY), and actin (Sigma). Immunoreactivity was detected by chemiluminescence. Specific bands on chemiluminescence films were quantified by densitometry with Sigma Gel Software (Jandel Scientific, Erkrath, Germany). To test the effect of phosphatase inhibitors, once transferred to a 24-well plate with mesh inserts in a humidified oxygenated chamber (95% O2, 5% CO2, 30°C), the slices were left to equilibrate with ACSF alone or together with calyculin A (200 nm) for an additional 90 minutes before proceeding with drug treatment (FSK with/without CX3CL1).
Cell transfection.
Human embryonic kidney 293 (HEK) cells were routinely grown in DMEM plus 10% fetal bovine serum. Cells were plated on poly-l-lysine-coated 35 mm dishes and transiently transfected 24 h later with cDNA encoding human GluR1 (flip-variant; ATCC, Manassas, VA) and human CX3CR1 (Limatola et al., 2005) using either a standard calcium-phosphate procedure or Lipofectamine 2000 (Invitrogen, Carlsbad, CA). Routinely, cells were used for electrophysiological experiments 48 h after transfection.
Hippocampal neuronal cultures.
Hippocampal neuronal cultures were prepared from 2-d-old [postnatal day 2 (P2)] Wistar rats. Briefly, after careful dissection from diencephalic structures, the meninges were removed and tissues were chopped and digested in 0.25% trypsin for 15 min at 37°C. Cells were dissociated and plated at a density of 4.5 × 105 in poly-l-lysine-coated plates in MEM, with Earl's salts and GLUTAMAX containing 10% dialysed and heat-inactivated fetal bovine serum, 100 μg/ml gentamycin, and 25 mm KCl, and maintained at 37°C in 5% CO2. After 24 h, cytosine-β-d-arabinofuranoside was added at a final concentration of 10 μm to prevent glial cell proliferation. Cells were used for experiments after 7–8 d.
Cell stimulation and Western blot analysis.
HEK cells were serum starved for 16 h, incubated in Locke's buffer (in mm: 154 NaCl, 5.6 KCl, 3.6 NaHCO3, 2.3 CaCl2, 1 MgCl2, 5.6 glucose, buffered with 5 HEPES, pH 7.4) for 2 h and stimulated for 30 min with FSK (5.2 μm) alone or together with CX3CL1 (100 nm). Cultured hippocampal neurons were incubated for 2 h in Locke's buffer and stimulated for 5 min with FSK (5.2 μm) alone or together with CX3CL1 (100 nm). When necessary, cells were pretreated with PTX (1 μg/ml; 2 h), BAPTA/AM (5 μm; 1 h), or cyclosporin A (1 μm; 1 h). Cells treated with BAPTA were rinsed twice in Ca2+-free Locke's buffer with 2 mm EGTA and 3 mm MgCl2, once in Ca2+-free Locke's buffer containing 3 mm MgCl2, and then stimulated in the last buffer containing 5 μm BAPTA/AM. Corresponding cell lysates were run on SDS-polyacrylamide gel and analyzed for GluR1 Ser845 phosphorylation as above.
Measurement of cAMP concentration.
Eight-day-old cultured hippocampal neurons were starved for 2 h in Locke's buffer, incubated for 30 min with IBMX (0.5 mm), and stimulated with FSK (5.2 μm) in the presence or the absence of CX3CL1 for different time, from 5 to 30 min. Incubation was terminated by adding a solution of ice-cold 30% trichloroacetic acid, and cells were scraped and centrifuged for 30 min at 15,000 × g. Supernatant was neutralized by three washes with water-saturated diethyl ether, and NaHCO3 addition, lyophilized and reconstituted with H2O for cAMP quantification with a commercial radioactive assay (GE Healthcare, Buckinghamshire, UK). PTX and BAPTA treatment were performed as described above.
Phosphatase assay.
Eight-day-old cultured hippocampal neurons were starved for 2 h in Locke's buffer and stimulated with FSK (5.2 μm) in the presence or in the absence of CX3CL1 for 15 min. Cells were lysed in buffer containing 20 mm HEPES, pH 7.4, 50 mm NaCl, 0.2 mm EGTA, 0.5 mm DTT, 0.3% Triton X-100, and protease inhibitors centrifuged for 1 h at 20,000 × g, and supernatant was analyzed for protein phosphatase 2B (PP2B) activity after removal of endogenous phosphate with a Sephadex G-25 resin column (Promega, Madison, WI). Phosphatase assay was performed on 100 μm phosphopeptide RRA(pT)VA in buffer containing the following: 50 mm imidazole, pH 7.2, 0.2 mm EGTA, 10 mm MgCl2, 1 mm NiCl2, 50 μg/ml calmodulin, and 0.02% β-mercaptoethanol for 30 min at 30°C, and reaction was blocked by a Molybdate solution and free phosphate quantified measuring absorbance at 630 nm with a plate reader.
Results
CX3CL1 reduces EPSC amplitude through the activation of CX3CR1
Whole-cell recordings of EPSCs were performed in CA1 pyramidal neurons from acute rat slices while repeatedly stimulating Schaffer collateral axons (0.25 Hz). Superfusion with CX3CL1 (6–12 min) caused a reduction in EPSC peak amplitude (Fig. 1A) without any obvious effect on resting membrane conductance or holding current. CX3CL1-induced EPSC amplitude depression developed within 5 min of CX3CL1 application with maximal depression (to 68 ± 8%; n = 9) observed at ∼10 min and was followed by partial recovery (to 85 ± 4% within 30 min of chemokine withdrawal) with a few exceptions, where full recovery was observed (Figs. 1A, 4B). This depression was induced by CX3CL1 at a concentration as low as 0.2 nm and was dose dependent (Fig. 1B).
Figure 1.
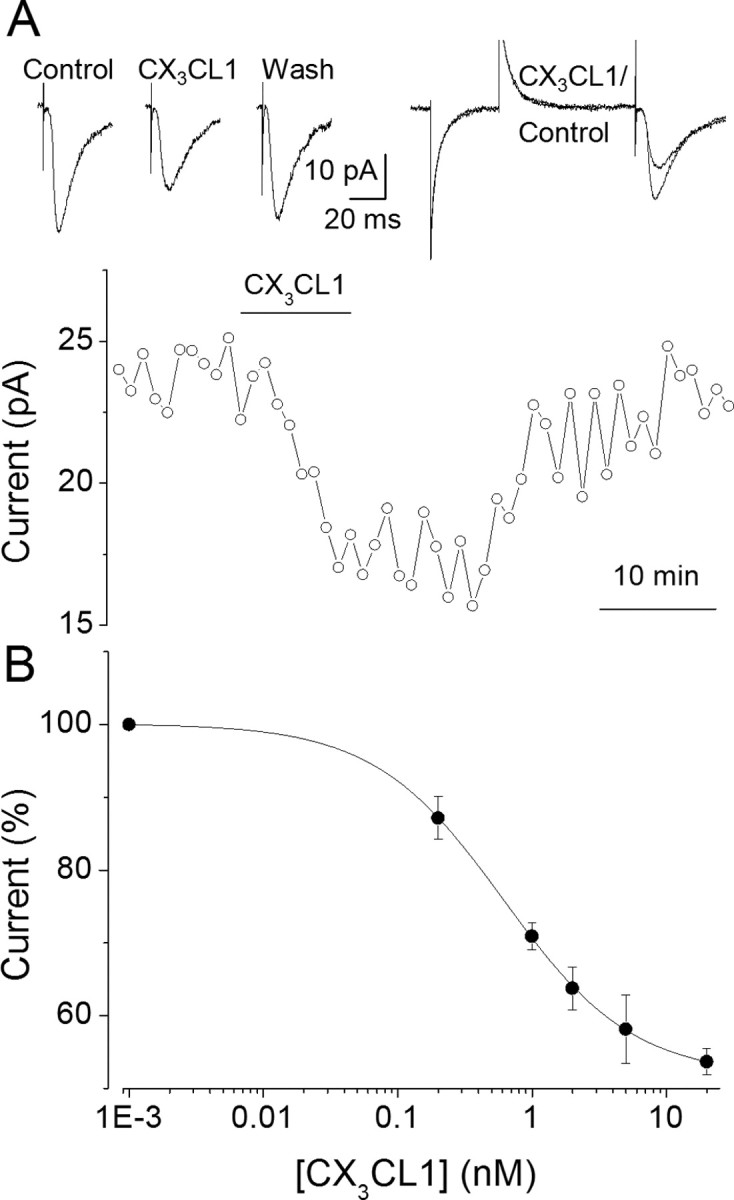
CX3CL1 reduces the amplitude of evoked EPSCs in CA1 pyramidal neurons. A, Top left, Sample traces of EPSCs (average of 15 events over 1 min) recorded before, after 10 min of chemokine application, and 30 min after washout, as indicated. A, Top right, Superimposed enlarged traces in control and CX3CL1; note the absence of changes in the input resistance. A, Bottom, Time course of the current peak amplitude in the same cell. Circles represent averages as indicated previously. Horizontal bar, Chemokine application. Note full recovery after washout. The concentration of CX3CL1 here and in subsequent figures, unless otherwise indicated, is 2 nm. B, CX3CL1 dose-EPSC amplitude relationship. The filled circles, here and in subsequent figures, refer to means ± SEM of single determinations on separate slices (n = 5–14). The calculated IC50 value for CX3CL1 is 1 nm with nH = 1.
Figure 4.
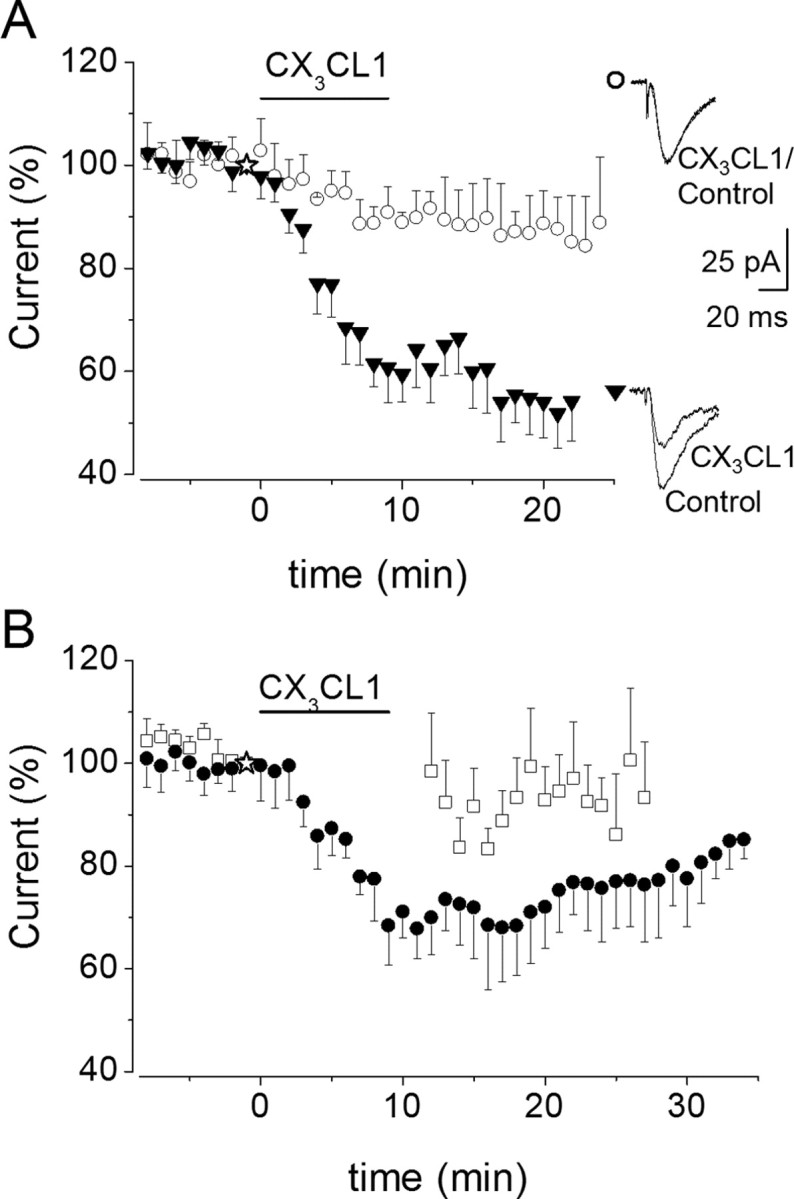
CX3CL1-induced EPSC depression is Ca2+ and activity dependent. A, Left, Time course of CX3CL1 effects on EPSCs in the presence of the Ca2+ chelator BAPTA at 20 mm (○) or 1 mm (▾) in the patch pipette solution, as indicated. Symbols represent mean values of four to nine experiments. A, Right, Sample EPSCs traces recorded in the presence of 20 mm (○) and 1 mm BAPTA (▾) before and 10 min after CX3CL1 application, as indicated. B, Time course of EPSCs after CX3CL1 application to stimulated (control, ●; n = 9) and nonstimulated (□; n = 9) pyramidal neurons. Application of CX3CL1 is as indicated.
We then examined whether the action of CX3CL1 on EPSC amplitude was effectively mediated through CX3CR1 by performing the same experiments in wt and CX3CR1 KO mice (Haskell et al., 2001). CX3CL1 reduced the average EPSC amplitude to 64 ± 12% (n = 6) (Fig. 2A,B) in pyramidal neurons of wt mice. Notably, the CX3CL1 effect was sustained considerably longer after washout in mouse slices compared with rat slices. The CX3CL1 effect was slightly attenuated in slices from heterozygous mice (to 77 ± 15%; p = 0.1; n = 7) (Fig. 2B) and absent in slices from CX3CR1-KO mice (to 100 ± 8%; p = 0.41; n = 9) (Fig. 2), indicating that CX3CL1-induced EPSCs depression is mediated through CX3CR1 activation.
Figure 2.

CX3CL1-induced EPSC depression is absent in CX3CR1-KO mice. A, Sample traces of EPSCs recorded in CX3CR1-KO mice (left, −/−) and wt littermates (right, +/+) before and after CX3CL1 application, as indicated. B, Time course of CX3CL1 effect on EPSC amplitude in hippocampal CA1 neurons from CX3CR1-KO mice (□; n = 9), wt (●; n = 6) and heterozygous littermates (half-filled circle; n = 7). Here and in subsequent figures, current amplitudes are normalized to current recorded before chemokine application. The star represents 100% current.
We also conducted experiments to see whether endogenous CX3CL1 could affect glutamatergic synaptic transmission in our slices. Western blot analysis revealed that CX3CL1 was expressed at levels comparable with those observed in cultured hippocampal neurons (data not shown) (Limatola et al., 2005). We then tested the effect of an anti-CX3CL1 antibody (Ab) on glutamatergic transmission. Pretreatment with anti-CX3CL1 Ab (TP 203, 2 μg/ml, 20 min superfusion; Torrey Pines Biolabs, Houston, TX) suppressed the effects of exogenous CX3CL1 on evoked synaptic transmission (n = 3) (data not shown) but did not produce any obvious change in the control evoked response (n = 4) (data not shown), suggesting that endogenous CX3CL1 does not contribute significantly to the modulation of glutamatergic neurotransmission under the conditions used in our studies.
CX3CL1 acts postsynaptically on pyramidal neurons
It has been reported previously that CX3CL1 modulates both glutamatergic spontaneous synaptic currents (Meucci et al., 1998) and AMPA-activated currents (AMPA currents) (Limatola et al., 2005) in cultured hippocampal neurons. To determine whether CX3CL1 modulates glutamatergic neurotransmission via presynaptic or postsynaptic mechanisms, we analyzed paired-pulse facilitation of synaptic responses and spontaneous excitatory events in rat pyramidal neurons from hippocampal slices.
First, we analyzed the paired-pulse ratio of the responses evoked by two closely spaced stimuli. Paired-pulse facilitation was 136 ± 20% (n = 7) and persisted after 10 min CX3CL1 application (139 ± 21%; n = 7; p > 0.05) before washout. Amplitudes of both EPSCs decreased to a similar degree (first EPSC, to 67 ± 6%, p < 0.01; second EPSC, to 68 ± 5%, p < 0.01) (Fig. 3A,B). These findings indicate that the presynaptic site is probably not involved in EPSC depression by CX3CL1 (Dobrunz and Stevens 1997). Next, we analyzed the distribution of spontaneous EPSC (sEPSC) amplitudes after chemokine treatment. sEPSCs exhibited a mean amplitude of −18.9 ± 6.1 pA and frequency of 2.7 ± 0.3 Hz (n = 7) and were glutamatergic in nature, being blocked by CNQX (10 μm) (data not shown). In four neurons, after CX3CL1 treatment (2 nm; 10 min), the mean amplitude of sEPSCs was significantly reduced from −15.2 ± 2.1 to −11.3 ± 1.4 pA (p < 0.05), the frequency (2.3 ± 0.3 Hz) remaining stable (data not shown). The analysis of sEPSC amplitude distribution showed a shift toward lower amplitude after CX3CL1 treatment (Fig. 3C,D). Together, these findings suggest that postsynaptic glutamatergic receptors are involved in the synaptic depression induced by CX3CL1 treatment. Interestingly, CX3CL1 was unable to reduce the sEPSC amplitude when the frequency of spontaneous sEPSCs was relatively low, because CX3CL1 did not alter either the frequency or the amplitude of sEPSCs in five neurons displaying a frequency <0.5 Hz (data not shown).
Figure 3.

CX3CL1-induced depression is postsynaptic. A, Superimposed traces (top) of EPSC responses (average as in Fig. 1A) evoked in a CA1 neuron by paired-pulse stimulation (50 ms interval) in control and after 10 min application of CX3CL1, as indicated. Scaled traces (bottom) in the presence of CX3CL1, showing no difference in paired-pulse ratio. B, Histogram of EPSC amplitude showing the CX3CL1-induced average depression of EPSCs evoked by the first (P1, white column) and the second (P2, gray column) pulse. C, Histogram of the spontaneous EPSC amplitudes recorded before (empty columns) and after (dashed columns) 10 min CX3CL1 treatment, as indicated by arrows, from a pyramidal neuron. Distributions fitted to Gaussian equations (control data, X0 = 21.5 pA, σ = 10.5; CX3CL1, X0 = 17.9 pA, σ = 8.2). Traces (inset) represent EPSCs (average of 30 events) before and at the end of CX3CL1 treatment, as indicated. D, Cumulative amplitude distribution of EPSCs in control and CX3CL1 from the same neuron as in C. Note the shift to lower amplitudes in the presence of CX3CL1.
CX3CL1-induced EPSC depression is modulated by Ca2+
It has been reported previously that CX3CL1 can elicit a transient increase in intracellular Ca2+ (Meucci et al., 2000) and that chelating intracellular Ca2+ in hippocampal cultures blocks CX3CL1-dependent inhibition of glutamatergic currents (Limatola et al., 2005). To investigate whether Ca2+ modulates CX3CL1-induced depression of EPSC amplitude, we performed experiments under various concentrations of the Ca2+ chelator BAPTA in the patch pipette recording solution. When BAPTA was increased from 5 mm (standard solution) to 20 mm, CX3CL1 failed to significantly affect EPSC amplitude (to 91 ± 3%, mean ± SEM; p = 0.61, one-way ANOVA; n = 4) (Fig. 4A, open circles). Conversely, when BAPTA was decreased to 1 mm, EPSC amplitude was reduced to 60 ± 3% before washout (Fig. 4A, black triangles) (p = 0.03, one-way ANOVA; n = 9), and this depression was maintained even after washout. These findings suggest that the strength of cytosolic Ca2+ buffering regulates the efficacy of CX3CL1 in depressing the EPSC amplitude.
To see whether the efficacy of CX3CL1 in depressing EPSCs could depend on the kinetics of Ca2+ buffering, we performed experiments with the slow Ca2+ chelator EGTA. CX3CL1 applied to pyramidal neurons dialyzed with EGTA-containing solution (EGTA 20 mm) reduced the EPSC amplitude during 10 min application (to 67 ± 9%; n = 4; p = 0.03; not illustrated), indicating that slow Ca2+ chelators were unable to regulate the efficacy of CX3CL1 in inducing synaptic depression.
To identify the source of Ca2+ involved in the CX3CL1-induced synaptic AMPA receptor modulation, we dialyzed neurons with the Ca2+-ATPase inhibitor thapsigargin (1 μm) to reduce cytosolic Ca2+ mobilization from intracellular stores. We found that, with thapsigargin in the patch pipette solution, CX3CL1 was still able to reduce EPSC amplitude with the same efficacy as in control neurons (to 75 ± 9%; n = 5; p = 0.05) (data not shown). Together, these findings suggest that Ca2+ entry rather than cytosolic Ca2+ mobilization is involved in the CX3CL1 effect observed.
Because Ca2+ entry at glutamatergic synapses is primarily mediated through NMDA receptors, we investigated whether the activation of NMDA receptors could contribute to the effects of CX3CL1 on EPSC amplitude. The NMDA receptor blocker d-APV (40 μm) was applied to pyramidal cells that were subsequently treated with CX3CL1. Pyramidal neurons exposed to d-APV were still responsive to CX3CL1 (average depression to 70 ± 7%; n = 6; p = 0.006; 10 min CX3CR1 treatment) (data not shown), indicating that the EPSC depression was not dependent on NMDA receptor function.
CX3CL1-induced EPSC depression is regulated by synaptic activity
Because under our experimental conditions excitatory synaptic activity modulates cytosolic Ca2+ level increasing Ca2+ influx at active synapses (Yuste et al., 1999), we investigated whether synaptic activity could influence the CX3CL1-induced EPSC depression. To test for an effect on synaptic activity, we first obtained stable EPSC responses from a pyramidal neuron, then we superfused the slice with CX3CL1-containing medium while Schaffer collateral stimulation was interrupted, and finally we resumed the electrical stimulation 2 min after chemokine withdrawal. Under these conditions, we found that CX3CL1 failed to inhibit EPSCs with the EPSC amplitude remaining unchanged (to 98 ± 11%; n = 9) (Fig. 4B, open squares) when compared with stimulated neurons (to 67 ± 6%; p = 0.04; n = 9) (Fig. 4B, closed circles). These findings indicate that CX3CL1 exerts an inhibitory action on hippocampal neurons in a manner depending on synaptic activity.
CX3CL1 inhibits AMPA currents
To gather additional evidence for CX3CL1 action at a postsynaptic site, we applied AMPA (10–100 μm) onto CA1 hippocampal pyramidal neurons, because under our experimental conditions, synaptic currents were mediated mainly through AMPA/KA receptors (Hestrin et al., 1990). As shown in Figure 5A, brief AMPA applications (10–100 ms) in the presence of TTX (1 μm) and bicuculline (10 μm) induced inward currents (range, 55–550 pA). These currents were completely abolished in the presence of CNQX (10 μm; n = 3) (data not shown) and thus mediated by the activation of postsynaptic AMPA/KA receptors. We found that the amplitude of AMPA currents was reversibly depressed by CX3CL1 application to 79 ± 5% (n = 11; p = 0.001) after 10 min CX3CL1 treatment and recovered to 98 ± 3% after washout (n = 11) (Fig. 5A). Interestingly, CX3CL1-induced inhibition of AMPA currents was dependent on the current amplitude, with larger currents more strongly depressed by CX3CL1 (Fig. 5B).
Figure 5.

CX3CL1 reduces AMPA current responses in CA1 neurons. A, Top, Current responses to AMPA applications (▾) in control, 10 min after CX3CL1 application, and 15 min after chemokine washout, as indicated. Arrowheads signal agonist applications (AMPA, 10 μm; 100 ms). A, Bottom, Time course of CX3CL1-induced depression of postsynaptic AMPA currents. B, Relationship between AMPA current peak amplitude and CX3CL1-induced depression. Data fitted to a single exponential, y = y0 + A(−x/τ). Note reduced CX3CL1-induced depression at lower AMPA current amplitudes. C, Time course of AMPA currents after CX3CL1 application to nonstimulated pyramidal neurons (n = 5). Application of CX3CL1, as indicated. The dashed line signals the 100% value. Note that the CX3CL1-induced depression of AMPA current is activity dependent.
Because the repetitive stimulation of synaptic AMPA receptors could influence CX3CL1-induced EPSC depression, being blocked by absence of synaptic activity (Fig. 4B), experiments were performed to determine whether the AMPA current amplitude depression could be influenced by repetitive AMPA receptor activation. It was found that CX3CL1 application to pyramidal neurons, when it was not accompanied by AMPA receptors stimulation with exogenous AMPA, was unable to depress AMPA currents at the end of CX3CL1 application. In fact, after CX3CL1 treatment and washout (2 min), the amplitude of AMPA responses was unchanged compared with the control (102 ± 17%; n = 5; p = 0.9) (Fig. 5C) with only a slight transient depression seen at ∼10 min washout.
Finally, a number of experiments were performed to see whether AMPA current depression was dependent on Ca2+. With 0.2 mm BAPTA in the recording patch pipette, AMPA currents ranging from −61 to −217 pA were significantly depressed by CX3CL1 application (10 nm) with an irreversible current rundown (Fig. 6, closed circles). Conversely, when similar AMPA applications were performed in a Ca2+-free solution (0 Ca2+/1 mm EGTA; current amplitudes from −70 to −240 pA), slice superfusion with CX3CL1 did not cause a reduction of AMPA currents (Fig. 6, open squares). Together, these findings indicate that Ca2+ entry is required for the CX3CL1-induced depression of AMPA currents generated by the activation of extrasynaptic AMPA receptors in analogy with findings reported above for synaptic AMPA receptors.
Figure 6.

CX3CL1-induced depression of AMPA currents is suppressed in Ca2+-free solution. Time course of the effect of CX3CL1 on the amplitude of AMPA-evoked currents (100 μm; 100 ms) recorded in CA1 pyramidal neurons (0.2 mm BAPTA in intracellular patch pipette solution). ●, AMPA applications in normal external solution (n = 11). □, Applications of AMPA dissolved in Ca2+-free solution (n = 8). Note that, in all experiments, CX3CL1 dissolved in standard ACSF.
CX3CL1-induced EPSC depression is dependent on Gi-mediated cAMP reduction
CX3CR1 is coupled to Gi- and Gz-proteins in natural killer cells (Al-Aoukaty et al., 1998), and CX3CL1 effects on Ca2+ transients and cell survival are abolished by PTX in hippocampal neurons (Meucci et al., 2000; Limatola et al., 2005). Thus, experiments were performed in hippocampal slices to determine whether PTX, a selective inhibitor of the αi subunits of Gi, was able to block CX3CL1-induced EPSC depression. It was found that, in cells internally dyalized with activated PTX (1 μg/ml), EPSC amplitude remained stable after CX3CL1 stimulation (15 min; 109 ± 19%; p = 0.66; n = 10; five rats), indicating that CX3CL1-induced EPSC depression was mediated through signaling pathways downstream of Gi activity (data not shown). To identify the signaling pathway involved, intracellular levels of cAMP were measured in hippocampal cultures exposed to the adenylate cyclase (AC) stimulator FSK (5 μm) in the presence or absence of CX3CL1 (100 nm) for different time, up to 30 min. Results, shown in Figure 7A (left), demonstrate that CX3CL1 significantly reduced FSK-induced cAMP accumulation, indicative of AC inhibition via Gi activation, and that this effect is already evident after 5 min stimulation. CX3CL1-mediated cAMP reduction was blocked by PTX treatment and was independent of Ca2+, because it was insensitive to treatment with BAPTA/AM (Fig. 7A, right).
Figure 7.

CX3CL1 reduces FSK-induced cAMP accumulation coupled to EPSC depression. A, Left, Time course of cAMP accumulation in cultured hippocampal neurons treated as indicated. Untreated neurons, after 30 min incubation, contained 0.17 pmol/μg proteins. Right, Accumulation of cAMP in cells treated for 15 min with FSK/CX3CL1 and pretreated as indicated. Data are expressed as a percentage of cAMP accumulation in cells stimulated with FSK alone. CX3CL1, 100 nm; FSK, 5.2 μm; pretreatment, BAPTA/AM, 5 μm, 1 h; PTX, 1 μg/ml, 2 h. B, Time course of the effect of CX3CL1 application (horizontal bar, as indicated) on the amplitude of EPSC recorded in CA1 pyramidal neurons in slices treated with FSK (●, 5 μm; n = 10) or 8Br-cAMP (□, 100 μm, n = 6). Either FSK or 8Br-cAMP superfusion started 20–40 min before applying CX3CL1.
To investigate whether the CX3CL1-induced reduction of intracellular cAMP levels could contribute to CX3CL1-induced EPSC depression, we performed electrophysiological recordings in the presence of either FSK or 8Br-cAMP, drugs that increase cAMP signaling by activating AC and mimicking cAMP, respectively. As expected, exposure of slices to FSK (5 μm; 20–40 min) caused an increase in the amplitude of evoked EPSCs (to 220 ± 20% after 40 min, n = 10) (Chavez-Noriega and Stevens, 1994; Sokolova et al., 2006). CX3CL1 caused a reversible decrease in EPSC amplitude that was significantly greater in the presence (to 52 ± 8%; n = 10) (Fig. 7B, closed circles) than in the absence of FSK (to 77 ± 4%; p = 0.04, one-way ANOVA; n = 6) (data not shown). As in the absence of FSK, this effect was not observed when Schaffer collateral stimulation was interrupted (n = 5) (data not shown).
Conversely, when slices were superfused with a stable membrane permeant cAMP analog, 8Br-cAMP (100 μm), and then cotreated with CX3CL1 (12 min), an analogous depression of EPSC was not observed (to 110 ± 8%; p = 0.61; n = 6) (Fig. 7B, open squares). Similar results were obtained in four cells internally perfused with 8Br-cAMP (100 μm) (data not shown). Together, these findings indicate that the effects of CX3CL1 on EPSC are stringently related to a Gi-dependent reduction of cAMP formation.
CX3CL1-induced EPSC depression is dependent on association of protein kinase A to GluR1
Because we observed that CX3CL1-mediated depression of EPSC relied on a Gi-dependent reduction of cAMP formation, we investigated whether protein kinase A (PKA) anchoring to the complex AKAP-GluR1 was involved in CX3CL1 action. Cells internally dialyzed with Ht31 (1 μm), a competitive antagonist of PKA anchoring (Tavalin et al., 2002), did not exhibit any obvious CX3CL1-mediated EPSC depression during 15 min treatment (98 ± 7%, p = 0.8; n = 6) (data not shown). In contrast, EPSC depression occurred normally when Ht31 was substituted by the inactive prolinated peptide (Ht-31p, 1 μm). We concluded that a physical association of PKA to GluR1 was required for the development of CX3CL1-mediated EPSC depression.
CX3CL1 stimulation reduces GluR1 phosphorylation at Ser845
The phosphorylation of GluR1 by cAMP-dependent PKA at Ser845 and by calcium calmodulin-dependent protein kinase II (CaMKII) and protein kinase C (PKC) at Ser831 has been associated with modulatory effects on AMPA receptor function. GluR1 phosphorylation at Ser845 increases channel opening probability (Banke et al., 2000), whereas phosphorylation at Ser831 enhances channel conductance (Derkach et al., 1999). In addition, changes in the phosphorylation level at both Ser845 and Ser831 are associated with the long-term modulation of synaptic AMPA receptors (Lee et al., 1998, 2000). Thus, we investigated whether CX3CL1-induced AMPA-current depression could be associated with a change in the phosphorylation of GluR1 at Ser845 and/or Ser831. We found that CX3CL1 clearly reduced Ser845 GluR1 phosphorylation in hippocampal slices stimulated with FSK (to 55.6 ± 2.2%; p < 0.001; n = 11) (Fig. 8A, left) to increase the basal level of GluR1 phosphorylation. No obvious effect of CX3CL1 was seen on basal Ser845 phosphorylation (data not shown). Interestingly, the phosphorylation of Ser831 induced by the protein kinase C activator, TPA, was not obviously reduced by CX3CL1, with even a slight increase being eventually observed (n = 3) (Fig. 8A, right). Analogous results were obtained in HEK cells transiently expressing GluR1 and CX3CR1 (to 62.4 ± 4%; p < 0.001; n = 10) (Fig. 8B, top, one representative experiment), where CX3CL1 (10 nm) was able to induce reversible AMPA current depression (to 60 ± 8%; p = 0.002; n = 10) (Fig. 8B, bottom) and in cultured rat hippocampal neurons (to 66.7 ± 3%; p < 0.01; n = 4) (Fig. 8C, top, one representative experiment), where CX3CL1 was able to reduce AMPA-mediated currents (Limatola et al., 2005).
Figure 8.

CX3CL1 reduces GluR1 phoshorylation at Ser845 but not at Ser 831. A, Cell lysates from rat hippocampal slices treated as indicated with FSK (15 min, 1 μm; left) or phorbol ester TPA (15 min, 1 μm; right) alone or together with CX3CL1 (100 nm), probed, respectively, with anti-phosphoSer845 or anti-phosphoSer831 GluR1 and anti-actin antibodies and analyzed by Western blot (representative of 11 and 3). B, Top, Cell lysates from HEK cells transiently expressing CX3CR1 and GluR1 analyzed as in A after 30 min of drug treatment (representative of 10). Film exposed for 30 s to avoid saturation signals. A longer exposure showing more pronounced basal level of Ser845 phosphorylation is not illustrated. Bottom, Glutamate current traces from HEK cells (representative of 10). Arrowheads, Glutamate (200 μm) application. A cell treated with CX3CL1 (as indicated) for 7 min is shown. C, Top, Cell lysates from rat hippocampal cell cultures treated as indicated with FSK (5 min, 5.2 μm) alone or together with CX3CL1 (100 nm) and analyzed as in A (representative of 4). Bottom, Histogram of Ser845 phosphorylation in hippocampal neurons stimulated for 15 min as indicated. Data are expressed as a percentage of Ser845 phosphorylation in cells stimulated with FSK/CX3CL1 versus FSK alone. CX3CL1, 100 nm; FSK, 5.2 μm; pretreatment, BAPTA/AM, 5 μm, 1 h; PTX, 1 μg/ml, 2 h.
We next investigated whether PTX or the intracellular Ca2+ chelator BAPTA/AM could modulate the CX3CL1-induced reduction of Ser845 phosphorylation, as was the case for the CX3CL1-induced EPSC depression. Hippocampal cultured neurons were exposed to BAPTA/AM (5 μm; 1 h) or PTX (1 μg/ml; 2 h) and then treated with FSK in the presence or absence of CX3CL1. No reduction of phosphorylation was observed in the presence of BAPTA-AM (to 131 ± 12.4%; p = 0.1; n = 3) or PTX (to 147 ± 27%; p = 0.2; n = 3), as shown in the histogram in Figure 8C (bottom). Together, these data suggest that CX3CL1-induced reduction of Ser845 phosphorylation is mediated by a Gi-dependent reduction of adenylyl cyclase activity and thereby PKA activity. In addition, CX3CL1-induced reduction of Ser845 phosphorylation is dependent on intracellular Ca2+.
Phosphatase inhibitors block both CX3CL1-induced EPSC depression and reduction of Ser845 phosphorylation
Because the phosphorylation state of Ser845 GluR1 results from a delicate balance between protein kinase A and phosphatase activity (Lee et al., 2000), we investigated the role of phosphatases in CX3CL1-mediated effects.
We performed several experiments in which ATPγS was substituted for ATP in the intracellular solution. ATPγS promotes irreversible substrate thio-phosphorylation (Eckstein, 1985) and blocked CX3CL1-induced EPSC depression (to 99 ± 12%; p = 0.96; n = 6) (data not shown), indicating that substrate dephosphorylation was necessary for EPSC depression. Subsequently, we found that calyculin A (100 nm in the patch pipette) and okadaic acid (1 μm, 1 h in the external solution or in the patch pipette), both potent blockers of PP1/2A (Bialojan and Takai, 1988; Herzig and Neumann, 2000; Winder and Sweatt, 2001), abolished CX3CL1-induced AMPA current depression (calyculin A, to 96 ± 2%; n = 5; p = 0.2) (Fig. 9) (okadaic acid, to 96 ± 4%, n = 8, p = 0.24 in the external solution and to 114 ± 12%, n = 4, p = 0.3, in the pipette) (data not shown). We found similar results with cyclosporin A, a specific PP2B/calcineurin inhibitor (Unno et al., 1999) (250 μm, 1 h superfusion, or 1 μm in the pipette), which fully inhibited CX3CL1-induced AMPA current amplitude depression (to 93 ± 4%, n = 8, p = 0.28, in external medium, to 104 ± 5%, n = 4; p = 0.4, in the pipette) (data not shown). Considered together, these findings indicate that functional protein phosphatases are required for CX3CL1-dependent AMPA current depression.
Figure 9.

Calyculin A blocks CX3CL1-induced reduction of AMPA currents in hippocampal slices. Time course of the effect of CX3CL1 on the amplitude of AMPA-evoked currents (>100 pA) recorded in CA1 pyramidal neurons with calyculin A (□, 100 nm, n = 5) in the recording pipette, compared with control (●, n = 18).
As expected, phosphatase inhibitors reduced the effects of CX3CL1 on Ser845 phosphorylation. After preincubation with calyculin A (200 nm; 90 min), hippocampal slices were treated for 15 min with FSK (1 μm) alone or together with CX3CL1 (100 nm) in the continuous presence of calyculin A. Under these conditions, CX3CL1 was less effective in reducing FSK-induced Ser845 phosphorylation (to 88.3 ± 1.7%; n = 5) when compared with untreated slices (to 55.6%; p < 0.01; n = 11). Analogous results were obtained in hippocampal cultures treated with cyclosporin A (1 μm, 60 min; to 100.5 ± 7.5%; n = 5), compared with cyclosporin A untreated cultures (52 ± 4%; n = 5) (data not shown).
Our findings obtained with phosphatase inhibitors, together with our data showing Ca2+ dependency of CX3CL1 effects, led us to investigate whether CX3CL1 might directly stimulate a Ca2+-dependent phosphatase activity. When hippocampal neurons were stimulated with CX3CL1 either in the presence or absence of FSK, no change in PP2B-specific phosphatase activity was observed (phosphatase assay, 2.52 ± 0.43 pmol phosphate/min/μg of protein in untreated vs 2.56 ± 0.37 in cells stimulated with CX3CL1; 100 nm, 15 min) (data not shown).
Considered together, our data indicate that several factors, including intracellular Ca2+ and Gi-dependent cAMP reduction, were involved in the modulation of the CX3CL1-induced decrease of Ser845 phosphorylation and EPSC and AMPA current depression. Importantly, our data are consistent with the effects of CX3CL1 being mediated by changes in PKA rather than phosphatase activity.
Discussion
We report the effects of CX3CL1 on glutamatergic synaptic transmission between Schaffer collaterals and CA1 pyramidal cells in rat hippocampal slices. CX3CL1 reduces the amplitude of both evoked whole-cell EPSCs and AMPA current responses by a mechanism dependent on the modulation of postsynaptic AMPA/KA receptors. CX3CL1 acts via a PTX-sensitive, Gi-dependent reduction of intracellular cAMP, which in turn weakens PKA-related phosphorylation of GluR1 Ser845, and favors the action of phosphatases on this residue, thus promoting current depression. Several pieces of experimental evidence support this hypothesis: (1) CX3CL1 reduces FSK-induced cAMP accumulation; (2) PTX abolishes CX3CL1 effects on EPSC amplitude, cAMP accumulation, and Ser845 GluR1 phosphorylation; and (3) both the displacement of PKA from AKAP79 with Ht31 and the overcoming of AC inhibition with 8-Br-cAMP suppress CX3CL1-mediated current depression. Furthermore, CX3CL1-induced effects are coupled to phosphatase activity, because blockers of protein phosphatases affect both CX3CL1-mediated current depression and the reduction of Ser845 phosphorylation. Finally, CX3CL1 action requires Ca2+ entry because EPSC depression and the reduction of Ser845 phosphorylation are absent when postsynaptic Ca2+ is tightly controlled by the fast Ca2+ chelator BAPTA and because the depression of AMPA current is blocked in Ca2+-free medium.
Postsynaptic site of EPSC depression
Our data support a postsynaptic site of action of CX3CL1 on EPSC depression based on two pieces of evidence: (1) the absence of changes in the paired-pulse ratio suggests an unchanged probability of transmitter release during chemokine application (Dobrunz and Stevens, 1997), and (2) the reduction of the amplitudes of spontaneous EPSCs and of AMPA currents by CX3CL1 treatment indicates decreased postsynaptic sensitivity. Our conclusion is in contrast to a previous report in cultured hippocampal neurons, suggesting a presynaptic site for CX3CL1-induced depression (Meucci et al., 1998). In addition, previous results from our laboratory, showing that glutamatergic currents and field EPSPs are depressed by CX3CL1 in hippocampal neurons (Limatola et al., 2005; Bertollini et al., 2006). It is worthy to note that the CX3CL1 doses used in our experiments (2–10 nm) are much lower than those used by Meucci et al. (1998) and Oh et al. (2002) (50–100 nm), leaving open the possibility that higher doses could modulate other ion channels.
Ca2+ and activity dependence of CX3CL1-induced EPSC depression
CX3CL1-induced EPSC depression observed at the Schaffer collateral-CA1 synapse is Ca2+ dependent, because it is suppressed when neurons are dialyzed with a high intracellular BAPTA concentration (Glitsch et al., 2000). In addition, at low BAPTA concentrations, EPSC and AMPA currents are depressed more efficiently by CX3CL1 and without recovery. Although it has been reported that CX3CL1 causes a transient intracellular Ca2+ rise in neurons (Meucci et al., 1998, 2000; Kansra et al., 2001; Deiva et al., 2004), our results indicate that Ca2+ release from intracellular stores does not play a major role in the CX3CL1 effect, because EPSCs are depressed by CX3CL1 when neurons are dialyzed with the Ca2+-ATPase inhibitor thapsigargin. Furthermore, CX3CL1-induced depression of AMPA currents generated by the activation of extrasynaptic receptors does not develop when Ca2+ is briefly withdrawn during 10–50 ms AMPA applications, suggesting that Ca2+ influx is necessary to activate current depression. Interestingly, the fact that the slow Ca2+ chelator EGTA does not interfere with CX3CL1 effects suggests that localized fast Ca2+ transients are required for AMPA receptor modulation (Dargan and Parker, 2003).
We report that CX3CL1-induced glutamatergic depression does not develop in the absence of coincident stimulation, and AMPA currents are depressed by CX3CL1 treatment only after repetitive stimulation of AMPA receptors. This is probably because of the fact that Ca2+ influx is necessary both for EPSC and AMPA current depression. In our experimental conditions, EPSCs still contain a small NMDA component (Hestrin et al., 1990) that can contribute substantially to Ca2+ influx (Garaschuk et al., 1996). Nevertheless, a major role of Ca2+ entry through NMDA receptors is unlikely, because the NMDA receptor blocker d-APV did not influence the CX3CL1-induced effects here reported.
Role of AC and phosphatases in CX3CL1-induced depression
Synaptic plasticity in the hippocampus requires a balance of protein kinase and phosphatase activities (Colbran, 2004). Because Ser845 GluR1 is a PKA target and CX3CR1 activates Gi (Roche et al., 1996; Al-Aoukaty et al., 1998), we speculate that CX3CL1 impairs Ser845 phosphorylation by reducing PKA activity. This mechanism is consistent with the observations that: (1) CX3CL1 reduces intracellular cAMP levels and Ser845 phosphorylation, and these effects are blocked by PTX; (2) PTX inhibits CX3CL1-mediated depression of EPSC; (3) CX3CL1 does not affect EPSC after slice treatment with the stable cAMP analog, 8-Br-cAMP, mimicking irreversible AC activation or by intracellular dialysis with Ht31, a peptide that detaches PKA from AKAP 79 (Tavalin et al., 2002; Snyder et al., 2005), preventing PKA-dependent Ser845 phosphorylation. We conclude that the reduction of PKA activity favors the action of phosphatases, leading to an impairment of GluR1 phosphorylation and current amplitudes (Banke et al., 2000; Winder and Sweatt, 2001).
The critical role of protein phosphatases in CX3CL1-induced current depression is further supported by the fact that EPSC depression is absent when dephosphorylation is prevented by ATPγS or phosphatase inhibitors. It is likely that phosphatase activity requires sustained synaptic Ca2+ entry, as suggested by EPSC depression blockade when intracellular Ca2+ is tightly buffered with BAPTA. It is unlikely that phosphatase activity is directly stimulated by CX3CL1, because no stimulation of PP2B enzymatic activity by CX3CL1 was observed. The observation that cell treatment with BAPTA-AM impairs the CX3CL1-mediated reduction of Ser845 phosphorylation, but is ineffective at reducing CX3CL1-mediated cAMP reduction, indicates that Ca2+-mediated basal phosphatase activity is necessary for this effect. This scenario, that couples the phosphorylation state of GluR1 Ser845 to CX3CL1-induced rundown of glutamate receptor current (Tavalin et al. 2002; Snyder et al., 2005), shares many properties with LTD (Lisman, 1989) and is supported by the occlusion of CX3CL1-induced synaptic depression by the induction of homosynaptic LTD (Bertollini et al., 2006). Based on the reported evidence, we propose that CX3CL1-induced signaling shifts the balance between kinases and phosphatases acting on Ser845 in favor of phosphatases by reducing PKA activity. Because the effects of CX3CL1 on current depression and phosphorylation levels of Ser 845 require coincident Ca2+ influx, we hypothesize that, physiologically, CX3CL1 behaves as a modulator of active glutamatergic synapses.
Physiological significance of CX3CR1 signaling in the hippocampus
Several studies have shown that both CX3CL1 and its receptor CX3CR1 are constitutively expressed in hippocampal neurons (Hughes at al., 2002; Tarozzo et al., 2003). In cultured hippocampal neurons, functional CX3CR1 is present (Meucci et al., 1998, 2000; Limatola et al., 2005), and its activation leads to G-protein-sensitive signaling (Meucci et al., 2000). The CX3CL1-induced synaptic modulation we report here is CX3CR1 dependent, because it is absent in CX3CR1 KO mice (Fig. 2). Furthermore, CX3CL1-induced synaptic modulation is likely to be physiologically relevant, because its EC50 value is compatible with the known chemotactic and neuroprotective effects of CX3CL1 (Limatola et al., 2005) and with the functional properties of cloned CX3CR1 (Combadiere et al., 1998). In addition, the localized synaptic effects of CX3CL1 are consistent with the diffuse expression of CX3CR1 on the soma (Hughes et al., 2002; Tarozzo et al., 2003) and dendrites of hippocampal neurons (Meucci et al., 2000; Limatola et al., 2005). Despite the known glial expression of CX3CR1 (Harrison et al., 1998; Nishiyori et al., 1998), we ruled out the possibility that exogenous CX3CL1 causes indirect EPSC modulation through the glial release of other mediators, because EPSC depression is exclusively postsynaptic and we did not observe significant changes in the membrane properties of CA1 pyramidal neurons, as would be predicted in the case of glia-released neurotransmitters (Limatola et al., 2000; Ragozzino et al., 2002). In support of this argument, we report that CX3CL1-induced current depression also occurs in a reconstituted cell expression system (Fig. 8B).
Our findings suggest that endogenous CX3CL1 is also likely to modulate glutamatergic transmission via similar signaling mechanisms. However, the failure of CX3CL1 antibody to modulate glutamatergic synaptic transmission suggests that the effect of endogenous CX3CL1 may be not detectable under our experimental conditions (although failure of the antibody to exhaustively scavenge endogenous chemokine cannot be ruled out) and disclosed only under conditions of physiological challenge, such as cellular or tissue stress. Consistently, in the brain, soluble CX3CL1 is generated on protease cleavage after multiple forms of stimulation (Chapman et al., 2000; Tsou et al., 2001). Furthermore, we have shown previously that, in cultured hippocampal neurons, CX3CL1 is released with a slow kinetics after prolonged exposure to glutamate and it protects neurons from excitotoxic cell death, suggesting a neuroprotective role for the chemokine (Limatola et al., 2005). Thus, the glutamatergic inhibition described here may contribute to CX3CL1 neuroprotective effects, providing a feedback mechanism against excessive glutamate exposure.
In conclusion, CX3CL1-dependent glutamatergic depression represents a novel form of synaptic modulation in hippocampal neurons. Fully understanding the impact of this modulation on brain function will require additional experiments aimed at the in vivo characterization of the functional effects of this chemokine and, in particular, at ascertaining the possible pathophysiological circumstances inducing its release.
Footnotes
This work was supported by a grant from the Ministero Università e Ricerca and by Fondo Integrativo Ricerca di Base Grants RBNE01NR34-003 (F.E.) and RBNE019J7C-003 (D.R.). We thank Drs. Clotilde Lauro and Myriam Catalano for helping in the determination of endogenous CX3CL1 levels and for hippocampal neuronal cultures, and Dr. Enrica Audero for animal breeding.
References
- Al-Aoukaty A, Rolstad B, Giaid A. MIP-3alpha, MIP-3beta and fractalkine induce the locomotion and the mobilization of intracellular calcium, and activate the heterotrimeric G proteins in human natural killer cells. Immunol. 1998;95:618–624. doi: 10.1046/j.1365-2567.1998.00603.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araujo DM, Cotman CV. Trophic effects of interleukin-4, -7 and -8 on hippocampal neuronal cultures: potential involvement of glial-derived factors. Brain Res. 1993;600:49–55. doi: 10.1016/0006-8993(93)90400-h. [DOI] [PubMed] [Google Scholar]
- Banke TG, Bowie D, Lee H, Huganir RL, Schousboe A, Traynelis SF. Control of GluR1 AMPA receptor function by cAMP-dependent protein kinase. J Neurosci. 2000;20:89–102. doi: 10.1523/JNEUROSCI.20-01-00089.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertollini C, Ragozzino D, Gross C, Limatola C, Eusebi F. Fractalkine/CX3CL1 depresses central synaptic transmission in mouse hippocampal slices. Neuropharmacol. 2006;51:816–821. doi: 10.1016/j.neuropharm.2006.05.027. [DOI] [PubMed] [Google Scholar]
- Bezzi P, Domercq M, Brambilla L, Galli R, Schols D, De Clercq E, Vescovi A, Bagetta G, Kollias G, Meldolesi J, Volterra A. CXCR4-activated astrocyte glutamate release via TNFα: amplification by microglia triggers neurotoxicity. Nat Neurosci. 2001;4:702–710. doi: 10.1038/89490. [DOI] [PubMed] [Google Scholar]
- Bialojan C, Takai A. Inhibitory effect of a marine-sponge toxin, okadaic acid, on protein phosphatases. Specificity and kinetics. Biochem J. 1988;256:283–290. doi: 10.1042/bj2560283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruno V, Copani A, Besong G, Scoto G, Nicoletti F. Neuroprotective activity of chemokines against N-methyl-d-aspartate or beta-amyloid-induced toxicity in culture. Eur J Pharmacol. 2000;399:117–121. doi: 10.1016/s0014-2999(00)00367-8. [DOI] [PubMed] [Google Scholar]
- Carroll RC, Lissin DV, von Zastrow M, Nicoll RA, Malenka RC. Rapid redistribution of glutamate receptors contributes to long-term depression in hippocampal cultures. Nat Neurosci. 1999;2:454–460. doi: 10.1038/8123. [DOI] [PubMed] [Google Scholar]
- Chapman GA, Moores K, Harrison D, Campbell CA, Steward BR, Strijbos PJLM. Fractalkine cleavage from neuronal membranes represents an acute event in the inflammatory response to excitotoxic brain damage. J Neurosci. 2000;20:RC87. doi: 10.1523/JNEUROSCI.20-15-j0004.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez-Noriega LE, Stevens CF. Increased transmitter release at excitatory synapses produced by direct activation of adenylate cyclase in rat hippocampal slices. J Neurosci. 1994;14:310–317. doi: 10.1523/JNEUROSCI.14-01-00310.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colbran RJ. Protein phosphatases and calcium/calmodulin-dependent protein kinase II-dependent synaptic plasticity. J Neurosci. 2004;24:8404–8409. doi: 10.1523/JNEUROSCI.3602-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combadiere C, Gao J, Tiffany HL, Murphy PM. Gene cloning, RNA distribution, and functional expression of mCX3CR1, a mouse chemotactic receptor for the CX3C chemokine fractalkine. Biochem Biophys Res Commun. 1998;253:728–732. doi: 10.1006/bbrc.1998.9849. [DOI] [PubMed] [Google Scholar]
- Dargan SL, Parker J. Buffer kinetics shape the spatiotemporal patterns of IP3-evoked Ca2+ signals. J Physiol (Lond) 2003;553:775–788. doi: 10.1113/jphysiol.2003.054247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deiva K, Geeraerts T, Salim H, Leclerc P, Hery C, Hugel B, Freyssinet JM, Tardieu M. Fractalkine reduces N-methyl-d-aspartate-induced calcium flux and apoptosis in human neurons through extracellular signal-regulated kinase activation. Eur J Neurosci. 2004;20:3222–3232. doi: 10.1111/j.1460-9568.2004.03800.x. [DOI] [PubMed] [Google Scholar]
- Derkach V, Barria A, Soderling TR. Ca2+/calmodulin-kinase II enhances channel conductance of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionate type glutamate receptors. Proc Natl Acad Sci USA. 1999;96:3269–3274. doi: 10.1073/pnas.96.6.3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrunz LE, Stevens CF. Heterogeneity of release probability, facilitation, and depletion at central synapses. Neuron. 1997;18:995–1008. doi: 10.1016/s0896-6273(00)80338-4. [DOI] [PubMed] [Google Scholar]
- Eckstein F. Nucleoside phosphorothioates. Annu Rev Biochem. 1985;54:367–402. doi: 10.1146/annurev.bi.54.070185.002055. [DOI] [PubMed] [Google Scholar]
- Garaschuk O, Schneggenburger R, Schirra C, Tempia F, Konnerth A. Fractional Ca2+ currents through somatic and dendritic glutamate receptor channels of rat hippocampal CA1 pyramidal neurones. J Physiol (Lond) 1996;491:757–772. doi: 10.1113/jphysiol.1996.sp021255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giovannelli A, Limatola C, Ragozzino D, Mileo AM, Ruggieri A, Ciotti MT, Mercanti D, Santoni A, Eusebi F. CXC chemokines interleukin-8 (IL-8) and growth-related gene product α (GROα) modulate Purkinje neuron activity in mouse cerebellum. J Neuroimmunol. 1998;92:122–132. doi: 10.1016/s0165-5728(98)00192-1. [DOI] [PubMed] [Google Scholar]
- Glitsch M, Parra P, Llano I. The retrograde inhibition of IPSCs in rat cerebellar purkinje cells is highly sensitive to intracellular Ca2+ Eur J Neurosci. 2000;12:987–993. doi: 10.1046/j.1460-9568.2000.00994.x. [DOI] [PubMed] [Google Scholar]
- Harrison JK, Jiang Y, Chen S, Xia Y, Maciejewski D, McNamara RK, Streit WJ, Salafranca MN, Adhikari S, Thompson DA, Botti P, Bacon KB, Feng L. Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Proc Natl Acad Sci USA. 1998;95:10896–10901. doi: 10.1073/pnas.95.18.10896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haskell CA, Hancock WW, Salant DJ, Gao W, Csizmadia V, Peters W, Faia K, Fituri O, Rottman JB, Charo IF. Targeted deletion of CX(3)CR1 reveals a role for fractalkine in cardiac allograft rejection. J Clin Invest. 2001;108:679–688. doi: 10.1172/JCI12976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatori K, Nagai A, Heisel R, Ryu JK, Kim SU. Fractalkine and fractalkine receptors in human neurons and glial cells. J Neurosci Res. 2002;69:418–426. doi: 10.1002/jnr.10304. [DOI] [PubMed] [Google Scholar]
- Herzig S, Neumann J. Effects of serine/threonine protein phosphatases on ion channels in excitable membranes. Physiol Rev. 2000;80:173–210. doi: 10.1152/physrev.2000.80.1.173. [DOI] [PubMed] [Google Scholar]
- Hestrin S, Nicoll RA, Perkel DJ, Sah P. Analysis of excitatory synaptic action in pyramidal cells using whole-cell recording from rat hippocampal slices. J Physiol (Lond) 1990;422:203–225. doi: 10.1113/jphysiol.1990.sp017980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes PM, Botham MS, Frentzel S, Mir A, Perry VH. Expression of fractalkine (CX3CL1) and its receptor, CX3CR1, during acute and chronic inflammation in the rodent CNS. Glia. 2002;37:314–327. [PubMed] [Google Scholar]
- Kameyama K, Lee HK, Bear M, Huganir RL. Involvement of a postsynaptic protein kinase A substrate in the expression of homosynaptic long-term depression. Neuron. 1998;21:1163–1175. doi: 10.1016/s0896-6273(00)80633-9. [DOI] [PubMed] [Google Scholar]
- Kansra V, Groves C, Gutierrez-Ramos JC, Polakiewicz RD. Phosphatidylinositol 3-kinase-dependent extracellular calcium influx is essential for CX(3)CR1-mediated activation of the mitogen-activated protein kinase cascade. J Biol Chem. 2001;276:31831–31838. doi: 10.1074/jbc.M009374200. [DOI] [PubMed] [Google Scholar]
- Krathwohl MD, Kaiser JL. Chemokines promote quiescence and survival of human neural progenitor cells. Stem Cells. 2004;22:109–118. doi: 10.1634/stemcells.22-1-109. [DOI] [PubMed] [Google Scholar]
- Kullmann DM, Asztely F, Walker MC. The role of mammalian ionotropic receptors in synaptic plasticity: LTP, LTD and epilepsy. Cell Mol Life Sci. 2000;57:1551–1561. doi: 10.1007/PL00000640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lax P, Limatola C, Fucile S, Trettel F, Di Bartolomeo S, Renzi M, Ragozzino D, Eusebi F. Chemokine receptor CXCR2 regulates the functional properties of AMPA-type glutamate receptor GluR1 in HEK cells. J Neuroimmunol. 2002;129:66–73. doi: 10.1016/s0165-5728(02)00178-9. [DOI] [PubMed] [Google Scholar]
- Lee HK, Kameyama K, Huganir RL, Bear MF. NMDA induces long-term synaptic depression and dephosphorylation of the GluR1 subunit of AMPA receptors in hippocampus. Neuron. 1998;21:1151–1162. doi: 10.1016/s0896-6273(00)80632-7. [DOI] [PubMed] [Google Scholar]
- Lee HK, Barbarosie M, Kameyama K, Bear MF, Huganir RL. Regulation of distinct AMPA receptor phopshorylation sites during bidirectional synaptic plasticity. Nature. 2000;405:955–959. doi: 10.1038/35016089. [DOI] [PubMed] [Google Scholar]
- Limatola C, Giovannelli A, Maggi L, Ragozzino D, Castellani L, Ciotti MT, Vacca F, Mercanti D, Santoni A, Eusebi F. SDF-1α-mediated modulation of synaptic transmission in rat cerebellum. Eur J Neurosci. 2000;12:2497–2504. doi: 10.1046/j.1460-9568.2000.00139.x. [DOI] [PubMed] [Google Scholar]
- Limatola C, Lauro C, Catalano M, Di Angelantonio S, Bertollini C, Ragozzino D, Eusebi F. Chemokine fractalkine reduces glutamate-mediated excitotoxic death of cultured hippocampal neurons. J Neuroimmunol. 2005;166:19–28. doi: 10.1016/j.jneuroim.2005.03.023. [DOI] [PubMed] [Google Scholar]
- Lin JY, Dubey R, Funk GD, Lipski J. Receptor subtype-specific modulation by dopamine of glutamatergic responses in striatal medium spiny neurons. Brain Res. 2003;959:251–262. doi: 10.1016/s0006-8993(02)03757-5. [DOI] [PubMed] [Google Scholar]
- Lisman JA. A mechanism for the Hebb and the anti-Hebb processes underlying learning and memory. Proc Natl Acad Sci USA. 1989;86:9574–9578. doi: 10.1073/pnas.86.23.9574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M, Grove EA, Miller RJ. Abnormal development of the hippocampal dentate gyrus in mice lacking the CXCR4 chemokine receptor. Proc Natl Acad Sci USA. 2002;99:7090–7095. doi: 10.1073/pnas.092013799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, Kauer JA, Perkel DJ, Mauk MD, Kelly PT, Nicoll RA, Waxham MN. An essential role for postsynaptic calmodulin and protein kinase activity in long-term potentiation. Nature. 1989;340:554–557. doi: 10.1038/340554a0. [DOI] [PubMed] [Google Scholar]
- Meucci O, Fatatis A, Simen AA, Bushell TJ, Gray PW, Miller RJ. Chemokines regulate hippocampal neuronal signaling and gp120 neurotoxicity. Proc Natl Acad Sci USA. 1998;95:14500–14505. doi: 10.1073/pnas.95.24.14500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meucci O, Fatatis A, Simen AA, Miller RJ. Expression of CX3CR1 chemokine receptors on neurons and their role in neuronal survival. Proc Natl Acad Sci USA. 2000;97:8075–8080. doi: 10.1073/pnas.090017497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulkey RM, Herron CE, Malenka RC. An essential role for protein phosphatases in hippocampal long-term depression. Science. 1993;261:1051–1055. doi: 10.1126/science.8394601. [DOI] [PubMed] [Google Scholar]
- Mulkey RM, Endo S, Shenolikar S, Malenka RC. Involvement of a calcineurin/inhibitor-1 phosphatase cascade in hippocampal long-term depression. Nature. 1994;369:486–488. doi: 10.1038/369486a0. [DOI] [PubMed] [Google Scholar]
- Nishiyori A, Minami M, Ohtani Y, Takami S, Yamamoto J, Kawaguchi N, Kume T, Akaike A, Satoh M. Localization of fractalkine and CX3CR1 mRNAs in rat brain: does fractalkine play a role in signaling from neuron to microglia? FEBS Lett. 1998;429:167–172. doi: 10.1016/s0014-5793(98)00583-3. [DOI] [PubMed] [Google Scholar]
- Oh SB, Endoh T, Simen AA, Ren D, Miller RJ. Regulation of calcium currents by chemokines and their receptors. J Neuroimmunol. 2002;123:66–75. doi: 10.1016/s0165-5728(01)00485-4. [DOI] [PubMed] [Google Scholar]
- Ragozzino D, Renzi M, Giovannelli A, Eusebi F. Stimulation of chemokine CXC receptor 4 induces synaptic depression of evoked parallel fibers inputs onto Purkinje neurons in mouse cerebellum. J Neuroimmunol. 2002;127:30–36. doi: 10.1016/s0165-5728(02)00093-0. [DOI] [PubMed] [Google Scholar]
- Robinson S, Tani M, Strieter RM, Ransohoff RM, Miller RH. The chemokine growth-related oncogene-α promotes spinal cord oligodendrocytes precursor proliferation. J Neurosci. 1998;18:10457–10463. doi: 10.1523/JNEUROSCI.18-24-10457.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche KW, O'Brien RJ, Mammer AL, Bernhardt J, Huganir RL. Characterization of multiple phosphorylation site on AMPA receptor GluR1. Neuron. 1996;16:1179–1188. doi: 10.1016/s0896-6273(00)80144-0. [DOI] [PubMed] [Google Scholar]
- Snyder EM, Colledge M, Crozier RA, Chen WS, Scott JD, Bear MF. Role for A kinase-anchoring proteins (AKAPS) in glutamate receptor trafficking and long term synaptic depression. J Biol Chem. 2005;280:16962–16968. doi: 10.1074/jbc.M409693200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokolova IV, Lester HA, Davison N. Postsynaptic mechanisms are essential for FSK-induced potentiation of synaptic transmission. J Neurophysiol. 2006;95:2570–2579. doi: 10.1152/jn.00617.2005. [DOI] [PubMed] [Google Scholar]
- Song I, Huganir RL. Regulation of AMPA receptors during synaptic plasticity. Trends Neurosci. 2002;11:578–588. doi: 10.1016/s0166-2236(02)02270-1. [DOI] [PubMed] [Google Scholar]
- Tarozzo G, Bortolazzi S, Crochemore C, Chen SC, Lira AS, Abrams JS, Beltramo M. Fractalkine protein localization and gene expression in mouse brain. J Neurosci Res. 2003;73:81–88. doi: 10.1002/jnr.10645. [DOI] [PubMed] [Google Scholar]
- Tavalin SJ, Colledge M, Hell JW, Langeberg LK, Huganir RL, Scott JD. Regulation of GluR1 by the A-kinase anchoring protein 79 (AKAP79) signaling complex shares properties with long-term depression. J Neurosci. 2002;22:3044–3051. doi: 10.1523/JNEUROSCI.22-08-03044.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran PB, Miller RJ. Chemokine receptors: signposts to brain development and disease. Nat Rev Neurosci. 2003;4:444–455. doi: 10.1038/nrn1116. [DOI] [PubMed] [Google Scholar]
- Tsou CL, Haskell CA, Charo IF. Tumor necrosis factor-α-converting enzyme mediates the inducible cleavage of fractalkine. J Biol Chem. 2001;276:44622–44626. doi: 10.1074/jbc.M107327200. [DOI] [PubMed] [Google Scholar]
- Unno T, Komori S, Ohashi H. Microtubule cytoskeleton involvement in muscarinic suppression of voltage-gated calcium channel current in guinea-pig ileal smooth muscle. Br J Pharmacol. 1999;127:1703–1711. doi: 10.1038/sj.bjp.0702711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winder DG, Sweatt JD. Roles of serine/threonine phosphatases in hippocampal synaptic plasticity. Nat Rev Neurosci. 2001;2:461–474. doi: 10.1038/35081514. [DOI] [PubMed] [Google Scholar]
- Wu K, Len GW, McAuliffe G, Ma C, Tai JP, Xu F, Black IB. Brain-derived neurotrophic factor acutely enhances tyrosine phosphorylation of the AMPA receptor subunit GluR1 via NMDA receptor-dependent mechanisms. Brain Res Mol Brain Res. 2004;130:178–186. doi: 10.1016/j.molbrainres.2004.07.019. [DOI] [PubMed] [Google Scholar]
- Xie CW, Lewis DV. Involvement of cAMP-dependent protein kinase in mu-opioid modulation of NMDA-mediated synaptic currents. J Neurophysiol. 1997;78:759–766. doi: 10.1152/jn.1997.78.2.759. [DOI] [PubMed] [Google Scholar]
- Yuste R, Majewska A, Cash SS, Denk W. Mechanisms of calcium influx into hippocampal spines: heterogeneity among spines, coincidence detection by NMDA receptors, and optical quantal analysis. J Neurosci. 1999;19:1976–1987. doi: 10.1523/JNEUROSCI.19-06-01976.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]