

Introduction
As neuroscientists, we can be so taken by the complexity and unique capabilities of the brain that we occasionally forget that it is part of a larger integrated biological system reliant on signaling and communication throughout the organism. The interactions between key organs that release hormones (e.g., gonads) and the nervous system are an excellent reflection of this “whole-brain, whole-body” level of integration. For example, effects of estrogens such as 17-β-estradiol (the major estrogen in most mammals, including women, referred to henceforward as estradiol) on a broad array of brain regions and the widespread distribution of estrogen receptors throughout the entire brain highlight the extraordinary integrative power of this hormone. An important concept in this interaction is that the brain both controls estrogen release through the hypothalamus–pituitary–gonadal (HPG) axis and responds to estrogen. Neuroendocrine function initiates in the hypothalamus (see below, section by A.C.G.) (Fig. 1), but the circuits that respond to estrogens go well beyond the hypothalamus to include neocortex, hippocampus, and brainstem. In addition, estradiol plays a key role in the neurobiology of aging, because endocrine and neural senescence overlap in time and are mechanistically intertwined in complex feedback loops. This aspect of aging is fundamental for humans, because all women will experience a dramatic drop in circulating estrogens if they live long enough to reach the menopause transition. Interestingly, at the turn of the past century, both life expectancy and the average age of onset of menopause for women in America was slightly over 50 years, whereas currently women can expect to live until 80 years of age, although the average age of menopause remains in the early 50s. Given the importance of the brain as a target organ for estrogen, it is not surprising that many of the complaints that prompt women to seek treatment related to menopause are neurological in origin. From this perspective, basic neuroscience has a great deal to contribute to the clinical issues surrounding estrogen, menopause, and the aging brain.
Figure 1.

Interactions between the brain and reproductive endocrine system. The hypothalamic–pituitary–gonadal axis of females is shown, with the three levels of regulation of reproductive function. GnRH neurons in the hypothalamus (shown in blue) release the decapeptide into the portal capillary vasculature, leading to the pituitary gland (shown in purple). Pituitary gonadotropes release the gonadotropins LH and FSH into the general circulation. LH and FSH act on their receptors on the ovary (shown in pink) to regulate sex steroid hormone production and release and folliculogenesis and ovulation. Sex steroids in turn are released into the circulation in which they exert effects on the body, and they also exert feedback actions on the hypothalamus and pituitary gland. In addition, steroids exert regulatory actions on receptors in nonreproductive brain regions, including (but not limited to) prefrontal cortex and hippocampus, thalamus, and brainstem. Additional communication between reproductive and nonreproductive brain regions occurs via neural circuitry linking the hypothalamus to other CNS systems (shown with light blue arrows). Some of this circuitry probably involves glutamate inputs to GnRH neurons, acting on NMDARs on GnRH neurons and their surrounding hypothalamic milieu. BS, Brainstem; CB, cerebellum; CC, corpus callosum; FC, frontal cortex; HIPPO, hippocampus; HYP, hypothalamus; PIT, pituitary; THAL, thalamus.
Over the past 20–30 years, a clinical body of work investigating the nature of neurologic changes that can accompany menopause has emerged. There has been a particular emphasis on whether or not cognitive function changes or the risk for depression increases with menopause and the degree to which such alterations, if present, respond to hormone treatment. Treatment commonly consists of either combined hormone treatment [estrogen plus a progestin (HT)] or unopposed estrogen treatment [estrogen alone (ET)]. Numerous studies have revealed beneficial effects on cognition across multiple HT/ET regimens, as reviewed recently by Sherwin (2006). Simultaneously, a large body of work on animal models emerged that provided compelling evidence for the potential of estrogen to alter synaptic circuitry in hypothalamus, hippocampus, and more recently neocortex (see section by J.H.M.), as well as its capacity to be neuroprotective (Bryant et al., 2006). In addition, animal studies have demonstrated positive effects of ET on cognitive behavior, particularly in nonhuman primates (NHPs), although the extent and nature of cognitive enhancement varies with age (Lacreuse, 2006).
Thus, a consensus began to emerge (although not without controversy) that HT/ET at the time of the menopause transition and afterward could have beneficial effects on several neurological symptoms. However, investigators in this arena received a very different message when the largest randomized clinical trial of HT ever conducted, the Women's Health Initiative (WHI) studies, began to publish their results on cognition in 2003 investigating the effects of daily administration of a popular combined HT. The WHI Memory Study (WHIMS) concluded that this HT regimen did not improve cognitive function (Rapp et al., 2003b) and “increased the risk for probable dementia in postmenopausal women aged 65 or older” (Shumaker et al., 2003b). Related WHIMS studies with unopposed estrogens (i.e., ET) also failed to protect against cognitive decline, although this regimen did not carry the same risk for increased incidence of dementia (Espeland et al., 2004; Shumaker et al., 2004). The public reaction to these studies permeated the media, in which the distinctions between the natural hormones estradiol and progesterone and the pharmaceutical formulations were rarely mentioned, yet there are critically important differences in their bioactivity (see section by R.D.B.). In addition, few people reading about the WHI studies appreciated the fact that the treatment had been initiated in women 65 years or older, which may be too late to reap certain of the neurological benefits of ET or combined HT (Maki, 2006; Manson et al., 2006). In the aftermath of the WHI studies, both the clinical and basic science communities were forced to try and make sense of these divergent results and take a careful look at the nature of HT and ET formulations and variables, such as the time of initiation of treatment and the age of the subjects (see section by P.J.S.). Although the WHI investigators appropriately tested the most commonly used combined HT regimen (i.e., Prempro), there are important disconnects between daily administration of this formulation to women and endocrine physiology as defined in both humans and animal models (Turgeon et al., 2004). In addition, although most women considering HT/ET are in the menopausal transition, until recently the animal literature was based almost exclusively on ovariectomy of young animals, followed by short-term treatments with estradiol alone or with progesterone. Thus, we are in the midst of an intense reevaluation of both the clinical studies and the preclinical studies, with an emerging consensus that each must inform the other in a more directed manner than in the past. From this perspective, the animal models of ET and/or HT have never been more important (Adams et al., 2001b; Gore, 2001; Bellino and Wise, 2003; Gibbs and Gabor, 2003; Turgeon et al., 2004; Brinton, 2005; Lacreuse, 2006). In the 36th Annual Meeting of the Society for Neuroscience presentations and the four sections below (designated by presenter/author initials), we have attempted to provide insight into the current clinical issues surrounding menopause and HT/ET with respect to the brain, as well as a preclinical perspective on hypothalamic control of reproductive aging, synaptic effects of estradiol related to cognitive performance, and analyses of the cellular mechanisms of ovarian hormones. In all sections, we have paid particular attention to the important interface between endocrine senescence and neural aging.
Reproductive aging, sex steroids, and behavior in women (by P.J.S.)
A burgeoning array of preclinical studies has documented the manifold regulatory actions of ovarian steroids on neuronal biochemistry, cellular metabolism, and behavior. Consistent with these findings were the ostensible beneficial effects of HT/ET on cognitive function and on the course of dementia, including Alzheimer's disease (AD), repeatedly reported in observational studies. In contrast, the impact of reproductive aging and HT/ET on either mood disorders or sexual function was considerably more controversial. Recently, the results of several randomized controlled trials of HT and ET have questioned previously held beliefs about the effects of ovarian steroids, both beneficial and not, on brain and behavior. Efforts to reconcile the discordance between studies have identified several important characteristics of the participants studied. These characteristics could contribute to several of the discrepancies in the literature and include age, stage of reproductive aging, duration of hypogonadism, and the presence of symptoms. A better understanding of the nature of these discrepancies will inform future studies of the clinical relevance of ovarian steroids and hormone therapies in women.
This section will first review the physiology of reproductive aging in women. Emphasis will be placed on the considerable variability in ovarian hormone secretion characterizing the menopause transition compared with the relatively stable endocrine milieu of the postmenopause (see section by A.C.G.). Second, studies will be described that have evaluated the effects of the menopause transition, hypogonadism, and HT/ET on measures of cognition, mood, and sexual function. Differences in observed outcomes in these measures will be identified. Finally, questions of clinical relevance will be presented that remain to be answered and that may help to reconcile some of the otherwise discordant findings in the current literature.
Characteristics of the menopause transition: a state of considerable variability in ovarian hormone secretion
The process of reproductive aging in women has several unique features that distinguish it from reproductive senescence in most animal species (described below in the section by A.C.G.). Although the average age of the menopause in women is 51 years, the transition from normal reproductive life to the last year after the final menstrual period (referred to as either the menopause transition or the perimenopause) may be 10–15 years in duration. The endocrinology of the menopause transition has yet to be fully characterized and represents a complex interplay of actions at all levels of the hypothalamic–pituitary–ovarian axis (Santoro, 2005). Nonetheless, evidence suggests that this phase of reproductive aging occurs in stages. The early menopause transition is associated with lower ovarian inhibin secretion, which in turn reduces the restraint on both the hypothalamus and pituitary and results in elevated pituitary gonadotropin [follicle-stimulating hormone (FSH)] secretion. In addition to reduced ovarian inhibin secretion, age-related increases in gonadotropin-releasing hormone (GnRH) production could contribute to the elevated pituitary gonadotropin levels (Maffucci and Gore, 2006; Yin and Gore, 2006). Importantly, ovarian estradiol secretion is normal or at times elevated during this early stage of the menopause transition (Santoro, 2005). As ovarian aging proceeds into the late menopause transition, despite occasional episodes of normal cycling, women are exposed to periods of estrogen withdrawal, fewer ovulatory cycles, and prolonged hypogonadism, ultimately leading to the last menstrual period. Five to 10 years after the menopause, the activity of the HPG axis becomes relatively more stable and is characterized by persistent hypogonadism and relatively tonic (not phasic) elevated gonadotropin secretion (Hall and Gill, 2001). In addition to changes in ovarian estradiol and progesterone secretion, production rates of several other hormones (e.g., androgens and growth factors) decline with aging and overlap with the menopause transition (Burger et al., 2000; Davison et al., 2005).
These findings emphasize that, if the impact of reproductive hormones on brain function and behavior during the menopause transition is to be examined, then it makes no sense to collapse the process of reproductive aging into a single physiologic event.
Cognitive function and dementia: discrepant findings and potential sources of variation
The evidence in women that ovarian steroids play a regulatory role in cognitive function, at first glance at least, has gone from all good for the brain to all bad. Both observational- and clinic-based studies have suggested that combined HT and unopposed ET in hypogonadal women improve cognition, lessen the risk for the development of dementia, and, possibly, improve the severity and course of dementia. Preclinical studies have identified biologically plausible mechanisms by which estrogen could enhance neuronal function, improve neuronal resilience, and serve as a neuroprotective agent (see below, sections by J.H.M. and R.D.B.).
Epidemiological surveys prospectively monitoring women as they progress through the menopause transition have suggested that self reports of decreased concentration and poor memory are frequent accompaniments of this phase of life and the postmenopause (Woods et al., 2000). Additionally, the possibility that declining ovarian estrogen secretion could exacerbate the age-related decrement in episodic memory (Maki et al., 2001; Henderson et al., 2003) was suggested by several clinic-based trials. These trials demonstrated the beneficial effects of short-term (i.e., 2–12 weeks) ET on measures of verbal learning and memory (Sherwin, 2003). However, to date, studies have not consistently identified deficits in performance corresponding to self reports of cognitive impairment in perimenopausal women. Relatively recent meta-analyses, examining the effects of ET on measures of cognitive function, conclude that HT/ET has no universal benefits on cognitive function in hypogonadal women (Yaffe et al., 1998; LeBlanc et al., 2001; Hogervorst et al., 2002). Nonetheless, the results of these meta-analyses did suggest that there were beneficial effects of HT/ET on several domains of cognitive function (i.e., verbal memory, vigilance, reasoning, and motor speed) limited to recently hypogonadal women. Specifically, cognitive benefits were observed in symptomatic perimenopausal women and in women in whom hypogonadism was recently induced by medical or surgical interventions.
In addition to the potential beneficial effects of estrogen on episodic memory, some evidence suggested that HT reduced the risks of both dementia (including AD) and mild cognitive impairment (MCI). MCI is a condition thought to represent a transitional state between normal cognition and dementia in some individuals, with a 12% conversion rate from MCI to dementia each year (Gauthier et al., 2006). Observational studies repeatedly document that women taking HT enjoy an ∼30% reduced risk for dementia compared with women not taking HT [odds ratio range, 0.30–0.66 (Yaffe et al., 1998; Hogervorst et al., 2002)]. Thus, observational studies suggest that declining reproductive function could be a modifiable risk factor for dementia or that HT/ET could serve a protective role against some of the risks for developing dementia. In contrast, several recent findings (Shumaker et al., 2003b; Resnick et al., 2006; Yaffe et al., 2006) have suggested that these observational studies contain sample selection biases and experimental confounds that spuriously increased the apparent beneficial effects of estrogen and failed to capture some potentially important adverse effects of HT/ET. For example, recent randomized placebo-controlled trials (RCTs) sponsored by the WHI investigated the effects of HT in community-dwelling women between the ages of 65 and 79 years (Rossouw et al., 2002; Anderson et al., 2004). HT was in the form of continuous conjugated equine estrogens (CEEs) (0.625 mg/d) with medroxyprogesterone acetate (MPA) (2.5 mg/d; Prempro) in women with a uterus. ET consisted of CEE (0.625 mg/d) alone in women without a uterus. After an average follow-up of 4–5 years, the WHIMS failed to observe any improvement in measures of global cognition or rates of either MCI or dementia in women taking HT/ET compared with placebo (Rapp et al., 2003b; Shumaker et al., 2003b; Espeland et al., 2004; Shumaker et al., 2004). Indeed, in contrast to the observational studies, the WHIMS observed an increased risk of dementia with HT [consistent with the WHI observations of increased rates of cerebrovascular disease and thrombotic phenomena related to combined HT (Rossouw et al., 2002)]. The WHI study was a large, multicenter, RCT and represents the “highest” level of medical evidence (Harris et al., 2001). The results of the observational studies, therefore, were essentially dismissed as being flawed by several confounds, including a healthy user bias and a masking of initial mortality rates attributable to the limited frequency of follow-ups in the early phases of the observational studies. Nevertheless, several recent observational studies have identified that the stage of reproductive aging at which HT/ET is started modifies the risk of dementia. In these studies, women who take HT/ET during the late menopause transition or early postmenopause have a lower risk of dementia than those starting HT/ET later (Zandi et al., 2002; Henderson et al., 2005). Thus, the timing of starting HT/ET relative to the menopause has been proposed to be one factor explaining the otherwise discordant observations between the observational studies and the RCTs (Resnick and Henderson, 2002; Manson et al., 2006). Recent preclinical studies reviewed below highlight the importance of timing of ET in this report.
Mood disorders: evidence of an important temporal link between the onset of depression and the menopause transition in a subgroup of women
Historically, there has been a presumption that declining reproductive function plays no role in the onset of mood disorders that occur during midlife in women. For example, depression that occurred during the menopause transition was presumed to be either secondary to the disruptive effects of hot flushes on sleep or a coincidental occurrence of an otherwise recurrent mood disorder. The symptoms of depression during the menopause transition also were assumed to be transient and of such minor severity that they were dismissed to be of little clinical consequence. Recent studies, however, suggest that these presumptions are incorrect. First, several community-based longitudinal studies have reported the relative independence of depressions during the menopause transition and hot flushes: both occur at this stage of life, but depression is not simply caused by hot flushes (Avis et al., 2001). Second, recent longitudinal studies that followed women with no past history of depression demonstrated an increased risk of first-onset depressions during the late menopause transition (Schmidt et al., 2004; Cohen et al., 2006; Freeman et al., 2006). Finally, both major and minor depressions are clinically significant to women at midlife, because both are associated with an increased risk for several other medical conditions (Wassertheil-Smoller et al., 2004) relevant to the health of women at midlife (e.g., cardiovascular disease, dementia, and the metabolic syndrome).
The majority of women do not develop depression during the menopause transition, and, therefore, reproductive aging is not uniformly associated with either depressive symptoms or the syndrome of depression. Nonetheless, despite numerous studies concluding that the menopause is not associated with an increased risk for developing depression in women, several other longitudinal, community-based studies reported an association between the menopause transition and an increased risk for depression (Schmidt, 2005). Indeed, five recent longitudinal studies all have documented an increased risk for depression during the menopause transition, with odds ratios ranging from 1.8 to 2.9 compared with the premenopause (Bromberger et al., 2001; Freeman et al., 2004, 2006; Schmidt et al., 2004; Cohen et al., 2006). In particular, two recent studies (Cohen et al., 2006; Freeman et al., 2006) observed a 2 to 2.5 times greater risk for the first onset of depression during the late menopause transition compared with the premenopause. These studies differ from previous surveys in their use of criteria to distinguish the menopause transition from either the premenopause or postmenopause. Additionally, several of these studies used syndromal measures of depression, including structured diagnostic interviews. It warrants emphasis that the majority of the women in these studies remained asymptomatic throughout the menopause transition. However, these data suggest that events surrounding the final menstrual period may predispose some women to develop clinically significant depressive illness. Although several factors could precipitate depression in these women, endocrine events are suggested by the stage of the menopause transition (i.e., late) during which depressions appeared. The late transition is characterized by more prolonged hypogonadism than the early perimenopause, during which estradiol secretion may be increased. Thus, the timing of appearance of the depressions observed suggest an endocrine mechanism related to the perimenopause (estradiol withdrawal and/or recent-onset of prolonged hypogonadism) in the pathophysiology of perimenopausal depression.
Efforts to investigate the potential role of declining ovarian hormone secretion in the onset of depression have examined the effects on mood of administering HT/ET in women with perimenopausal and postmenopausal depression. The antidepressant efficacy of estradiol has been examined in three relatively recent RCTs of women meeting standardized diagnostic criteria for major and minor depression, who were randomly assigned to enter double-blind, placebo-controlled trials (Schmidt et al., 2000; Soares et al., 2001; Morrison et al., 2004). In perimenopausal women, short-term administration (3–8 weeks) of estradiol significantly decreased depression scores compared with both baseline and placebo conditions. In one study, a full or partial therapeutic response was seen in 80% of perimenopausal women on estradiol compared with 22% of those on placebo (Schmidt et al., 2000). The efficacy of ET in perimenopausal depression is consistent with the observed effect size (0.69) in a recent meta-analysis of studies examining the effects of estrogen on mood (Zweifel and O'Brien, 1997). The therapeutic response to estradiol was observed in both major and minor depression as well as in women with and without hot flushes. Thus, the efficacy of ET in perimenopausal depression is not solely a product of its ability to reduce the distress of hot flushes. In contrast to these studies in perimenopausal depression, the administration of estradiol under similar conditions failed to improve mood in depressed women who were 5–10 years postmenopause (Morrison et al., 2004). Thus, the effects of estradiol on depression may be limited to perimenopausal women. Additionally, as with the potential effects of estrogen on the course of dementia, the stage of reproductive aging at which women present and/or commence ET might modify the observed outcomes.
In summary, the majority of women do not develop depression during or after the menopause transition. Nevertheless, recent prospective studies monitoring both reproductive status and mood have documented that, for some women, perimenopause-related events increase the risk for the onset of depression. The role of ovarian function in these episodes of depression is suggested, albeit indirectly, by both the timing of their onset relative to the last menstrual period and the antidepressant efficacy of short-term ET.
Sexual function: impact of deficient ovarian steroid secretion is variably expressed in women
In addition to the role of ovarian steroids in cognitive function and affective adaptation, another behavior, sexual function, is highly valued, intricately related to social behavior, and presumed to be regulated by ovarian steroids. Indeed, recent community-based studies have documented that, after natural or surgically induced menopause, ∼9 and 26% of women, respectively, will report a clinically significant loss of sexual desire (Leiblum et al., 2006). Preclinical studies have clearly documented both the neurocircuitry of sexual behavior and the important regulatory role estrogen serves in many aspects of this behavior (Pfaff, 1980). Although several small RCTs of HT/ET in postmenopausal women reported beneficial effects in several domains of sexual functioning (Dennerstein et al., 1980; Sherwin, 1991; Nathorst-Boos et al., 1993b; Wiklund et al., 1993; Sarrel et al., 1998), more recent trials were unable to confirm the initial positive results (Sherwin, 1985; Myers et al., 1990; Nathorst-Boos et al., 1993a). A recent study evaluated the effects of GnRH agonist-induced hypogonadism on sexual functioning in 20 young healthy women (Schmidt et al., 2006). Many (35%) of the women reported a clinically significant decline in libido (defined by at least a 50% decline in rating scale scores relative to baseline); however, estradiol failed to restore sexual functioning to that reported at baseline. Several recent RCTs reported improvements in libido after testosterone therapy in hypogonadal women (Shifren et al., 2000; Simon et al., 2005). Thus, it is likely that androgen secretion, which was partially suppressed by the GnRH agonist, accounted for the reported decline in libido. Interestingly, baseline level of sexual function, but not hormone levels, predicted the response to hypogonadism. Those women with the highest libido at baseline experienced the greatest decline during hypogonadism. Overall, the observed effects of hypogonadism on sexual function in our study, as well as in community-based studies, suggest that ovarian steroids are critical determinants of behavior in only some women. Thus, as with mood disorders and, possibly, cognitive function, there is not a simple relationship between declining ovarian function and loss of sexual function. Moreover, the impact of declining reproductive function on sexual function is variably expressed in women.
Conclusions
Before concluding that the basic neuroscience data documenting the important and beneficial effects of ovarian steroids on many of the processes of brain aging are irrelevant to all women, several remaining observations must be reconciled. First, an excessive amount of variance in outcomes could be introduced by failing to appreciate the considerable variability in the endocrinology of the menopause transition. As originally demonstrated in NHPs by Mikkola and Clarkson (2002) and in rats by Gibbs (2000), the duration of hypogonadism before the commencement of HT or ET is critical to the observed outcome, with earlier intervention (i.e., in recently hypogonadal animals) resulting in benefit. Second, even within a uniformly defined period of reproductive function, there can be significant variability in the response characteristics to removal or introduction of reproductive steroids. For example, the meta-analyses by Yaffe et al. (1998) and LeBlanc et al. (2001) suggested that the overall beneficial effects of HT/ET were secondary to the concurrent improvement in the perimenopausal symptoms that some study subjects experience. Additionally, observational studies document that women who started early and remained on HT/ET had lower risks of dementia. These women constitute a unique group in many aspects, including being healthier, more educated, and more compliant, as suggested by Matthews et al. (1996). However, these women also are distinguished from the majority of women to whom HT/ET was prescribed because they remained on HT/ET, whereas many women stopped after 1 year of therapy (i.e., 50–75% in some studies) (Eiken and Kolthoff, 1995; Ettinger and Pressman, 1999). Thus, early initiation and adherence to treatment could be related to the ongoing relief of the symptoms with which they originally presented for treatment. If we dismiss the variability in observed behavioral outcomes related to the menopause transition or HT/ET, then we could conclude incorrectly that declining ovarian steroids play no role in brain aging in any woman. We would, by so doing, eliminate an important conceptual tool for understanding the regulation of behavior and an equally powerful therapeutic tool for reducing symptoms in those women for whom ovarian steroids are critically involved in the regulation of affective, cognitive, and behavioral adaptation to aging.
Hypothalamic control of reproductive aging (by A.C.G.)
Although the hallmark of menopause is a loss of ovarian follicles, this process needs to be considered in the context of the three levels of the reproductive axis, comprising hypothalamus, pituitary, and ovary. During most of the reproductive life cycle, the hypothalamus provides the driving force on reproductive function. Therefore, the idea that menopause is attributable entirely to an ovarian change exclusive of input from the hypothalamus and/or pituitary gland is counterintuitive to this concept. Both animal models and studies in women indicate a causal role for the hypothalamus in reproductive failure. It has been extremely difficult to dissect out the relative role of each level of the HPG axis, but our interpretation is that each level of the HPG axis plays a unique role, both independently and also interactively, with the other levels in a potentially additive or synergistic manner to result in reproductive failure. These points will be discussed in detail below.
Virtually all mammals and many other vertebrates undergo declines in reproductive capacity during the aging process (Maffucci and Gore, 2006). However, menopause is unique to species with menstrual cycles such as women and higher primates. Ultimately, the loss of a woman's limited pool of ovarian follicles causes declines in circulating levels of the steroid hormone estradiol. Many of the major postmenopausal symptoms reported in women, such as osteoporosis, vasomotor symptoms, and vaginal dryness, are directly attributable to low circulating estradiol concentrations. As discussed above (see section by P.J.S.), other neurological symptoms may also relate to the menopausal decline in estradiol.
As mentioned above, the reproductive system comprises three inter-related levels that undergo both independent and interdependent age-related changes during the menopausal transition (Maffucci and Gore, 2006). These HPG components are as follows (Fig. 1): (1) the hypothalamus at the base of the brain, which secretes the neuropeptide GnRH from ∼1000 neurons into a portal capillary system; (2) the anterior pituitary gland, in which the GnRH decapeptide binds to its receptor on a subset of cells called gonadotropes, resulting in the synthesis of the gonadotropins, luteinizing hormone (LH), and FSH and their release into the general circulation; and (3) the gonad (the ovary in females), which responds to LH and FSH with the synthesis and release of sex steroid hormones such as estrogens, progestins, and androgens, and peptide hormones such as inhibin and activin. Follicular development and ovulation are also stimulated by actions of FSH and LH in the ovary. Although the primary signal for reproductive function is generated in the hypothalamic GnRH neurons, ovarian steroid hormones exert feedback regulation at both the brain and the pituitary to regulate output of GnRH and the gonadotropins (Fig. 1).
Patterns of reproductive senescence differ across species
Studies on the process of reproductive senescence in nonprimate species such as rodents, which do not menstruate but rather have estrous cycles, have shed considerable light on a causal brain role. The female rat is an important animal model of reproductive aging because it is big enough to perform reproductive physiology measures (e.g., hormone sampling), small enough to house cheaply, its genome is known, and available antibodies and probes enable investigators to study molecular and cellular mechanisms of reproductive aging (Maffucci and Gore, 2006; Yin and Gore, 2006). Like primates, rats have reproductive cycles, but, rather than the 28 d menstrual cycle, young adult female rats experience 4 d estrous cycles. Beginning in middle age (between ∼9 and 12 months of age), estrous cycles begin to become irregular and eventually cease with further aging. This transitional process is quite comparable with the menopausal transition in women, also characterized by increasingly irregular cycles and an ultimate cessation of menses (see above, section by P.J.S.). However, an important distinction must be drawn between rodent “estropause” and primate menopause. The loss of reproductive cycles in middle-aged rats does not occur concurrently with follicular atresia and its concomitant decline in estradiol concentrations. Rather, middle-aged acyclic rats are in a state of persistent estrus, with chronically high estradiol concentrations. It is important to keep this consideration in mind when using rats as models for reproductive aging. For this reason, many laboratories, including our own (J.H.M., A.C.G.) often turn to the ovariectomized (OVX) rat model. Using age-appropriate rats at young, middle-aged, and old ages, we ovariectomize the animals, wait 1 month, and then give ET (i.e., estradiol), with or without progestins, in physiologically relevant dosages (Gore et al., 2002; Yin and Gore, 2006). Another important difference between primates and rodents is that postmenopausal increases in GnRH or surrogates for GnRH (e.g., gonadotropins or gonadotropin-free a subunit) occur during the perimenopause (Hall and Gill, 2001; Maffucci and Gore, 2006), whereas in rodents, GnRH neurosecretion and the preovulatory GnRH/LH surge declines at estropause (Rubin and Bridges, 1989).
The circuitry driving GnRH neurons undergoes age-related changes
We have begun to elucidate the mechanisms for the primary hypothalamic change in GnRH neurons and their regulatory factors. Evidence for this role was provided by the following observations. (1) An ovary transplanted from an old acyclic rat to a young rat begins to show follicular development and ovulation, suggesting that the ovary is not a limiting factor for acyclicity in old rats (Krohn, 1955). (2) Acyclic old rats can be induced to ovulate by neurochemical, hormonal, or social stimuli (for review, see Gore, 2001). (3) Pulsatile GnRH release decreases in middle-aged rats (Rubin and Bridges, 1989) and increases in perimenopausal rhesus monkeys (Gore et al., 2004). Although there are species differences in these phenomena, both of these changes precede actual reproductive failure. (4) Activation of GnRH neurons during the preovulatory GnRH/LH surge, as indicated by GnRH gene expression or the immediate early gene fos expression in GnRH neurons, declines before reproductive senescence (Lloyd et al., 1994; Rubin et al., 1994; Gore et al., 2000b).
GnRH cell numbers do not change appreciably with aging so this is probably not the causal event for these alterations documented above (for review, see Yin and Gore, 2006). Rather, inputs to GnRH neurons from other neurotransmitters and glia in the CNS appear to be very important in causing age-associated changes in GnRH output (Gore, 2004). There are numerous factors that regulate GnRH neurons, both directly on GnRH cell bodies and terminals and indirectly via other inputs to GnRH cells (Gore, 2002). Among these many regulatory substances, our laboratory (A.C.G.) has been studying the role of glutamate, acting via its NMDA receptor (NMDAR), in modulating GnRH output during reproductive aging. The rationale for these studies from a neuroendocrine perspective was that NMDAR agonists stimulate, and antagonists inhibit, GnRH release, gene expression, and reproductive function (Bonavera et al., 1998; Gore et al., 2000a, 2002; Gore, 2001; Brann and Mahesh, 2005; Yin and Gore, 2006). In addition, age-related changes in NMDARs are observed in, and contribute to, other nonhypothalamic CNS dysfunctions during aging, including the normal age-related declines in cognitive function (Burke and Barnes, 2006). Therefore, it is reasonable to predict that changes in NMDARs in the hypothalamus may contribute to a loss of output from hypothalamic GnRH cells that may ultimately be manifested as alterations in ovarian function. These studies on NMDARs provide an important example of how one neurotransmitter system and its receptor (among many) contribute to the reproductive aging process.
The actions of glutamate on its NMDAR are exerted directly on GnRH cell bodies, localized in the rostral hypothalamus–preoptic area, as well as on GnRH neuroterminals in the median eminence, the region in which the portal capillary vasculature carries the GnRH decapeptide to the anterior pituitary gland. We have proposed that these are two distinct and potentially differential, albeit interrelated, functional units of the GnRH cell, based on differing effects of pharmacologic agents on GnRH perikarya and terminals (Yin and Gore, 2006). The large distance between GnRH cell bodies and their nerve terminals, as well as the lack of strong evidence for connectivity between GnRH perikarya, further supports this concept for local, and potentially differential, actions of glutamate. At the GnRH perikarya, glutamate and other neurotransmitters regulate GnRH gene expression and biosynthesis, and we showed that this changes with aging: when an NMDAR agonist was administered to female rats, it significantly stimulated GnRH mRNA levels in young rats but decreased levels in middle-aged rats (Gore et al., 2000a). We also found that GnRH somata coexpress NR1 and NR2B subunits of the NMDAR, and we specifically detected age-related changes in expression of the NR2B subunit in GnRH perikarya (Miller and Gore, 2002). This finding suggests alterations in NMDAR stoichiometry or in other functional properties of the NMDAR that may explain the opposite response of GnRH neurons to NMDAR agonists in young and middle-aged rats. Moreover, findings that both aging and estradiol alter the subcellular localization of the NR1 and NR2B subunits in hippocampus (see below, section by J.H.M.) highlight the importance of performing careful, region-specific analyses of aging-related changes in key neurotransmitter systems.
NMDARs are also detected in the median eminence, a region at the base of the hypothalamus in which hypothalamic releasing hormones are secreted from nerve terminals into the blood system, leading to the anterior pituitary gland (Fig. 1). GnRH secretion from the median eminence is regulated by NMDAR agonists even when the GnRH perikarya are not present (Purnelle et al., 1997; Kawakami et al., 1998b), suggesting a direct site of action on GnRH terminals. One laboratory demonstrated that GnRH terminals express NMDARs (Kawakami et al., 1998a). Preliminary evidence from our laboratory (A.C.G.) indicates that NMDARs may be localized within GnRH terminals on secretory vesicles and that this localization is influenced by both hormones and aging (Yin et al., 2002). This is an exciting new area of investigation for understanding how GnRH release may change and be regulated by NMDARs during reproductive aging.
Estradiol, NMDA receptors, and GnRH neurons interact to result in reproductive aging
Effects of glutamate in the hypothalamus may also occur on NMDARs on non-GnRH cells that are part of the neural network forming inputs to the GnRH neurosecretory system (Fig. 1). This concept is particularly important because most gonadal steroid feedback does not occur on GnRH perikarya, because estrogen receptor α (ERα), progestin receptors, and androgen receptors are lacking in GnRH nuclei (Shivers et al., 1983; Herbison et al., 1996) (for review, see Gore, 2002). Although GnRH neurons coexpress the ERβ (Hrabovszky et al., 2000), this appears to be at low levels and does not appear to be necessary for fertility (Kudwa et al., 2006). Instead, hypothalamic cells that provide inputs to GnRH neurons express nuclear hormone receptors, and this appears to be the most important mechanism for steroid feedback onto HPG function. For example, we found that, in the anteroventral periventricular nucleus (AVPV), a preoptic nucleus that is crucial for ovulatory function and makes direct projections to GnRH neurons (Wiegand et al., 1978; Simerly and Swanson, 1987), a substantial population of NMDAR-positive neurons also coexpresses ERα (Chakraborty et al., 2003a), and many cells in the rat AVPV coexpress NMDARs and ERβ (Chakraborty et al., 2003b). We also found that numbers of ERα-positive cells (Chakraborty et al., 2003c) and the coexpression of ERs and NMDARs change with aging in female rats (Chakraborty et al., 2003b). Our finding that ERs and NMDARs are extensively colocalized in the interneurons that regulate GnRH function probably represents a key mechanism for how steroids and neurotransmitters interact to influence GnRH gene expression and release and their changes during aging.
Implications of age-related changes in estradiol extend beyond the hypothalamus
Menopause in humans is characterized by a plummeting of circulating estradiol levels back to levels comparable with prepubertal girls. Although this is not a pathological event, it can result in pathologies and quality-of-life changes that necessitate medical interventions. Many of these effects go far beyond reproductive symptoms (see above, section by P.J.S.), and this emphasizes the important concept that any change in HPG activity, and hence hormonal output, will affect other, nonhypothalamic brain systems that respond to hormones. Reports that steroid hormone receptors such as ERs are abundant throughout the entire CNS (Simerly et al., 1990; Shughrue and Merchenthaler, 2000; Mitra et al., 2003) are consistent with diverse aging-related consequences of altered estradiol concentrations on neural function on both hypothalamic and other neural systems. Moreover, hormone feedback is not the only way the hypothalamus communicates with other CNS regions. Direct and indirect pathways convey information from the hypothalamus to cortex, thalamus, amygdala, brainstem, cerebellum, and most other brain regions (Fig. 1). Therefore, age-related changes in hypothalamic function may affect CNS function by these neural routes.
Conclusion
This section has provided evidence for changes in the hypothalamic level of the HPG axis that may play a causal role in the transition to reproductive senescence. Some of these hypothalamic changes are attributable to properties inherent to GnRH neurons themselves, such as gene expression and release. In addition, other changes are probably attributable to age-related alterations in neural inputs to GnRH cells from neurotransmitters such as glutamate and others, as well as their interactions with estradiol through ERs. Because the hypothalamus provides the primary signal driving the HPG axis, the consequence of a functional decline in the GnRH neurosecretory system is a loss of stimulation on the pituitary and the ovary. Any subsequent alteration in levels or the pattern of estradiol secretion from the ovary would then exert additional feedback actions on the brain via ERs. At the hypothalamic level, these effects are manifested by additional alterations in output from GnRH neurons and may extend to dysfunctions in sexual behavior and/or libido (e.g., see above, section by P.J.S.). In other brain regions such as hippocampus and frontal cortex, the loss or change in estradiol signaling may be manifested by some of the morphological and behavioral changes that are reported in animal models and in humans (see section above by P.J.S. and section below by J.H.M.) and in cell lines (see below, section by R.D.B.). As a whole, it is critical to take this whole-brain, whole-body approach to reproductive failure to understand the causes and consequences of this important aspect of normal aging in females.
Synaptic effects of estrogen in hippocampus and prefrontal cortex: implications for cognitive aging (by J.H.M.)
The series of papers from McEwen's laboratory in the early 1990s regarding the impact of estradiol on hippocampal synapses profoundly altered the prevailing views on cortical circuitry and plasticity. Most neuroscientists had never considered that estradiol, a gonadal steroid, could increase CA1 synapse number in a matter of days (Gould et al., 1990; Woolley et al., 1990; Woolley and McEwen, 1992, 1993). These experiments from the McEwen laboratory were all done in young adult female rats, but they provided the framework to extend such analyses to investigate estrogen-induced synaptic alterations in the context of aging in female rats and, more recently, NHPs. This section will review synaptic effects of estradiol on hippocampus in female rat and NHP models, with particular attention to the interactive effects of aging on the effect of ET. In addition, both behavioral and synaptic effects of estradiol implicating dorsolateral prefrontal cortex (dlPFC) in aged NHPs will be presented and discussed, particularly as they relate to issues relevant to women, menopause, and cognitive aging. The goal with respect to neurobiology of aging is to delineate the elements of neural aging that alter the response of the brain to endocrine influences, identify potential interactions between neural and endocrine senescence that augment functional decline, and provide translational insights for interventions that will sustain cognitive performance and be useful clinically.
Estrogen modulates synaptic circuitry of CA1 in the hippocampus
McEwen, Gould, Woolley, and colleagues demonstrated that spine and synapse density fluctuate with variations in ovarian steroid levels across the 4 d estrous cycle in young rats (Woolley et al., 1990; Woolley and McEwen, 1992), and ET in OVX female rats increases dendritic spine density (Gould et al., 1990; Woolley and McEwen, 1993) and synapses (Woolley et al., 1996) by 30% on CA1 pyramidal cells. These morphologic enhancements of CA1 excitatory synapses occur via a mechanism that is NMDAR dependent (Woolley and McEwen, 1994; Murphy and Segal, 1996), as reflected by enhanced NMDAR levels (Weiland, 1992; Gazzaley et al., 1996) and synaptic transmission (Woolley and McEwen, 1993; Foy et al., 1999). Thus, there appear to be interacting morphologic and neurochemical effects of estradiol in the hippocampus that augment NMDAR-mediated synaptic transmission in key hippocampal circuits subserving memory. Effects of estradiol on GABAergic (Murphy et al., 1998; Rudick and Woolley, 2001) and cholinergic (Gibbs, 2002; Gabor et al., 2003; Rudick et al., 2003; Tinkler et al., 2004) systems have been described that would likely augment the glutamatergic impact on hippocampal function. In addition, ERα is present in CA1 spines and synapses (Milner et al., 2001; Adams et al., 2002), as is ERβ (Milner et al., 2005), and all of the enzymatic machinery required to synthesize estradiol from cholesterol to provide local (non-ovarian) sources of estradiol is present in CA1 spines as well (Hojo et al., 2004). In addition, estradiol rapidly stimulates signaling cascades such as the mitogen-activated protein (MAP) kinase family (Toran-Allerand et al., 1999) and the phosphotidylinositol 3-kinase (PI3K), pathway leading to the phosphorylation of Akt, a key signaling molecule, and this could promote local protein synthesis related to the formation of new spines through a nongenomic mechanism (Akama and McEwen, 2003; Znamensky et al., 2003). Furthermore, phosphorylated Akt (pAkt) is present in CA1 dendrites, spines, and synapses and is increased by the presence of estradiol (Znamensky et al., 2003). All of these data have been generated from young female rats with short-term exposure to estradiol, yet as discussed below, age alters this scenario in fundamental ways.
The synaptic effects and behavioral impact of estrogen in CA1 differ in young and aged female rats
As recently as 6 years ago, there were very few cellular, synaptic, or behavioral data available comparing the effects of estradiol on the hippocampus in young versus old animals, although it is now clear that the aged rat reacts differently to estradiol. Markowska and Savonenko demonstrated an age-related attenuation of the beneficial cognitive effects of estrogen manipulations in rats (Markowska and Savonenko, 2002; Savonenko and Markowska, 2003). Rodent studies have also suggested that there is a “window of opportunity” related to age and duration of loss of estrogen, after which ET is less effective (Gibbs, 2000; Adams et al., 2001b). Several studies in the rat have now been completed that provide critically important synaptic data on the relevance of aging to ET. For example, whereas young animals displayed a 30% increase in axospinous synapse density in CA1, as has been reported previously (Gould et al., 1990; Woolley and McEwen, 1993), estradiol did not induce such an increase in synapse number in aged rats (Adams et al., 2001a). However, estradiol did increase the synaptic representation of NR1, the obligatory subunit for the NMDAR, in aged rats, and the increase effectively restored synaptic NR1 to that of CA1 axospinous synapses in young females (Adams et al., 2001a). In addition, the same analysis showed that NR2B increased in the lateral portion of the synapse, suggesting that estradiol induced a lateral insertion of NR1/NR2B receptors, which has interesting implications for the mechanism of synaptic plasticity and estradiol-induced functional alterations in NMDARs that might lead to a more youthful receptor profile (Adams et al., 2004).
What is it about aged CA1 axospinous synapses in female rats that makes them less responsive to estradiol? Some clues have emerged from ultrastructural studies targeting estrogen receptors, ERα and ERβ, and potential activated signaling molecules such as pAkt, which is present in CA1 dendrites and spines (Milner et al., 2001, 2005; Znamensky et al., 2003) and hypothesized to play a critical role in estradiol-induced synaptic alterations through nongenomic mechanisms (Toran-Allerand et al., 1999; McEwen, 2002). For example, the percentage of ERα-labeled synapses decreases with age, independent of estrogen status, by ∼50% (Adams et al., 2002), which might contribute to age-related decreases in the capacity to form additional spines and synapses in response to estradiol. In addition, synaptic pAkt thought to be activated by ERα (Znamensky et al., 2003) is also decreased dramatically in aged CA1 axospinous synapses (Yildirim et al., 2006), as is ERβ (Janssen et al., 2006), suggesting that several key players in the local synaptic response to estradiol are compromised with age in female rats.
Estrogen increases axospinous synapses in CA1 of young and aged rhesus monkeys
Recent interdisciplinary studies using an NHP model of ET have revealed compelling cognitive and neurobiological effects of ET in the context of aging. Such studies are particularly relevant to menopausal women, because the reproductive physiology and pattern of menopause in rhesus monkeys is very similar to women (see above, section by A.C.G.). These studies used a regimen of cyclic exposure to estradiol in an attempt to replicate a pulsatile peak that would be more physiological than daily exposure to the same dose. Initially, the total number of spines in CA1 stratum radiatum (CA1sr) was assessed in young and aged female ovariectomized monkeys with and without short-term ET (i.e., two treatments 21 d apart). In CA1sr of both young and aged monkeys, estradiol led to a 35% increase in total spines, an increase that amounted to more that 1 billion additional spines in this layer of CA1 (Hao et al., 2003). Unlike the female rat, the aged female monkey retained the capacity for such an increase in spines, which is particularly relevant to women, given the similarities in patterns of endocrine senescence between humans and macaques and the fact that these aged monkeys (20–24 years old) were perimenopausal when ovariectomized, modeling early initiation of ET for menopausal women. Ongoing studies using long-term cyclical ET (2–3 years) have revealed the same synaptic effect in CA1 of aged rhesus monkeys as with short-term ET (Tang et al., 2004a), and estradiol also enhanced performance on a hippocampus-dependent memory task in the same monkeys (Rapp et al., 2003a).
Long-term cyclical ET enhances cognitive performance and spine density in dlPFC of aged rhesus monkeys
It has been suggested that complex cognitive functions such as those mediated by dlPFC require an increase in spine number (Elston, 2003). However, spine number and morphology are also an important reflections of the capacity for youthful adaptiveness and plasticity (Kasai et al., 2003). Thus, our view is that estradiol-induced increases in spine number can be viewed as a reflection of a very healthy ability of the system to respond to estrogen through appropriate synaptic plasticity and remodeling rather than just an increase in synapses. This perspective is particularly relevant to recent analyses of the effect of ET on synaptic organization of dlPFC. A recent study of short-term cyclical ET demonstrated that this regimen increased spine density dramatically in dlPFC of young female monkeys yet had no effect on primary visual cortex (Tang et al., 2004b). This finding opened up an entirely new target for modulation of cognition by estradiol beyond hippocampus and led to a focus on area 46 of dlPFC in studies of behaviorally characterized monkeys as well. Recent behavioral studies tested whether long-term cyclical estrogen treatment after surgical menopause affects normal cognitive aging (Rapp et al., 2003a). This ET regimen substantially reversed the marked age-related impairment that untreated ovariectomized monkeys exhibited on a delayed response (DR) test of working memory, a test sensitive to dlPFC integrity (Fig. 2). Also, age appears to be an important contributor in that parallel research in young adult monkeys has reported more subtle, task-selective effects of estradiol (Voytko, 2002; Lacreuse, 2006), and our ongoing analyses of young animals with the same long-term-cyclic ET as these aged animals are not showing the same dramatic difference between treated and untreated groups in prefrontal tasks (Hao et al., this meeting).
Figure 2.

Behavioral and morphological effects of cyclical estradiol treatment in aged female rhesus monkeys. A, Neuropsychological performance in aged OVX plus vehicle (OVX + VEH) and OVX plus estradiol (OVX + E) monkeys in DR (for details, see text), showing the mean percentage correct across delays of 5–60 s. Note enhanced performance of OVX + E group at all delays (Rapp et al., 2003a). B, Histogram showing quantitative data on spine density (spine number per micrometer of dendritic length) in layer III pyramidal neurons from area 46. Note that the OVX + E group has significantly higher spine density in both apical and basal dendritic arbors and throughout all branch orders in the apical tree. C, High-resolution examples of apical and basal dendritic segments from a filled neuron from both OVX + E- and OVX + VEH-treated monkeys. Scale bar, 5 μm. **p < 0.01. Error bars indicate SEM.
Detailed quantitative morphometric analyses of neurons in layer III of area 46 in the same aged ovariectomized female rhesus monkeys that were behaviorally assessed have now been completed, and parallel data on the young monkeys will be presented at the 36th Annual Meeting of the Society for Neuroscience. These neurons were targeted because area 46 has been shown to be critically important for DR in rhesus monkeys (Goldman-Rakic, 1988), the precise task that was so sensitive to ET in aged monkey (Rapp et al., 2003a). The morphometric analyses showed that there were no differences in total dendritic length or total branch numbers across treatment groups of aged monkeys. Against this background of stability in gross dendritic architecture, studies were done to test whether ET might regulate spine number and morphology in area 46. Quantitative three-dimensional analyses revealed that long-term cyclical ET substantially increased spine density across all dendritic branch orders (Fig. 2). In addition, spine size was determined, and it was revealed that spines induced by ET are predominantly those with a relatively small spine head, a morphological distinction linked to enhanced plasticity over larger spines (Kasai et al., 2003). These data support the hypothesis that age-related decline in dlPFC function may be a result of compromised plasticity in the context of spine loss and highlight spine morphology as a critical reflection of sustained capacity for plasticity required for learning.
Conclusions
The findings from the NHP model are particularly important for their translational impact. Although previous studies in rodents and monkeys had demonstrated an estrogen-induced increase in spines in the hippocampus, these data are the first demonstration of estrogen-induced synaptic enhancement of pyramidal neurons in the dlPFC. The dramatic effect of ET on dlPFC is of great interest because this neocortical region has also been implicated as responsive to ET in humans (Berman et al., 1997; Keenan et al., 2001). dlPFC mediates the highest levels of cognitive function and is perhaps the single most important cortical region for goal-directed behavior, attention, planning, and formulating complex responses. This cortical region is very well developed in primates in general and uniquely well developed in humans. Unfortunately, it is also highly vulnerable in aging and AD, yet as these data show, estrogen may have a role in sustaining or recovering youthful synaptic structure and cognitive function dependent on dlPFC.
The findings in dlPFC as well as hippocampus in aged female rhesus monkeys are also important in that they demonstrate that these rhesus monkeys, which correspond in chronological age and endocrine status to early perimenopausal women, retain a vibrant synaptic response to estrogen in brain regions linked to learning and memory. This is encouraging because previous studies in young and aged rats demonstrated that hippocampus from aged female rats did not increase spine/synapse density in response to estrogen, and, as described above, this may be attributable to multiple age-related changes in the molecular profile of axospinous CA1 synapses that blunts the synaptic response to estrogen. The degree to which aged subjects respond to estrogen may vary across species but is also likely linked to the nature of the estrogen delivery, the duration of time without estrogen, and the age of the subject when estrogen treatment is initiated. In fact, studies have shown that beneficial cognitive effects of estrogen are decreased in aged female rats, and this is exacerbated if the time frame without estrogen before treatment is extended, suggesting that a window of opportunity may exist during which estrogen is most effective (Gibbs and Gabor, 2003). Current thinking is that such a window of opportunity may exist for menopausal women as well (Maki, 2006).
Perhaps most importantly, the NHP results show that the precise cognitive behaviors and related circuits that are prone to age-related decline are, in fact, responsive to therapy. The therapy used in this case was obviously hormonal, and, over the next 5 years, this model will be expanded to test the interactive effects of progesterone with cyclical and chronic estradiol HT regimens to achieve even greater direct relevance to choices available to menopausal women. However, these data have general implications for cognitive aging in that they demonstrate a resiliency and capacity for rehabilitation within these circuits that leaves us optimistic with respect to treatment of age-related cognitive decline on a much broader scale.
Strategies for predicting clinical outcome in women: implications for hormone therapy development for the brain (by R.D.B.)
The goal of this section is to address controversies emerging from clinical trials of estrogen and hormone therapy by explicating the cellular conditions that determine estrogen-inducible outcomes in brain and the cellular mechanisms of estrogen action that predict these outcomes. First, evidence for a healthy cell bias of estrogen action is presented. Subsequently, a brief analysis of the signaling cascades required for estrogen-inducible neuroprotective, neurotrophic, and neurogenic actions is presented. Estrogen-induced calcium signaling pathways both promote neuronal function and could exacerbate neuronal demise in neurodegenerative disease states. Second, the penultimate sections link findings derived from basic science analyses with results of clinical studies. Finally, insights gained from basic and clinical investigations of estrogen action in brain provides the foundation for development of novel neuro-selective estrogen receptor modulator (NeuroSERM) and phyto-selective estrogen receptor modulator (PhytoSERM) molecules as safe and efficacious estrogen alternatives for the brain to sustain neurological function and prevent neurodegenerative disease.
Evidence for a healthy cell bias of estrogen benefit
Multiple factors have been hypothesized to contribute to the disparity between basic science, observational, and small clinical trials of ET on cognitive function and large randomized clinical trials of ET and HT on risk and progression of AD. Among those factors were the formulation of HT, the route of administration, years since menopause before ET or HT was initiated, and the health status of the brain (Resnick and Henderson, 2002; Breitner and Zandi, 2003; Brinton and Nilsen, 2003; Yaffe, 2003; Brinton, 2005).
To address controversies of ET, in vitro models of perimenopausal estrogen exposure were developed and used to assess the neuroprotective efficacy of estradiol against β-amyloid 1–42 (Aβ1–42)-induced neurodegeneration in rat primary hippocampal neurons (Chen et al., 2006). Results of these analyses indicated that low estradiol (10 ng/ml) exposure exerted neuroprotection in each of the perimenopausal temporal patterns: acute, continuous, and intermittent. In contrast, high estradiol (200 ng/ml) was ineffective at inducing neuroprotection regardless of temporal pattern of exposure. Whereas high estradiol alone was not toxic, neurons pretreated with 200 ng/ml estradiol resulted in greater Aβ1–42-induced neurodegeneration. In prevention versus treatment simulations, low estradiol was most effective when present before and during Aβ1–42 insult. In contrast, estradiol treatment after Aβ1–42 exposure was ineffective at reversing Aβ-induced degeneration and exacerbated Aβ1–42-induced cell death when administered after Aβ1–42 insult. The mechanism by which high estradiol exacerbated Aβ1–42-induced neurodegeneration was addressed by investigating the impact of low versus high estradiol on Aβ1–42-induced dysregulation of calcium homeostasis. Results of these analyses indicated that low estradiol significantly prevented Aβ1–42-induced rise in intracellular calcium, whereas high estradiol significantly increased intracellular calcium and did not prevent Aβ1–42-induced calcium dysregulation. Thus, therapeutic benefit resulted only from low-dose estradiol exposure before, but not after, Aβ1–42-induced neurodegeneration.
How do these in vitro results compare with those of observational and clinical trials? In basic science and observational trials, ET or HT was administered to healthy neurons, animals, or middle-aged women (for review, see Brinton, 2005). In clinical treatment trials investigating impact on AD, unopposed ET was administered to aged women typically 15+ years postmenopause with AD (Henderson et al., 2000; Mulnard et al., 2000), or, as in WHIMS, HT or ET were administered to women with a mixed health profile (Shumaker et al., 2003a,b, 2004). Results of these trials indicated that ET was without benefit in the long-term AD treatment trials (Henderson et al., 2000; Mulnard et al., 2000). In the WHIMS trial, women on unopposed ET showed no reduction in risk, whereas women on combined HT showed a twofold risk of developing dementia (Shumaker et al., 2003a, 2004). Results of our in vitro analyses, indicating that estrogen treatment of hippocampal neurons before Aβ insult protects neurons against Aβ-induced neuron death, are remarkably consistent with epidemiological analyses indicating that women who receive ET at the time of menopause, in a prevention mode well before age-associated degeneration is rampant, have a lower risk of developing AD than women who have never received ET or HT (Tang et al., 1996; Kawas et al., 1997; Zandi et al., 2002). In contrast, in vitro exposure of neurons to estrogen after Aβ insult was of no benefit and, if the dose is high enough, can exacerbate degeneration, which is consistent with clinical studies with negative outcomes regarding ET or HT and AD (Henderson et al., 2000; Mulnard et al., 2000; Shumaker et al., 2004).
Estrogen mechanisms of action: a window through which to view clinical outcomes
By understanding the mechanisms of estrogen and progestin action in brain, it is possible to predict the efficacy of ET and HT to promote neurological function and prevent neurodegenerative disease. In hippocampal and cortical neurons, estradiol and other estrogenic ligands bind to membrane-associated, mitochondrial, and nuclear estrogen receptors (Fig. 3A) (McEwen et al., 2001; Toran-Allerand, 2004; Wu et al., 2004; Milner et al., 2005). In select cell types, the membrane estrogen receptor complex can bind to and activate PI3K (Simoncini et al., 2000; Mannella and Brinton, 2006), which in turn activates a Ca2+ independent PKC isoform (Cordey et al., 2003; Setalo et al., 2005) (Fig. 3A). Activated PKC phosphorylates the L-type Ca2+ channel, followed by Ca2+ influx and activation of conventional Ca2+-dependent PKCs, which then phosphorylates Src kinase to activate the MAP kinase kinase (MEK)/extracellular signal-regulated kinase 1/2 (ERK1/2) pathway, followed by translocation of activated ERK into the nucleus (Singh et al., 2000; Nilsen and Brinton, 2003). Estradiol activation of the Src/ERK signaling cascade also leads to potentiation of glutamate-activated NMDA receptors, enhanced long-term potentiation, and neuronal morphogenesis (Brinton et al., 1997; Woolley et al., 1997; Foy et al., 1999; Bi et al., 2000; Nilsen et al., 2002). Translocated pERK phosphorylates cAMP response element-binding protein (CREB) to increase transcription of the morphogenesis-related gene spinophilin (Choi et al., 2003; Zhao et al., 2005b) and the antiapoptotic genes Bcl-2 (B-cell CLL/lymphoma) and Bcl-xl (Garcia-Segura et al., 1998; Pike, 1999; Wu et al., 2005).Thus, CREB appears to be a downstream convergence point for key estradiol-inducible outcomes.
Figure 3.
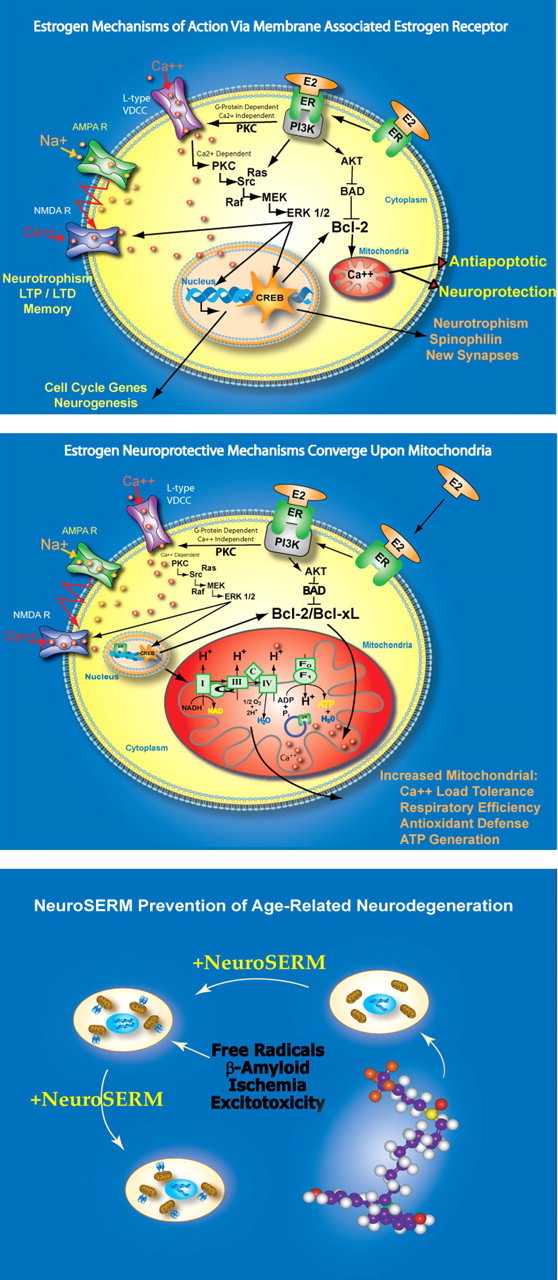
Estrogen mechanisms of action that lead to neurotrophic and neuroprotective outcomes. Top, 17-β-Estradiol (E2) acting via a membrane-associated site (mER) activates a cascade required for multiple responses that lead to enhanced neural plasticity, morphogenesis, neurogenesis, and neural survival. The signaling sequence induced by E2 at the membrane site is as follows: (1) E2 binding to mER, (2) E2–mER complexes with p85 to activate PI3K, (3) activating calcium-independent PKC, (4) phosphorylating the L-type calcium channel, (5) inducing calcium influx, (6) activating calcium-dependent PKCs, (7) activating Src kinase, (8) activating the MEK/ERK1/2 pathway, (9) ERK translocates to the nucleus, (10) activating and phosphorylating CREB, (11) enhancing transcription of antiapoptotic genes Bcl-2 and Bcl-xl, which enhance mitochondrial vitality, and spinophilin, which encourages synaptic growth, (12) simultaneously, estrogen activation of PI3K leads to activation of Akt, which phosphorylates and inhibits the proapoptotic protein BAD. Middle, Estrogen-induced neuroprotective mechanisms converge on mitochondria. Estrogen-activated cellular signaling cascade promotes enhanced mitochondrial function, leading to increased calcium load tolerance, enhanced electron transport chain efficiency, and promotion of antioxidant defense mechanisms. These actions are mediated by the regulation of both nuclear and mitochondrial encoded genes initiated by the activation of second-messenger signaling cascades. Bottom, Conceptual schematic of NeuroSERM design and therapeutic use. Consistent with the healthy cell bias of estrogen benefit hypothesis, NeuroSERM or PhytoSERM molecules would be administered before neurodegenerative insult while neurons are still healthy. NeuroSERM exposure would lead to enhanced neural survival mechanisms, represented as mitochondria with Bcl-2 additions, that promote neural defense against neurodegenerative insults associated with age-associated diseases such as Alzheimer's and Parkinson's. Designer NeuroSERM molecules target the membrane site of estrogen action, whereas PhytoSERM molecules preferentially target estrogen receptor β. AMPAR, AMPA receptor; C, cytochrome oxidase; F0, F1, ATPase subunits; LTD, long-term depression; LTP, long-term potentiation; NAD, nicotinamide adenine dinucleotide; NADH, nicotinamide adenine dinucleotide; VDCC, voltage-dependent calcium channel.
Estrogen receptor interaction with p85/PI3K also enhances pAkt, which phosphorylates the proapoptotic protein Bcl-2-associated death protein (BAD) to prevent heterodimerization with, and inactivation of, Bcl-2 (Singh, 2001; Mannella and Brinton, 2006). In cortical neurons, estradiol induced pAkt translocation to the nucleus (Mannella and Brinton, 2006). Recent analyses indicate that estradiol, via the PI3K signaling pathway, activates both the Akt and the ERK1/2 cascades in the same population of cortical and hippocampal neurons (Mannella and Brinton, 2006). Simultaneous activation of two pathways that prevent mitochondria from activating cell-death cascades is likely to promote neuron survival.
Interestingly, estrogen-inducible neuroprotective mechanisms converge onto mitochondria, which are pivotal to sustaining calcium homeostasis and cell survival (Nilsen and Brinton, 2004; Simpkins et al., 2005). Estradiol increased mitochondrial sequestration of Ca2+ and protected neurons against adverse consequences of excess cytoplasmic Ca2+ and subsequent dysregulation of Ca2+ homeostasis (Nilsen et al., 2006a). Despite an increased mitochondrial Ca2+ load, estradiol preserved mitochondrial respiratory capacity (Nilsen et al., 2006a). Subsequent analyses demonstrated that estradiol regulated calcium homeostasis in healthy neurons derived from middle-aged and aged rat hippocampus as well as those derived from embryonic rat hippocampus (Brewer et al., 2006).
Our most recent analyses indicate that estradiol promotes mitochondrial respiration and hence ATP generation and antioxidant enzymes that offset the increase in free radical generation induced by increased respiration (Nilsen et al., 2006a) (Fig. 3B). Our data and those of the Simpkins group demonstrate that estradiol significantly reduces mitochondrial lipid peroxidation, although our effects appear to be through an estrogen receptor-dependent mechanism (Nilsen et al., 2006a), whereas the Simpkins findings are mediated through estrogenic accumulation in the lipid in which they act as oxidant substrates (Singh et al., 2006). Together, the constellation of mitochondrial responses suggests that estradiol enhances expression of critical determinants of neuronal survival (Fig. 3B).
As indicated above, calcium dynamics play a pivotal and obligatory role in the estradiol-inducible cascade that leads to neurotrophic and neuroprotective benefit. Dynamics of Ca2+ homeostasis are tightly regulated in healthy neurons and dysfunctional in degenerating neurons (Khachaturian, 1994; Landfield, 1994; LaFerla, 2002; Chen et al., 2006). Therein lies the Achilles' heel of estrogen action, in which increasing Ca2+ influx and Ca2+ sequestration into cytoplasmic and mitochondrial compartments of neurons unable to maintain Ca2+ homeostasis leads to exacerbation of Ca2+-dependent degenerative insults (Chen et al., 2006).
Clinical implications of cellular effects of HT components
The issue of ET or HT formulation as a determinant of the adverse outcomes of treatment trials has been hotly debated. Results of in vitro analyses indicate that the complex formulation of CEEs (Premarin) often used clinically promoted morphogenesis and neuroprotection against neurodegenerative insults (Brinton et al., 2000). Additional analyses indicated that the neurotrophic and neuroprotective effects were mediated by some but not all estrogens contained within CEEs (Zhao and Brinton, 2006b). In fact, the complex formulation of CEEs and select estrogens within this mixture were as effective as estradiol, with a dose–response profile that would predict a wider therapeutic range than estradiol alone (Brinton et al., 2000). These data were consistent with the epidemiological studies in which Premarin was the most frequently used ET regimen (Brinton, 2004).
However, the results are strikingly different in studies investigating the impact of a frequently prescribed combined estrogen/progestin formulation, CEE plus MPA (Depo-Provera; PremPro). These in vitro analyses indicated that MPA was the best antagonist to estrogen action in neurons that had thus far been identified in this model. Unlike progesterone, which was neuroprotective alone and synergistic with estradiol, MPA was neither neuroprotective nor did it synergize with estradiol. In fact, MPA antagonized estradiol-induced neuroprotection (Nilsen and Brinton, 2003; Nilsen et al., 2005). Moreover, MPA exacerbated glutamate excitotoxic-induced neuron death (Nilsen et al., 2006b). Results of the WHIMS trial in which the hormone therapy group (CEE plus MPA) had a twofold greater risk of developing dementia strongly suggest that the addition of MPA in vivo or in vitro has deleterious outcomes for the brain (Shumaker et al., 2003b). Overall, relatively little is known regarding progesterone or progestin action in brain or the neurobiological consequences of estrogen plus progesterone or progestins on outcomes relevant to neurodegenerative disease. An intensive effort to develop progesterone neurobiology in the hippocampus and cortex is currently underway to determine the efficacy of clinically relevant progestins to promote beneficial effects of progesterone in these brain regions.
Translating basic neuroscience insights into safe and efficacious estrogen therapy for the brain
Multiple groups in academia are developing estrogen therapeutics to target the brain. Simpkins and colleagues have led the field in developing nonfeminizing estrogens (Simpkins et al., 2005), and the chemical synthesis efforts of Katzenellebogen and colleagues has been instrumental in developing ERα- and ERβ-selective ligands (Katzenellenbogen et al., 2000; Meyers et al., 2001). In the industrial sector, Wyeth (Princeton, NJ), Eli Lilly & Company (Indianapolis, IN), Pfizer (New York, NY), and others all have ET or HT development efforts that are now conscious of the importance of the neural effects of these therapeutics (Zhao et al., 2005a; Ariazi et al., 2006). The preclinical research plan of our laboratory is a hybrid of both discovery and translational research to develop a brain SERM that selectively targets either the membrane site of estrogen action or ERβ . We have termed such molecules NeuroSERMs to denote their preferential selectivity for activating estrogen mechanisms in brain or PhytoSERMs to denote their natural source origins with preferential targeting of ERβ . As part of this endeavor, the efficacy of existing SERMs, tamoxifen and raloxifene, to promote estrogen outcomes in hippocampal and cortical neurons has been investigated. Results of these analyses indicate that both SERMS are weak estrogen agonists with neuroprotective effects but modest to no neurotrophic efficacy (Zhao et al., 2005a), which is consistent with clinical observations (Yaffe et al., 2001, 2005). Importantly, previous analyses demonstrated that both ERα and ERβ agonists can promote neuronal survival by activating estrogen mechanisms of action in rat hippocampal neurons (Zhao and Brinton, 2006a). The current strategy in our laboratory to develop NeuroSERMS and PhytoSERMs is threefold: (1) determine the target of estrogen action in brain required for the neurotrophic, neurogenic, and neuroprotective actions of estrogen; (2) develop NeuroSERM and PhytoSERM candidate molecules using three in silico discovery and design strategies; and (3) determine the neurotrophic and neuroprotective efficacy of candidate molecules using neuronal responses predictive of clinical efficacy (Fig. 3C). Novel NeuroSERMs will act as estrogen receptor agonists at the membrane site of estrogen action in brain but will act as an estrogen receptor antagonist in breast and uterus. PhytoSERM formulations will capitalize on our finding that activation of ERβ is equally neuroprotective as ERα (Zhao and Brinton, 2006a). The goal is to promote estrogen action in the brain while not activating the breast or uterus.
Conclusions
The demographics suggest that we face a devastating increase in the prevalence of AD, reinforcing the immediate need for basic and translational neuroscience to develop safe and efficacious ET and HT regimens for the brain. Of those affected with AD, 68% are female and 32% are male (Brookmeyer et al., 1998). Because women have a longer life expectancy than men, the absolute number of women with AD exceeds that of men. However, a double danger exists for women. Results of a meta-analysis of seven sex-specific studies concluded that women are 1.5 times more likely to develop AD than age-matched men (Gao et al., 1998), which was supported by the Cache County analysis that showed a clear female gender increase in the incidence of AD (Zandi et al., 2002).
At the turn of the new millennium in the United States, there were nearly 42 million women over the age of 50 years and, of these, >31 million women were over the age of 55 years (North American Menopause Society, 2004). Worldwide, there are currently >470 million women aged 50 years or older, and 30% of those are projected to live into their 80s (North American Menopause Society, 2004). These women can anticipate spending one-third to one-half of their lifetime in the menopausal state. Reports on prevalence of AD vary, but of the 18 million American women in their mid to late 70s, as many as 5–10% may suffer from AD, and this figure increases dramatically at older ages (Brookmeyer et al., 1998). The projected exponential increase in the prevalence of AD, along with the anticipated impact on families and society, highlights the imperative for developing strategies to prevent or delay the onset of AD sooner rather than later.
Future directions
Perhaps a guiding principle for future work in this arena is to bear in mind that the brain is the key target organ for ovarian steroids, and the brain being targeted in the context of HT is one that has likely begun to exhibit age-related alterations in its relevant circuitry and cellular responses to estrogen and progesterone. Furthermore, the clinical and basic neuroscience investigators need to move forward in a coordinated manner that both informs each arena and helps guide the key questions to be asked and hopefully answered.
With respect to clinical studies, we need to address the issue of heterogeneity among women in the menopause transition. How many women experience negative neurologic symptoms and which symptoms (e.g., hot flushes, depression, and cognitive alterations) are most debilitating? What are the risk factors for the various symptoms and are there premenopausal and/or perimenopausal endocrine patterns that are predictive of behavioral alterations? In turn, which women will react well to HT/ET and which will not benefit or may react negatively? What are the important health patterns or comorbidities that predict positive or negative reactions to HT and are their genetic and identifiable environmental contributions to both vulnerability to symptoms and response to treatment? In addition, to what degree should HT formulations be guided by physiological patterns of exposure (i.e., cyclical rather than chronic) and demonstrated neuronal responses (i.e., progesterone vs MPA) and what new formulations should be tried based on the pharmacology of estrogen and progesterone receptors? Finally, how do we more accurately define the postulated window of opportunity for HT/ET and use it to optimize the clinical outcome?
The preclinical studies also have to take into consideration that endocrine senescence in the form of menopause is occurring against a background of brain aging. Although the hypothalamus drives steroid release through the HPG axis, it also responds to estradiol. How does brain aging impact the regulation of release through the HPG axis and just as importantly, what age-related changes have occurred in the hypothalamic circuits that alter the response to endocrine feedback? These issues regarding the aging hypothalamus may be particularly relevant to predicting vulnerability to the menopause transition, because they are likely to play a critically important role early in the process. Also, animal studies need to be designed to inform issues revolving around time of initiation of treatment, importance of age of the subject, and duration, dose, and formulation of the ET or HT, because these variables are so easily controlled in animal models. Animal data on exposure after long-term absence of estradiol and long-term exposure to HT/ET are particularly important, because such studies will inform the basis of the window of opportunity and reveal the cellular basis of the transition from beneficial to harmful results of exposure to estrogens. In addition, we need more specific and detailed information on the neurons and circuits that are the targets of estradiol and progesterone, how their response changes with aging, and how those alterations translate into the neurologic events that occur during the menopause transition.
Finally, we need to design and execute in vitro and in vivo paradigms that reveal the different roles and contributions of ERα, ERβ, and progesterone receptors with respect to neuronal targets and which responses to each are mediated through membrane receptors, particularly those at the synapse. This information will allow for much more targeted pharmacology and, in turn, more specific therapeutic agents that take advantage of our increasing knowledge of the signaling cascades that are likely initiated by membrane receptors as opposed to nuclear receptors. Armed with such data, novel HT/ET agents could be developed. The coordinated movement from in vitro tests through rodent models to the NHP models should be placed at a high priority and designed with the explicit goal of developing more useful HT and ET regimens for women.
Footnotes
This work was supported by National Institute of Mental Health Intramural Research Program (P.J.S.) and Grant MH67159 (R.D.B.), National Institute of Aging Grants AG06647 (J.H.M.), AG16765 (J.H.M., A.C.G.), and AG14751 and AG026572 (R.D.B.), and the Kenneth T. and Eileen L. Norris Foundation (R.D.B.). We are very grateful to the members of the Morrison, Brinton, Gore, and Schmidt laboratories and numerous collaborating teams and colleagues for their insights and contributions to the work presented in this article and at the 36th Annual Meeting of the Society for Neuroscience.
References
- Adams MM, Shah RA, Janssen WG, Morrison JH. Different modes of hippocampal plasticity in response to estrogen in young and aged female rats. Proc Natl Acad Sci USA. 2001a;98:8071–8076. doi: 10.1073/pnas.141215898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams MM, Oung T, Morrison JH, Gore AC. Length of postovariectomy interval and age, but not estrogen replacement, regulate N-methyl-d-aspartate receptor mRNA levels in the hippocampus of female rats. Exp Neurol. 2001b;170:345–356. doi: 10.1006/exnr.2001.7716. [DOI] [PubMed] [Google Scholar]
- Adams MM, Fink SE, Shah RA, Janssen WG, Hayashi S, Milner TA, McEwen BS, Morrison JH. Estrogen and aging affect the subcellular distribution of estrogen receptor-alpha in the hippocampus of female rats. J Neurosci. 2002;22:3608–3614. doi: 10.1523/JNEUROSCI.22-09-03608.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams MM, Fink SE, Janssen WG, Shah RA, Morrison JH. Estrogen modulates synaptic N-methyl-d-aspartate receptor subunit distribution in the aged hippocampus. J Comp Neurol. 2004;474:419–426. doi: 10.1002/cne.20148. [DOI] [PubMed] [Google Scholar]
- Akama KT, McEwen BS. Estrogen stimulates postsynaptic density-95 rapid protein synthesis via the Akt/protein kinase B pathway. J Neurosci. 2003;23:2333–2339. doi: 10.1523/JNEUROSCI.23-06-02333.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson GL, Limacher M, Assaf AR, Bassford T, Beresford SA, Black H, Bonds D, Brunner R, Brzyski R, Caan B, Chlebowski R, Curb D, Gass M, Hays J, Heiss G, Hendrix S, Howard BV, Hsia J, Hubbell A, Jackson R. Effects of conjugated equine estrogen in postmenopausal women with hysterectomy: the Women's Health Initiative randomized controlled trial. JAMA. 2004;291:1701–1712. doi: 10.1001/jama.291.14.1701. [DOI] [PubMed] [Google Scholar]
- Ariazi EA, Ariazi JL, Cordera F, Jordan VC. Estrogen receptors as therapeutic targets in breast cancer. Curr Top Med Chem. 2006;6:181–202. [PubMed] [Google Scholar]
- Avis NE, Stellato R, Crawford S, Bromberger J, Ganz P, Cain V, Kagawa-Singer M. Is there a menopausal syndrome? Menopausal status and symptoms across racial/ethnic groups. Soc Sci Med. 2001;52:345–356. doi: 10.1016/s0277-9536(00)00147-7. [DOI] [PubMed] [Google Scholar]
- Bellino FL, Wise PM. Nonhuman primate models of menopause workshop. Biol Reprod. 2003;68:10–18. doi: 10.1095/biolreprod.102.005215. [DOI] [PubMed] [Google Scholar]
- Berman KF, Schmidt PJ, Rubinow DR, Danaceau MA, Van Horn JD, Esposito G, Ostrem JL, Weinberger DR. Modulation of cognition-specific cortical activity by gonadal steroids: a positron-emission tomography study in women. Proc Natl Acad Sci USA. 1997;94:8836–8841. doi: 10.1073/pnas.94.16.8836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi R, Broutman G, Foy MR, Thompson RF, Baudry M. The tyrosine kinase and mitogen-activated protein kinase pathways mediate multiple effects of estrogen in hippocampus. Proc Natl Acad Sci USA. 2000;97:3602–3607. doi: 10.1073/pnas.060034497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonavera JJ, Swerdloff RS, Hikim APS, Lue YH, Wang C. Aging results in attenuated gonadotropin releasing hormone-luteinizing hormone axis responsiveness to glutamate receptor agonist N-methyl-d-aspartate. J Neuroendocrinol. 1998;10:93–99. doi: 10.1046/j.1365-2826.1998.00177.x. [DOI] [PubMed] [Google Scholar]
- Brann DW, Mahesh VB. The aging reproductive neuroendocrine axis. Steroids. 2005;70:273–283. doi: 10.1016/j.steroids.2004.12.008. [DOI] [PubMed] [Google Scholar]
- Breitner JCS, Zandi PP. Effects of estrogen plus progestin on risk of dementia. JAMA. 2003;290:1706–1707. doi: 10.1001/jama.290.13.1706-b. [DOI] [PubMed] [Google Scholar]
- Brewer GJ, Reichensperger JD, Brinton RD. Prevention of age-related dysregulation of calcium dynamics by estrogen in neurons. Neurobiol Aging. 2006;27:306–317. doi: 10.1016/j.neurobiolaging.2005.01.019. [DOI] [PubMed] [Google Scholar]
- Brinton RD. Impact of estrogen therapy on Alzheimer's disease: a fork in the road? CNS Drugs. 2004;18:405–422. doi: 10.2165/00023210-200418070-00001. [DOI] [PubMed] [Google Scholar]
- Brinton RD. Investigative models for determining hormone therapy-induced outcomes in brain: evidence in support of a healthy cell bias of estrogen action. Ann NY Acad Sci. 2005;1052:57–74. doi: 10.1196/annals.1347.005. [DOI] [PubMed] [Google Scholar]
- Brinton RD, Nilsen J. Effects of estrogen plus progestin on risk of dementia. JAMA. 2003;290:1706. doi: 10.1001/jama.290.13.1706-a. author reply 1707–1708. [DOI] [PubMed] [Google Scholar]
- Brinton RD, Proffitt P, Tran J, Luu R. Equilin, a principal component of the estrogen replacement therapy premarin, increases the growth of cortical neurons via an NMDA receptor-dependent mechanism. Exp Neurol. 1997;147:211–220. doi: 10.1006/exnr.1997.6619. [DOI] [PubMed] [Google Scholar]
- Brinton RD, Chen S, Montoya M, Hsieh D, Minaya J, Kim J, Chu HP. The women's health initiative estrogen replacement therapy is neurotrophic and neuroprotective. Neurobiol Aging. 2000;21:475–496. doi: 10.1016/s0197-4580(00)00109-3. [DOI] [PubMed] [Google Scholar]
- Bromberger JT, Meyer PM, Kravitz HM, Sommer B, Cordal A, Powell L, Ganz PA, Sutton-Tyrrell K. Psychologic distress and natural menopause: a multiethnic community study. Am J Public Health. 2001;91:1435–1442. doi: 10.2105/ajph.91.9.1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brookmeyer R, Gray S, Kawas C. Projections of Alzheimer's disease in the United States and the public health impact of delaying disease onset. Am J Public Health. 1998;88:1337–1342. doi: 10.2105/ajph.88.9.1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant DN, Sheldahl LC, Marriott LK, Shapiro RA, Dorsa DM. Multiple pathways transmit neuroprotective effects of gonadal steroids. Endocrine. 2006;29:199–207. doi: 10.1385/ENDO:29:2:199. [DOI] [PubMed] [Google Scholar]
- Burger HG, Dudley EC, Cui J, Dennerstein L, Hopper JL. A prospective longitudinal study of serum testosterone, dehydroepiandrosterone sulfate, and sex hormone-binding globulin levels through the menopause transition. J Clin Endocrinol Metab. 2000;85:2832–2838. doi: 10.1210/jcem.85.8.6740. [DOI] [PubMed] [Google Scholar]
- Burke SN, Barnes CA. Neural plasticity in the ageing brain. Nat Rev Neurosci. 2006;7:30–40. doi: 10.1038/nrn1809. [DOI] [PubMed] [Google Scholar]
- Chakraborty TR, Ng L, Gore AC. Colocalization and hormone regulation of estrogen receptor alpha and NMDA receptor in the hypothalamus of female rats. Endocrinology. 2003a;144:299–305. doi: 10.1210/en.2002-220749. [DOI] [PubMed] [Google Scholar]
- Chakraborty TR, Ng L, Gore AC. Age-related changes in estrogen receptor beta in rat hypothalamus: a quantitative analysis. Endocrinology. 2003b;144:4164–4171. doi: 10.1210/en.2003-0052. [DOI] [PubMed] [Google Scholar]
- Chakraborty TR, Hof PR, Ng L, Gore AC. Stereologic analysis of estrogen receptor alpha (ERα) expression in rat hypothalamus and its regulation by aging and estrogen. J Comp Neurol. 2003c;466:409–421. doi: 10.1002/cne.10906. [DOI] [PubMed] [Google Scholar]
- Chen S, Nilsen J, Brinton R. Dose and temporal pattern of estrogen exposure determines neuroprotective outcome in hippocampal neurons: therapeutic implications. Endocrinology. 2006 doi: 10.1210/en.2006-0495. in press. [DOI] [PubMed] [Google Scholar]
- Choi JM, Romeo RD, Brake WG, Bethea CL, Rosenwaks Z, McEwen BS. Estradiol increases pre- and post-synaptic proteins in the CA1 region of the hippocampus in female rhesus macaques (Macaca mulatta) Endocrinology. 2003;144:4734–4738. doi: 10.1210/en.2003-0216. [DOI] [PubMed] [Google Scholar]
- Cohen LS, Soares CN, Vitonis AF, Otto MW, Harlow BL. Risk for new onset of depression during the menopausal transition: the Harvard study of moods and cycles. Arch Gen Psychiatry. 2006;63:385–390. doi: 10.1001/archpsyc.63.4.385. [DOI] [PubMed] [Google Scholar]
- Cordey M, Gundimeda U, Gopalakrishna R, Pike CJ. Estrogen activates protein kinase C in neurons: role in neuroprotection. J Neurochem. 2003;84:1340–1348. doi: 10.1046/j.1471-4159.2003.01631.x. [DOI] [PubMed] [Google Scholar]
- Davison SL, Bell R, Donath S, Montalto JG, Davis SR. Androgen levels in adult females: changes with age, menopause, and oophorectomy. J Clin Endocrinol Metab. 2005;90:3847–3853. doi: 10.1210/jc.2005-0212. [DOI] [PubMed] [Google Scholar]
- Dennerstein L, Burrows GD, Wood C, Hyman G. Hormones and sexuality: effect of estrogen and progestogen. Obstet Gynecol. 1980;56:316–322. [PubMed] [Google Scholar]
- Eiken P, Kolthoff N. Compliance with long-term oral hormonal replacement therapy. Maturitas. 1995;22:97–103. doi: 10.1016/0378-5122(95)00921-7. [DOI] [PubMed] [Google Scholar]
- Elston GN. Cortex, cognition and the cell: new insights into the pyramidal neuron and prefrontal function. Cereb Cortex. 2003;13:1124–1138. doi: 10.1093/cercor/bhg093. [DOI] [PubMed] [Google Scholar]
- Espeland MA, Rapp SR, Shumaker SA, Brunner R, Manson JE, Sherwin BB, Hsia J, Margolis KL, Hogan PE, Wallace R, Dailey M, Freeman R, Hays J. Conjugated equine estrogens and global cognitive function in postmenopausal women: Women's Health Initiative Memory Study. JAMA. 2004;291:2959–2968. doi: 10.1001/jama.291.24.2959. [DOI] [PubMed] [Google Scholar]
- Ettinger B, Pressman A. Continuation of postmenopausal hormone replacement therapy in a large health maintenance organization: transdermal matrix patch versus oral estrogen therapy. Am J Manag Care. 1999;5:779–785. [PubMed] [Google Scholar]
- Foy MR, Xu J, Xie X, Brinton RD, Thompson RF, Berger TW. 17beta-estradiol enhances NMDA receptor-mediated EPSPs and long-term potentiation. J Neurophysiol. 1999;81:925–929. doi: 10.1152/jn.1999.81.2.925. [DOI] [PubMed] [Google Scholar]
- Freeman EW, Sammel MD, Liu L, Gracia CR, Nelson DB, Hollander L. Hormones and menopausal status as predictors of depression in women in transition to menopause. Arch Gen Psychiatry. 2004;61:62–70. doi: 10.1001/archpsyc.61.1.62. [DOI] [PubMed] [Google Scholar]
- Freeman EW, Sammel MD, Lin H, Nelson DB. Associations of hormones and menopausal status with depressed mood in women with no history of depression. Arch Gen Psychiatry. 2006;63:375–382. doi: 10.1001/archpsyc.63.4.375. [DOI] [PubMed] [Google Scholar]
- Gabor R, Nagle R, Johnson DA, Gibbs RB. Estrogen enhances potassium-stimulated acetylcholine release in the rat hippocampus. Brain Res. 2003;962:244–247. doi: 10.1016/s0006-8993(02)04053-2. [DOI] [PubMed] [Google Scholar]
- Gao S, Hendrie HC, Hall KS, Hui S. The relationships between age, sex, and the incidence of dementia and Alzheimer disease: a meta-analysis. Arch Gen Psychiatry. 1998;55:809–815. doi: 10.1001/archpsyc.55.9.809. [DOI] [PubMed] [Google Scholar]
- Garcia-Segura LM, Cardona-Gomez P, Naftolin F, Chowen JA. Estradiol upregulates Bcl-2 expression in adult brain neurons. NeuroReport. 1998;9:593–597. doi: 10.1097/00001756-199803090-00006. [DOI] [PubMed] [Google Scholar]
- Gauthier S, Reisberg B, Zaudig M, Petersen RC, Ritchie K, Broich K, Belleville S, Brodaty H, Bennett D, Chertkow H, Cummings JL, de Leon M, Feldman H, Ganguli M, Hampel H, Scheltens P, Tierney MC, Whitehouse P, Winblad B. Mild cognitive impairment. Lancet. 2006;367:1262–1270. doi: 10.1016/S0140-6736(06)68542-5. [DOI] [PubMed] [Google Scholar]
- Gazzaley AH, Weiland NG, McEwen BS, Morrison JH. Differential regulation of NMDAR1 mRNA and protein by estradiol in the rat hippocampus. J Neurosci. 1996;16:6830–6838. doi: 10.1523/JNEUROSCI.16-21-06830.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs RB. Long-term treatment with estrogen and progesterone enhances acquisition of a spatial memory task by ovariectomized aged rats. Neurobiol Aging. 2000;21:107–116. doi: 10.1016/s0197-4580(00)00103-2. [DOI] [PubMed] [Google Scholar]
- Gibbs RB. Basal forebrain cholinergic neurons are necessary for estrogen to enhance acquisition of a delayed matching-to-position T-maze task. Horm Behav. 2002;42:245–257. doi: 10.1006/hbeh.2002.1825. [DOI] [PubMed] [Google Scholar]
- Gibbs RB, Gabor R. Estrogen and cognition: applying preclinical findings to clinical perspectives. J Neurosci Res. 2003;74:637–643. doi: 10.1002/jnr.10811. [DOI] [PubMed] [Google Scholar]
- Goldman-Rakic PS. Topography of cognition: parallel distributed networks in primate association cortex. Annu Rev Neurosci. 1988;11:137–156. doi: 10.1146/annurev.ne.11.030188.001033. [DOI] [PubMed] [Google Scholar]
- Gore AC. Gonadotropin-releasing hormone neurons, NMDA receptors, and their regulation by steroid hormones across the reproductive life cycle. Brain Res Rev. 2001;37:235–248. doi: 10.1016/s0165-0173(01)00121-7. [DOI] [PubMed] [Google Scholar]
- Gore AC. GnRH: the master molecule of reproduction. Norwell, MA: Kluwer Academic; 2002. [Google Scholar]
- Gore AC. GnRH neurons: multiple inputs, multiple outputs. Endocrinology. 2004;145:4016–4017. doi: 10.1210/en.2004-0855. [DOI] [PubMed] [Google Scholar]
- Gore AC, Yeung G, Morrison JH, Oung T. Neuroendocrine aging in the female rat: the changing relationship of hypothalamic gonadotropin-releasing hormone neurons and N-methyl-d-aspartate receptors. Endocrinology. 2000a;141:4757–4767. doi: 10.1210/endo.141.12.7841. [DOI] [PubMed] [Google Scholar]
- Gore AC, Oung T, Yung S, Flagg RA, Woller MJ. Neuroendocrine mechanisms for reproductive senescence in the female rat: gonadotropin-releasing hormone neurons. Endocrine. 2000b;13:315–323. doi: 10.1385/ENDO:13:3:315. [DOI] [PubMed] [Google Scholar]
- Gore AC, Oung T, Woller MJ. Age-related changes in hypothalamic gonadotropin-releasing hormone (GnRH) and NMDA receptor gene expression and their regulation by estrogen in the female rat. J Neuroendocrinol. 2002;14:300–309. doi: 10.1046/j.1365-2826.2002.00777.x. [DOI] [PubMed] [Google Scholar]
- Gore AC, Windsor-Engnell BM, Terasawa E. Menopausal increases in pulsatile gonadotropin-releasing hormone (GnRH) release in a non-human primate (Macaca mulatta) Endocrinology. 2004;145:4653–4659. doi: 10.1210/en.2004-0379. [DOI] [PubMed] [Google Scholar]
- Gould E, Woolley CS, Frankfurt M, McEwen BS. Gonadal steroids regulate dendritic spine density in hippocampal pyramidal cells in adulthood. J Neurosci. 1990;10:1286–1291. doi: 10.1523/JNEUROSCI.10-04-01286.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall JE, Gill S. Neuroendocrine aspects of aging in women. Endocrinol Metab Clin North Am. 2001;30:631–646. doi: 10.1016/s0889-8529(05)70205-x. [DOI] [PubMed] [Google Scholar]
- Hao J, Janssen WG, Tang Y, Roberts JA, McKay H, Lasley B, Allen PB, Greengard P, Rapp PR, Kordower JH, Hof PR, Morrison JH. Estrogen increases the number of spinophilin-immunoreactive spines in the hippocampus of young and aged female rhesus monkeys. J Comp Neurol. 2003;465:540–550. doi: 10.1002/cne.10837. [DOI] [PubMed] [Google Scholar]
- Harris RP, Helfand M, Woolf SH, Lohr KN, Mulrow CD, Teutsch SM, Atkins D. Current methods of the US Preventive Services Task Force: a review of the process. Am J Prev Med. 2001;20:21–35. doi: 10.1016/s0749-3797(01)00261-6. [DOI] [PubMed] [Google Scholar]
- Henderson VW, Paganini-Hill A, Miller BL, Elble RJ, Reyes PF, Shoupe D, McCleary CA, Klein RA, Hake AM, Farlow MR. Estrogen for Alzheimer's disease in women: randomized, double-blind, placebo-controlled trial. Neurology. 2000;54:295–301. doi: 10.1212/wnl.54.2.295. [DOI] [PubMed] [Google Scholar]
- Henderson VW, Guthrie JR, Dudley EC, Burger HG, Dennerstein L. Estrogen exposures and memory at midlife: a population-based study of women. Neurology. 2003;60:1369–1371. doi: 10.1212/01.wnl.0000059413.75888.be. [DOI] [PubMed] [Google Scholar]
- Henderson VW, Benke KS, Green RC, Cupples LA, Farrer LA. Postmenopausal hormone therapy and Alzheimer's disease risk: interaction with age. J Neurol Neurosurg Psychiatry. 2005;76:103–105. doi: 10.1136/jnnp.2003.024927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbison AE, Skinner DC, Robinson JE, King IS. Androgen receptor-immunoreactive cells in ram hypothalamus: distribution and co-localization patterns with gonadotropin-releasing hormone, somatostatin and tyrosine hydroxylase. Neuroendocrinology. 1996;63:120–131. doi: 10.1159/000126948. [DOI] [PubMed] [Google Scholar]
- Hogervorst E, Yaffe K, Richards M, Huppert F. Hormone replacement therapy for cognitive function in postmenopausal women. Cochrane Database Syst Rev. 2002 doi: 10.1002/14651858.CD003122. CD003122. [DOI] [PubMed] [Google Scholar]
- Hojo Y, Hattori TA, Enami T, Furukawa A, Suzuki K, Ishii HT, Mukai H, Morrison JH, Janssen WG, Kominami S, Harada N, Kimoto T, Kawato S. Adult male rat hippocampus synthesizes estradiol from pregnenolone by cytochromes P45017alpha and P450 aromatase localized in neurons. Proc Natl Acad Sci USA. 2004;101:865–870. doi: 10.1073/pnas.2630225100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrabovszky E, Shughrue PJ, Merchenthaler I, Hajszan T, Carpenter CD, Liposits Z, Petersen SL. Detection of estrogen receptor-b messenger ribonucleic acid and 125I-estrogen binding in luteinizing hormone-releasing hormone neurons of the rat brain. Endocrinology. 2000;141:3506–3509. doi: 10.1210/endo.141.9.7788. [DOI] [PubMed] [Google Scholar]
- Janssen WG, Yildirim M, Lou WWY, McEwen BS, Milner TA, Morrison JH. Estrogen receptor β is affected by estrogen and aging in CA1sr region of female rat hippocampus. Soc Neurosci Abstr. 2006;32:659.17. [Google Scholar]
- Kasai H, Matsuzaki M, Noguchi J, Yasumatsu N, Nakahara H. Structure-stability-function relationships of dendritic spines. Trends Neurosci. 2003;26:360–368. doi: 10.1016/S0166-2236(03)00162-0. [DOI] [PubMed] [Google Scholar]
- Katzenellenbogen BS, Choi I, Delage-Mourroux R, Ediger TR, Martini PG, Montano M, Sun J, Weis K, Katzenellenbogen JA. Molecular mechanisms of estrogen action: selective ligands and receptor pharmacology. J Steroid Biochem Mol Biol. 2000;74:279–285. doi: 10.1016/s0960-0760(00)00104-7. [DOI] [PubMed] [Google Scholar]
- Kawakami S, Hirunagi K, Ichikawa M, Tsukamura H, Maeda K-I. Evidence for terminal regulation of GnRH release by excitatory amino acids in the median eminence in female rats: a dual immunoelectron microscopic study. Endocrinology. 1998a;139:1458–1461. doi: 10.1210/endo.139.3.5979. [DOI] [PubMed] [Google Scholar]
- Kawakami S, Ichikawa M, Murahashi K, Hirunagi K, Tsukamura H, Maeda K. Excitatory amino acids act on the median eminence nerve terminals to induce gonadotropin-releasing hormone release in female rats. Gen Comp Endocrinol. 1998b;112:372–382. doi: 10.1006/gcen.1998.7140. [DOI] [PubMed] [Google Scholar]
- Kawas C, Resnick S, Morrison A, Brookmeyer R, Corrada M, Zonderman A, Bacal C, Lingle DD, Metter E. A prospective study of estrogen replacement therapy and the risk of developing Alzheimer's disease: the Baltimore Longitudinal Study of Aging. Neurology. 1997;48:1517–1521. doi: 10.1212/wnl.48.6.1517. [DOI] [PubMed] [Google Scholar]
- Keenan PA, Ezzat WH, Ginsburg K, Moore GJ. Prefrontal cortex as the site of estrogen's effect on cognition. Psychoneuroendocrinology. 2001;26:577–590. doi: 10.1016/s0306-4530(01)00013-0. [DOI] [PubMed] [Google Scholar]
- Khachaturian ZS. Calcium hypothesis of Alzheimer's disease and brain aging. Ann NY Acad Sci. 1994;747:1–11. doi: 10.1111/j.1749-6632.1994.tb44398.x. [DOI] [PubMed] [Google Scholar]
- Krohn PL. Ovarian homotransplantation. Ann NY Acad Sci. 1955;59:443–447. doi: 10.1111/j.1749-6632.1955.tb45958.x. [DOI] [PubMed] [Google Scholar]
- Kudwa AE, Michopoulos V, Gatewood JD, Rissman EF. Roles of estrogen receptors alpha and beta in differentiation of mouse sexual behavior. Neuroscience. 2006;138:921–928. doi: 10.1016/j.neuroscience.2005.10.018. [DOI] [PubMed] [Google Scholar]
- Lacreuse A. Effects of ovarian hormones on cognitive function in nonhuman primates. Neuroscience. 2006;138:859–867. doi: 10.1016/j.neuroscience.2005.09.006. [DOI] [PubMed] [Google Scholar]
- LaFerla FM. Calcium dyshomeostasis and intracellular signalling in Alzheimer's disease. Nat Rev Neurosci. 2002;3:862–872. doi: 10.1038/nrn960. [DOI] [PubMed] [Google Scholar]
- Landfield PW. Increased hippocampal Ca2+ channel activity in brain aging and dementia. Hormonal and pharmacologic modulation. Ann NY Acad Sci. 1994;747:351–364. doi: 10.1111/j.1749-6632.1994.tb44422.x. [DOI] [PubMed] [Google Scholar]
- LeBlanc ES, Janowsky J, Chan BK, Nelson HD. Hormone replacement therapy and cognition: systematic review and meta-analysis. JAMA. 2001;285:1489–1499. doi: 10.1001/jama.285.11.1489. [DOI] [PubMed] [Google Scholar]
- Leiblum SR, Koochaki PE, Rodenberg CA, Barton IP, Rosen RC. Hypoactive sexual desire disorder in postmenopausal women: US results from the Women's International Study of Health and Sexuality (WISHeS) Menopause. 2006;13:46–56. doi: 10.1097/01.gme.0000172596.76272.06. [DOI] [PubMed] [Google Scholar]
- Lloyd JM, Hoffman GE, Wise PM. Decline in immediate early gene expression in gonadotropin-releasing hormone neurons during proestrus in regularly cycling, middle-aged rats. Endocrinology. 1994;134:1800–1805. doi: 10.1210/endo.134.4.8137745. [DOI] [PubMed] [Google Scholar]
- Maffucci JA, Gore AC. Age-related changes in hormones and their receptors in animal models of female reproductive senescence. In: Conn PM, editor. Handbook of models for human aging. New York: Elsevier; 2006. pp. 533–552. [Google Scholar]
- Maki PM. Hormone therapy and cognitive function: is there a critical period for benefit? Neuroscience. 2006;138:1027–1030. doi: 10.1016/j.neuroscience.2006.01.001. [DOI] [PubMed] [Google Scholar]
- Maki PM, Zonderman AB, Resnick SM. Enhanced verbal memory in nondemented elderly women receiving hormone-replacement therapy. Am J Psychiatry. 2001;158:227–233. doi: 10.1176/appi.ajp.158.2.227. [DOI] [PubMed] [Google Scholar]
- Mannella P, Brinton RD. Estrogen receptor protein interaction with PI3K leads to activation of pAkt and pERK in the same population of cortical neurons: a unified mechanism of estrogen action. J Neurosci. 2006;26:9439–9447. doi: 10.1523/JNEUROSCI.1443-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manson JE, Bassuk SS, Harman SM, Brinton EA, Cedars MI, Lobo R, Merriam GR, Miller VM, Naftolin F, Santoro N. Postmenopausal hormone therapy: new questions and the case for new clinical trials. Menopause. 2006;13:139–147. doi: 10.1097/01.gme.0000177906.94515.ff. [DOI] [PubMed] [Google Scholar]
- Markowska AL, Savonenko AV. Effectiveness of estrogen replacement in restoration of cognitive function after long-term estrogen withdrawal in aging rats. J Neurosci. 2002;22:10985–10995. doi: 10.1523/JNEUROSCI.22-24-10985.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews KA, Kuller LH, Wing RR, Meilahn EN, Plantinga P. Prior to use of estrogen replacement therapy, are users healthier than nonusers? Am J Epidemiol. 1996;143:971–978. doi: 10.1093/oxfordjournals.aje.a008678. [DOI] [PubMed] [Google Scholar]
- McEwen B. Estrogen actions throughout the brain. Recent Prog Horm Res. 2002;57:357–384. doi: 10.1210/rp.57.1.357. [DOI] [PubMed] [Google Scholar]
- McEwen B, Akama K, Alves S, Brake WG, Bulloch K, Lee S, Li C, Yuen G, Milner TA. Tracking the estrogen receptor in neurons: implications for estrogen-induced synapse formation. Proc Natl Acad Sci USA. 2001;98:7093–7100. doi: 10.1073/pnas.121146898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers MJ, Sun J, Carlson KE, Marriner GA, Katzenellenbogen BS, Katzenellenbogen JA. Estrogen receptor-beta potency-selective ligands: structure-activity relationship studies of diarylpropionitriles and their acetylene and polar analogues. J Med Chem. 2001;44:4230–4251. doi: 10.1021/jm010254a. [DOI] [PubMed] [Google Scholar]
- Mikkola TS, Clarkson TB. Estrogen replacement therapy, atherosclerosis, and vascular function. Cardiovasc Res. 2002;53:605–619. doi: 10.1016/s0008-6363(01)00466-7. [DOI] [PubMed] [Google Scholar]
- Miller BH, Gore AC. N-methyl-d-aspartate receptor subunit expression in gonadotropin-releasing hormone neurons changes during reproductive senescence in the female rat. Endocrinology. 2002;143:3568–3574. doi: 10.1210/en.2002-220346. [DOI] [PubMed] [Google Scholar]
- Milner TA, McEwen BS, Hayashi S, Li CJ, Reagan LP, Alves SE. Ultrastructural evidence that hippocampal alpha estrogen receptors are located at extranuclear sites. J Comp Neurol. 2001;429:355–371. [PubMed] [Google Scholar]
- Milner TA, Ayoola K, Drake CT, Herrick SP, Tabori NE, McEwen BS, Warrier S, Alves SE. Ultrastructural localization of estrogen receptor beta immunoreactivity in the rat hippocampal formation. J Comp Neurol. 2005;491:81–95. doi: 10.1002/cne.20724. [DOI] [PubMed] [Google Scholar]
- Mitra SW, Hoskin E, Yudkovitz J, Pear L, Wilkinson HA, Hayashi S, Pfaff DW, Ogawa S, Rohrer SP, Schaeffer JM, McEwen BS, Alves SE. Immunolocalization of estrogen receptor β in the mouse brain: comparison with estrogen receptor α. Endocrinology. 2003;144:2055–2067. doi: 10.1210/en.2002-221069. [DOI] [PubMed] [Google Scholar]
- Morrison MF, Kallan MJ, Ten Have T, Katz I, Tweedy K, Battistini M. Lack of efficacy of estradiol for depression in postmenopausal women: a randomized, controlled trial. Biol Psychiatry. 2004;55:406–412. doi: 10.1016/j.biopsych.2003.08.011. [DOI] [PubMed] [Google Scholar]
- Mulnard RA, Cotman CW, Kawas C, van Dyck CH, Sano M, Doody R, Koss E, Pfeiffer E, Jin S, Gamst A, Grundman M, Thomas R, Thal LJ. Estrogen replacement therapy for treatment of mild to moderate Alzheimer disease: a randomized controlled trial. Alzheimer's Disease Cooperative Study. JAMA. 2000;283:1007–1015. doi: 10.1001/jama.283.8.1007. [DOI] [PubMed] [Google Scholar]
- Murphy DD, Segal M. Regulation of dendritic spine density in cultured rat hippocampal neurons by steroid hormones. J Neurosci. 1996;16:4059–4068. doi: 10.1523/JNEUROSCI.16-13-04059.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy DD, Cole NB, Greenberger V, Segal M. Estradiol increases dendritic spine density by reducing GABA neurotransmission in hippocampal neurons. J Neurosci. 1998;18:2550–2559. doi: 10.1523/JNEUROSCI.18-07-02550.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers LS, Dixen J, Morrissette D, Carmichael M, Davidson JM. Effects of estrogen, androgen, and progestin on sexual psychophysiology and behavior in postmenopausal women. J Clin Endocrinol Metab. 1990;70:1124–1131. doi: 10.1210/jcem-70-4-1124. [DOI] [PubMed] [Google Scholar]
- Nathorst-Boos J, von Schoultz B, Carlstrom K. Elective ovarian removal and estrogen replacement therapy: effects on sexual life, psychological well-being and androgen status. J Psychosom Obstet Gynaecol. 1993a;14:283–293. doi: 10.3109/01674829309084451. [DOI] [PubMed] [Google Scholar]
- Nathorst-Boos J, Wiklund I, Mattsson LA, Sandin K, von Schoultz B. Is sexual life influenced by transdermal estrogen therapy? A double blind placebo controlled study in postmenopausal women. Acta Obstet Gynecol Scand. 1993b;72:656–660. doi: 10.3109/00016349309021160. [DOI] [PubMed] [Google Scholar]
- Nilsen J, Brinton RD. Divergent impact of progesterone and medroxyprogesterone acetate (Provera) on nuclear mitogen-activated protein kinase signaling. Proc Natl Acad Sci USA. 2003;100:10506–10511. doi: 10.1073/pnas.1334098100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsen J, Brinton RD. Mitochondria as therapeutic targets of estrogen action in the central nervous system. Curr Drug Targets CNS Neurol Disord. 2004;3:297–313. doi: 10.2174/1568007043337193. [DOI] [PubMed] [Google Scholar]
- Nilsen J, Chen S, Brinton RD. Dual action of estrogen on glutamate-induced calcium signaling: mechanisms requiring interaction between estrogen receptors and src/mitogen activated protein kinase pathway. Brain Res. 2002;930:216–234. doi: 10.1016/s0006-8993(02)02254-0. [DOI] [PubMed] [Google Scholar]
- Nilsen J, Deng J, Brinton RD. Impact of clinically relevant progestins on the neural effects of estradiol and the signaling pathways involved. Drug News Perspect. 2005;18:545–553. doi: 10.1358/dnp.2005.18.9.948450. [DOI] [PubMed] [Google Scholar]
- Nilsen J, Irwin RW, Brinton RD. Brain mitochondria as therapeutic target for the prevention of Alzheimer's pathology. Alzheimer's and Dementia. 2006a;2([Suppl 3]):S632–S633. [Google Scholar]
- Nilsen J, Morales A, Brinton RD. Medroxyprogesterone acetate exacerbates glutamate excitotoxicity. Gynecol Endocrinol. 2006b;22:355–361. doi: 10.1080/09513590600863337. [DOI] [PubMed] [Google Scholar]
- North American Menopause Society. The menopause practice: a clinician's guide. 2004. http://www.menopause.org/aboutmeno/overview.htm.
- Pfaff DW. Estrogens and brain function: neural analysis of a hormone-controlled mammalian reproductive behavior. New York: Springer; 1980. [Google Scholar]
- Pike CJ. Estrogen modulates neuronal Bcl-xL expression and beta-amyloid-induced apoptosis: relevance to Alzheimer's disease. J Neurochem. 1999;72:1552–1563. doi: 10.1046/j.1471-4159.1999.721552.x. [DOI] [PubMed] [Google Scholar]
- Purnelle G, Gerard A, Czajkowski V, Bourguignon JP. Pulsatile secretion of gonadotropin-releasing hormone by rat hypothalamic explants without cell bodies of GnRH neurons. Neuroendocrinology. 1997;66:305–312. doi: 10.1159/000127253. [DOI] [PubMed] [Google Scholar]
- Rapp PR, Morrison JH, Roberts JA. Cyclic estrogen replacement improves cognitive function in aged ovariectomized rhesus monkeys. J Neurosci. 2003a;23:5708–5714. doi: 10.1523/JNEUROSCI.23-13-05708.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapp SR, Espeland MA, Shumaker SA, Henderson VW, Brunner RL, Manson JE, Gass ML, Stefanick ML, Lane DS, Hays J, Johnson KC, Coker LH, Dailey M, Bowen D. Effect of estrogen plus progestin on global cognitive function in postmenopausal women: the Women's Health Initiative Memory Study: a randomized controlled trial. JAMA. 2003b;289:2663–2672. doi: 10.1001/jama.289.20.2663. [DOI] [PubMed] [Google Scholar]
- Resnick SM, Henderson VW. Hormone therapy and risk of Alzheimer disease: a critical time. JAMA. 2002;288:2170–2172. doi: 10.1001/jama.288.17.2170. [DOI] [PubMed] [Google Scholar]
- Resnick SM, Maki PM, Rapp SR, Espeland MA, Brunner R, Coker LH, Granek IA, Hogan P, Ockene JK, Shumaker SA. Effects of combination estrogen plus progestin hormone treatment on cognition and affect. J Clin Endocrinol Metab. 2006;91:1802–1810. doi: 10.1210/jc.2005-2097. [DOI] [PubMed] [Google Scholar]
- Rossouw JE, Anderson GL, Prentice RL, LaCroix AZ, Kooperberg C, Stefanick ML, Jackson RD, Beresford SA, Howard BV, Johnson KC, Kotchen JM, Ockene J. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results from the Women's Health Initiative randomized controlled trial. JAMA. 2002;288:321–333. doi: 10.1001/jama.288.3.321. [DOI] [PubMed] [Google Scholar]
- Rubin BS, Bridges RS. Alterations in luteinizing hormone-releasing hormone release from the mediobasal hypothalamus of ovariectomized, steroid-primed middle-aged rats as measured by push-pull perfusion. Neuroendocrinology. 1989;49:225–232. doi: 10.1159/000125121. [DOI] [PubMed] [Google Scholar]
- Rubin BS, Lee CE, King JC. A reduced proportion of luteinizing hormone (LH)-releasing hormone neurons express Fos protein during the preovulatory or steroid-induced LH surge in middle-aged rats. Biol Reprod. 1994;51:1264–1272. doi: 10.1095/biolreprod51.6.1264. [DOI] [PubMed] [Google Scholar]
- Rudick CN, Woolley CS. Estrogen regulates functional inhibition of hippocampal CA1 pyramidal cells in the adult female rat. J Neurosci. 2001;21:6532–6543. doi: 10.1523/JNEUROSCI.21-17-06532.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudick CN, Gibbs RB, Woolley CS. A role for the basal forebrain cholinergic system in estrogen-induced disinhibition of hippocampal pyramidal cells. J Neurosci. 2003;23:4479–4490. doi: 10.1523/JNEUROSCI.23-11-04479.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro N. The menopausal transition. Am J Med. 2005;118:8–13. doi: 10.1016/j.amjmed.2005.09.008. [DOI] [PubMed] [Google Scholar]
- Sarrel P, Dobay B, Wiita B. Estrogen and estrogen-androgen replacement in postmenopausal women dissatisfied with estrogen-only therapy. Sexual behavior and neuroendocrine responses. J Reprod Med. 1998;43:847–856. [PubMed] [Google Scholar]
- Savonenko AV, Markowska AL. The cognitive effects of ovariectomy and estrogen replacement are modulated by aging. Neuroscience. 2003;119:821–830. doi: 10.1016/s0306-4522(03)00213-6. [DOI] [PubMed] [Google Scholar]
- Schmidt PJ. Mood, depression, and reproductive hormones in the menopausal transition. Am J Med. 2005;118:54–58. doi: 10.1016/j.amjmed.2005.09.033. [DOI] [PubMed] [Google Scholar]
- Schmidt PJ, Nieman L, Danaceau MA, Tobin MB, Roca CA, Murphy JH, Rubinow DR. Estrogen replacement in perimenopause-related depression: a preliminary report. Am J Obstet Gynecol. 2000;183:414–420. doi: 10.1067/mob.2000.106004. [DOI] [PubMed] [Google Scholar]
- Schmidt PJ, Haq N, Rubinow DR. A longitudinal evaluation of the relationship between reproductive status and mood in perimenopausal women. Am J Psychiatry. 2004;161:2238–2244. doi: 10.1176/appi.ajp.161.12.2238. [DOI] [PubMed] [Google Scholar]
- Schmidt PJ, Palladino-Negro P, Haq N, Gibson C, Rubinow DR. Pharmacologically-induced hypogonadism and sexual function in healthy young women and men. Endocr Soc Abstr. 2006;3:553. doi: 10.1038/npp.2008.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Setalo G, Jr, Singh M, Nethrapalli IS, Toran-Allerand CD. Protein kinase C activity is necessary for estrogen-induced Erk phosphorylation in neocortical explants. Neurochem Res. 2005;30:779–790. doi: 10.1007/s11064-005-6871-y. [DOI] [PubMed] [Google Scholar]
- Sherwin BB. Changes in sexual behavior as a function of plasma sex steroid levels in post-menopausal women. Maturitas. 1985;7:225–233. doi: 10.1016/0378-5122(85)90044-1. [DOI] [PubMed] [Google Scholar]
- Sherwin BB. The impact of different doses of estrogen and progestin on mood and sexual behavior in postmenopausal women. J Clin Endocrinol Metab. 1991;72:336–343. doi: 10.1210/jcem-72-2-336. [DOI] [PubMed] [Google Scholar]
- Sherwin BB. Estrogen and cognitive functioning in women. Endocr Rev. 2003;24:133–151. doi: 10.1210/er.2001-0016. [DOI] [PubMed] [Google Scholar]
- Sherwin BB. Estrogen and cognitive aging in women. Neuroscience. 2006;138:1021–1026. doi: 10.1016/j.neuroscience.2005.07.051. [DOI] [PubMed] [Google Scholar]
- Shifren JL, Braunstein GD, Simon JA, Casson PR, Buster JE, Redmond GP, Burki RE, Ginsburg ES, Rosen RC, Leiblum SR, Caramelli KE, Mazer NA. Transdermal testosterone treatment in women with impaired sexual function after oophorectomy. N Engl J Med. 2000;343:682–688. doi: 10.1056/NEJM200009073431002. [DOI] [PubMed] [Google Scholar]
- Shivers BD, Harlan RE, Morrell JI, Pfaff DW. Absence of oestradiol concentration in cell nuclei of LHRH-immunoreactive neurons. Nature. 1983;304:345–347. doi: 10.1038/304345a0. [DOI] [PubMed] [Google Scholar]
- Shughrue PJ, Merchenthaler I. Estrogen is more than just a “sex hormone”: novel sites for estrogen action in the hippocampus and cerebral cortex. Front Neuroendocrinol. 2000;21:95–101. doi: 10.1006/frne.1999.0190. [DOI] [PubMed] [Google Scholar]
- Shumaker SA, Rapp SR, Espeland MA, Wallace RB, Hendrix SL, Henderson VW, Gass ML, Thal L. Effects of estrogen plus progestin on risk of dementia—reply. JAMA. 2003a;290:1707–1708. [Google Scholar]
- Shumaker SA, Legault C, Rapp SR, Thal L, Wallace RB, Ockene JK, Hendrix SL, Jones BN, III, Assaf AR, Jackson RD, Kotchen JM, Wassertheil-Smoller S, Wactawski-Wende J. Estrogen plus progestin and the incidence of dementia and mild cognitive impairment in postmenopausal women: the Women's Health Initiative Memory Study: a randomized controlled trial. JAMA. 2003b;289:2651–2662. doi: 10.1001/jama.289.20.2651. [DOI] [PubMed] [Google Scholar]
- Shumaker SA, Legault C, Kuller L, Rapp SR, Thal L, Lane DS, Fillit H, Stefanick ML, Hendrix SL, Lewis CE, Masaki K, Coker LH. Conjugated equine estrogens and incidence of probable dementia and mild cognitive impairment in postmenopausal women: Women's Health Initiative Memory Study. JAMA. 2004;291:2947–2958. doi: 10.1001/jama.291.24.2947. [DOI] [PubMed] [Google Scholar]
- Simerly RB, Swanson LW. The distribution of neurotransmitter-specific cells and fibers in the anteroventral periventricular nucleus: Implications for the control of gonadotropin secretion in the rat. Brain Res. 1987;400:11–34. doi: 10.1016/0006-8993(87)90649-4. [DOI] [PubMed] [Google Scholar]
- Simerly RB, Chang C, Muramatsu M, Swanson LW. Distribution of androgen and estrogen receptor mRNA-containing cells in the rat brain: an in situ hybridization study. J Comp Neurol. 1990;294:76–95. doi: 10.1002/cne.902940107. [DOI] [PubMed] [Google Scholar]
- Simon J, Braunstein G, Nachtigall L, Utian W, Katz M, Miller S, Waldbaum A, Bouchard C, Derzko C, Buch A, Rodenberg C, Lucas J, Davis S. Testosterone patch increases sexual activity and desire in surgically menopausal women with hypoactive sexual desire disorder. J Clin Endocrinol Metab. 2005;90:5226–5233. doi: 10.1210/jc.2004-1747. [DOI] [PubMed] [Google Scholar]
- Simoncini T, Hafezi-Moghadam A, Brazil DP, Ley K, Chin WW, Liao JK. Interaction of oestrogen receptor with the regulatory subunit of phosphatidylinositol-3-OH kinase. Nature. 2000;407:538–541. doi: 10.1038/35035131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpkins JW, Wang J, Wang X, Perez E, Prokai L, Dykens JA. Mitochondria play a central role in estrogen-induced neuroprotection. Curr Drug Targets CNS Neurol Disord. 2005;4:69–83. doi: 10.2174/1568007053005073. [DOI] [PubMed] [Google Scholar]
- Singh M. Ovarian hormones elicit phosphorylation of Akt and extracellular-signal regulated kinase in explants of the cerebral cortex. Endocrine. 2001;14:407–415. doi: 10.1385/ENDO:14:3:407. [DOI] [PubMed] [Google Scholar]
- Singh M, Setalo G, Jr, Guan X, Frail DE, Toran-Allerand CD. Estrogen-induced activation of the mitogen-activated protein kinase cascade in the cerebral cortex of estrogen receptor-alpha knock-out mice. J Neurosci. 2000;20:1694–1700. doi: 10.1523/JNEUROSCI.20-05-01694.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh M, Dykens JA, Simpkins JW. Novel mechanisms for estrogen-induced neuroprotection. Exp Biol Med (Maywood) 2006;231:514–521. doi: 10.1177/153537020623100505. [DOI] [PubMed] [Google Scholar]
- Soares CN, Almeida OP, Joffe H, Cohen LS. Efficacy of estradiol for the treatment of depressive disorders in perimenopausal women: a double-blind, randomized, placebo-controlled trial. Arch Gen Psychiatry. 2001;58:529–534. doi: 10.1001/archpsyc.58.6.529. [DOI] [PubMed] [Google Scholar]
- Tang MX, Jacobs D, Stern Y, Marder K, Schofield P, Gurland B, Andrews H, Mayeux R. Effect of oestrogen during menopause on risk and age at onset of Alzheimer's disease. Lancet. 1996;348:429–432. doi: 10.1016/S0140-6736(96)03356-9. [DOI] [PubMed] [Google Scholar]
- Tang Y, Minwalla L, Janssen WGM, Adams MM, McKay H, Roberts JA, Hof PR, Rapp PR, Morrison JH. Effects of estrogen on excitatory synapses in hippocampal CA1 of aged female rhesus monkeys. Soc Neurosci Abstr. 2004a;30:757.16. [Google Scholar]
- Tang Y, Janssen WG, Hao J, Roberts JA, McKay H, Lasley B, Allen PB, Greengard P, Rapp PR, Kordower JH, Hof PR, Morrison JH. Estrogen replacement increases spinophilin-immunoreactive spine number in the prefrontal cortex of female rhesus monkeys. Cereb Cortex. 2004b;14:215–223. doi: 10.1093/cercor/bhg121. [DOI] [PubMed] [Google Scholar]
- Tinkler GP, Tobin JR, Voytko ML. Effects of two years of estrogen loss or replacement on nucleus basalis cholinergic neurons and cholinergic fibers to the dorsolateral prefrontal and inferior parietal cortex of monkeys. J Comp Neurol. 2004;469:507–521. doi: 10.1002/cne.11028. [DOI] [PubMed] [Google Scholar]
- Toran-Allerand CD. Minireview: a plethora of estrogen receptors in the brain: where will it end? Endocrinology. 2004;145:1069–1074. doi: 10.1210/en.2003-1462. [DOI] [PubMed] [Google Scholar]
- Toran-Allerand CD, Singh M, Setalo G., Jr Novel mechanisms of estrogen action in the brain: new players in an old story. Front Neuroendocrinol. 1999;20:97–121. doi: 10.1006/frne.1999.0177. [DOI] [PubMed] [Google Scholar]
- Turgeon JL, McDonnell DP, Martin KA, Wise PM. Hormone therapy: physiological complexity belies therapeutic simplicity. Science. 2004;304:1269–1273. doi: 10.1126/science.1096725. [DOI] [PubMed] [Google Scholar]
- Voytko ML. Estrogen and the cholinergic system modulate visuospatial attention in monkeys (Macaca fascicularis) Behav Neurosci. 2002;116:187–197. doi: 10.1037//0735-7044.116.2.187. [DOI] [PubMed] [Google Scholar]
- Wassertheil-Smoller S, Shumaker S, Ockene J, Talavera GA, Greenland P, Cochrane B, Robbins J, Aragaki A, Dunbar-Jacob J. Depression and cardiovascular sequelae in postmenopausal women. The Women's Health Initiative (WHI) Arch Intern Med. 2004;164:289–298. doi: 10.1001/archinte.164.3.289. [DOI] [PubMed] [Google Scholar]
- Weiland NG. Estradiol selectively regulates agonist binding sites on the N-methyl-d-aspartate receptor complex in the CA1 region of the hippocampus. Endocrinology. 1992;131:662–668. doi: 10.1210/endo.131.2.1353442. [DOI] [PubMed] [Google Scholar]
- Wiegand SJ, Terasawa E, Bridson WE. Persistent estrus and blockade of progesterone-induced LH release follows lesions which do not damage the suprachiasmatic nucleus. Endocrinology. 1978;102:1645–1648. doi: 10.1210/endo-102-5-1645. [DOI] [PubMed] [Google Scholar]
- Wiklund I, Karlberg J, Mattsson LA. Quality of life of postmenopausal women on a regimen of transdermal estradiol therapy: a double-blind placebo-controlled study. Am J Obstet Gynecol. 1993;168:824–830. doi: 10.1016/s0002-9378(12)90828-5. [DOI] [PubMed] [Google Scholar]
- Woods NF, Mitchell ES, Adams C. Memory functioning among midlife women: observations from the Seattle Midlife Women's Health Study. Menopause. 2000;7:257–265. [PubMed] [Google Scholar]
- Woolley CS, McEwen BS. Estradiol mediates fluctuation in hippocampal synapse density during the estrous cycle in the adult rat. J Neurosci. 1992;12:2549–2554. doi: 10.1523/JNEUROSCI.12-07-02549.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolley CS, McEwen BS. Roles of estradiol and progesterone in regulation of hippocampal dendritic spine density during the estrous cycle in the rat. J Comp Neurol. 1993;336:293–306. doi: 10.1002/cne.903360210. [DOI] [PubMed] [Google Scholar]
- Woolley CS, McEwen BS. Estradiol regulates hippocampal dendritic spine density via an N-methyl-d-aspartate receptor-dependent mechanism. J Neurosci. 1994;14:7680–7687. doi: 10.1523/JNEUROSCI.14-12-07680.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolley CS, Gould E, Frankfurt M, McEwen BS. Naturally occurring fluctuation in dendritic spine density on adult hippocampal pyramidal neurons. J Neurosci. 1990;10:4035–4039. doi: 10.1523/JNEUROSCI.10-12-04035.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolley CS, Wenzel HJ, Schwartzkroin PA. Estradiol increases the frequency of multiple synapse boutons in the hippocampal CA1 region of the adult female rat. J Comp Neurol. 1996;373:108–117. doi: 10.1002/(SICI)1096-9861(19960909)373:1<108::AID-CNE9>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Woolley CS, Weiland NG, McEwen BS, Schwartzkroin PA. Estradiol increases the sensitivity of hippocampal CA1 pyramidal cells to NMDA receptor-mediated synaptic input: correlation with dendritic spine density. J Neurosci. 1997;17:1848–1859. doi: 10.1523/JNEUROSCI.17-05-01848.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T, Chen S, Brinton RD. Estrogen membrane receptor imaging coupled with estradiol activation of intracellular calcium rise and ERK activation in single neurons. Soc Neurosci Abstr. 2004;30:659.20. [Google Scholar]
- Wu TW, Wang JM, Chen S, Brinton RD. 17beta-estradiol induced Ca2+ influx via L-type calcium channels activates the Src/ERK/cyclic-AMP response element binding protein signal pathway and BCL-2 expression in rat hippocampal neurons: a potential initiation mechanism for estrogen-induced neuroprotection. Neuroscience. 2005;135:59–72. doi: 10.1016/j.neuroscience.2004.12.027. [DOI] [PubMed] [Google Scholar]
- Yaffe K. Hormone therapy and the brain: deja vu all over again? JAMA. 2003;289:2717–2719. doi: 10.1001/jama.289.20.2717. [DOI] [PubMed] [Google Scholar]
- Yaffe K, Sawaya G, Lieberburg I, Grady D. Estrogen therapy in postmenopausal women: effects on cognitive function and dementia. JAMA. 1998;279:688–695. doi: 10.1001/jama.279.9.688. [DOI] [PubMed] [Google Scholar]
- Yaffe K, Krueger K, Sarkar S, Grady D, Barrett-Connor E, Cox DA, Nickelsen T. Cognitive function in postmenopausal women treated with raloxifene. N Engl J Med. 2001;344:1207–1213. doi: 10.1056/NEJM200104193441604. [DOI] [PubMed] [Google Scholar]
- Yaffe K, Krueger K, Cummings SR, Blackwell T, Henderson VW, Sarkar S, Ensrud K, Grady D. Effect of raloxifene on prevention of dementia and cognitive impairment in older women: the Multiple Outcomes of Raloxifene Evaluation (MORE) randomized trial. Am J Psychiatry. 2005;162:683–690. doi: 10.1176/appi.ajp.162.4.683. [DOI] [PubMed] [Google Scholar]
- Yaffe K, Vittinghoff E, Ensrud KE, Johnson KC, Diem S, Hanes V, Grady D. Effects of ultra-low-dose transdermal estradiol on cognition and health-related quality of life. Arch Neurol. 2006;63:945–950. doi: 10.1001/archneur.63.7.945. [DOI] [PubMed] [Google Scholar]
- Yildirim M, Janssen WG, Lou WWY, McEwen BS, Milner TA, Morrison JH. Effects of estrogen and aging on distribution of pAkt in CA1 of rat hippocampus. Soc Neurosci Abstr. 2006;32:659.1. [Google Scholar]
- Yin W, Gore AC. Neuroendocrine control of reproductive aging: roles of GnRH neurons. Reproduction. 2006;131:403–414. doi: 10.1530/rep.1.00617. [DOI] [PubMed] [Google Scholar]
- Yin W, Oung T, Ng CL, Janssen WGM, Morrison JH, Gore AC. Novel subcellular localization of NMDA receptors (NMDARs) within neuroendocrine GnRH terminals. Soc Neurosci Abstr. 2002;28:368.10. [Google Scholar]
- Zandi PP, Carlson MC, Plassman BL, Welsh-Bohmer KA, Mayer LS, Steffens DC, Breitner JC. Hormone replacement therapy and incidence of Alzheimer disease in older women: the Cache County Study. JAMA. 2002;288:2123–2129. doi: 10.1001/jama.288.17.2123. [DOI] [PubMed] [Google Scholar]
- Zhao L, Brinton R. Estrogen receptor beta as a therapeutic target for promoting neurogenesis and preventing neurodegeneration. Drug Dev Res. 2006a;66:103–117. [Google Scholar]
- Zhao L, Brinton R. Select estrogens within the complex formulation of conjugated equine estrogens (Premarin) are protective against neurodegenerative insults: implications for a composition of estrogen therapy to promote neuronal function and prevent Alzheimer's disease. BMC Neurosci. 2006b;7:24. doi: 10.1186/1471-2202-7-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, O'Neill K, Diaz Brinton R. Selective estrogen receptor modulators (SERMs) for the brain: current status and remaining challenges for developing NeuroSERMs. Brain Res Brain Res Rev. 2005a;49:472–493. doi: 10.1016/j.brainresrev.2005.01.009. [DOI] [PubMed] [Google Scholar]
- Zhao L, Chen S, Wang JM, Brinton RD. 17beta-estradiol induces Ca2+ influx, dendritic and nuclear Ca2+ rise and subsequent cyclic AMP response element-binding protein activation in hippocampal neurons: a potential initiation mechanism for estrogen neurotrophism. Neuroscience. 2005b;132:299–311. doi: 10.1016/j.neuroscience.2004.11.054. [DOI] [PubMed] [Google Scholar]
- Znamensky V, Akama KT, McEwen BS, Milner TA. Estrogen levels regulate the subcellular distribution of phosphorylated Akt in hippocampal CA1 dendrites. J Neurosci. 2003;23:2340–2347. doi: 10.1523/JNEUROSCI.23-06-02340.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zweifel JE, O'Brien WH. A meta-analysis of the effect of hormone replacement therapy upon depressed mood. Psychoneuroendocrinology. 1997;22:189–212. doi: 10.1016/s0306-4530(96)00034-0. [DOI] [PubMed] [Google Scholar]