

Abstract
The neuron-specific K+–Cl− cotransporter KCC2 extrudes Cl− and renders GABA and glycine action hyperpolarizing. Thus, it plays a pivotal role in neuronal inhibition. Development-dependent KCC2 activation is regulated at the transcriptional level and by unknown posttranslational mechanisms. Here, we analyzed KCC2 activation at the protein level in the developing rat lateral superior olive (LSO), a prominent auditory brainstem structure. Electrophysiology demonstrated ineffective KCC2-mediated Cl− extrusion in LSO neurons at postnatal day 3 (P3). Immunohistochemical analyses by confocal and electron microscopy revealed KCC2 signals at the plasma membrane in the somata and dendrites of both immature and mature neurons. Biochemical analysis demonstrated mature glycosylation pattern of KCC2 at both stages. Immunoblot analysis of the immature brainstem demonstrated mainly monomeric KCC2. In contrast, three KCC2 oligomers with molecular masses of ∼270, ∼400, and ∼500 kDa were identified in the mature brainstem. These oligomers were sensitive to sulfhydryl-reducing agents and resistant to SDS, contrary to the situation seen in the related Na+–(K+)–Cl− cotransporter. In HEK-293 cells, coexpressed hemagglutinin-tagged KCC2 assembled with histidine-tagged KCC2, demonstrating formation of homomers. Based on these findings, we conclude that the oligomers represent KCC2 dimers, trimers, and tetramers. Finally, immunoblot analysis identified a development-dependent increase in the oligomer/monomer ratio from embryonic day 18 to P30 throughout the brain that correlates with KCC2 activation. Together, our data indicate that the developmental shift from depolarization to hyperpolarization can be determined by both increased gene expression and KCC2 oligomerization.
Keywords: auditory brainstem, brain development, chloride regulation, synaptic inhibition, oligomerization, cation-chloride cotransporters
Introduction
Fast inhibitory neurotransmission in the CNS is mediated by GABA and glycine via ligand-gated Cl− channels. In the vast majority of mature neurons, a low intracellular chloride concentration ([Cl−]i) causes Cl− influx during channel opening, resulting in hyperpolarization (Eccles, 1966; Kaila, 1994). In contrast, a high [Cl−]i in immature neurons results in Cl− efflux at the resting membrane potential (Zhang et al., 1991; Wang et al., 1994; Obrietan and van den Pol, 1995; Ehrlich et al., 1999; Kakazu et al., 1999). This efflux depolarizes the neurons and can even elicit action potentials. In all CNS regions analyzed so far, the neuron-specific K+–Cl− cotransporter KCC2 (Payne et al., 1996) is responsible for Cl− extrusion and, hence, for the generation of the Cl−-mediated hyperpolarizing currents prevalent in mature neurons (Rivera et al., 1999; Hübner et al., 2001; Balakrishnan et al., 2003; Zhu et al., 2005). Recently, it was also shown that KCC2 is not only necessary but sufficient to cause hyperpolarizing GABA currents (Chudotvorova et al., 2005; Fiumelli et al., 2005; Lee et al., 2005). In agreement with its essential role for inhibitory synapses, KCC2 knock-out mice die perinatally (Hübner et al., 2001).
KCC2 belongs to the cation–chloride cotransporter (CCC) family, which comprises the Na+–(K+)–Cl− cotransporters [N(K)CCs] NCC, NKCC1, and NKCC2, as well as the K+–Cl− cotransporters (KCCs) KCC1, KCC2, KCC3, and KCC4 (Delpire and Mount, 2002; Payne et al., 2003). All members likely contain 12 transmembrane domains (Gerelsaikhan and Turner, 2000). Biochemical studies have demonstrated a homodimeric organization of NKCC1 (Moore-Hoon and Turner, 2000), NCC (de Jong et al., 2003), and NKCC2 (Starremans et al., 2003). A homo-oligomeric organization was also proposed for KCCs, because a truncated KCC1 form was found to be a dominant-negative inhibitor of KCCs (Casula et al., 2001).
Recent results point to two rate-limiting steps of KCC2 activation during development. In the rat hippocampus, the pure increase in the protein amount renders GABAergic action hyperpolarizing (Rivera et al., 1999). This is in contrast to the lateral superior olive (LSO), a conspicuous auditory structure in the medullary brainstem. Expression analysis demonstrated abundant KCC2 protein in immature LSO neurons without an substantial increase in KCC2 during maturation (Balakrishnan et al., 2003). However, a genetic knockdown of the KCC2 gene SLC12A5 (solute carrier family 12, member 5) only affects [Cl−]i in mature, but not in immature, LSO neurons (Balakrishnan et al., 2003). This implies the presence of transport-inactive KCC2 protein during early development. A transport-inactive form of KCC2 was also reported by other groups (Kelsch et al., 2001; Vale et al., 2003; Woodin et al., 2003; Fiumelli et al., 2005; Khirug et al., 2005). To characterize the underlying mechanism that leads to transport-inactive KCC2 protein, we here analyzed KCC2 in the developing brainstem, beginning with electrophysiological and immunohistochemical approaches. To gain insight into the structure–function relationship of KCC2, we further analyzed the structural organization of transport-active and transport-inactive KCC2 in the mature and immature brainstem, respectively. This analysis led us to propose a novel posttranslational regulatory mechanism for KCC2.
Materials and Methods
Animals were bred and housed in our animal facilities and treated in compliance with the current national Animal Protection Law. All protocols were approved by the regional animal care and use committees and adhered to the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Slice preparation for electrophysiology.
Rats between postnatal day 3 (P3) and P12 were deeply anesthetized with pentobarbital (30–40 mg/kg body weight, i.p.). Brains were removed in ice-cold standard physiological solution containing 124 mm NaCl, 3 mm KCl, 2 mm CaCl2, 25 mm NaHCO3, 1.1 mm NaH2PO4, 2 mm MgSO4, and 10 mm d-glucose, equilibrated with 95% O2 and 5% CO2, pH 7.4, and 300-μm-thick coronal brainstem slices were cut with a VT 1000S vibrating blade microtome (Leica, Wetzlar, Germany). Slices were allowed to recover for at least 1 h at 32°C in standard physiological solution (bubbled with 95% O2/5% CO2).
Assay of Cl− extrusion capacity in brainstem slices.
The electrophysiological experiments were performed as described previously (Khirug et al., 2005). In brief, somatic recordings of LSO neurons were performed in standard physiological solution at room temperature (22–25°C) in the whole-cell voltage-clamp configuration using an EPC 10 patch-clamp amplifier (HEKA Elektronik, Lambrecht, Germany). Patch pipettes were fabricated from borosilicate glass (Harvard Apparatus, Edenbridge, UK), and their resistances were 5–7.5 MΩ. Composition of the patch-pipette solution was as follows (in mm): 18 KCl, 111 K-gluconate, 0.5 CaCl2, 2 NaOH, 10 d-glucose, 10 HEPES, 2 Mg-ATP, and 5 BAPTA, pH 7.3 adjusted with KOH. In some experiments, the Cl− concentration was increased by using 40 mm KCl and 79 mm K-gluconate. The membrane potential was clamped at −70 mV, and all potential values were corrected for a liquid junction potential of 10 mV. Only cells with a resting membrane potential more negative than −50 mV were included in the analysis. The reversal potential of GABAergic currents (EGABA) was determined from the current–voltage (I–V) relationship obtained by stepwise clamping the membrane potential at different holding values.
α-Carboxy-2-nitrobenzyl-caged GABA (Invitrogen, Karlsruhe, Germany) was diluted in the standard physiological solution (5 mm final concentration) and delivered to the vicinity of the patch-clamped cell via a quartz syringe tip (100 μm internal diameter) using an UltraMicroPump II syringe pump (World Precision Instruments, Sarasota, FL) at a flow rate of 1–5 μl/min. For local photolysis of caged GABA, the 351–364 nm output of a continuous emission argon–ion laser (Enterprise 653; Coherent, Palo Alto, CA) was delivered via a multimode optical fiber through an Olympus Optical (Tokyo, Japan) LUMPlanFl 60× water-immersion objective. The diameter of the uncaging spot was 10 μm (Khirug et al., 2005). An electronic shutter (Uniblitz, Puchheim, Germany) was used to set the light pulse duration at 15–25 ms. To visualize the dendrites, a fluorescent indicator (Alexa Fluor 488, 50 μm; Invitrogen) was dialyzed via the patch pipette, and dendrites were traced using a confocal microscope (Radiance 2100; Bio-Rad, Herts, UK).
To assess the efficacy of Cl− extrusion, 19 or 41 mm Cl− was added to the pipette solution. These concentrations result in EGABA values of −50 or −30 mV, respectively, calculated on the basis of the Nernst equation. EGABA values were compared for currents evoked at the soma (EGABA,soma) and at the dendrite (EGABA,dendrite). If robust Cl− extrusion occurs, i.e., if KCC2 activity is strong, this leads to more negative dendritic EGABA compared with the somatic EGABA, which is clamped by the Cl− load via the patch pipette.
Statistical analysis and curve fitting were performed using Origin software (Microcal Software, Northampton, MA). Averaged data are presented as mean ± SD. Statistical significance was assessed using a Student's unpaired t test.
Production of a rabbit N-terminal KCC2 polyclonal antibody.
A plasmid was constructed that encodes a fusion protein of the maltose binding protein (New England Biolabs, Frankfurt, Germany) and the N-terminal residues 1–93 of the rat KCC2 (Payne et al., 1996). The fusion protein was produced in the Escherichia coli strain BL21 (Stratagene, Heidelberg, Germany), purified with an amylose resin column, and injected subcutaneously into rabbits to generate polyclonal sera. Polyclonal anti-N-terminal KCC2 polyclonal (nKCC2) antibodies were affinity purified by linking the fusion protein to Affi-Gel 15 support (Bio-Rad, München, Germany), following the protocol of the manufacturer. Specific antibody was eluted with 0.1 m Na-citrate, pH 3.1, and neutralized with NaOH. After purification, anti-nKCC2 was concentrated in PBS (in mm: 130 NaCl, 7 Na2HPO4, and 3 NaH2PO4, pH 7.4) using a 30 kDa cutoff vivaspin column (Vivascience, Hannover, Germany) and stored in aliquots at −20°C.
Immunohistochemistry.
Experiments were performed on Sprague Dawley rats of both genders, aged P0–P60, and on P12 wild-type and KCC2 knockdown mice (Woo et al., 2002). The genotyping of KCC2 knockdown mice was performed according to the protocol described by Woo et al. (2002). Animals were deeply anesthetized with chloral hydrate (700 mg/kg body weight, i.p.) and perfused transcardially with PBS, followed by Zamboni's solution (2% paraformaldehyde and 15% picric acid in 0.1 m phosphate buffer, pH 7.4). Brains were removed and stored in fixative overnight in the refrigerator. After incubation in 30% sucrose/PBS for cryoprotection, 30-μm-thick coronal sections were cut through the brainstem, collected in 15% sucrose/PBS, thoroughly rinsed in PBS, and blocked for 1 h in 3% bovine serum albumin (BSA), 10% goat serum, and 0.3% Triton X-100 in Tris-buffered saline, pH 7.4. In addition to the novel anti-nKCC2 antibody, a commercially available rabbit anti-cKCC2 antibody (raised against residues 932–1043 of the rat KCC2; catalog #07-432; Upstate Biotechnology, Hamburg, Germany) was used in some experiments. This antibody corresponds to the antibody published previously by Williams et al. (1999). No difference between the two KCC2 antibodies was noticed throughout the experiments. The monoclonal mouse anti-MAP2 antibody against the microtubule-associated protein 2 was obtained from Sigma (Taufkirchen, Germany) and used to identify neuronal somata and dendrites. Antibodies were diluted 1:500 and added to the blocking solution. Incubation with agitation was performed in a refrigerator for 24 h. After several rinses in PBS, sections were transferred to carrier solution (0.3% Triton X-100, 1% BSA, and 1% goat serum) and treated with the secondary antibodies, goat anti-rabbit conjugated to Alexa Fluor 488, and goat anti-mouse conjugated to Alexa Fluor 546 (both diluted 1:1000; Invitrogen). Sections were incubated overnight with agitation in a refrigerator, rinsed again, mounted on slides, and air dried, and coverslips were mounted with Vectashield H-1400 anti-fade mounting medium (Vector Laboratories, Burlingame, CA). Control experiments were performed by omitting primary antibodies and resulted in the absence of immunosignals. Photomicrographs of sagittal sections from P4 and P12 mouse brains were taken with an Axioskop (Zeiss, Oberkochen, Germany) with a 5× objective and mounted together with Axio Vision software 4.2.
Confocal fluorescence microscopy.
Images were taken with a confocal laser scanning microscope (LSM 510; Zeiss) equipped with argon (488 nm) and helium–neon (543 nm) lasers and appropriate excitation and emission filters for maximum separation of Alexa Fluor 488 (488 nm excitation; bandpass filter 505–550) and Alexa Fluor 546 (543 nm excitation; long-pass filter 560). This configuration prevented bleed-through. A 10× objective lens (Plan-Neofluar, 10×/numerical aperture 0.3; Zeiss) and a 40× oil-immersion objective lens (Plan-Neofluar, 40×/numerical aperture 1.3 oil; Zeiss) were used. A pinhole size of 67 μm resulted in optical sections of <10 μm thickness (10× objective lens) and <1 μm (40× objective lens). Images of 2048 × 2048 pixels were obtained and further processed with Zeiss LSM Image browser software 2.80. Figures were prepared on a personal computer running Adobe Photoshop 5.5 (Adobe Systems, San Jose, CA).
Electron microscopy.
Preembedding immunoelectron microscopy was performed as follows: P4 and P12 rats were deeply anesthetized by a mixture of 1.2% ketamine 10 and 0.13% Rompun in physiological salt solution (100 μl/g body weight) and then perfused through the heart with 4% paraformaldehyde and 0.5% glutaraldehyde in 0.1 m phosphate buffer. Coronal vibratome sections, 500-μm-thick were prepared at the level of the LSO. Sections containing the LSO were cryoprotected through an ascending series of sucrose solution (0.5, 1, 1.5, and 2 m) and then subjected to two cycles of freeze–thaw in liquid nitrogen to aid the penetration of immunoreagents into the cells. The sections were blocked for 1 h in 10% normal goat serum and 0.2% BSA in PBS. Incubation of primary antibody (1:200; rabbit anti-KCC2) (Hübner et al., 2001) was done in carrier (PBS with 1% serum and 0.2% BSA) at 4°C overnight. The secondary antibody (1:100, goat anti-rabbit nanogold; Nanoprobes, Stony Brook, NY) was applied for 2 h and subsequently enhanced with the HQ silver enhancement kit (Nanoprobes), followed by gold toning with 0.05% gold chloride (Sigma) in 150 mm sodium acetate buffer. After fixation with 1% osmium tetroxide, sections were dehydrated in an ascending series of ethanol and embedded in Epon (Roth, Karlsruhe, Germany). The semithin sections were finally examined with a Zeiss EM 902.
Tissue RNA isolation and reverse transcription-PCR.
Sprague Dawley rats at P3 or P12 were deeply anesthetized with chloral hydrate and killed by decapitation. Their brains were rapidly removed, and the brainstems were dissected and stored at −80°C. Total RNA was isolated from brainstems by the guanidine thiocyanate method (Chomczynski and Sacchi, 1987). The quality and quantity of the RNA samples were assessed by gel electrophoresis and optical density measurements. Reverse transcription of total RNA (20 μg) was performed using standard protocols with random hexanucleotide priming and SuperScript II as enzyme (Invitrogen, Karlsruhe, Germany). The open reading frame of the KCC2 gene SLC12A5 was amplified in nine overlapping fragments, using the primers listed in supplemental Table S1 (available at www.jneurosci.org as supplemental material). PCR was performed for 35 cycles in a total volume of 50 μl. Denaturing was at 94°C for 30 s, annealing was at 60°C for 30 s, and elongation was at 72°C for 1 min. Ten microliters of each reaction were loaded onto a high-resolution 5% polyacrylamide gel. After electrophoresis, the gel was stained with silver nitrate and scanned using a GS-800 calibrated densitometer (Bio-Rad, München, Germany). Single-strand conformational polymorphism (SSCP) analysis was performed as described previously (Orita et al., 1989). In brief, PCR products were denatured for 10 min at 95°C in SSCP loading buffer (95% formamide and 0.2% xylene cyanol), chilled on ice, and then loaded onto 5% polyacrylamide gels with or without 10% glycerol. After electrophoresis, gels were silver stained and scanned.
Immunoblot analysis.
Proteins were resolved by linear 3–8% Tris–acetate gel or 4–12% Bis–Tris gel systems (NuPAGE; Invitrogen, Karlsruhe, Germany). Treatment of samples with sulfhydryl-reducing agents or detergents was for 5 min at 40°C, if not indicated otherwise. Gel loading was done in a buffer containing 0.5% lithium dodecyl sulfate (LDS) to solubilize KCC2. Electrophoretic separation lasted either 1.5 (normal run) or 3 h (long run). Proteins were electrophoretically transferred to polyvinylidene difluoride (PVDF) membranes (Roth) in NuPAGE transfer buffer (in mm: 25 Bicine, 25 Bis-Tris, 1 EDTA, and 0.05 chlorobutanol) using the NuPAGE system. PVDF-bound proteins were visualized by staining with ponceau S. Membranes were incubated in TTBS/milk (20 mm Tris, 150 mm NaCl, 0.1% Tween 20, and 5% nonfat dry milk, pH 7.5) with the respective antiserum overnight in the refrigerator, followed by 2 h at room temperature. Anti-nKCC2 was used for KCC2 and the monoclonal antibody T4 against NKCC1 (Lytle et al., 1995) and α5 against the α-subunit of the Na+/K+-ATPase (Lebovitz et al., 1989). T4 and α5 were obtained from the Developmental Studies Hybridoma Bank of the University of Iowa (Iowa City, IA). Anti-hemagglutinin (HA) was from Covance (Princeton, NJ) and anti-histidine (His) from Santa Cruz Biotechnology (Heidelberg, Germany). Both antibodies were used at a 1:1000 dilution. After four washes in TTBS, the secondary antibody (1:5000, goat anti-rabbit IgG or anti-mouse IgG horseradish peroxidase conjugated; Amersham Biosciences, Freiburg, Germany) was applied for 1 h in TTBS/milk. After four washes in TTBS, bound antibodies were detected using an enhanced chemiluminescence kit and a VersaDoc 3000 documentation system (Bio-Rad, München, Germany). Quantification of the volume (i.e., the product of area and intensity) of different KCC2-immunoreactive bands on immunoblots was performed using the Quantity One software (Bio-Rad, München, Germany). To calculate the relative amount of monomeric or oligomeric KCC2, the volume of the monomeric structure or the sum of the volumes of the three oligomeric structures, respectively, was divided by the sum of the volumes of all KCC2-immunoreactive bands and multiplied by 100. To get the monomer/oligomer ratio, the volume of the monomeric structure was divided by the sum of the volumes of the three oligomeric structures. Averaged data are presented as mean ± SD. Statistical significance was assessed using a Student's unpaired t test.
Deglycosylation with endoglycosidase H and N-glycosidase F.
Deglycosylation with endoglycosidase H (endo H) and N-glycosidase F (PGNase F) (New England Biolabs) was performed with purified membrane protein fractions according to the protocol provided by the manufacturer. Membrane proteins were first incubated for 10 min at 100°C in glycoprotein denaturing buffer (0.5% SDS and 1% β-mercaptoethanol) and then incubated for 60 min at 37°C in either 50 mm sodium citrate, pH 5.5, at 37°C supplemented with 1250 U of endo H or in 50 mm sodium phosphate, pH 7.5, 1% NP-40, supplemented with 1250 U of PGNase. After incubation, samples were separated on a 4–12% Bis–Tris NuPAGE system.
DNA constructs and His tag pull-down.
The full-length cDNA encoding the rat KCC2 was subcloned into the pENTR/D-TOPO gateway vector (Invitrogen, Karlsruhe, Germany). After sequence confirmation, the clone was transferred to the pCDNA3 vector (Invitrogen, Karlsruhe, Germany) to express wild-type KCC2 or to the pMTH2SM–HA clone (Kaufman et al., 1989) to obtain HA-tagged KCC2. To generate His10-KCC2, the open reading frame of KCC2 was amplified using a forward primer encoding a start methionin, His10, and amino acids 2–6 of KCC2 (CACCATGCATCATCACCATCATCACCATCATCACCATCTCAACAACCTGACGGACTG) and a reverse KCC2 primer encoding the last six amino acids and the stop codon and cloned in the pENTR/D-TOPO gateway vector. After sequence confirmation, the clone was transferred to the pCDNA3 vector containing a gateway cassette.
HEK-293 cells were grown to 30% confluency in 100 mm dishes and transiently transfected using Fugene (Roche, Mannheim, Germany). After 36 h in culture, cells were harvested and lysed in 500 μl per dish of 50 mm NaH2PO4, 300 mm NaCl, 10 mm imidazol, and 0.5% Triton X-100, pH 8.0, for 10 min at 4°C. For pull-down experiments, the lysates were cleared by centrifugation at 10,000 × g for 10 min, and the supernatant was added to 10 μl of Ni2+ resin (Qiagen, Hilden, Germany). After 2 h at 4°C, the mixture was pelleted and the resin were washed three times in 50 mm NaH2PO4, 300 mm NaCl, 20 mm imidazol, and 0.5% Triton X-100, pH 8.0, according to the protocol of the manufacturer. Elution was in 30 μl of 50 mm NaH2PO4, 300 mm NaCl, 250 mm imidazol, and 0.5% Triton X-100, and the sample was separated by electrophoresis.
Results
Cl− extrusion capacity in immature and mature LSO neurons
The activity of KCC2 during early postnatal development of LSO neurons was addressed previously by applying the perforated patch-clamp technique to wild-type and KCC2 knockdown mice (Balakrishnan et al., 2003). The 95% reduction of the KCC2 level in the knockdown mice (Woo et al., 2002) affected [Cl−]i in LSO neurons at P12 yet not at P3. To assess the KCC2 extrusion activity more directly, we performed whole-cell patch-clamp recordings from rat LSO neurons. Under a constant Cl− load via a somatic patch pipette, transport-active KCC2 generates a declining somatodendritic Cl− gradient (Jarolimek et al., 1999; Kelsch et al., 2001; Khirug et al., 2005). We determined EGABA at the soma and at proximal dendrites, 50 μm away from the soma, by focal photolysis of caged GABA (Khiroug et al., 2003; Khirug et al., 2005). We observed declining somatodendritic Cl− gradients at P12 yet not at P3 (n = 5 for each age group). Figure 1, A and B, illustrates examples of GABA-evoked currents and the resulting I–V relationships at P3 and P12, respectively. At both ages, EGABA,soma was close to −50 mV, as expected from the Nernst equation for a constant load with 19 mm [Cl−] via the patch pipette. EGABA,dendrite was −50 mV for the P3 neuron but more negative (−65 mV) for the P12 neuron. Statistical analysis resulted in EGABA,soma values of −54.2 ± 4.2 mV at P3 and −56.4 ± 2.3 mV at P12. EGABA,dendrite was −56.2 ± 5.0 mV at P3 and −69.0 ± 6.2 mV at P12 (Fig. 1C). Statistically significant differences between EGABA,soma and EGABA,dendrite occurred only in the P12 neurons (p < 0.01; n = 5 for each age group). The results show effective Cl− extrusion activity at P12 and ineffective activity at P3. The decrease in the amplitude of GABA-evoked currents between P3 and P12 is consistent with a developmental decrease in GABAergic transmission in the superior olivary complex (SOC) (Kotak et al., 1998; Nabekura et al., 2004).
Figure 1.

Analysis of Cl− extrusion capacity in the developing LSO. A, Current traces obtained from a P3 LSO neuron by focal GABA uncaging at the soma and at a proximal dendrite at different holding potentials. Horizontal bars indicate the time of the uncaging UV flash. Peak amplitudes of the GABA-evoked currents were used to generate the I–V relationship (right). EGABA was −48 and −50 mV at the soma and at the dendrite, respectively. B, Current traces (left) and EGABA determination (right) for a P12 LSO neuron. Horizontal bars indicate the time of the GABA uncaging UV flash. In contrast to the P3 neuron (A), EGABA,dendrite (−65 mV) was clearly more negative than EGABA,soma (−56 mV). C, At P3 (n = 5), EGABA,soma (−54.6 ± 4.2 mV) was not different from EGABA,dendrite (−56.2 ± 5.0 mV; p > 0.05). Under the same conditions, EGABA,soma at P12 (−56.4 ± 2.3 mV) differed significantly from EGABA,dendrite (−69.0 ± 6.2 mV; p < 0.01; n = 5). D, When the Cl− load was increased by raising the Cl− concentration in the patch pipette to 41 mm, this resulted in an EGABA,soma of approximately −30 mV. Bumetanide was added to the bath (10 μm final concentration) to exclude an influence of NKCC1 on EGABA. However, under these conditions, there was no difference between EGABA,soma (−29.2 ± 2.4 mV) and −EGABA,dendrite (−29.7 ± 2.9 mV; n = 10; p > 0.05) at P3. E, Schematic drawing of the experimental setup. Focal photolytic release of caged GABA was used to determine EGABA,dendrite and EGABA,soma under constant Cl− load via the patch pipette. Flashes indicate sites of GABA uncaging. If EGABA,dendrite < EGABA,soma, an effective Cl− extrusion mechanism is present.
NKCC1 is considered to be a major source of Cl− import into immature neurons (Sung et al., 2000; Yamada et al., 2004) and, if also present in LSO neurons, might mask KCC2 activity at P3 in these cells. To exclude this possibility, we added bumetanide to the bath at a concentration of 10 μm, known to specifically block NKCC1 (Russell, 2000). In addition, we raised the Cl− concentration in the patch pipettes to 41 mm (corresponding to an ECl− of −30 mV) to impose the neurons to the high Cl− load observed in immature LSO neurons (Balakrishnan et al., 2003). Both values, EGABA,soma of −29.2 ± 2.4 mV and EGABA,dendrite of −29.7 ± 2.9 mV, were close to the calculated value of −30 mV and not statistically different at P3 (Fig. 1 D) (p > 0.05; n = 10). Under the same conditions, however, the difference between EGABA,soma and EGABA,dendrite was 12.7 ± 4.0 mV in P12 LSO neurons, indicating robust Cl− extrusion at this age (n = 3; data not shown). These results strongly imply transport-inactive KCC2 in immature LSO neurons.
Polyclonal antibody production and characterization
To be able to analyze KCC2 during development by biochemical and immunohistochemical assays, we generated a novel antibody (anti-nKCC2) against the 93 amino acids at the N terminus of rat KCC2. We chose the N-terminal region because the available antibodies against KCC2 were directed against the C terminus (anti-cKCC2) (Lu et al., 1999; Williams et al., 1999). We tested the specificity of the antibody by immunohistochemistry and immunoblot analysis on P12 wild-type and KCC2 knockdown mice. In wild-type mice, strong labeling was obtained in the entire brainstem, including the SOC nuclei (Fig. 2A). At high magnification, strong KCC2 immunoreactivity (IR) outlined the somata and proximal dendrites and resulted in diffuse labeling of the neuropil (Fig. 2C). In contrast, in KCC2 knockdown mice, only a faint, punctuated labeling was present (Fig. 2B), making it impossible to identify individual neurons (Fig. 2D). Identical results were obtained when we used the anti-cKCC2 antibody (Fig. 2E,F). The faint immunoreactivity observed with both antibodies in the KCC2 knockdown mice likely represents the residual 5% KCC2 protein amount in these animals, because no labeling was obtained when the anti-KCC2 antibodies were omitted (Fig. 2G). Immunoblot analysis confirmed the specificity of the anti-nKCC2 antibody. A strong signal at the correct molecular weight range of 130–140 kDa was present in the brain membrane fraction of wild-type animals, whereas the signal was very faint in the membrane fraction of KCC2 knockdown mice (Fig. 2H). A weak signal at a lower molecular weight (86 kDa) was detected in both wild-type and knockdown mice, indicating a slight cross-reactivity of the anti-nKCC2 antibody. These results demonstrate that, despite a slight cross-reactivity, the novel antibody anti-nKCC2 is well suited for immunohistochemical KCC2 analysis.
Figure 2.

Analysis of anti-KCC2 antibody specificity. A–F, Anti-KCC2 antibodies directed against the N terminus (anti-nKCC2; A–D) or the C terminus (anti-cKCC2; E, F) were applied to coronal brainstem sections of P12 wild-type (A, C, E) or KCC2 knockdown (B, D, F) mice. A, For anti-nKCC2 in wild-type mice, a moderate IR was observed in sections containing the medial nucleus of the trapezoid body (MNTB) and the LSO, two of the main SOC nuclei. B, Only faint IR was present in KCC2 knockdown mice. C, E, High-magnification confocal images illustrated a similar labeling pattern for anti-nKCC2 and anti-cKCC2 in +/+ mice. D, F, Labeling of the presumed surface of LSO neuron somata (n) and transversely cut dendrites in the neuropil (np) is indicated by arrows and arrowheads, respectively. Both antibodies revealed only faint IR in the LSO of −/− mice. G, No immunoreactive signal was present in the LSO of wild-type mice incubated with secondary antibodies alone. Scale bar: A, B, 200 μm; C–G, 10 μm. H, Immunoblot analysis of anti-nKCC2. Membrane fractions of brains from wild-type (left) or KCC2 knockdown (right) mice were probed with anti-nKCC2. A strong signal in the range of the expected molecular weight was observed in +/+ mice but not in −/− mice. In addition, a weak signal was observed at lower molecular weight, independent of the genotype. 7n, Facial nerve; d, dorsal; l, lateral; Mr, relative molecular mass; +/+, wild-type mice; −/−, KCC2 knockdown mice.
KCC2 immunoreactivity in developing LSO neurons
To analyze age-dependent changes in the amount and distribution of KCC2 at the cellular level, we focused on rat LSO neurons and analyzed KCC2-IR at P0, P4, P12, and P60, using anti-nKCC2. Already at P0, i.e., well before Cl− extrusion is effective, KCC2-IR densely outlined the somata and proximal dendrites. A strong and diffuse labeling pattern was also present in the remaining neuropil (Fig. 3A). At P4, the labeling pattern was similar in that the somata and proximal dendrites were again outlined (Fig. 3B). However, the diffuse neuropil labeling was reduced, leading to the detection of KCC2-IR at the membrane of transversely cut dendrites. The reduction in diffuse neuropil labeling proceeded with additional development, resulting in clearly outlined somata and dendrites by P12 and P60 (Fig. 3C,D). Throughout development, the soma interior was weakly labeled (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). Indistinguishable results were obtained with the anti-cKCC2 antibody, i.e., there was no difference in the intensity of KCC2-IR at the soma surface between immature and mature neurons (data not shown).
Figure 3.
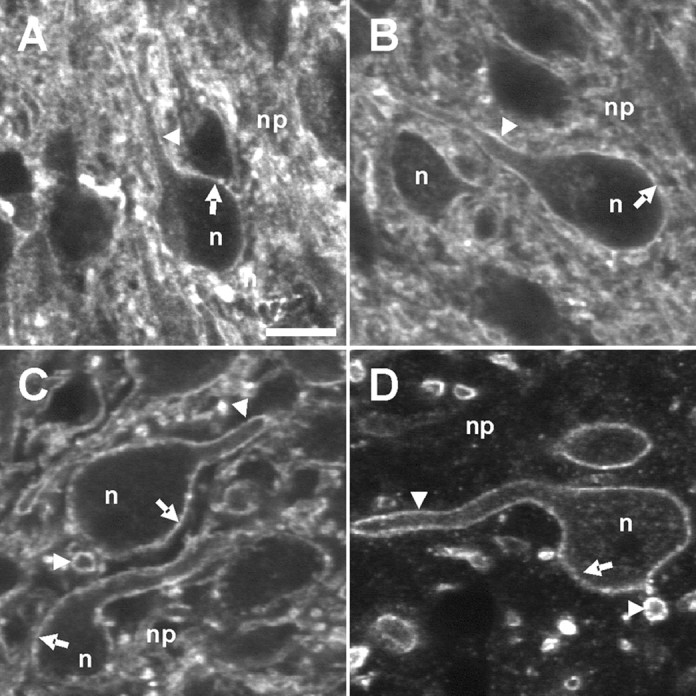
KCC2-IR in rat LSO neurons during maturation. A–D, Strong KCC2-IR was present in LSO neurons (n) at P0 (A), P4 (B), P12 (C), and P60 (D) using anti-nKCC2. Both the plasma membrane of somata (arrows) and the plasma membrane of transversely or longitudinally cut dendrites (arrowheads) were strongly KCC2 immunoreactive. The density of KCC2-IR in the neuropil (np) decreased with age. A weak labeling in the soma interior was obvious at all ages. Scale bar: (in A) AA–D, 10 μm.
The age-related reduction of KCC2-IR in the LSO neuropil may reflect pruning of dendrites (Rietzel and Friauf, 1998) and/or the confinement of KCC2 to dendritic subregions in mature LSO neurons. To distinguish between these two possibilities, we performed double-immunofluorescent labeling for KCC2 and the dendritic marker MAP2. At all ages, dendrites identified by MAP2 were also KCC2 immunoreactive (supplemental Fig. S2A,B, available at www.jneurosci.org as supplemental material). Thus, the reduction of KCC2-IR in the LSO neuropil during maturation is most likely obtained via pruning of dendrites. To corroborate the localization of KCC2 at the plasma membrane, we performed double-labeling studies with KCC2 and markers of the biosynthetic–secretory pathway. At both P4 and P12, we observed no colocalization of KCC2 with either protein disulfide isomerase, a marker for the endoplasmatic reticulum (Vaux et al., 1990), or with the Golgi matrix protein 130, a marker for the Golgi apparatus (Nakamura et al., 1995) (supplemental Fig. S2C–F, available at www.jneurosci.org as supplemental material). Only the vesicle-associated membrane protein 2, a marker for several types of vesicles (Jahn and Sudhof, 1999; Carvalho et al., 2004), revealed a weak colocalization in the neuropil at both ages (supplemental Fig. S2G,H, available at www.jneurosci.org as supplemental material).
Together, these immunohistochemical data demonstrate that KCC2 proteins are abundant and located in the plasma membrane, or in close proximity to it, in LSO neurons already at a time when Cl− extrusion is not yet effective. This implies that KCC2 transport activity in developing LSO neurons is not regulated via an intracellular retention mechanism.
Ultrastructural analysis of KCC2 location
To corroborate the data obtained by confocal microscopy, we next analyzed the location of KCC2 in immature and mature LSO neurons at the ultrastructural level. Using electron microscopy and the preembedding immunogold technique, the density and distribution of KCC2 was found to be similar to the labeling pattern observed at the light microscopic level. In semithin sections, we observed KCC2-IR at somatic and dendritic plasma membranes at both P4 and P12 (Fig. 4A,B). In high-magnification electron micrographs, we localized gold particles along the cytoplasmic side of the dendritic plasma membranes at both ages (Fig. 4C,D). The distribution of the gold particles was homogenous at extrasynaptic and perisynaptic sites of excitatory and inhibitory synapses (Fig. 4E–H). Only the postsynaptic thickening was almost devoid of KCC2 molecules. The only age-related change appeared to be a higher number of gold particles inside LSO dendrites at P4 compared with P12 (Fig. 4C,D). The localization of the antibody, which was raised against the N terminus of KCC2 (Hübner et al., 2001), at the intracellular surface of the plasma membrane, is in line with the model predicting an intracellular position of both protein termini (Payne et al., 1996).
Figure 4.
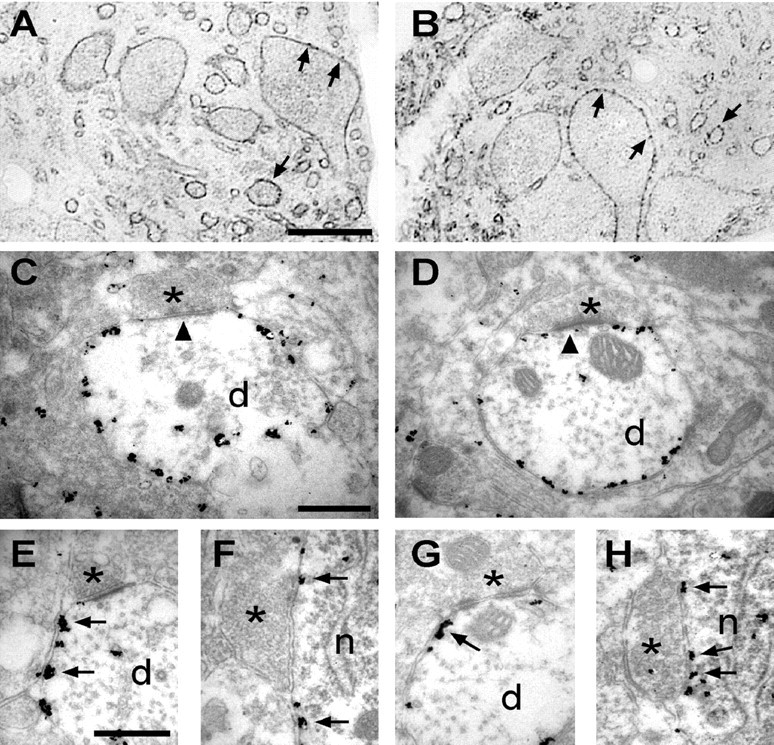
Ultrastructural localization of KCC2 in LSO neurons during maturation. Location of KCC2 molecules in the LSO of P4 (A, C, E, F) and P12 (B, D, G, H) rats, revealed with the preembedding immunogold technique. A, B, Semithin sections with labeled neurons in the LSO. Silver-intensified gold particles were found at the presumed surface of somatic and dendritic membranes (arrows). C, D, High-magnification electron micrographs illustrated labeled dendritic profiles with boutons (asterisks) making asymmetrical synapses (arrowheads) onto the dendrite. KCC2 was localized homogenously along the perisynaptic and extrasynaptic membranes at both ages. The postsynaptic thickening was almost devoid of KCC2 (arrowheads). E–H, High-magnification ultrastructural analysis showed even distribution of gold particles at perisynaptic sites (arrows) of excitatory (E, G) and inhibitory (F, H) synapses (asterisks). n, Neuronal soma; d, dendrite. Scale bars: (in A) A, B, 30 μm; (in C) C–H, 500 μm.
Glycosylation pattern of KCC2
The KCC2-IR results obtained at the light and electron microscopic levels, together with our electrophysiological data, suggested the presence of transport-inactive KCC2 molecules at the plasma membrane before Cl− extrusion becomes effective. CCCs are glycoproteins (Payne, 1997; Williams et al., 1999; de Jong et al., 2002). To determine whether transport-inactive and transport-active KCC2 forms differ in their glycosylation pattern, we treated isolated membrane fractions from P2 and P30 rats with the glycosidases endo H and PGNase F. Endo H cleaves only high-mannose glycosylated forms and some hybrid oligosaccharides from N-linked glycoproteins (Maley et al., 1989). In contrast, PGNase F is an amidase that cleaves high-mannose, hybrid, and complex oligosaccharides from N-linked glycoproteins (Maley et al., 1989). Treatment with PGNase resulted in a molecular mass shift from ∼130–140 to ∼120 kDa for transport-inactive and transport-active KCC2 protein (Fig. 5A). In contrast, the ∼130–140 kDa protein was insensitive to endo H at both ages (Fig. 5B). These data demonstrate that transport-inactive KCC2 displays a mature glycosylation pattern. Thus, age-related differences in glycosylation are unlikely to cause KCC2 activation.
Figure 5.
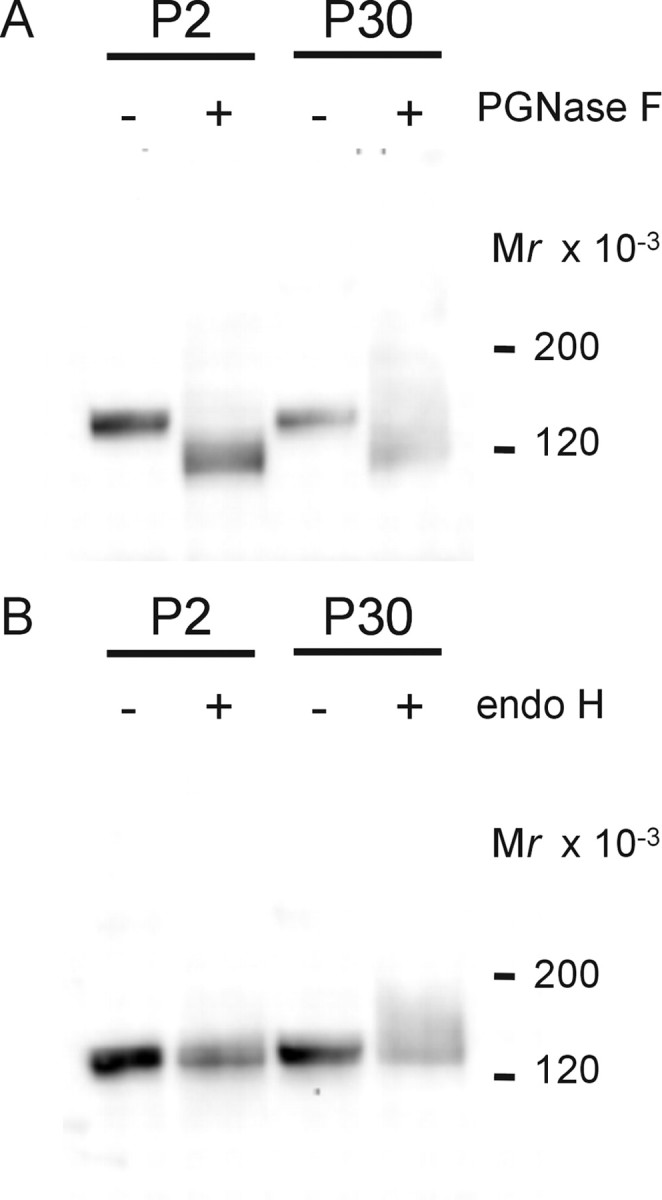
A, B, Glycosylation pattern of KCC2 during maturation. Membrane fractions from P2 or P30 rat brainstems were left untreated or treated with PGNase F (A) or endo H (B) and then separated under reducing conditions by a linear 4–12% Bis–Tris NuPAGE system for 1.5 h. At both ages, KCC2 was PGNase sensitive and endo H insensitive.
Analysis for developmentally regulated KCC2 splice variants
Transport-inactive and transport-active KCC2 forms may be generated via the expression of age-dependent splice variants. To address this possibility, we amplified the open reading frame of the KCC2 gene SLC12A5 in nine overlapping fragments from cDNA of P3 and P12 rat brainstem. Separation of the PCR products on a high-resolution polyacrylamide gel revealed no age-related difference in length (supplemental Fig. S3, available at www.jneurosci.org as supplemental material). The products showed a migration pattern that fits to the calculated lengths. In a next step, we analyzed whether small variations in the transcripts occurred that escaped detection under conditions of normal PAGE. We performed a SSCP analysis, which is capable of detecting single nucleotide exchanges or minimal alterations in length (Orita et al., 1989). We found no difference for the nine fragments amplified from P3 or P12 brainstem cDNA (data not shown).
Structural organization of KCC2
We next investigated the structural organization of KCC2, with the aim to further explore whether the functionally relevant changes with age occur at the protein level. Previous biochemical analyses had demonstrated a dimeric organization of NKCC1 (Moore-Hoon and Turner, 2000), NCC (de Jong et al., 2003), and NKCC2 (Starremans et al., 2003). These dimers became destabilized by high concentrations of the non-ionic detergent Triton X-100 (≥0.4%) or the anionic detergent SDS (≥0.01%). We performed an immunoblot analysis of the membrane fraction from P30 rat brainstem, separated by electrophoresis in a 4–12% Bis–Tris gel in the absence of sulfhydryl-reducing agents. It revealed most KCC2-IR above 200 kDa (Fig. 6), although 0.5% LDS was present in the loading buffer. Only a minor signal was detected at ∼130–140 kDa, which is the molecular mass of the monomeric protein (Lu et al., 1999; Williams et al., 1999; Hübner et al., 2001). In experiments involving additional treatment of the membrane fraction with 4% Triton X-100 or 0.5% SDS for 5 min at 40°C before electrophoresis, most KCC2-IR was still detected at molecular masses higher than the monomer, with no apparent difference to the control (Fig. 6). Under the same conditions, however, NKCC1 was present as a monomer (data not shown). Preincubation of the membrane fraction in 1% of the non-ionic detergent N-dodecyl-β-d-maltoside or in 0.5% of the anionic detergent N-laurylsarcosine also caused no increase in the monomeric KCC2 fraction (Fig. 6). Together, these data indicate that KCC2 is present in the form of higher-molecular-mass complexes in the mature brainstem (P30) that are resistant to detergents. Monomeric KCC2 might represent unassembled protein, because the membrane fraction analyzed contains all compartments of the biosynthetic–secretory pathway (Guillemin et al., 2005). Furthermore, the data show that the detection of mainly monomeric KCC2 in previous reports (Lu et al., 1999; Williams et al., 1999; Hübner et al., 2001; Balakrishnan et al., 2003) is likely attributable to the presence of sulfhydryl-reducing agents and not to SDS in the commonly used Laemmli sample buffer.
Figure 6.

Effects of detergents on KCC2. Membranes from P30 rat brainstems were treated for 5 min at 40°C with no detergent (control), 4% Triton X-100, 0.5% SDS, 1% N-dodecyl-β-d-maltoside, or 0.5% N-laurylsarcosine. After adding sample buffer containing 0.5% LDS, samples were separated by a linear 4–12% Bis–Tris NuPAGE system. Immunoblot analysis revealed that, under all conditions, KCC2 was present mainly in molecular masses higher than that expected for the monomer. Asterisk denotes monomeric KCC2, and the bracket depicts high-molecular-mass KCC2-IR.
To examine the effect of sulfhydryl-reducing agents on KCC2 oligomers, we incubated the membrane fraction at increasing concentrations of β-mercaptoethanol. We improved the resolution of higher-molecular-mass KCC2 molecules by performing the electrophoretic separation in a 3–8% Tris–acetate gel for 3 h. In the absence of reducing agents, three additional distinct KCC2-immunoreactive bands were detected, at ∼270, ∼400, and ∼500 kDa (Fig. 7A). This fits very well with the calculated mass of 270, 405, and 540 kDa for dimeric, trimeric, and tetrameric KCC2 molecules, respectively (considering 135 kDa for the KCC2 monomer). Increasing the concentration of β-mercaptoethanol (from 0.005 to 0.1%) resulted in the disassembly of the three oligomeric structures (Fig. 7A). Nevertheless, even at high concentrations of reducing agents (3% β-mercaptoethanol and 1% SDS), we observed some residual oligomeric KCC2 forms (Fig. 7A). In the absence of reducing agents, all three higher-molecular-mass bands were resistant to 1% SDS (Fig. 7A).
Figure 7.

Effect of sulfhydryl-reducing agents on KCC2 oligomers. A, Membranes from P30 rat brainstem were treated for 5 min at 40°C with increasing concentrations of β-mercaptoethanol, separated by a linear 3–8% Tris–acetate NuPAGE system for 3 h, and analyzed by immunoblot. In the absence of reducing agents, four distinct KCC2-immunoreactive bands were present, with molecular masses of ∼130–140, ∼270, ∼400, and ∼500 kDa. Asterisk denotes the monomeric KCC2, and arrows point to KCC2 oligomers. All KCC2 oligomers were β-mercaptoethanol sensitive yet resistant to 1% SDS. B, Analysis of oligomeric structures for the comigration with the Na+/K+-ATPase α subunit. According to A, P30 rat brainstem was analyzed for immunoreactivity against the Na+/K+-ATPase α subunit. Under all conditions, only monomeric Na+/K+-ATPase α subunits were observed, with a molecular mass of ∼100 kDa.
We reasoned that some of the KCC2-IR that we observed at the high molecular mass could have been attributable to a possible interaction of KCC2 with the Na+/K+-ATPase α subunit (Ikeda et al., 2004). To address this possibility, we probed the immunoblot with an antibody against this subunit (Lebovitz et al., 1989). All immunoreactivity against the subunit was detected at ∼100 kDa, which corresponds to the molecular mass of Na+/K+-ATPase α monomers (Fig. 7B). Therefore, we reject the idea that KCC2 monomers had formed hetero-oligomers with Na+/K+-ATPase α subunits under the conditions used.
Next, we examined whether KCC2 forms homo-oligomeric complexes in HEK-293 cells. To this end, KCC2 was first heterologously expressed in HEK-293 cells and oligomerization was assessed. After immunoblot analysis, four KCC2-immunoreactive bands were detected with similar sizes as in the mature brainstem, i.e., at ∼135, ∼270, ∼400, and ∼500 kDa (Fig. 8A). No KCC2-IR was observed in untransfected HEK-293 cells. This result shows that KCC2 can form homo-oligomers in heterologous expression systems or that HEK-293 cells express the interacting protein partners required for forming KCC2 hetero-oligomers. To probe directly for KCC2 homo-oligomers, we tagged KCC2 proteins with the His peptide (His–KCC2) or the HA peptide (HA–KCC2) and coexpressed the corresponding constructs in HEK-293 cells. As a control, HA–KCC2 was coexpressed with untagged KCC2. Purification of His–KCC2 by Ni2+ resin resulted in the copurification of HA–KCC2 only when His–KCC2 was coexpressed (Fig. 8B,C). When substituting His–KCC2 by untagged KCC2, HA–KCC2 could not be isolated by Ni2+ beads (Fig. 8B,C). Together, these data demonstrate that KCC2 can indeed form homo-oligomers. Thus, the observed KCC2 oligomers in the brainstem likely represent homomeric complexes, ranging from dimers to tetramers.
Figure 8.

Physical association between differentially tagged KCC2 proteins. A, Wild-type KCC2 was transfected in HEK-293 cells. After expression, the membrane fraction was isolated and separated by a 3–8% Tris–acetate NuPAGE system for 3 h and analyzed by immunoblot. Four distinct KCC2-immunoreactive bands were present, with molecular masses of ∼130–140, ∼270, ∼400, and ∼500 kDa, similar to the KCC2 oligomers in the P30 brainstem. No KCC2-IR was observed in untransfected HEK-293. B1, Protein lysates from HEK-293 cells expressing either HA–KCC2 and His–KCC2 (lanes 1, 2) or HA–KCC2 with untagged KCC2 (lanes 3, 4) were subjected to purification by Ni2+ beads and resolved by 8% SDS-PAGE. Proteins were detected by immunoblotting with anti-HA or anti-His6 antibody. HA–KCC2 can be isolated by Ni2+ beads only in the presence of coexpressed His–KCC2. Asterisk denotes the monomeric KCC2, and arrowheads indicates high-molecular-mass KCC2 immunoreactivity. Control staining with His tag demonstrates presence of His–KCC2 only in the Ni2+ eluate of HA–KCC2/His–KCC2 double-transfected cells. B2, Schematic drawing of the experimental design. If HA–KCC2 form dimers with His–KCC2, both will be purified by Ni2+ beads. C, Immunoblot of HEK-293 cells transfected with either His–KCC2 or HA–KCC2. Anti-KCC2 detects both fusion proteins, and anti-HA detects only HA–KCC2 and not His–KCC2. This demonstrates that the antibody is not cross-reacting.
Age-dependent oligomerization of KCC2 in the brainstem
We next set out to explore whether oligomerization correlates with KCC2 transport activation in the developing brainstem. To do so, we obtained the membrane fraction from P1/P2 rat brainstem and separated the sample under nonreducing conditions by electrophoresis. Because we assumed highly reactive thiol groups in the KCC2 protein, based on the residual KCC2 oligomers observed in the presence of 3% β-mercaptoethanol, we prepared membranes in the presence of the alkylating agent iodoacetamide to prevent disulfide bonding during lysis, membrane preparation, and electrophoresis. When we did so, quantitative immunoblot analysis revealed 87 ± 4% of KCC2 as monomers at P1/P2, whereas the dimeric, trimeric, and tetrameric structures summed up to 13 ± 4% of the total KCC2 amount (Fig. 9A,B). In contrast, when we used membranes from mature brainstem (P25–P60) prepared in the presence of iodoacetamide, 68 ± 1% of KCC2 was present as oligomers (Fig. 9A,B). Thus, the monomer/oligomer ratio decreased from 7.10 ± 2.60 at P1/P2 to 0.5 ± 0.03 at P25 or older (Fig. 9C). The omission of iodoacetamide from the sample at P1/P2 led to an increased amount of oligomeric KCC2, with molecular masses identical to KCC2 dimer, trimer, and tetramer (Fig. 9A). In contrast, we observed no change at P25 or older for KCC2 oligomerization when iodoacetamide was omitted (Fig. 9A). These data indicate that KCC2 oligomerization in the immature brainstem is prevented by as yet unknown mechanisms, leading to transport-inactive KCC2.
Figure 9.

KCC2 oligomerization during brainstem development. A, Membranes isolated from P2 or P30 rat brainstem were prepared in the presence (+) or absence (−) of 8% iodoacetamide. After separation by a linear 3–8% Tris–acetate NuPAGE system for 3 h, immunoblot analysis was performed. In the presence of iodoacetamide, a prominent monomeric structure and low-abundant oligomeric structures were detected at P2. At P30, oligomeric structures were dominant. In the absence of iodoacetamide, oligomeric KCC2 structures became more prominent at P2 but not at P30. The mass shift induced by iodoacetamide is attributable to its binding to KCC2 thiol groups. Images show representative examples of four independent experiments. B, Relative abundance of the monomeric and the oligomeric structures compared with the whole amount of KCC2 at P1/P2 and in rats aged between P30 and P60. The optical density of the four KCC2-immunoreactive bands was quantified from densitometric scans of eight lanes from three independent preparations for rats aged P1/P2 and six lanes from two independent preparations for rats aged between P25 and P60. Both the decrease in the monomer fraction (87 ± 4 to 32 ± 1%) and the increase in the oligomer fraction (13 ± 4 to 68 ± 1%) were highly significant (p < 0.0001). C, The monomer/oligomer ratio decreased from 7.10 ± 2.60 at P1/P2 to 0.5 ± 0.03 in animals aged between P25 and P60.
KCC2-IR throughout the developing brain
Finally, we addressed the question of whether the abundance of transport-inactive KCC2 protein during the depolarizing phase is limited to the LSO or a common phenomenon. To do so, we studied KCC2-IR throughout the brain at P4 and P12 (Fig. 10). This time window was chosen because [Cl−]i is still high in most brain areas at P4 yet has become low by P12 [neocortex (Owens et al., 1996), hippocampus (Ben Ari et al., 1989; Rivera et al., 1999), hypothalamus (Wang et al., 2001), ventral tegmental area (Ye, 2000), cerebellum (Eilers et al., 2001), SOC (Ehrlich et al., 1999; Kakazu et al., 1999; Löhrke et al., 2005)]. At P4, moderate to strong KCC2-IR was restricted to some brain regions, namely the glomeruli and mitral cell layer of the olfactory bulb, the rostrodorsal region of the caudate–putamen, the thalamus, the hypothalamus, the superior colliculus, the inferior colliculus, the pons, and the medulla oblongata, including the SOC (Fig. 10A). In the cortex and the cerebellum, KCC2-IR was present in dorsocaudal and ventrocaudal regions, respectively. In contrast, a rather uniform level of KCC2-IR was observed at P12 throughout the brain (Fig. 10B). In the regions displaying a high KCC2-IR already at P4, there was no additional increase in the signal, whereas in regions with weak KCC2-IR at P4, e.g., the hippocampus and main parts of the cortex, the signal increased until P12 (Fig. 10A,B). This suggests that transport-inactive KCC2 is not limited to the brainstem but might be also present in other brain regions during early development.
Figure 10.

KCC2-IR throughout the developing brain. A, Montage forming a sagittal section of a P4 rat brain. Moderate to strong KCC2-IR was observed in various brain regions, including the olfactory bulb (OB), the dorsorostral border of the caudate–putamen (CPu), the thalamus (Th), the hypothalamus (Hy), the superior colliculus (SC), the inferior colliculus (IC), the pons (Po), and the medulla oblongata (MO). A dorsocaudal region of the cortex (Cx) showed also KCC2-IR. In the cerebellum (Cb), KCC2-IR is only weak in the rostral region but moderate in the ventrocaudal region. B, At P12, KCC2-IR was moderate throughout the brain, including the hippocampus (Hp) and the cortex, which showed only faint IR at P4. Signal intensity in the cerebellum was stronger than at P4, but a rostrocaudal gradient was still obvious. Scale bar: A, B, 1 mm. 7, Facial nucleus; MNTB, medial nucleus of the trapezoid body; PN, pontine nuclei.
To address whether KCC2 oligomerization is associated with this process, we assessed the oligomerization status of KCC2 in various CNS regions during development [at embryonic day 18 (E18), P1, and P30]. In all eight regions analyzed, namely the olfactory bulb, the cortex/hippocampus, the ventral telencephalon, the diencephalon, the tectum, the cerebellum, the brainstem, and the spinal cord, KCC2 was already detected in immunoblots at E18 (Fig. 11). The highest amount was observed in the caudal regions (brainstem and spinal cord), whereas the lowest amount was detected in the cortex/hippocampus. This conforms well with embryonic KCC2 expression reported recently (Hübner et al., 2001; Li et al., 2002; Stein et al., 2004; Wojcik et al., 2006). In each CNS region analyzed, monomeric KCC2 was prevailing; oligomeric KCC2 was restricted to the olfactory bulb, the cerebellum, and the brainstem (Fig. 11). At P1, the prevailing form was still monomeric KCC2, but now, KCC2 oligomers were clearly present in all regions except the cortex/hippocampus, which, moreover, again displayed the lowest KCC2 abundance among the various regions. Finally, at P30, all regions displayed similar KCC2 abundance with more oligomeric than monomeric KCC2. Together, these data demonstrate that development-dependent KCC2 oligomerization is not restricted to the brainstem but that it occurs throughout the CNS.
Figure 11.
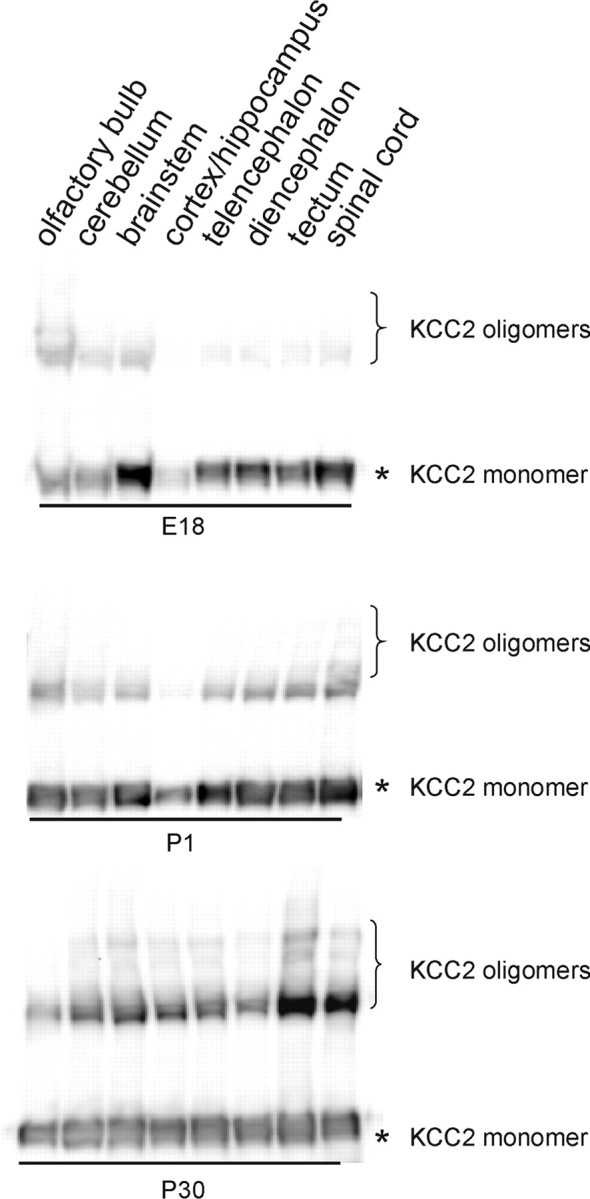
KCC2 oligomerization during development of the nervous system. Indicated brain areas were dissected at E18, P1, or P30, and the membrane fraction was prepared. After separation by a linear 3–8% Tris–acetate NuPAGE for 3 h, immunoblot analysis was performed. KCC2 is detected as early as E18 in all brain areas. During development, the monomer/oligomer ratio decreases in all areas analyzed. Asterisk denotes the monomeric KCC2, and the bracket indicates high-molecular-mass KCC2 immunoreactivity.
Discussion
There are three main conclusions of the present study: (1) functional KCC2 consists of disulfide-bonded homo-oligomers, (2) age-dependent KCC2 oligomerization occurs throughout the nervous system, and (3), both transport-inactive and transport-active KCC2 are present at the plasma membrane.
Oligomeric organization of KCC2
Our conclusion that the structural core unit of transport-active KCC2 is a homo-oligomer is based on evidence obtained from a series of experiments. First, differentially tagged KCC2 fusion proteins physically interact in a heterologous expression system. Second, the molecular masses of 270, ∼400, and ∼500 kDa at which we detected the majority of KCC2-IR in the mature nervous system correspond very well with the calculated values for multimeric KCC2. Third, all three oligomeric KCC2 structures were disulfide bonded and resistant to detergents, thus displaying similar biochemical properties among each other. A homo-oligomeric organization of transport-active KCC2 is in agreement with previous results obtained with a KCC1 dominant mutant form, which reduced KCC1, KCC2, or KCC3 transport activity in Xenopus oocytes by at least two-thirds (Casula et al., 2001). These data suggest that monomeric KCCs are transport inactive and require oligomerization to become transport active. Coimmunoprecipitation studies indeed confirmed a physical association between KCC1 and the mutant form. Moreover, modeling of the oligomeric stoichiometry of interaction between mutant and wild-type KCC1, based on relative mole fractions of injected cRNAs for both proteins, was compatible with a dimeric or tetrameric state (Casula et al., 2001). The requirement of an oligomeric KCC structure for transport activity is supported by our finding that the monomer/oligomer ratio of KCC2 decreases during maturation of the nervous system.
Additional evidence for KCC homo-oligomerization stems from a recent analysis of KCC3. In extracts of Xenopus oocytes expressing a KCC3Q mutant, which lacks the last 338 amino acids, oligomeric KCC3Q was detected with approximately twice the molecular weight of monomeric truncated KCC3Q (Howard et al., 2002). Finally, a homomeric organization of CCCs was also reported for NKCC1 (Moore-Hoon and Turner, 2000), NCC (de Jong et al., 2003), and NKCC2 (Starremans et al., 2003). Together, these data suggest that CCCs form homo-oligomers similar to other transporters such as the organic anion transporter OAT1 (Hong et al., 2005) and the excitatory amino acid transporters EAAT1 and EAAT2 (Dehnes et al., 1998).
The sensitivity of KCC2 oligomers to reducing agents and their resistance to 4% Triton X-100 or 1% SDS demonstrates disulfide bonding as the major type of interaction between the monomers. This contrasts with N(K)CCs, in which dimerization involves noncovalent interactions that are sensitive to detergents such as the non-ionic detergent Triton X-100 (≥0.4%) or the anionic detergent SDS (≥0.01%) (Moore-Hoon and Turner, 2000; de Jong et al., 2003). In addition, we noted a population of KCC2 that was resistant to 3% β-mercaptoethanol and 1% SDS. The resistance may be caused by enzymes such as transglutaminases, which can generate γ-glutamyl isopeptide cross-links between proteins (Nemes et al., 2005). Similar observations were made for the cytoskeleton protein ezrin (Berryman et al., 1995). Thus, different types of interaction are involved in the oligomerization of CCCs.
Oligomerization of KCC2 correlates with onset of Cl− extrusion in the brainstem
We previously reported transport-inactive KCC2 in the rat auditory brainstem at P3 (Balakrishnan et al., 2003). In the present study, we used a distinct electrophysiological approach based on the observation that a somatodendritic EGABA gradient is a useful indicator of efficient KCC2 transport (Jarolimek et al., 1999; Kelsch et al., 2001; Khirug et al., 2005). The results of this approach, together with those from immunohistochemistry, confirmed the presence of transport-inactive KCC2 in immature LSO neurons. Focal GABA uncaging under a constant somatic Cl− load demonstrated detectable dendritic Cl− extrusion only in P12 LSO neurons. P3 LSO neurons did not reveal a somatodendritic EGABA gradient, implying that KCC2 is not involved in Cl− homeostasis at this early age and thus is transport inactive. Immunohistochemistry, using confocal and electron microscopy, confirmed a high abundance of KCC2 at all ages. In addition, immunoblot analysis demonstrated no developmental increase in KCC2 protein. This conforms well with the abundance of KCC2 protein already at E15.5, with no additional increase beyond P3 in the mouse brainstem (Stein et al., 2004). These data are in contrast to those from a recent study that suggested that an increase in KCC2 causes the shift from depolarizing to hyperpolarizing action of glycine in LSO neurons (Shibata et al., 2004).
Our biochemical analysis revealed that the monomer/oligomer ratio decreases from ∼7 to ∼0.5 during maturation of the brainstem. The oligomeric KCC2-immunoreactive signals observed at P1/P2 (13%) are likely attributable to some brainstem regions already containing transport-active KCC2 and displaying effective Cl− extrusion, such as the superior paraolivary nucleus (Löhrke et al., 2005) or the respiratory center (Ikeda et al., 2004). Because functional CCCs are most likely oligomers, the lack of effective KCC2 transport activity in immature neurons can be explained by the prevailing monomeric form at this stage (Fig. 12). This conclusion is supported by our KCC2 expression analysis in other areas of the CNS. Immunohistochemistry analysis detected KCC2 in several CNS areas during early development. More importantly, the monomer/oligomer ratio decreased in all regions analyzed. This demonstrates that development-dependent oligomerization is a widespread phenomenon in the CNS. Furthermore, these data are in line with oligomerization being a rate-limiting step of KCC2 activation during development.
Figure 12.

Model of KCC2 activation in the LSO. In immature LSO neurons, monomeric KCC2 is present in the plasma membrane. Because the monomeric KCC2 is transport inactive, LSO neurons exhibit a relative high [Cl−]i. Development-dependent oligomerization activates KCC2, thereby lowering [Cl−]i. This change in [Cl−]i results in a developmental shift from depolarization to hyperpolarization (D/H-shift). Hatched ovals represent glycine receptors (GlyR), and filled ovals represent individual KCC2 polypeptides.
Transport-inactive KCC2 is localized at the plasma membrane
The mechanisms that give rise to predominantly monomeric KCC2 in the immature brainstem are unknown. Preliminary immunohistochemical analysis suggested the retention of KCC2 in the biosynthetic–secretory pathway in immature LSO neurons (Balakrishnan et al., 2003). However, confocal and electron microscopy clearly demonstrate KCC2 labeling at the plasma membrane of somata and dendrites in both immature and mature LSO neurons. This is in agreement with our deglycosylation experiments that identified an endo H-resistant, mature glycosylation pattern of KCC2 already at P2. The presence of KCC2 at the plasma membrane in immature auditory brainstem neurons is in accordance with recent studies in other auditory brainstem centers, which also detected KCC2-IR at the plasma membrane in immature and mature neurons (Löhrke et al., 2005; Vale et al., 2005). Thus, localization of the protein at the plasma membrane appears to be an insufficient criterion to distinguish transport-active from transport-inactive KCC2.
KCC2 oligomerization may be prevented by development-dependent splice variants. However, even the highly sensitive SSCP analysis did not detect any RNA variants. This is in line with our observation that monomeric KCC2 is oligomerization competent, because the absence of the alkylating agent iodoacetamide resulted in oligomerization of KCC2 in isolated P1/P2 membrane fractions. These data suggest that oligomerization is the default state that requires a specific suppression mechanism. Another mechanism to regulate KCC2 transport activity might be its development-dependent presence in detergent-resistant membranes (DRMs). However, a biochemical analysis using Triton X-100-prepared DRMs did not reveal a redistribution of KCC2 among plasma membrane microdomains. At both P0–P2 and P30, most KCC2-IR was present in the non-DRM fraction (T. M. Kranz and H. G. Nothwang, unpublished data).
Another possibility to regulate transport activity is the developmentally related (de-)phosphorylation of KCC2. This mechanism has already been associated with changes in KCC2 transport activity (Kelsch et al., 2001; Kahle et al., 2005; Khirug et al., 2005; de Los et al., 2006; Gagnon et al., 2006). In fact, immunoblot analysis in the cochlear nucleus revealed an age-dependent increase in tyrosine phosphorylation of KCC2 (Vale et al., 2005). Finally, it cannot be excluded that auxiliary proteins are involved in the stabilization of monomeric and/or the formation of oligomeric KCC2 forms (Green, 1999).
Role of transport-inactive KCC2
What may be the function of transport-inactive KCC2 at the plasma membrane? Obviously, this situation allows rapid activation of the transporter. However, the developmental shift from depolarizing to hyperpolarizing action of inhibitory neurotransmitters occurs over several days and thus does not require rapid activation. KCC2 may, therefore, add to the growing list of bifunctional proteins, also referred to as moonlighting proteins (Jeffery, 2003; Sriram et al., 2005). Oligomerization, for instance, changes the activity of the 37 kDa subunit of glyceraldehyde-3-phosphate dehydrogenase. As a tetramer, this protein converts glyceraldehyde-3-phosphate to 1,3-diphosphoglycerate, whereas the monomer acts as uracil-DNA glycosylase (Meyer-Siegler et al., 1991). The cystic fibrose gene product, besides being a Cl− channel itself, regulates the activity of other ion channels, such as the outwardly rectifying Cl− channel ORCC (Gabriel et al., 1993; Stutts et al., 1995) and the Na+ channel ENaC (Johnson et al., 1995). Therefore, a role of KCC2 aside from K+–Cl− cotransport activity is feasible. Indeed, such a function of KCC2 has been proposed recently (Chudotvorova et al., 2005).
In summary, our results reveal that oligomers are the dominant structural unit of KCC2 in the plasma membrane of the mature brainstem. In contrast to N(K)CCs, oligomerization of KCC2 involves disulfide bonds. In the nervous system, oligomerization is age dependent, thereby adding a novel facet to the regulation of KCC2 at the protein level. The localization of transport-inactive KCC2 at the plasma membrane in immature neurons suggests a role in addition to transport activity and points to a double life of this protein.
Footnotes
This work was supported by Deutsche Forschungsgemeinschaft Grants GRK 845/1 and 428/2-1. We thank Dr. W. Nastainczyk for generating the rabbit anti-nKCC2 antibody, Dr. D. Bruns for the generous gift of the anti-vesicle-associated membrane protein 2 antibody, Dr. C. Hübner for providing anti-KCC2 antibody, S. Khirug for assistance at the uncaging setup, Drs. J. W. Deitmer and C. Lohr for assistance with confocal microscopy, and Drs. R. Zimmermann, K. Kaila, and C. Rivera for helpful discussions. K. Ociepka is acknowledged for expert technical assistance and X. Fütterer for help with the HEK-293 cell experiments. The monoclonal antibodies T4 from Dr. C. Lytle and α5 from Dr. Fambrough were obtained through the Developmental Studies Hybridoma Bank, developed under the auspices of the National Institute of Child Health and Human Development and maintained by The University of Iowa, Department of Biological Sciences, Iowa City, IA.
References
- Balakrishnan V, Becker M, Löhrke S, Nothwang HG, Güresir E, Friauf E. Expression and function of chloride transporters during development of inhibitory neurotransmission in the auditory brainstem. J Neurosci. 2003;23:4134–4145. doi: 10.1523/JNEUROSCI.23-10-04134.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben Ari Y, Cherubini E, Corradetti R, Gaiarsa JL. Giant synaptic potentials in immature rat CA3 hippocampal neurones. J Physiol (Lond) 1989;416:303–325. doi: 10.1113/jphysiol.1989.sp017762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berryman M, Gary R, Bretscher A. Ezrin oligomers are major cytoskeletal components of placental microvilli: a proposal for their involvement in cortical morphogenesis. J Cell Biol. 1995;131:1231–1242. doi: 10.1083/jcb.131.5.1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho E, Schellhorn SE, Zabolotny JM, Martin S, Tozzo E, Peroni OD, Houseknecht KL, Mundt A, James DE, Kahn BB. GLUT4 overexpression or deficiency in adipocytes of transgenic mice alters the composition of GLUT4 vesicles and the subcellular localization of GLUT4 and insulin-responsive aminopeptidase. J Biol Chem. 2004;279:21598–21605. doi: 10.1074/jbc.M312269200. [DOI] [PubMed] [Google Scholar]
- Casula S, Shmukler BE, Wilhelm S, Stuart-Tilley AK, Su WF, Chernova MN, Brugnara C, Alper SL. A dominant negative mutant of the KCC1K-Cl cotransporter: both N- and C-terminal cytoplasmic domains are required for K-Cl cotransport activity. J Biol Chem. 2001;276:41870–41878. doi: 10.1074/jbc.M107155200. [DOI] [PubMed] [Google Scholar]
- Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- Chudotvorova I, Ivanov A, Rama S, Hubner CA, Pellegrino C, Ben Ari Y, Medina I. Early expression of KCC2 in rat hippocampal cultures augments expression of functional GABA synapses. J Physiol (Lond) 2005;566:671–679. doi: 10.1113/jphysiol.2005.089821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehnes Y, Chaudhry FA, Ullensvang K, Lehre KP, Storm-Mathisen J, Danbolt NC. The glutamate transporter EAAT4 in rat cerebellar Purkinje cells: a glutamate-gated chloride channel concentrated near the synapse in parts of the dendritic membrane facing astroglia. J Neurosci. 1998;18:3606–3619. doi: 10.1523/JNEUROSCI.18-10-03606.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jong JC, Van Der Vliet WA, van den Heuvel LP, Willems PH, Knoers NV, Bindels RJ. Functional expression of mutations in the human NaCl cotransporter: evidence for impaired routing mechanisms in Gitelman's syndrome. J Am Soc Nephrol. 2002;13:1442–1448. doi: 10.1097/01.asn.0000017904.77985.03. [DOI] [PubMed] [Google Scholar]
- de Jong JC, Willems PH, Mooren FJ, van den Heuvel LP, Knoers NV, Bindels RJ. The structural unit of the thiazide-sensitive NaCl cotransporter is a homodimer. J Biol Chem. 2003;278:24302–24307. doi: 10.1074/jbc.M303101200. [DOI] [PubMed] [Google Scholar]
- de Los HP, Kahle KT, Rinehart J, Bobadilla NA, Vazquez N, San Cristobal P, Mount DB, Lifton RP, Hebert SC, Gamba G. WNK3 bypasses the tonicity requirement for K-Cl cotransporter activation via a phosphatase-dependent pathway. Proc Natl Acad Sci USA. 2006;103:1976–1981. doi: 10.1073/pnas.0510947103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delpire E, Mount DB. Human and murine phenotypes associated with defects in cation-chloride cotransport. Annu Rev Physiol. 2002;64:803–843. doi: 10.1146/annurev.physiol.64.081501.155847. [DOI] [PubMed] [Google Scholar]
- Eccles JC. The ionic mechanisms of excitatory and inhibitory synaptic action. Ann NY Acad Sci. 1966;137:473–494. doi: 10.1111/j.1749-6632.1966.tb50176.x. [DOI] [PubMed] [Google Scholar]
- Ehrlich I, Löhrke S, Friauf E. Shift from depolarizing to hyperpolarizing glycine action in rat auditory neurons is due to age-dependent Cl− regulation. J Physiol (Lond) 1999;520:121–137. doi: 10.1111/j.1469-7793.1999.00121.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eilers J, Plant TD, Marandi N, Konnerth A. GABA-mediated Ca2+ signalling in developing rat cerebellar Purkinje neurones. J Physiol (Lond) 2001;536:429–437. doi: 10.1111/j.1469-7793.2001.0429c.xd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiumelli H, Cancedda L, Poo MM. Modulation of GABAergic transmission by activity via postsynaptic Ca2+-dependent regulation of KCC2 function. Neuron. 2005;48:773–786. doi: 10.1016/j.neuron.2005.10.025. [DOI] [PubMed] [Google Scholar]
- Gabriel SE, Clarke LL, Boucher RC, Stutts MJ. CFTR and outward rectifying chloride channels are distinct proteins with a regulatory relationship. Nature. 1993;363:263–268. doi: 10.1038/363263a0. [DOI] [PubMed] [Google Scholar]
- Gagnon KB, England R, Delpire E. Volume sensitivity of cation-Cl− cotransporters is modulated by the interaction of two kinases: Ste20-related proline-alanine-rich kinase and WNK4. Am J Physiol Cell Physiol. 2006;290:C134–C142. doi: 10.1152/ajpcell.00037.2005. [DOI] [PubMed] [Google Scholar]
- Gerelsaikhan T, Turner RJ. Transmembrane topology of the secretory Na+-K+-2Cl− cotransporter NKCC1 studied by in vitro translation. J Biol Chem. 2000;275:40471–40477. doi: 10.1074/jbc.M007751200. [DOI] [PubMed] [Google Scholar]
- Green WN. Ion channel assembly: creating structures that function. J Gen Physiol. 1999;113:163–170. doi: 10.1085/jgp.113.2.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillemin I, Becker M, Ociepka K, Friauf E, Nothwang HG. A subcellular prefractionation protocol for minute amounts of mammalian cell cultures and tissue. Proteomics. 2005;5:35–45. doi: 10.1002/pmic.200400892. [DOI] [PubMed] [Google Scholar]
- Hong M, Xu W, Yoshida T, Tanaka K, Wolff DJ, Zhou F, Inouye M, You G. Human organic anion transporter hOAT1 forms homooligomers. J Biol Chem. 2005;280:32285–32290. doi: 10.1074/jbc.M501447200. [DOI] [PubMed] [Google Scholar]
- Howard HC, Mount DB, Rochefort D, Byun N, Dupre N, Lu JM, Fan XM, Song LY, Riviere JB, Prevost C, Horst J, Simonati A, Lemcke B, Welch R, England R, Zhan FQ, Mercado A, Siesser WB, George AL, McDonald MP, Bouchard JP, Mathieu J, Delpire E, Rouleau GA. The K-Cl cotransporter KCC3 is mutant in a severe peripheral neuropathy associated with agenesis of the corpus callosum. Nat Genet. 2002;32:384–392. doi: 10.1038/ng1002. [DOI] [PubMed] [Google Scholar]
- Hübner CA, Stein V, Hermans-Borgmeyer I, Meyer T, Ballanyi K, Jentsch TJ. Disruption of KCC2 reveals an essential role of K-Cl cotransport already in early synaptic inhibition. Neuron. 2001;30:515–524. doi: 10.1016/s0896-6273(01)00297-5. [DOI] [PubMed] [Google Scholar]
- Ikeda K, Onimaru H, Yamada J, Inoue K, Ueno S, Onaka T, Toyoda H, Arata A, Ishikawa TO, Taketo MM, Fukuda A, Kawakami K. Malfunction of respiratory-related neuronal activity in Na+, K+-ATPase α2 subunit-deficient mice is attributable to abnormal Cl− homeostasis in brainstem neurons. J Neurosci. 2004;24:10693–10701. doi: 10.1523/JNEUROSCI.2909-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahn R, Sudhof TC. Membrane fusion and exocytosis. Annu Rev Biochem. 1999;68:863–911. doi: 10.1146/annurev.biochem.68.1.863. [DOI] [PubMed] [Google Scholar]
- Jarolimek W, Lewen A, Misgeld U. A furosemide-sensitive K+-Cl− cotransporter counteracts intracellular Cl− accumulation and depletion in cultured rat midbrain neurons. J Neurosci. 1999;19:4695–4704. doi: 10.1523/JNEUROSCI.19-12-04695.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffery CJ. Moonlighting proteins: old proteins learning new tricks. Trends Genet. 2003;19:415–417. doi: 10.1016/S0168-9525(03)00167-7. [DOI] [PubMed] [Google Scholar]
- Johnson LG, Boyles SE, Wilson J, Boucher RC. Normalization of raised sodium absorption and raised calcium-mediated chloride secretion by adenovirus-mediated expression of cystic fibrosis transmembrane conductance regulator in primary human cystic fibrosis airway epithelial cells. J Clin Invest. 1995;95:1377–1382. doi: 10.1172/JCI117789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahle KT, Rinehart J, de Los HP, Louvi A, Meade P, Vazquez N, Hebert SC, Gamba G, Gimenez I, Lifton RP. WNK3 modulates transport of Cl− in and out of cells: implications for control of cell volume and neuronal excitability. Proc Natl Acad Sci USA. 2005;102:16783–16788. doi: 10.1073/pnas.0508307102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaila K. Ionic basis of GABAA receptor channel function in the nervous system. Prog Neurobiol. 1994;42:489–537. doi: 10.1016/0301-0082(94)90049-3. [DOI] [PubMed] [Google Scholar]
- Kakazu Y, Akaike N, Komiyama S, Nabekura J. Regulation of intracellular chloride by cotransporters in developing lateral superior olive neurons. J Neurosci. 1999;19:2843–2851. doi: 10.1523/JNEUROSCI.19-08-02843.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman RJ, Davies MV, Pathak VK, Hershey JW. The phosphorylation state of eucaryotic initiation factor 2 alters translational efficiency of specific mRNAs. Mol Cell Biol. 1989;9:946–958. doi: 10.1128/mcb.9.3.946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelsch W, Hormuzdi S, Straube E, Lewen A, Monyer H, Misgeld U. Insulin-like growth factor 1 and a cytosolic tyrosine kinase activate chloride outward transport during maturation of hippocampal neurons. J Neurosci. 2001;21:8339–8347. doi: 10.1523/JNEUROSCI.21-21-08339.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khiroug L, Giniatullin R, Klein RC, Fayuk D, Yakel JL. Functional mapping and Ca2+ regulation of nicotinic acetylcholine receptor channels in rat hippocampal CA1 neurons. J Neurosci. 2003;23:9024–9031. doi: 10.1523/JNEUROSCI.23-27-09024.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khirug S, Huttu K, Ludwig A, Smirnov S, Voipio J, Rivera C, Kaila K, Khiroug L. Distinct properties of functional KCC2 expression in immature mouse hippocampal neurons in culture and in acute slices. Eur J Neurosci. 2005;21:899–905. doi: 10.1111/j.1460-9568.2005.03886.x. [DOI] [PubMed] [Google Scholar]
- Kotak VC, Korada S, Schwartz IR, Sanes DH. A developmental shift from GABAergic to glycinergic transmission in the central auditory system. J Neurosci. 1998;18:4646–4655. doi: 10.1523/JNEUROSCI.18-12-04646.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebovitz RM, Takeyasu K, Fambrough DM. Molecular characterization and expression of the (Na+ + K+)-ATPase alpha-subunit in Drosophila melanogaster. EMBO J. 1989;8:193–202. doi: 10.1002/j.1460-2075.1989.tb03364.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Chen CX, Liu YJ, Aizenman E, Kandler K. KCC2 expression in immature rat cortical neurons is sufficient to switch the polarity of GABA responses. Eur J Neurosci. 2005;21:2593–2599. doi: 10.1111/j.1460-9568.2005.04084.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Tornberg J, Kaila K, Airaksinen MS, Rivera C. Patterns of cation-chloride cotransporter expression during embryonic rodent CNS development. Eur J Neurosci. 2002;16:2358–2370. doi: 10.1046/j.1460-9568.2002.02419.x. [DOI] [PubMed] [Google Scholar]
- Löhrke S, Srinivasan G, Oberhofer M, Doncheva E, Friauf E. Shift from depolarizing to hyperpolarizing glycine action occurs at different perinatal ages in superior olivary complex nuclei. Eur J Neurosci. 2005;22:2708–2722. doi: 10.1111/j.1460-9568.2005.04465.x. [DOI] [PubMed] [Google Scholar]
- Lu J, Karadsheh M, Delpire E. Developmental regulation of the neuronal-specific isoform of K-Cl cotransporter KCC2 in postnatal rat brains. J Neurobiol. 1999;39:558–568. [PubMed] [Google Scholar]
- Lytle C, Xu JC, Biemesderfer D, Forbush B., III Distribution and diversity of Na-K-Cl cotransport proteins: a study with monoclonal antibodies. Am J Physiol. 1995;269:C1496–C1505. doi: 10.1152/ajpcell.1995.269.6.C1496. [DOI] [PubMed] [Google Scholar]
- Maley F, Trimble RB, Tarentino AL, Plummer TH., Jr Characterization of glycoproteins and their associated oligosaccharides through the use of endoglycosidases. Anal Biochem. 1989;180:195–204. doi: 10.1016/0003-2697(89)90115-2. [DOI] [PubMed] [Google Scholar]
- Meyer-Siegler K, Mauro DJ, Seal G, Wurzer J, Deriel JK, Sirover MA. A human nuclear uracil DNA glycosylase is the 37-kDa subunit of glyceraldehyde-3-phosphate dehydrogenase. Proc Natl Acad Sci USA. 1991;88:8460–8464. doi: 10.1073/pnas.88.19.8460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore-Hoon ML, Turner RJ. The structural unit of the secretory Na+-K+-2Cl− cotransporter (NKCC1) is a homodimer. Biochemistry. 2000;39:3718–3724. doi: 10.1021/bi992301v. [DOI] [PubMed] [Google Scholar]
- Nabekura J, Katsurabayashi S, Kakazu Y, Shibata S, Matsubara A, Jinno S, Mizoguchi Y, Sasaki A, Ishibashi H. Developmental switch from GABA to glycine release in single central synaptic terminals. Nat Neurosci. 2004;7:17–23. doi: 10.1038/nn1170. [DOI] [PubMed] [Google Scholar]
- Nakamura N, Rabouille C, Watson R, Nilsson T, Hui N, Slusarewicz P, Kreis TE, Warren G. Characterization of a cis-Golgi matrix protein, GM130. J Cell Biol. 1995;131:1715–1726. doi: 10.1083/jcb.131.6.1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemes Z, Petrovski G, Fesus L. Tools for the detection and quantitation of protein transglutamination. Anal Biochem. 2005;342:1–10. doi: 10.1016/j.ab.2004.10.015. [DOI] [PubMed] [Google Scholar]
- Obrietan K, van den Pol AN. GABA neurotransmission in the hypothalamus: developmental reversal from Ca2+ elevating to depressing. J Neurosci. 1995;15:5065–5077. doi: 10.1523/JNEUROSCI.15-07-05065.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orita M, Suzuki Y, Sekiya T, Hayashi K. Rapid and sensitive detection of point mutations and DNA polymorphisms using the polymerase chain reaction. Genomics. 1989;5:874–879. doi: 10.1016/0888-7543(89)90129-8. [DOI] [PubMed] [Google Scholar]
- Owens DF, Boyce LH, Davis MB, Kriegstein AR. Excitatory GABA responses in embryonic and neonatal cortical slices demonstrated by gramicidin perforated-patch recordings and calcium imaging. J Neurosci. 1996;16:6414–6423. doi: 10.1523/JNEUROSCI.16-20-06414.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne JA. Functional characterization of the neuronal-specific K-Cl cotransporter: implications for [K+]o regulation. Am J Physiol. 1997;273:C1516–C1525. doi: 10.1152/ajpcell.1997.273.5.C1516. [DOI] [PubMed] [Google Scholar]
- Payne JA, Stevenson TJ, Donaldson LF. Molecular characterization of a putative K-Cl cotransporter in rat brain. A neuronal-specific isoform. J Biol Chem. 1996;271:16245–16252. doi: 10.1074/jbc.271.27.16245. [DOI] [PubMed] [Google Scholar]
- Payne JA, Rivera C, Voipio J, Kaila K. Cation-chloride co-transporters in neuronal communication, development and trauma. Trends Neurosci. 2003;26:199–206. doi: 10.1016/S0166-2236(03)00068-7. [DOI] [PubMed] [Google Scholar]
- Rietzel H-J, Friauf E. Neuron types in the rat lateral superior olive and developmental changes in the complexity of their dendritic arbors. J Comp Neurol. 1998;390:20–40. [PubMed] [Google Scholar]
- Rivera C, Voipio J, Payne JA, Ruusuvuori E, Lahtinen H, Lamsa K, Pirvola U, Saarma M, Kaila K. The K+/Cl− co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature. 1999;397:251–255. doi: 10.1038/16697. [DOI] [PubMed] [Google Scholar]
- Russell JM. Sodium-potassium-chloride cotransport. Physiol Rev. 2000;80:211–276. doi: 10.1152/physrev.2000.80.1.211. [DOI] [PubMed] [Google Scholar]
- Shibata S, Kakazu Y, Okabe A, Fukuda A, Nabekura J. Experience-dependent changes in intracellular Cl− regulation in developing auditory neurons. Neurosci Res. 2004;48:211–220. doi: 10.1016/j.neures.2003.10.011. [DOI] [PubMed] [Google Scholar]
- Sriram G, Martinez JA, McCabe ER, Liao JC, Dipple KM. Single-gene disorders: what role could moonlighting enzymes play? Am J Hum Genet. 2005;76:911–924. doi: 10.1086/430799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starremans PG, Kersten FF, van den Heuvel LP, Knoers NV, Bindels RJ. Dimeric architecture of the human bumetanide-sensitive Na-K-Cl co-transporter. J Am Soc Nephrol. 2003;14:3039–3046. doi: 10.1097/01.asn.0000097370.29737.5b. [DOI] [PubMed] [Google Scholar]
- Stein V, Hermans-Borgmeyer I, Jentsch TJ, Hubner CA. Expression of the KCl cotransporter KCC2 parallels neuronal maturation and the emergence of low intracellular chloride. J Comp Neurol. 2004;468:57–64. doi: 10.1002/cne.10983. [DOI] [PubMed] [Google Scholar]
- Stutts MJ, Canessa CM, Olsen JC, Hamrick M, Cohn JA, Rossier BC, Boucher RC. CFTR as a cAMP-dependent regulator of sodium channels. Science. 1995;269:847–850. doi: 10.1126/science.7543698. [DOI] [PubMed] [Google Scholar]
- Sung KW, Kirby M, McDonald MP, Lovinger DM, Delpire E. Abnormal GABAA receptor-mediated currents in dorsal root ganglion neurons isolated from Na–K–2Cl cotransporter null mice. J Neurosci. 2000;20:7531–7538. doi: 10.1523/JNEUROSCI.20-20-07531.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vale C, Schoorlemmer J, Sanes DH. Deafness disrupts chloride transporter function and inhibitory synaptic transmission. J Neurosci. 2003;23:7516–7524. doi: 10.1523/JNEUROSCI.23-20-07516.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vale C, Caminos E, Martinez-Galan JR, Juiz JM. Expression and developmental regulation of the K+-Cl− cotransporter KCC2 in the cochlear nucleus. Hear Res. 2005;206:107–115. doi: 10.1016/j.heares.2005.03.012. [DOI] [PubMed] [Google Scholar]
- Vaux D, Tooze J, Fuller S. Identification by anti-idiotype antibodies of an intracellular membrane protein that recognizes a mammalian endoplasmic reticulum retention signal. Nature. 1990;345:495–502. doi: 10.1038/345495a0. [DOI] [PubMed] [Google Scholar]
- Wang J, Reichling DB, Kyrozis A, Macdermott AB. Developmental loss of GABA- and glycine-induced depolarization and Ca2+ transients in embryonic rat dorsal horn neurons in culture. Eur J Neurosci. 1994;6:1275–1280. doi: 10.1111/j.1460-9568.1994.tb00317.x. [DOI] [PubMed] [Google Scholar]
- Wang YF, Gao XB, van den Pol AN. Membrane properties underlying patterns of GABA-dependent action potentials in developing mouse hypothalamic neurons. J Neurophysiol. 2001;86:1252–1265. doi: 10.1152/jn.2001.86.3.1252. [DOI] [PubMed] [Google Scholar]
- Williams JR, Sharp JW, Kumari VG, Wilson M, Payne JA. The neuron-specific K-Cl cotransporter, KCC2. Antibody development and initial characterization of the protein. J Biol Chem. 1999;274:12656–12664. doi: 10.1074/jbc.274.18.12656. [DOI] [PubMed] [Google Scholar]
- Wojcik SM, Katsurabayashi S, Guillemin I, Friauf E, Rosenmund C, Brose N, Rhee JS. A shared vesicular carrier allows synaptic corelease of GABA and glycine. Neuron. 2006;50:575–587. doi: 10.1016/j.neuron.2006.04.016. [DOI] [PubMed] [Google Scholar]
- Woo NS, Lu JM, England R, McClellan R, Dufour S, Mount DB, Deutch AY, Lovinger DM, Delpire E. Hyperexcitability and epilepsy associated with disruption of the mouse neuronal-specific K-Cl cotransporter gene. Hippocampus. 2002;12:258–268. doi: 10.1002/hipo.10014. [DOI] [PubMed] [Google Scholar]
- Woodin MA, Ganguly K, Poo MM. Coincident pre- and postsynaptic activity modifies GABAergic synapses by postsynaptic changes in Cl− transporter activity. Neuron. 2003;39:807–820. doi: 10.1016/s0896-6273(03)00507-5. [DOI] [PubMed] [Google Scholar]
- Yamada J, Okabe A, Toyoda H, Kilb W, Luhmann HJ, Fukuda A. Cl− uptake promoting depolarizing GABA actions in immature rat neocortical neurones is mediated by NKCC1. J Physiol (Lond) 2004;557:829–841. doi: 10.1113/jphysiol.2004.062471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye JH. Physiology and pharmacology of native glycine receptors in developing rat ventral tegmental area neurons. Brain Res. 2000;862:74–82. doi: 10.1016/s0006-8993(00)02073-4. [DOI] [PubMed] [Google Scholar]
- Zhang L, Spigelman I, Carlen PL. Development of GABA-mediated, chloride-dependent inhibition of CA1 pyramidal neurones of immature rat hippocampal slices. J Physiol (Lond) 1991;444:25–49. doi: 10.1113/jphysiol.1991.sp018864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L, Lovinger D, Delpire E. Cortical neurons lacking KCC2 expression show impaired regulation of intracellular chloride. J Neurophysiol. 2005;93:1557–1568. doi: 10.1152/jn.00616.2004. [DOI] [PubMed] [Google Scholar]