

Abstract
The expression of the neuron-specific K+/Cl− cotransporter (KCC2) is restricted to the CNS and is strongly upregulated during neuronal maturation, yielding a low intracellular chloride concentration that is required for fast synaptic inhibition in adult neurons. To elucidate the mechanisms of KCC2 gene regulation, we analyzed the KCC2 (alias Slc12a5) promoter and proximal intron-1 regions and revealed 10 candidate transcription factor binding sites that are highly conserved in mammalian KCC2 genes. Here we focus on one of these factors, early growth response 4 (Egr4), which shows a similar developmental upregulation in CNS neurons as KCC2. KCC2 luciferase reporter constructs containing the Egr4 site (Egr4KCC2) were strongly induced by Egr4 overexpression in neuro-2a neuroblastoma cells and in cultured neurons. Egr4-mediated induction was decreased significantly by point-mutating the Egr4KCC2. Insertion of Egr4KCC2 into the KCC2 basal promoter in the endogenous reverse, but not in the opposite, orientation reestablished Egr4-mediated induction. Electrophoretic mobility shift assay confirmed specific Egr4 binding to Egr4KCC2. Interference RNA-mediated knock-down of Egr4 and a dominant-negative isoform of Egr4 significantly inhibited KCC2 reporter induction and endogenous KCC2 expression in cultured neurons. Together, the results indicate an important role for Egr4 in the developmental upregulation of KCC2 gene expression.
Keywords: chloride cotransporter, GABA, Egr4, promoter regulation, activity-dependent gene expression, electrophoretic mobility shift assay
Introduction
The neuron-specific K+/Cl− cotransporter (KCC2), a member of the cation chloride cotransporter family (for review, see Payne et al., 2003; Mercado et al., 2004), is necessary for fast synaptic inhibition, i.e., for maintaining a low intracellular chloride concentration that is required for the hyperpolarizing actions of the inhibitory neurotransmitters GABA and glycine (Rivera et al., 1999; Hubner et al., 2001). KCC2 mRNA is expressed exclusively in mature CNS neurons (Payne et al., 1996; Rivera et al., 1999; Kanaka et al., 2001). During embryonic development KCC2 mRNA expression follows neuronal maturation, first becoming detectable in the postmitotic neurons of the brainstem and spinal cord and then gradually increasing in higher brain structures (Li et al., 2002; Stein et al., 2004). In mature animals the KCC2 mRNA level is downregulated after axotomy (Nabekura et al., 2002), sciatic nerve lesion (Coull et al., 2003), and kindling seizures (Rivera et al., 2002), leading to depolarizing GABAergic responses. KCC2 protein levels quickly follow the mRNA changes because of unusually rapid protein turnover (Rivera et al., 2004), consistent with the proposed role of KCC2 regulation in the plasticity of inhibitory synapses under physiological or pathological conditions (Fiumelli et al., 2005). Thus characterization of the molecular mechanisms that regulate KCC2 gene expression may contribute to the understanding of neuropsychiatric disorders, including epilepsy (Cohen et al., 2002) and neuropathic pain (Coull et al., 2003).
The early growth response (Egr) family of immediate early genes encodes four zinc finger transcription factors (TFs): Egr1 [alias nerve growth factor-induced-A (NGFI-A), Krox24, zinc finger-interacting 268 (zif268), Tis8, and zif268/Egr1/NGFI-A/Krox24 (ZENK)], Egr2 (alias Krox20), Egr3 (alias Pilot), and Egr4 (alias NGFI-C) (for review, see O'Donovan et al., 1999). They all contain three zinc fingers and bind a guanine-cytosine-rich (GC-rich) Egr-responsive element located in the promoter region of target genes (Swirnoff and Milbrandt, 1995). The optimal sequence for binding Egr proteins is at least 10 nucleotides and varies somewhat among the members (Swirnoff and Milbrandt, 1995). Egr proteins are induced by neuronal activity and have an overlapping expression pattern in adult brain (for review, see Beckmann and Wilce, 1997). Of the four Egr proteins, Egr4 has the most neural-specific pattern of expression, and in rats its expression increases gradually during the first 3 postnatal weeks of development (Crosby et al., 1992). However, in contrast to other Egr proteins like Egr1, which is known to be involved in many processes of neuronal plasticity (Knapska and Kaczmarek, 2004; Li et al., 2005) and in the regulation of several neural-specific genes (James et al., 2005), the role of Egr4 in the regulation of neuronal genes remains elusive (Tourtellotte et al., 1999, 2000).
Our previous study (Uvarov et al., 2005) demonstrated that a 1.4 kb promoter region is mostly sufficient to mediate neuron-specific expression of KCC2 in transgenic mice. Here we have characterized the 1.4 kb mouse KCC2 promoter and identified multiple TF binding sites that potentially regulate KCC2 promoter activity. We found that Egr4 binds to one of these sites and strongly upregulates the activity of the mouse KCC2 gene in neuronal cells.
Materials and Methods
Sequence analysis.
Mouse, rat, human, and chimpanzee KCC2 (alias Slc12a5) genomic sequences [obtained from the National Center for Biotechnology Information (Bethesda, MD) genome database], corresponding to a 1.4 kb sequence upstream of the transcription start site as well as a proximal part of intron-1, were aligned by the ClustalW program (Chenna et al., 2003). Putative TF binding sites within this region were identified by using MatInspector software (Genomatix Software, Munich, Germany). TF sites that are highly conserved in all four species were considered likely true-binding sites and are reported here (see Fig. 1).
Figure 1.
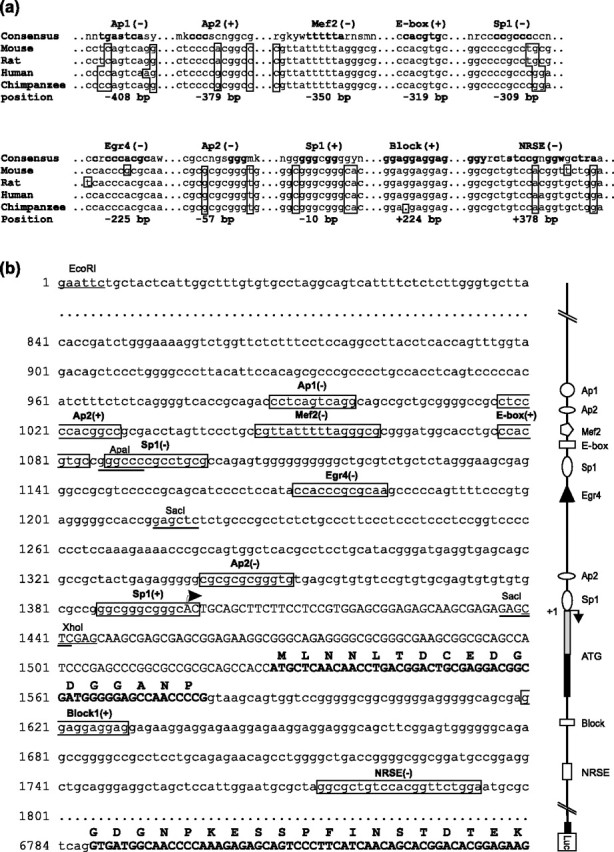
Analysis of conserved TF binding sites in the mouse KCC2 regulatory region. a, Shown are 10 TF binding sites that are highly conserved in the mouse, rat, human, and chimpanzee KCC2 regulatory region: two each of the Sp1 and AP2 sites and one site each of AP1, Mef2, E-box, Egr4, NRSE, and Block. The positions of the consensus sequences are indicated relative to the previously characterized transcription start site (+1). Nucleotides that differ from the consensus sequences are shown in boxes. Highly conserved nucleotides according to the matrix of TF binding sites appear in bold. Abbreviations for nucleotides in consensus sequences include n, for any nucleotide; s, for g/c; w, for a/t; y, for c/t; r, for a/g; m, for a/c; k, for g/t. b, Single-stranded (forward) DNA sequence of the mouse KCC2 promoter region, exon-1, part of intron-1, and exon-2 fused to the firefly luciferase cDNA. A corresponding schematic drawing of the promoter is depicted on the right. The previously identified consensus TF binding sites, with their locations on either the sense (+) or the antisense (−) DNA strand, are boxed. The position for the previously identified transcription start site is indicated with an arrow. KCC2 exon-1 and exon-2 sequences are shown in capital letters; coding regions also are in bold, and the translated protein sequence is presented above the corresponding nucleotide strand.
Western blotting.
Animal experiments were approved by the local ethics committee for animal research at the University of Helsinki. Animals were killed by CO2 and cervical dislocation. Mouse hippocampal and cerebellar samples (taken on the 2nd, 15th, and 30th d of postnatal development) were homogenized in a lysis buffer (50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 1% Triton X-100, 0.5% deoxycholic acid, 0.1% SDS). Protein concentrations were determined by using a DC Protein Assay kit (Bio-Rad, Hercules, CA). Protein samples (20 μg) were separated by using 7.5% SDS-PAGE gels and transferred onto a Hybond-ECL nitrocellulose membrane (Amersham Biosciences, Buckinghamshire, UK). Blots were probed with antibodies against KCC2 (Ludwig et al., 2003), neuronal class III β-tubulin (BabCO, Berkeley, CA), and Egr4 (Zipfel et al., 1997), were developed with Amersham Biosciences ECL Plus, and were visualized with the luminescent image analyzer LAS-3000 (Fuji Photo Film, Tokyo, Japan). Optical densities of the bands were analyzed with Advanced Image Data Analysis (AIDA) imaging software (Raytest, Straubenhardt, Germany).
KCC2 luciferase reporters and Egr expression constructs.
KCC2(−1398/+42), a luciferase reporter that includes 1398 bp of an upstream regulatory region as well as 42 bp of the 5′-untranslated region (5′-UTR) of the mouse KCC2 gene cloned into the pGL3-Basic vector, has been described by Uvarov et al. (2005) (see Fig. 3a). KCC2(−1398/+5445), another luciferase reporter described by Uvarov et al. (2005), contains 6843 bp of a KCC2 genomic sequence comprising 1440 bp of the upstream regulatory region, exon-1, intron-1, and part of exon-2 (see Figs. 1b, 3a). The KCC2(−309/+42) construct (see Fig. 3a) was obtained by cutting KCC2(−1398/+42) with NheI (in polylinker) and ApaI (in KCC2 promoter), filling in the protruding ends with Klenow polymerase, and self-circularizing with T4 DNA ligase (Fermentas, Vilnius, Lithuania). The KCC2(−180/+42) construct (see Fig. 3a) was produced by inserting a (−180/+42) SacI fragment of KCC2 promoter into pGL3-Basic. The KCC2(−309/+42)Egr4mut construct was prepared by replacing two critical dG nucleotides with two dTs in the Egr4KCC2 sequence of KCC2(−309/+42) (see Fig. 5a,b). Site-directed mutagenesis was accomplished by PCR with the use of the following primers (Egr4 consensus is underlined; mutated nucleotides are shown in a small case letters): Egr4mut-sense 5′-CATCCCCTCCATACCAaaCGCGCAAGCCCCCAG-3′ and Egr4mut-antisense 5′-CTGGGGGCTTGCGCGttTGGTATGGAGGGGATG-3′. KCC2(−180/+42)2xEgr4(−) and KCC2(−180/+42)2xEgr4(+) luciferase reporter constructs carry two copies of the Egr4KCC2 binding site upstream of the (−180/+42) basal promoter fragment in endogenous [(−), reverse] or opposite [(+), forward] orientation, respectively (see Fig. 5c). They were produced by annealing two oligonucleotides (KpnI-compatible ends are in bold; Egr4KCC2 sites are underlined): 5′-TTGCGCGGGTGGATTGCGCGGGTGGGTAC-3′ and 5′-CCACCCGCGCAATCCACCCGCGCAAGTAC-3′. Then the annealed, double-stranded oligonucleotide was inserted into a KpnI site in the KCC2(−180/+42) polylinker that resulted in the two constructs, depending on the orientation of the incorporated Egr4KCC2 sites.
Figure 3.
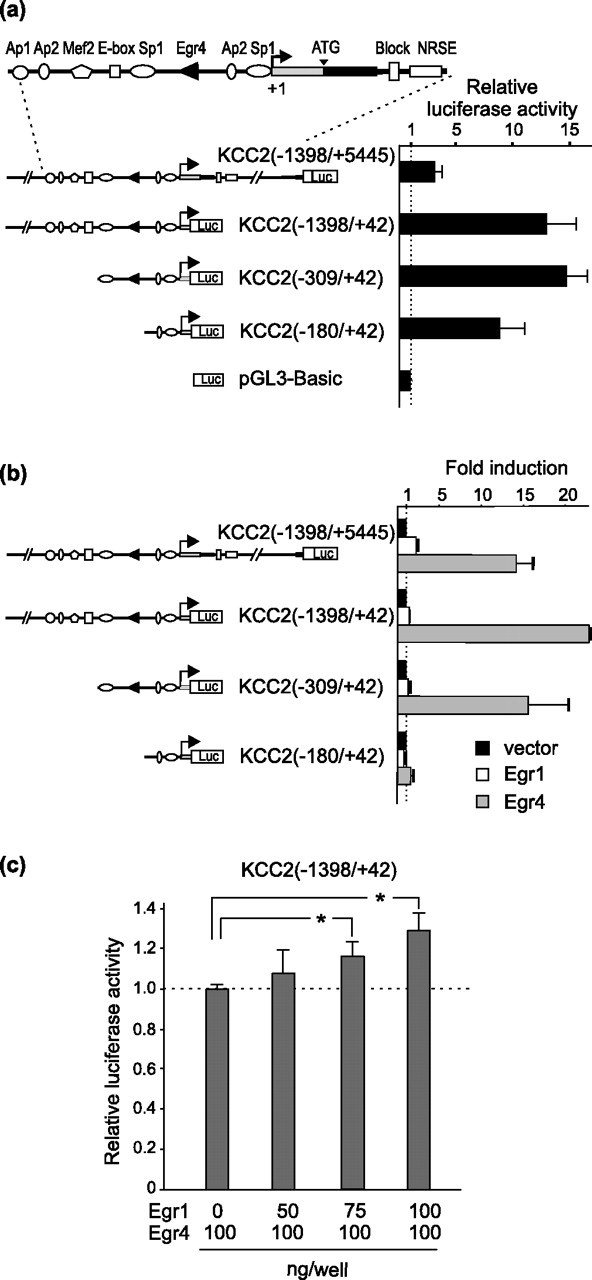
Egr4, but not Egr1, strongly induces the activity of KCC2 reporter constructs containing the Egr4KCC2 site. a, Basal activity of KCC2 promoter constructs in N2a cells. Activity of the KCC2(−1398/+42) construct, containing a 1.4 kb KCC2 promoter sequence upstream of the transcription start site, is ∼12 times stronger than the activity of the promoterless pGL3-Basic vector. Activities of KCC2(−1398/+42) and KCC2(−309/+42) constructs are similar, implying that the proximal promoter region is mostly sufficient to provide a basal level of KCC2 promoter activity. Note the significantly lower activity of the KCC2(−1398/+5445) construct, which may be explained by the presence of unknown repressor elements in the proximal part of intron-1 (Uvarov et al., 2005). b, Egr4 overexpressed in N2a cells strongly (from 15- to 22-fold) induces luciferase activity of previously described KCC2 reporter constructs (gray bars). Egr4 does not induce activity of the KCC2(−180/+42) construct lacking the Egr4KCC2 site. Egr1 expression (open bars) moderately upregulates activity of the KCC2(−1398/+5445) construct but has no significant effect on the activity of other KCC2 reporters. To take into account different basal levels of activity for KCC2 reporter constructs, we present the results as fold inductions of Egr4 or Egr1 overexpression in N2a cells in comparison with empty vector transfection. c, Simultaneous coexpression of Egr1 and Egr4 does not prevent induction of the KCC2(−1398/+42) construct. However, activity of the KCC2(−1398/+42) construct is upregulated slightly by increasing amounts of Egr1 plasmid transfected into N2a cells (*p < 0.05; Student's t test; n = 3). Error bars indicate SEM.
Figure 5.
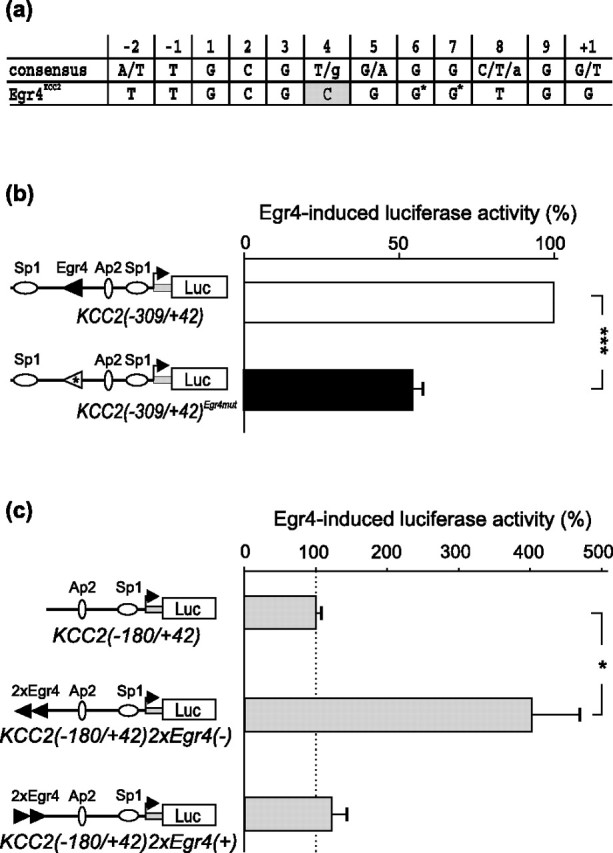
Mutagenesis of the Egr4KCC2 element decreases Egr4-mediated induction of the KCC2(−309/+42) reporter construct in N2a cells. a, The C nucleotide at position 4 in Egr4KCC2 that deviates from the Egr consensus sequence is shown in gray. Two G nucleotides modified by site-directed mutagenesis are indicated with an asterisk. b, Mutagenesis of the two G critical nucleotides in Egr4KCC2 significantly prevents Egr4-induced activation of the KCC2(−309/+42)Egr4mut reporter. c, Insertion of two Egr4KCC2 sites in the endogenous (reverse), but not in the opposite (forward), orientation just upstream of the minimal KCC2(−180/+42) promoter enables the reporter to be activated by Egr4 (*p < 0.05; ***p < 0.001; Student's t test; n = 4). Error bars indicate SEM.
Rat Egr4 and Egr1 expression vectors [under the cytomegalovirus (CMV) promoter] were a kind gift from Dr. J. Milbrandt (Washington University, St. Louis, MO). A dominant-negative Egr4 (DN-Egr4) expression vector was designed to contain the C-terminal part and all three zinc fingers, but not the N-terminal part (see Fig. 6a), based on a similar approach with Egr3 (Levkovitz et al., 2001). The 352–478 aa region of rat Egr4 was amplified by PCR with dominant-negative forward primer (Kozak sequence is underlined), 5′-CAGCTGCAGCCACCATGGGACGCCGGGGCGGCAAGTG-3′, and DN-reverse primer (ClaI site is in bold), 5′-GATATCGATGACCTACGGAGCCAGCTCCGGCTACAG-3′, and ligated into pGEM-T Easy vector (Promega, Madison, WI). After being cut with EcoRI (in polylinker) and ClaI, the fragment was ligated into the corresponding fragment of the Egr4 expression vector backbone (containing the CMV promoter). Because the cloning strategy resulted in a deletion of ∼140 bp of the Egr4 5′-UTR, which may influence the mRNA stability of DN-Egr4 (as compared with Egr4), a relatively high ratio of DN-Egr4 to Egr4 expression plasmids was used in the experiments. The correctness of all cloning procedures was verified by sequencing.
Figure 6.
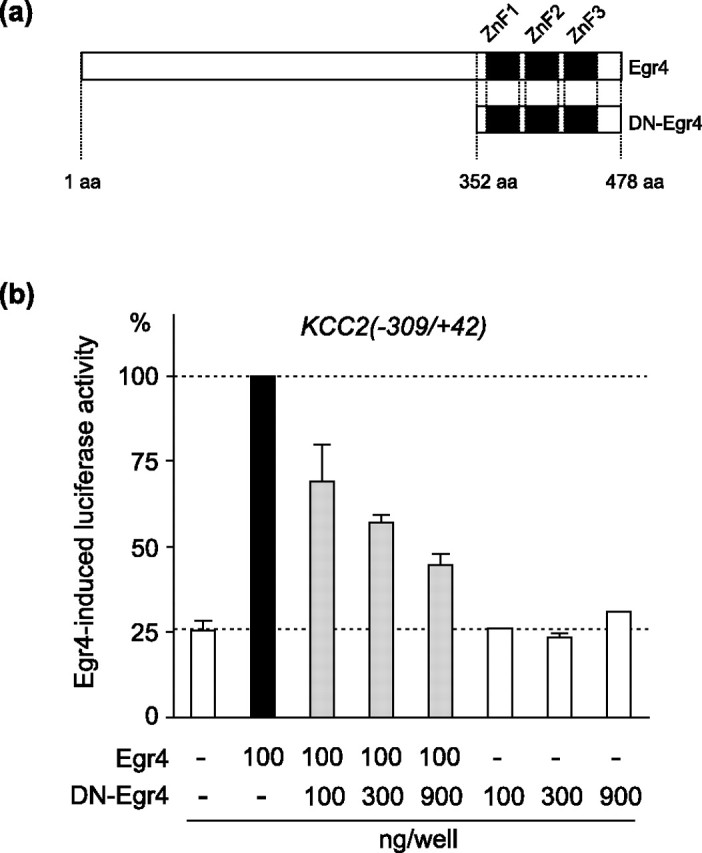
The dominant-negative isoform of Egr4 inhibits Egr4-mediated induction of KCC2(−309/+42) reporter activity in N2a cells. a, Schematic drawing of the Egr4 and DN-Egr4 isoforms. DN-Egr4 retains the DNA binding domain containing three zinc fingers, whereas the N-terminal regulatory region of Egr4 is deleted. b, Increasing the ratio of DN-Egr4 to Egr4 effectively inhibits Egr4-mediated induction of the KCC2(−309/+42) reporter, whereas even the highest amount of DN-Egr4 used has no effect alone on basal reporter activity. Error bars indicate SEM.
Cell culture, transfection, luciferase assay, and immunocytochemistry.
Mouse neuroblastoma neuro-2a (N2a) cells (number CCL-131; American Type Culture Collection, Manassas, VA) (Klebe and Ruddle, 1969), which express a low level of KCC2 and no Egr4 (data not shown), were cultured and transfected; luciferase activity was measured as described previously (Uvarov et al., 2005). Standard dissociated hippocampal neuronal cultures were prepared from embryonic day 17 (E17) mice as described previously (Banker and Goslin, 1998), with small modifications. Briefly, hippocampal cells were dissociated by enzymatic treatment (0.25% trypsin for 15 min at 37°C) and plated on poly-d,l-ornithine-coated coverslips (2000–5000 cells/cm2). The cells were grown in Neurobasal medium containing B27 supplement (Invitrogen, Carlsbad, CA) that had been preincubated on astroglial cultures for 24 h. Astroglial cultures were prepared according to Banker and Goslin (1998) and were maintained in DMEM supplemented with 10% fetal calf serum, 100 U/ml penicillin, and 100 μg/ml streptomycin. After 7 d in vitro (7 DIV) the neurons were cotransfected with the DN-Egr4 (or empty control) vector and a green fluorescent protein (GFP) expression plasmid (to allow for identification of transfected cells), using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol. To avoid cytotoxicity, we used relatively low amounts of the expression plasmids (0.5 μg per 1-cm-diameter well). Then 2 d after transfection the cultures were fixed with 4% formaldehyde in PBS, permeabilized with 0.5% Triton X-100, and blocked in 10% sheep serum plus 0.2% Triton X-100. The cells were incubated in 2% sheep serum containing 1:5000 diluted anti-KCC2 rabbit polyclonal antibody overnight at 4°C (Ludwig et al., 2003) and then with cyanine 3-conjugated donkey anti-rabbit secondary antibody (1:1000) (Jackson ImmunoResearch, West Grove, PA). Processed coverslips were mounted in Gelvatol (Biomedia, Foster City, CA). Optical densities for the neuronal cytoplasmic staining were analyzed with AIDA imaging software (Raytest).
Standard dissociated embryonic cortical cultures (2 × 105 cells/cm2) were prepared from E18 rats by using a procedure similar to the one described for mouse hippocampal cultures (see above). Neurons were plated onto 24-well plates coated with poly-l-lysine (Sigma, St. Louis, MO) and were grown in Neurobasal medium containing B27 supplement. After 3 or 8 DIV the neurons were cotransfected with the luciferase reporter constructs and either DN-Egr4- or Egr4-expressing plasmids (see Fig. 8); luciferase activity was measured 2 d later.
Figure 8.
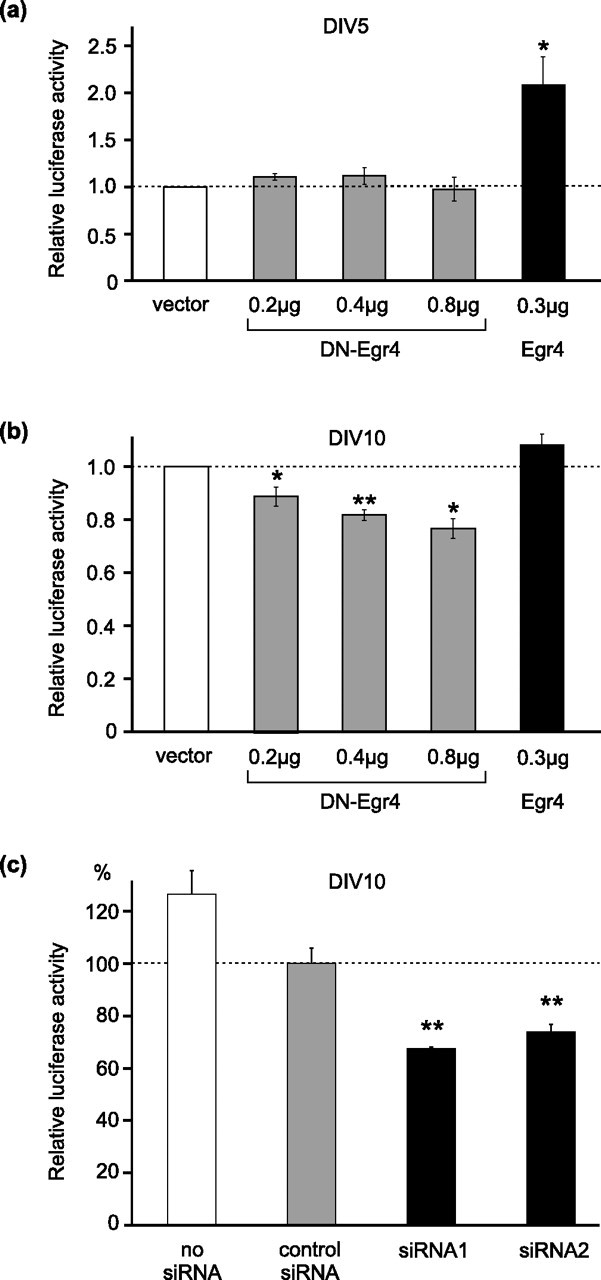
Knock-down of Egr4 inhibits KCC2 promoter activity in mature neurons. a, In 5 DIV neurons exogenously expressed Egr4 increased the activity of the KCC2(−309/+42) luciferase reporter (by twofold), whereas DN-Egr4 failed to affect the reporter activity. b, In 10 DIV neurons overexpressed DN-Egr4 suppressed the KCC2(−309/+42) reporter activity in a dose-dependent manner, whereas overexpression of Egr4 failed to increase the KCC2 promoter activity. c, Synthetic Egr4 siRNA duplexes transfected into 8 DIV neurons significantly suppressed (by 26–33% compared with the negative control) the luciferase activity of the KCC2(−309/+42) reporter in 10 DIV neurons (*p < 0.05; **p < 0.01; Student's t test). Error bars indicate SEM.
Electrophoretic mobility shift assay.
Nuclear extracts of N2a cells, transfected with the Egr4 expression vector, were purified according to a standard protocol (Sambrook and Russell, 2001). Briefly, the cells were washed several times with rinse buffer (40 mm Tris-HCl, pH 7.4, 1 mm EDTA, and 0.15 m NaCl) and scraped into 1 ml of the buffer. The cells were pelleted by centrifuging at 13,000 × g for 2 min. The pellet was resuspended in resuspension buffer [40 mm HEPES-KOH, pH 7.9, 0.4 m KCl, 1 mm DTT, 10% (v/v) glycerol, protease inhibitor mixture (Roche Molecular Biochemicals, Mannheim, Germany), and 0.1 mm PMSF] and subjected to three cycles of freezing/thawing, followed by centrifuging at 13,000 × g for 5 min at 4°C. For electrophoretic mobility shift assay (EMSA) fractions of the nuclear extract (20 μg) were preincubated with 1 μg of poly(dI-dC) in EMSA binding buffer [containing the following (in mm): 7.8 HEPES, pH 7.9, 52 KCl, 3.3 MgCl2, 0.4 EDTA, and 0.4 DTT plus 65 μg/ml BSA and 7.8% (v/v) glycerol] for 30 min at 30°C. Some fractions included unlabeled competitor oligonucleotides (for competition assay) or Egr4 antibodies (Zipfel et al., 1997) (for supershift assay). Then the extracts were incubated with l.5 ng of 32P-labeled oligonucleotide probe for 30 min at 30°C. To produce 32P-labeled Egr4KCC2 probes corresponding to the original Egr4KCC2 or to the mutated Egr4KCC2 site, we annealed the following pairs of PAGE-purified oligonucleotides (Egr-binding sites are underlined; mutated nucleotides are shown in small case letters): Egr4KCC2 sense 5′-CCATACCACCCGCGCAA-3′ and Egr4KCC2 antisense 5′-GGGGCTTGCGCGGGTGGT-3′; Egr4KCC2mut sense 5′-CCATACCAaaCGCGCAA-3′ and Egr4KCC2mut antisense 5′-GGGGCTTGCGCGttTGGT-3′. The 3′ ends of annealed oligonucleotides were filled in with a mixture of unlabeled dATP, deoxythymidine triphosphate (dTTP), and dGTP and labeled [α-32P]deoxycytidine triphosphate (dCTP), using Klenow polymerase (Fermentas). Unlabeled double-stranded oligonucleotides for a competition assay were obtained in the same way but with the use of unlabeled dCTP instead of [α-32P]dCTP. The probes were purified with a QIAquick Nucleotide Removal kit (Qiagen, Hilden, Germany).
Real-time PCR analysis of KCC2 and Egr4 levels.
Quantification of KCC2 mRNA levels in the hippocampal cultures was performed as described previously (Uvarov et al., 2005). The following primers were used for real-time PCR Egr4 quantification: Egr4-forward, 5′-TCTCTCCAAGCCCACCGAAG-3′, and Egr4-reverse, 5′-AACCGCCTGG-ATGAAGAAGC-3′.
RNAi assay.
Two predesigned small-interfering RNA (siRNA) duplexes targeting Egr4 were ordered from Ambion (Austin, TX): siRNA1 (catalog #16708A, ID #200866) 5′-GGCGAUACCUUAGGACUGAtt-3′ (sense) and 5′-UCAGUCCUAAGGUAUCGCCtg-3′ (antisense) and siRNA2 (catalog #16708A, ID #200867) 5′-GCAAGCCUUUGGAUGUGAUtt-5′ (sense) and 5′-AUCACAUCCAAAGGCUUGCtg-3′ (antisense). Silencer Negative Control #1 siRNA (catalog #4611; Ambion) was used as a negative control (see Fig. 8c). Another negative control siRNA (AlexaFluor546-labeled, catalog #1027098; Qiagen) was used to estimate siRNA transfection efficiency into 8 DIV neurons and produced results similar to Silencer Negative Control #1 siRNA (data not shown). The siRNA duplexes (100 nm final concentration) were cotransfected with KCC2(−309/+42) and pRL-TK luciferase reporters into 8 DIV rat cortical neurons with Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol. As a control of unspecific effects, a part of the neurons was cotransfected without siRNA (no siRNA). Luciferase activity was measured after 2 d as described above.
Results
Identification of putative TF binding sites in the KCC2 gene promoter
Our previous study demonstrated that the genomic fragment bearing a 1.4 kb sequence upstream of the transcription start site is mostly sufficient for mediating CNS-specific expression of the KCC2 gene (Uvarov et al., 2005). To identify possible TFs controlling KCC2 gene expression, we analyzed mouse, rat, human, and chimpanzee KCC2 genomic sequences corresponding to the 1.4 kb upstream promoter and proximal intron-1 regions, using a MatInspector software tool, which uses a large library of matrix descriptions for TF binding sites (Cartharius et al., 2005). We identified each consensus of 10 TF binding sites that were highly conserved in all four species (Fig. 1a,b): two sites for specificity protein 1 (Sp1) (Kadonaga et al., 1987) and activating enhancer-binding protein 2 (AP2) (Williams and Tjian, 1991) and one site for AP1 (Lee et al., 1987), myocyte-enhancing factor 2 (Mef2) (Gossett et al., 1989), E-box (Sawadogo, 1988), Egr4 (Swirnoff and Milbrandt, 1995), neuron-restrictive silencer element (NRSE) (Schoenherr et al., 1996), and Block (Collart et al., 1991). Eight of the 10 sites are located within the −415/+9 region, which previously was predicted to be the putative core promoter of the KCC2 gene (Uvarov et al., 2005) and has a GC content above 70%. The region just upstream of the KCC2 transcription start site (−50/−20) contains multiple guanine-thymine (GT) repeats of unknown function, as also found in other neuron-specific genes (Sakimura et al., 1987; Sudhof, 1990; Nedivi et al., 1992; Petersohn et al., 1995). No typical TATA or CAAT boxes are present in the KCC2 promoter region.
Increase in KCC2 and Egr4 expression in the postnatal mouse brain
The predicted Egr4 binding site (Egr4KCC2) in the KCC2 promoter (Fig. 1) and a previous report of rat Egr4 mRNA expression being neural-specific and increasing during postnatal development (Crosby et al., 1992) suggest that Egr4 could be involved in the developmental upregulation of KCC2 gene expression. Consistent with this idea, Western blot analysis showed a synchronous upregulation of KCC2 and Egr4 protein expression in the mouse hippocampus and cerebellum during postnatal development (Fig. 2).
Figure 2.

KCC2 and Egr4 proteins are upregulated synchronously in the hippocampus (Hc) and cerebellum (Cb) during postnatal development. KCC2 and Egr4 protein levels in mouse hippocampus and cerebellum were measured by Western blot (top) at P2, P15, and P30 and are shown relative to β-tubulin (bottom; n = 3 mice for each time point).
Delineation of the KCC2 basal promoter region
To confirm whether the predicted core promoter is indeed sufficient for basal activity in neuronal cells, we produced a series of deletion constructs in which firefly luciferase reporter was placed under the control of different KCC2 gene regulatory regions, and we measured the reporter activity in transiently transfected mouse neuroblastoma N2a cells (Fig. 3a). As shown previously (Uvarov et al., 2005), the activity of the KCC2(−1398/+5445) construct was inhibited in the cells, presumably by the presence of unknown repressor(s) in KCC2 intron-1. The basal activity of KCC2(−309/+42) was as high as that of KCC2(−1398/+42). Moreover, the basal activity of the KCC2(−180/+42) construct was slightly, but not significantly, lower than the activity of the KCC2(−1398/+42) construct (Fig. 3a). These results indicate that, at least in the N2a cells, all KCC2 basal promoter activity lies within the −309/+42 region, and most of the basal activity is defined by the −180/+42 region, which bears only the transcription start site and Sp1 and AP2 consensus sequences.
Egr4 strongly activates the KCC2 promoter in N2a cells
To determine whether Egr4 (or other members of the Egr family) can regulate KCC2 promoter activity via the Egr4KCC2 site, we cotransfected an Egr4 expression vector with each of the above deletion constructs in N2a cells, which do not express endogenous Egr4 (data not shown). Egr4 induced a robust (from 15- to 22-fold) upregulation of luciferase activity for all constructs containing the Egr4KCC2 element, whereas no induction was observed for the KCC2(−180/+42) construct lacking Egr4KCC2 (Fig. 3b). In contrast, overexpression of Egr1, which binds a sequence closely related to the Egr4 consensus (Swirnoff and Milbrandt, 1995), did not induce significant luciferase activity of the KCC2 promoter constructs containing Egr4KCC2 [except for KCC2(−1398/+5445)] (Fig. 3b). To test additionally whether Egr1 might be able to bind the Egr4KCC2 site, albeit with a lower efficacy than Egr4, we used increasing amounts of Egr1 to compete with Egr4 for the binding site, which should lead to a decrease in reporter activity. However, by our keeping the Egr4 expression level constant, increasing the amount of Egr1 expression led to a small upregulation of reporter activity (Fig. 3c). Together, these results imply that Egr4 effectively can upregulate promoter activity of KCC2 reporter constructs containing the Egr4KCC2 site, whereas Egr1, which has a consensus sequence very similar to Egr4, cannot activate the KCC2 promoter via Egr4KCC2 or compete with Egr4 for the site.
Egr4 protein specifically binds to the Egr4KCC2 element in vitro
To test Egr4 protein binding to the Egr4KCC2 element, we used EMSA. Nuclear extracts of N2a cells were transfected transiently with the Egr4 expression plasmid and assayed for the proteins that specifically bind labeled, double-stranded oligonucleotides bearing the Egr4KCC2 site (Fig. 4a,b). Three specific complexes (I, II, and III) were detected. Complex I was most intensive and competed effectively by cold Egr4KCC2, but not by mutant Egr4KCC2 (Egr4KCC2mut) oligonucleotides, in which two dG nucleotides critical for binding of all Egr family members were substituted by two dT nucleotides. Furthermore, complex I specifically was supershifted by increasing amounts of the Egr4 antibody. These results suggest a specific interaction of the Egr4 protein and the Egr4KCC2 site in complex I. In contrast, the two other complexes (II and III) had a non-Egr4 origin. Competition of complex II with a 15-fold excess of cold Egr4KCC2 had no effect, and even a 150-fold excess did not substitute labeled Egr4KCC2 oligonucleotides completely. Moreover, Egr4 antibody failed to supershift complex II. On the other hand, complex III formation was competed by both wild-type and mutant Egr4KCC2 oligonucleotides, also suggesting a non-Egr4 origin. Interestingly, the intensity of complex III was enhanced strongly by the addition of Egr4 antibody (Fig. 4a). One reason could be that the Egr4 antibody, by partially preventing the Egr4 protein from binding to the Egr4KCC2 site, allowed another competing nuclear protein to bind a sequence close to the Egr4KCC2 in the oligonucleotide used for EMSA. Together, the EMSA demonstrated effective binding of Egr4 to the Egr4KCC2 element in vitro.
Figure 4.
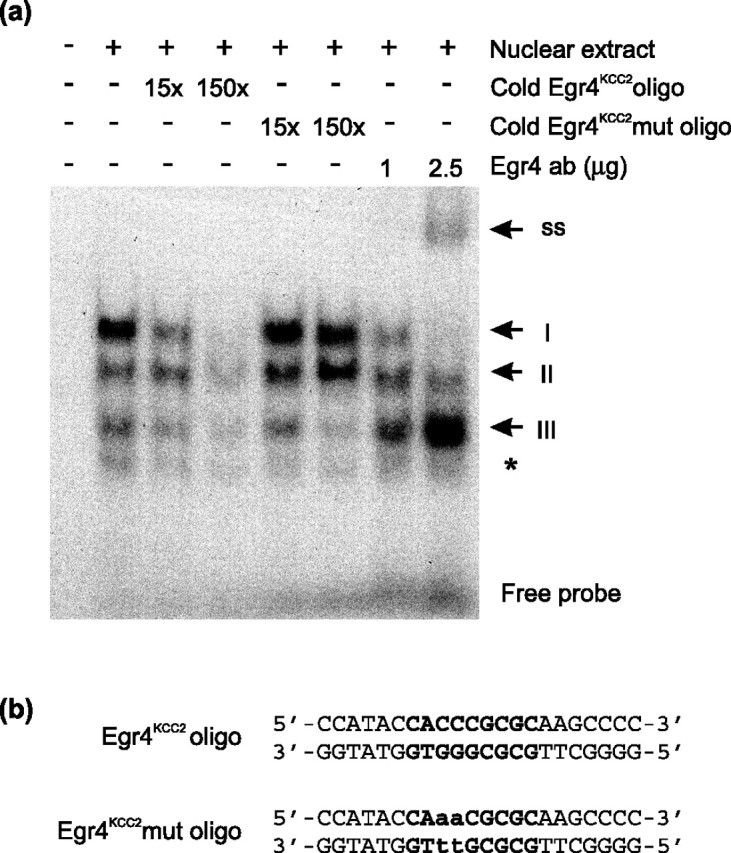
Egr4 binds the Egr4KCC2 element in vitro. a, Three specific complexes (I–III) are detected in EMSA, using labeled oligonucleotides bearing a single copy of the Egr4KCC2 element and nuclear extracts of N2a cells transfected with Egr4 expression plasmid. Complex I is competed effectively by cold Egr4KCC2 oligonucleotides. In contrast, unlabeled Egr4KCC2mut oligonucleotides do not influence the formation of complex I. The addition of Egr4 antibody prevents complex I formation in a dose-dependent manner and results in the formation of a new supershifted complex (indicated by ss). Complex II is affected less by Egr4KCC2 oligonucleotides and not at all by Egr4KCC2mut oligonucleotides. Complex III is competed by both wild-type and mutant Egr4KCC2 oligonucleotides. Note the significant increase in complex III intensity after the addition of the Egr4 antibody. Nonspecific binding is indicated by an asterisk. b, Sequences of Egr4KCC2 and Egr4KCC2mut oligonucleotides used in EMSA. The Egr4KCC2 sequence is highlighted in bold. Mutated nucleotides are shown in small case letters.
Disruption of the Egr4KCC2 site decreases Egr4-mediated induction of the KCC2 promoter in N2a cells
To reveal whether Egr4KCC2 is required for Egr4-mediated upregulation of the KCC2 promoter, we created a KCC2(−309/+42)Egr4mut construct that differed from the previously described KCC2(−309/+42) reporter (Fig. 3b) by only a 2 bp mutation in the Egr4KCC2 site. This mutagenesis substituted two critical dG nucleotides at positions 6 and 7 of the Egr consensus for two dTs (Fig. 5a). As shown previously (Christy and Nathans, 1989; Swirnoff and Milbrandt, 1995; O'Donovan and Baraban, 1999), even a single nucleotide mutation in position 6 of the Egr consensus drastically reduces the binding affinity of the Egr site; moreover, EMSA confirmed that this 2 bp mutation completely prevents Egr4 binding to the Egr4KCC2 sequence (Fig. 4a). KCC2(−309/+42) and KCC2(−309/+42)Egr4mut reporters were cotransfected either with an Egr4-overexpressing plasmid or with an empty vector into N2a cells, and Egr4-induced promoter activity for these constructs was measured. As a result, Egr4-mediated induction was ∼50% lower for the KCC2(−309/+42)Egr4mut construct than for the KCC2(−309/+42) construct (Fig. 5b). This experiment indicates that the Egr4KCC2 element is necessary to mediate the direct Egr4 effect on KCC2 promoter activity.
One reason that disruption of Egr4KCC2 did not prevent completely the Egr4-mediated induction of the KCC2(−309/+42)Egr4mut construct might be the presence of less stringent Egr-binding sites that were excluded in the initial analysis. We reanalyzed the −309/−180 bp fragment of the mouse KCC2 promoter sequence that was sufficient to mediate Egr4-induced upregulation of the reporter (Fig. 3b). We found an Egr-like site at position −305/−294 bp that differed from the Egr consensus at positions −1 and +4 and was not conserved in KCC2 promoters of other mammals. Mutagenesis of the Egr-like site did not alter further the Egr4-mediated induction of the KCC2(−309/+42) construct (data not shown). Thus the residual induction of KCC2(−309/+42)Egr4mut by Egr4 is likely to be indirect.
Egr4KCC2 element is sufficient for Egr4-mediated induction of the KCC2 promoter and is orientation-specific
To determine whether the Egr4KCC2 element is sufficient to mediate induction of the KCC2 promoter by Egr4, we inserted two copies of this element upstream of the basal KCC2 promoter in the KCC2(−180/+42) construct, which was not induced by Egr4 in N2a cells (Fig. 3b). In KCC2(−180/+42)2xEgr4(−) the two copies of Egr4KCC2 had the same orientation as the endogenous Egr4KCC2 (on the reverse DNA strand), whereas in KCC2(−180/+42)2xEgr4(+) the two Egr4KCC2 sites had the opposite orientation (on the forward DNA strand) (Fig. 5c). The parent KCC2(−180/+42) reporter, as well as the two KCC2(−180/+42)2xEgr4(+/−) constructs, were cotransfected with the Egr4 (or empty) expression vector into N2a cells, and Egr4-induced luciferase activity for these constructs was assayed. A strong (fourfold) upregulation of the reporter activity was observed for the KCC2(−180/+42)2xEgr4(−) construct, but not for the KCC2(−180/+42) construct (Fig. 5c). Surprisingly, the KCC2(−180/+42)2xEgr4(+) reporter, containing Egr4KCC2 elements on the forward DNA strand (in an orientation opposite to the endogenous Egr4KCC2), was not activated by Egr4 (Fig. 5c). The results show that the Egr4KCC2 element is sufficient to mediate Egr4-induced activation of the KCC2 promoter. This experiment also reveals the importance of correct orientation of Egr4 TF for efficient regulation of the KCC2 promoter.
Dominant-negative isoform of Egr4 prevents Egr4-mediated induction of the KCC2 reporter construct in N2a cells
As a tool to study additionally the role of Egr4 in KCC2 gene regulation, we produced DN-Egr4 that contained the C-terminal DNA binding part of Egr4 but lacked the N-terminal regulatory part (Fig. 6a). Increasing amounts of the DN-Egr4-expression plasmid effectively inhibited up to 70% of Egr4-mediated induction of KCC2(−309/+42) reporter activity (Fig. 6b). In contrast, even the highest amount of the DN-Egr4 plasmid had no significant effect on the basal activity of the KCC2(−309/+42) reporter (Fig. 6b). This experiment established that DN-Egr4 is able to prevent the Egr4-mediated activation of the KCC2 promoter in N2a cells effectively.
DN-Egr4 downregulates endogenous KCC2 expression in cultured hippocampal neurons
KCC2 expression is known to be increased during neuronal maturation also in dissociated CNS neurons as they form synapses in vitro (Ludwig et al., 2003). To address the role of Egr4 in KCC2 upregulation in primary neurons, we first established that the developmental upregulation of both KCC2 and Egr4 in mouse brain during postnatal development (Fig. 2) was recapitulated in cultured mouse hippocampal neurons (Fig. 7a). KCC2 mRNA was detectable already by 4 DIV and increased up to 11 DIV. Egr4 mRNA expression was also detectable on 4 DIV and, similar to KCC2, increased during the subsequent days in culture at least up to 11 DIV (Fig. 7a).
Figure 7.
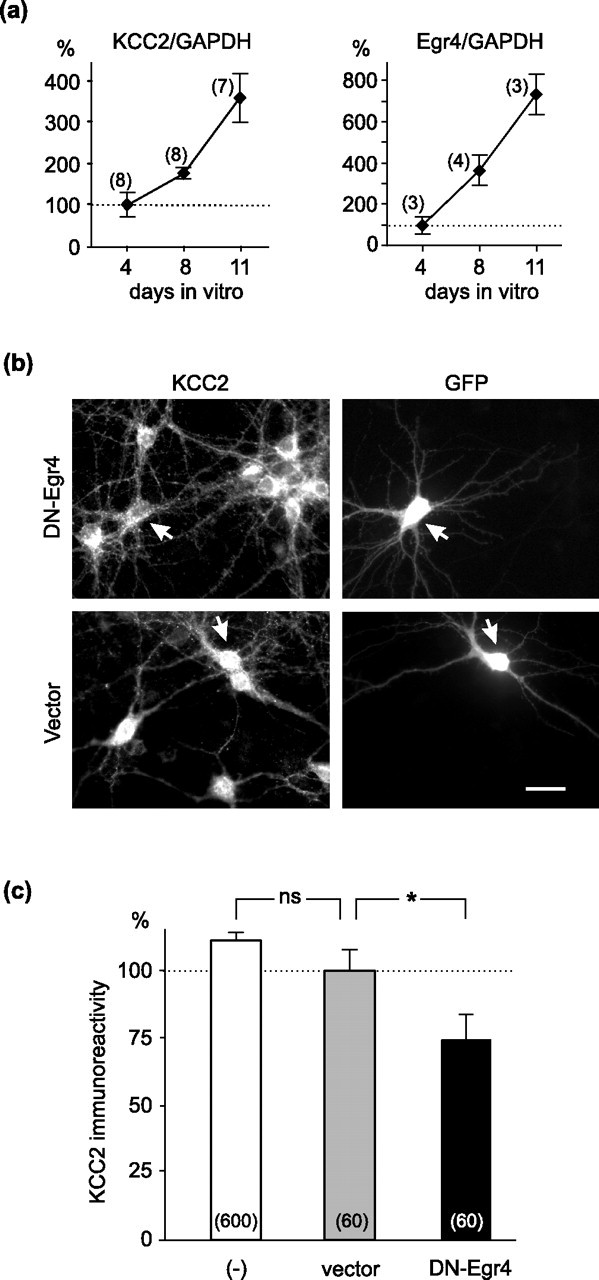
DN-Egr4 downregulates endogenous KCC2 expression in neurons. a, KCC2 mRNA expression is already detectable in mouse hippocampal neurons cultured at 4 DIV and is increased gradually up to 11 DIV. Egr4 mRNA also is detected in the cultures on day 4 and is increased approximately sevenfold by 11 DIV. Quantification of KCC2 and Egr4 mRNA expression was performed by using real-time reverse transcription-PCR and normalized to GAPDH (glyceraldehyde phosphate dehydrogenase). The number of independent experiments for each point is indicated in parentheses. b, Hippocampal neurons were cotransfected at 7 DIV with a GFP expression vector and either the DN-Egr4 or the control empty expression plasmid. After 2 d the cultures were fixed and assayed for KCC2 immunoreactivity. Images of GFP-positive neurons (indicated with arrows) coexpressing either DN-Egr4 or control vector plasmids are shown. Scale bar, 20 μm. c, The endogenous KCC2 expression clearly is reduced in neurons transfected with DN-Egr4, but not with the empty vector. KCC2 immunoreactivity is similar between nontransfected neurons (−) and neurons transfected with the empty vector. The number of cells analyzed in each group is indicated in parentheses. ns, Not significant (*p < 0.05; Student's t test). Error bars indicate SEM.
To determine whether Egr4 is responsible for the increase in KCC2 expression in cultured neurons, we used DN-Egr4 to prevent binding of endogenous Egr4 to the KCC2 promoter. The neurons were cotransfected with the DN-Egr4 (or empty) vector and a GFP expression plasmid at 7 DIV, and KCC2 immunoreactivity of GFP-positive neurons was assayed at 9 DIV (Fig. 7b). KCC2 immunoreactivity was significantly (∼25%) lower in neurons expressing the DN-Egr4 plasmid than in neurons transfected with the vector only (Fig. 7c). Importantly, KCC2 immunoreactivity in nontransfected (GFP-negative) neurons did not differ from neurons transfected with the empty vector. Thus the dominant-negative isoform of Egr4 downregulates endogenous KCC2 expression in cultured hippocampal neurons, presumably by preventing the binding of the endogenous Egr4 to the Egr4KCC2 site.
Ability of exogenous Egr4 to upregulate and DN-Egr4 to downregulate KCC2 reporter activity in cultured neurons may depend on endogenous Egr4 levels
We compared the ability of exogenous Egr4 to increase and DN-Egr4 to suppress the KCC2(−309/+42) reporter activity in rat cortical neuron cultures at two different time points. The KCC2(−309/+42) reporter and either DN-Egr4 or Egr4 expression constructs were cotransfected into 3 and 8 DIV rat cortical neurons, and luciferase activity was measured 48 h later (at 5 and 10 DIV, respectively). As in the cultured mouse hippocampal neurons (Fig. 7a) the expression of endogenous Egr4 mRNA in the rat cortical neurons shows a several-fold increase during this period of maturation in vitro (data not shown). Overexpression of Egr4 increased the KCC2 promoter activity by ∼100% in the 5 DIV neurons (Fig. 8a), confirming the ability of the KCC2 promoter to be induced by Egr4 in the primary neurons also. In contrast, the same amount of exogenous Egr4 failed to increase the luciferase activity of the KCC2(−309/+42) reporter in 10 DIV neurons (Fig. 8b) presumably because of the already high level of endogenous Egr4. On the other hand, expression of DN-Egr4 had no significant effect on KCC2(−309/+42) activity in 5 DIV neurons (Fig. 8a), presumably because of the low level of endogenous Egr4, whereas DN-Egr4 suppressed the reporter activity in 10 DIV neurons in a dose-dependent manner (Fig. 8b), resulting in ∼25% suppression with the use of the highest DN-Egr4 amount (0.8 μg/well).
RNAi-mediated knock-down of Egr4 in neurons confirms the role of endogenous Egr4 in KCC2 gene upregulation
Although the ability of DN-Egr4 to suppress endogenous KCC2 expression and KCC2 promoter activity in neurons suggests that the effect most likely is mediated via endogenous Egr4, DN-Egr4 overexpression theoretically could influence other factors. On the other hand, the amount of DN-Egr4 in neurons may be limiting (because of the relatively small amount of DN-Egr4 plasmid used in neurons to avoid cytotoxicity and the relatively high resistance of mature neurons to transfection) and thus underestimate the contribution of Egr4 to KCC2 gene upregulation. To evaluate additionally the specific contribution of endogenous Egr4 to regulate the KCC2 promoter activity in neurons, we used an RNAi knock-down approach. Egr4-specific siRNA duplexes suppressed KCC2(−309/+42) reporter activity by ∼35% (siRNA1) and ∼25% (siRNA2) compared with negative siRNA control (Fig. 8c). Similar results were obtained when neurons were transfected at 6 or 7 DIV and analyzed at 9 DIV (data not shown). This result indicates a specific contribution of endogenous Egr4 to KCC2 promoter upregulation in neurons.
Egr4KCC2 is necessary for the upregulation of KCC2 in neurons
Although the ability of DN-Egr4 and Egr4 siRNAs to suppress endogenous KCC2 expression and KCC2 promoter activity in neurons indicates that the effect is mediated via endogenous Egr4, part of the Egr4 effect could be indirect (as in N2a cells). To estimate the specific contribution of the Egr4KCC2 site to KCC2 gene regulation in neurons, we compared the fold induction of the KCC2(−309/+42) and KCC2(−309/+42)Egr4mut constructs in the rat cortical neurons between 5 and 10 DIV. The activity of the KCC2(−309/+42) reporter increased 4.2-fold (± 0.2) during this period of time. In contrast, the activity of the KCC2(−309/+42)Egr4mut reporter, bearing the mutated Egr4KCC2 site, increased only 2.7-fold (± 0.3) during the same period. This is ∼35% less compared with the KCC2(−309/+42) construct (p < 0.05; Student's t test). Together, the results indicate a major role for the Egr4KCC2 site in regulating KCC2 gene expression in neurons.
Discussion
In this study we have described putative TF binding sites within the core promoter of the neuronal K+/Cl− cotransporter KCC2 gene and demonstrated that Egr4 binds to one of these sites and activates the KCC2 gene transcription in neuronal cells.
Our previous study (Uvarov et al., 2005) showed that a 1.4 kb genomic fragment upstream of the main KCC2 transcription start site is mostly sufficient to mediate the CNS neuron-specific expression of KCC2, whereas the NRSE in intron-1 of the KCC2 gene is dispensable. Here we subjected the 1.4 kb KCC2 promoter and proximal intron-1 sequences to detailed analysis for the presence of possible regulatory sites. This search resulted in 10 putative TF binding sites that were conserved among the corresponding genomic sequences of mouse, rat, human, and chimpanzee KCC2: two each for Sp1 and AP2 as well as one each for AP1, Mef2, E-box, Egr4, NRSE, and Block.
We focused on one of these factors, Egr4, because its mRNA levels gradually increased during the first weeks of postnatal development in rats (Crosby et al., 1992) and mice (this study), resembling the KCC2 expression pattern over the course of postnatal development (Lu et al., 1999). Moreover, Egr4 (like other Egr proteins) is induced by neurotrophins in vitro (O'Donovan et al., 1999), whereas KCC2 is induced by BDNF in vivo (Aguado et al., 2003). Thus we considered Egr4 as a candidate TF involved in the developmental upregulation of KCC2.
Egr4 overexpression in N2a neuroblastoma cells resulted in a strong (∼25-fold) induction of luciferase activity for the KCC2(−1398/+42) reporter. Similar induction was obtained for other short KCC2 promoter constructs containing the Egr4KCC2 element, but not for KCC2(−180/+42) lacking this site. The activation of KCC2 constructs by Egr4 was approximately as high as the activation of the reporter constructs carrying a single Egr consensus binding site (GCGGGGGCG) after induction by Egr4 or Egr1 (Crosby et al., 1991). Although a reported binding consensus for Egr1 is very similar to that for Egr4 (Swirnoff and Milbrandt, 1995), overexpression of Egr1 failed to induce the KCC2 reporter constructs containing the Egr4KCC2 site, and coexpression of Egr4 and Egr1 revealed no competition between these proteins. Egr4 binding to the Egr4KCC2 site was confirmed in vitro by EMSA. Labeled Egr4KCC2 oligonucleotides formed three complexes (I–III) with nuclear proteins, only the most intense of which (complex I) could be considered a result of specific Egr4 binding to the Egr4KCC2 site.
Mutagenesis in the sixth position of the Egr consensus has been reported to reduce drastically the binding affinity of the Egr site (Christy and Nathans, 1989; O'Donovan and Baraban, 1999). We found that substitution of two critical dG nucleotides in the Egr4KCC2 element (positions 6 and 7 in the consensus) (Fig. 5a) by two dTs prevented ∼50% of the Egr4-mediated induction of the KCC2(−309/+42) reporter. Because mutation of Egr4KCC2 failed to block the Egr4-mediated induction completely, we tested the possible involvement of an Egr-like site present within the −309/+42 fragment of the mouse KCC2 promoter. However, mutagenesis of the Egr-like site did not affect the Egr4-mediated induction of KCC2 promoter activity, consistent with its lower similarity to the Egr4 consensus sequence and its absence in other mammals (data not shown). Thus Egr4 regulates the KCC2 promoter both directly (via the Egr4KCC2 site) as well as indirectly (via an unknown mechanism).
To demonstrate sufficiency of the Egr4KCC2 element for KCC2 promoter induction by Egr4 in neuronal cells, we used the KCC2(−180/+42) construct (which does not respond to Egr4) with two copies of the Egr4KCC2 element inserted into the reverse DNA promoter strand (i.e., the same orientation as the endogenous Egr4KCC2). The resulting construct, KCC2(−180/+42)2xEgr4(−), was induced by fourfold with Egr4 overexpression. Surprisingly, the similar KCC2(−180/+42)2xEgr4(+) construct, containing two copies of Egr4KCC2 but positioned on the forward DNA strand (i.e., opposite orientation to the endogenous Egr4KCC2 site), was not activated by Egr4. To our knowledge this is the first demonstration of orientation specificity for the Egr site. Members of the Egr family bind the recognition element as monomers in an anti-parallel configuration (Pavletich and Pabo, 1991); thus the position of the Egr4KCC2 element defines the orientation of the Egr4 protein relative to the promoter. Moreover, Egr proteins can cooperate functionally with other TFs, binding a promoter nearby, and in this way induce a promoter activity synergistically (Decker et al., 2003). This suggests that the functional activity of the Egr4 bound to the Egr4KCC2 site might depend on interactions with other TFs. Future studies will address whether any of the other TFs that bind to the KCC2 promoter form complexes with Egr4.
Inhibition of Egr-mediated transcription by dominant-negative isoforms of Egr proteins has been a successful approach to demonstrate a role for endogenous Egr family members in gene regulation (Levkovitz et al., 2001; Roberts et al., 2005). We cloned a dominant-negative isoform of the Egr4 protein, which contained only the DNA binding domain of Egr4, and showed that it effectively suppressed Egr4-mediated induction of the KCC2 luciferase construct in N2a cells (by >70%), but it did not affect the basal level of KCC2 promoter activity. Cultured mouse hippocampal neurons transfected with the DN-Egr4 plasmid demonstrated a ∼25% decrease in KCC2 protein expression, compared with neurons transfected with an empty vector. Moreover, DN-Egr4 suppressed KCC2(−309/+42) reporter activity (by 23%) in mature (10 DIV) rat cortical neurons, whereas exogenous Egr4 induced the reporter activity by twofold in immature (5 DIV) neurons. Finally, RNAi-mediated knock-down of Egr4 suppressed reporter activity by ∼35% in the mature neurons. Together, these experiments indicate that endogenous Egr4 is important for the KCC2 upregulation in primary neurons. The clear but partial (25–35%) suppression of KCC2 protein expression and promoter activity in the neurons by Egr4 knock-down suggests that factors other than Egr4 also play a role in KCC2 upregulation. To address the specific contribution of Egr4KCC2 in KCC2 gene regulation in primary neurons, we compared the developmental upregulation of the KCC2(−309/+42) versus KCC2(−309/+42)Egr4mut reporter activities in cultured neurons (between 5 and 10 DIV). Mutagenesis of the Egr4KCC2 site blocked ∼35% of the developmental induction of the KCC2 promoter. This result additionally confirms the important role of Egr4KCC2 on KCC2 gene upregulation in primary CNS neurons.
In conclusion, we have characterized the regulatory region of the mouse neuronal K+/Cl− cotransporter gene, determined putative TF binding sites inside the core KCC2 promoter, and demonstrated that the Egr4 TF induces KCC2 gene activity in neuronal cells and primary neurons via the Egr4KCC2 site. More experiments are needed to confirm the role of Egr4 in KCC2 gene regulation in vivo. One approach would be analysis of KCC2 expression in Egr4-deficient mice. Despite the mostly neural expression pattern of Egr4, Egr4-deficient mice exhibit no identifiable behavioral or obvious phenotypic abnormalities in the nervous system (Tourtellotte et al., 1999). In contrast, KCC2-deficient and KCC2-hypomorphic mice, depending on the level of retained KCC2 protein, either die at birth (Hubner et al., 2001) or exhibit spontaneous seizures (Woo et al., 2002) or other milder but still obvious behavioral defects (Tornberg et al., 2005). Because mice that retain 30–40% of the normal KCC2 protein level in the brain no longer exhibit obvious behavioral abnormalities (our unpublished data), the absence of identifiable behavioral abnormalities in Egr4-deficent mice might be attributed to a moderate impact of Egr4 on KCC2 promoter regulation in vivo. On the other hand, possible compensation, e.g., by other Egr family members (Tourtellotte et al., 1999, 2000), may be an obstacle in attempts to study KCC2 regulation in vivo with the use of Egr4-deficent mice. Transgenic mice carrying a reporter gene under control of the KCC2 promoter region with either an intact or a mutated Egr4KCC2 element might be another, more powerful means of investigating Egr4-mediated induction of KCC2 promoter activity in vivo.
Footnotes
This work was supported by grants from the Academy of Finland and the Sigrid Jusélius Foundation (M.S.A.). We thank Jeffrey Milbrandt for the Egr1 and Egr4 expression plasmids, Christine Skerka for the Egr4 antibodies, and Seija Lehto and Outi Nikkilä for the neuronal cultures.
References
- Aguado F, Carmona MA, Pozas E, Aguilo A, Martinez-Guijarro FJ, Alcantara S, Borrell V, Yuste R, Ibanez CF, Soriano E. BDNF regulates spontaneous correlated activity at early developmental stages by increasing synaptogenesis and expression of the K+/Cl− co-transporter KCC2. Development. 2003;130:1267–1280. doi: 10.1242/dev.00351. [DOI] [PubMed] [Google Scholar]
- Banker G, Goslin K. Ed 2. Cambridge, MA: MIT; 1998. Culturing nerve cells. [Google Scholar]
- Beckmann AM, Wilce PA. Egr transcription factors in the nervous system. Neurochem Int. 1997;31:477–510. doi: 10.1016/s0197-0186(96)00136-2. [DOI] [PubMed] [Google Scholar]
- Cartharius K, Frech K, Grote K, Klocke B, Haltmeier M, Klingenhoff A, Frisch M, Bayerlein M, Werner T. MatInspector and beyond: promoter analysis based on transcription factor binding sites. Bioinformatics. 2005;21:2933–2942. doi: 10.1093/bioinformatics/bti473. [DOI] [PubMed] [Google Scholar]
- Chenna R, Sugawara H, Koike T, Lopez R, Gibson TJ, Higgins DG, Thompson JD. Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 2003;31:3497–3500. doi: 10.1093/nar/gkg500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christy B, Nathans D. DNA binding site of the growth factor-inducible protein Zif268. Proc Natl Acad Sci USA. 1989;86:8737–8741. doi: 10.1073/pnas.86.22.8737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen I, Navarro V, Clemenceau S, Baulac M, Miles R. On the origin of interictal activity in human temporal lobe epilepsy in vitro. Science. 2002;298:1418–1421. doi: 10.1126/science.1076510. [DOI] [PubMed] [Google Scholar]
- Collart MA, Tourkine N, Belin D, Vassalli P, Jeanteur P, Blanchard JM. c-fos gene transcription in murine macrophages is modulated by a calcium-dependent block to elongation in intron-1. Mol Cell Biol. 1991;11:2826–2831. doi: 10.1128/mcb.11.5.2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coull JAM, Boudreau D, Bachand K, Prescott SA, Nault F, Sik A, De Koninck P, De Koninck Y. Trans-synaptic shift in anion gradient in spinal lamina I neurons as a mechanism of neuropathic pain. Nature. 2003;424:938–942. doi: 10.1038/nature01868. [DOI] [PubMed] [Google Scholar]
- Crosby SD, Puetz JJ, Simburger KS, Fahrner TJ, Milbrandt J. The early response gene NGFI-C encodes a zinc finger transcriptional activator and is a member of the GCGGGGGCG (GSG) element-binding protein family. Mol Cell Biol. 1991;11:3835–3841. doi: 10.1128/mcb.11.8.3835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crosby SD, Veile RA, Doniskeller H, Baraban JM, Bhat RV, Simburger KS, Milbrandt J. Neural-specific expression, genomic structure, and chromosomal localization of the gene encoding the zinc-finger transcription factor NGFI-C. Proc Natl Acad Sci USA. 1992;89:4739–4743. doi: 10.1073/pnas.89.10.4739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decker EL, Nehmann N, Kampen E, Eibel H, Zipfel PF, Skerka C. Early growth response proteins (EGR) and nuclear factors of activated T-cells (NFAT) form heterodimers and regulate proinflammatory cytokine gene expression. Nucleic Acids Res. 2003;31:911–921. doi: 10.1093/nar/gkg186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiumelli H, Cancedda L, Poo MM. Modulation of GABAergic transmission by activity via postsynaptic Ca2+-dependent regulation of KCC2 function. Neuron. 2005;48:773–786. doi: 10.1016/j.neuron.2005.10.025. [DOI] [PubMed] [Google Scholar]
- Gossett LA, Kelvin DJ, Sternberg EA, Olson EN. A new myocyte-specific enhancer-binding factor that recognizes a conserved element associated with multiple muscle-specific genes. Mol Cell Biol. 1989;9:5022–5033. doi: 10.1128/mcb.9.11.5022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubner CA, Stein V, Hermans-Borgmeyer I, Meyer T, Ballanyi K, Jentsch TJ. Disruption of KCC2 reveals an essential role of K-Cl cotransport already in early synaptic inhibition. Neuron. 2001;30:515–524. doi: 10.1016/s0896-6273(01)00297-5. [DOI] [PubMed] [Google Scholar]
- James AB, Conway AM, Morris BJ. Genomic profiling of the neuronal target genes of the plasticity-related transcription factor—Zif268. J Neurochem. 2005;95:796–810. doi: 10.1111/j.1471-4159.2005.03400.x. [DOI] [PubMed] [Google Scholar]
- Kadonaga JT, Carner KR, Masiarz FR, Tijian R. Isolation of cDNA-encoding transcription factor Sp1 and functional analysis of the DNA-binding domain. Cell. 1987;51:1079–1090. doi: 10.1016/0092-8674(87)90594-0. [DOI] [PubMed] [Google Scholar]
- Kanaka C, Ohno K, Okabe A, Kuriyama K, Itoh T, Fukuda A, Sato K. The differential expression patterns of messenger RNAs encoding K-Cl cotransporters (KCC1, 2) and Na-K-2Cl cotransporter (NKCC1) in the rat nervous system. Neuroscience. 2001;104:933–946. doi: 10.1016/s0306-4522(01)00149-x. [DOI] [PubMed] [Google Scholar]
- Klebe RJ, Ruddle FH. Neuroblastoma: cell culture analysis of a differentiating stem cell system. J Cell Biol. 1969;43:69a. [Google Scholar]
- Knapska E, Kaczmarek L. A gene for neuronal plasticity in the mammalian brain: Zif268/Egr-1/NGFI-A/Krox-24/TIS8/ZENK? Prog Neurobiol. 2004;74:183–211. doi: 10.1016/j.pneurobio.2004.05.007. [DOI] [PubMed] [Google Scholar]
- Lee W, Mitchell P, Tjian R. Purified transcription factor AP-1 interacts with TPA-inducible enhancer elements. Cell. 1987;49:741–752. doi: 10.1016/0092-8674(87)90612-x. [DOI] [PubMed] [Google Scholar]
- Levkovitz Y, O'Donovan KJ, Baraban JM. Blockade of NGF-induced neurite outgrowth by a dominant-negative inhibitor of the Egr family of transcription regulatory factors. J Neurosci. 2001;21:45–52. doi: 10.1523/JNEUROSCI.21-01-00045.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Tornberg J, Kaila K, Airaksinen MS, Rivera C. Patterns of cation-chloride cotransporter expression during embryonic rodent CNS development. Eur J Neurosci. 2002;16:2358–2370. doi: 10.1046/j.1460-9568.2002.02419.x. [DOI] [PubMed] [Google Scholar]
- Li L, Carter J, Gao X, Whitehead J, Tourtellotte WG. The neuroplasticity-associated arc gene is a direct transcriptional target of early growth response (Egr) transcription factors. Mol Cell Biol. 2005;25:10286–10300. doi: 10.1128/MCB.25.23.10286-10300.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Karadsheh M, Delpire E. Developmental regulation of the neuronal-specific isoform of K-Cl cotransporter KCC2 in postnatal rat brains. J Neurobiol. 1999;39:558–568. [PubMed] [Google Scholar]
- Ludwig A, Li H, Saarma M, Kaila K, Rivera C. Developmental up-regulation of KCC2 in the absence of GABAergic and glutamatergic transmission. Eur J Neurosci. 2003;18:3199–3206. doi: 10.1111/j.1460-9568.2003.03069.x. [DOI] [PubMed] [Google Scholar]
- Mercado A, Mount DB, Gamba G. Electroneutral cation-chloride cotransporters in the central nervous system. Neurochem Res. 2004;29:17–25. doi: 10.1023/b:nere.0000010432.44566.21. [DOI] [PubMed] [Google Scholar]
- Nabekura J, Ueno T, Okabe A, Furuta A, Iwaki T, Shimizu-Okabe C, Fukuda A, Akaike N. Reduction of KCC2 expression and GABAA receptor-mediated excitation after in vivo axonal injury. J Neurosci. 2002;22:4412–4417. doi: 10.1523/JNEUROSCI.22-11-04412.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedivi E, Basi GS, Akey IV, Skene JHP. A neural-specific GAP-43 core promoter located between unusual DNA elements that interact to regulate its activity. J Neurosci. 1992;12:691–704. doi: 10.1523/JNEUROSCI.12-03-00691.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Donovan KJ, Baraban JM. Major Egr3 isoforms are generated via alternate translation start sites and differ in their abilities to activate transcription. Mol Cell Biol. 1999;19:4711–4718. doi: 10.1128/mcb.19.7.4711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Donovan KJ, Tourtellotte WG, Milbrandt J, Baraban JM. The EGR family of transcription-regulatory factors: progress at the interface of molecular and systems neuroscience. Trends Neurosci. 1999;22:167–173. doi: 10.1016/s0166-2236(98)01343-5. [DOI] [PubMed] [Google Scholar]
- Pavletich NP, Pabo CO. Zinc finger-DNA recognition: crystal structure of a Zif268–DNA complex at 2.1 A. Science. 1991;252:809–817. doi: 10.1126/science.2028256. [DOI] [PubMed] [Google Scholar]
- Payne JA, Stevenson TJ, Donaldson LF. Molecular characterization of a putative K-Cl cotransporter in rat brain. A neuronal-specific isoform. J Biol Chem. 1996;271:16245–16252. doi: 10.1074/jbc.271.27.16245. [DOI] [PubMed] [Google Scholar]
- Payne JA, Rivera C, Voipio J, Kaila K. Cation-chloride co-transporters in neuronal communication, development, and trauma. Trends Neurosci. 2003;26:199–206. doi: 10.1016/S0166-2236(03)00068-7. [DOI] [PubMed] [Google Scholar]
- Petersohn D, Schoch S, Brinkmann DR, Thiel G. The human synapsin-II gene promoter. Possible role for the transcription factors Zif268/Egr-1, polyoma enhancer activator-3, and AP2. J Biol Chem. 1995;270:24361–24369. doi: 10.1074/jbc.270.41.24361. [DOI] [PubMed] [Google Scholar]
- Rivera C, Voipio J, Payne JA, Ruusuvuori E, Lahtinen H, Lamsa K, Pirvola U, Saarma M, Kaila K. The K+/Cl− co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature. 1999;397:251–255. doi: 10.1038/16697. [DOI] [PubMed] [Google Scholar]
- Rivera C, Li H, Thomas-Crusells J, Lahtinen H, Viitanen T, Nanobashvili A, Kokaia Z, Airaksinen MS, Voipio J, Kaila K, Saarma M. BDNF-induced TrkB activation down-regulates the K+-Cl− cotransporter KCC2 and impairs neuronal Cl− extrusion. J Cell Biol. 2002;159:747–752. doi: 10.1083/jcb.200209011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera C, Voipio J, Thomas-Crusells J, Li H, Emri Z, Sipila S, Payne JA, Minichiello L, Saarma M, Kaila K. Mechanism of activity-dependent downregulation of the neuron-specific K-Cl cotransporter KCC2. J Neurosci. 2004;24:4683–4691. doi: 10.1523/JNEUROSCI.5265-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts DS, Raol YH, Bandyopadhyay S, Lund IV, Budreck EC, Passini MJ, Wolfe JH, Brooks-Kayal AR, Russek SJ. Egr3 stimulation of GABRA4 promoter activity as a mechanism for seizure-induced up-regulation of GABAA receptor α4 subunit expression. Proc Natl Acad Sci USA. 2005;102:11894–11899. doi: 10.1073/pnas.0501434102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakimura K, Kushiya E, Takahashi Y, Suzuki Y. The structure and expression of neuron-specific enolase gene. Gene. 1987;60:103–113. doi: 10.1016/0378-1119(87)90218-6. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Russell DW. Ed 3. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 2001. Molecular cloning: a laboratory manual. [Google Scholar]
- Sawadogo M. Multiple forms of the human gene-specific transcription factor USF. II. DNA binding properties and transcriptional activity of the purified HeLa USF. J Biol Chem. 1988;263:11994–12001. [PubMed] [Google Scholar]
- Schoenherr CJ, Paquette AJ, Anderson DJ. Identification of potential target genes for the neuron-restrictive silencer factor. Proc Natl Acad Sci USA. 1996;93:9881–9886. doi: 10.1073/pnas.93.18.9881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein V, Hermans-Borgmeyer I, Jentsch TJ, Hubner CA. Expression of the KCl cotransporter KCC2 parallels neuronal maturation and the emergence of low intracellular chloride. J Comp Neurol. 2004;468:57–64. doi: 10.1002/cne.10983. [DOI] [PubMed] [Google Scholar]
- Sudhof TC. The structure of the human synapsin-I gene and protein. J Biol Chem. 1990;265:7849–7852. [PubMed] [Google Scholar]
- Swirnoff AH, Milbrandt J. DNA-binding specificity of NGFI-A and related zinc finger transcription factors. Mol Cell Biol. 1995;15:2275–2287. doi: 10.1128/mcb.15.4.2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tornberg J, Voikar V, Savilahti H, Rauvala H, Airaksinen MS. Behavioural phenotypes of hypomorphic KCC2-deficient mice. Eur J Neurosci. 2005;21:1327–1337. doi: 10.1111/j.1460-9568.2005.03959.x. [DOI] [PubMed] [Google Scholar]
- Tourtellotte WG, Nagarajan R, Auyeng A, Mueller C, Milbrandt J. Infertility associated with incomplete spermatogenic arrest and oligozoospermia in Egr4-deficient mice. Development. 1999;126:5061–5071. doi: 10.1242/dev.126.22.5061. [DOI] [PubMed] [Google Scholar]
- Tourtellotte WG, Nagarajan R, Bartke A, Milbrandt J. Functional compensation by Egr4 in Egr1-dependent luteinizing hormone regulation and Leydig cell steroidogenesis. Mol Cell Biol. 2000;20:5261–5268. doi: 10.1128/mcb.20.14.5261-5268.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uvarov P, Pruunsild P, Timmusk T, Airaksinen MS. Neuronal K+/Cl− co-transporter (KCC2) transgenes lacking neurone restrictive silencer element recapitulate CNS neurone-specific expression and developmental up-regulation of endogenous KCC2 gene. J Neurochem. 2005;95:1144–1155. doi: 10.1111/j.1471-4159.2005.03434.x. [DOI] [PubMed] [Google Scholar]
- Williams T, Tjian R. Characterization of a dimerization motif in AP-2 and its function in heterologous DNA-binding proteins. Science. 1991;251:1067–1071. doi: 10.1126/science.1998122. [DOI] [PubMed] [Google Scholar]
- Woo NS, Lu JM, England R, McClellan R, Dufour S, Mount DB, Deutch AY, Lovinger DM, Delpire E. Hyperexcitability and epilepsy associated with disruption of the mouse neuronal-specific K-Cl cotransporter gene. Hippocampus. 2002;12:258–268. doi: 10.1002/hipo.10014. [DOI] [PubMed] [Google Scholar]
- Zipfel PF, Decker EL, Holst C, Skerka C. The human zinc finger protein EGR-4 acts as autoregulatory transcriptional repressor. Biochim Biophys Acta. 1997;1354:134–144. doi: 10.1016/s0167-4781(97)00084-5. [DOI] [PubMed] [Google Scholar]