

Abstract
What determines where synapses will form along an axon or how proteins are deposited at nascent synapses remains unknown. Here, we show that the initial formation of presynaptic terminals occurs preferentially at predefined sites within the axons of cortical neurons. Time-lapse imaging of synaptic vesicle protein transport vesicles (STVs) indicates that STVs pause repeatedly at these sites, even in the absence of neuronal or glial contact. Contact with a neuroligin-expressing non-neuronal cell induces formation of presynaptic terminals specifically at these STV pause sites. Remarkably, formation of stable contacts with dendritic filopodia also occurs selectively at STV pause sites. Although it is not yet known which molecules comprise the predefined sites, STV pausing is regulated by cues that affect synaptogenesis. Overall, these data are consistent with the hypothesis that regulation of STV pausing might be an important mechanism for accumulation of presynaptic proteins at nascent synapses and support a new model in which many en passant synapses form specifically at predefined sites in young axons.
Keywords: synapse formation, synaptogenesis, VAMP–EGFP, time-lapse imaging, visual cortex, development
Introduction
Synaptogenesis in the CNS requires coordinated assembly of the protein complexes and specialized membrane domains required for synaptic transmission. Although some of the early events in synaptogenesis have been identified (Scheiffele, 2003; Waites et al., 2005), a number of issues remain unresolved, such as how synaptic proteins accumulate at sites of axodendritic contact or what determines where along axons synapses form.
During synaptogenesis, presynaptic proteins are transported along axons in at least two types of organelles: piccolo transport vesicles (PTVs) and synaptic vesicle (SV) protein transport vesicles (STVs), respectively (Ahmari et al., 2000; Zhai et al., 2001). PTVs transport active zone proteins and are biochemically and morphologically distinct from STVs (Zhai et al., 2001). STVs, the focus of the present study, carry many SV-associated proteins (Ahmari et al., 2000; Zhai et al., 2001) and exhibit morphologies ranging from pleiomorphic, tubulovesicular organelles (Ahmari et al., 2000) to clusters of SVs (Kraszewski et al., 1995).
Presynaptic proteins accumulate at new synapses by two distinct mechanisms. In immature neurons, presynaptic proteins accumulate via recruitment of STVs and PTVs to axodendritic adhesions (Ahmari et al., 2000; Friedman et al., 2000; Washbourne et al., 2002; Shapira et al., 2003). In more mature neurons, portions of existing terminals (“orphans”), containing both SVs and an active zone, mobilize to form new terminals (Krueger et al., 2003). Orphans and STVs are functionally distinct because orphan cycling is sensitive to tetanus toxin but not brefeldin-A, whereas STV cycling is sensitive to brefeldin-A but not tetanus toxin (Verderio et al., 1999; Krueger et al., 2003).
Although synapse formation is often initiated by contact between dendritic filopodia and axons (Dailey and Smith, 1996; Ziv and Smith, 1996; Fiala et al., 1998; Jontes and Smith, 2000), it remains unknown what determines where along axons those contacts are stabilized and synapses are formed. The entire length of axon could be equally capable of forming presynaptic terminals, and synapses could form wherever dendritic filopodia touch an axon. Alternatively, synaptogenesis could be restricted to particular sites along the axon, and only dendritic filopodia that contact those sites would be stabilized. Because most contacts between dendritic filopodia and axons are not stabilized, unidentified factors within axons might determine where axodendritic contacts stabilize and presynaptic terminals form.
Here, time-lapse imaging of developing rodent cortical neurons was used to analyze quantitatively STV transport and recruitment to sites of presynaptic terminal formation. Stable STV accumulation and formation of presynaptic terminals induced by neuroligin-expressing non-neuronal cells was restricted to particular sites within the axon. Although it is unclear which molecules comprise these sites, STVs paused repeatedly at these predefined sites, even in the absence of contact with dendrites or glia. Pausing was regulated by signals that influence synaptogenesis and may result in glutamate release. Remarkably, stabilization of contacts with dendritic filopodia occurred selectively at pause sites, suggesting a new model in which many en passant presynaptic terminals form specifically at STV pause sites in young axons.
Materials and Methods
All studies were conducted with an approved protocol from the University of California, Davis Animal Care and Use Committee, in compliance with National Institutes of Health guidelines for the care and use of experimental animals.
Neuronal cultures.
Cultures were prepared from postnatal rat visual cortex essentially as described previously (Washbourne et al., 2002; Sabo and McAllister, 2003). Neurons were either grown directly on a monolayer of rat cortical astrocytes or on poly-l-lysine-coated coverslips inverted over astrocyte monolayers. For synapsin triple knock-out mice (STKO), timed-pregnant knock-out mice and wild-type control mice were obtained from H. T. Kao and Paul Greengard at Rockefeller University (New York, NY) (Gitler et al., 2004). Neurons were cultured from postnatal cortices of these mice as above for rat neuronal cultures.
Transfection.
Neurons were transfected with Lipofectamine 2000 (Invitrogen, Carlsbad, CA) 24–48 h before imaging essentially as described previously (Washbourne et al., 2002; Sabo and McAllister, 2003). Transfection with small amounts of DNA (∼4–7 ng/mm2) and short expression times (typically 16–24 h) yielded relatively low expression levels and expression patterns that appeared identical to endogenous proteins in neighboring axons (Sabo and McAllister, 2003). With double transfection, localization of each protein appeared similar when expressed alone. The distribution of vesicle-associated membrane protein 2 (VAMP2)–Discosoma red (DsRed)–labeled STVs appeared similar to that of VAMP2–enhanced green fluorescent protein (EGFP)-labeled STVs. After transfection, neurons were healthy and their growth cones were active.
Live imaging.
At 3–5 d in vitro (DIV), axons were imaged with a Zeiss (Thornwood, NY) Pascal or an Olympus Optical (Center Valley, PA) Fluoview confocal system using a 40× Plan Neofluor 0.75 numerical aperture objective. Lasers were 488 nm argon and 543 nm helium–neon. Detection filters were bandpass 505–530 nm for EGFP and long-pass 560 nm for DsRed and HcRed (a far-red fluorescent protein modified from the reef coral Heteractis crispa). Laser power was kept low (6% or less for 488 nm argon and 18% or less for 543 nm helium–neon) to avoid phototoxicity. Typically, images were collected every 15 s, with scan times of 1.9 s for single-channel imaging. This imaging interval was selected as a compromise between having a high temporal resolution and minimizing the time the neurons were exposed to the laser to avoid any toxicity and photobleaching. In some cases, images were collected with 2× Kalman averaging. Imaging was conducted with constant perfusion with artificial CSF (ACSF) (120 mm NaCl, 3 mm KCl, 2 mm CaCl2, 2 mm MgCl2, 30 mm glucose, 0.2% sorbitol, and 20 mm HEPES, pH 7.2). ACSF temperatures were kept consistent within an experiment. Initially, experiments were performed at 30–35°C. When necessary, experiments were performed at ambient temperature (as indicated in the descriptions of those individual experiments). STV mobility was not reduced and there was no apparent difference in STV behavior when experiments were performed at 25°C. Calcium-free ACSF included the following modifications: 0 mm CaCl2, 4 mm MgCl2, and 1 mm EGTA. Axons were identified using morphological criteria, as described previously (Sabo and McAllister, 2003). For dual-color imaging for colocalization, channels were collected sequentially to eliminate bleed-through. For imaging of double-transfected neurons, neurons were imaged in which expression levels of both fusion proteins appeared comparable. Controls for the accurate localization of VAMP2–DsRed have been described previously (Sabo and McAllister, 2003). To disrupt actin polymerization, neurons were treated with latrunculin A (5 μm for at least 1 h at 37°C). Neurons were perfused with ACSF containing latrunculin A at 25°C during imaging. The disappearance or immobility of filopodia and altered glial morphology were used as an indication that the drug had been effective. Control neurons were treated with vehicle only (0.1% DMSO). When imaging contact formation, neurons were imaged at ambient temperature, which yielded a more stable focal plane.
Immunofluorescence.
Neurons were fixed for 10–15 min in 4% paraformaldehyde in PBS containing 4% sucrose, permeabilized for 5 min with 0.2–0.25% Triton X-100, and blocked with 10% BSA. Primary antibodies were as follows: VAMP (1:200; Synaptic Systems, Gottingen, Germany); EGFP (rabbit 1:2000, mouse 1:500; Invitrogen); rSec6 (an exocyst complex component; 1:25; Abcam, Cambridge, MA); hemagglutinin HA.11 (1:1000); βIII tubulin (TUJ-1; 1:1000; Abcam), and vesicular glutamate transporter 1 (VGlut1; 1:3000; Chemicon, Temecula, CA). Secondary antibodies were Alexa 488 or 546 (1:200; Invitrogen) or cyanine 2 (Cy2), Cy3, or Cy5-tagged (1:200; Jackson ImmunoResearch, West Grove, PA) antibodies. Phalloidin-568 (1:40; Invitrogen) was used to label actin filaments. Coverslips were mounted in Fluoromount (Fisher Scientific, Pittsburgh, PA) containing DABCO (1,4-diazabicyclo-[2.2.2]octane) (Sigma, St. Louis, MO). For retrospective immunocytochemistry, gridded coverslips were fixed and labeled immediately after live imaging. The same neuron that had been imaged live was located using the grid, and images were aligned in Image Pro (Media Cybernetics, Silver Spring, MD) by comparing live EGFP and fixed anti-EGFP antibody signals.
FM4-64 loading and unloading.
Vesicles were loaded with FM4-64 [N-(3-triethylammoniumpropyl)-4-(6-(4-(diethylamino)phenyl)hexatrienyl)pyridinium dibromide] (Invitrogen) by incubating neurons in high-K+ ACSF (38 mm NaCl, 85 mm KCl, 2 mm CaCl2, 2 mm MgCl2, 30 mm glucose, 0.2% sorbitol, and 20 mm HEPES, pH 7.2) containing 10 or 15 μm FM4-64. In some cases, neurons were subsequently incubated with FM dye in normal ACSF for 1 min. Images were collected after extensive washing (>10 min) with ACSF in the absence of FM dye.
Presynaptic terminal induction.
To induce formation of presynaptic terminals, methods were adapted from Scheiffele et al. (2000). HEK-293 cells were cotransfected with HA-tagged neuroligin and HcRed (or DsRed) 1 d after plating. HEK-293 cells that expressed HcRed reliably coexpressed neuroligin (92.5% of cells; n = 146), as confirmed by immunostaining with anti-HA tag antibodies. Approximately 24–48 h after transfection, cells were dissociated by incubation in HBSS (without calcium and magnesium; Invitrogen) and trituration. Cells were collected by centrifugation and resuspended in a small volume of ACSF. To identify pause sites, 5–7 DIV neurons transfected with VAMP2–EGFP were imaged for at least 10 min before addition of HEK-293 cells. A concentrated suspension of HEK-293 cells was added dropwise to the neurons during imaging. Imaging was continued for at least 20 min after addition of non-neuronal cells, but typically on the order of 1–2 h, to determine whether VAMP became stably accumulated at sites of contact with transfected HEK-293 cells. Imaging was performed at ambient temperature without perfusion because perfusion with warmed ACSF would move the HEK-293 cells. Vesicles were defined as stably anchored if they became trapped at a given site for at least 5 min and through the end of imaging. This duration is larger than the values obtained for the mean + 2 SD during analysis of typical pause duration in the absence of HEK-293 cells. Pause sites were identified while blind to the location of stable clusters at the end of the movie. Conversely, stable clusters were identified without knowledge of the location of pause sites. Pause sites were defined as the site ± the approximate diffraction limited width of the pausing STV (typically no more than 1.5 μm). Based on this and the average distance between pause sites (∼4 μm) in the regions eventually contacted by HEK-293 cells, the likelihood that a site of stable accumulation would randomly appear to be a pause site was estimated to be 38%. It is likely that most of the pause sites were identified during imaging because multiple pauses occurred at nearly half of the sites. For clarity, only novel accumulations that became stabilized after contact with neuroligin were included in the analysis. Often, adhesion to a neuroligin-expressing HEK-293 cell induced a change in the morphology of the axon or caused the axon to move. In these cases, the axons were omitted from the analysis.
Analysis and statistics.
Image manipulations were restricted to Gaussian filtering and linear scaling of intensities. Colocalization was determined by analyzing line scans along the axon length for peaks that lay above the mean + SD intensity for that line or by identifying puncta on images corresponding to each fluorochrome while blind to the other signals and then determining where the selections coincided. The probability that sec6 randomly colocalizes with a vesicle pausing at random locations was determined by assuming that, if sec6 is distributed at ∼3 μm intervals along the axon, a vesicle pausing every 5 μm would fall in the same 1 μm bin as a sec6 punctum every third pause, yielding a predicted random colocalization of ∼33%. This calculation, based on the reported distribution of sec6 in hippocampal neurons (Hazuka et al., 1999), predicts an upper limit for random colocalization of sec6 and paused STVs because, in the cortical neurons studied here, sec6 puncta did not typically appear as densely spaced as reported by Hazuka and colleagues.
STV movements were tracked in Image Pro (Media Cybernetics). To restrict the quantification of time-lapse movies to healthy neurons, axons were included in the analysis only if at least 1 vesicle moved within the field of view. Regions of axon with an intermediate STV density were imaged to allow us to track STVs reliably. During long imaging periods or when manipulations were made to the axons during imaging, axons sometimes appeared to shift laterally in the field of view as a result of factors such as small movements of the microscope stage. Therefore, when localizing STV pause sites for comparison to the locations of stabilized STVs and stabilized filopodia or retrospective immunocytochemistry labels, the accurate registration of the images was ensured by aligning the frames of each movie and image based on landmarks (e.g., axon branches and bends or stable STVs) before analysis.
For quantification of pausing, rate data were imported into Matlab (Mathworks, Natick, MA), in which additional analyses were performed. Given the pixel density and average vesicle size, a vesicle was typically imaged in ∼8–30 ms, permitting reliable tracking of STVs based on their apparent size, shape, and intensity, with relatively low influence of vesicle movements on these parameters. The fastest STV movements, in the range of 1–2 μm/s [comparable with the fastest velocities that have been reported for STV movements (Ahmari et al., 2000; Kaether et al., 2000)], resulted in movements of ∼15 μm during the imaging interval and were easily measured. Given the length of axon imaged and the typical 15 s imaging interval, the fastest movements that could be tracked were ∼4 μm/s. If vesicles were moving faster than this velocity, they would be impossible to track; however, there is little evidence in the literature for such high velocities. In fact, the velocities reported for KIF1a (kinesin superfamily member 1a), the fastest motor that is thought to transport SV proteins, are from 0.5 to 1.2 μm/s (Okada et al., 1995; Zhou et al., 2001; Lee et al., 2003; Vale, 2003). Finally, although there are likely to be small, dimly fluorescent vesicles that could not be followed in this study, the imaging parameters used here were comparable with those used to demonstrate that STVs are recruited to sites of new synapse formation and incorporated into developing synapses (Ahmari et al., 2000). Therefore, it is likely that a significant population of the vesicles that are incorporated into nascent synapses were reliably tracked.
Vesicles that never moved were not included in the analysis of pausing. A pause was defined as a period greater than at least 20 s, during which the velocity of the vesicle went to 0 ± 0.1 μm/s. The cutoff zero velocity (0.1 μm/s) was chosen based on histograms of vesicle velocities and the maximum velocities obtained when tracking a particle that clearly did not move. Ends of images were included in the analysis; therefore, very long duration pauses may be underestimated.
The statistical test used was ANOVA factorial with Fisher's PLSD using StatView (SAS, Cary, NC). For post hoc testing, the α value was 5%. Data were fitted by Gaussian curves in StatView. All data are presented as mean ± SEM. Normalizations are to mean control values.
Results
Contact with neuroligin-expressing cells induces STV deposition at restricted, predefined sites
To begin to address whether presynaptic terminals can form at any site along the axon or only at restricted locations, the recruitment of STVs to sites of de novo presynaptic terminal formation was visualized. Because the formation of presynaptic terminals between neighboring neurons is an infrequent event during the hour in which the neurons are imaged, the formation of bona fide presynaptic terminals was induced on axons of young neurons using a more reliable assay initially described by Scheiffele et al. (2000). In this assay, presynaptic terminals are induced along axons through contact with non-neuronal cells expressing the synaptogenic adhesion molecule neuroligin. Contact between axons and neuroligin-expressing non-neuronal cells is sufficient to induce differentiation of presynaptic terminals that are ultrastructurally and functionally indistinguishable from those found at synapses between axons and dendrites (Scheiffele et al., 2000). Although this approach has been used to induce presynaptic terminal formation (Scheiffele et al., 2000; Graf et al., 2004), time-lapse imaging of the dynamics of STV recruitment to sites of contact with neuroligin-expressing cells has not yet been reported.
In the experiments described here, 3–5 DIV postnatal rodent cortical neurons were first transfected with a construct encoding the SV-associated protein VAMP2 (also known as synaptobrevin) fused to EGFP to label STVs. Consistent with previous reports (Ahmari et al., 2000; Sabo and McAllister, 2003), VAMP2–EGFP was distributed in axons in a pleiomorphic, punctate pattern mirroring endogenous VAMP and appearing in 87 ± 5% of clusters containing endogenous VAMP (n = 14 neurons). It has been shown previously that VAMP2–EGFP is targeted properly to synapses in which SV fusion occurs normally (Miesenbock et al., 1998; Nonet, 1999; Estes et al., 2000; Alsina et al., 2001; Sankaranarayanan and Ryan, 2001) and that VAMP2–EGFP is recruited rapidly to sites of axodendritic contact during synaptogenesis (Ahmari et al., 2000; Washbourne et al., 2002). To initiate presynaptic terminal formation, neuroligin-expressing HEK-293 cells were then dropped onto the neuronal culture, inducing contact between transfected axons and neuroligin-expressing cells. By imaging VAMP2–EGFP continuously before, during, and after contact, it was determined whether STVs accumulated at specific sites or along the entire length of the contact with the neuroligin-expressing cells. When STVs became anchored at sites of contact between an axon and a neuroligin-expressing HEK-293 cell, novel accumulations were typically stabilized at only one or two foci (∼1–1.5 μm in length) rather than along the entire length of the contact region (Fig. 1), which was typically between 5 and 15 μm long. Interestingly, even when stable STV accumulations occurred, many other STVs often passed by contact sites without becoming tethered to them or without noticeably altered behavior. This suggests that STVs comprise a heterogeneous pool with regard to responsiveness to synaptogenic signals.
Figure 1.

Contact with neuroligin-expressing HEK-293 cells preferentially induces accumulation of STVs at pause sites. Time-lapse images of VAMP2–EGFP-labeled STVs (green) in an axon. White arrows indicate the position at which an STV paused during the frames indicated by the red bars to the left of the images, before contact. After the pause site was identified, an HEK-293 cell expressing neuroligin and HcRed (red) was dropped onto the axon. Once contact was made, another STV moved in (yellow arrows) and became stably anchored at the previously identified pause site (white arrows). STVs were not stabilized at sites of contact with the neuroligin-expressing cell that were not previously defined pause sites. The time during imaging that each frame was captured is indicated in seconds. Scale bar, 5 μm.
Given that STVs were stabilized only at restricted sites along the area of contact with the neuroligin-expressing cell, the next question asked was whether the sites at which STVs accumulated were determined by the neuroligin-expressing cell or were intrinsic to the axon. To test this, the behavior of STVs was examined in the eventual contact region. Before contact was made with a neuroligin-expressing cell, STVs moved along the axon in both the anterograde and retrograde directions, often switching between the two. Surprisingly, even before contact, many STVs appeared to pause transiently at the sites at which STVs eventually stably accumulated in response to contact (Fig. 1). An STV was considered to pause if it did not move more than its own diffraction-limited width between consecutive frames. Of nine sites of contact resulting in stable anchoring of STVs, 88.9% were independently identified as sites of previous STV pausing from previous VAMP2–EGFP imaging. This result is substantially higher than that predicted by chance (see Materials and Methods). Thus, although most of the axonal contact area with neuroligin-expressing cells did not contain pause sites, STVs accumulated specifically at the small areas that were predefined STV pause sites. In the one example in which an STV stably accumulated in a location not identified as a pause site, the STV density was low; therefore, it remains possible that, in this case, all pause sites might not have been revealed before contact. Because the density of pause sites was high enough that each of the imaged contacts with the relatively large HEK-293 cells contained at least one pause site and most contacts with neuroligin-transfected cells did not lead to stable accumulation of STVs, it was not possible to determine whether neuroligin-expressing cells lead to stable accumulation of STVs when contacting regions of the axon that do not contain pause sites. In summary, these data strongly suggest that presynaptic terminals form selectively at predefined sites intrinsic to axons. They further imply that these preferred sites for SV protein accumulation are sites at which, in the absence of contact, STVs pause.
STVs pause repeatedly at defined sites along naive axons
Because STV pause sites were specialized sites for presynaptic terminal formation, STV pausing at these sites was characterized next in axons of young cortical neurons before substantial contact with other neurons. Although saltatory movements of STVs have been anecdotally reported (Kraszewski et al., 1995; Dai and Peng, 1996; Nakata et al., 1998; Ahmari et al., 2000; Kaether et al., 2000), pausing behavior has been essentially unexamined. To study pausing, STV dynamics were examined using time-lapse confocal microscopy of VAMP2–EGFP in young axons (3–5 DIV), when synapses were just beginning to form and dendritic contacts remained fairly sparse. Similar to previous reports (Ahmari et al., 2000), VAMP2–EGFP clusters moved rapidly along axons in both anterograde and retrograde directions, frequently switching between the two. Nearly all VAMP2-containing vesicles that moved also paused (Fig. 2). Importantly, STVs paused repeatedly at the same sites rather than at random locations (Fig. 2 A–C). In some cases, a site was visited multiple times by one STV (Fig. 2 A,B). In other cases, multiple STVs paused at the same site (Fig. 2 C). Because pauses occurred during transport via both anterograde and retrograde motors, pausing did not appear to be a property of a particular motor. This is the first demonstration to our knowledge that STVs pause at stable, defined sites along the axon in the absence of contact with a neighboring neuron and before most synapses have formed.
Figure 2.

As STVs are transported along immature axons, they pause repeatedly at the same sites. A–C, Time-lapse images of neurons transfected with VAMP2–EGFP showing that STVs paused frequently with varying durations (A, B) and repeatedly at the same site (B, C). Red arrows in A and B indicate the position of the same STV in each frame. Green arrows to the left in B indicate a site at which the same STV paused multiple times. Yellow arrows in C indicate a site at which multiple STVs paused. Red bars to the left of each set of images indicate the frames during which the indicated STVs were paused. The time during imaging that each frame was captured is indicated in seconds. Scale bars, 10 μm. D, Quantification of pause behavior in 3–5 DIV neurons. Histograms of pause frequency (n = 47 vesicles), pause duration (n = 185 pauses), and pause probability, a measure of the likelihood an STV is paused at any point in time (n = 47 vesicles). Means are indicated by yellow arrows in D. The data were essentially normally distributed, as shown by the Gaussian fits overlaid on each histogram.
Next, various aspects of pausing were quantified that might be relevant to STV recruitment during synaptogenesis. During 300 s of imaging at 30–35°C, 62 ± 3% of VAMP2–EGFP puncta were mobile (n = 7 axons) (Table 1). Only mobile STVs were included in the remainder of the analysis. Pause behavior was quantified using three measures: pause frequency, duration, and probability. The mean pause frequency, calculated as the average number of pause events per STV per second, was 0.008 ± 0.001 Hz (n = 47 vesicles) (Fig. 2 D, Table 1). The mean pause duration, a measure of how long an STV remained at a pause site during a pause event, was 72.9 ± 6.1 s (n = 185 pauses) (Fig. 2 D, Table 1). The shortest pauses that could be observed given the imaging parameters were 20 s. Although it is likely that there are shorter pauses that were missed, these imaging intervals were comparable with those used in previous studies of STV recruitment (see Materials and Methods) and were sufficient to allow reliable identification of the pause sites that contributed to the STV accumulation described above. The mean pause probability, representing the likelihood that any given STV is paused at any point in time, was 0.61 ± 0.03 (Fig. 2 D, Table 1), indicating that, at steady state, ∼61% of mobile STVs (or 38% of all VAMP clusters) were paused.
Table 1.
Temporal characteristics of pausing vary with neuronal maturity
| Measure | Mean ± SE |
|
|---|---|---|
| 3–4 DIV | 10 DIV | |
| Pause probability | 0.61 ± 0.03 | 0.84 ± 0.08 |
| Pause frequency (Hz) | 0.008 ± 0.001 | 0.006 ± 0.001 |
| Pause duration (s) | 72.9 ± 6.1 | 155.7 ± 24.9 |
Movement and pause parameters are compared for STVs transported along axons of neurons cultured for 3–4 DIV, when synapses are only beginning to form, and for 10 DIV, when many synapses have already formed. Imaging was performed at 30–35°C.
As neurons mature and the number of axodendritic contacts increases, pausing could more frequently lead to stable incorporation of STVs into nascent synapses. This should result in altered pause behavior. To test whether pause characteristics change as neurons mature and synapses form, pausing behavior was also quantified at 10 DIV. At 10 DIV, ∼65% of SV protein clusters were found at synapses, while new synaptic connections were still forming. In these more mature axons, there were no differences in apparent sizes or shapes of mobile VAMP clusters when compared with those observed in 3–5 DIV axons, suggesting that STVs are composed of similar heterogeneous transport vesicles at both ages. As predicted, the mean pause probability (0.84 ± 0.08; n = 16 axons) and pause duration (155.7 ± 24.9 s; n = 21 pauses) were higher in 10 DIV neurons than in 3–4 DIV neurons (Table 1). Maturation was also accompanied by changes in the pause probability density function: whereas the distribution was Gaussian at 3–4 DIV, the 10 DIV distribution was bimodal (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). A large fraction of STVs in 10 DIV neurons had high pause probabilities, possibly attributable to an increased chance of becoming stably anchored at nascent synaptic sites. Overall, these results are consistent with the hypothesis that pausing of STVs can lead to their stable accumulation at sites that are or will become synapses.
Pausing results in stable accumulation of SV proteins
Because STVs accumulated at predefined sites along the axon to form bona fide presynaptic terminals when induced by neuroligin-expressing cells, the question addressed next was whether STVs could stably accumulate at predefined pause sites in the absence of induced contact. When STVs were imaged for 25–30 min, 28.9% of moving STVs became stabilized at pause sites for at least the last 240 s of imaging (2 SD above the mean pause duration; n = 11 axons) (Fig. 3). STVs frequently paused at sites that had already stably accumulated STVs, and some of these pauses resulted in stable accumulation of STVs at those sites (Fig. 3 B). Further examination revealed an additional, novel mechanism for deposition of synaptic proteins: an STV could stably deposit a portion of its material at pause sites, leaving that material behind, whereas the remainder of the STV continued its movement along the axon (Fig. 3 C). Recently, it has been suggested that PTVs accumulate in a quantal manner to constitute a single synapse (Shapira et al., 2003); however, it was not known whether the same is true for STVs. Although the heterogeneity of STV sizes is inconsistent with the kind of quantal accumulation described for PTVs (Shapira et al., 2003), aggregation of multiple STVs to contribute material to a new presynaptic terminal could be observed in the present experiments. Thus, the data suggest a cumulative, but not quantal, model in which multiple STVs deposit all or part of their contents to contribute to stable accumulation at a given pause site.
Figure 3.

Pausing provides at least three novel mechanisms for depositing and accumulating synaptic material at specialized sites in axons. A, De novo stable anchoring: an entire STV (white arrows) becomes stably anchored at a pause site that had not previously accumulated STV proteins. B, Stable anchoring at existing accumulations: STVs often paused at sites that had already accumulated some presynaptic material. Here, an STV (white arrows) becomes stably incorporated into one of those sites (white circle). C, Partial deposition: after pausing, an STV (red arrows) leaves a portion of its material at the pause site (yellow arrows). Red bars next to each set of images indicate the frames during which the indicated STVs were paused. Scale bars, 10 μm.
Pausing occurs independent of adhesion to glia and dendrites
In the brain, neurons physically interact with both glia and other neurons. Because recent reports suggest that adhesion to non-neuronal cells can determine where presynaptic terminals form (Shen and Bargmann, 2003; Shen et al., 2004) and transient synapses can form between neurons and glia (Vaughn, 1989), it seemed possible that contact with other cells might be necessary for establishing pause sites. The above experiments were performed using neurons cultured on top of a monolayer of astrocytes; therefore, it was important to determine whether pause sites were induced by the interaction of axons with glia. To test this, neurons were grown in physical isolation from glia, on poly-l-lysine-coated glass coverslips. When these neurons were transfected with VAMP2–EGFP or loaded with FM4-64 dye, which labels cycling STVs, STVs still paused. The observed pausing was not qualitatively different from pausing observed in neurons grown in contact with glia (Fig. 4).
Figure 4.

Pause sites are formed within axons, independent of contact with glia and dendrites of neighboring neurons. Neurons (4 DIV) grown in the absence of glial contact and transfected with VAMP2–EGFP were imaged. A, Low-magnification image showing the axonal arbor of an individual transfected neuron. B, Transmitted light image of the same field. The axon is free of contact with either glia or dendrites. C, Time-lapse images of VAMP2–EGFP-labeled STVs from the regions of the axon outlined in A and B. Yellow and white arrows illustrate the movement and pausing of an STV from the yellow and white boxes, respectively. Red bars to the left of each set of images indicate the frames during which an STV is paused. The STV indicated by the yellow arrow was stabilized for the remainder of imaging (at least 225 s). Scale bar, 10 μm.
Another possibility is that adhesion of axons to dendrites could have induced pause sites. Axons and dendrites in young cortical cultures can be distinguished using morphological criteria (Sabo et al., 2003). Low plating densities also allow the identification of long stretches of axon that are physically isolated from dendritic contact in 3–4 DIV cultures. When naive axons that were free of dendritic contact were imaged, STVs still paused in a qualitatively similar manner to those in neurons grown on glial monolayers (Fig. 4). Recently, Krueger et al. (2003) demonstrated that functional presynaptic terminals can exist in the absence of dendritic contact; however, it could not be determined from their study whether dendritic contact is required for de novo accumulation of synaptic proteins. As illustrated in Figure 4 C (left), STVs not only paused in the absence of dendritic contact, but paused STVs could also become stably anchored at sites that were free of dendritic contact. These data indicate that pause sites can be formed independent of the interaction of axons with other cells.
Signals involved in synaptogenesis regulate STV pausing
Although axonal contact with their targets or neighboring glia is not required for the formation of pause sites, axon–target interactions would be expected to regulate STV behavior at those sites if they are indeed sites at which synapses will form. Current models for synapse formation propose that initial axodendritic contact initiates a cascade of intracellular events within the axon that alters the velocity or direction of STV transport, leading to the active recruitment of STVs to the site of contact. However, the observation that presynaptic terminals form selectively at predefined pause sites, and not at non-pause sites, suggests an equally plausible alternative model in which STV pausing could be regulated to allow for STV accumulation at nascent synapses. In this alternate model, signals that alter STV pausing could increase the likelihood that STVs become stabilized at the right place at the right time. If this model is correct, then signals that are known to regulate synaptogenesis should also regulate pausing. To test this hypothesis, the effects of the following manipulations on STV transport and pausing were investigated: (1) a second messenger, calcium, (2) a cytoskeletal component, actin, and (3) a synaptic vesicle-associated protein, synapsin.
Because calcium is an important component of many signal transduction cascades, axonal calcium transients are induced by axodendritic contact (Funte and Haydon, 1993; Dai and Peng, 1998), and calcium influx regulates trafficking of SVs at mature synapses (Murthy and De Camilli, 2003), experiments were performed to test whether calcium regulates STV pausing. When neurons imaged in calcium-free or normal ACSF were compared (Fig. 5 A), depletion of extracellular calcium decreased the pause probability to 56.7 ± 3.4% of control (p < 0.0001; n = 194 vesicles). This appeared to be the result of a decrease in pause duration in the absence of calcium to 50.3 ± 2.5% of control (p < 0.0001; n = 543 pauses) without a significant change in pause frequency (p = 0.0721; n = 194 vesicles). The fraction of vesicles that were mobile (p = 0.676; n = 7 axons) was unchanged by depletion of calcium. Thus, extracellular calcium regulates the amount of time STVs spend paused at a given site.
Figure 5.
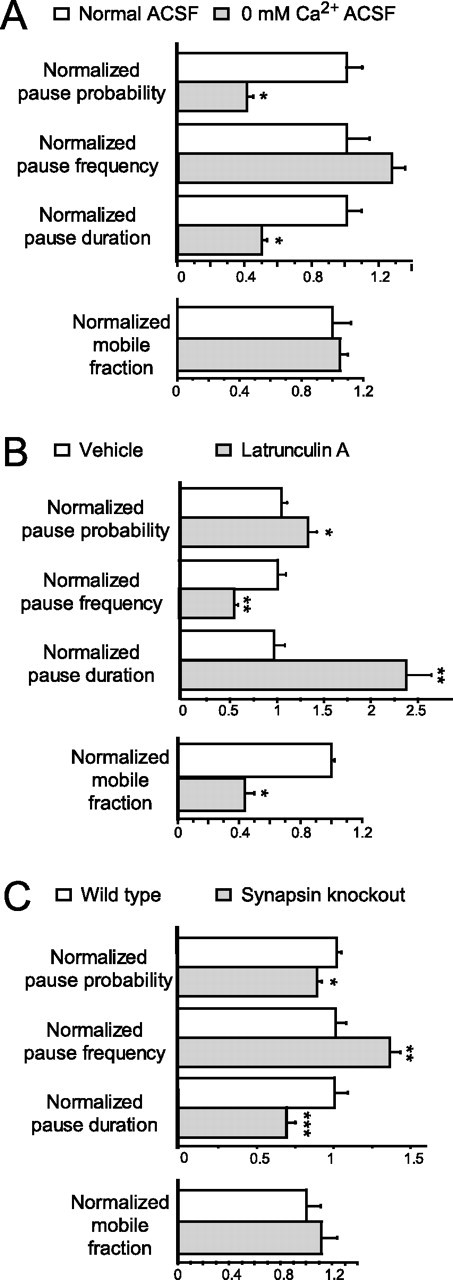
Synapsins, actin, and calcium differentially regulate STV pausing. A–C, Plots of the mean ± SE of pause probability, pause frequency, pause duration, and the fraction of STVs that were mobile with (white bars) and without (gray bars) extracellular calcium (*p < 0.001; n = 194 vesicles, 543 pauses) (A), with (gray bars) and without (white bars) depolymerization of actin filaments with latrunculin A (*p = 0.0148; **p < 0.0001; n = 43 vesicles, 108 pauses) (B), and in wild-type neurons (white bars) and neurons derived from mice with all three synapsin genes knocked out (gray bars) (*p = 0.010; **p = 0.001; ***p = 0.002; n = 126 vesicles, 653 pauses) (C). All data were normalized to mean values derived from the appropriate control for each manipulation.
The changes in STV pausing after elimination of extracellular calcium could be attributable to a lack of calcium influx into the cell through calcium-permeable channels at the surface or from inhibition of calcium-dependent interactions on the extracellular face of the neuron. If the effects seen were attributable to altered calcium influx through channels, buffering of intracellular calcium should yield similar results. To test whether this is the case, VAMP2–EGFP-transfected neurons were loaded with BAPTA-AM, a calcium chelator that crosses the plasma membrane and then becomes trapped inside the cell when esterases cleave off the AM group. Imaging of STV movements revealed that BAPTA-dependent buffering of intracellular calcium yielded similar effects on STV pausing (supplemental Fig. 2, available at www.jneurosci.org as supplemental material): pause probability and duration were decreased to 69.4 ± 14.5% (p = 0.0305; n = 24 vesicles) and 48.3 ± 4.5% (p < 0.001; n = 82 pauses) of control values, respectively, whereas pause frequency remained unchanged (p = 0.5375; n = 24). Together, the data suggest that calcium influx regulates STV pausing.
Next, it was determined whether actin plays a role in STV pausing. Vesicles at mature synapses appear to be tethered to actin (Doussau and Augustine, 2000), and it has been proposed that interaction of vesicles with actin-based motors removes them from their microtubule tracks (Langford, 2002). In addition, actin is important for establishment of cell-matrix adhesion sites, which could define pause sites. Actin also concentrates in developing presynaptic terminals (Dai and Peng, 1996; Zhang and Benson, 2002) and is essential for synaptic development (Zhang and Benson, 2001). Finally, association with or rearrangement of the actin cytoskeleton is predicted to be downstream of the engagement of axonal and dendritic adhesion molecules (Scheiffele, 2003; Waites et al., 2005). To test the role of actin filaments in STV transport, movements were quantified in axons treated with latrunculin A, which sequesters actin monomers and disrupts the actin network (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). STVs appeared normally spaced and sized and still paused in the absence of actin filaments; however, STV behavior was quantitatively altered (Fig. 5 B). Surprisingly, actin disruption increased the pause duration by 2.39 ± 0.26-fold (n = 108 pauses; p < 0.0001), suggesting that actin might be important for resuming movement after pausing rather than for initiating pauses. Consistent with this idea, only 26.2 ± 8.7% of STVs were mobile after latrunculin A treatment (n = 10 axons). Concomitant with the increased pause duration, the pause probability increased to 1.26 ± 0.089-fold of control (n = 43 vesicles; p = 0.0148), and pause frequency was decreased to 55.6 ± 4.0% of control (n = 54; p < 0.0001). These data suggest that actin filaments might help mobilize STVs after pausing.
Finally, the effects of the elimination of synapsins on STV transport and pausing were determined. Synapsins are SV proteins involved in neurotransmitter release and regulation of synapse formation (Hilfiker et al., 1999; Ferreira and Rapoport, 2002). To test whether synapsins regulate STV pausing, STV movements were examined in neurons derived from either wild-type mice or STKO mice, from which all three synapsin genes had been deleted (Fig. 5 C). STVs appeared indistinguishable in wild-type and STKO neurons and moved similarly. The fraction of vesicles that were mobile was not significantly different (p = 0.563; n = 12 axons). In contrast, when STV pause behavior was compared in wild-type and STKO axons, the mean pause duration was significantly decreased in STKO neurons (to 69.6 ± 5.0% of control; n = 296 pauses for wild type and 357 for STKO; p = 0.002). This ∼30% decrease in pause duration was offset by a 1.33-fold increase in pause frequency (±0.071; n = 53 and 73 vesicles; p = 0.001), yielding a small change in the pause probability (87.9 ± 3.0% of control; p = 0.010; n = 53 and 99 vesicles). The observed shortening of pauses in the absence of synapsins suggests that synapsins are important for keeping STVs at pause sites; these data are consistent with a role for synapsins in tethering STVs to pause sites, similar to their proposed role in tethering SVs to the cytoskeleton in mature synapses.
Although each of these manipulations of molecules that regulate synaptogenesis altered specific aspects of pausing behavior, none of these manipulations altered the average instantaneous velocity of STV transport. STVs moved at a mean instantaneous velocity of 0.29 ± 0.02 μm/s in untreated neurons, with equal instantaneous velocities in both anterograde and retrograde directions [0.29 ± 0.02 μm/s for anterograde (n = 277) and 0.28 ± 0.02 μm/s for retrograde (n = 211) movements, respectively; p = 0.837]. The average instantaneous velocities of STVs were unchanged in the absence of calcium (p = 0.559; n = 923 movements), after depolymerization of actin (p = 0.069; n = 69 movements), or in the absence of all three synapsins (p = 0.266; n = 1418 movements). In addition, after each of these manipulations, STVs continued to move in both directions, with no apparent change in directional bias (data not shown). Although the existence of subtle changes in movement that are beyond resolution cannot be ruled out, the results presented indicate that pausing behavior is the aspect of STV transport that is most dramatically regulated by these molecules that influence synapse formation.
Pause sites contain exocytic and endocytic markers
If pause sites are favored sites of synaptogenesis, there should be a signal that encourages presynaptic terminal formation at those sites. Because STVs cycle with the plasma membrane (Matteoli et al., 1992; Kraszewski et al., 1995; Dai and Peng, 1998), vesicle fusion at pause sites could lead to release of diffusible factors that encourage contacts to form or stabilize at pause sites. First, the presence of STV cycling in immature cortical axons in the absence of contact with dendrites was confirmed. FM4-64, a lipophilic dye that cannot cross the lipid bilayer, has been used extensively to label cycling SVs at mature synapses (Cochilla et al., 1999). When immature neurons were briefly depolarized in the presence of FM4-64, STVs were labeled throughout the axons, including long stretches that were free of dendritic contact (Fig. 6 A). FM4-64-labeled STVs appeared similar to those labeled with VAMP2–EGFP. Importantly, when neurons transfected with VAMP2–EGFP were depolarized in the presence of FM4-64, VAMP-labeled STVs could be loaded with FM4-64 dye (data not shown). Similar to VAMP2–EGFP, FM-labeled vesicles moved both anterogradely and retrogradely and paused at defined sites along the axon (Fig. 6 A). Although the age and origin of these cycling vesicles is unknown, the above data combined with the young age of the cultures strongly suggest that the FM-labeled vesicles were STVs rather than mature terminals. Their apparent heterogeneity is similar to that of VAMP2–EGFP-labeled STVs, suggesting that these cycling vesicles might also range morphologically from relatively immature forms, such as the tubulovesiclular organelles described by Ahmari et al. (2000), to clusters of bona fide SVs, as described by Kraszewski et al. (1995). Labeled STVs appear to undergo some spontaneous fusion with the plasma membrane because a significant amount of FM dye is lost from vesicles during imaging (p = 0.0145; data not shown). This loss cannot be attributed solely to photobleaching or focal drift because neighboring background fluorescence did not change during the same imaging periods.
Figure 6.

During transport, STVs cycle with the plasma membrane at pause sites. A, Time-lapse images of STVs in 4 DIV neurons labeled with FM4-64 by high-K+ depolarization. Arrows, STVs that resumed movement after loading. Each color arrow tracks a separate STV. B, Localization of rSec6, a component of the exocyst complex, at both stable (white arrows) and transient (red arrows) pause sites by live imaging of STV movement (top 5 panels), followed by retrospective immunocytochemistry for rSec6 (yellow arrows, bottom panel). Red bars indicate the frames during which the indicated STV (red arrows) was paused. C, Localization of clathrin–EGFP (green) at pause sites, identified by live imaging of VAMP2–DsRed (red) movement. Plots show line intensity profiles along the axon during movement and pausing. Images at the top correspond to the three frames during the pause (red bars) in the bottom three line scans. Clathrin can associate with (yellow arrows, middle panel and penultimate line scan) and dissociate from (right panel and last line scan) pause sites. D, VAMP2 is highly colocalized with vGlut1, the vesicular glutamate transporter, in young neurons as shown by immunocytochemistry. Colocalization is indicated by yellow in the overlay (bottom panel). Scale bars, 10 μm.
If cycling occurs at pause sites, then proteins involved in exocytosis and endocytosis should localize to pause sites. In contrast to cycling at mature synapses (Andrews et al., 2002; Murthy et al., 2003), vesicle targeting and fusion in young axons has been proposed to depend on components of the exocyst complex (Hazuka et al., 1999). Therefore, pause sites were identified by time-lapse imaging of VAMP2–EGFP and then the same neurons were immediately fixed and immunolabeled for sec6, a component of the exocyst (Fig. 6 B). A total of 69.2% of pause sites contained sec6 (n = 13), a substantially higher percentage than is predicted for a random localization of pausing and sec6 puncta (for details, see Materials and Methods). sec6 was found at pause sites regardless of whether STVs remained at those sites, suggesting that sec6 associates with the plasma membrane at pause sites rather than with STVs themselves. To determine whether endocytosis occurs during pausing, pause sites were tested for the presence of the endocytic coat protein clathrin. Because clathrin transiently associates with endocytosing vesicles, an approach was used that permitted observation of both clathrin and STV dynamics: neurons were cotransfected with VAMP2–DsRed and clathrin–EGFP and then imaged live (Fig. 6 C). [It has been demonstrated previously that VAMP2–DsRed localizes properly in axons and that the dynamics of VAMP2–DsRed-labeled STVs are similar to those labeled by VAMP2–EGFP (Sabo and McAllister, 2003)]. Pause sites and clathrin-containing sites were identified independently. Overlays then showed where clathrin overlapped with pause sites. When pausing did not result in STV stabilization, 92.4 ± 5.5% of pause sites contained clathrin–EGFP at some point (n = 6 axons). For stable STV sites, 89.7 ± 5.4% of sites contained clathrin–EGFP (n = 9 axons). Interestingly, 90.4 ± 2.6% of clathrin sites contained VAMP2–DsRed at some point during imaging. Sometimes clathrin moved with STVs for short distances, presumably when newly endocytosed vesicles moved away from the site of endocytosis before the coat dissociated. Overall, these data demonstrate a strong correlation between sites of exocytosis and endocytosis and sites of STV pausing, supporting the hypothesis that cycling occurs at pause sites.
STVs likely release glutamate at pause sites
It is possible that localized release of glutamate from pause sites encourages filopodial stabilization and synapse formation at those predefined axonal sites because glutamate is a potent regulator of dendritic filopodial motility (Wong and Wong, 2001; Tashiro et al., 2003) and many synaptogenic contacts are initiated by dendritic filopodia (Saito et al., 1992; Dailey and Smith, 1996; Ziv and Smith, 1996; Fiala et al., 1998; Jontes and Smith, 2000; Niell et al., 2004). To determine whether STVs contain neurotransmitter, 4 DIV neurons were immunolabeled for VGlut1 and VAMP. The presence of the vesicular glutamate transporter VGlut1 is a reliable indicator of whether vesicles are loaded with glutamate (Bellocchio et al., 2000; Takamori et al., 2000). VGlut1 colocalized with SV markers even before synapses were formed (Fig. 6 D), indicating the presence of VGlut1 in STVs. In axons of excitatory neurons, 89.1 ± 5.0% of VAMP puncta also contained VGlut1 (n = 256 puncta in 5 axons). Conversely, 90.6 ± 2.2% of VGlut1 puncta contained VAMP2. These results suggest that glutamate could be released from STVs on fusion with the plasma membrane: this glutamate release could occur preferentially at pause sites, potentially influencing the motility of dendritic filopodia selectively in the vicinity of the pause site.
Axodendritic contacts are exclusively stabilized at STV pause sites
If synapses form specifically at pause sites, then contacts made by dendritic filopodia should be selectively stabilized at predefined pause sites on axons of young cortical neurons. To test this, neurons were loaded with FM4-64 to label STVs at 5–7 DIV, an age at which axodendritic contacts are made relatively frequently. Fluorescence and differential interference contrast (DIC) images were captured simultaneously, allowing imaging of both STVs and dendritic filopodia. Because the percentage of axons labeled using FM dye is higher than the percentage transfected with VAMP2–EGFP, use of FM4-64 labeling maximized the likelihood of observing initial contact between dendrites and labeled axons.
From fluorescence images, pause sites were identified before contact between axons and dendritic filopodia. Contact was analyzed using DIC images, while blind to where STVs had paused. Contacts initiated by dendritic filopodia were mostly transient, often lasting only a frame or two. However, when contacts between dendritic filopodia and axons became stabilized (i.e., persisted at least 10 min), they were always stabilized at sites at which STVs had paused previously and never at non-pause sites (n = 6 of 6) (Fig. 7) (supplemental movie, available at www.jneurosci.org as supplemental material). When a dendritic filopodium made its initial contact with the axon at a site other than the site of eventual stabilization, the filopodium could be observed scanning along the axon before stabilizing at the pause site (supplemental movie, available at www.jneurosci.org as supplemental material). Interestingly, STVs could often be observed at pause sites within a few frames surrounding the time of contact (n = 4 of 5 contacts), making it plausible that factors released by paused STVs (such as glutamate) could contribute to stabilization of contacts. In some cases, anchoring of STVs at contact sites was clearly observed (n = 2) (Fig. 7 A), but loss of fluorescence attributable to photobleaching and exocytosis of FM4-64 dye prevented reliable determination of whether STVs always accumulated at stable contacts. Because synapse formation is an unsynchronized and protracted process, occurring over a period of several weeks, and contacts are made and stabilized relatively infrequently over the time an individual neuron could be imaged, it was not possible to quantify the proportion of STV pause sites that are eventually converted into presynaptic terminals; however, it is clear that dendritic filopodia are selectively stabilized at these predefined sites. Similar contacts initiated by dendritic filopodia on axon shafts have been shown previously to reliably result in the formation of nascent synapses (Ahmari et al., 2000). Thus, the selective stabilization of dendritic filopodial contacts at STV pause sites strongly suggests that en passant presynaptic terminals form specifically at predefined pause sites during the initial stages of synapse formation between young neurons.
Figure 7.

Dendritic filopodial contacts are selectively stabilized at previously defined STV pause sites. A, B, Time-lapse transmitted light images of initiation of contact between axons and dendritic growth cones and fluorescence images showing STV pauses. The time during imaging of each frame is indicated in seconds. A, The left column of images shows a dendritic growth cone (located on the far right sides of the images) making contact with an axon (located on the left sides of the images). The tip of the filopodium that made the stable contact is indicated by green arrows. The series of images in the right column are a higher magnification of the contact region (black dashed box) and are overlaid with fluorescence images of STVs in the axon, as visualized by loading with FM4-64 dye (red). In the frames indicated by the red bars, an STV was paused at the site indicated by the white arrows. Yellow arrows track the movement of the STV. The dendritic growth cone filopodium (dashed white line) stabilized at the previously defined pause site (white arrows). In this example, an FM4-64-labeled STV accumulated at the site of stabilization of the filopodium. Scale bars: left, 10 μm; right, 5 μm. B, STVs were labeled with FM4-64 (white) and are shown in the left panel of each pair, and the transmitted light image from the same frame is shown in the right panel of each pair. The region shown in the fluorescence panels corresponds to the region indicated (first panel, black dashed box). Before contact, an STV (yellow arrows) paused at the site (red arrows) at which the stable contact was subsequently made. Red bars indicate the frames during which an STV was paused at the contact site. The white dashed line traces the distal end of the dendritic filopodium that made the stable contact. Scale bar, 10 μm.
Discussion
A new model: formation of presynaptic terminals at predefined sites
Although dendritic filopodia frequently initiate synaptogenic contact (Dailey and Smith, 1996; Ziv and Smith, 1996; Jontes and Smith, 2000), it is unknown what determines where along axons contacts are stabilized and synapses formed. The entire axon could be equally competent for synaptogenesis, and synapse formation could occur wherever dendrites contact axons. Alternatively, synaptogenic interactions could be stabilized only at restricted locations along axons. The data presented here support a version of the latter model, in which de novo synaptogenesis between dendritic filopodia and axon shafts of young neurons occurs preferentially at predefined sites in axons: STV pause sites.
If this model is correct, contacts should stabilize and STVs accumulate preferentially at those predefined sites. Consistent with this, when presynaptic terminal formation was induced by contact with neuroligin-expressing cells, STVs specifically aggregated at pause sites but not along the rest of the axon that contacted neuroligin-expressing cells. Furthermore, contacts initiated by dendritic filopodia were stabilized at pause sites but never at sites that were not pause sites.
Although it is unclear what percentage of stabilized contacts eventually became functional synapses, it is likely that synapses form at the majority of stabile axodendritic contacts for a number of reasons. First, when FM-labeled STVs remained visible throughout imaging, STVs stably accumulated opposite stabilized dendritic filopodia. Second, synapses in young neurons are often found associated with dendritic filopodia (Fiala et al., 1998). Third, stabilized axodendritic contact results in recruitment of both presynaptic and postsynaptic components and development of functional synapses (Ahmari et al., 2000; Friedman et al., 2000; Okabe et al., 2001; Washbourne et al., 2002). Finally, the density of pause sites correlates well with the density of synapses observed in these neurons at 8–10 DIV (∼0.17–0.19 synapses/μm). Together, these data are consistent with conversion of transient STV pause sites into stable accumulations of presynaptic protein and eventually presynaptic terminals. Notably, recent reports using nonmammalian systems, including studies of zebrafish Mauthner cell (Jontes et al., 2000) and Caenorhabditis elegans HSNL neuron (Shen and Bargmann, 2003) synaptogenesis, have demonstrated the utilization of predefined, reproducible sites for presynaptic terminal formation in vivo.
These predefined sites might be intrinsic to axons because STV pause sites are established before dendritic contact and in the absence of glia. In the absence of glia and dendrites, interactions between axons and their substrate could have induced pause sites; however, pause sites cannot be merely an artifact of adhesion to poly-l-lysine because they were present when neurons were grown on glia and not in contact with poly-l-lysine. To determine whether pause sites are intrinsic to axons, it will be important to define which molecules comprise these sites. This information would also permit determination of whether pause sites are unique for STVs or can also be used by other organelles. Cytoskeletal specializations could define pause sites; however, pause sites do not appear to correspond to foci of actin enrichment because disruption of actin filaments did not prevent pausing. Moreover, phalloidin-labeled actin filaments were found at 66.7% of pause sites at which STVs remained paused or anchored, but only 33.3% of pause sites displayed significant phalloidin labeling when STVs were no longer at the site (data not shown; n = 9 pause sites). Whichever proteins or lipids comprise these sites, pause sites appear to be used in a remarkable way to determine where presynaptic terminals initially form.
A recent report showed that functional presynaptic terminals not associated with dendrites (orphans) are present in relatively mature axons (Krueger et al., 2003). Because orphan terminals derive from mobilization of all or part of existing synapses that had active zones and postsynaptic densities, it remained unclear whether de novo accumulation of presynaptic proteins depends on axodendritic interactions. In the present study, STV accumulation was observed in the absence of dendritic contact even in young neurons that had relatively few existing synapses; therefore, the high number of STVs observed could not have come from those synapses and likely represent bona fide de novo accumulations.
Because contacts are stabilized specifically at STV pause sites, there should be a mechanism to encourage dendritic contact at those sites. Localized release of diffusible signals could influence the likelihood of formation or stabilization of contacts at pause sites. In support of this hypothesis and consistent with previous reports (Matteoli et al., 1992; Kraszewski et al., 1995; Dai and Peng, 1996), STVs must cycle with the axonal surface because they can be loaded with and release FM dye. Moreover, because STVs contain VGlut1, glutamate may be released when STVs fuse. Although the role of synaptic activity in synaptogenesis has been questioned (Rao and Craig, 1997; Verhage et al., 2000; Varoqueaux et al., 2002; Bouwman et al., 2004; Harms and Craig, 2005), glutamate potently regulates filopodial motility (Zheng et al., 1996; Chang and De Camilli, 2001; Wong and Wong, 2001; Tashiro et al., 2003). Thus, glutamate release from STVs could influence the likelihood and location of formation and stabilization of early synaptic contacts.
Although the initial complement of en passant synapses appears to form at STV pause sites in young neurons, this mechanism cannot completely account for the high density of synapses in the adult brain. Synaptogenesis can also be initiated by axonal growth cones (Ahmari et al., 2000; Washbourne et al., 2002), which may use separate mechanisms. In addition, presynaptic proteins might be recruited to sites of preexisting clusters of postsynaptic scaffolding protein complexes (Gerrow et al., 2006); however, it is unclear whether these sites of postsynaptic protein accumulation occurred at contacts with predefined axonal STV pause sites. Finally, at later stages of synaptogenesis (at approximately 16 DIV as opposed to the 4–7 DIV stage studied here), new presynaptic boutons can be formed as a result of an existing functional bouton splitting into multiple new terminals (Krueger et al., 2003). Obviously splitting of existing terminals cannot account for formation of the first set of terminals from which the next wave derives. Therefore, presynaptic terminal formation on axon shafts may occur in at least two phases: an early “accumulation phase” described here, in which many synapses are formed at pause sites, and a later “splitting phase” characterized by Krueger et al. (2003), in which synapse density is increased by division of existing terminals.
How are STVs stabilized at sites of axodendritic contact?
How synaptogenic signals recruit synaptic proteins to new synapses is unknown. Axodendritic contact is thought to initiate a cascade of intracellular events within the axon that alters STV transport, recruiting STVs to the contact. Because accumulation of synaptic proteins at sites of synapse formation requires moving precursors to stop and presynaptic terminals appear to form preferentially at STV pause sites, regulation of pausing may be the primary mechanism by which synaptogenic signals induce stable accumulation of STVs at nascent presynaptic terminals.
In support of this hypothesis, calcium, synapsins, and actin (all signals that regulate synapse formation) potently regulate STV pausing but do not appear to affect STV velocity or directionality of transport. None of the tested manipulations eliminated pausing; therefore, these molecules appear to serve regulatory roles downstream of the initial capturing of STVs. Because each manipulation affected the anchoring of STVs to pause sites, synaptogenic cues might alter intracellular signals, such as calcium influx, synapsin phosphorylation, or actin dynamics, to permanently tether paused STVs at sites of presynaptic terminal formation. For example, calcium influx after axodendritic contact (Funte and Haydon, 1993; Dai and Peng, 1998) could increase the likelihood that STVs become stably anchored at contact sites by increasing how long vesicles remain paused. Similarly, modification of synapsins may increase tethering of STVs to pause sites, analogous to their proposed role at mature synapses (Hilfiker et al., 1999). Finally, synaptogenic signals could locally limit growth of actin filaments or inhibit myosin activity, trapping STVs at pause sites.
It will be important to determine the mechanisms by which these molecules regulate STV pausing in future experiments. Delineating the function of actin at pause sites will be especially interesting. Actin is thought to act as a scaffold at adhesion sites and mature nerve terminals (Allison et al., 2000; Doussau and Augustine, 2000; Halpain, 2003; Sankaranarayanan et al., 2003), and actin-dependent myosin motors can remove vesicles from microtubules (Langford, 2002); therefore, one would have predicted that actin filaments would either tether STVs to pause sites or cause a pause by inducing removal of STVs from microtubules. Instead, inhibiting actin polymerization doubled pause duration and increased pause probability, suggesting that actin polymerization or actin-dependent motors drive small, local movements that transfer STVs from pause site anchors to microtubules, in which long-range transport resumes. Consistent with this idea, actin mobilizes SVs within mature nerve terminals (Cole et al., 2000; Shupliakov et al., 2002). Actin does not appear to regulate STV pausing through binding to synapsins because the effects of actin disruption and elimination of synapsins were opposite one another. In summary, the results presented here suggest that regulation of STV pausing is an important aspect of STV trafficking that is modulated by molecules that influence early stages of synaptogenesis.
What are the implications of presynaptic terminal formation at predefined sites?
What broader physiological role might the formation of presynaptic terminals at predefined sites play in the development of the CNS? In vivo imaging in zebrafish demonstrated that synaptogenesis stabilizes dendritic filopodia, which then mature into dendritic branches (Niell et al., 2004). If developing synapses serve a similar function in the mammalian cortex, synaptogenesis at distributed predefined pause sites could guide the initial stages of dendritic growth, yielding the extended, sparse dendritic arbors that characterize young cortical neurons. Subsequent synapse formation from splitting of preexisting terminals (Krueger et al., 2003) might then guide the filling in and extensive higher-order branching that occurs at later stages of dendritic growth. In the future, it will be important to investigate whether and how STV pause sites in naive axons define the mechanistic relationship between synaptogenesis and dendritic arborization.
Footnotes
This work was supported by the Alfred P. Sloan Foundation (A.K.M.), the PEW Charitable Trusts (A.K.M.), the March of Dimes (A.K.M.), National Eye Institute (A.K.M.), National Institute of Neurological Disorders and Stroke (S.L.S.), National Institute of Mental Health (S.L.S.), and Fundacao para a Ciencia e Tecnologia (R.A.G.). R.A.G. is a scholar from Programa Gulbenkian de Doutoramento em Biomedicina. We thank E. Diaz and P. Greengard for helpful reading of this manuscript. We thank Faten El-Sabeawy, Jennie Bennett, and Jeremy Lathrop for technical assistance. We also thank H. T. Kao and P. Greengard for the synapsin triple knock-out and control mice, R. Scheller for VAMP2–EGFP, P. Scheiffele for HA–neuroligin, and T. Kirchhausen for clathrin–EGFP fusion constructs.
References
- Ahmari SE, Buchanan J, Smith SJ. Assembly of presynaptic active zones from cytoplasmic transport packets. Nat Neurosci. 2000;3:445–451. doi: 10.1038/74814. [DOI] [PubMed] [Google Scholar]
- Allison DW, Chervin AS, Gelfand VI, Craig AM. Postsynaptic scaffolds of excitatory and inhibitory synapses in hippocampal neurons: maintenance of core components independent of actin filaments and microtubules. J Neurosci. 2000;20:4545–4554. doi: 10.1523/JNEUROSCI.20-12-04545.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alsina B, Vu T, Cohen-Cory S. Visualizing synapse formation in arborizing optic axons in vivo: dynamics and modulation by BDNF. Nat Neurosci. 2001;4:1093–1101. doi: 10.1038/nn735. [DOI] [PubMed] [Google Scholar]
- Andrews HK, Zhang YQ, Trotta N, Broadie K. Drosophila sec10 is required for hormone secretion but not general exocytosis or neurotransmission. Traffic. 2002;3:906–921. doi: 10.1034/j.1600-0854.2002.31206.x. [DOI] [PubMed] [Google Scholar]
- Bellocchio EE, Reimer RJ, Fremeau RT, Jr, Edwards RH. Uptake of glutamate into synaptic vesicles by an inorganic phosphate transporter. Science. 2000;289:957–960. doi: 10.1126/science.289.5481.957. [DOI] [PubMed] [Google Scholar]
- Bouwman J, Maia AS, Camoletto PG, Posthuma G, Roubos EW, Oorschot VM, Klumperman J, Verhage M. Quantification of synapse formation and maintenance in vivo in the absence of synaptic release. Neuroscience. 2004;126:115–126. doi: 10.1016/j.neuroscience.2004.03.027. [DOI] [PubMed] [Google Scholar]
- Chang S, De Camilli P. Glutamate regulates actin-based motility in axonal filopodia. Nat Neurosci. 2001;4:787–793. doi: 10.1038/90489. [DOI] [PubMed] [Google Scholar]
- Cochilla AJ, Angleson JK, Betz WJ. Monitoring secretory membrane with FM1-43 fluorescence. Annu Rev Neurosci. 1999;22:1–10. doi: 10.1146/annurev.neuro.22.1.1. [DOI] [PubMed] [Google Scholar]
- Cole JC, Villa BR, Wilkinson RS. Disruption of actin impedes transmitter release in snake motor terminals. J Physiol (Lond) 2000;525:579–586. doi: 10.1111/j.1469-7793.2000.t01-2-00579.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai Z, Peng HB. Dynamics of synaptic vesicles in cultured spinal cord neurons in relationship to synaptogenesis. Mol Cell Neurosci. 1996;7:443–452. doi: 10.1006/mcne.1996.0032. [DOI] [PubMed] [Google Scholar]
- Dai Z, Peng HB. Fluorescence microscopy of calcium and synaptic vesicle dynamics during synapse formation in tissue culture. Histochem J. 1998;30:189–196. doi: 10.1023/a:1003247403685. [DOI] [PubMed] [Google Scholar]
- Dailey ME, Smith SJ. The dynamics of dendritic structure in developing hippocampal slices. J Neurosci. 1996;16:2983–2994. doi: 10.1523/JNEUROSCI.16-09-02983.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doussau F, Augustine GJ. The actin cytoskeleton and neurotransmitter release: an overview. Biochimie. 2000;82:353–363. doi: 10.1016/s0300-9084(00)00217-0. [DOI] [PubMed] [Google Scholar]
- Estes PS, Ho GL, Narayanan R, Ramaswami M. Synaptic localization and restricted diffusion of a Drosophila neuronal synaptobrevin–green fluorescent protein chimera in vivo. J Neurogenet. 2000;13:233–255. doi: 10.3109/01677060009084496. [DOI] [PubMed] [Google Scholar]
- Ferreira A, Rapoport M. The synapsins: beyond the regulation of neurotransmitter release. Cell Mol Life Sci. 2002;59:589–595. doi: 10.1007/s00018-002-8451-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiala JC, Feinberg M, Popov V, Harris KM. Synaptogenesis via dendritic filopodia in developing hippocampal area CA1. J Neurosci. 1998;18:8900–8911. doi: 10.1523/JNEUROSCI.18-21-08900.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman HV, Bresler T, Garner CC, Ziv NE. Assembly of new individual excitatory synapses: time course and temporal order of synaptic molecule recruitment. Neuron. 2000;27:57–69. doi: 10.1016/s0896-6273(00)00009-x. [DOI] [PubMed] [Google Scholar]
- Funte LR, Haydon PG. Synaptic target contact enhances presynaptic calcium influx by activating cAMP-dependent protein kinase during synaptogenesis. Neuron. 1993;10:1069–1078. doi: 10.1016/0896-6273(93)90055-v. [DOI] [PubMed] [Google Scholar]
- Gerrow K, Romorini S, Nabi SM, Colicos MA, Sala C, El Husseini A. A preformed complex of postsynaptic proteins is involved in excitatory synapse development. Neuron. 2006;49:547–562. doi: 10.1016/j.neuron.2006.01.015. [DOI] [PubMed] [Google Scholar]
- Gitler D, Xu Y, Kao HT, Lin D, Lim S, Feng J, Greengard P, Augustine GJ. Molecular determinants of synapsin targeting to presynaptic terminals. J Neurosci. 2004;24:3711–3720. doi: 10.1523/JNEUROSCI.5225-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graf ER, Zhang X, Jin SX, Linhoff MW, Craig AM. Neurexins induce differentiation of GABA and glutamate postsynaptic specializations via neuroligins. Cell. 2004;119:1013–1026. doi: 10.1016/j.cell.2004.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halpain S. Actin in a supporting role. Nat Neurosci. 2003;6:101–102. doi: 10.1038/nn0203-101. [DOI] [PubMed] [Google Scholar]
- Harms KJ, Craig AM. Synapse composition and organization following chronic activity blockade in cultured hippocampal neurons. J Comp Neurol. 2005;490:72–84. doi: 10.1002/cne.20635. [DOI] [PubMed] [Google Scholar]
- Hazuka CD, Foletti DL, Hsu SC, Kee Y, Hopf FW, Scheller RH. The sec6/8 complex is located at neurite outgrowth and axonal synapse-assembly domains. J Neurosci. 1999;19:1324–1334. doi: 10.1523/JNEUROSCI.19-04-01324.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilfiker S, Pieribone VA, Czernik AJ, Kao HT, Augustine GJ, Greengard P. Synapsins as regulators of neurotransmitter release. Philos Trans R Soc Lond B Biol Sci. 1999;354:269–279. doi: 10.1098/rstb.1999.0378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jontes JD, Smith SJ. Filopodia, spines, and the generation of synaptic diversity. Neuron. 2000;27:11–14. doi: 10.1016/s0896-6273(00)00003-9. [DOI] [PubMed] [Google Scholar]
- Jontes JD, Buchanan J, Smith SJ. Growth cone and dendrite dynamics in zebrafish embryos: early events in synaptogenesis imaged in vivo. Nat Neurosci. 2000;3:231–237. doi: 10.1038/72936. [DOI] [PubMed] [Google Scholar]
- Kaether C, Skehel P, Dotti CG. Axonal membrane proteins are transported in distinct carriers: a two-color video microscopy study in cultured hippocampal neurons. Mol Biol Cell. 2000;11:1213–1224. doi: 10.1091/mbc.11.4.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraszewski K, Mundigl O, Daniell L, Verderio C, Matteoli M, De Camilli P. Synaptic vesicle dynamics in living cultured hippocampal neurons visualized with CY3-conjugated antibodies directed against the lumenal domain of synaptotagmin. J Neurosci. 1995;15:4328–4342. doi: 10.1523/JNEUROSCI.15-06-04328.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger SR, Kolar A, Fitzsimonds RM. The presynaptic release apparatus is functional in the absence of dendritic contact and highly mobile within isolated axons. Neuron. 2003;40:945–957. doi: 10.1016/s0896-6273(03)00729-3. [DOI] [PubMed] [Google Scholar]
- Langford GM. Myosin-V, a versatile motor for short-range vesicle transport. Traffic. 2002;3:859–865. doi: 10.1034/j.1600-0854.2002.31202.x. [DOI] [PubMed] [Google Scholar]
- Lee JR, Shin H, Ko J, Choi J, Lee H, Kim E. Characterization of the movement of the kinesin motor KIF1A in living cultured neurons. J Biol Chem. 2003;278:2624–2629. doi: 10.1074/jbc.M211152200. [DOI] [PubMed] [Google Scholar]
- Matteoli M, Takei K, Perin MS, Sudhof TC, De Camilli P. Exo-endocytotic recycling of synaptic vesicles in developing processes of cultured hippocampal neurons. J Cell Biol. 1992;117:849–861. doi: 10.1083/jcb.117.4.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miesenbock G, De Angelis DA, Rothman JE. Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature. 1998;394:192–195. doi: 10.1038/28190. [DOI] [PubMed] [Google Scholar]
- Murthy M, Garza D, Scheller RH, Schwarz TL. Mutations in the exocyst component Sec5 disrupt neuronal membrane traffic, but neurotransmitter release persists. Neuron. 2003;37:433–447. doi: 10.1016/s0896-6273(03)00031-x. [DOI] [PubMed] [Google Scholar]
- Murthy VN, De Camilli P. Cell biology of the presynaptic terminal. Annu Rev Neurosci. 2003;26:701–728. doi: 10.1146/annurev.neuro.26.041002.131445. [DOI] [PubMed] [Google Scholar]
- Nakata T, Terada S, Hirokawa N. Visualization of the dynamics of synaptic vesicle and plasma membrane proteins in living axons. J Cell Biol. 1998;140:659–674. doi: 10.1083/jcb.140.3.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niell CM, Meyer MP, Smith SJ. In vivo imaging of synapse formation on a growing dendritic arbor. Nat Neurosci. 2004;7:254–260. doi: 10.1038/nn1191. [DOI] [PubMed] [Google Scholar]
- Nonet ML. Visualization of synaptic specializations in live C. elegans with synaptic vesicle protein-GFP fusions. J Neurosci Methods. 1999;89:33–40. doi: 10.1016/s0165-0270(99)00031-x. [DOI] [PubMed] [Google Scholar]
- Okabe S, Miwa A, Okado H. Spine formation and correlated assembly of presynaptic and postsynaptic molecules. J Neurosci. 2001;21:6105–6114. doi: 10.1523/JNEUROSCI.21-16-06105.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada Y, Yamazaki H, Sekine-Aizawa Y, Hirokawa N. The neuron-specific kinesin superfamily protein KIF1A is a unique monomeric motor for anterograde axonal transport of synaptic vesicle precursors. Cell. 1995;81:769–780. doi: 10.1016/0092-8674(95)90538-3. [DOI] [PubMed] [Google Scholar]
- Rao A, Craig AM. Activity regulates the synaptic localization of the NMDA receptor in hippocampal neurons. Neuron. 1997;19:801–812. doi: 10.1016/s0896-6273(00)80962-9. [DOI] [PubMed] [Google Scholar]
- Sabo SL, McAllister AK. Mobility and cycling of synaptic protein-containing vesicles in axonal growth cone filopodia. Nat Neurosci. 2003;6:1264–1269. doi: 10.1038/nn1149. [DOI] [PubMed] [Google Scholar]
- Saito Y, Murakami F, Song WJ, Okawa K, Shimono K, Katsumaru H. Developing corticorubral axons of the cat form synapses on filopodial dendritic protrusions. Neurosci Lett. 1992;147:81–84. doi: 10.1016/0304-3940(92)90779-7. [DOI] [PubMed] [Google Scholar]
- Sankaranarayanan S, Ryan TA. Calcium accelerates endocytosis of vSNAREs at hippocampal synapses. Nat Neurosci. 2001;4:129–136. doi: 10.1038/83949. [DOI] [PubMed] [Google Scholar]
- Sankaranarayanan S, Atluri PP, Ryan TA. Actin has a molecular scaffolding, not propulsive, role in presynaptic function. Nat Neurosci. 2003;6:127–135. doi: 10.1038/nn1002. [DOI] [PubMed] [Google Scholar]
- Scheiffele P. Cell-cell signaling during synapse formation in the CNS. Annu Rev Neurosci. 2003;26:485–508. doi: 10.1146/annurev.neuro.26.043002.094940. [DOI] [PubMed] [Google Scholar]
- Scheiffele P, Fan J, Choih J, Fetter R, Serafini T. Neuroligin expressed in nonneuronal cells triggers presynaptic development in contacting axons. Cell. 2000;101:657–669. doi: 10.1016/s0092-8674(00)80877-6. [DOI] [PubMed] [Google Scholar]
- Shapira M, Zhai RG, Dresbach T, Bresler T, Torres VI, Gundelfinger ED, Ziv NE, Garner CC. Unitary assembly of presynaptic active zones from Piccolo-Bassoon transport vesicles. Neuron. 2003;38:237–252. doi: 10.1016/s0896-6273(03)00207-1. [DOI] [PubMed] [Google Scholar]
- Shen K, Bargmann CI. The immunoglobulin superfamily protein SYG-1 determines the location of specific synapses in C. elegans . Cell. 2003;112:619–630. doi: 10.1016/s0092-8674(03)00113-2. [DOI] [PubMed] [Google Scholar]
- Shen K, Fetter RD, Bargmann CI. Synaptic specificity is generated by the synaptic guidepost protein SYG-2 and its receptor, SYG-1. Cell. 2004;116:869–881. doi: 10.1016/s0092-8674(04)00251-x. [DOI] [PubMed] [Google Scholar]
- Shupliakov O, Bloom O, Gustafsson JS, Kjaerulff O, Low P, Tomilin N, Pieribone VA, Greengard P, Brodin L. Impaired recycling of synaptic vesicles after acute perturbation of the presynaptic actin cytoskeleton. Proc Natl Acad Sci USA. 2002;99:14476–14481. doi: 10.1073/pnas.212381799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamori S, Rhee JS, Rosenmund C, Jahn R. Identification of a vesicular glutamate transporter that defines a glutamatergic phenotype in neurons. Nature. 2000;407:189–194. doi: 10.1038/35025070. [DOI] [PubMed] [Google Scholar]
- Tashiro A, Dunaevsky A, Blazeski R, Mason CA, Yuste R. Bidirectional regulation of hippocampal mossy fiber filopodial motility by kainate receptors: a two-step model of synaptogenesis. Neuron. 2003;38:773–784. doi: 10.1016/s0896-6273(03)00299-x. [DOI] [PubMed] [Google Scholar]
- Vale RD. The molecular motor toolbox for intracellular transport. Cell. 2003;112:467–480. doi: 10.1016/s0092-8674(03)00111-9. [DOI] [PubMed] [Google Scholar]
- Varoqueaux F, Sigler A, Rhee JS, Brose N, Enk C, Reim K, Rosenmund C. Total arrest of spontaneous and evoked synaptic transmission but normal synaptogenesis in the absence of Munc13-mediated vesicle priming. Proc Natl Acad Sci USA. 2002;99:9037–9042. doi: 10.1073/pnas.122623799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughn JE. Fine structure of synaptogenesis in the vertebrate central nervous system. Synapse. 1989;3:255–285. doi: 10.1002/syn.890030312. [DOI] [PubMed] [Google Scholar]
- Verderio C, Coco S, Bacci A, Rossetto O, De Camilli P, Montecucco C, Matteoli M. Tetanus toxin blocks the exocytosis of synaptic vesicles clustered at synapses but not of synaptic vesicles in isolated axons. J Neurosci. 1999;19:6723–6732. doi: 10.1523/JNEUROSCI.19-16-06723.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhage M, Maia AS, Plomp JJ, Brussaard AB, Heeroma JH, Vermeer H, Toonen RF, Hammer RE, van den Berg TK, Missler M, Geuze HJ, Sudhof TC. Synaptic assembly of the brain in the absence of neurotransmitter secretion. Science. 2000;287:864–869. doi: 10.1126/science.287.5454.864. [DOI] [PubMed] [Google Scholar]
- Waites CL, Craig AM, Garner CC. Mechanisms of vertebrate synaptogenesis. Annu Rev Neurosci. 2005;28:251–274. doi: 10.1146/annurev.neuro.27.070203.144336. [DOI] [PubMed] [Google Scholar]
- Washbourne P, Bennett JE, McAllister AK. Rapid recruitment of NMDA receptor transport packets to nascent synapses. Nat Neurosci. 2002;5:751–759. doi: 10.1038/nn883. [DOI] [PubMed] [Google Scholar]
- Wong WT, Wong RO. Changing specificity of neurotransmitter regulation of rapid dendritic remodeling during synaptogenesis. Nat Neurosci. 2001;4:351–352. doi: 10.1038/85987. [DOI] [PubMed] [Google Scholar]
- Zhai RG, Vardinon-Friedman H, Cases-Langhoff C, Becker B, Gundelfinger ED, Ziv NE, Garner CC. Assembling the presynaptic active zone: a characterization of an active one precursor vesicle. Neuron. 2001;29:131–143. doi: 10.1016/s0896-6273(01)00185-4. [DOI] [PubMed] [Google Scholar]
- Zhang W, Benson DL. Stages of synapse development defined by dependence on F-actin. J Neurosci. 2001;21:5169–5181. doi: 10.1523/JNEUROSCI.21-14-05169.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Benson DL. Developmentally regulated changes in cellular compartmentation and synaptic distribution of actin in hippocampal neurons. J Neurosci Res. 2002;69:427–436. doi: 10.1002/jnr.10313. [DOI] [PubMed] [Google Scholar]
- Zheng JQ, Wan JJ, Poo MM. Essential role of filopodia in chemotropic turning of nerve growth cone induced by a glutamate gradient. J Neurosci. 1996;16:1140–1149. doi: 10.1523/JNEUROSCI.16-03-01140.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou HM, Brust-Mascher I, Scholey JM. Direct visualization of the movement of the monomeric axonal transport motor UNC-104 along neuronal processes in living Caenorhabditis elegans . J Neurosci. 2001;21:3749–3755. doi: 10.1523/JNEUROSCI.21-11-03749.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziv NE, Smith SJ. Evidence for a role of dendritic filopodia in synaptogenesis and spine formation. Neuron. 1996;17:91–102. doi: 10.1016/s0896-6273(00)80283-4. [DOI] [PubMed] [Google Scholar]