

Abstract
Disruptions in brain-derived neurotrophic factor (BDNF) expression are proposed to contribute to the molecular pathogenesis of Rett syndrome (RTT), a severe neurological disorder caused by loss-of-function mutations in methyl-CpG-binding protein-2 (MeCP2). Although MeCP2 is a transcriptional regulator of BDNF, it is unknown how MeCP2 mutations affect transynaptic BDNF signaling. Our findings demonstrate an early, abnormal neurosecretory phenotype in MeCP2-deficient neurons characterized by significant increases in the percentage of cellular BDNF content available for release. However, loss of MeCP2 also results in deficits in total cell BDNF content that are developmentally regulated in a cell-type-specific manner. Thus, the net effect of MeCP2 loss on absolute BDNF secretion changes with age and is determined by both the amount of BDNF available for release and progressive declines in total cellular BDNF. We propose, therefore, that loss of MeCP2 function disrupts transynaptic BDNF signaling by perturbing the normal balance between BDNF protein levels and secretion. However, mutant neurons are capable of secreting wild-type levels of BDNF in response to high-frequency electrical stimulation. In addition, we found elevated exocytic function in Mecp2 −/y adrenal chromaffin cells, indicating that the Mecp2 null mutation is associated with alterations of neurosecretion that are not restricted to BDNF. These findings are the first examples of abnormal neuropeptide and catecholamine secretion in a mouse model of RTT.
Keywords: Rett syndrome, BDNF, nodose ganglion, chromaffin cells, dense-core vesicles, readily releasable pool
Introduction
Rett syndrome (RTT) is a progressive X-linked-dominant disorder caused by loss-of-function mutations in the gene encoding methyl-CpG-binding protein-2 (MeCP2) (Amir et al., 1999) and is characterized by severe mental retardation and somatomotor, respiratory, and autonomic dysfunction. Abnormal expression of brain-derived neurotrophic factor (BDNF) has been highlighted as a possible cause of neurological dysfunction in RTT because (1) BDNF is a transcriptional target of MeCP2 (Chen et al., 2003; Martinowich et al., 2003); (2) BDNF protein levels in various brain regions are depressed in Mecp2 null (Mecp2 −/y) mice (Chang et al., 2006); and (3) genetic overexpression of BDNF can ameliorate some of the functional deficits in Mecp2 −/y mice (Chang et al., 2006). However, no studies have examined how genetic loss of MeCP2 affects transynaptic BDNF signaling, a regulated process that requires tight coupling between activity-dependent presynaptic BDNF expression and secretion. To approach this issue, we compared BDNF secretion from wild-type and Mecp2 null (Chen et al., 2001) neurons using ELISA in situ, a method that permits detection of native BDNF release in vitro (Balkowiec and Katz, 2000). To determine whether perturbations are specific to BDNF-releasing neurons, or more global, we also compared exocytic function and catecholamine release in wild-type and MeCP2 null adrenal chromaffin cells. Our data reveal qualitatively similar neurosecretory phenotypes in both systems characterized by enhanced availability and/or transmitter release.
Materials and Methods
Animals.
Mecp2 null mice (Chen et al., 2001) were bred from founder animals from the Mutant Mouse Regional Resource Center (University of California, Davis, CA). All experimental procedures were approved by the Institutional Animal Care and Use Committee at Case Western Reserve University.
Primary culture of nodose neurons.
Wild-type and Mecp2 −/y null mice were killed with CO2 on postnatal day 35 (P35). The nodose ganglia (NGs) were removed, digested in 0.1% collagenase (Sigma, St. Louis, MO) in Earle’s balanced salt solution (Invitrogen, San Diego, CA) for 70 min at 37°C, triturated in culture medium (see below) containing 0.15% BSA, and plated at a density of one NG per well into 96-well flat-bottom ELISA plates coated with poly-d-lysine and BDNF monoclonal antibody (Promega, Madison, WI). Cultures were grown for 3 d in DMEM-F-12 medium supplemented with 5% fetal bovine serum (Invitrogen) and 1% penicillin–streptomycin–neomycin. Newborn (P0) NG cultures were grown as described previously (Balkowiec and Katz, 2000).
BDNF immunoassay.
BDNF release was measured with a modified sandwich ELISA, termed ELISA in situ. This technique measures release of native BDNF at levels below the sensitivity of standard ELISA (Balkowiec and Katz, 2000; Wang et al., 2006). Standard curves and blank values were generated using the same serum-containing medium as the cultured cells. BDNF in lysates was detected by standard ELISA. For brain lysates, total protein concentration was measured using Coomassie Plus Protein Assay reagent (Pierce, Rockford, IL), and 150 μg of protein from each sample was loaded in duplicate for ELISA.
Electrical field stimulation.
After 3 d in culture, wild-type or Mecp2 −/y NG neurons were stimulated with concentric platinum wire electrodes (Goodfellow-Cambridge Limited, Huntingdon, UK) connected to a Master-8 Programmable Pulse Generator (AMP Instruments, Jerusalem, Israel). Control wells received electrodes not connected to the stimulator. Neurons were stimulated once every 20 s with 2 s trains of biphasic rectangular pulses (pulse duration, 10 ms; current per well, 0.2 mA) delivered at 100 Hz.
NGs total cell counts.
Total cell counts were performed on cresyl violet-stained cryostat sections as described previously (Erickson et al., 1996).
Determination of neuron viability.
Cell viability was assessed either by counting β-tubulin-immunoreactive cells after fixation with 95% methanol/5% glacial acetic acid (Fisher Scientific, Houston, TX) at −20°C (cortical cultures) or by counting unstained live neurons (NG cultures).
Adrenal slices.
Adult C57BL/6 wild-type and Mecp2 −/y mice were anesthetized with halothane (Sigma) and killed by decapitation, and adrenal tissue slices were prepared as described previously (Chan et al., 2005).
BDNF immunohistochemistry.
Animals were killed with CO2 and perfused with 4% paraformaldehyde, and their brains were cut at 30 μm with a vibratome. Sections were stained with rabbit polyclonal anti-BDNF (a gift from Amgen Pharmaceuticals, Thousand Oaks, CA) as described previously (Brady et al., 1999).
Electrophysiology.
Adrenal slices were constantly superfused during recording with normal-calcium bicarbonate-buffered saline containing the following (in mm): 140 NaCl, 2 KCl, 3 CaCl2, 2 MgCl2, 26 NaHCO3, and 10 glucose, gassed with a 95% O2/5% CO2 mix. The osmolarity was ∼310 mOsm. The volume of the recording chamber was ∼1.5 ml, and the buffer flow was set at 1 ml/min and visualized with a 40× water-immersion objective.
Borosilicate glass patch pipettes (resistance, 4–5 MΩ) were partially coated with molten dental wax and polished with a microforge (Narashige, Tokyo, Japan). Recordings were performed in perforated-patch configuration. The pipette solution contained (in mm) 135 Cs-glutamate, 10 HEPES (free acid), 9.5 NaCl, 0.5 tetraethylammonium-Cl, and 0.53 amphotericin B, prepared as described previously (Chan et al., 2005). The pH was adjusted to 7.3, and the osmolarity was 310 mOsm. Cells were perforated to a series resistance of <30 MΩ (mean, 18.2 ± 6.15 MΩ; n = 13) and held at −80 mV. Leak currents were not corrected. Voltage-clamp recordings were acquired as described previously (Chan et al., 2005). Nonsecretory capacitance signals were eliminated from dual-pulse recordings as described in the literature (Moser and Neher, 1997). Only cells with less than a −60 pA leak current were analyzed.
Electrochemical recordings.
Carbon fiber electrodes (ALA Scientific, Longneck, NY) of 5 μm tip diameter were used for amperometric catecholamine detection. A +650 mV potential was placed on the carbon fiber once it was in the bath, and the background current was allowed to relax to a steady value. Recording conditions and digitization were as described previously (Chan et al., 2005).
Statistical analysis.
Data were analyzed by Student’s t test or ANOVA with Tukey’s multiple comparison post hoc analysis. A p value of <0.05 was considered statistically significant.
Results
BDNF protein expression in wild-type and Mecp2 −/y mice
To test the hypothesis that genetic loss of MeCP2 disrupts BDNF-dependent signaling, initial studies analyzed levels of BDNF protein in several populations of neurons previously shown to secrete and/or express BDNF, including the NG, brainstem, hippocampus, and cortex. Tissues from wild-type and Mecp2 −/y mice were compared at P0 and P35. At P0, BDNF levels were not significantly different between wild-type and MeCP2 null tissues, although a trend toward lower values in mutant NGs and the brainstem was evident (Fig. 1 A). At P35, BDNF levels in the NGs and brainstem were significantly lower in Mecp2 −/y animals compared with wild-type controls (Fig. 1 A). The difference in NG BDNF protein levels was accompanied by 50–60% lower level of total BDNF mRNA in the mutant ganglia as determined by quantitative real-time PCR. These differences were not attributable to neuronal loss after birth, because quantitative morphometric analysis demonstrated no significant difference in ganglion cell number between wild-type and mutant ganglia at either P0 or P35 (P0 wild-type NG, 3711 ± 804 vs P0 Mecp2 −/y NG, 3722 ± 340; P35 wild-type NG, 2104 ± 96 vs P35 Mecp2 −/y NG, 1733 ± 30). BDNF protein levels in the hippocampus and cortex were not significantly different from wild-type samples at either age.
Figure 1.
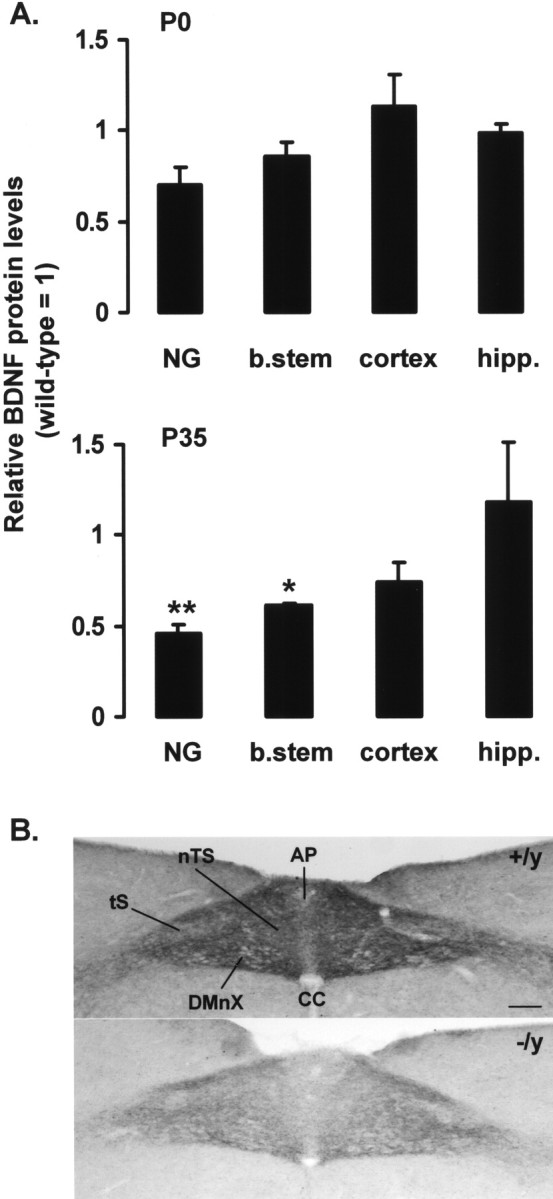
Mecp2 −/y mice exhibit progressive deficits in BDNF in vivo. A, BDNF protein levels were compared in P0 and P35 wild-type and Mecp2 −/y NGs, brainstem (b.stem), hippocampus (hipp.), and cortex by ELISA. Mutant values are expressed as a fraction of the corresponding wild-type control (wild type = 1). Each bar represents the mean ± SEM from at least three animals. *p < 0.05, **p < 0.01 (Student’s t test). B, BDNF immunostaining is depleted in the terminal field of NG neurons in the brainstem nTS in P35 Mecp2 −/y mice (−/y) compared with Mecp2 +/y controls (+/y). CC, Central canal; DMnX, dorsal motor nucleus of the vagus nerve; tS, solitary tract. Scale bar, 100 μm.
To further characterize the loss of BDNF protein in the NGs and brainstem, we compared BDNF immunostaining in the nucleus tractus solitarius (nTS), the target of afferent projections from NG sensory neurons, in P35 wild-type and Mecp2 −/y animals (Fig. 1 B). In wild-type animals, there is a dense neuropil of BDNF-positive fibers in the nTS and adjacent area postrema (AP), the distribution of which parallels the pattern of primary afferent projections from the NG. In contrast, mutant animals exhibit a marked decrease in the intensity of BDNF fiber staining in the nTS and AP. Because Mecp2 −/y animals have normal numbers of NG neurons, these data are consistent with a decrease in BDNF content in the afferent terminal field.
Spontaneous BDNF secretion from Mecp2 −/y neurons
To determine how changes in BDNF expression in Mecp2 −/y mice might influence BDNF secretion, we initially compared spontaneous secretion from P0 wild-type and Mecp2 −/y NGs and cortical neurons in dissociated culture by ELISA in situ. Cultures were grown for 3 d under control conditions and assayed for the total amount of BDNF released during the culture period. These experiments revealed that P0 and P35 Mecp2 −/y NG neurons released a significantly larger proportion of their BDNF content than wild-type controls (Fig. 2 A). Thus, at P0, the absolute amount of BDNF released by Mecp2 −/y NG neurons (Fig. 2 B) and cortical neurons (Fig. 2 C) was higher than in wild-type control cultures. However, because P35 Mecp2 −/y NG neurons express markedly less BDNF than wild-type controls in vitro (wild type, 251.1 ± 14.1 pg/ml vs Mecp2 −/y, 112.7 ± 4.4 pg/ml; p < 0.01), the absolute amount of BDNF released was actually less than in wild-type control cultures at this age (Fig. 2 B). These differences were not associated with any change in neuronal survival (P0 wild-type cortical neurons, 19,387 ± 694 vs P0 Mecp2 −/y cortical neurons, 20,014 ± 1025; P0 wild-type NGs, 3088 ± 62 vs P0 Mecp2 −/y NGs, 3064 ± 66; P35 wild-type NGs, 1967 ± 118 vs P35 Mecp2 −/y NGs, 1964 ± 99). Moreover, levels of spontaneous secretion were not significantly affected by the addition of 1.5 μm tetrodotoxin (TTX) to the culture medium (in picograms BDNF per milliliter: P35 wild-type NG control, 43.2 ± 3.2 vs P35 wild-type NG plus TTX, 37.7 ± 2.4; P35 Mecp2 −/y NG control, 32.5 ± 1.8 vs P35 Mecp2 −/y NG plus TTX, 30.5 ± 2.2).
Figure 2.

Mecp2 −/y neurons exhibit age-dependent defects in spontaneous BDNF release. Spontaneous BDNF release from dissociated cultures of wild-type and Mecp2 −/y NGs and cortical neurons was measured during 3 d in culture using ELISA in situ. A, The amount of BDNF released as a percentage of total BDNF content. B, C, The absolute amount of BDNF released. Each bar represents the mean ± SEM from at least three independent experiments. *p < 0.05, **p < 0.01 (Student’s t test).
Evoked BDNF secretion from P35 Mecp2 −/y NG neurons
To determine whether or not reduced BDNF protein levels are associated with alterations in evoked BDNF secretion, we compared BDNF release from cultured P35 NG neurons in response to patterned electrical field stimulation. Cultures of P35 wild-type and Mecp2 −/y NG neurons were stimulated once every 20 s, with 2 s trains of biphasic pulses delivered at 100 Hz for a total of 30 min, and assayed for the total amount of BDNF released as well as cellular BDNF content. The absolute amount of BDNF released during stimulation was not different between wild-type and Mecp2 −/y cultures (Fig. 3 A), despite the fact that total BDNF content was markedly depressed in P35 cells (Fig. 3 B). Thus, as with spontaneous release, stimulated Mecp2 −/y neurons secreted a greater percentage of their total BDNF content compared with wild-type controls (Fig. 3 C). Under the conditions of these experiments, therefore, the Mecp2 −/y secretory phenotype was sufficient to compensate for the deficit in cellular BDNF content in these cells.
Figure 3.

Evoked BDNF release in Mecp2 −/y NG neurons. BDNF release evoked by patterned electrical field stimulation was compared in dissociate cultures of P35 wild-type and Mecp2 −/y NG neurons. Cultures were stimulated once every 20 s, with 2 s trains of biphasic pulses at 100 Hz, and assayed for released BDNF by ELISA in situ and for cellular BDNF content by standard ELISA. A, Wild-type and Mecp2 −/y neurons released the same absolute amount of BDNF during the period of stimulation (stimulated release = total release after stimulation − unstimulated release as measured in control cultures). However, as shown in B, cellular BDNF content was significantly lower in mutant neurons compared with wild-type cells, as in vivo. Therefore, as shown in C, mutant neurons released a significantly greater percentage of total BDNF content during stimulation than did wild-type cells. Each bar represents the mean ± SEM from at least three independent experiments. *p < 0.05 (ANOVA).
Elevated exocytic function in Mecp2 −/y chromaffin cells
To investigate whether loss of MeCP2 results in similar alterations in other neurosecretory systems, we examined secretory granule fusion and catecholamine release in Mecp2 −/y adrenal chromaffin cells. Chromaffin cells and neurons exhibit common neurosecretory control mechanisms, including clostridial toxin-sensitive and SNARE (soluble N-ethylmaleimide-sensitive factor attachment receptor) protein-dependent vesicle release (Penner et al., 1986; Xu et al., 1998). Moreover, adrenal catecholamines are stored in large, dense-core vesicles, similar to BDNF in sensory neurons (Luo et al., 2001).
We used a cell capacitance-based, dual-pulse protocol (Gillis et al., 1996) to compare the size of the release-ready granule pool in wild-type and Mecp2 −/y chromaffin cells in an in vitro slice preparation of the adrenal medulla. The dual-pulse protocol separates Ca2+ influx from the secretion process because the Ca2+ influx is sufficiently large and, therefore, is not the limiting factor for secretion; rather, the number of granules defines the magnitude of the response. Briefly, two 100 ms square pulse stimuli were delivered at a 100 ms interval, and their amplitudes were adjusted to evoke equivalent amounts of Ca2+ entry (Fig. 4 Ai). The first Ca2+ influx causes secretion (a jump in cell capacitance) (Fig. 4 Ai, bottom trace) and depletion of the ready releasable granule pool (RRP), leaving fewer granules for release during the second Ca2+ influx. This results in a depletion-dependent secretory depression in response to the second pulse. The evoked capacitance jumps for each pulse were measured, and their magnitudes were used to calculate the total number of release-ready granules [see Fig. 4 legend and the study by Gillis et al. (1996) for a full description of the technique]. The results from this analysis are provided in Figure 4 Aii and show that the RRP is significantly larger in the Mecp2 −/y mice than in wild types.
Figure 4.

Mecp2 −/y adrenal chromaffin cells exhibit an increase in the catecholamine RRP and evoked release. The RRP was quantified in wild-type and Mecp2 −/y chromaffin cells as described previously (Gillis et al., 1996). Ai, Cells were voltage clamped at −80 mV and stimulated with a pair of 100 ms depolarizations, first to 0 mV and then to +5 mV, to balance Ca2+ influx. The top trace shows evoked current influx (IMon.), and the bottom trace shows cell capacitance (Cm). In this example, the first pulse resulted in a greater capacitance increase than the second pulse (Cm1 and Cm2, respectively), reflecting a consumption of release-ready granules by the first pulse, leaving few for the second pulse to access. Formally, the RRP can thus be quantified as follows: RRP = S/(1 − R 2), where S is the sum of Cm1 and Cm2 and R is the ratio of Cm2 to Cm1. Aii, This dual-pulse protocol was repeated on MeCP2+/y and MeCP2−/y cells. The quantified RRP sizes from Mecp2 +/y and Mecp2 −/y records (n = 36 and 65, respectively) are summarized and show that the RRP was significantly larger in the Mecp2 −/y (*p < 0.02, paired Student’s t test). Bi, To correlate the increased RRP to evoked catecholamine release, the amperometric (I Amp.) current was measured from chromaffin cells in control Ringer’s solution and during nicotine stimulation. A representative record from Mecp2 +/+ chromaffin tissue is plotted. Bii, Amperometric currents (ΣAmp.) recorded under this protocol from Mecp2 +/y and Mecp2 −/y cells were integrated to allow relative comparison of catecholamine release (n = 8 and 10 for Mecp2 +/y and Mecp2 −/y cells, respectively). These data show significant increases in both spontaneous and evoked catecholamine release from Mecp2 −/y cells compared with Mecp2 +/y (*p < 0.02, paired Student’s t test).
One implication of these findings is that regulated secretion of catecholamines from Mecp2 −/y chromaffin cells in response to acute secretagogic stimuli will be larger than in wild-type cells. Chromaffin cells receive sympathetic input through the splanchnic nerve, and cholinergic stimulation results in catecholamine release into the circulation. We stimulated chromaffin cells in situ by puffing nicotine onto their surface and measured catecholamine output by electrochemical amperometry (Chow et al., 1992). As shown in Figure 4 Bi, this technique results in a robust release of catecholamine reported as spike-like events in the amperometric record. Each spike reflects the detection of a single granule quantum of catecholamine. With heavy catecholamine release (Fig. 4 Bi), quanta also sum to provide an increase in the baseline amperometric current. This protocol was repeated on wild-type and Mecp2 −/y mice; data were pooled for each genotype, and the results are shown in Figure 4 Bii. These data demonstrate that mutant mice exhibit a greater spontaneous rate of catecholamine release and also respond to nicotine stimulation with a significantly larger rate of evoked catecholamine release. Together, data from both protocols reflect a greater number of releasable secretory granules in the Mecp2 −/y mice compared with wild-type controls. Although not directly addressed by our studies, it is possible that catecholamine content per granule is also increased in the mutants.
Discussion
Our results demonstrate that genetic loss of MeCP2 is associated not only with deficits in neuronal BDNF content but also with a significant increase in the percentage of BDNF content available for release. Because deficits in BDNF protein levels are temporally regulated in a cell-type- or region-specific manner, the net effect of the Mecp2 −/y mutation on BDNF release will vary among neuronal populations and with age. The fact that newborn Mecp2 −/y neurons release abnormally high levels of BDNF raises the possibility that BDNF-dependent regulation of neural development is disrupted in mutant mice in vivo. For example, hypersecretion of BDNF, particularly by newborn Mecp2 −/y neurons at rest, could derange developmental processes that depend on tight coupling between neuronal activity and BDNF release, such as activity-dependent refinement of synaptic connections (Lein and Shatz, 2000). Additional studies are required to determine how the Mecp2 −/y mutation affects evoked release at different stimulus frequencies.
In view of the fact that neuronal activity appears to be depressed in Mecp2 −/y mice (Dani et al., 2005; Chang et al., 2006), it is possible that both reduced BDNF expression and increased BDNF availability for release are consequences of decreased neuronal activation. Activity-dependent BDNF expression is well established (Thoenen, 2000; Lu, 2003). In addition, however, the static size of the readily releasable pool has been shown to be inversely proportional to neuronal activity, such that neurons grown under chronic activity blockade exhibit more docked synaptic vesicles and greater transmitter release (Murthy et al., 2001). Whether or not the proportion of cellular BDNF content available for release is similarly regulated remains to be defined.
Abnormal expression and secretion of BDNF in Mecp2 −/y NG neurons may be particularly relevant to respiratory and autonomic dysfunction in RTT. NG neurons are the major source of visceral afferent input to the nTS, the primary site of visceral sensory integration in the brainstem, and thereby play pivotal roles in respiratory and autonomic reflex control. RTT patients suffer from autonomic disturbances, including prolonged QT interval, diminished cardiac vagal tone, and reduced cardiac baroreflex sensitivity that may result from defects in reflex integration of vagal and sympathetic cardiac rhythm control in the nTS (Julu et al., 2001). The possibility that perturbations of visceral afferent signaling contribute to autonomic disturbances in RTT is particularly compelling because BDNF can potently modulate glutamatergic transmission by inhibiting AMPA receptor currents at primary afferent nTS synapses (Balkowiec et al., 2000).
Our studies identify a secretory phenotype in Mecp2 −/y adrenal chromaffin cells characterized by an increase in RRP size and evoked catecholamine release. This enhancement of exocytic function is qualitatively similar to the BDNF secretory phenotype in Mecp2 −/y NG neurons (i.e., increased availability for release). Although additional studies are required to define underlying mechanisms, this similarity may be related to the fact that BDNF in sensory neurons (Luo et al., 2001) and adrenal catecholamines are both stored in large dense-core vesicles. It will be important to determine whether or not other systems involving large-dense core vesicle release, including peptidergic neurons (Zupanc, 1996), hypothalamic neurosecretory cells (Gainer and Chin, 1998), pancreatic β-cells (Lang, 1999), and atrial cardiac myocytes (Peters et al., 2006), are also affected by loss of MeCP2. On the other hand, the size of the RRP underlying spontaneous excitatory transmission in hippocampal cultures, which is reflective of the pool of glutamatergic small, clear vesicles, appears to be unaffected by loss of MeCP2 (Nelson et al., 2006). It is possible, therefore, that loss of MeCP2 may differentially affect dense-core and clear vesicle pools, respectively. However, mice carrying a truncation mutation in MeCP2 (Mecp2 308/y) exhibit a higher density of docked clear vesicles at active zones in some hippocampal synapses (Moretti et al., 2006).
In summary, our data indicate that the net effect of the Mecp2 −/y mutation on BDNF release is determined both by progressive, region-specific changes in total BDNF content after birth and by an increase in the percentage of BDNF available for release. The fact that adult mutant neurons isolated in vitro express significantly less BDNF than wild-type cells, as in vivo, suggests that the BDNF expression phenotype may be cell autonomous. In addition, the fact that altered regulation of neurosecretion is not restricted to BDNF indicates that loss of MeCP2 is likely to affect secretion of multiple transynaptic signaling molecules in the central and peripheral nervous systems. This pleiotropy may contribute to the diversity of neural effector systems disrupted in RTT.
Footnotes
This work was supported by grants from the National Heart, Lung, and Blood Institute and the Rett Syndrome Research Foundation (D.M.K.). We thank Dr. Diana Kunze for her critical review of this manuscript.
References
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- Balkowiec A, Katz DM. Activity-dependent release of endogenous brain-derived neurotrophic factor from primary sensory neurons detected by ELISA in situ. J Neurosci. 2000;20:7417–7423. doi: 10.1523/JNEUROSCI.20-19-07417.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balkowiec A, Kunze DL, Katz DM. Brain-derived neurotrophic factor acutely inhibits AMPA-mediated currents in developing sensory relay neurons. J Neurosci. 2000;20:1904–1911. doi: 10.1523/JNEUROSCI.20-05-01904.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady R, Zaidi SI, Mayer C, Katz DM. BDNF is a target-derived survival factor for arterial baroreceptor and chemoafferent primary sensory neurons. J Neurosci. 1999;19:2131–2142. doi: 10.1523/JNEUROSCI.19-06-02131.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan SA, Polo-Parada L, Landmesser LT, Smith C. Adrenal chromaffin cells exhibit impaired granule trafficking in NCAM knockout mice. J Neurophysiol. 2005;94:1037–1047. doi: 10.1152/jn.01213.2004. [DOI] [PubMed] [Google Scholar]
- Chang Q, Khare G, Dani V, Nelson S, Jaenisch R. The disease progression of Mecp2 mutant mice is affected by the level of BDNF expression. Neuron. 2006;49:341–348. doi: 10.1016/j.neuron.2005.12.027. [DOI] [PubMed] [Google Scholar]
- Chen RZ, Akbarian S, Tudor M, Jaenisch R. Deficiency of methyl-CPG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat Genet. 2001;27:327–331. doi: 10.1038/85906. [DOI] [PubMed] [Google Scholar]
- Chen WG, Chang Q, Lin Y, Meissner A, West AE, Griffith EC, Jaenisch R, Greenberg ME. Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science. 2003;302:885–889. doi: 10.1126/science.1086446. [DOI] [PubMed] [Google Scholar]
- Chow RH, von Ruden L, Neher E. Delay in vesicle fusion revealed by electrochemical monitoring of single secretory events in adrenal chromaffin cells. Nature. 1992;356:60–63. doi: 10.1038/356060a0. [DOI] [PubMed] [Google Scholar]
- Dani VS, Chang Q, Maffei A, Turrigiano GG, Jaenisch R, Nelson SB. Reduced cortical activity due to a shift in the balance between excitation and inhibition in a mouse model of Rett syndrome. Proc Natl Acad Sci USA. 2005;102:12560–12565. doi: 10.1073/pnas.0506071102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson JT, Conover JC, Borday V, Champagnat J, Barbacid M, Yancopoulos G, Katz DM. Mice lacking brain-derived neurotrophic factor exhibit visceral sensory neuron losses distinct from mice lacking NT4 and display a severe developmental deficit in control of breathing. J Neurosci. 1996;16:5361–5371. doi: 10.1523/JNEUROSCI.16-17-05361.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gainer H, Chin H. Molecular diversity in neurosecretion: reflections on the hypothalamo-neurohypophysial system. Cell Mol Neurobiol. 1998;18:211–230. doi: 10.1023/A:1022568904002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillis KD, Mossner R, Neher E. Protein kinase C enhances exocytosis from chromaffin cells by increasing the size of the readily releasable pool of secretory granules. Neuron. 1996;16:1209–1220. doi: 10.1016/s0896-6273(00)80147-6. [DOI] [PubMed] [Google Scholar]
- Julu PO, Kerr AM, Apartopoulos F, Al-Rawas S, Engerstrom IW, Engerstrom L, Jamal GA, Hansen S. Characterisation of breathing and associated central autonomic dysfunction in the Rett disorder. Arch Dis Child. 2001;85:29–37. doi: 10.1136/adc.85.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang J. Molecular mechanisms and regulation of insulin exocytosis as a paradigm of endocrine secretion. Eur J Biochem. 1999;259:3–17. doi: 10.1046/j.1432-1327.1999.00043.x. [DOI] [PubMed] [Google Scholar]
- Lein ES, Shatz CJ. Rapid regulation of brain-derived neurotrophic factor mRNA within eye-specific circuits during ocular dominance column formation. J Neurosci. 2000;20:1470–1483. doi: 10.1523/JNEUROSCI.20-04-01470.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu B. BDNF and activity-dependent synaptic modulation. Learn Mem. 2003;10:86–98. doi: 10.1101/lm.54603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo XG, Rush RA, Zhou XF. Ultrastructural localization of brain-derived neurotrophic factor in rat primary sensory neurons. Neurosci Res. 2001;39:377–384. doi: 10.1016/s0168-0102(00)00238-8. [DOI] [PubMed] [Google Scholar]
- Martinowich K, Hattori D, Wu H, Fouse S, He F, Hu Y, Fan G, Sun YE. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science. 2003;302:890–893. doi: 10.1126/science.1090842. [DOI] [PubMed] [Google Scholar]
- Moretti P, Levenson JM, Battaglia F, Atkinson R, Teague R, Antalffy B, Armstrong D, Arancio O, Sweatt JD, Zoghbi HY. Learning and memory and synaptic plasticity are impaired in a mouse model of Rett syndrome. J Neurosci. 2006;26:319–327. doi: 10.1523/JNEUROSCI.2623-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser T, Neher E. Rapid exocytosis in single chromaffin cells recorded from mouse adrenal slices. J Neurosci. 1997;17:2314–2323. doi: 10.1523/JNEUROSCI.17-07-02314.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murthy VN, Schikorski T, Stevens CF, Zhu Y. Inactivity produces increases in neurotransmitter release and synapse size. Neuron. 2001;32:673–682. doi: 10.1016/s0896-6273(01)00500-1. [DOI] [PubMed] [Google Scholar]
- Nelson ED, Kavalali ET, Monteggia LM. MeCP2-dependent transcriptional repression regulates excitatory neurotransmission. Curr Biol. 2006;16:710–716. doi: 10.1016/j.cub.2006.02.062. [DOI] [PubMed] [Google Scholar]
- Penner R, Neher E, Dreyer F. Intracellularly injected tetanus toxin inhibits exocytosis in bovine adrenal chromaffin cells. Nature. 1986;324:76–78. doi: 10.1038/324076a0. [DOI] [PubMed] [Google Scholar]
- Peters CG, Miller DF, Giovannucci DR. Identification, localization and interaction of SNARE proteins in atrial cardiac myocytes. J Mol Cell Cardiol. 2006;40:361–374. doi: 10.1016/j.yjmcc.2005.12.007. [DOI] [PubMed] [Google Scholar]
- Thoenen H. Neurotrophins and activity-dependent plasticity. Prog Brain Res. 2000;128:183–191. doi: 10.1016/S0079-6123(00)28016-3. [DOI] [PubMed] [Google Scholar]
- Wang H, Ward N, Boswell M, Katz DM. Secretion of brain-derived neurotrophic factor from brain microvascular endothelial cells. Eur J Neurosci. 2006;23:1665–1670. doi: 10.1111/j.1460-9568.2006.04682.x. [DOI] [PubMed] [Google Scholar]
- Xu T, Binz T, Niemann H, Neher E. Multiple kinetic components of exocytosis distinguished by neurotoxin sensitivity. Nat Neurosci. 1998;1:192–200. doi: 10.1038/642. [DOI] [PubMed] [Google Scholar]
- Zupanc GK. Peptidergic transmission: from morphological correlates to functional implications. Micron. 1996;27:35–91. doi: 10.1016/0968-4328(95)00028-3. [DOI] [PubMed] [Google Scholar]