

Abstract
We studied the cellular response to orexin type 1 receptor (OX1R) stimulation in differentiated IMR-32 neuroblastoma cells. In vitro differentiation of IMR-32 cells with 5-bromo-2′-deoxyuridine leads to a neuronal phenotype with long neurite extensions and an upregulation of mainly N-type voltage-gated calcium channels. Transduction of differentiated IMR-32 cells with baculovirus harboring an OX1R–green fluorescent protein cDNA fusion construct resulted in appearance of fluorescence that was confined mainly to the plasma membrane in the cell body and to neurites. Application of orexin-A to fluorescent cells led to an increase in intracellular free Ca2+ concentration, [Ca2+]i. At low nanomolar concentrations of orexin-A, the response was reversibly attenuated by removal of extracellular Ca2+, by application of a high concentration (10 mm) of Mg2+, and by the pharmacological channel blocker dextromethorphan. A diacylglycerol, dioctanoylglycerol, but not thapsigargin or depolarization with potassium, mimicked the OX1R response with regard to Mg2+ sensitivity. A reverse transcription-PCR screening identified mRNAs for all transient receptor potential canonical (TRPC) channels, including TRPC3, TRPC6, and TRPC7, which are known to be activated by diacylglycerol. Expression of a dominant-negative TRPC6 channel subunit blunted the responses to both dioctanoylglycerol and OX1R stimulation. The results suggest that the OX1R activates a Ca2+ entry pathway that involves diacylglycerol-activated TRPC channels in neuronal cells.
Keywords: baculovirus, calcium, differentiation, neuroblastoma, orexin, TRP channel
Introduction
Orexins/hypocretins are peptide transmitters synthesized by neurons in lateral hypothalamus (de Lecea et al., 1998; Sakurai et al., 1998). These neurons project to multiple other brain loci and to the spinal cord (Peyron et al., 1998; van den Pol et al., 1998), with particularly dense innervation of areas involved in arousal, such as locus ceruleus (Horvath et al., 1999), raphe nucleus (Date et al., 1999; Liu et al., 2002), and tuberomammillary nucleus (Peyron et al., 1998; Eriksson et al., 2001). Evidence for orexin involvement in arousal and sleep/wake regulation has come from studies on animals with disrupted or modified orexin signaling system (Chemelli et al., 1999; Lin et al., 1999; Hara et al., 2001), which in some cases leads to the sleeping disorder narcolepsy. Orexins are also implicated in other physiological functions, such as regulation of food intake and metabolic processes and neuroendocrine functions (for review, see Kukkonen et al., 2002).
A central question is how orexins exert their actions at the cellular and molecular level. Orexins excite neurons by activating either or both of two identified G-protein-coupled orexin receptors, OX1R and OX2R (Sakurai et al., 1998). Investigations of the intracellular mechanisms for excitation have generated several plausible signaling pathways (for review, see Kukkonen et al., 2002; Ferguson and Samson, 2003). A useful indicator for OXR activation is an increase in intracellular Ca2+ concentration, [Ca2+]i. The [Ca2+]i can increase in cells by a variety of mechanisms, including depolarization-induced opening of voltage-gated calcium channels (VGCCs), opening of nonselective cation channels, release from intracellular stores, as well as reversal of electrogenic Na+/Ca2+ exchanger. An increase in [Ca2+]i during OXR stimulation is evident in neurons (van den Pol et al., 1998; Uramura et al., 2001; Kohlmeier et al., 2004), and it is also observable in different cell types used for heterologous expression (Sakurai et al., 1998; Smart et al., 1999; Holmqvist et al., 2002).
[Ca2+]i measurements combined with patch-clamp recordings have demonstrated that the OX1R activates a pathway for Ca2+ entry, which closely follows an inward current and depolarization, in Chinese hamster ovary (CHO) cells (Lund et al., 2000; Larsson et al., 2005). This Ca2+ entry pathway is well separated from store release at subnanomolar to low nanomolar concentrations of orexin-A. Inactivation of transient receptor potential canonical (TRPC) 1 and TRPC3 channels with dominant-negative (DN) constructs indicates involvement of these channels in the Ca2+ entry pathway in nonexcitable cells (Larsson et al., 2005).
Most TRP ion channels are Ca2+-permeable, nonselective cation channels (for review, see Minke and Cook, 2002). The TRPC subfamily, TRPC1–TRPC7, has mostly been implicated in regulation by G-proteins and metabolites of phosphoinositide hydrolysis. TRPC channels are widely expressed in different tissues, including brain, and single cell reverse transcription (RT)-PCR in different brain loci has demonstrated that TRPC channels and OXRs are coexpressed in neurons (Sergeeva et al., 2003).
To characterize the OX1R-induced Ca2+ entry in excitable cells, we have in this study used an in vitro differentiated neuroblastoma cell line, IMR-32, and a calcium imaging approach.
Materials and Methods
Materials.
Orexin-A was from Bachem (St. Helens, UK). 5-Bromo-2′-deoxyuridine (BrdU), d-3-methoxy-N-methylmorphine (dextromethorphan), 1,2-dioctanoyl-sn-glycerol (DOG), and 12-O-tetradecanoyl phorbol-13-acetate (TPA) were from Sigma-Aldrich (Helsinki, Finland). Fura-2 AM was from Invitrogen (Paisley, UK). ω-Conotoxin GVIA (ωCTx) and 2-[2-[4-(4-nitrobenzyloxy)phenyl]ethyl]isothiourea methanesulfonate (KB-R7943) were from Tocris Cookson (Bristol, UK). Thapsigargin and N,N,N,-trimethyl-4-(2-oxo-1-pyrrolidinyl)-2-butyn-1-ammonium iodide (oxotremorine-M) was from Research Biochemicals International (Natick, MA) and bisindolylmaleimide I (GF109203X) was from Calbiochem (San Diego, CA).
Cell cultures.
The human neuroblastoma IMR-32 cell line (Tumilowicz et al., 1970) was obtained from American Type Culture Collection (Manassas, VA) and grown in 80 cm2 cell culture flasks (Nunc, Roskilde, Denmark) at 37°C in a humidified atmosphere (95% air/5% CO2). The cells were cultured in standard MEM culture medium (Invitrogen) supplemented with 10% heat-inactivated fetal bovine serum (Invitrogen) and 100 U/ml penicillin–streptomycin (Invitrogen). The continuous cell culture (passages 53–63) was grown until a confluent monolayer appeared, then divided 1:8, and reseeded in new flasks. Cells for experiments were divided 1:3 or 1:4 and seeded onto coverslips kept in tissue culture dishes (35 mm diameter; Nunc). The following day, 5 μm BrdU was added, and thereafter the medium was exchanged with fresh medium containing BrdU every 2 or 3 d. After 5–8 d, the cells were transduced with baculovirus, and experiments were performed 1 or 2 d later.
Baculovirus and cell transduction.
For mammalian cell transduction, we first designed a baculovirus construct to drive the expression of enhanced green fluorescent protein (EGFP). An AseI (blunted)–NotI fragment from pEGFP–N1 (BD Biosciences Clontech, Palo Alto, CA), including the cytomegalovirus (CMV) promoter and the gene for GFP, was subcloned into an SnaBI–NotI gap in pFastBac1 (Invitrogen), removing the polyhedrin promoter. The resultant vector was called pFastBac–CMV–GFP. The human OX1R cDNA in pcDNA3 (Invitrogen) was a gift from Dr. M. Detheux (Euroscreen SA, Bruxelles, Belgium). The OX1R cDNA was processed by PCR to remove the stop codon and subsequently subcloned into pEGFP–N3 (BD Biosciences Clontech). The OX1R cDNA fused to the cDNA for GFP was then transferred to pFastBac–CMV–GFP as an EcoRI–NotI fragment. An untagged OX1R construct was generated by subcloning the whole coding sequence of OX1R cDNA into pFastBac–CMV–GFP, the GFP from vector being cut out. For plasma membrane localization of the red fluorescent protein (RFP) Discosoma red (DsRed)-Monomer, a CAAX motif from K-ras, KKKKKSKTKCVIM, was added to the EcoRI–BamHI gap of pDsRed-Monomer-C1 (BD Biosciences Clontech) by ligation of two complementary oligonucleotides, and subsequently the RFP fused to CAAX was transferred to pFastBac–CMV–GFP (GFP cut out) with BshTI and SphI. The enhanced yellow fluorescent protein (EYFP)-tagged TRPC3DN was constructed by subcloning a 1620 bp BamHI-StuI (partial digest) fragment of hTRPC3 cDNA (Hofmann et al., 1999) (gift from C. Harteneck, Institute for Pharmacology, Freie Universität Berlin, Berlin, Germany) into pEYFP–C1 (BD Biosciences Clontech). The fused YFP–TRPC3DN was then transferred to pFastBac—CMV–GFP, and the GFP cDNA was replaced by complementary oligonucleotides encoding a V5 epitope, followed by a STOP codon. The hTRPC6DN fused to YFP in pcDNA3 (Hofmann et al., 2002) (gift from T. Gudermann, Institute for Pharmacology and Toxicology, Phillips Universität, Marburg, Germany) was subcloned into pFastBac–CMV–GFP with BamHI and XbaI (GFP removed). All recombinant baculoviruses were obtained using the Bac-to-Bac expression system (Invitrogen).
For transient expression in IMR-32 cells, 0.5 ml of a high titer virus stock (107 pfu/ml), originating from Sf9 cell infection, was spun down in a microcentrifuge 12,000 rpm for 30 min. The pelleted viruses were resuspended in IMR-32 cell culture medium, added back to dishes with coverslips, and incubated until experimental use.
Confocal microscopy.
Transduced cells were washed once with PBS and fixed for 30 min in 4% paraformaldehyde containing PBS. After removal of fixative, cells were washed four times with PBS, and then coverslips were mounted on glass slides. Confocal images were obtained using a Nikon (Tokyo, Japan) 100× (1.30 numerical aperture) Plan Fluor oil immersion objective with an Eclipse TE300 inverted microscope (Nikon) equipped with a Radiance 2100 confocal scanner (Bio-Rad, Hertfordshire, UK) under the control of LaserSharp 2000 software (Bio-Rad). GFP was excited at 488 nm with an argon laser, and RFP was excited at 543 nm with a green helium–neon laser. Images were acquired using a Kalman filter (n = 8).
Fura-2 imaging.
Fura-2 AM was dissolved in dimethylsulfoxide to a concentration of 4 mm. Cells on coverslips were loaded with 4 μm fura-2 AM for 20 min and subsequently transferred to a perfusion chamber. The imaging experiments were performed using an InCyt2 fluorescence imaging system (Intracellular Imaging, Cincinnati, OH) essentially as described previously (Larsson et al., 2005). The perfusion HEPES-buffered Na+ medium (HBM) consisted of the following (in mm): 137 NaCl, 5 KCl, 1 CaCl2, 0.44 KH2PO4, 4.2 NaHCO3, 10 glucose, 20 HEPES, and 1.2 MgCl2, pH adjusted to 7.4 with NaOH. In the nominally Ca2+-free HBM, no CaCl2 was added. In the high K+ HBM, NaCl was replaced by KCl, and the medium was diluted with HBM to achieve the desired K+ concentration. The cells were perfused in HBM at 37°C and excited by alternating wavelengths of 340 and 380 nm using narrow band excitation filters. Fluorescence was measured through a 430 nm dichroic mirror and a 510 nm barrier filter with a Cohu (San Diego, CA) CCD camera. One ratioed image was acquired per second.
Identification of TRP channel mRNA.
Total RNA was extracted from differentiated cells using TRIzol (Invitrogen). Cells differentiated for 6, 8, and 10 d were used. Total RNA (5 μg) was used to make cDNA using SuperscriptII and oligo-dT (Invitrogen). An aliquot of the first-strand cDNA template (approximately the equivalent of 250 ng total RNA) was amplified with an annealing temperature of 55°C for 30 cycles with Dynazyme II (Finnzymes, Espoo, Finland) using specific primers (Table 1). The PCR reactions were electrophoretically analyzed on 2% agarose gels and stained with ethidium bromide, and images were collected using a GelDoc imaging system (Bio-Rad). Amplified DNA fragments were gel purified and ligated into pGemTeasy (Promega, Madison, WI) and sequenced. Sequences were identified using the BLAST (basic local alignment search tool) program (Altschul et al., 1997).
Table 1.
Primer pairs used for detection of TRPC channel transcripts
| Channel subtype | Primer sequence | Expected size | Intron in amplicon | Gene location |
|---|---|---|---|---|
| TRPC1–5′ | GGGTCCATTACAGATTTCAA | 207 bp | 1 | chr 3 |
| TRPC1–3′ | AAGCAGGTGCCAATGAACGA | |||
| TRPC3–5′ | GTATGTGGACAGTTACGTC | 553 bp | 2 | chr 4 |
| TRPC3–3′ | CTACATCACTGTCATCCTC | |||
| TRPC4–5′ | TGGGATGGCGGACTTCAG | 391 bp | 1 | chr 13 |
| TRPC4–3′ | ATGCCTTTGCAGGTTAACCC | |||
| TRPC5–5′ | GTGGAGAAGGGGGACTATGC | 525 bp | 1 | chr X |
| TRPC5–3′ | CCTCACTTGATAAGGCAATG | |||
| TRPC6–5′ | CTCTGAAGGTCTTTATGC | 428 bp | 1 | chr 11 |
| TRPC6–3′ | TCATCCTCAATTTCCTGG | |||
| TRPC7–5′ | AACCCAGCGTTTACAACG | 361 bp | 1 | chr 5 |
| TRPC7–3′ | ATGAGGCACATCTTGATTC |
Primers were designed according to sequences available from the European Molecular Biology Laboratory database. At least one intron region was included in the amplicons to avoid amplification of genomic DNA and unprocessed RNA. Both 5′ primers and 3′ primers are written in 5′ to 3′ direction. chr, Chromosome.
Data analysis.
Fura-2 imaging data were analyzed with Microcal Software (Northampton, MA) Origin 6.0 and given as absolute [Ca2+] levels, ([Ca2+]i), or as changes in [Ca2+] levels (Δ[Ca2+]i). Traces show recordings from imaging of multiple cells simultaneously, and vertical lines indicate SDs. Every third or fifth SD is shown. Statistical significance between groups was determined with the unpaired Student's t test. Significance is depicted as *p < 0.05 or **p < 0.01 [not significant (ns), p > 0.05].
Results
Baculovirally expressed OX1 receptors in differentiated IMR-32 cells
For this study, we sought a cell line that would resemble mature neurons and that would be prone to take up foreign DNA for OX1R expression. The neuroblastoma cell line IMR-32 is an adrenergic human cell line that can be differentiated in vitro to extend long axon-like processes with numerous growth cones (Clementi et al., 1986; Carbone et al., 1990). After ∼1 week of treatment of IMR-32 cell cultures with BrdU, the cell proliferation had ceased and an extensive network of processes was visible (data not shown). We found that the differentiated cells were very susceptible to baculovirus transduction. When transduced with the OX1R–GFP baculovirus, the receptor colocalized with the CAAX motif fused to red fluorescent protein in the plasma membrane (Fig. 1a–c). Some GFP fluorescence was found in submembranous vesicle-like compartments, likely representing receptors being transported to or from the plasma membrane. The receptor fusion protein was also localized in cellular processes (Fig. 1a). The fluorescence observed when GFP was expressed alone was confined to the cytosol (data not shown).
Figure 1.

Expression of the OX1R–GFP fusion protein in IMR-32 cells. a–c, Confocal images of fusion proteins in IMR-32 cells. Cells were treated with BrdU and then transduced with recombinant baculovirus. One day after transduction, the cells were fixed and mounted for confocal imaging. a, OX1R–GFP fluorescence in differentiated cells with associated neurites. b, Image of a cell that is not fully differentiated and thereby better illustrates the plasma membrane localization of OX1R–GFP fluorescence. c, The same cell as in b showing RFP fluorescence from RFP–CAAX. d, e, [Ca2+]i increases in response to activation of the OX1R 2 d after transduction. IMR-32 cells were perfused at 37°C with HBM. Where indicated, the cells were challenged with 1 nm (d) or 3 nm (e) orexin-A (Ox-A) in a nominally Ca2+-free (−Ca2+) or Ca2+-containing HBM. The Ca2+ was removed simultaneously with application of orexin-A. The data represent averaged ± SD responses from 18 (d) and 36 (e) cells from single experiments. f, Dose–response curves in normal conditions (filled triangles) and in nominally Ca2+-free conditions (open triangles). The data represent averaged ± SEM responses from four experiments. g, The percentages ± SD of fura-2-loaded cells responding to orexin-A applications in three to eight batches of cells.
Intracellular [Ca2+] elevation in response to OX1R stimulation
Orexin-A application to transduced cells resulted in an elevation of [Ca2+]i as determined with fura-2 (Fig. 1d,e). The [Ca2+]i increase was dose dependent and consisted of store release as well as Ca2+ influx at orexin-A concentrations ≥3 nm (Fig. 1e,f). At 1 nm orexin-A, mainly an extracellular Ca2+-dependent elevation was seen (Fig. 1d,f). The viral transduction efficiency was estimated by counting how many cells responded with a [Ca2+]i elevation to orexin-A application. The efficiency reached >80% when tested with 100 nm (Fig. 1g). Of nontransduced or GFP-transduced cells, ∼2% (6 of 303 cells) responded with a [Ca2+]i elevation to 100 nm orexin-A. None responded to 10 nm (80 cells) or lower (220 cells).
Because our main interest lies on the Ca2+ entry activated by OXRs, we chose to use 1 nm orexin-A in subsequent experiments unless otherwise stated. Stimulation with 1 nm orexin-A resulted in most cells in a stable elevation of [Ca2+]i (Fig. 2a). In some cells, this was preceded by a transient spike, indicating that some store release may occur also at this concentration. It should be taken into account that, whereas few cells showed an initial spiking at this agonist concentration, the absolute [Ca2+]i increase in these cells was high enough to produce a spike in the average response in many experiments. The sustained [Ca2+]i elevation lasted for >10 min. In some experiments, a decline of the sustained response over time was seen.
Figure 2.

Effect of Mg2+ and dextromethorphan on the orexin-A-stimulated Ca2+ influx. Experimental conditions were as in Figure 1. a, Cells (average ± SD of 32 cells) were continuously challenged with 1 nm orexin-A (Ox-A) to illustrate the stable [Ca2+]i elevation. b, A trace (average ± SD of 23 cells) of cells stimulated with 1 nm orexin-A and the effect of 10 mm Mg2+, 100 μm dextromethorphan (Dex), or a nominally Ca2+-free HBM (−Ca2+). c, The averages ± SEM from six experiments measured under similar conditions as in b. d, The dose–response relationships in the presence (open triangles) or absence (filled triangles) of high [Mg2+]. The Δ[Ca2+]i increase after removal of 10 mm Mg2+ is additionally plotted (open squares). Data represent averages ± SEM from three to five experiments. e, Areas containing only processes were monitored under similar conditions as in b with a subsequent depolarization with 70 mm K+. Note that the regions of interest were larger than the areas of the processes and therefore the magnitude of the response is underestimated.
Previous studies on the OX1R expressed in nonexcitable cells have shown that the activated Ca2+ entry and the inward Ca2+-dependent current are significantly inhibited by high extracellular [Mg2+] and by dextromethorphan, a rather nonspecific calcium channel blocker (Larsson et al., 2005). To investigate whether the [Ca2+]i elevation observed in IMR-32 cells was similar to that seen in CHO cells, cells were challenged with orexin-A in the presence of 10 mm Mg2+ (Fig. 2b). Only a small and transient response was observed under these conditions. Reduction of extracellular Mg2+ to 1.2 mm in the continued presence of orexin-A caused a sustained elevation of [Ca2+]i. This [Ca2+]i elevation was reversibly blocked by 10 mm Mg2+ and by 100 μm dextromethorphan, as well as by removal of extracellular Ca2+ (Fig. 2b). Statistical analysis of experiments similar to Figure 2b shows that high [Mg2+] and dextromethorphan almost completely block the sustained response to orexin-A (Fig. 2c). Similar types of experiments performed with higher doses of orexin-A indicated that the influx phase is partially Mg2+ sensitive at concentrations leading to store release as well (Fig. 2d) (see also Fig. 7c). Because the OX1R–GFP receptor also was detected in neurites, it was of interest to test the response to orexin-A in these structures. Figure 2e shows a fura-2 recording from varicose-like structures on neurites. An Mg2+- and dextromethorphan-sensitive [Ca2+]i elevation was also apparent here.
Figure 7.

PKC-mediated inhibition of OX1R response. Experimental conditions were as in Figure 1. a, b, The effect of DOG and GF109203X on orexin-A (1 nm) response. b, Bar graph presentation of average ± SEM responses from three to four experiments. The Δ[Ca2+]i denotes changes from basal level or from the DOG response level when DOG was included. c, TPA at 100 nm was used to test the effect of PKC activation on the sustained response to 1 nm orexin-A (average ± SD of 15 cells). d, Statistical analysis was performed on data from three experiments.
Role of VGCCs in orexin-A-stimulated Ca2+ elevation
Orexins have been reported to depolarize cells via several different mechanisms (for review, see Kukkonen et al., 2002; Ferguson and Samson, 2003). Depolarization and a subsequent opening of VGCCs could potentially account for the OX1R-mediated Ca2+ entry in IMR-32 cells. We thus performed a series of experiments that ought to reveal whether such mechanisms were involved. It is known that dextromethorphan also acts on VGCCs (Shariatmadari et al., 2001). We tested the effect of high [Mg2+] on the depolarization-induced Ca2+ influx (Fig. 3a). We could not detect any difference in the magnitude of [Ca2+]i elevation when compared with control. It is of course possible that a minor component of VGCCs, which would not be detectable because of the robust [Ca2+]i increase, could be blocked by high [Mg2+]. Differentiation of IMR-32 cells leads to an upregulation of mainly N-type VGCCs, which can be blocked with ωCTx (Carbone et al., 1990). The depolarization-induced Ca2+ influx was to a large extent abolished in the presence of 0.5 μm ωCTx (Fig. 3b). ωCTx had no effect on the OX1R-mediated Ca2+ influx (Fig. 3c,d). The residual depolarization-induced Ca2+ influx in the presence of ωCTx was partially attributable to nimodipine-sensitive VGCCs (presumably of L-type). Because nimodipine block of this residual Ca2+ influx was inconsistent and appeared to decrease with increased differentiation times, we chose to study the effect of orexin-A in ωCTx-treated cells that also received a depolarizing buffer before and during orexin-A application (Fig. 3c). The OX1R response was still present and not apparently different from control, suggesting that the OX1R response is not attributable to depolarization-induced opening of VGCCs.
Figure 3.

VGCCs in differentiated IMR-32 cells. Cells treated with BrdU were perfused at 37°C with HBM. Depolarization-induced [Ca2+]i elevation with 70 mm K+ (using an isotonic K+-based HBM) in the presence or absence of 10 mm Mg2+ (a) and in the presence or absence of 0.5 μm ωCTx (b). Cells were pretreated with ωCTx for 20 min before the experiment. c, Transduced cells were challenged with 1 nm orexin-A (Ox-A) in the presence or absence of 0.5 μm ωCTx. In the presence of ωCTx, the depolarizing buffer was used to activate residual VGCCs with a subsequent 1 nm orexin-A application. The data in a–c represent averaged ± SD responses from single representative experiments (10–24 cells). d, Bar graph (averages ± SEM; n = 4) on the effect of ωCTx on orexin-A response.
Role of store release and Na+/Ca2+ exchange in the OX1R response
Ca2+ entry in response to depletion of intracellular Ca2+ stores occurs via the store-operated channels (for review, see Parekh and Putney, 2005). To test whether this type of channel is involved in Ca2+ elevation caused by orexin-A, cells were treated with thapsigargin to discharge Ca2+ from intracellular stores. Application of 100 nm thapsigargin caused a slowly developing, sustained elevation of [Ca2+]i (Fig. 4a). Application of high [Mg2+] had no effect on the [Ca2+]i, suggesting an entirely different Ca2+ entry channel for capacitative store refilling compared with the OX1R-activated channel. Dextromethorphan had a small inhibitory effect on the thapsigargin signal, and, as expected, removal of extracellular Ca2+ reversibly lowered the [Ca2+]i (Fig. 4a,b).
Figure 4.
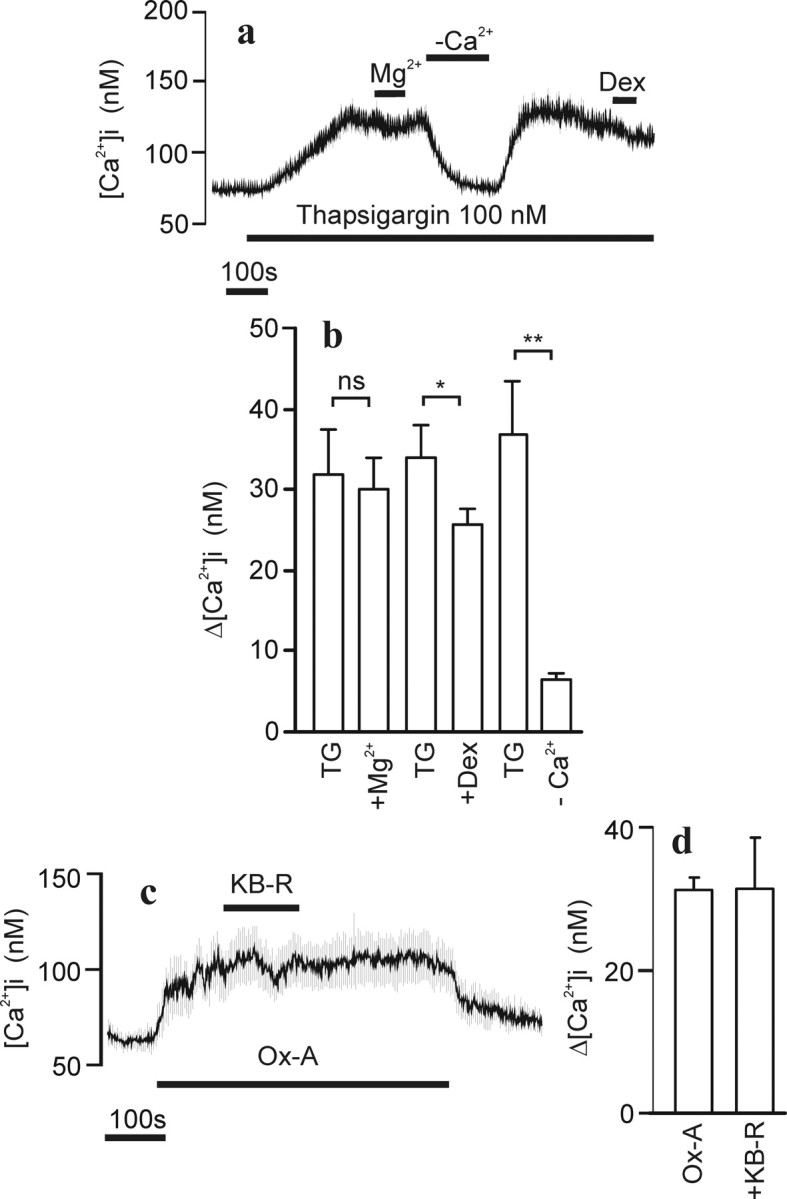
Ca2+ store release and Na+/Ca2+ exchange in relation to orexin-A responses. Experimental conditions were as in Figure 1. a, Cells (average ± SD of 40 cells) were challenged with 100 nm thapsigargin (TG), 10 mm Mg2+, a nominally Ca2+-free HBM (−Ca2+), and 100 μm dextromethorphan (Dex) where indicated. b, The averages ± SEM from four experiments treated under similar conditions as in a. c, Cells (average ± SD of 23 cells) were challenged with 1 nm Ox-A and 10 μm KB-R7943 (KB-R) transiently added as indicated. d, Bar graph presentation of the average ± SEM effect of KB-R from three experiments.
The electrogenic Na+/Ca2+ exchanger has been implicated in OXR-mediated neuronal excitation (Eriksson et al., 2001; Burdakov et al., 2003; Wu et al., 2004). It has also been shown to associate with TRPC3, a putative effector channel of OX1R (Larsson et al., 2005), and to increase [Ca2+]i as a consequence of TRPC3 activation attributable to the reverse mode of action (Rosker et al., 2004). A potent blocker of the reverse operation mode of Na+/Ca2+ exchange, KB-R7943 (Iwamoto et al., 1996), did not significantly alter the response to orexin-A in IMR-32 cells when averages of cells were plotted and compared (Fig. 4c,d). It should be noted, however, that when analyzed on a single-cell level, there appeared to be three populations of responding cells: one (∼10%) that showed a decrease in [Ca2+]i when KB-R7943 was applied during the stable phase of OX1R activation, a second population with unaltered response, and a third population (∼15%) that showed an increase in [Ca2+]i with KB-R7943 (data not shown). The reason for this variability is unknown at present, but, based on the average effect of KB-R7943, we do not consider the exchange mechanism as a main contributor to the Ca2+ influx pathway under investigation in the present study.
Diacylgycerol-activated [Ca2+]i elevation and protein kinase C
Because TRPC channels were implicated in the OX1R response in a previous study (Larsson et al., 2005), we turned our focus to these. Messenger RNAs for all human TRPC-type channels were detected in these cells as determined using RT-PCR (Fig. 5). The TRPC3/6/7 subfamily can be activated with DAG (Hofmann et al., 1999; Tesfai et al., 2001; Jung et al., 2002). Application of 30 μm DOG, a membrane-permeant DAG analog, caused a small irregular [Ca2+]i elevation in a large proportion of the differentiated cells (Fig. 6a). In many of the cells, transient spiky [Ca2+]i elevations were also seen (data not shown). The DOG-induced [Ca2+]i elevation was sensitive to high [Mg2+] (Fig. 6b) and to dextromethorphan (Fig. 6c). Because DAGs also activate protein kinase C (PKC), which is known to inhibit several TRPC channel subtypes (Trebak et al., 2003; Venkatachalam et al., 2003), the effect of a PKC inhibitor, GF109203X, was tested. The inhibitor significantly enhanced the response to DOG. Data for DOG effects is compiled in Figure 6d.
Figure 5.

Identification of TRPC1–TRPC7 channel mRNAs in differentiated IMR-32 cells. The PCR-amplified DNA was separated on a 2% agarose gel and stained with ethidium bromide. Bands were visualized by UV transillumination and imaged using Bio-Rad Gel Doc 2000. The 200 and 500 bp bands of the 100 bp DNA size ladder are indicated.
Figure 6.

DOG-stimulated [Ca2+]i elevation. Experimental conditions were as in Figure 1. a, Cells (average ± SD of 33 cells) were challenged with 30 μm DOG, and, in b and c, the effect of 10 mm Mg2+ and 100 μm dextromethorphan (Dex) is shown, respectively. d, Statistical analysis (averages ± SEM; n = 3–4) of similar experiments as in a–c. The DOG response varied somewhat between cell batches and was therefore always analyzed separately for each batch.
Stimulation of the OX1R during DOG application caused only a small, additional elevation of [Ca2+]i (Fig. 7a,b) or, in a significant number of cells, no additional elevation at all (data not shown). In the presence of GF109203X, the OX1R response was restored and even enhanced when compared with control conditions (Fig. 7a,b).
The results above suggested that the OX1R response is linked to the DOG-activated channels and that these channels are inhibited by activated PKC. To activate PKC, without a concurrent [Ca2+]i elevation, we used TPA, a mimic of DAG in activation of PKC. The OX1R-activated Ca2+ entry was considerably reduced by TPA application, and the reduction was partially reversed by GF109203X (Fig. 7c,d).
Dominant-negative inhibition of the OX1R response
To get additional evidence for TRPC channel involvement in the OX1R response, we coexpressed a truncated dominant-negative TRPC3 (C3DN) construct, shown previously to inhibit OX1R-mediated Ca2+ entry in CHO cells (Larsson et al., 2005). Because we found it more important to have a tagged C3DN construct rather than the receptor, we expressed an untagged OX1R for these experiments. There was no observable difference between the tagged and untagged receptor at the Ca2+ signaling level (data not shown). Quantitative determination of the effects of DN constructs on [Ca2+]i elevation in differentiated IMR-32 cells was complicated by several factors. First, in coexpression experiments, only a proportion of the cells that responded to orexin-A expressed sufficient amount of the DN construct as determined by YFP fluorescence. Second, during differentiation, the cells tend to form aggregates whereupon the responses of neighboring cells, not expressing DN constructs, contaminated the observed response (as seen from the response profile). For that reason, we performed measurements only from areas containing individual cells. We also chose an experimental approach enabling measurement of the Ca2+ response sensitive to 10 mm Mg2+ to exclude signals attributable to Ca2+ discharge or capacitative entry. In addition, we set up two criteria to be fulfilled in the data processing: (1) the response to oxotremorine-M, acting on endogenous muscarinic receptors (Kukkonen et al., 1992), should exceed 300 nm [Ca2+]i as a measure of healthy, viable cells, and (2) the peak response, arising from store release and not influenced by DN constructs, to 10 nm orexin-A should exceed 250 nm [Ca2+]i as a measure of sufficiently high receptor expression to yield a “normal” response to 1 nm orexin-A. When these criteria were met, the C3DN had only a minor inhibitory effect on the response to 1 nm orexin-A (Δ[Ca2+]i of 68 ± 8 nm in control versus 48 ± 9 nm with C3DN; p > 0.05).
The dominant-negative strategy is based on incorporation of nonfunctional channel subunits into either homomeric or heteromeric channel complexes. This is governed, at least chemically, by the affinity between subunits and the availability of proteins. Given that the DAG-activated TRPC3/6/7 subfamily are able to form heteromeric complexes with each other (Hofmann et al., 2002), one would expect a DN construct from this subfamily to distort all DAG-activated channel complexes. Because the C3DN had so small an effect on the OX1R response, we reasoned that this could be attributable to too low expression level or the inability of this truncated construct to interact sufficiently well with the endogenous related channel subunits. We therefore tested a full-length, triple-mutated TRPC6 dominant-negative construct (C6DN) (Hofmann et al., 2002). Using the same criteria as above, we found that the C6DN almost totally abolished the response to DOG (Fig. 8a,b,e). When tested with orexin-A applications, the response to 1 nm was significantly inhibited, whereas the 10 nm peak response was unaltered (Fig. 8c,d,f). The 10 nm steady-state response was also slightly inhibited, indicating, as shown previously, that the Mg2+-sensitive Ca2+ entry is inherent to the OX1R response regardless of agonist concentration.
Figure 8.

Effect of dominant-negative TRPC6 channel subunit expression. Experimental conditions were as in Figure 1 except that 10 μm GF109203X was included in HBM in all experiments. a, b, Representative traces of the effect of 60 μm DOG and 10 mm Mg2+ in control cells (a, average ± SD of 24 cells) and cells transduced with C6DN (b, average ± SD of 14 cells). e, Compiled data from three experiments. c, d, Representative traces of cells challenged with orexin-A (Ox-A) and oxotremorine M (Oxo-M) in batches transduced with the OX1R (c, average ± SD of 21 cells) or with the OX1R and C6DN (d, average ± SD of 14 cells). f, Bar graph presentation of data from 10–12 experiments. Data for 1 nm orexin-A (Ox-A 1) was collected from peak responses after removal of high [Mg2+]. Data for 10 nm orexin-A was collected from both peak response in the presence of high [Mg2+] (Ox-A 10 peak) and steady-state responses after removal of high [Mg2+] (Ox-A 10 ss). Peak responses were collected for 30 μm Oxo-M.
Discussion
The IMR-32 neuroblastoma cells used in this study undergo a striking functional and morphological differentiation to mature neuron-like cells when treated with BrdU (Carbone et al., 1990). We found that the differentiated cells were surprisingly susceptible to baculovirus-mediated gene expression. It has been reported previously that baculovirus can enter mammalian cells, including undifferentiated neuroblastoma cells (Shoji et al., 1997; Sarkis et al., 2000; Tani et al., 2003), to drive expression of recombinant proteins (for review, see Kost and Condreay, 2002). It has also been demonstrated that baculovirus enters mature neurons in vivo, albeit with low efficiency (Sarkis et al., 2000). To our knowledge, this is the first report on baculovirus-transduced differentiated neuroblastoma cells. Although we did not perform a systematic study on the transduction efficiency, it was evident that, with an estimated 5–10 plaque forming units per cell, a large proportion of cells (≥80%) expressed the recombinant OX1R–GFP protein as determined by counting of fluorescent cells and by counting of cells responding to orexin-A application.
When challenged with orexin-A, the OX1R–GFP-transduced cells responded with an increase in [Ca2+]i. At low agonist concentrations, a measurable Ca2+ release from intracellular stores was essentially absent. This is in agreement with previous studies (van den Pol et al., 1998; van den Pol, 1999; Lund et al., 2000; Uramura et al., 2001; Holmqvist et al., 2002; Willie et al., 2003; Kohlmeier et al., 2004) and indicates Ca2+ entry as a primary OX1R response. In neuronal cells, a Ca2+ elevation in response to G-protein-coupled receptors could be the consequence of several different mechanisms, including activation of VGCC, reversal of electrogenic Na+/Ca2+ exchange, or phospholipase C-linked Ca2+ store discharge with a consequent activation of store-operated channels. We excluded the involvement of N-type VGCC by the lack of an effect of ωCTx on the OX1R response. An effect on the residual non-N-type VGCC seems unlikely because orexin-A still activated Ca2+ entry during depolarization in the presence of ωCTx. Conversely, elevated [Mg2+], which strongly blocked the response to orexin-A, did not affect the response to elevated [K+]. OX1R-activated Ca2+ entry was also apparent in the presence of 10 μm KB-R7943, a concentration at which this compound should selectively block the reverse mode of Na+/Ca2+ exchange (Iwamoto et al., 1996). Ca2+ entry has been shown previously to be separated from release-induced Ca2+ entry at low agonist concentrations in CHO cells (Lund et al., 2000; Larsson et al., 2005). High Mg2+ concentrations or dextromethorphan inhibited the OX1R-mediated Ca2+ entry in IMR-32 cells, whereas there was no or only a weak effect on thapsigargin-stimulated Ca2+ entry. This indicates that Ca2+ is entering through separate entities in the two cases.
The TRP channel family, in particular the TRPC channels, represents the most likely candidate as entities for Ca2+ entry activated by G-protein-coupled receptors (for review, see Minke and Cook, 2002; Clapham, 2003). In differentiated IMR-32 cells, we found transcripts of all members of the TRPC subfamily. An involvement of TRPC channels in the OX1R response was indicated by the reduced Mg2+-sensitive Ca2+ entry at low orexin-A concentrations on expression of the C6DN construct. In the same cells, the Mg2+-insensitive [Ca2+]i elevation (mainly Ca2+ release from stores) as well as [Ca2+]i elevation by muscarinic receptor stimulation were essentially unaffected. Several other features of the Ca2+ response to orexin-A observed in this study suggest an involvement of TRP channels. The sensitivity to block by high [Mg2+] has been demonstrated for several TRPC channels (Schaefer et al., 2000; Larsson et al., 2005). Dextromethorphan, an NMDA receptor channel blocker, which also blocks a variety of other Ca2+-permeable channels (Shariatmadari et al., 2001), including overexpressed TRPC3 channels (Larsson et al., 2005), also blocked the response to orexin-A in IMR-32 cells. DAG is a known activator of the TRPC3, TRPC6, and TRPC7 channel subunits (Hofmann et al., 1999; Tesfai et al., 2001; Jung et al., 2002). Functional evidence for the presence of these channels in differentiated IMR-32 cells comes from the DOG-induced [Ca2+]i elevation, which was nearly abolished in C6DN-expressing cells. The DOG-induced [Ca2+]i elevation was also sensitive to high [Mg2+] and to dextromethorphan in a manner similar to the response to low concentrations of orexin-A. The TRPC channels have been suggested to form both homotetrameric and heterotetrameric channel complexes, and the heteromeric complexes would be confined within certain subfamilies, such as TRPC3/6/7 (Hofmann et al., 2002). In agreement with this, the C6DN has been demonstrated to inhibit ion fluxes through both TRPC6 and TRPC3 channels (Hofmann et al., 2002). It thus appears likely that the TRPC3/6/7 subfamily makes up channel complexes engaged in the OX1R-mediated Ca2+ influx in IMR-32 cells.
If the DAG-activated channels were the target for the OX1R, one would expect DAG to modify the response to orexin-A. DAGs are, in addition to their ability to activate TRP channels, endogenous activators of PKC. TRP channels, in particular TRPC3, TRPC5, TRPC6, and TRPC7, are inhibited by activation of PKC (Trebak et al., 2003, 2005; Venkatachalam et al., 2003). This is considered to represent a feedback mechanism to fine tune the magnitude of the Ca2+ elevation and to prevent Ca2+ overload (Trebak et al., 2003). The reduced responsiveness to orexin-A in the presence of DOG and the enhanced response of DOG in the presence of the PKC inhibitor GF109203X are in agreement with this. A similar enhancement of DAG-stimulated TRPC3 channels with GF109203X has been demonstrated previously (Venkatachalam et al., 2003). In addition, TPA, an activator of PKC, which does not activate TRPC3/6/7 nor by itself increase [Ca2+]i in IMR-32 cells, inhibited the response to 1 nm orexin-A, and this inhibition was partially reversed by GF109203X. Surprisingly, the magnitude of the response to orexin-A in the presence of DOG and GF109203X was actually larger than that seen under control conditions, indicating that DOG potentiates the receptor response. This suggests that DAG is not necessarily the sole signal for channel activation but may rather function as a coactivator in addition to other more specific receptor-generated signals.
How does our finding relate to other studies performed on neurons? A [Ca2+]i elevation in response to orexins has been described in several instances (van den Pol et al., 1998; van den Pol, 1999; Uramura et al., 2001; Kohlmeier et al., 2004; Muroya et al., 2004). One of the first reports on orexin action, which was performed on hypothalamic neurons, described an activated Ca2+ entry with no measurable membrane current (van den Pol et al., 1998). A difference between the Ca2+ entry in hypothalamic neurons and the Ca2+ entry evoked in IMR-32 cells is the apparent opposite regulation by PKC. van den Pol et al. (1998) found that the same inhibitor of PKC that we used totally blocked the [Ca2+]i increase. In several other studies, PKC has been suggested to be a crucial intermediate effector in orexin action, often with downstream effects linked to specific ion channels (Uramura et al., 2001; Xu et al., 2002; Kohlmeier et al., 2004) or less well defined ion conductances (Yang et al., 2003). PKC has long been known to modulate VGCCs (Yang and Tsien, 1993) and K+ conductances (Henry et al., 1996). In our current study, the pharmacological tools used to probe for PKC action all appeared to indicate a blocking role for PKC on the OX1R-mediated Ca2+ entry. The PKC- and Mg2+-sensitive Ca2+ entry shown here and previously in nonexcitable cells precedes other actions of orexin from a dose–response point of view and is thus expected to be a primary response (Kukkonen et al., 2002). It is possible that this signaling pathway is activated in parallel with VGCCs in native neurons as well but may be masked because of its sensitivity to PKC activity.
At orexin-A concentrations ≥3 nm, there was a clear release of Ca2+ from internal stores in IMR-32 cells. This is frequently observed in heterologous expression systems with both OX1R and OX2R (Smart et al., 1999; Holmqvist et al., 2002; Ammoun et al., 2003) and is likely to be a consequence of activation of phospholipase C (PLC) and the generation of IP3. Some studies on native receptors in neurons have demonstrated sensitivity to inhibitors of phosphatidylinositol-specific PLC (Zhu et al., 2003; Muroya et al., 2004), suggesting that some store release may take place. Conversely, a Ca2+ release from intracellular stores has not been demonstrated in native neurons although explicitly tested for (van den Pol et al., 1998; Kohlmeier et al., 2004). It is possible that the OXRs are highly compartmentalized in native neurons and therefore the Ca2+ release may occur only in localized sparks, as would be the case also with the release-independent Ca2+ entry.
In conclusion, the primary response of the OX1R at low concentrations of orexin-A in differentiated IMR-32 cells is dependent on the activation of a Ca2+-permeable channel with many properties such as those described for defined TRPC channel subtypes, including block by Mg2+, potentiation by DAG, and regulation by PKC. Higher agonist concentrations additionally activate Ca2+ store release with a subsequent pharmacologically different store-operated Ca2+ entry.
Footnotes
This study was supported by European Union Contracts ERBBIO4CT960699 and QLG3-CT-2002-00826, the Academy of Finland, the Sigrid Jusélius Foundation, and the Magnus Ehrnrooth Foundation. We thank Dr. M. Detheux for the OX1R cDNA, Dr. C. Harteneck for the TRPC3 cDNA, and Dr. T. Gudermann for the dominant-negative TRPC6. Veera Pevgonen is acknowledged for the laboratory assistance.
References
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ammoun S, Holmqvist T, Shariatmadari R, Oonk HB, Detheux M, Parmentier M, Akerman KE, Kukkonen JP. Distinct recognition of OX1 and OX2 receptors by orexin peptides. J Pharmacol Exp Ther. 2003;305:507–514. doi: 10.1124/jpet.102.048025. [DOI] [PubMed] [Google Scholar]
- Burdakov D, Liss B, Ashcroft FM. Orexin excites GABAergic neurons of the arcuate nucleus by activating the sodium–calcium exchanger. J Neurosci. 2003;23:4951–4957. doi: 10.1523/JNEUROSCI.23-12-04951.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbone E, Sher E, Clementi F. Ca currents in human neuroblastoma IMR32 cells: kinetics, permeability and pharmacology. Pflügers Arch. 1990;416:170–179. doi: 10.1007/BF00370239. [DOI] [PubMed] [Google Scholar]
- Chemelli RM, Willie JT, Sinton CM, Elmquist JK, Scammell T, Lee C, Richardson JA, Williams SC, Xiong Y, Kisanuki Y, Fitch TE, Nakazato M, Hammer RE, Saper CB, Yanagisawa M. Narcolepsy in orexin knockout mice: molecular genetics of sleep regulation. Cell. 1999;98:437–451. doi: 10.1016/s0092-8674(00)81973-x. [DOI] [PubMed] [Google Scholar]
- Clapham DE. TRP channels as cellular sensors. Nature. 2003;426:517–524. doi: 10.1038/nature02196. [DOI] [PubMed] [Google Scholar]
- Clementi F, Cabrini D, Gotti C, Sher E. Pharmacological characterization of cholinergic receptors in a human neuroblastoma cell line. J Neurochem. 1986;47:291–297. doi: 10.1111/j.1471-4159.1986.tb02861.x. [DOI] [PubMed] [Google Scholar]
- Date Y, Ueta Y, Yamashita H, Yamaguchi H, Matsukura S, Kangawa K, Sakurai T, Yanagisawa M, Nakazato M. Orexins, orexigenic hypothalamic peptides, interact with autonomic, neuroendocrine and neuroregulatory systems. Proc Natl Acad Sci USA. 1999;96:748–753. doi: 10.1073/pnas.96.2.748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lecea L, Kilduff TS, Peyron C, Gao X, Foye PE, Danielson PE, Fukuhara C, Battenberg EL, Gautvik VT, Bartlett FS, II, Frankel WN, van den Pol AN, Bloom FE, Gautvik KM, Sutcliffe JG. The hypocretins: hypothalamus-specific peptides with neuroexcitatory activity. Proc Natl Acad Sci USA. 1998;95:322–327. doi: 10.1073/pnas.95.1.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson KS, Sergeeva O, Brown RE, Haas HL. Orexin/hypocretin excites the histaminergic neurons of the tuberomammillary nucleus. J Neurosci. 2001;21:9273–9279. doi: 10.1523/JNEUROSCI.21-23-09273.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson AV, Samson WK. The orexin/hypocretin system: a critical regulator of neuroendocrine and autonomic function. Front Neuroendocrinol. 2003;24:141–150. doi: 10.1016/s0091-3022(03)00028-1. [DOI] [PubMed] [Google Scholar]
- Hara J, Beuckmann CT, Nambu T, Willie JT, Chemelli RM, Sinton CM, Sugiyama F, Yagami K, Goto K, Yanagisawa M, Sakurai T. Genetic ablation of orexin neurons in mice results in narcolepsy, hypophagia, and obesity. Neuron. 2001;30:345–354. doi: 10.1016/s0896-6273(01)00293-8. [DOI] [PubMed] [Google Scholar]
- Henry P, Pearson WL, Nichols CG. Protein kinase C inhibition of cloned inward rectifier (HRK1/KIR2.3) K+ channels expressed in Xenopus oocytes. J Physiol (Lond) 1996;495:681–688. doi: 10.1113/jphysiol.1996.sp021625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann T, Obukhov AG, Schaefer M, Harteneck C, Gudermann T, Schultz G. Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature. 1999;397:259–263. doi: 10.1038/16711. [DOI] [PubMed] [Google Scholar]
- Hofmann T, Schaefer M, Schultz G, Gudermann T. Subunit composition of mammalian transient receptor potential channels in living cells. Proc Natl Acad Sci USA. 2002;99:7461–7466. doi: 10.1073/pnas.102596199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmqvist T, Åkerman KE, Kukkonen JP. Orexin signaling in recombinant neuron-like cells. FEBS Lett. 2002;526:11–14. doi: 10.1016/s0014-5793(02)03101-0. [DOI] [PubMed] [Google Scholar]
- Horvath TL, Peyron C, Diano S, Ivanov A, Aston-Jones G, Kilduff TS, van Den Pol AN. Hypocretin (orexin) activation and synaptic innervation of the locus coeruleus noradrenergic system. J Comp Neurol. 1999;415:145–159. [PubMed] [Google Scholar]
- Iwamoto T, Watano T, Shigekawa M. A novel isothiourea derivative selectively inhibits the reverse mode of Na+/Ca2+ exchange in cells expressing NCX1. J Biol Chem. 1996;271:22391–22397. doi: 10.1074/jbc.271.37.22391. [DOI] [PubMed] [Google Scholar]
- Jung S, Strotmann R, Schultz G, Plant TD. TRPC6 is a candidate channel involved in receptor-stimulated cation currents in A7r5 smooth muscle cells. Am J Physiol Cell Physiol. 2002;282:C347–C359. doi: 10.1152/ajpcell.00283.2001. [DOI] [PubMed] [Google Scholar]
- Kohlmeier KA, Inoue T, Leonard CS. Hypocretin/orexin peptide signaling in the ascending arousal system: elevation of intracellular calcium in the mouse dorsal raphe and laterodorsal tegmentum. J Neurophysiol. 2004;92:221–235. doi: 10.1152/jn.00076.2004. [DOI] [PubMed] [Google Scholar]
- Kost TA, Condreay JP. Recombinant baculoviruses as mammalian cell gene-delivery vectors. Trends Biotechnol. 2002;20:173–180. doi: 10.1016/s0167-7799(01)01911-4. [DOI] [PubMed] [Google Scholar]
- Kukkonen J, Ojala P, Näsman J, Hämäläinen H, Heikkilä J, Åkerman KEO. Muscarinic receptor subtypes in human neuroblastoma cell lines SH-SY5Y and IMR-32 as determined by receptor binding, Ca2+ mobilization and northern blotting. J Pharmacol Exp Ther. 1992;263:1487–1493. [PubMed] [Google Scholar]
- Kukkonen JP, Holmqvist T, Ammoun S, Åkerman KE. Functions of the orexinergic/hypocretinergic system. Am J Physiol Cell Physiol. 2002;283:C1567–C1591. doi: 10.1152/ajpcell.00055.2002. [DOI] [PubMed] [Google Scholar]
- Larsson KP, Peltonen HM, Bart G, Louhivuori LM, Penttonen A, Antikainen M, Kukkonen JP, Åkerman KE. Orexin-A-induced Ca2+ entry: evidence for involvement of trpc channels and protein kinase C regulation. J Biol Chem. 2005;280:1771–1781. doi: 10.1074/jbc.M406073200. [DOI] [PubMed] [Google Scholar]
- Lin L, Faraco J, Li R, Kadotani H, Rogers W, Lin X, Qiu X, de Jong PJ, Nishino S, Mignot E. The sleep disorder canine narcolepsy is caused by a mutation in the hypocretin (orexin) receptor 2 gene. Cell. 1999;98:365–376. doi: 10.1016/s0092-8674(00)81965-0. [DOI] [PubMed] [Google Scholar]
- Liu RJ, van den Pol AN, Aghajanian GK. Hypocretins (orexins) regulate serotonin neurons in the dorsal raphe nucleus by excitatory direct and inhibitory indirect actions. J Neurosci. 2002;22:9453–9464. doi: 10.1523/JNEUROSCI.22-21-09453.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund PE, Shariatmadari R, Uustare A, Detheux M, Parmentier M, Kukkonen JP, Akerman KE. The orexin OX1 receptor activates a novel Ca2+ influx pathway necessary for coupling to phospholipase C. J Biol Chem. 2000;275:30806–30812. doi: 10.1074/jbc.M002603200. [DOI] [PubMed] [Google Scholar]
- Minke B, Cook B. TRP channel proteins and signal transduction. Physiol Rev. 2002;82:429–472. doi: 10.1152/physrev.00001.2002. [DOI] [PubMed] [Google Scholar]
- Muroya S, Funahashi H, Yamanaka A, Kohno D, Uramura K, Nambu T, Shibahara M, Kuramochi M, Takigawa M, Yanagisawa M, Sakurai T, Shioda S, Yada T. Orexins (hypocretins) directly interact with neuropeptide Y, POMC and glucose-responsive neurons to regulate Ca2+ signaling in a reciprocal manner to leptin: orexigenic neuronal pathways in the mediobasal hypothalamus. Eur J Neurosci. 2004;19:1524–1534. doi: 10.1111/j.1460-9568.2004.03255.x. [DOI] [PubMed] [Google Scholar]
- Parekh AB, Putney JW., Jr Store-operated calcium channels. Physiol Rev. 2005;85:757–810. doi: 10.1152/physrev.00057.2003. [DOI] [PubMed] [Google Scholar]
- Peyron C, Tighe DK, van den Pol AN, de Lecea L, Heller HC, Sutcliffe JG, Kilduff TS. Neurons containing hypocretin (orexin) project to multiple neuronal systems. J Neurosci. 1998;18:9996–10015. doi: 10.1523/JNEUROSCI.18-23-09996.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosker C, Graziani A, Lukas M, Eder P, Zhu MX, Romanin C, Groschner K. Ca2+ signaling by TRPC3 involves Na+ entry and local coupling to the Na+/Ca2+ exchanger. J Biol Chem. 2004;279:13696–13704. doi: 10.1074/jbc.M308108200. [DOI] [PubMed] [Google Scholar]
- Sakurai T, Amemiya A, Ishii M, Matsuzaki I, Chemelli RM, Tanaka H, Williams SC, Richardson JA, Kozlowski GP, Wilson S, Arch JR, Buckingham RE, Haynes AC, Carr SA, Annan RS, McNulty DE, Liu WS, Terrett JA, Elshourbagy NA, Bergsma DJ, Yanagisawa M. Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell. 1998;92:573–585. doi: 10.1016/s0092-8674(00)80949-6. [DOI] [PubMed] [Google Scholar]
- Sarkis C, Serguera C, Petres S, Buchet D, Ridet JL, Edelman L, Mallet J. Efficient transduction of neural cells in vitro and in vivo by a baculovirus-derived vector. Proc Natl Acad Sci USA. 2000;97:14638–14643. doi: 10.1073/pnas.260472897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer M, Plant TD, Obukhov AG, Hofmann T, Gudermann T, Schultz G. Receptor-mediated regulation of the nonselective cation channels TRPC4 and TRPC5. J Biol Chem. 2000;275:17517–17526. doi: 10.1074/jbc.275.23.17517. [DOI] [PubMed] [Google Scholar]
- Sergeeva OA, Korotkova TM, Scherer A, Brown RE, Haas HL. Co-expression of non-selective cation channels of the transient receptor potential canonical family in central aminergic neurones. J Neurochem. 2003;85:1547–1552. doi: 10.1046/j.1471-4159.2003.01806.x. [DOI] [PubMed] [Google Scholar]
- Shariatmadari R, Lund PE, Krijukova E, Sperber GO, Kukkonen JP, Åkerman KE. Reconstitution of neurotransmission by determining communication between differentiated PC12 pheochromocytoma and HEL 92.1.7 erythroleukemia cells. Pflügers Arch. 2001;442:312–320. doi: 10.1007/s004240000513. [DOI] [PubMed] [Google Scholar]
- Shoji I, Aizaki H, Tani H, Ishii K, Chiba T, Saito I, Miyamura T, Matsuura Y. Efficient gene transfer into various mammalian cells, including non-hepatic cells, by baculovirus vectors. J Gen Virol. 1997;78:2657–2664. doi: 10.1099/0022-1317-78-10-2657. [DOI] [PubMed] [Google Scholar]
- Smart D, Jerman JC, Brough SJ, Rushton SL, Murdock PR, Jewitt F, Elshourbagy NA, Ellis CE, Middlemiss DN, Brown F. Characterization of recombinant human orexin receptor pharmacology in a Chinese hamster ovary cell-line using FLIPR. Br J Pharmacol. 1999;128:1–3. doi: 10.1038/sj.bjp.0702780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tani H, Limn CK, Yap CC, Onishi M, Nozaki M, Nishimune Y, Okahashi N, Kitagawa Y, Watanabe R, Mochizuki R, Moriishi K, Matsuura Y. In vitro and in vivo gene delivery by recombinant baculoviruses. J Virol. 2003;77:9799–9808. doi: 10.1128/JVI.77.18.9799-9808.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesfai Y, Brereton HM, Barritt GJ. A diacylglycerol-activated Ca2+ channel in PC12 cells (an adrenal chromaffin cell line) correlates with expression of the TRP-6 (transient receptor potential) protein. Biochem J. 2001;358:717–726. doi: 10.1042/0264-6021:3580717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trebak M, Vazquez G, Bird GS, Putney JW., Jr The TRPC3/6/7 subfamily of cation channels. Cell Calcium. 2003;33:451–461. doi: 10.1016/s0143-4160(03)00056-3. [DOI] [PubMed] [Google Scholar]
- Trebak M, Hempel N, Wedel BJ, Smyth JT, Bird GS, Putney JW., Jr Negative regulation of TRPC3 channels by protein kinase C-mediated phosphorylation of serine 712. Mol Pharmacol. 2005;67:558–563. doi: 10.1124/mol.104.007252. [DOI] [PubMed] [Google Scholar]
- Tumilowicz JJ, Nichols WW, Cholon JJ, Greene AE. Definition of a continuous human cell line derived from neuroblastoma. Cancer Res. 1970;30:2110–2118. [PubMed] [Google Scholar]
- Uramura K, Funahashi H, Muroya S, Shioda S, Takigawa M, Yada T. Orexin-a activates phospholipase C- and protein kinase C-mediated Ca2+ signaling in dopamine neurons of the ventral tegmental area. NeuroReport. 2001;12:1885–1889. doi: 10.1097/00001756-200107030-00024. [DOI] [PubMed] [Google Scholar]
- van den Pol AN. Hypothalamic hypocretin (orexin): robust innervation of the spinal cord. J Neurosci. 1999;19:3171–3182. doi: 10.1523/JNEUROSCI.19-08-03171.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Pol AN, Gao XB, Obrietan K, Kilduff TS, Belousov AB. Presynaptic and postsynaptic actions and modulation of neuroendocrine neurons by a new hypothalamic peptide, hypocretin/orexin. J Neurosci. 1998;18:7962–7971. doi: 10.1523/JNEUROSCI.18-19-07962.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatachalam K, Zheng F, Gill DL. Regulation of canonical transient receptor potential (TRPC) channel function by diacylglycerol and protein kinase C. J Biol Chem. 2003;278:29031–29040. doi: 10.1074/jbc.M302751200. [DOI] [PubMed] [Google Scholar]
- Willie JT, Chemelli RM, Sinton CM, Tokita S, Williams SC, Kisanuki YY, Marcus JN, Lee C, Elmquist JK, Kohlmeier KA, Leonard CS, Richardson JA, Hammer RE, Yanagisawa M. Distinct narcolepsy syndromes in Orexin receptor-2 and Orexin null mice: molecular genetic dissection of Non-REM and REM sleep regulatory processes. Neuron. 2003;38:715–730. doi: 10.1016/s0896-6273(03)00330-1. [DOI] [PubMed] [Google Scholar]
- Wu M, Zaborszky L, Hajszan T, van den Pol AN, Alreja M. Hypocretin/orexin innervation and excitation of identified septohippocampal cholinergic neurons. J Neurosci. 2004;24:3527–3536. doi: 10.1523/JNEUROSCI.5364-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu R, Wang Q, Yan M, Hernandez M, Gong C, Boon WC, Murata Y, Ueta Y, Chen C. Orexin-A augments voltage-gated Ca2+ currents and synergistically increases growth hormone (GH) secretion with GH-releasing hormone in primary cultured ovine somatotropes. Endocrinology. 2002;143:4609–4619. doi: 10.1210/en.2002-220506. [DOI] [PubMed] [Google Scholar]
- Yang B, Samson WK, Ferguson AV. Excitatory effects of orexin-A on nucleus tractus solitarius neurons are mediated by phospholipase C and protein kinase C. J Neurosci. 2003;23:6215–6222. doi: 10.1523/JNEUROSCI.23-15-06215.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Tsien RW. Enhancement of N- and L-type calcium channel currents by protein kinase C in frog sympathetic neurons. Neuron. 1993;10:127–136. doi: 10.1016/0896-6273(93)90305-b. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Miwa Y, Yamanaka A, Yada T, Shibahara M, Abe Y, Sakurai T, Goto K. Orexin receptor type-1 couples exclusively to pertussis toxin-insensitive G-proteins, while orexin receptor type-2 couples to both pertussis toxin-sensitive and -insensitive G-proteins. J Pharmacol Sci. 2003;92:259–266. doi: 10.1254/jphs.92.259. [DOI] [PubMed] [Google Scholar]