

Abstract
The neurobiological mechanisms by which high-frequency stimulation of the subthalamic nucleus (STN–HFS) alleviates the motor symptoms of Parkinson's disease (PD) remain unclear. In this study, we analyzed the effects of STN–HFS on motor behavior in intact or hemiparkinsonian rats (6-hydroxydopamine lesion of the substantia nigra pars compacta) and investigated the correlation between these effects and extracellular glutamate (Glu) and GABA levels, assessed by intracerebral microdialysis in the substantia nigra pars reticulata (SNr). STN–HFS at an intensity corresponding to the threshold inducing contralateral forelimb dyskinesia, increased Glu levels in the SNr of both intact and hemiparkinsonian rats. In contrast, STN–HFS at half this intensity did not affect Glu levels in the SNr in intact or hemiparkinsonian rats but increased GABA levels in hemiparkinsonian rats only. STN–HFS-induced forelimb dyskinesia was blocked by microinjection of the Glu receptor antagonist kynurenate into the SNr and facilitated by microinjection of a mixture of the Glu receptor agonists AMPA and NMDA into the SNr. These new neurochemical data suggest that STN–HFS-induced forelimb dyskinesia is mediated by glutamate, probably via the direct activation of STN axons, shedding light on the mechanisms of STN–HFS in PD.
Keywords: subthalamic nucleus, substantia nigra pars reticulata, glutamate, GABA, high-frequency deep brain stimulation, Parkinson's disease, dyskinesia
Introduction
The subthalamic nucleus (STN) plays a key role in controlling the output nuclei of the basal ganglia (BG), the substantia nigra pars reticulata (SNr), and the internal segment of the globus pallidus (GPi). It therefore plays an important role in controlling movement (Kitai and Deniau, 1981; Kita and Kitai, 1987; Smith et al., 1998; Nambu, 2004). It receives its main inputs from the cortex, thalamus, and brainstem (Kita, 1994) and, via its glutamatergic projections, excites the GABAergic neurons of the SNr and GPi, thereby reinforcing the inhibitory effects of the BG on the thalamic and brainstem premotor networks (Albin et al., 1989; Alexander and Crutcher, 1990; Chevalier and Deniau, 1990). Pathological or experimental lesions of the STN result in dyskinesia or hemiballism (Whittier and Mettler, 1949; Carpenter et al., 1950; Hammond et al., 1979; Dewey and Jankovic, 1989). STN neurons have higher firing rates and burst activity in Parkinson's disease (PD) patients and animal models of PD than in normal subjects (Miller and DeLong, 1987; Bergman et al., 1994; Hassani et al., 1996; Hutchison et al., 1998; Bevan et al., 2002; Levy et al., 2002). The STN thus constitutes a strategic target for the neurosurgical treatment of PD (Bergman et al., 1990, Limousin et al., 1998; Benabid, 2003).
High-frequency stimulation (HFS) of the STN is a powerful approach for treatment of parkinsonian motor syndrome. It has been reported to result in clinical improvement in both PD patients and experimental animal models, further reducing levodopa requirements and therefore levodopa-induced dyskinesia (Benazzouz et al., 1993; Limousin et al., 1995; Krack et al., 2003). However, the mechanisms underlying the effects of STN–HFS remain unclear (Benabid et al., 2002; Dostrovsky and Lozano, 2002; McIntyre et al., 2004a). Because STN–HFS effects have been shown to be functionally equivalent to those of STN lesions (Bergman et al., 1990; Aziz et al., 1991; Guridi et al., 1996; Alvarez et al., 2005), it has been suggested that STN–HFS silences STN neurons (Benazzouz et al., 1993). However, recent modeling and in vivo studies have shown that additional mechanisms, such as activation of the STN afferent and efferent pathways, may be involved, leading to distant synaptic inhibitory and excitatory effects in BG output nuclei (Windels et al., 2000, Windels et al., 2003, Windels et al., 2005; Salin et al., 2002; Hashimoto et al., 2003; Maurice et al., 2003; McIntyre et al., 2004b; Kita et al., 2005).
We investigated the mechanisms involved further by studying STN stimulation in rats in conditions comparable with those used in clinical practice: parameters, monopolar stimulation, and awake animals. We also analyzed the effects of various HFS parameters on motor behavior in freely moving intact and hemiparkinsonian rats. We then used intracerebral microdialysis to assess variations of extracellular glutamate (Glu) and GABA levels in the SNr during STN–HFS, according to whether the intensity of stimulation used did or did not evoke forelimb dyskinesia. We also investigated the effects of local injections of active Glu compounds during STN–HFS in hemiparkinsonian animals to determine whether SNr Glu transmission mediated STN–HFS-induced forelimb dyskinesia.
Materials and Methods
Animals.
We used 69 adult male Sprague Dawley rats (Janvier, Le Genest St. Isle, France), weighing 280 to 350 g, housed under standard laboratory conditions (12 h light/dark cycle) with food and water provided ad libitum. Protocols conformed to the National Institutes of Health Guide for the Care and Use of Laboratory Animals (publication 865-23) and French Ministry of Agriculture regulations (authorization number 38-R 1001).
Lesion procedure.
For substantia nigra pars compacta (SNc) lesioning, all animals were anesthetized with chloral hydrate (400 mg/kg, i.p.) and secured in a Kopf stereotaxic apparatus (Phymep, Paris, France). We treated 43 animals with desipramine (25 mg/kg, s.c.), to protect noradrenergic neurons, and then injected 9 μg of 6-hydroxydopamine (6-OHDA) (Sigma, St. Quentin-Fallavier, France) dissolved in 3 μl of sterile 0.9% NaCl and 0.2% ascorbic acid into the left SNc of these animals, at a flow rate of 0.5 μl/min. The stereotaxic coordinates of the injection site relative to the bregma were as follows: anteroposterior (AP), −5.3 mm; lateral (L), +2.35 mm; and dorsoventral (DV), 7.5 mm, with the incisor bar at 3.3 mm below the interaural plane. All stereotaxic coordinates cited here are according to the stereotaxic atlas of Paxinos and Watson, (1982). Animals were kept warm after the injections and allowed to recover from anesthesia. They were returned to the animal facility for 3 weeks to allow the degeneration of DA neurons induced by the neurotoxin to stabilize and were then processed for microdialysis experiments.
Implantation of guide cannula and microdialysis probe, injection cannula, and stimulation electrode.
For microdialysis experiments, normal (n = 20) and 6-OHDA-lesioned (n = 20) rats were anesthetized by inhalation (1 L/min) of a 5% halothane/air mixture (22% O2, 78% N2) and mounted in a stereotaxic frame (David Kopf Instruments, Tujunga, CA). Anesthesia was maintained with an inhaled 1% halothane/air mixture (1 L/min). The dorsal skull was exposed, and holes were drilled to facilitate the unilateral (left side, i.e., ipsilateral to 6-OHDA injection) implantation of the guide cannula, through which the dialysis probe was lowered into the SNr, and of the stimulation electrode in the STN. The guide cannula, which consisted of a stainless steel tube (outer diameter, 0.60 mm; inner diameter, 0.48 mm) was inserted obliquely (17° from the vertical) such that its tip was located at the edge of the SNr. The stereotaxic coordinates used were (expressed relative to bregma): (1) guide cannula: AP, −5.3 mm; L, 5 mm; V, −7.3 mm; (2) microdialysis probe (into SNr): AP, −5.3 mm; L, 5 mm; V, −8.5 mm; (3) stimulation electrode (into STN): AP, −3.7 mm; L, 2.4 mm; V, −7.8 mm.
For the local injection of drugs, stainless steel cannulas (outer diameter, 0.28 mm; inner diameter, 0.18 mm) were inserted bilaterally into the SNr in 6-OHDA-lesioned rats only (n = 17). The stereotaxic coordinates of the injection site relative to bregma were as follows: AP, −5.3 mm; L, 2.4 mm; DV, 8.2 mm. The guide and injection cannulas, together with the stimulation electrode, were fixed onto the skull with dental cement (Methax, Perrigot, France). During surgery, body temperature was maintained at 37°C with a feedback-controlled heating pad (Harvard Apparatus, Edenbridge, UK). Animals were allowed to recover from surgery for 3 d before microdialysis experiments, which were performed in awake animals.
Electrical stimulation.
Monopolar electrodes consisting of platinum–iridium wire insulated with Teflon but with an exposed end (wire diameter, 110 μm insulated, 76 μm bare) (Phymep) were implanted unilaterally into 43 6-OHDA-lesioned rats and 26 intact animals. The electrode was implanted such that the exposed end (length, 400 μm) was inserted into the sensorimotor part of the STN. The exposed end of the electrode served as the negative stimulation pole, with a screw fixed on the skull used as the positive pole. Stimuli were delivered with a World Precision Instruments (Stevenage, UK) acupulser and a stimulus isolation unit generating rectangular pulses.
For behavioral studies, two series of stimulation experiments were performed in intact (n = 6) and 6-OHDA-injected (n = 6) rats. In the first series, frequency and pulse width were set at 130 Hz and 60 μs, respectively, for the entire stimulation period, for all stimulated animals. The behavioral effects of increasing the stimulation intensity from 0 to 350 μA were analyzed in each animal during the first 2 min of stimulation. In particular, we determined whether the STN–HFS applied triggered dyskinetic movements similar to those induced after levodopa treatment, i.e., orofacial, axial, or forelimb dyskinesia (Winkler et al., 2002), as well as rotational behavior.
In the second series, STN stimulations were performed with various frequencies (10, 60, 130, 150, and 200 Hz) and pulse widths (20, 40, 60, 80, or 100 μs). Pulse width was set at 60 μs for experiments involving frequency variation, whereas the frequency was set at 130 Hz for experiments involving pulse width variation. In both cases, the STN was stimulated electrically by gradually increasing the intensity of stimulation until dyskinetic movement of the contralateral forelimb was observed, as described by Salin et al., (2002).
For microdialysis and microinjection experiments, frequency and pulse width values were kept constant throughout the stimulation period (1 h) for all stimulated animals and were similar to those used routinely in clinical practice (130 Hz and 60 μs, respectively). Two different intensities were tested: I1, corresponding to the threshold value inducing contralateral forelimb dyskinesia, ranging from 60 to 200 μA, depending on the animal considered; I2, a value below this threshold value, arbitrarily fixed at half I1, varying with the individual and often corresponding to values <60 μA.
At the end of each experiment, an electrical lesion was created in the STN so that the position of the electrode could be checked postmortem.
Microdialysis experiments.
Homemade microdialysis probes were prepared and used as described previously (Windels et al., 2000; Bruet et al., 2003). They consisted of a concentric arrangement of stainless steel tube (outer diameter, 0.4 mm) (Phymep), and polyethylene tubing (outer diameter, 1.09 mm; inner diameter, 0.38 mm) (Phymep) into which we inserted a piece of silica tubing (outer diameter, 150 μm; inner diameter, 75 μm) (Phymep). The silica tubing extended beyond the distal end of the steel tube and was covered with a cuprophan tubular dialysis membrane (Hospal, Lyon, France), sealed at the bottom with epoxy glue. The length of the dialysis membrane was adapted for the rat brain nucleus studied (1 mm for the SNr).
The perfusion liquid flowed out of the distal end of the steel tube, passing proximally between the tube and the membrane (Tossman and Ungerstedt, 1986). The probes were perfused with artificial CSF (in mm: 149 NaCl, 2.8 KCl, 1.2 MgCl2, 1.2 CaCl2, and 5.4 glucose, pH 7.3) at a flow rate of 1 μl/min. Before implantation, each probe was tested in vitro in a standard amino acid solution, to determine amino acid recovery (Tossman and Ungerstedt, 1986).
The dialysis probe was implanted via the guide cannula and dialysates were collected at 15 min intervals, for 5 h, in awake animals. The first eight fractions were discarded to prevent effects caused by parenchymal disturbance and to obtain an approximate steady state. The next eight fractions (120 min) were collected for basal value determination, and four fractions were then collected during STN–HFS. An additional eight fractions were collected to estimate poststimulation effects over a 2 h period. Dialysates were collected automatically with a refrigerated autosampler (Univentor, Zejton, Malta) and stored at −80°C until analysis.
Glutamate and GABA assays.
Glu and GABA concentrations in the dialysates were determined by HPLC, with laser-induced fluorescence detection. Briefly, 2 μl of sample or standard was derivatized with naphthalene-2,3-dicarboxaldehyde. The resulting mixture was automatically loaded onto a Symmetry Shield-C18 reverse-phase column (100 × 2.1 mm, 3.5 μm particle size; Waters, Milford, MA), using a refrigerated Triathlon autoinjector (Polymer Laboratories, Marseille, France). The mobile phase consisted of 0.04 m NaH2PO4, pH 6, in a 3–50% acetonitrile gradient. The flow rate was 0.35 ml/min, maintained with two Shimadzu (Kyoto, Japan) LC 10AT pumps. Amino acid peaks were identified based on retention time. Extracellular amino acid concentrations were estimated by rationing peak areas of each amino acid and their respective external standard (analytical software class LC10; Shimadzu). The running time for each determination was 12 min.
Local drug injections.
We applied Glu and GABA agonists or antagonists bilaterally to the SNr of 17 freely moving 6-OHDA-lesioned rats through a stainless steel cannula, implanted as described above. Bilateral injections were used to prevent crossed effects of drugs. The cannula was connected to a 10 μl syringe (Hamilton, Bonaduz, Switzerland) via a piece of polyethylene tubing (outer diameter, 1.09 mm; inner diameter, 0.38 mm) (Phymep). The syringes contained a mixture of Glu agonists (AMPA and NMDA), a Glu antagonist (kynurenic acid), or a GABAA agonist (muscimol). These drugs were purchased from Sigma and dissolved in saline. We injected 0.5 μl of solution into both sides of the SNr, at a rate of 0.25 μl/min, using a micropump (CMA/100; Phymep).
Ten, 20, and 30 min after local drug injection, STN–HFS with an intensity of I1 or I2 (as determined for each animal as described above), was applied for 2 min, and effects on motor behavior were analyzed. If an intensity of I1 was used for STN–HFS, kynurenic acid (n = 5) or muscimol (n = 5) was injected. If an intensity of I2 was used, only the AMPA/NMDA mixture was injected (n = 5). We tested the effects of two or three doses of each drug for each application to the SNr: 1 or 2 nmol AMPA/NMDA; 0.5 or 1 nmol kynurenic acid; and 0.8, 1.5, or 3 nmol muscimol. Higher concentrations of Glu agonists were not used to prevent depolarization block phenomena attributable to excessive excitatory stimulation. Each animal always received the same drug, and injections of different concentrations were administered 48 h apart.
Histology.
At the end of the microdialysis or microinjection experiments, unlesioned rats (n = 26) were killed by decapitation under deep anesthesia; their brains were quickly removed from the skull and frozen in isopentane at −30°C. Lesioned animals (n = 43) were perfused transcardially, under chloral hydrate anesthesia, with 200 ml of 0.9% NaCl, pH 7.2, followed by 300 ml of 4% paraformaldehyde in 0.1 m PBS, pH 7.4 (in mm: 2.6 KCl, 1.4 KH2PO4, 136 NaCl, and 83 NaH2PO4). Brains were quickly removed and immersed overnight in 20% sucrose in 0.1 phosphate buffer, pH 7.4, frozen in cooled (−40°C) isopentane, and stored at −30°C. Serial frontal sections (20 μm) were cut on a cryostat (Microm HM 500; Microm, Francheville, France). The correct locations of the microdialysis probe, injection cannula, and stimulation electrode were checked by collecting several nigral and subthalamic tissue sections (Paxinos and Watson, 1982) and counterstained with cresyl violet. These histological controls were systematically performed for all of the animals in each experimental group. All animals presenting internal bleeding around the microdialysis probes or electrodes were excluded to prevent microdialysate contamination (n = 2), as were animals with incorrectly positioned microdialysis probes (n = 4), injection cannulas (n = 2), or stimulation electrodes (n = 5). We assessed the dopaminergic denervation induced by nigral 6-OHDA injection by tyrosine hydroxylase (TH) immunostaining of striatal and nigral sections from the fixed brains of lesioned animals. Free-floating sections were thoroughly washed with TBS and incubated for 1 h in 0.3% Triton X-100 in TBS (TBST) and 3% normal goat serum (Sigma). They were then incubated with primary antisera diluted in TBST containing 1% normal goat serum for 24 h, with shaking, at 4°C. The antiserum was diluted 1:500 for TH staining (mouse monoclonal antibody; Chemicon, Temecula, CA). Antibody binding was detected with the avidin–biotin–peroxidase conjugate (Vectastain ABC Elite; Vector Laboratories, Burlingame, CA) using 3,3′-diaminobenzidine as chromogen. It was applied to the sections for 2–5 min, as described previously (Guesdon et al., 1979). Sections were dehydrated through graded ethanol solutions, cleared in xylene, mounted in DPX (DBH Laboratory Supplies, Poole, UK), and covered with a coverslip for microscopy.
Data and statistical analysis.
For behavioral and microinjection experiments, results are expressed as the mean ± SEM. Mann–Whitney and Wilcoxon's tests were used to analyze data from behavioral and local injection experiments, respectively.
For microdialysis experiments, the basal levels of the measured substances are expressed as concentrations in dialysates. Basal amino acid levels in the SNr were analyzed, for the various experimental groups, with a Mann–Whitney U test. For data expressed as a percentage of the control, the mean concentration of the four samples preceding STN–HFS was set at 100%. The effects of STN–HFS on extracellular Glu and GABA levels were analyzed by one-way repeated-measures ANOVA over time (Windels et al., 2005). Dunnett's or Games–Howell test was used for comparisons with prestimulation baseline levels. Values of p < 0.05 were considered statistically significant.
Results
Control of electrode and microdialysis probe location and of the extent of the dopamine lesion
Three weeks after the unilateral injection of 6-OHDA (n = 41), a massive loss of TH immunolabeling was observed in the SNc (Fig. 1A) and in the striatum (caudate–putamen nucleus) (Fig. 1B) on the lesioned side. Dense TH immunolabeling was detected throughout the SNc, the ventral tegmental area, the striatum, the nucleus accumbens, and the olfactory tubercles, on the intact side, in 6-OHDA-injected rats (Fig. 1A,B). Figure 1 illustrates the correct implantation of the microdialysis probe in the parenchyma of the SNr (Fig. 1D) and of the stimulation electrode in the STN (Fig. 1F). Figure 1G shows, at a higher magnification, the small electrical lesion created at the end of the experiment.
Figure 1.
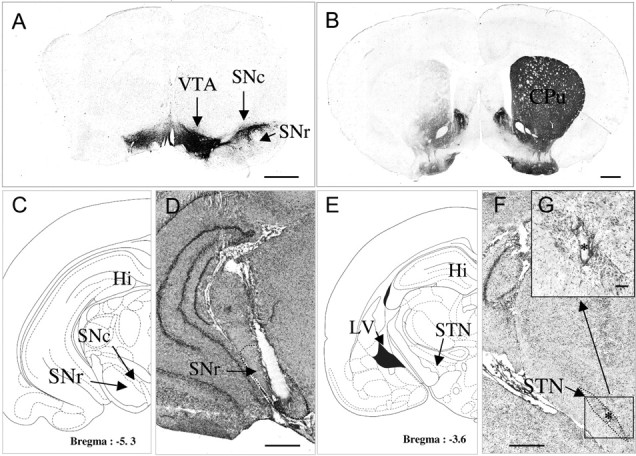
A, B, D, F, G, Photographs of TH-immunostained coronal rat brain sections at the nigral (A) and striatal (B) levels and of cresyl violet-stained coronal rat brain sections at the nigral (D) and subthalamic (F, G) levels in 6-OHDA-lesioned rats. C, E, Schematic diagrams adapted from the stereotaxic atlas of Paxinos and Watson, (1982). A, B, Note on the lesioned side (left) the loss of dopaminergic cells in the SNc (A) and the loss of dopaminergic terminals in the striatum (B). D, F, G, Note also the correct implantation of the microdialysis probe in the SNr (D) and of the stimulation electrode within the STN (F, G). F, The asterisk indicates the point stimulated. Scale bar, 1 mm. G, Coronal section, at a higher magnification, through the subthalamus (corresponding to the area limited by a square in F) in SNc-lesioned animals on the lesioned side. Scale bars, 0.1 mm. CPu, Caudate–putamen; Hi, hippocampus; LV, lateral ventricle; VTA, ventral tegmental area.
Basal extracellular Glu and GABA levels in the SNr are higher in awake hemiparkinsonian rats
Lesioning the SNc with 6-OHDA dramatically increased the concentration of Glu in the SNr (Table 1) ipsilaterally to the lesion. In the SNr of SNc-lesioned rats, mean Glu concentration was 1.88 ± 0.7 μm (n = 17), 20 times (p < 0.05) higher than basal levels in intact control animals (0.09 ± 0.01 μm; n = 14). Treatment with 6-OHDA also tripled GABA levels in the SNr, from the basal level of 0.006 ± 0.0007 μm in intact animals (n = 14) to 0.02 ± 0.004 μm in SNc-lesioned animals (n = 17; p < 0.05) (Table 1).
Table 1.
Extracellular levels of glutamate and GABA (expressed as a micromolar concentration) measured in the SNr (n = 5–6) on the stimulated side of intact and SNc-lesioned rats in basal conditions and evoked by STN high (I1)- or low (I2)-intensity stimulation
| Intact |
SNc-lesioned |
|||||
|---|---|---|---|---|---|---|
| Basal | STN–HFS I1 | STN–HFS I2 | Basal | STN–HFS I1 | STN–HFS I2 | |
| Glu (μm) | 0.09 ± 0.014 | 0.13 ± 0.008 (+44%; p < 0.001) | 0.09 ± 0.004 (No effect) | 1.88 ± 0.708 | 3.18 ± 0.225 (+69%; p < 0.001) | 1.54 ± 0.034 (No effect) |
| GABA (μm) | 0.006 ± 0.0007 | 0.009 ± 0.0006 (+50%; p < 0.001) | 0.006 ± 0.0002 (No effect) | 0.020 ± 0.004 | 0.019 ± 0.002 (No effect) | 0.028 ± 0.002 (+40%; p < 0.001) |
Motor behavior observations
Dyskinesia is induced by STN–HFS in an intensity-dependent manner
A gradual increase in stimulation intensity (from 0 to 350 μA) led to the induction of a motor sequence that appeared similar in freely moving intact and 6-OHDA-lesioned animals (Fig. 2A). This motor behavioral sequence was characterized by (1) orofacial dyskinesia followed by (2) axial dyskinesia, consisting of contralateral positioning of the head and trunk, (3) contralateral forelimb dyskinesia and a strong contralateral bias in head position, and finally, (4) contralateral rotation. The stimulation intensity required to produce these sequential motor effects was similar in intact and lesioned animals. Figure 2A illustrates the range of stimulation intensity threshold values corresponding to each behavioral effect observed when frequency and pulse width were set at 130 Hz and 60 μs, respectively: orofacial dyskinesias, 15–50 μA, corresponding to a frequency of 69 ± 12 movements/min; axial dyskinesias, 65–110 μA, with a continuous effect; contralateral forelimb dyskinesias, 60–200 μA corresponding to 87 ± 15 movements/min; contralateral rotations, ≥190 μA with a frequency of 6 ± 0.5 rotations/min. I1, the stimulation intensity used in the first set of microdialysis experiments, corresponded to the intensity threshold generating contralateral forelimb dyskinesia.
Figure 2.

Graphic representation of the motor behavior observed in intact and 6-OHDA-lesioned rats in response to electrical stimulation of the STN. A, STN–HFS-induced movements (frequency, 130 Hz; pulse width, 60 μs; intensity, 0–350 μA) in intact (n = 5) or 6-OHDA-lesioned (n = 5) rats. Each symbol indicates the threshold value for the stimulation intensity triggering dyskinesia. The horizontal bar indicates the mean intensity value inducing each specific movement, as observed for all animals (intact and hemiparkinsonian) considered together (n = 10). B, C, Determination of the threshold value for stimulation intensity inducing contralateral forelimb dyskinesia under STN stimulation at various frequencies and constant pulse width (60 μs) (B) or with various pulse width values and constant frequency (130 Hz) (C). Note that the threshold intensity value was higher, in both normal and 6-OHDA-lesioned rats, only at low frequency (10 and 60 Hz) or at short pulse width (20 and 40 μs). For short pulse width, the intensity threshold values were higher for 6-OHDA-lesioned rats than for normal rats. For frequency values over 130 Hz (B) or pulse width values over 60 μs (C), this threshold was similar in both groups, at ∼90–100 μA. *p < 0.05 versus normal rats.
Influence of frequency or pulse width on behavioral effects
The intensity required to induce contralateral forelimb dyskinesia depended on the frequency (Fig. 2B) and pulse width (Fig. 2C). The stimulation intensity required to produce these effects was higher for fixed frequencies of 10 or 60 Hz or fixed pulse widths of 20 or 40 μs than for fixed frequencies 130 or 200 Hz or fixed pulse widths of 60 or 100 μs. Stimulation intensity was also significantly higher (p < 0.05) in lesioned than in intact stimulated rats, indicating a lower sensitivity to the stimulation. However, this did not hold for frequency values of 130 Hz and over (Fig. 2B) or for pulse width values >60 μs (Fig. 2C), for which an intensity of ∼90–100 μA was required for dyskinetic movements.
STN–HFS-induced forelimb dyskinesia in hemiparkinsonian rats is linked to an increase in Glu levels in the SNr, with no change in GABA levels
In intact (n = 7) and 6-OHDA-treated (n = 8) rats, STN–HFS at intensity I1 (60–200 μA, the intensity threshold value triggering contralateral forelimb dyskinesia) triggered a gradual increase in extracellular Glu level in the ipsilateral SNr during the 1 h stimulation period (Fig. 3A, fractions 9–12). This increase was maximal in the last 15 min of this period: +80 ± 24% for intact rats (p < 0.01; n = 7) and +97 ± 37% for hemiparkinsonian animals (p < 0.01; n = 8). In both intact and 6-OHDA-treated rats, Glu levels remained high during the poststimulation period (fractions 13–19), decreasing in the last fraction collected (Fig. 3A). However, changes in Glu levels were observed smaller in hemiparkinsonian than in intact control rats during this period, possibly because Glu levels had already been strongly increased by the dopaminergic lesion. Interestingly, forelimb dyskinesias induced by STN stimulation were always paralleled by increases in extracellular Glu levels in the SNr.
Figure 3.

Extracellular glutamate (A) and GABA (B) levels, determined at 15 min intervals, in the SNr ipsilateral to the stimulation in intact and 6-OHDA-lesioned rats under basal conditions and during 1 h of STN–HFS with an intensity inducing forelimb dyskinesia (I1, >60 μA). The prestimulation period (PRE-STIM) corresponds to fractions 1–8 of the dialysates; the stimulation period (STIM), indicated by the black horizontal bar, corresponds to fractions 9–12 (4 fractions) and the poststimulation period (POST-STIM) corresponds to fractions 13–20 (8 fractions). The mean ± SEM of the eight PRE-STIM dialysates, collected before STN–HFS, was used to determine baseline levels. Results are expressed as a percentage of variation of this baseline value. Each percentage corresponds to the mean ± SEM variations calculated for five to eight animals. Note that Glu levels increased in both intact and 6-OHDA-lesioned rats, whereas GABA levels increased only in intact animals. *p < 0.05, **p < 0.01 versus baseline values. Error bars correspond to the SEM.
In contrast, STN–HFS had a very different effect on GABA levels. STN–HFS did not affect extracellular GABA levels in the ipsilateral SNr of 6-OHDA-treated rats during the 1 h stimulation period (Fig. 3B, fractions 9–12). Conversely, in intact rats, STN–HFS increased extracellular GABA levels in the ipsilateral SNr. This increase was maximal in the last fraction collected (+90 ± 20%; p < 0.01; fraction 12; n = 5) and remained high throughout most of the poststimulation period. This effect of stimulation is consistent with previous reports (Windels et al., 2000) but was of lower amplitude in awake than in anesthetized intact rats.
STN–HFS at an intensity below the threshold value triggering forelimb dyskinesia increases GABA levels, but not Glu levels, in the SNr of hemiparkinsonian rats
STN–HFS at intensity I2 (below the threshold value evoking contralateral forelimb dyskinesia) did not affect extracellular Glu level in the ipsilateral SNr in intact (n = 6) or 6-OHDA-treated rats (n = 9). Extracellular Glu levels remained stable around baseline throughout the microdialysis experiment (Fig. 4A).
Figure 4.

Extracellular glutamate (A) and GABA (B) levels determined, at 15 min intervals, in the SNr ipsilateral to the stimulation in intact and 6-OHDA-lesioned rats, under basal conditions and during 1 h of STN–HFS with an intensity below the threshold for forelimb dyskinesia (I2). The prestimulation period (PRE-STIM) corresponds to fractions 1–8 of the dialysates; the stimulation period (STIM), indicated by the black horizontal bar, corresponds to fractions 9–12 (4 fractions), and the poststimulation period (POST-STIM) corresponds to fractions 13–20 (8 fractions). The mean ± SEM of the eight PRE-STIM dialysates, collected before STN–HFS, was used to determine baseline levels. Results are expressed as a percentage of variation of this baseline value. Each percentage corresponds to the mean ± SEM variations calculated for six to nine animals. Note that Glu levels in the SNr were not significantly affected by STN–HFS in the SNr in either intact or 6-OHDA-lesioned rats, whereas GABA levels increased only in 6-OHDA-lesioned animals. *p < 0.05, **p < 0.01 versus baseline values. Error bars indicate the SEM.
In contrast to what was observed for Glu levels, STN–HFS at intensity I2 increased extracellular GABA levels in the ipsilateral SNr during the 1 h stimulation period (Fig. 4B, fractions 9–12) in hemiparkinsonian rats only. This increase reached +69 ± 21% (p < 0.01; fraction 10; n = 8) during the stimulation period and remained high throughout the poststimulation period, peaking at +108 ± 25% (p < 0.05; fraction 16; n = 8) (Fig. 4B).
Modulation of STN–HFS-induced forelimb dyskinesia by the nigral injection of Glu antagonists or agonist in hemiparkinsonian rats
We investigated the possible involvement of increases in extracellular Glu levels in the SNr of hemiparkinsonian rats in the induction of forelimb dyskinesia by high-intensity STN–HFS by analyzing the effects of bilateral SNr injections of kynurenic acid. All animals subjected to high-intensity STN–HFS (I1) presented contralateral forelimb dyskinesia. These motor effects were totally prevented by the bilateral nigral injection of kynurenate at a dose of 1 nmol, 10 min before STN–HFS (n = 5) (Fig. 5A). A similar effect was obtained with a dose of 0.5 nmol. This effect of kynurenate persisted over 30 min under STN–HFS. No motor effects were detected when these doses of kynurenate were injected alone into the SNr of 6-OHDA-lesioned animals, without STN stimulation.
Figure 5.
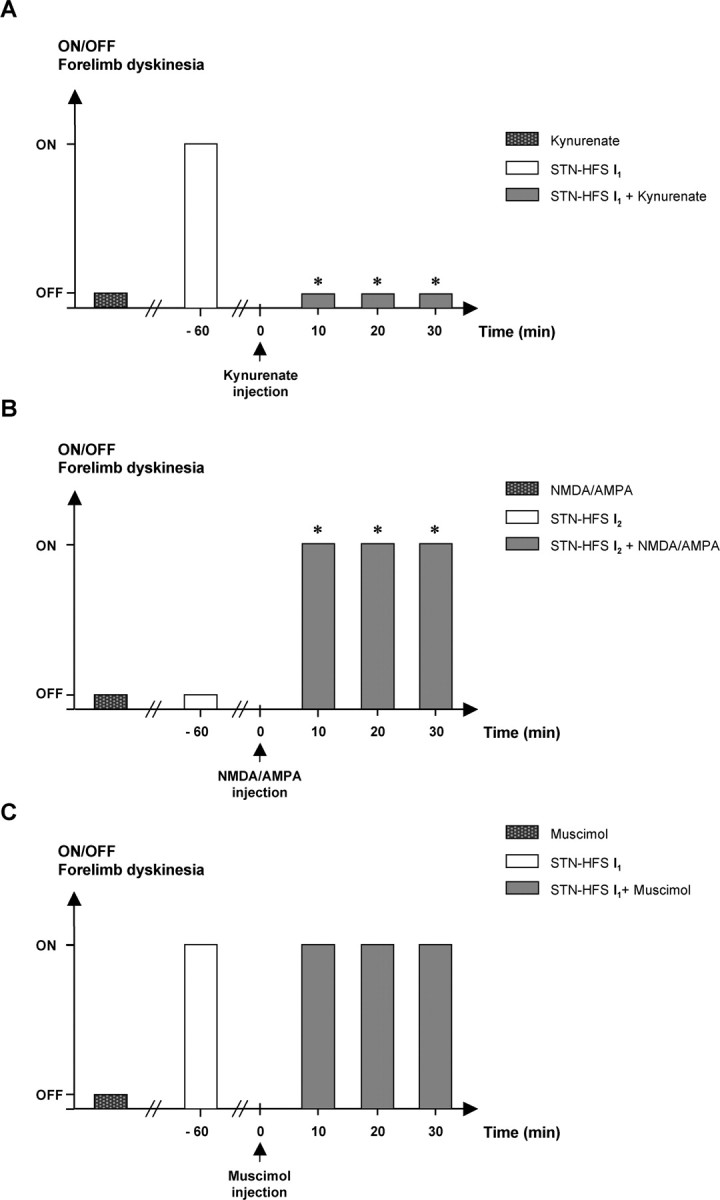
Effects of bilateral nigral injections of kynurenic acid (A), NMDA plus AMPA (B), and muscimol (C) on STN–HFS-induced contralateral forelimb dyskinesia in 6-OHDA-lesioned rats. STN stimulation was applied before and after drug injection. Note that, in A, kynurenate (1 nmol/side) alone did not provoke dyskinesia but, at the same dose, did prevent those induced by STN–HFS at intensity I1 (n = 5). *p < 0.05 versus 6-OHDA-lesioned rats plus STN–HFS at I1. In B, STN stimulation was applied at intensity I2, resulting in dyskinesia only if NMDA plus AMPA (1 nmol/side) was injected (n = 5) during STN–HFS. *p < 0.05 versus 6-OHDA-lesioned rats plus NMDA/AMPA or versus 6-OHDA-lesioned rats plus STN–HFS at I2. Note in C that muscimol (3 nmol/side; n = 5) did not prevent the dyskinesia induced by STN–HFS at intensity I1.
When STN–HFS was applied at an intensity of I2, eliciting no forelimb dyskinesia, the bilateral injection of a mixture of Glu agonists (NMDA plus AMPA) into the SNr facilitated dyskinetic movements (n = 5) similar to those observed under STN–HFS at an intensity of I1, whatever the dose used (1 or 2 nmol) (Fig. 5B). However, 2 nmol doses of these compounds led to slight paralysis of the hindquarters and akinesia of the forelimbs. As in experiments with kynurenate, the injection of these substances (NMDA plus AMPA) alone did not induce dyskinesia in hemiparkinsonian rats in the absence of stimulation.
In contrast, dyskinetic movements induced by STN–HFS at intensity I1 were not blocked by the intranigral injection of muscimol (a GABAA receptor agonist), whatever the dose administered (0.8, 1.5, or 3 nmol) (Fig. 5C).
Discussion
The mechanisms by which STN–HFS affects the BG output nuclei remain unclear. We show here that unilateral STN–HFS induces stimulation intensity-dependent dyskinesias in awake intact or hemiparkinsonian rats. High-intensity STN stimulation provokes contralateral forelimb dyskinesia and increases Glu levels in the SNr, probably attributable to subthalamonigral pathway activation, increases of Glu levels being detectable from the first sample collected during stimulation. Low-intensity stimulation did not trigger forelimb dyskinesia but increased GABA levels in the SNr. STN–HFS-induced forelimb dyskinesia was blocked by intranigral microinjection of a Glu antagonist in hemiparkinsonian rats. Subthalamonigral pathway activation, modulating SNr activity, may therefore be involved in forelimb dyskinesia, providing insight into the mechanisms of STN–HFS in PD.
Changes in Glu and GABA levels in awake rats after SNc lesion
The large increase in basal Glu levels in the SNr in awake hemiparkinsonian rats is consistent with electrophysiological data showing that SNc lesion or neuroleptic treatment increases STN neurons discharge (Bergman et al., 1994; Hassani et al., 1996; Degos et al., 2005). This extracellular Glu may come from STN axon terminals, glial cells, or both (Danbolt, 2001; Parpura et al., 2004). However, STN lesions strongly decrease basal Glu levels in the SNr (Rosales et al., 1997) and prevent their increase under STN–HFS (M. Savasta and F. Windels, unpublished observations). Most of the extracellular Glu in the SNr is therefore probably linked to STN neuron activity, confirming the hyperactivity of the subthalamonigral pathway after dopamine lesioning in freely moving 6-OHDA-lesioned rats and consistent with data for anesthetized animals (Windels et al., 2005). SNr GABA levels also increased hemiparkinsonian rats but less so than Glu levels. Some of this nigral GABA may result from an increase in globus pallidus (GP) neuron activity attributable to STN hyperactivity, itself resulting from dopaminergic denervation (Hassani et al., 1996). However, additional GABA sources, such as neuron collaterals from the SNr or striatum, may also be involved. The increase in basal nigral Glu and GABA levels after SNc lesioning was larger in awake than in anesthetized animals (Windels et al., 2005), consistent with anesthetic effects on amino acid levels (Rozza et al., 2000; Windels and Kiyatkin, 2006).
Effects of STN stimulation parameters on motor behavior
STN–HFS in normal monkeys induces contralateral dyskinesia (Beurrier et al., 1997) resembling human and primate hemiballism after STN lesioning (Hamada and DeLong, 1992; Lee and Marsden, 1994). We found that unilateral STN–HFS in awake intact or hemiparkinsonian rats induced contralateral forelimb dyskinesia and even contralateral rotation, depending on stimulation parameters and intensity, as observed after STN lesioning (Henderson et al., 1999). Rotational effects were observed at intensities exceeding 190 μA and were preceded by orofacial, axial, and forelimb dyskinesias resembling l-3,4-dihydroxyphenylalanine (l-DOPA)-induced dyskinesias in hemiparkinsonian rats (Winkler et al., 2002; Sgambato-Faure et al., 2005). Higher-intensity stimulation was required to elicit dyskinesias in SNc-lesioned than in intact rats, but only for frequencies <130 Hz or pulse widths <60 μs. Sensitivity to STN–HFS therefore differed between the parkinsonian and normal states, as predicted after STN lesioning (Guridi and Obeso, 2001) and consistent with previous studies (Salin et al., 2002). No differential effect was observed, in any group, if STN–HFS parameters were set so as to induce dyskinesia (Chang et al., 2003). We determined the threshold value for forelimb dyskinesia, for investigation of the neurochemical changes induced by STN stimulation in normal and hemiparkinsonian rats at two different intensities, only one of which induced forelimb dyskinesia.
Changes in nigral Glu and GABA levels induced by high (I1)- or low (I2)-intensity STN stimulation
The neurochemical changes in the SNr depended on STN–HFS intensity. In normal and hemiparkinsonian rats, high-intensity STN–HFS increased Glu concentration in the SNr, whereas lower-intensity stimulation not inducing forelimb dyskinesia increased GABA levels in 6-OHDA-lesioned rats only. The excitatory responses to high-intensity STN stimulation in the SNr observed in electrophysiological studies probably result from specific activation of the glutamatergic subthalamonigral pathway (Hammond et al., 1978; Maurice et al., 2003), because the latency of the excitatory responses evoked in nigral cells corresponds to the conduction time for this pathway (Kitai and Deniau, 1981). STN–HFS at higher frequencies systematically led to forelimb dyskinesia in both normal and hemiparkinsonian rats, probably by increasing the rate of SNr cell discharge through glutamatergic transmission (Hashimoto et al., 2003; Kita et al., 2005). These findings are consistent with reports of hemiballism in PD patients during STN–HFS at voltages higher than used for chronic treatment (Limousin et al., 1996; Moro et al., 2002). This seems to conflict with data reporting a direct relationship between hemiballism and STN lesions in humans and experimental models (Whittier and Mettler, 1949; Carpenter et al., 1950; Hammond et al., 1979; Hamada and DeLong, 1992; Guridi and Obeso, 2001). Indeed, even discrete STN lesions in primates, probably linked to a decrease in the Glu outflow, have been reported to produce hemiballism. Our data linking forelimb dyskinesia and an increase in Glu outflow under STN–HFS seem to conflict with these previous observations. However, pharmacological STN activation has also been reported to produce hemiballism (Crossman et al., 1980, Crossman et al., 1984; Perier et al., 2002), so the mechanisms involved remain unclear and are still under debate. The STN thus forms part of a complex network. The impact of STN–HFS should therefore not be considered as a simple “functional lesion” of the STN, because it may have multiple effects on synapses (Shen et al., 2003) and neurotransmitter release.
The forelimb dyskinesia induced by high-intensity STN stimulation in hemiparkinsonian rats was prevented by nigral injection of the broad-spectrum Glu antagonist kynurenic acid, demonstrating that the hyperactive Glu transmission induced in the SNr by STN–HFS probably causes the observed forelimb dyskinesia. Glu transmission and forelimb dyskinesia are clearly linked because direct infusion of NMDA plus AMPA into the SNr triggered forelimb dyskinesia in hemiparkinsonian rats under low-intensity STN–HFS. Neither low-intensity STN–HFS nor local Glu agonist injection without STN–HFS caused forelimb dyskinesia, suggesting synergistic action. The lack of forelimb dyskinesia after Glu agonist injection alone suggests that STN–HFS may induce forelimb dyskinesia by activating/modulating several sites simultaneously and that SNr activation alone may be insufficient. Furthermore, the doses of Glu agonists used here may have been too low and/or may not have reached the effective synaptic sites within the SNr neuronal network. The GABAA agonist (muscimol) may have been unable to prevent dyskinetic movements for the same reason. The action of muscimol on presynaptic and postsynaptic GABAA receptors in the SNr may also account for complex opposite cellular interactions within the nigral network and related structures. However, regardless of the functional effects of high- or low-intensity STN–HFS, our data suggest that Glu transmission is involved in generating of forelimb dyskinesias after STN stimulation. It would be interesting to identify the subtypes of Glu receptors involved in generating STN–HFS-induced forelimb dyskinesia, because recent studies in animal models of l-DOPA-induced dyskinesia have implicated the NMDA Glu receptor and/or the metabotropic Glu receptor 5 (Papa and Chase, 1996; Nash et al., 2004; Dekundy et al., 2006). However, it is unclear whether l-DOPA therapy and high-intensity STN–HFS induced forelimb dyskinesia in similar ways.
Orofacial dyskinesia may also be induced by low-intensity STN–HFS but with no Glu concentration increase in the SNr. We cannot exclude the possibility that this type of dyskinesia results from preferential Glu release in an unsampled part of the SNr or from Glu release in quantities too small for detection by the method used. The mechanisms underlying orofacial dyskinesia may therefore differ from those underlying forelimb dyskinesia under STN–HFS. This is consistent with data showing that l-DOPA-induced dyskinesias involve different BG structures/pathways (Sharp et al., 1987; Winkler et al., 2002).
GABA release in the SNr after high-intensity STN stimulation in intact rats or low-intensity stimulation in hemiparkinsonian rats probably inhibits SNr cell activity, consistent with electrophysiological data showing that SNr inhibition is abolished by the iontophoretic application of bicuculline, a GABAA receptor antagonist (Maurice et al., 2003). This inhibitory effect results principally from pallidonigral fiber activation through axon collaterals, because GABAergic pallidosubthalamic neurons send an axon collateral to the SNr (Kita and Kitai, 1994). We also demonstrated that, in anesthetized hemiparkinsonian animals, GP lesions abolish the increase in SNr GABA levels induced by STN–HFS (Windels et al., 2005). However, SNr inhibition may also result from activation of the intranigral axon collateral network of GABAergic SNr cells (Mailly et al., 2003). According to the “classical” model, direct striatonigral GABAergic pathway activation inhibits the tonically active GABAergic neurons of the SNr, abolishing target nucleus inhibition in premotor structures in the thalamus and brainstem (Chevalier and Deniau, 1990; DeLong, 1990). This process is crucial in BG physiology (Mink and Thach, 1993). In PD patients and experimental models, dopaminergic neuron degeneration leads to STN neuron hyperactivity increasing the tonic discharge of BG GABAergic output structures (Bergman et al., 1994; Levy et al., 2002) and to imbalance between the direct and indirect striatonigral pathways, inducing motor disturbances (Chesselet and Delfs, 1996; Obeso et al., 1997). Our neurochemical data show that motor function recovery in PD patients probably requires low-intensity STN–HFS (1) to abolish the excitatory influence of the indirect striatonigral pathway on SNr cells and other BG output structures (Vitek, 2002; McIntyre et al., 2004a,b) and/or (2) to modify the spatiotemporal pattern of neuronal discharge in STN and BG output nuclei (Hashimoto et al., 2003).
In conclusion, STN–HFS-induced forelimb dyskinesia is mediated by Glu, probably through STN axon activation, in awake hemiparkinsonian rats. During STN–HFS at intensities below the threshold for forelimb dyskinesia, GABA may be involved in the functional disinhibition by which the BG organize normal movement and in the mechanisms of STN–HFS in PD.
Footnotes
This work was supported by the Institut National de la Santé et de la Recherche Médicale, Ministère de la Recherche et des Nouvelles Technologies (Agence Nationale pour la Recherche Grant ANR-05-NEUR-013-01), Région Rhône-Alpes (Cluster 11), and the Association France Parkinson. We thank Drs. M. Albrieux, G. Chouvet, and P. Krack for critical reading of this manuscript.
References
- Albin RL, Young AB, Penney JB. The functional anatomy of basal ganglia disorders. Trends Neurosci. 1989;12:366–375. doi: 10.1016/0166-2236(89)90074-x. [DOI] [PubMed] [Google Scholar]
- Alexander GE, Crutcher MD. Functional architecture of basal ganglia circuits: neural substrates of parallel processing. Trends Neurosci. 1990;13:266–271. doi: 10.1016/0166-2236(90)90107-l. [DOI] [PubMed] [Google Scholar]
- Alvarez L, Macias R, Lopez G, Alvarez E, Pavon N, Rodriguez-Oroz MC, Juncos JL, Maragoto C, Guridi J, Litvan I, Tolosa ES, Koller W, Vitek J, DeLong MR, Obeso JA. Bilateral subthalamotomy in Parkinson's disease: initial and long-term response. Brain. 2005;128:570–583. doi: 10.1093/brain/awh397. [DOI] [PubMed] [Google Scholar]
- Aziz TZ, Peggs D, Sambrook MA, Crossman AR. Lesion of the subthalamic nucleus for the alleviation of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced parkinsonism in the primate. Mov Disord. 1991;6:288–292. doi: 10.1002/mds.870060404. [DOI] [PubMed] [Google Scholar]
- Benabid AL. Deep brain stimulation for Parkinson's disease. Curr Opin Neurobiol. 2003;13:696–706. doi: 10.1016/j.conb.2003.11.001. [DOI] [PubMed] [Google Scholar]
- Benabid AL, Benazzouz AL, Pollak P. Mechanisms of deep brain stimulation. Mov Disord. 2002;3:S73–S74. doi: 10.1002/mds.10145. [DOI] [PubMed] [Google Scholar]
- Benazzouz A, Gross C, Féger J, Boraud T, Bioulac B. Reversal of rigidity and improvement in motor performance by subthalamic high-frequency stimulation in MPTP-treated monkey. Eur J Neurosci. 1993;5:382–389. doi: 10.1111/j.1460-9568.1993.tb00505.x. [DOI] [PubMed] [Google Scholar]
- Bergman H, Wichmann T, DeLong MR. Reversal of experimental parkinsonism by lesions of the subthalamic nucleus. Science. 1990;249:1436–1438. doi: 10.1126/science.2402638. [DOI] [PubMed] [Google Scholar]
- Bergman H, Wichmann T, Karmon B, DeLong MR. The primate subthalamic nucleus. II. Neuronal activity in the MPTP model of parkinsonism. J Neurophysiol. 1994;72:507–520. doi: 10.1152/jn.1994.72.2.507. [DOI] [PubMed] [Google Scholar]
- Beurrier C, Bezard E, Bioulac B, Gross C. Subthalamic stimulation elicits hemiballismus in normal monkey. NeuroReport. 1997;8:1625–1629. doi: 10.1097/00001756-199705060-00014. [DOI] [PubMed] [Google Scholar]
- Bevan MD, Magill PJ, Terman D, Bolam JP, Wilson CJ. Move to the rhythm: oscillations in the subthalamic nucleus-external globus pallidus network. Trends Neurosci. 2002;25:525–531. doi: 10.1016/s0166-2236(02)02235-x. [DOI] [PubMed] [Google Scholar]
- Bruet N, Windels F, Carcenac C, Feuerstein C, Bertrand A, Poupard A, Savasta M. Neurochemical mechanisms induced by high frequency stimulation of the subthalamic nucleus: increase of extracellular striatal glutamate and GABA in normal and hemiparkinsonian rats. J Neuropathol Exp Neurol. 2003;62:1228–1240. doi: 10.1093/jnen/62.12.1228. [DOI] [PubMed] [Google Scholar]
- Carpenter MB, Whittier JR, Mettler FA. Analysis of choreoid hyperkinesia in the rhesus monkey: surgical and pharmacological analysis of hyperkinesia resulting from lesions in the subthalamic nucleus of Luys. J Comp Neurol. 1950;92:293–332. doi: 10.1002/cne.900920303. [DOI] [PubMed] [Google Scholar]
- Chang JY, Shi LH, Luo F, Woodward DJ. High frequency stimulation of the subthalamic nucleus improves treadmill locomotion in unilateral 6-hydroxydopamine lesioned rats. Brain Res. 2003;983:174–184. doi: 10.1016/s0006-8993(03)03053-1. [DOI] [PubMed] [Google Scholar]
- Chesselet MF, Delfs JM. Basal ganglia and movement disorders: an update. Trends Neurosci. 1996;19:417–422. doi: 10.1016/0166-2236(96)10052-7. [DOI] [PubMed] [Google Scholar]
- Chevalier G, Deniau JM. Disinhibition as a basic process in the expression of striatal functions. Trends Neurosci. 1990;13:277–280. doi: 10.1016/0166-2236(90)90109-n. [DOI] [PubMed] [Google Scholar]
- Crossman AR, Sambrook MA, Jackson A. Experimental hemiballismus in the baboon produced by injection of a gamma-aminobutyric acid antagonist into the basal ganglia. Neurosci Lett. 1980;20:369–372. doi: 10.1016/0304-3940(80)90176-7. [DOI] [PubMed] [Google Scholar]
- Crossman AR, Sambrook MA, Jackson A. Experimental hemichorea/hemiballismus in the monkey. Studies on the intracerebral site of action in a drug-induced dyskinesia. Brain. 1984;107:579–596. doi: 10.1093/brain/107.2.579. [DOI] [PubMed] [Google Scholar]
- Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001;65:1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- Degos B, Deniau JM, Thierry AM, Glowinski J, Pezard L, Maurice N. Neuroleptic-induced catalepsy: electrophysiological mechanisms of functional recovery induced by high-frequency stimulation of the subthalamic nucleus. J Neurosci. 2005;25:7687–7696. doi: 10.1523/JNEUROSCI.1056-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekundy A, Malgorzata P, Schaefer D, Cenci MA, Danysz W. Effects of group I metabotropic glutamate receptors blockade in experimental models of Parkinson's disease. Brain Res Bull. 2006;69:318–326. doi: 10.1016/j.brainresbull.2005.12.009. [DOI] [PubMed] [Google Scholar]
- DeLong MR. Primate models of movement disorders of basal ganglia origin. Trends Neurosci. 1990;13:281–285. doi: 10.1016/0166-2236(90)90110-v. [DOI] [PubMed] [Google Scholar]
- Dewey RB, Jankovic J. Hemiballism-hemichorea. Clinical and pharmacologic findings in 21 patients. Arch Neurol. 1989;46:862–867. doi: 10.1001/archneur.1989.00520440044020. [DOI] [PubMed] [Google Scholar]
- Dostrovsky JO, Lozano AM. Mechanisms of deep brain stimulation. Mov Disord. 2002;17:S63–S68. doi: 10.1002/mds.10143. [DOI] [PubMed] [Google Scholar]
- Guesdon JL, Ternynck T, Avrameas S. The use of avidin-biotin interaction in immunoenzymatic techniques. J Histochem Cytochem. 1979;27:1131–1139. doi: 10.1177/27.8.90074. [DOI] [PubMed] [Google Scholar]
- Guridi J, Herrero MT, Luquin MR, Guillen J, Ruberg M, Laguna J, Vila M, Javoy-Agid F, Agid Y, Hirsch E, Obeso JA. Subthalamotomy in parkinsonian monkeys. Behavioral and biochemical analysis. Brain. 1996;119:1717–1727. doi: 10.1093/brain/119.5.1717. [DOI] [PubMed] [Google Scholar]
- Guridi J, Obeso JA. The subthalamic nucleus, hemiballismus and Parkinson's disease: reappraisal of a neurosurgical dogma. Brain. 2001;124:5–19. doi: 10.1093/brain/124.1.5. [DOI] [PubMed] [Google Scholar]
- Hamada I, DeLong MR. Excitotoxic acid lesions of the primate subthalamic nucleus result in transient dyskinesias of the contralateral limbs. J Neurophysiol. 1992;68:1850–1858. doi: 10.1152/jn.1992.68.5.1850. [DOI] [PubMed] [Google Scholar]
- Hammond C, Deniau JM, Rizk A, Féger J. Electrophysiological demonstration of an excitatory subthalamonigral pathway in the rat. Brain Res. 1978;151:235–244. doi: 10.1016/0006-8993(78)90881-8. [DOI] [PubMed] [Google Scholar]
- Hammond C, Féger J, Bioulac B, Souteyrand JP. Experimental hemiballism in the monkey produced by unilateral kainic acid lesion in corpus Luysii. Brain Res. 1979;171:577–580. doi: 10.1016/0006-8993(79)91066-7. [DOI] [PubMed] [Google Scholar]
- Hashimoto T, Elder C, Okun MS, Patrick SK, Vitek JL. Stimulation of the subthalamic nucleus changes the firing pattern of pallidal neurons. J Neurosci. 2003;23:1916–1923. doi: 10.1523/JNEUROSCI.23-05-01916.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassani OK, Mouroux M, Féger J. Increased subthalamic neuronal activity after nigral dopaminergic lesion independent of disinhibition via the globus pallidus. Neuroscience. 1996;72:105–115. doi: 10.1016/0306-4522(95)00535-8. [DOI] [PubMed] [Google Scholar]
- Henderson JM, Annett LE, Ryan LJ, Chiang W, Hidaka S, Torres EM, Dunnett SB. Subthalamic nucleus lesions induce deficits as well as benefits in the hemiparkinsonian rat. Eur J Neurosci. 1999;11:2749–2757. doi: 10.1046/j.1460-9568.1999.00692.x. [DOI] [PubMed] [Google Scholar]
- Hutchison WD, Allan RJ, Opitz H, Levy R, Dostrovsky JO, Lang AE, Lozano AM. Neurophysiological identification of the subthalamic nucleus in surgery for Parkinson's disease. Ann Neurol. 1998;44:622–628. doi: 10.1002/ana.410440407. [DOI] [PubMed] [Google Scholar]
- Kita H. Physiology of two disynaptic pathways from the sensorimotor cortex to the basal ganglia output nuclei. In: Percheron G, McKenzie JS, Féger J, editors. The basal ganglia IV. New York: Plenum; 1994. pp. 263–276. [Google Scholar]
- Kita H, Kitai ST. Efferent projections of the subthalamic nucleus in the rat: light and electron microscopic analysis with the PHA-L method. J Comp Neurol. 1987;260:435–452. doi: 10.1002/cne.902600309. [DOI] [PubMed] [Google Scholar]
- Kita H, Kitai ST. The morphology of globus pallidus projection neurons in the rat: an intracellular staining study. Brain Res. 1994;636:308–319. doi: 10.1016/0006-8993(94)91030-8. [DOI] [PubMed] [Google Scholar]
- Kita H, Tachibana Y, Nambu A, Chicken S. Balance of monosynaptic excitatory and disynaptic inhibitory responses of the globus pallidus induced after stimulation of the subthalamic nucleus in the monkey. J Neurosci. 2005;25:8611–8619. doi: 10.1523/JNEUROSCI.1719-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitai ST, Deniau JM. Cortical inputs to the subthalamus: intracellular analysis. Brain Res. 1981;214:411–415. doi: 10.1016/0006-8993(81)91204-x. [DOI] [PubMed] [Google Scholar]
- Krack P, Batir A, Van Blercom N, Chabardes S, Fraix V, Ardouin C, Koudsie A, Dowsey Limousin P, Benazzouz A, Lebas JF, Benabid AL, Pollak P. Five-year follow-up bilateral stimulation of the subthalamic nucleus in advanced Parkinson's disease. N Eng J Med. 2003;349:1925–1934. doi: 10.1056/NEJMoa035275. [DOI] [PubMed] [Google Scholar]
- Lee MS, Marsden CD. Movement disorders following lesions of the thalamus or subthalamic region. Mov Disord. 1994;9:493–507. doi: 10.1002/mds.870090502. [DOI] [PubMed] [Google Scholar]
- Levy R, Ashby P, Hutchinson WD, Lang AE, Lozano AM, Dostrovsky JO. Dependence of subthalamic nucleus oscillations on movement and dopamine in Parkinson's disease. Brain. 2002;125:1196–1209. doi: 10.1093/brain/awf128. [DOI] [PubMed] [Google Scholar]
- Limousin P, Pollak P, Benazzouz A, Hoffman D, Broussolle E, Perret JE, Benaabid Bilateral subthalamic nucleus stimulation for severe Parkinson's disease. Mov Disord. 1995;10:672–674. doi: 10.1002/mds.870100523. [DOI] [PubMed] [Google Scholar]
- Limousin P, Pollak P, Hoffmann D, Benazzouz A, Perret JE, Benabid AL. Abnormal involuntary movements induced by subthalamic stimulation in parkinsonian patients. Mov Disord. 1996;11:231–235. doi: 10.1002/mds.870110303. [DOI] [PubMed] [Google Scholar]
- Limousin P, Krack P, Pollak P, Benazzouz A, Ardouin C, Hoffmann D, Benabid AL. Electrical stimulation of the subthalamic nucleus in advanced Parkinson's disease. N Engl J Med. 1998;339:1105–1111. doi: 10.1056/NEJM199810153391603. [DOI] [PubMed] [Google Scholar]
- Mailly P, Charpier S, Menetrey A, Deniau JM. Three-dimensional organization of the recurrent axon collateral network of the substantia nigra pars reticulata neurons in the rat. J Neurosci. 2003;23:5247–5257. doi: 10.1523/JNEUROSCI.23-12-05247.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurice N, Thierry AM, Glowinski J, Deniau JM. Spontaneous and evoked activity of substantia nigra pars reticulata neuron during high-frequency stimulation of the subthalamic nucleus. J Neurosci. 2003;23:9929–9936. doi: 10.1523/JNEUROSCI.23-30-09929.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntyre CC, Savasta M, Kerkerian-Legoff L, Vitek JL. Uncovering the mechanism(s) of action of deep brain stimulation: activation, inhibition, or both. Clin Neurophysiol. 2004a;115:1239–1248. doi: 10.1016/j.clinph.2003.12.024. [DOI] [PubMed] [Google Scholar]
- McIntyre CC, Savasta M, Walter B, Vitek JL. How does deep brain stimulation work? Present understanding and future questions. J Clin Neurophysiol. 2004b;21:1–11. doi: 10.1097/00004691-200401000-00006. [DOI] [PubMed] [Google Scholar]
- Miller WC, DeLong MR. Altered tonic activity in the globus pallidus and subthalamic nucleus in the primate MPTP model parkinsonian. In: Carpenter MB, Jayaraman A, editors. The basal ganglia II. New York: Plenum; 1987. pp. 415–427. Structure and function: current concepts. [Google Scholar]
- Mink JW, Thach WT. Basal ganglia intrinsic circuits and their role in behavior. Curr Opin Neurobiol. 1993;3:950–957. doi: 10.1016/0959-4388(93)90167-w. [DOI] [PubMed] [Google Scholar]
- Moro E, Esselink RJA, Xie J, Hommel M, Benabid AL, Pollak P. The impact on Parkinson's disease of electrical parameter settings in STN stimulation. Neurology. 2002;59:1–7. doi: 10.1212/wnl.59.5.706. [DOI] [PubMed] [Google Scholar]
- Nambu A. A new dynamic model of the cortico-basal ganglia loop. Prog Brain Res. 2004;143:461–466. doi: 10.1016/S0079-6123(03)43043-4. [DOI] [PubMed] [Google Scholar]
- Nash JE, Ravenscroft P, McGuire S, Crossman AR, Menniti FS, Brotchie JM. The NR2B-selective NMDA receptor antagonist CP-101,606 exacerbates l-DOPA-induced dyskinesia and provides mild potentiation of anti-parkinsonian effects of l-DOPA in the MPTP-lesioned marmoset model of Parkinson's disease. Exp Neurol. 2004;188:471–479. doi: 10.1016/j.expneurol.2004.05.004. [DOI] [PubMed] [Google Scholar]
- Obeso JA, Rodriguez MC, DeLong MR. Basal ganglia pathophysiology. A critical review. Adv Neurol. 1997;74:3–18. [PubMed] [Google Scholar]
- Papa SM, Chase TN. Levodopa induced dyskinesias improved by a glutamate antagonist in parkinsonian monkeys. Ann Neurol. 1996;39:574–578. doi: 10.1002/ana.410390505. [DOI] [PubMed] [Google Scholar]
- Parpura V, Scemes E, Spray DC. Mechanisms of glutamate release from astrocytes: gap junction “hemichannels,” purinergic receptors and exocytotic release. Neurochem Int. 2004;45:259–264. doi: 10.1016/j.neuint.2003.12.011. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates, Ed 4. San Diego: Academic; 1982. [Google Scholar]
- Perier C, Tremblay L, Feger J, Hirsch EC. Behavioral consequences of bicuculline injection in the subthalamic nucleus and the zona incerta in rat. J Neurosci. 2002;22:8711–8719. doi: 10.1523/JNEUROSCI.22-19-08711.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosales MG, Martinez-Fong D, Morales R, Nunez A, Flores G, Gongora-Alfaro JL, Floran B, Aceves J. Reciprocal interaction between glutamate and dopamine in the pars reticulata of the rat substantia nigra: a microdialysis study. Neuroscience. 1997;80:803–810. doi: 10.1016/s0306-4522(97)00160-7. [DOI] [PubMed] [Google Scholar]
- Rozza A, Masoero E, Favalli L, Lanza E, Govoni S, Rizzo V, Montalbetti L. Influence of different anaesthetics on extracellular aminoacids in rat brain. J Neurosci Methods. 2000;101:165–169. doi: 10.1016/s0165-0270(00)00266-1. [DOI] [PubMed] [Google Scholar]
- Salin P, Manrique C, Forni C, Kerkerian-Le Goff L. High-frequency stimulation of the subthalamic nucleus selectively reverses dopamine denervation induced cellular defects in the output structures of the basal ganglia in the rat. J Neurosci. 2002;22:5137–5148. doi: 10.1523/JNEUROSCI.22-12-05137.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sgambato-Faure V, Buggia V, Gilbert F, Lévesque D, Benabid AL, Berger F. Coordinated and spatial up regulation of Arc in striato-nigral neurons correlates with l-Dopa-induced-behavioral sensitization in dyskinetic rats. J Neuropathol Exp Neurol. 2005;64:936–947. doi: 10.1097/01.jnen.0000186922.42592.b7. [DOI] [PubMed] [Google Scholar]
- Sharp T, Zetterstrom T, Ljunberg T, Ungerstedt U. A direct comparison of amphetamine-induced behaviours and regional brain dopamine release in the rat using intracerebral dialysis. Brain Res. 1987;401:322–330. doi: 10.1016/0006-8993(87)91416-8. [DOI] [PubMed] [Google Scholar]
- Shen K, Zhu ZT, Munhall A, Johnson SW. Synaptic plasticity in rat subthalamic induced by high frequency stimulation. Synapse. 2003;50:314–319. doi: 10.1002/syn.10274. [DOI] [PubMed] [Google Scholar]
- Smith Y, Shink E, Sidibe M. Neuronal circuitry and synaptic connectivity of the basal ganglia. Neurosurg Clin N Am. 1998;9:203–222. [PubMed] [Google Scholar]
- Tossman U, Ungerstedt U. Microdialysis in the study of extracellular levels of amino acids in the rat brain. Acta Physiol Scand. 1986;128:9–14. doi: 10.1111/j.1748-1716.1986.tb07943.x. [DOI] [PubMed] [Google Scholar]
- Vitek JL. Mechanisms of deep brain stimulation: excitation or inhibition. Mov Disord. 2002;17:S69–S72. doi: 10.1002/mds.10144. [DOI] [PubMed] [Google Scholar]
- Whittier JR, Mettler FA. Studies of the subthalamus of the rhesus monkey. II. Hyperkinesia and other physiologic effects of subthalamic lesions with special references to the subthalamic nucleus of Luys. J Comp Neurol. 1949;90:319–372. doi: 10.1002/cne.900900304. [DOI] [PubMed] [Google Scholar]
- Windels F, Kiyatkin EA. General anesthesia as a factor affecting impulse activity and neuronal responses to putative neurotransmitters. Brain Res. 2006;1086:104–116. doi: 10.1016/j.brainres.2006.02.064. [DOI] [PubMed] [Google Scholar]
- Windels F, Bruet N, Poupard A, Urbain N, Chouvet G, Feuerstein C, Savasta M. Effects of high frequency stimulation of subthalamic nucleus on extracellular glutamate and GABA in substantia nigra and globus pallidus in the normal rat. Eur J Neurosci. 2000;12:4141–4146. doi: 10.1046/j.1460-9568.2000.00296.x. [DOI] [PubMed] [Google Scholar]
- Windels F, Bruet N, Poupard A, Feuerstein C, Bertrand A, Savasta M. Influence of the frequency parameter on extracellular glutamate and gamma-aminobutyric acid in substantia nigra and globus pallidus during electrical stimulation of subthalamic nucleus in rats. J Neurosci Res. 2003;72:259–267. doi: 10.1002/jnr.10577. [DOI] [PubMed] [Google Scholar]
- Windels F, Carcenac C, Poupard A, Savasta M. Pallidal origin of GABA release within the substantia nigra pars reticulata during high frequency stimulation of the subthalamic nucleus. J Neurosci. 2005;25:5079–5086. doi: 10.1523/JNEUROSCI.0360-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler C, Kirik D, Bjorklund A, Cenci MA. L-DOPA induced dyskinesia in the intrastriatal 6-hydroxydopamine model of Parkinson's disease: relation to motor and cellular parameters of nigrostriatal function. Neurobiol Dis. 2002;10:165–186. doi: 10.1006/nbdi.2002.0499. [DOI] [PubMed] [Google Scholar]