

Abstract
The identities of some of the constituents of the hair-cell transduction apparatus have been elucidated only recently. The molecular motor myosin-1c (Myo1c) functions in adaptation of the hair-cell response to sustained mechanical stimuli and is therefore an integral part of the transduction complex. Recent data indicate that Myo1c interacts in vitro with two other molecules proposed to be important for transduction: cadherin 23 (Cdh23), a candidate for the stereociliary tip link, and phosphatidylinositol 4,5-bisphosphate (PIP2), which is abundant in the membranes of hair-cell stereocilia. It is not known, however, whether these interactions occur in hair cells. Using an in situ binding assay on saccular hair cells, we demonstrated previously that Myo1c interacts with molecules at stereociliary tips, the site of transduction, through sequences contained within its calmodulin (CaM)-binding neck domain, which can bind up to four CaM molecules. In the current study, we identify the second CaM-binding IQ domain as a region of Myo1c that mediates CaM-sensitive binding to stereociliary tips and to PIP2 immobilized on a solid support. Binding of Myo1c to stereociliary tips of cochlear and vestibular hair cells is disrupted by treatments that break tip links. In addition, Myo1c does not bind to stereocilia from mice whose hair cells lack Cdh23 protein despite the presence of PIP2 in the stereociliary membranes. Collectively, our data suggest that Myo1c and Cdh23 interact at the tips of hair-cell stereocilia and that this interaction is modulated by CaM.
Keywords: myosin 1c; hair cell; calmodulin; cadherin 23; phosphatidylinositol 4,5-bisphosphate; IQ domain
Introduction
In auditory and vestibular end organs, transduction of physical stimuli into electrical signals occurs in hair cells and is critically dependent on the hair bundle, which protrudes from the hair-cell apical surface and is comprised of actin-filled stereocilia arranged in rows by ascending height (Hudspeth, 1989). Stereocilia are interconnected by several types of extracellular linkages (Goodyear et al., 2005) including the tip link, which connects the top of a shorter stereocilium to the side of its tallest neighbor (Pickles et al., 1984). Cadherin 23 (Cdh23) has been proposed to be a tip-link component (Siemens et al., 2004; Söllner et al., 2004); however, this designation is controversial (Lagziel et al., 2005; Michel et al., 2005; Rzadzinska et al., 2005). In the prevailing model of hair-cell transduction, bundle deflection tenses tip links and opens mechanically gated, cation-selective transduction channels (Pickles et al., 1984; Howard and Hudspeth, 1987). During prolonged stimuli, transduction-current magnitude reduces within tens of microseconds (for review, see Gillespie and Cyr, 2004). This adaptation occurs at two rates (Wu et al., 1999; Holt et al., 2002; Stauffer et al., 2005) and involves the motor protein myosin-1c (Myo1c) (Holt et al., 2002; Stauffer et al., 2005), an unconventional myosin comprised of three domains: a mechanochemical head, a neck containing three to four calmodulin (CaM)-binding IQ domains, and a tail (see Fig. 1A). CaM binding to Myo1c IQ domains is favored in low Ca2+ and modulates both motor activity and binding to intracellular components (for review, see Gillespie and Cyr, 2004).
Figure 1.

Recombinant Myo1c proteins. A, Construct maps. Wild-type Myo1c includes a catalytic head domain, a neck composed of four tandem IQ domains that bind CaM, and a tail region. The recombinant protein Myo1c-T701 encompasses the Myo1c neck and tail domains. Myo1c-T701IQ2Mut is identical to Myo1c-T701, with the exception of mutations made to the first two amino acids of the IQ consensus sequence of the second IQ domain: I728A and Q729A. Myo1c-N2 consists of only IQ2. Purification and detection tags (H, His6-tag; S, S-tag; X, Xpress-tag) have been appended to the N terminus of all recombinant constructs and are indicated by the light gray box. Myo1c-T701IQ1RG1Mut is identical to Myo1c-T701, with the exception of mutations that made four amino acids of the IQ consensus sequence of the first IQ domain: L706A, Q707A, R711A, and G712A. The diagram is not drawn to scale. B, C, Coomassie blue-stained SDS-PAGE gels of recombinant Myo1c protein fragments Myo1c-T701 and Myo1c-T701IQ2Mut purification. HSS, High-speed supernatant; FT, flow through from Ni2+-NTA column; Wash, Ni2+-NTA column wash; Eluate, protein eluted from Ni2+-NTA column. Myo1c-T701 and Myo1c-T701IQ2Mut migrate at ∼34 kDa (*). CaM (#) copurifies with each Myo1c fragment and migrates at ∼16 kDa. D, Purification of recombinant Myo1c protein fragment N2. Lanes are labeled as in B. In addition, Gel filtration shows the Myo1c-N2 protein further purified by size-exclusion chromatography. CaM is purified bovine CaM. Note that although CaM is coexpressed with Myo1c-N2, CaM does not copurify with the Myo1c-N2 protein. E, Coomassie blue-stained SDS-PAGE gel of Myo1c-T701IQ1RG1Mut protein purification. Lanes are labeled as indicated in B. Molecular weight markers (kDa) are indicated to the left of each gel.
Slow adaptation is predicted to require, at a minimum, the transduction channel; the tip link, which may be, in part, comprised of Cdh23 (Siemens et al., 2004; Söllner et al., 2004); the adaptation motor, Myo1c (Holt et al., 2002); and phosphatidylinositol 4,5-bisphosphate (PIP2) (Hirono et al., 2004). To generate a functional transduction complex, these molecules are expected to interact with one another and perhaps other unidentified transduction-apparatus constituents. To date, Myo1c has been shown to bind, in vitro, to both PIP2 and Cdh23 (Hirono et al., 2004; Siemens et al., 2004; Hokanson and Ostap, 2006); however, such interactions have not been demonstrated in hair cells.
To examine Myo1c interactions with transduction components, we developed an in situ binding assay to visualize Myo1c binding to hair-cell constituents, which we term “Myo1c receptors” (Cyr et al., 2002). Using this assay, we demonstrated that the Myo1c neck region binds to available receptors at stereociliary tips, the site of transduction; in the pericuticular necklace, a vesicle-rich region encircling the cuticular plate; in the kinocilium, a cilium located at the tall edge of the hair bundle; and also within the hair-cell soma (Cyr et al., 2002). With the exception of the kinocilium, Myo1c binding is modulated by CaM, which implicated the CaM-binding IQ domains in receptor interactions (Cyr et al., 2002). In the current study, we determine that the second IQ domain (IQ2) of Myo1c mediates interactions with stereociliary receptors. In addition, these interactions are sensitive to Ca2+ chelation, a procedure that breaks tip links, and do not occur in hair cells lacking Cdh23.
Materials and Methods
Animals.
All animal procedures followed National Institutes of Health guidelines and were approved by either the Animal Care and Use Committee at West Virginia University School of Medicine or the United Kingdom Home Office and local ethical committee.
Myo1c constructs.
To generate Myo1c-T701IQ2Mut (see Fig. 1A), a construct with mutations in conserved residues in the second consensus IQ domain of Myo1c, the first two amino acids, Ile728 and Gln729, were each mutated to Ala in the context of the bullfrog Myo1c-T701 construct (Cyr et al., 2002), using the Quick-Change site-directed mutagenesis system (Stratagene, La Jolla, CA) and the oligonucleotides 5′-ATTCCGCGGTAGAGGCCGCGTCGTGGTGGCGTGG-3′ and 5′-CCACGCCACCACGACGCGGCCTCTACCGCGGAAAT-3′. A construct with mutations in the first IQ domain of bullfrog Myo1c (IQ1), Myo1c-T701IQ1RG1Mut, was generated by site-directed mutagenesis using the oligonucleotide primers 5′-GGGTAGCATAGCAACATTCGCCGCGGCTAGGTGGAGAGG-3′ and 5′-CCTCTCCACCTAGCCGCGGCGAATGTTGCTATGCTACCC -3′ to mutate Leu706 and Gln707 to Ala residues. The residues Arg711 and Gly712 were each also mutated to Ala using the primers 5′-GCAACATTCCTCCAGGCTAGGTGGGCGGCCTATCATCAACGACAG-3′ and 5′-CTGTCGTTGATGATAGGCCGCCCACCTAGCCTGGAGGAATGTTGC-3′, resulting in a construct in which four of the five residues of the IQ consensus sequence of IQ1 were altered. The mutated Myo1c sequences were ligated into the pFastbacI expression vector (Invitrogen, Carlsbad, CA) and sequenced. A construct encoding the second IQ domain of Myo1c, Myo1c-N2 (bullfrog Myo1c amino acids 721–744), was generated by PCR using pGus-T701 (Cyr et al., 2002) as the template and the oligonucleotides 5′-CATGCCATGGGTCACATGAAACATTCCGCGGTAG-3′ and 5′-CCGGAATTCTCACTACTCATTCTCAGTGCAACGGGG-3′. The amplified product was ligated into the EcoRI and NcoI sites of pBBHis2A-N123 (Cyr et al., 2002) and reamplified using the primers 5′-TTGGCGCGCCTATAAATTGCCGCGGGGTTC-3′ and 5′-CCGGAATTCTCACTACTCATTCTCAGTGCAACGGGG-3′. The PCR product was ligated into the BssHI and EcoRI sites of the pFastBacI vector and sequenced.
Recombinant Myo1c protein purification.
Recombinant bullfrog Myo1c protein fragments were coexpressed with CaM in Sf9 insect cells using baculovirus infection as described previously (Gillespie et al., 1999). Sf9 cells expressing recombinant protein were sedimented and stored at −80°C. Protein was purified from the frozen pellets using a Ni2+-nitrilotriacetic acid chromatography column (Qiagen, Valencia, CA) as outlined by Cyr et al. (2002). Eluted fractions exhibiting a high-protein concentration were pooled and stored on ice at 4°C. Eluates were analyzed by SDS-PAGE and gel-filtration chromatography on a 25 ml Superdex 200 HR 10/30 or Superdex 200 10/300 GL column (GE Healthcare, Piscataway, NJ) in 400 mm KCl, 0.1 mm EGTA, and 15 mm HEPES, pH 7.5, at room temperature. Protein concentrations were determined using the Bradford assay with bovine serum albumin (BSA) as a standard (Bradford, 1976).
CaM purification.
Bovine CaM was purified from brain tissue using a phenyl-Sepharose affinity column (Gopalakrishna and Anderson, 1982) and was quantified assuming 330 μm/A276nm.
Myo1c fragment:CaM stoichiometry.
Gel filtration-purified Myo1c-T701, Myo1c-T701IQ2Mut, and Myo1c-T701IQ1RG1Mut protein fragments were each concentrated by trichloroacetic acid (TCA) precipitation (8% v/v) and resuspended in NuPage lithium dodecyl sulfate sample buffer (Invitrogen). Resuspended TCA precipitates were separated by electrophoresis on 12% Bis-Tris NuPage gels in NuPage MOPS SDS running buffer (Invitrogen) and stained with colloidal blue (Invitrogen). Destained gels were scanned using a flatbed scanner and analyzed with Scion Image Beta software (v4.03; Scion Corporation, Frederick, MD). The ratio of mutated Myo1c heavy-chain fragments to CaM was calculated using the wild-type Myo1c-T701 protein complex as a standard. Under our purification conditions, Myo1c-T701 binds 1.8 ± 0.2 CaM molecules per Myo1c polypeptide (Gillespie and Cyr, 2002). Three separate preparations of each purified recombinant protein were analyzed in duplicate on two separate gels. Values are represented as the mean ± SD.
Cochlear cultures.
Cochlear cultures were prepared from postnatal day 0 (P0) to P2 Cdh23v2J and Myo7a6J mice as described previously (Russell and Richardson, 1987). Briefly, cochleas were dissected in HBSS buffered with 10 mm HEPES, pH 7.2 (HBHBSS). Cochlear coils were plated onto a hydrated collagen gel on a glass coverslip and maintained in sealed Maximow slide assemblies containing ∼50 μl of medium (93% DMEM/F12, 7% fetal calf serum, 10 μg/ml ampicillin). Cultures were grown for 1–2 d in vitro at 37°C, after which they were fixed in 3% paraformaldehyde (Agar Scientific, Essex, UK) and stored in PBS (137 mm NaCl, 2.7 mm KCl, 4.3 mm Na2HPO4, and 1.5 mm KH2PO4, pH 7.4) at 4°C. To assess the dependence of Myo1c binding on intact tip links, tips links were broken in a subset of the cultures before fixation by incubation for 15–30 min at room temperature in either a solution containing 5 mm EGTA or 5 mm BAPTA in HBHBSS supplemented with 0.5 mm MgCl2 and 0.4 mm MgSO4 or a solution containing 5 mm LaCl3 in 155 mm NaCl, 6 mm KCl, 3 mm glucose, 4 mm CaCl2, and 10 mm HEPES, pH 7.2.
In situ binding assay.
In situ binding assays were performed as described previously (Cyr et al., 2002) on bullfrog (Rana catesbeina) sacculi or mouse cochlear cultures. Briefly, the samples were fixed in 3% paraformaldehyde in PBS, washed in PBS, and permeabilized with 0.1% (w/v) Sarkosyl for 1 h. After a PBS wash, nonspecific binding sites were blocked with 5 mg/ml BSA (fraction V; Calbiochem, La Jolla, CA) in 25 mm HEPES, pH 7.5, and the samples were incubated overnight at room temperature with purified Myo1c fragments at 20 μg/ml [corresponding to either 230 nm for Myo1c-T701, Myo1c-T701IQ2Mut (n = 10) and Myo1c-T701IQ1RG1Mut (n = 9), or 1.65 μm for Myo1c-N2 (n = 4)] in 5 mg/ml BSA, 25 mm HEPES, pH 7.5, 1 mm EGTA, and 400 mm NaCl. After washing, bound Myo1c fragments were detected by virtue of the Xpress tag located at the N terminus of the Myo1c constructs, using the anti-Xpress antibody (5 μg/ml; Invitrogen). After three washes, bound antibody was detected with Alexa-488 goat anti-mouse secondary antibody (13 μg/ml; Invitrogen) in 25 mm HEPES, pH 7.5, 1 mm EGTA, and 5 mg/ml BSA. Filamentous actin was labeled with 33 nm Alexa 568-phalloidin (Invitrogen) present during the secondary antibody incubation. All aforementioned washes were performed using 25 mm HEPES, pH 7.5, 1 mm EGTA, and 0.1% Tween 20. Samples were then washed in 25 mm HEPES, pH 7.5, 1 mm EGTA, mounted using Vectashield (Vector Laboratories, Burlingame, CA), and imaged on a LSM-510 Meta confocal microscope (Zeiss, Thornwood, NY). In some experiments, 16 μm bovine brain CaM was added during the recombinant protein incubation step (for coincubations with Myo1c-T701IQ2Mut, n = 9; with Myo1c-T701IQ1RG1Mut, n = 4). To break tip links, bullfrog sacculi were treated for 30 min at room temperature with either 5 mm EGTA (n = 8) in low-Ca2+ saline (110 mm NaCl, 2 mm KCl, 2 mm MgCl2, 0.1 mm CaCl2, 3 mm d-glucose, 10 mm HEPES, pH 7.2) or 5 mm LaCl3 (n = 5) in low-Ca2+ saline without MgCl2. To assess the efficacy of these treatments, sacculi were subjected in parallel to a 5 s incubation with 3 μm AM1–43 (Biotium, Hayward, CA), washed, fixed, mounted, and visualized by confocal microscopy. Images of Myo1c binding to pretreated tissues are gain-matched to control conditions except for Figure 8, C and D, where the gains for Myo1c-T701 binding are 558 and 565, respectively.
Figure 8.
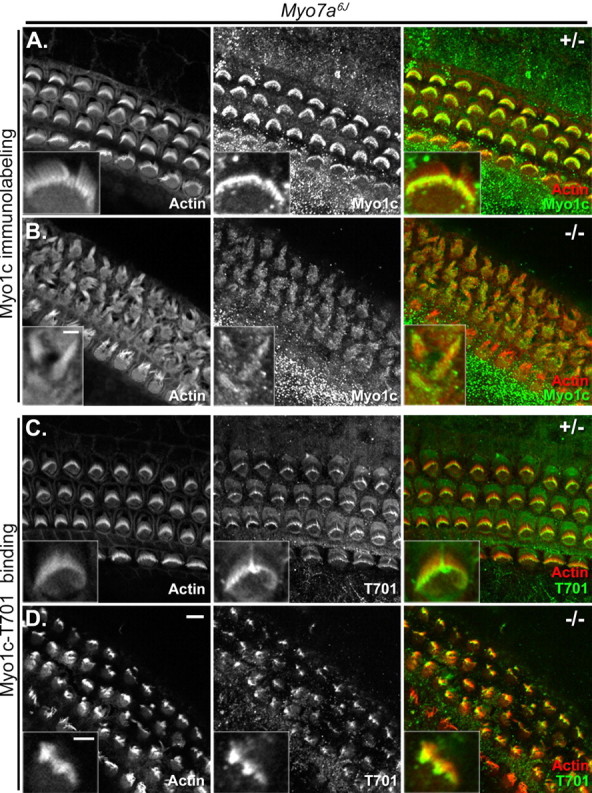
Myo1c and Myo1c receptor localization in cochlear hair cells of Myo7a6J mice. Cochlear cultures from Myo7a6J heterozygous (+/−) or homozygous (−/−) mice (P2–P4) are shown in each panel. The left column of each panel shows phalloidin labeling of filamentous actin, and the middle column of each panel shows Myo1c antibody or Myo1c-T701 binding alone. The right column of each panel shows the overlay of phalloidin labeling (red) and the binding of either a Myo1c antibody (green; A, B) or the Myo1c-T701 fragment (green; C, D). A, B, Endogenous Myo1c is present in the stereocilia of both Myo7a6J heterozygous and homozygous mice. C, D, The Myo1c receptor is detected in the hair cells of Myo7a6J heterozygous and homozygous mice. All insets are the hair bundles of outer hair cells except for that in A, which is an inner hair cell. Scale bars: (in D) 5 μm; B, inset, 2 μm; (in D, inset) A, C, D, insets, 2 μm.
PIP-strip binding assay.
PIP-strips (Invitrogen and Echelon Biosciences, Salt Lake City, UT) were incubated in blocking solution (1:10 dilution of Liquid Block in PBS; GE Healthcare) to reduce nonspecific binding. This and subsequent steps were performed at room temperature. Purified recombinant Myo1c protein was added at a concentration of 1 μg/ml (corresponding to either 11.5 nm for Myo1c-T701 and Myo1c-T701IQ2Mut or 82.5 nm Myo1c-N2) with or without 460 nm CaM to blocking solution containing 1 mm EGTA and incubated for 2 h. After three 10 min washes with PBS/0.1%Tween 20 (PBS-T), the membranes were incubated with 1 μg/ml anti-Xpress antibody in blocking solution for 1 h, washed in PBS-T, and incubated with 170 ng/ml goat anti-mouse alkaline phosphatase antibody (Jackson ImmunoResearch, West Grove, PA) in blocking solution for 1 h. The PIP strips were then washed three times for 10 min with PBS-T and two times for 5 min in alkaline phosphatase buffer (100 mm Tris, pH 9.5, 50 mm MgCl2, 100 mm NaCl) and developed using 1-step NBT/BCIP (Pierce, Rockford, IL) for 5–15 min.
Myo1c immunolabeling.
Cochlear cultures from Cdh23v2J or Myo7a6J mice were fixed with cold 3% paraformaldehyde in PBS for 25 min. Samples were washed and stored in PBS at 4°C until they were processed for immunocytochemistry, at which time the tissue was permeabilized with 0.2% saponin in PBS for 1 h. This and subsequent steps were performed at room temperature unless otherwise indicated. Nonspecific binding sites were blocked using 5% goat serum, 1% BSA, and 0.1% saponin in PBS for 1 h. The tissue was incubated overnight at 4°C with 3.4 μg/ml anti-Myo1c antibody (R2652; gift from P. Gillespie, Oregon Health and Science University, Portland, OR) (Dumont et al., 2002) in blocking solution. Samples were washed three times for 10 min in PBS-T and incubated with 13 μg/ml Alexa 488 goat anti-rabbit antibody and 33 nm Alexa-568 phalloidin in PBS containing 1% BSA and 0.1% saponin for 1.5 h. Samples were washed three times for 10 min in PBS-T followed by one time for 10 min in PBS. Samples were mounted in Vectashield and imaged by confocal microscopy. Immunolabeled images acquired for treated samples are gain-matched to control conditions. Ten apical and 10 basal cochlear coils from five heterozygous Cdh23v2J mice were examined, and 16 apical and 16 basal cochlear coils from eight homozygous Cdh23v2J animals were examined. Likewise, four apical and four basal cochlear coils from two heterozygous Myo7a6J mice and nine apical coils and 10 basal cochlear coils from five homozygous Myo7a6J mice were analyzed.
Cdh23 immunolabeling.
Cdh23 immunolabeling was performed on cochlear cultures from wild-type mice, acutely dissected vestibular organs (P3 and P6) from wild-type mice, or acutely dissected bullfrog sacculi with anti-Cdh23 antiserum (T8; gift from T. Friedman, National Institute on Deafness and Other Communication Disorders, Bethesda, MD) (Lagziel et al., 2005) as outlined for Myo1c immunolabeling, except the tissue was permeabilized for 30 min with 0.5% Triton X-100 in PBS and the blocking solution was 2% BSA, 5% goat serum in PBS. To break tip links, samples were subjected to the designated treatments before fixation as indicated above for cochlear cultures or as outlined in the in situ binding assay for bullfrog sacculi. Four apical and four basal mouse cochlear turns, five mouse utricules, and at least three frog sacculi were examined for each treatment. Immunolabeled images acquired for treated samples are gain-matched to control conditions.
PIP2 immunolabeling.
Mouse cochlear cultures (P0 plus 1 d in vitro) from Cdh23v2J animals were fixed in 4% paraformaldehyde (diluted from ampules of a 16% stock solution) in 0.1 m sodium phosphate pH 7.2, washed in PBS, and stored at 4°C overnight. The tissue was permeabilized with 1% (w/v) Sarkosyl in PBS for 1 h, blocked for 1 h with 5 mg/ml BSA in Tris-buffered saline (TBS; 10 mm Tris-HCl, pH 7.4, 150 mm NaCl), and incubated with 10 μg/ml anti-PIP2 monoclonal antibody (clone 2C11; Invitrogen) in blocking solution overnight at 4°C. The tissue was washed with 5 mg/ml BSA in TBS, incubated with FITC-conjugated goat anti-mouse IgM secondary antibody (1:200 dilution; Sigma Aldrich) and Texas Red-conjugated phalloidin (1U/ml, Invitrogen) in blocking solution for 2–3 h, washed, mounted, and visualized by confocal microscopy. All steps were performed at room temperature unless otherwise noted. The gain setting for the heterozygous image presented in Figure 9C is 590 and for the homozygous image in Figure 9D is 540. A total of 10 apical and 10 basal cochlear turns were examined from five homozygous Cdh23v2J mice; eight apical and eight basal cochlear coils from four heterozygous Cdh23v2J mice were examined.
Figure 9.

PIP2 is present in the stereocilia of Cdh23v2J mice. A, B, Localization of PIP2 in a cochlear apical coil from a heterozygous (A) and a homozygous (B) Cdh23v2J mouse using the PH domain of PLCδ1 conjugated to EGFP as a fluorescent indicator of PIP2 localization. C, D, Immunolocalization of PIP2 in a cochlear basal coil from a heterozygous (C) and a homozygous (D) Cdh23v2J mouse. All images are single confocal sections. In the merged image to the left, phalloidin-labeled filamentous actin is depicted in red, and PLCδ1PH-EGFP or an anti-PIP2 antibody is green. In the image on the right, PLCδ1PH-EGFP or the antibody labeling alone is shown. The arrowhead in B denotes a fluorescent mutant hair bundle. The arrows in D denote microvilli labeling. Scale bars: (in B) A, B, 5 μm; (in D) C, D, 5 μm.
Biolistic transfection of hair cells with the enhanced green fluorescent protein-tagged pleckstrin homology domain of PLCδ1.
A mammalian expression vector containing the N-terminal pleckstrin homology (PH) domain of phospholipase Cδ1 (PLCδ1) ligated in frame with the enhanced green fluorescent protein (EGFP) [PLCδ1PH-EGFP (Watt et al., 2002)] was a kind gift from Peter Downes (University of Dundee, Dundee, UK). Gold microcarrier particles (1 μm diameter) were coated with plasmid DNA and used to transfect cochlear cultures prepared from 1- to 2-d-old Cdh23v2J mouse pups using a Helios Gene Gun (Bio-Rad, Hercules, CA) (Schneider et al., 2002; Rzadzinska et al., 2004). After an additional 20–24 h in vitro, cultures were fixed with 3.7% formaldehyde in 0.1 m sodium phosphate buffer, pH 7.4, for 1 h, preblocked, and permeabilized with 10% horse serum in TBS containing 0.1% Triton X-100, and stained with rabbit anti-GFP antibody (4 μg/ml; Invitrogen) followed by FITC-conjugated swine anti-rabbit Ig (4 μg/ml; Dako, High Wycombe, UK) and Texas Red-conjugated phalloidin (1 U/ml; Invitrogen). Samples were mounted and imaged by scanning confocal microscopy. A total of 17 transfected hair cells from five heterozygous Cdh23v2J cultures and 37 hair cells from nine homozygous Cdh23v2J cultures were analyzed.
Results
The second IQ domain of Myo1c is necessary, but not sufficient, for interaction with stereociliary receptors
To evaluate the role of the Myo1c IQ domains in binding to stereociliary receptors, we exploited two previous observations: (1) CaM blocks the binding of Myo1c-T701, a fragment of Myo1c containing the neck and tail regions (Fig. 1A), to the tips of stereocilia (Fig. 2A,B) (Cyr et al., 2002); and (2) amino acid sequences in the second Myo1c IQ domain (IQ2) are highly conserved across species, suggesting that this domain may be involved in receptor interactions (Cyr et al., 2002). We hypothesized that if IQ2 is the receptor-binding site of Myo1c, then mutations that prevent CaM binding to IQ2 would abolish the ability of CaM to block interactions between Myo1c and its receptors. Because CaM binding to IQ domains critically depends on the IQ consensus sequence IQX3RGX3R [single amino acid code where X is any amino acid (Rhoads and Friedberg, 1997)], we sought to mutate the Myo1c IQ2 consensus sequence to prevent CaM interaction but retain the ability of the domain to bind to stereociliary receptors. Accordingly, we changed the first two amino acids of the bullfrog Myo1c IQ2 consensus sequence, isoleucine 728 and glutamine 729, to alanine residues. These mutations, I728A and Q729A, were made in the context of Myo1c-T701, resulting in a recombinant Myo1c fragment referred to as Myo1c-T701IQ2Mut (Fig. 1A).
Figure 2.
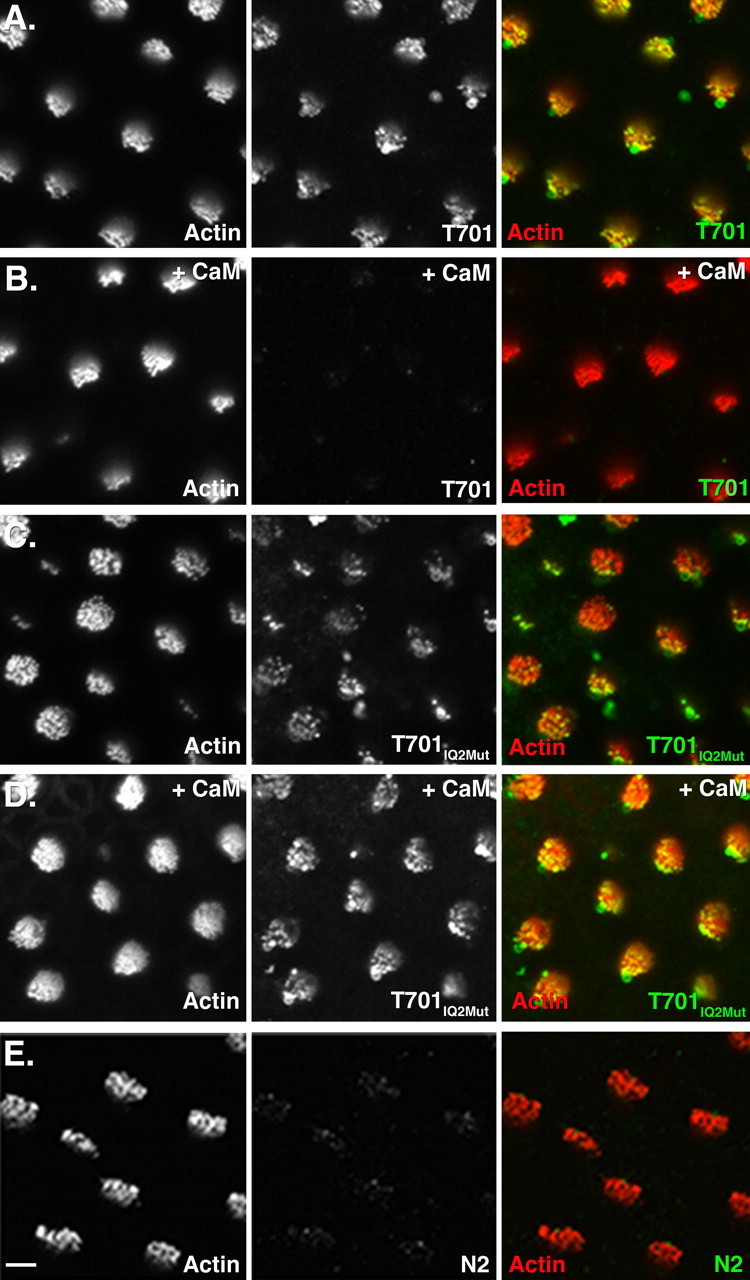
Binding of Myo1c fragments to stereociliary receptors. Each panel shows a single confocal section containing ∼10 hair bundles of a whole-mount bullfrog sacculus. Each group of three panels depicts the following: left, fluorescently conjugated phalloidin labeling of filamentous actin; middle, bound recombinant Myo1c fragments; right, overlay of actin (red) and Myo1c binding (green). A, Binding of Myo1c-T701 to receptors at the tips of stereocilia. B, Excess CaM abolishes Myo1c-T701 binding to stereociliary receptors. C, Myo1c-T701IQ2Mut binding to stereociliary receptors. D, Excess CaM does not block interactions of Myo1c-T701IQ2Mut with stereociliary receptors. E, Myo1c-N2 does not bind to stereociliary receptors. Scale bar: (in E) 5 μm.
Using baculovirus infection, Myo1c-T701IQ2Mut was coexpressed with CaM and purified by virtue of an N-terminal His6 tag (Fig. 1C). As with the purification of wild-type Myo1c-T701, CaM copurifies with the Myo1c-T701IQ2Mut protein indicating that CaM is bound to one or more of the Myo1c IQ domains (Fig. 1B,C). To verify that the mutations made in IQ2 disrupt CaM binding, the complex of Myo1c-T701IQ2Mut and CaM was further purified by gel-filtration chromatography, and the amount of CaM copurifying with the mutated Myo1c fragment was compared with that which copurifies with the wild-type Myo1c-T701 fragment using densitometry. Gel-filtration chromatography and sucrose-density centrifugation indicate that an average of 1.8 ± 0.2 CaM molecules are bound per wild-type Myo1c-T701 under identical purification conditions (Gillespie and Cyr, 2002). However, it is not known which IQ domains are bound by CaM in the wild-type protein. Analysis of Myo1c-T701IQ2Mut reveals that one CaM (1.0 ± 0.3; n = 3) is bound per Myo1c-T701IQ2Mut molecule, confirming that the mutations in IQ2 were successful in disrupting CaM interactions.
The ability of Myo1c-T701IQ2Mut to bind to Myo1c receptors in hair cells was confirmed using our in situ binding assay. Like the wild-type Myo1c-T701, Myo1c-T701IQ2Mut bound to receptors located at the tips of stereocilia (Fig. 2A,C), in the vesicle-rich pericuticular necklace and also in the hair-cell soma (data not shown). This interaction profile is indistinguishable from that of the wild-type protein (Fig. 2A), demonstrating that the IQ2 mutations did not adversely affect the ability of the molecule to bind to stereociliary receptors. To ascertain whether IQ2 is the site of CaM-sensitive interaction between Myo1c and its receptors, we tested the ability of excess CaM to block the observed interactions. As reported previously, wild-type Myo1c-T701 binding to stereociliary tips is blocked in the presence of excess CaM (Fig. 2B) (Cyr et al., 2002). Conversely, Myo1c-T701IQ2Mut binding is not altered despite the presence of excess CaM (Fig. 2D). These data confirm that the second IQ domain comprises a vital component of the interaction site between Myo1c and its receptors and that when this domain is occupied by CaM, binding is prevented.
To determine whether the Myo1c IQ2 domain is sufficient for receptor interaction, a protein fragment containing only the second IQ domain of Myo1c, termed Myo1c-N2, was generated (Fig. 1A). This Myo1c fragment was coexpressed with CaM, purified (Fig. 1D), and tested for its ability to bind stereociliary receptors. If the receptor-interaction domain of Myo1c is composed solely of IQ2, Myo1c-N2 would be expected to bind to stereociliary receptors in a manner similar to Myo1c-T701. However, purified Myo1c-N2 protein did not bind to stereocilia (Fig. 2E) or the pericuticular necklace region (data not shown), suggesting that IQ2 is insufficient for binding. These data support previous results in which the addition of excess IQ2 peptide did not block the interaction of Myo1c-T701 and Myo1c receptors (Cyr et al., 2002).
The presence of one CaM bound to Myo1c-T701IQ2Mut indicated that CaM is bound to one of the other three Myo1c IQ domains. The weak IQ consensus sequence and low apparent affinity of CaM for IQ4 (Gillespie and Cyr, 2002) suggested that the IQ domain occupied by CaM was either IQ1 or IQ3. To determine whether CaM is bound at IQ1 and if so, whether this CaM is important for receptor interactions, we mutated the IQ1 consensus sequence to prevent CaM binding. The Myo1c-T701IQ1RG1Mut protein was expressed, purified (Fig. 1E), and tested for receptor binding in our in situ binding assay (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Despite the simultaneous alteration of four of the five residues of the IQ1 consensus sequence (L706A, Q707A, R711A, and G712A), no loss of CaM was detected from the purified protein (1.5 ± 0.4 CaMs bound per T701IQ1RG1Mut; n = 3), and binding to stereociliary receptors was only slightly reduced compared with the control (supplemental Fig. 1A,B, available at www.jneurosci.org as supplemental material). In addition, as seen with the wild-type Myo1c-T701, incubation with excess CaM blocked the interaction of Myo1c-T701IQ1RG1Mut with stereociliary receptors (supplemental Fig. 1C, available at www.jneurosci.org as supplemental material). These data suggest that CaM is not bound to IQ1 in the Myo1c-T701 construct and therefore is likely bound at IQ3.
The second IQ domain of Myo1c mediates CaM-sensitive binding to PIP2 in vitro
Recent data demonstrate that PIP2, which is present in stereociliary membranes and plays a role in hair-cell transduction, binds to Myo1c in vitro (Hirono et al., 2004; Hokanson and Ostap, 2006). The region of Myo1c that binds to PIP2, however, is not clear; both the Myo1c neck and tail regions have been identified as sites of interaction (Hirono et al., 2004; Hokanson and Ostap, 2006). Using a nitrocellulose membrane-based assay and PIP2-containing vesicles, Hirono et al. (2004) determined that the neck region of Myo1c interacts with PIP2. In addition, Myo1c interactions with PIP2 vesicles were blocked by CaM in the presence of EGTA or Ca2+ (Hirono et al., 2004). We sought to further characterize Myo1c and PIP2 interactions and to explore the possibility that PIP2 may represent the Myo1c receptors observed in our in situ binding assay. To do so, we examined whether Myo1c-T701, Myo1c-T701IQ2Mut, and Myo1c-N2 bound to PIP2 in a membrane-based assay in a manner similar to their binding to stereociliary receptors. In 1 mm EGTA, Myo1c-T701 bound to PIP2, and this binding was blocked by the presence of excess CaM (Fig. 3A,B). Similar to the results seen in our in situ binding assay, Myo1c-T701IQ2Mut bound to PIP2 and this interaction was not blocked by the addition of excess CaM (Fig. 3C,D). In addition, Myo1c-N2, which did not bind to stereociliary receptors in the in situ binding assay (Fig. 2E), did not bind to PIP2 in the membrane-based assay (Fig. 3E). Finally, coincubation of Myo1c-T701 with an excess amount of a synthetic peptide encoding IQ2 did not block binding to PIP2 (Fig. 3F). These data demonstrate that, in an in vitro assay, Myo1c interacts with PIP2 and the stereociliary receptors in a similar manner. Although PIP2 binds to Myo1c in a membrane-based assay, additional evidence (detailed below) suggests that PIP2 is not the receptor detected in our in situ binding assay.
Figure 3.

Binding of Myo1c fragments to anionic phospholipids. A–F, Each panel shows one PIP-strip, a nitrocellulose membrane, which has been spotted with anionic lipids, that has been incubated with the indicated Myo1c fragment. Bound Myo1c fragments were detected by virtue of the N-terminal Xpress epitope tag. The diagram to the right indicates the location and identity of the anionic lipids. A, T701-Myo1c binds weakly to several of the anionic lipids and strongly to PI(4,5)P2 (PIP2). B, The addition of excess CaM abolishes Myo1c-T701 interactions with anionic lipids. C, Myo1c-T701IQ2Mut binds to the same subset of anionic lipids as Myo1c-T701. D, Excess CaM does not block the interactions of Myo1c-T701IQ2Mut with anionic lipids. E, Myo1c-N2 does not bind anionic lipids. F, Excess IQ2 peptide, when incubated with Myo1c-T701, does not block the binding of Myo1c-T701. G, Antibody only control.
Tip-link loss abolishes Myo1c binding to stereociliary receptors
Myo1c has been shown to interact with Cdh23, a proposed component of the tip link, when the proteins are coexpressed in tissue culture cells (Siemens et al., 2004). To examine the requirement for intact tip links on Myo1c interactions with its receptors, we exposed bullfrog saccular hair cells to either lanthanide ions or calcium chelators, agents which break tip links and eradicate mechanotransduction (Baumann and Roth, 1986; Assad et al., 1991; Michel et al., 2005), before fixation and Myo1c-T701 binding. Treatment with 5 mm La3+ or 5 mm EGTA successfully broke tip links as assayed by either a complete, or near-complete, loss of AM1–43 loading into hair cells through transduction channels (Fig. 4A,B,E,F). Moreover, pretreatment with either La3+ or EGTA eliminated virtually all Myo1c-T701 binding at both the hair-cell stereociliary tips and pericuticular necklace (Fig. 4D,H) (data not shown) in the bullfrog sacculus.
Figure 4.
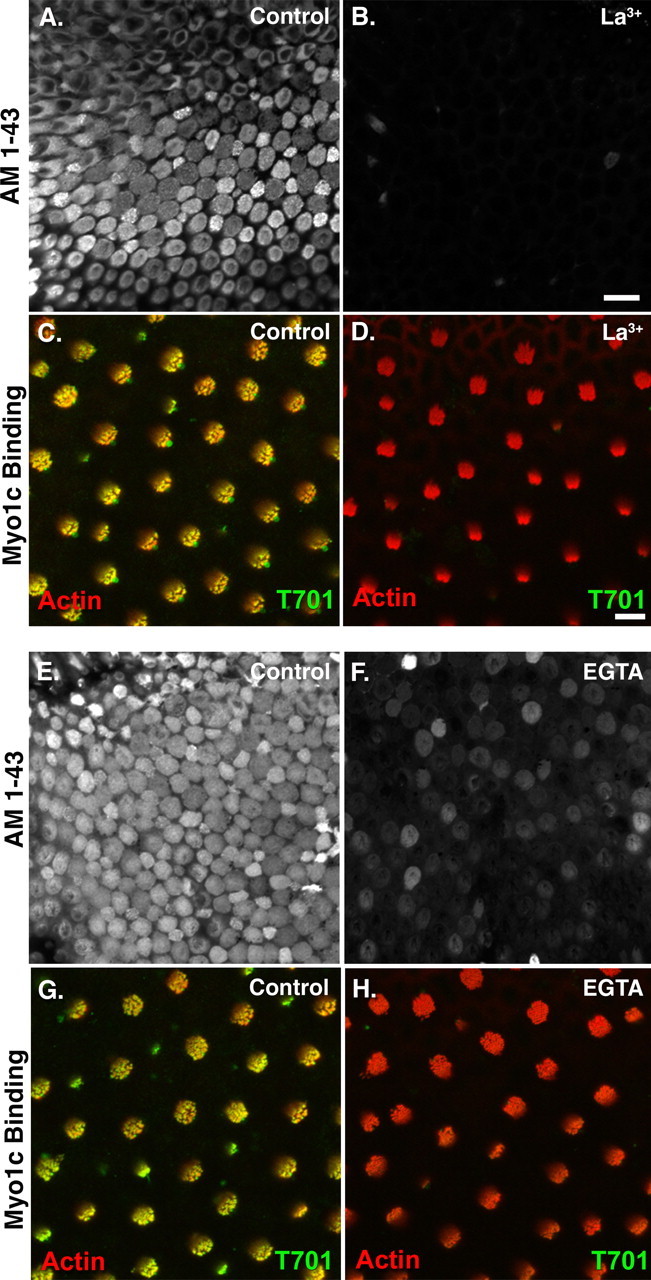
Myo1c binding to stereociliary receptors in the bullfrog sacculus requires intact tip links. A, Sensory epithelium of a bullfrog sacculus showing loading of hair cells with AM1–43, which enters through open transduction channels. B, Treatment of a sacculus with 5 mm La3+ breaks tip links as assayed by the loss of AM1–43 entry into hair cells. C, D, Treatment with 5 mm La3+ results in the loss of Myo1c-T701 binding to stereociliary tips. E, Bullfrog sacculus showing AM1–43 loading of hair cells. F, Treatment with 5 mm EGTA breaks tip links as assayed by the marked reduction of AM1–43 entry into hair cells. G, H, Treatment with 5 mm EGTA results in the loss of Myo1c-T701 binding to stereociliary tips. All images are single confocal sections. Scale bars: (in B) A, B, E, F, 20 μm; (in D) C, D, G, H, 5 μm. AM-143 uptake is white, phalloidin labeling of filamentous actin is red, and Myo1c-T701 binding is green.
To assess whether intact tip links are also needed for Myo1c binding in the mouse cochlea, we broke tip links with either 5 mm EGTA, BAPTA or La3+ and determined whether Myo1c bound to stereociliary tips after these treatments. In untreated cochlear hair cells, Myo1c-T701 bound at the tips of stereocilia as seen in the bullfrog sacculus (Fig. 5A). Pretreatment of cochleas with BAPTA eliminated all binding of Myo1c to stereocilia (Fig. 5B), whereas La3+ pretreatment reduced, but did not eliminate, Myo1c binding (Fig. 5D). EGTA pretreatment gave variable results ranging from a slight reduction in binding (data not shown) to a loss of binding (Fig. 5C); this variability could be found within a single cochlea and did not correspond to location of the hair cells along the length of the organ.
Figure 5.
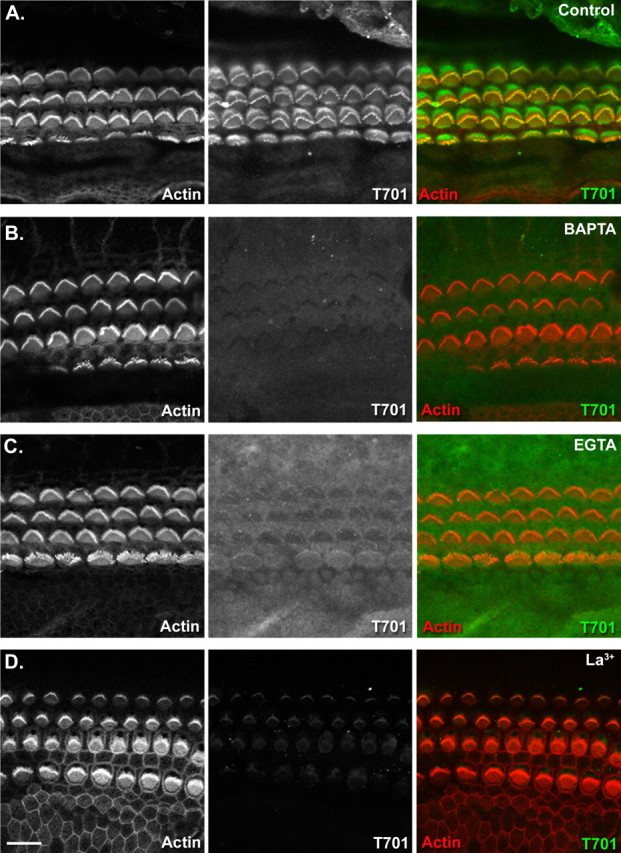
Myo1c binding to stereociliary receptors in the mouse organ of Corti requires intact tip links. Each panel shows a single confocal section containing inner and outer hair cells from a whole-mount mouse organ of Corti. Left column, Fluorescently conjugated phalloidin labeling of filamentous actin; middle column, bound recombinant Myo1c fragments; right column, overlay of actin (red) and Myo1c binding (green). A, Binding of Myo1c-T701 to mouse cochlear hair cells. B–D, Treatment of the organ of Corti with 5 mm BAPTA, EGTA, or La3+ results in the loss or marked reduction of Myo1c-T701 binding to stereociliary tips. Scale bar: (in D) 10 μm.
Effects of tip-link loss on Cdh23 immunolocalization in bullfrog sacculi and mouse cochlea
The elimination of Myo1c binding to stereociliary tips after tip-link loss led us to revisit the question of Cdh23 localization in stereocilia after such treatments in both bullfrogs and mice. To date, seemingly contradictory results have been reported regarding the effects of tip-link loss on Cdh23 localization in hair cells (Seimens et al., 2004; Michel et al., 2005). Siemens et al. (2004) demonstrate that EGTA treatment results in a loss of Cdh23 immunolocalization in bullfrog saccular hair bundles and an appearance of immunoreactivity observed at the pericuticular necklace, a vesicle-rich region surrounding the cuticular plate. However, in the mouse cochlea, similar treatments result neither in a loss of Cdh23 immunoreactivity in stereocilia nor the appearance of immunoreactivity in the pericuticular necklace (Michel et al., 2005). These ostensibly disparate results were performed using different antibodies against Cdh23 and may reflect differences in the antibodies used, species, or type of hair cell. To distinguish between these possibilities, we examined the immunolocalization of Cdh23 in the hair cells of the bullfrog sacculus, mouse utricle, and mouse cochlea after tip-link loss using an antiserum raised against the cytoplasmic domain of Cdh23 [T8 antiserum (Lagziel et al., 2005)]. In the bullfrog sacculus, this antibody detects Cdh23 at the tall edge of the hair bundle, presumably between the tallest stereocilia and the kinocilium as reported previously (Fig. 6A) (Lagziel et al., 2005). Similar to the results of Siemens et al. (2004), treatment of bullfrog saccular hair cells with La3+ did not abolish immunolabeling (Fig. 6B); however, treatment with BAPTA (Fig. 6C) or EGTA (Fig. 6D) eliminated Cdh23 immunolabeling. The loss of labeling was observed throughout the hair cells of the sacculus, including the immature cells located at the periphery of the sensory epithelium (data not shown). Unlike the results of Siemens et al. (2004), we did not detect Cdh23 immunoreactivity in the pericuticular necklace after EGTA treatment (data not shown). Moreover, as reported by Michel et al. (2005) using two different antibodies to the cytoplasmic domain of cadherin 23, we observed that treatment of mouse cochlear hair cells with La3+, BAPTA, or EGTA or had no detectable effect on Cdh23 immunolocalization (Fig. 6E–H). These data suggest that reported differences in Cdh23 immunolocalization after tip-link loss in bullfrog sacculus (Siemens et al., 2004) and mouse cochlea (Michel et al., 2005) are not attributable to the use of different antibodies.
Figure 6.
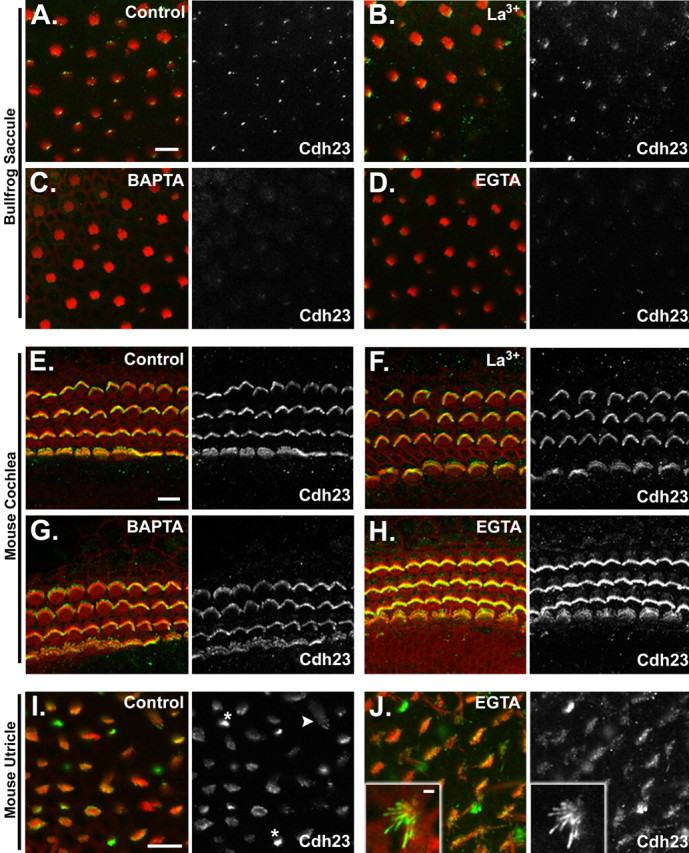
Cdh23 immunolocalization in hair cells after loss of tip links. Each panel depicts a single confocal section of whole mounted bullfrog saccule, mouse cochlea, or mouse utricle. The left panel for each group shows the merged image of actin (red) and Cdh23 immunolocalization (green). The right panel depicts the Cdh23 immunolabeling channel alone. A, Cdh23 immunolocalization in the bundles of bullfrog saccular hair cells. B–D, Cdh23 immunolocalization in the bundles of bullfrog saccular hair cells after treatment with 5 mm La3+, BAPTA, or EGTA, respectively. E, Cdh23 immunolocalization in the bundles of mouse cochlear hair cells (P1 plus 1 d in vitro). F–H, Cdh23 immunolocalization in the bundles of mouse cochlear hair cells after treatment with 5 mm La3+, BAPTA, or EGTA, respectively. I, Cdh23 immunolocalization in the bundles of mouse (P6) utricular hair cells. Asterisks demark representative small hair bundles, which are intensely labeled. An arrowhead denotes an example of stereociliary tip labeling. J, Cdh23 immunolocalization in the bundles of mouse utricular hair cells (P6) after treatment with 5 mm EGTA. Inset, A small hair bundle with labeling along the length of the stereocilium. This example is from a P3 mouse utricule treated with EGTA at 4°C. Similar results were seen with P6 utricles treated at room temperature. Scale bars: A, E, I, (for A–D, E–H, I–J), 10μm; J, inset, 2 μm.
To determine whether the effects of tip-link loss on Cdh23 immunolocalization differs in auditory and vestibular hair cells within a single species, we examined Cdh23 immunolabeling in mouse vestibular hair cells after treatment with EGTA (Fig. 6I,J). In untreated samples, Cdh23 was present in hair bundles (Fig. 6I) and occasionally could be seen at stereociliary tips (Fig. 6I, arrowhead). Smaller utricular hair bundles were much more intensely labeled than larger hair bundles (Fig. 6I, asterisk). In contrast to that observed in bullfrog saccular hair cells, tip-link loss in mouse utricular hair cells did not coincide with a loss of Cdh23 immunoreactivity (Fig. 6J). Hair bundles remained immunolabeled for Cdh23 to varying degrees. Noteably, smaller hair bundles remained intensely labeled after EGTA treatment, and often the immunolabeling was observed along the length of stereocilia rather than concentrated at stereociliary tips (Fig. 6J, inset). Such a Cdh23 distribution after Ca2+ chelation was observed when the EGTA incubation was done at both room temperature and 4°C (Fig. 6J, inset) (data not shown). These data suggest that the differences seen in Cdh23 immunolocalization after tip-link loss are species dependent and do not reflect a distinction between auditory and vestibular hair cells. We did, however, observe a slight difference in immunolabeling in the mouse utricle compared with the sacculus after EGTA treatment. Saccular hair cells exhibited reduced labeling after treatment, although labeling was still apparent (data not shown).
The molecular basis for the observed differences in Cdh23 immunoreactivity after Ca2+ chelation in the mouse and frog inner ear is unknown but suggest that the properties of Cdh23 differ between species or within a given species. It is possible that within a given species, there exist subpopulations of Cdh23 that differ in their clearance from the stereociliary membrane, their conformational state, or their interaction with other molecules after Ca2+ chelation. Indeed, multiple isoforms of Cdh23 have been reported in the mouse inner ear, which may reflect such Cdh23 subpopulations (Lagziel et al., 2005).
Interaction of Myo1c-T701 and stereociliary receptors does not occur in Cdh23v2J mice
The loss of Myo1c binding to the tips stereocilia after tip-link loss implicates Cdh23 as a stereociliary interaction partner for Myo1c. Although mouse cochlear hair cells retain Cdh23 immunoreactivity after tip-link loss, we are unable to determine whether a small fraction of the Cdh23 immunoreactivity, perhaps that located at the very tips of stereocilia, which binds to Myo1c, is eliminated by treatment with EGTA, BAPTA, or La3+ (Fig. 6E–H). We, therefore further explored whether the Myo1c receptors detected in our in situ binding assay are Cdh23. To do so, we examined the binding of Myo1c-T701 to stereocilia of mice lacking the Cdh23 protein. Immunolocalization studies using antibodies directed against the N-terminal and C-terminal domains of Cdh23 show that Cdh23v2J homozygous mice have no Cdh23 protein present in stereocilia (Michel et al., 2005) (A. Lagziel and T. Friedman, personal communication). Moreover, other antibodies against the C terminus do not detect Cdh23 protein in cochleas of Cdh23v2J homozygous mice by immunoblotting (Rzadzinska et al., 2005). To further characterize the hair bundles of the Cdh23v2J, we first examined the localization of endogenous Myo1c in cochleas of both Cdh23v2J heterozygous and homozygous mice. In heterozygous mice, which have normal hair-bundle morphology, Myo1c was located along the length of stereocilia in both inner and outer hair cells; however, immunolabeling was more prevalent in the outer hair cells (Fig. 7A, inset). Although the hair bundles of Cdh23v2J homozygous mice are drastically malformed, Myo1c is also present in the mutant hair bundles of both inner and outer hair cells (Fig. 7B, inset). As seen with bullfrog saccular hair cells (Cyr et al., 2002), Myo1c-T701 binds to the tips of stereocilia and to the kinocilium in cochlear hair cells from heterozygous Cdh23v2J mice (Fig. 7C, inset). In contrast, no binding was detected in hair cells of homozygous Cdh23v2J animals (Fig. 7D, inset).
Figure 7.
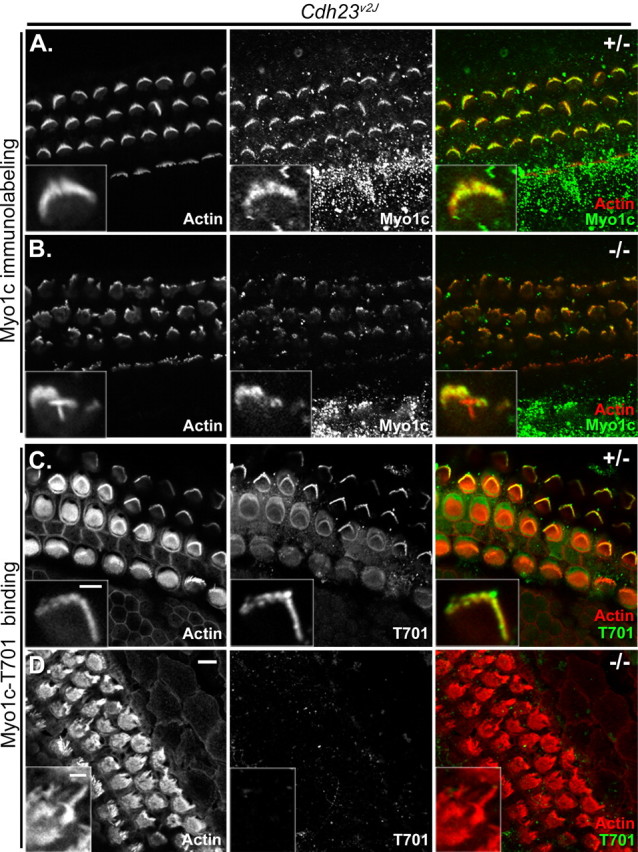
Myo1c and Myo1c receptor localization in cochlear hair cells of Cdh23v2J mice. Each panel shows three rows of outer and one row of inner hair cells of heterozygous (+/−) or homozygous (−/−) Cdh23v2J mutant mice (P2–P4). The left column of each panel shows phalloidin labeling alone, and the middle column of each panel shows the bound Myo1c antibody or Myo1c-T701 binding alone. The right column of each panel shows the overlay of phalloidin-labeled filamentous actin (red) and the binding of either a Myo1c antibody (green; A, B) or the Myo1c-T701 fragment (green; C, D). A–D, Cochlear cultures from Cdh23v2J mice. A, B, Endogenous Myo1c is present in stereocilia of both the Cdh23v2J heterozygous and homozygous mice. C, The Myo1c receptor is detected in the stereocilia of the Cdh23v2J heterozygous mice. D, Myo1c receptor is undetectable in the stereocilia of Cdh23v2J homozygous mice. Insets depict the hair bundles of outer hair cells. Scale bar: (in D) 5 μm; (in C, inset), A, B, C, insets, 2 μm; D, inset, 2 μm.
Interaction of Myo1c-T701 and stereociliary receptors occurs in Myo7a6J mice
The loss of tip links resulting from exogenous treatments is expected to result in transduction-channel closure, which could alter the available binding sites for our recombinant Myo1c probes. To examine whether transduction-channel closure prevents Myo1c binding in our in situ binding assay, we examined the ability of Myo1c-T701 to bind to the stereocilia in cochleas from Myo7a6J mice, which harbor a mutation in myosin VIIa resulting in bundle abnormalities and transduction channels that are closed at rest (Richardson et al., 1997; Self et al., 1998; Gale et al., 2001; Kros et al., 2002). Cochlear hair cells from heterozygous Myo7a6J animals exhibit normal bundle morphology; however, those from homozygous Myo7a6J mice have severely disorganized hair bundles that are fragmented into several smaller units each comprised of variable numbers of individual stereocilia (Fig. 8B,D). As demonstrated by immunolabeling, endogenous Myo1c is present in the stereocilia of both heterozygous and homozygous animals (Fig. 8A,B, insets). Unlike the Cdh23v2J homozygotes (Fig. 7D), however, Myo1c-T701 binds to receptors in Myo7a6J homozygotes as well as the heterozygotes (Fig. 8C,D). Binding to homozygous hair bundles was often less obvious than that seen in the heterozygous samples but was above background levels. Collectively, these data suggest that neither aberrant bundle morphology nor channel closure affect the availability of Myo1c binding sites in our assay.
PIP2 is present in Cdh23v2J hair bundles
In bullfrog saccular hair bundles, PIP2 is present in stereociliary membranes and is important for transduction (Hirono et al., 2004). The binding of Myo1c fragments to PIP2 in vitro (Fig. 3) (Hirono et al., 2004) and the loss of Myo1c-T701 binding to hair cells in Cdh23v2J homozygous animals (Fig. 7) led us to examine whether Cdh23v2J homozygous animals have PIP2 within their stereociliary membranes. To examine PIP2 localization, an expression construct comprised of the PLCδ1 pleckstrin homology domain coupled with EGFP was transiently transfected into cochlear hair cells of homozygous and heterozygous Cdh23v2J animals. PLCδ1PH-EGFP binds to PIP2 providing a fluorescent reporter of PIP2 localization in the membranes of living cells (Downes et al., 2005). Between 20 and 24 h after transfection, inner and outer hair cells of both Cdh23v2J heterozygous and homozygous mice show robust fluorescence of their stereociliary membranes verifying the presence of PIP2 in the hair bundles of living cells (Fig. 9A,B). We then examined whether PIP2 remains in the stereociliary membrane after fixation and extraction with Sarkosyl, the detergent used to permeabilize the tissue in our in situ binding assay. Using a monoclonal antibody against PIP2, we found PIP2 in the stereociliary membrane and concentrated at stereociliary tips in both Cdh23v2J heterozygous and homozygous cochlear hair cells even after extraction of the tissue with 1% Sarkosyl for 1 h (Fig. 9C,D). Similar results were obtained in the wild-type bullfrog sacculus (data not shown). In addition, PIP2 immunolabeling was detected in hair cell microvilli, which are very abundant at the apical surface of the cell (Fig. 9D, arrow). Thus, the lack of Myo1c-T701 binding to stereociliary receptors in homozygous Cdh23v2J hair cells is not attributable to the absence of stereociliary PIP2.
Discussion
The second IQ domain of Myo1c mediates binding to intracellular receptors
We established previously the importance of the Myo1c neck domain in interactions of the adaptation motor with its receptors at stereociliary tips (Cyr et al., 2002). This interaction is blocked by excess CaM, indicating that binding is modulated by the CaM occupancy of the Myo1c IQ domains, which in turn is modulated by Ca2+. In the present study, we demonstrate that mutations in IQ2 that are predicted to prevent CaM binding to this IQ domain also prevent the CaM modulation of Myo1c binding to its receptors. The second IQ domain of Myo1c, which is highly conserved across species (Cyr et al., 2002), therefore, mediates CaM-sensitive interactions of the adaptation motor at the site of transduction.
Although a CaM-free IQ2 domain is necessary for receptor interactions, it may not be sufficient for binding. Myo1c-N2, which contains only the Myo1c IQ2 domain and detection tags, does not bind to stereociliary components. These data are consistent with previous results showing an excess of the IQ2 peptide does not block Myo1c-T701 binding at stereociliary tips (Cyr et al., 2002) and support our previous observations that micromolar Ca2+ concentrations, which are expected to remove CaM from neighboring IQ domains, can prevent Myo1c-T701 binding to stereociliary receptors (Cyr et al., 2002). Collectively, these results suggest that additional portions of the Myo1c protein, perhaps CaM molecules bound at neighboring IQ domains, are also required for binding.
Sucrose-density centrifugation and gel-filtration chromatography demonstrates that ∼2 CaMs are bound to Myo1c-T701 after purification (Gillespie and Cyr, 2002). The weak affinity of IQ4 for CaM (Gillespie and Cyr, 2002) suggests that these CaMs are bound at two of the first three IQ domains. Moreover, the loss of a CaM from Myo1c-T701 after mutations in IQ2 suggests that one of the two CaMs that copurify with Myo1c-T701 is bound at IQ2. In order for Myo1c-T701 to bind to receptors, therefore, CaM must be displaced from IQ2 during our assay, perhaps because of competition, thereby unmasking IQ2 and allowing receptor interactions. Simultaneous mutation of four of the five residues comprising the CaM-binding consensus sequence of IQ1, however, did not result in a loss of a CaM molecule from Myo1c-T701. These results suggest that the second CaM that copurifies with Myo1c-T701 is not bound at IQ1 and therefore is likely bound at IQ3.
Myo1c binding to PIP2
PIP2, which is abundant in stereociliary membranes, is important for hair-cell transduction and binds to the Myo1c neck in vitro (Hirono et al., 2004). Using a nitrocellulose-based assay, we determined that CaM occupancy of the second IQ domain of Myo1c also modulates the interaction of the motor protein with PIP2 in vitro. However, as evidenced in cochleas from homozygous Cdh23v2J mice, which have stereociliary PIP2 but no Myo1c binding, our in situ binding assay does not detect Myo1c interactions with PIP2, suggesting that the conditions of the assay are not favorable for detecting this interaction. Nevertheless, the in situ binding assay enables the examination and characterization of Myo1c interactions with other stereociliary components.
Which molecules are the observed Myo1c receptors?
Our results allow us to eliminate several candidate molecules as the receptors visualized in our in situ binding assay. As mentioned above, whereas Myo1c may interact with immobilized PIP2 in vitro, we do not detect Myo1c interactions with PIP2 in our in situ binding assay. Similarly, because stereocilia of homozygous Cdh23v2J mice contain endogenous Myo1c but do not bind recombinant Myo1c probes, the observed Myo1c receptors are not endogenous Myo1c molecules. Another candidate Myo1c receptor is pleckstrin homology domain retinal protein 1 (PHR1), which binds to a Myo1c fragment containing IQ4 and the tail (Etournay et al., 2005). Because a Myo1c fragment containing only IQs 1–3 is sufficient for binding to stereociliary tips (Cyr et al., 2002), it is unlikely that the PHR1 molecules are the receptors observed in our assay, unless Myo1c has two independent PHR1 interaction domains. Our data do not preclude the possibility that PHR1 and Myo1c interact in hair bundles. Indeed, molecular motors often interact with more than one molecule.
Treatment of bullfrog vestibular or mouse cochlear hair cells with agents that break tip links including EGTA, BAPTA, and La3+ eliminates or greatly reduces Myo1c binding. The effects of tip-link loss on Myo1c binding implicate a component of the tip-link complex as the binding partner for Myo1c in our assay. As a candidate tip-link molecule, Cdh23, therefore, became an obvious contender for the Myo1c receptor. To examine the role of Cdh23 in the interactions of Myo1c with its receptors in hair cells, we made use of the homozygous Cdh23v2J mouse, which does not express Cdh23 protein in hair cells, has gross hair-bundle abnormalities, and exhibits both auditory and vestibular defects (Di Palma et al., 2001; Lagziel et al., 2005; Michel et al., 2005; Rzadzinska et al., 2005). Although Myo1c binds to the hair bundles from heterozygous Cdh23v2J cochleas, no binding is detected in the cochleas from homozygous animals. This lack of binding is unlikely to be simply caused by hair-bundle disarray or transduction-channel closure, because Myo1c binds to stereociliary tips of homozygous Myo7a6J cochlear hair cells.
Our data suggest that the lack of Myo1c binding to hair bundles with broken tip links and in homozygous Cdh23v2J cochleas reflects a perturbation of molecular interactions that involve both Cdh23 and Myo1c. It is formally possible that these interactions are not direct and that the loss of tip links and Cdh23 from hair bundles results in the loss of another molecule, which directly interacts with Myo1c. However, the interaction of Cdh23 and Myo1c in tissue culture cells (Siemens et al., 2004) lends support to the conclusion that these molecules also interact in hair cells.
The immunolocalization of Cdh23 in stereocilia of mouse cochlear hair cells after treatment with agents that break tip links is not incongruous with our hypothesis. We propose that the structural alterations of the Cdh23 ectodomain attributable to Ca2+ chelation result in a conformational change in the intracellular domain of the protein, thereby altering its interaction with other molecules. In support of this hypothesis, in bullfrog saccular hair cells, distinct immunolabeling patterns for Cdh23 after EGTA treatment have been obtained using different antisera raised against the Cdh23 cytoplasmic domain (Siemens et al., 2004; our study). Specifically, Seimens et al. (2004) report an appearance of Cdh23 immunoreactivity in the pericuticular necklace after EGTA treatment. However, using a different antibody against the Cdh23 cytoplasmic domain, we do not detect Cdh23 in this subcellular domain. These differences in labeling patterns suggest that the cytoplasmic domain of the protein undergoes conformational changes, perhaps by binding to or dissociating from other molecules, which render the epitopes recognized by certain antibodies inaccessible. Similar types of intramolecular or intermolecular changes in Cdh23 might also alter the ability of Myo1c probes to interact with this domain in our in situ binding assay. Thus, after Ca2+ chelation, Cdh23 may be present, but unable to bind to exogenously applied Myo1c fragments.
Myo1c interactions with transduction components
Our data demonstrate that the second IQ domain of Myo1c is important for Myo1c interactions with PIP2 in vitro and at stereociliary tips in situ. In hair cells, the later interaction requires intact tip links and is absent in hair cells lacking Cdh23, a proposed component of the tip link (Siemens et al., 2004; Söllner et al., 2004). The ability of IQ2 to bind CaM, which blocks the interaction, suggests that the local Ca2+ concentration plays a critical role in determining which molecule is bound to the Myo1c neck at a given point in time (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Because of the high degree of identity of IQ2 across species, the ability of a Myo1c fragment containing only IQs 1–3 to bind to stereociliary tips (Cyr et al., 2002), and the effects of mutations in IQ2 on the ability of CaM to block stereociliary interactions, we propose that the domain of Myo1c that modulates interactions with stereociliary receptors, which are likely to be Cdh23, is IQ2. We predict, therefore, that interactions of Myo1c and Cdh23 will depend both on the CaM occupancy of the Myo1c IQ domains and the conformation of the Cdh23 cytoplasmic domain (supplemental Fig. 2, available at www.jneurosci.org as supplemental material).
Accordingly, large increases in the Ca2+ concentration, perhaps immediately after channel opening and before fast adaptation has occurred to reduce channel open probability, should favor the loss of CaM from the Myo1c IQ domains and reduce the interaction of Myo1c with Cdh23. As the stereociliary plasma membrane Ca2+-ATPase begins to reduce the Ca2+ concentration, CaM would reassociate with the high-affinity IQ domains (IQ3, and perhaps IQ1), and Cdh23 binding would be favored. It would be predicted that interactions with Cdh23 would be maintained unless both the local Ca2+ concentration and the relative affinity for CaM favors CaM reassociation at IQ2 in vivo or hair cells are exposed to Ca2+ chelators in vitro (supplemental Fig. 2, available at www.jneurosci.org as supplemental material).
Footnotes
This work was supported by National Institutes of Health (NIH)–National Institute on Deafness and Other Communication Disorders Grants R01DC006402 (J.L.C.) and F31DC007558 (K.R.P.), a grant from American Hearing Research Foundation (J.L.C.), NIH–National Center for Research Resources Centers of Biomedical Research Excellence Grant P20RR015574-06 (to the Sensory Neuroscience Research Center), and Wellcome Trust Grant 071394/Z/03/Z (G.P.R.). We thank Gwenaelle Geleoc, Jeffrey Holt, and Eric Stauffer for tutorials on cochlear dissections, Peter Gillespie for the anti-Myo1c antibody, Thomas Freidman for the anti-Cdh23 antibody, Peter Downes for the PLCδ1PH-GFP construct, Corné Kros for mutant mice, Bill Wonderlin and Paul Fuchs for experimental suggestions, Matt Roberts for technical help, and George Spirou and Peter Mathers for critical reading of this manuscript.
References
- Assad JA, Shepherd GMG, Corey DP. Tip-link integrity and mechanical transduction in vertebrate hair cells. Neuron. 1991;7:985–994. doi: 10.1016/0896-6273(91)90343-x. [DOI] [PubMed] [Google Scholar]
- Baumann A, Roth A. The Ca++ permeability of the apical membrane in neuromast hair cells. J Comp Physiol. 1986;158:681–688. doi: 10.1007/BF00603825. [DOI] [PubMed] [Google Scholar]
- Boeda B, El-Amraoui A, Bahloul A, Goodyear R, Daviet L, Blanchard S, Perfettini I, Fath KR, Shorte S, Reiners J, Houdusse A, Legrain P, Wolfrum U, Richardson G, Petit C. Myosin VIIa, harmonin and cadherin 23, three Usher I gene products that cooperate to shape the sensory hair cell bundle. EMBO J. 2002;21:6689–6699. doi: 10.1093/emboj/cdf689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Cyr JL, Dumont RA, Gillespie PG. Myosin-1c interacts with hair-cell receptors through its calmodulin-binding IQ domains. J Neurosci. 2002;22:2487–2495. doi: 10.1523/JNEUROSCI.22-07-02487.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Palma F, Holme RH, Bryda EC, Belyantseva IA, Pellegrino R, Kachar B, Steel KP, Noben-Trauth K. Mutations in Cdh23, encoding a new type of cadherin, cause stereocilia disorganization in waltzer, the mouse model for Usher syndrome type 1D. Nat Genet. 2001;27:103–107. doi: 10.1038/83660. [DOI] [PubMed] [Google Scholar]
- Downes CP, Gray A, Lucocq JM. Probing phosphoinositide functions in signaling and membrane trafficking. Trends Cell Biol. 2005;15:259–268. doi: 10.1016/j.tcb.2005.03.008. [DOI] [PubMed] [Google Scholar]
- Dumont RA, Zhao YD, Holt JR, Bahler M, Gillespie PG. Myosin-I isozymes in neonatal rodent auditory and vestibular epithelia. J Assoc Res Otolaryngol. 2002;3:375–389. doi: 10.1007/s101620020049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etournay R, El-Amraoui A, Bahloul A, Blanchard S, Roux I, Pezeron G, Michalski N, Daviet L, Hardelin JP, Legrain P, Petit C. PHR1, an integral membrane protein of the inner ear sensory cells, directly interacts with myosin 1c and myosin VIIa. J Cell Sci. 2005;118:2891–2899. doi: 10.1242/jcs.02424. [DOI] [PubMed] [Google Scholar]
- Gale JE, Marcotti W, Kennedy HJ, Kros CJ, Richardson GP. FM1–43 dye behaves as a permeant blocker of the hair-cell mechanotransducer channel. J Neurosci. 2001;21:7013–7025. doi: 10.1523/JNEUROSCI.21-18-07013.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie PG, Cyr JL. Calmodulin binding to recombinant myosin-1c and myosin-1c IQ peptides. BMC Biochem. 2002;3:31. doi: 10.1186/1471-2091-3-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie PG, Cyr JL. Myosin-1c, the hair cell's adaptation motor. Annu Rev Physiol. 2004;66:521–545. doi: 10.1146/annurev.physiol.66.032102.112842. [DOI] [PubMed] [Google Scholar]
- Gillespie PG, Gillespie SK, Mercer JA, Shah K, Shokat KM. Engineering of the myosin-Ib nucleotide-binding pocket to create selective sensitivity to N6-modified ADP analogs. J Biol Chem. 1999;274:31373–31381. doi: 10.1074/jbc.274.44.31373. [DOI] [PubMed] [Google Scholar]
- Goodyear RJ, Marcotti W, Kros CJ, Richardson GP. Development and properties of stereociliary link types in hair cells of mouse cochlea. J Comp Neurol. 2005;485:75–85. doi: 10.1002/cne.20513. [DOI] [PubMed] [Google Scholar]
- Gopalakrishna R, Anderson WB. Ca2+-induced hydrophobic site on calmodulin: application for purification of calmodulin by phenyl-Sepharose affinity chromatography. Biochem Biophys Res Commun. 1982;104:830–836. doi: 10.1016/0006-291x(82)90712-4. [DOI] [PubMed] [Google Scholar]
- Hirono M, Denis CS, Richardson GP, Gillespie PG. Hair cells require phosphatidylinositol 4,5-bisphosphate for mechanical transduction and adaptation. Neuron. 2004;44:309–320. doi: 10.1016/j.neuron.2004.09.020. [DOI] [PubMed] [Google Scholar]
- Hokanson DE, Ostap EM. Myo1c binds tightly and specifically to PIP2 and InsP3. Proc Natl Acad Sci USA. 2006;103:3118–3123. doi: 10.1073/pnas.0505685103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt JR, Gillespie SK, Provance DW, Shah K, Shokat KM, Corey DP, Mercer JA, Gillespie PG. A chemical-genetic strategy implicates myosin-1c in adaptation by hair cells. Cell. 2002;108:371–381. doi: 10.1016/s0092-8674(02)00629-3. [DOI] [PubMed] [Google Scholar]
- Howard J, Hudspeth AJ. Mechanical relaxation of the hair bundle mediates adaptation in mechanoelectrical transduction by the bullfrog's saccular hair cell. Proc Natl Acad Sci USA. 1987;84:3064–3068. doi: 10.1073/pnas.84.9.3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudspeth AJ. How the ear's works work. Nature. 1989;341:397–404. doi: 10.1038/341397a0. [DOI] [PubMed] [Google Scholar]
- Kros CJ, Marcotti W, van Netten SM, Self TJ, Libby RT, Brown SD, Richardson GP, Steel KP. Reduced climbing and increased slipping adaptation in cochlear hair cells of mice with Myo7a mutations. Nat Neurosci. 2002;5:41–47. doi: 10.1038/nn784. [DOI] [PubMed] [Google Scholar]
- Lagziel A, Ahmed ZM, Schultz JM, Morell RJ, Belyantseva IA, Friedman TB. Spatiotemporal pattern and isoforms of cadherin 23 in wild type and waltzer mice during inner ear hair cell development. Dev Biol. 2005;280:295–306. doi: 10.1016/j.ydbio.2005.01.015. [DOI] [PubMed] [Google Scholar]
- Michel V, Goodyear RJ, Weil D, Marcotti W, Perfettini I, Wolfrum U, Kros CJ, Richardson GP, Petit C. Cadherin 23 is a component of the transient lateral links in the developing hair bundles of cochlear sensory cells. Dev Biol. 2005;280:281–294. doi: 10.1016/j.ydbio.2005.01.014. [DOI] [PubMed] [Google Scholar]
- Pickles JO, Comis SD, Osborne MP. Cross-links between stereocilia in the guinea pig organ of Corti and their possible relation to sensory transduction. Hearing Res. 1984;15:103–112. doi: 10.1016/0378-5955(84)90041-8. [DOI] [PubMed] [Google Scholar]
- Rhoads AR, Friedberg F. Sequence motifs for calmodulin recognition. FASEB J. 1997;11:331–340. doi: 10.1096/fasebj.11.5.9141499. [DOI] [PubMed] [Google Scholar]
- Richardson GP, Forge A, Kros CJ, Fleming J, Brown SD, Steel KP. Myosin VIIA is required for aminoglycoside accumulation in cochlear hair cells. J Neurosci. 1997;17:9506–9519. doi: 10.1523/JNEUROSCI.17-24-09506.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell IJ, Richardson GP. The morphology and physiology of hair cells in organotypic cultures of the mouse cochlea. Hearing Res. 1987;31:9–24. doi: 10.1016/0378-5955(87)90210-3. [DOI] [PubMed] [Google Scholar]
- Rzadzinska AK, Schneider ME, Davies C, Riordan GP, Kachar B. An actin molecular treadmill and myosins maintain stereocilia functional architecture and self-renewal. J Cell Biol. 2004;164:887–897. doi: 10.1083/jcb.200310055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rzadzinska AK, Derr A, Kachar B, Noben-Trauth K. Sustained cadherin 23 expression in young and adult cochlea of normal and hearing-impaired mice. Hear Res. 2005;208:114–121. doi: 10.1016/j.heares.2005.05.008. [DOI] [PubMed] [Google Scholar]
- Schneider ME, Belyantseva IA, Azevedo RB, Kachar B. Rapid renewal of auditory hair bundles. Nature. 2002;418:837–838. doi: 10.1038/418837a. [DOI] [PubMed] [Google Scholar]
- Self T, Mahony M, Fleming J, Walsh J, Brown SD, Steel KP. Shaker-1 mutations reveal roles for myosin VIIA in both development and function of cochlear hair cells. Development. 1998;125:557–566. doi: 10.1242/dev.125.4.557. [DOI] [PubMed] [Google Scholar]
- Siemens J, Lillo C, Dumont RA, Reynolds A, Williams DS, Gillespie PG, Müller U. Cadherin 23 is a component of the tip link in hair-cell stereocilia. Nature. 2004;428:950–955. doi: 10.1038/nature02483. [DOI] [PubMed] [Google Scholar]
- Söllner C, Rauch GJ, Siemens J, Geisler R, Schuster SC, Müller U, Nicolson T Tübingen 2000 Screen Consortium. Mutations in cadherin 23 affect tip links in zebrafish sensory hair cells. Nature. 2004;428:955–959. doi: 10.1038/nature02484. [DOI] [PubMed] [Google Scholar]
- Stauffer EA, Scarborough JD, Hirono M, Miller ED, Shah K, Mercer JA, Holt JR, Gillespie PG. Fast adaptation in vestibular hair cells requires Myosin-1c activity. Neuron. 2005;47:541–553. doi: 10.1016/j.neuron.2005.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt SA, Kular G, Fleming IN, Downes CP, Lucocq JM. Subcellular localisation of phophatidylinositol 4,5-bisphosphate using the pleckstrin homology domain of phospholipase Cd1. Biochem J. 2002;363:657–666. doi: 10.1042/0264-6021:3630657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu YC, Ricci AJ, Fettiplace R. Two components of transducer adaptation in auditory hair cells. J Neurophysiol. 1999;82:2171–2181. doi: 10.1152/jn.1999.82.5.2171. [DOI] [PubMed] [Google Scholar]