

Abstract
In traumatic brain injury (TBI), neurons surviving the primary insult may succumb through poorly understood secondary mechanisms. In vitro, cortical neurons exposed to stretch injury exhibited enhanced vulnerability to NMDA, apoptotic-like DNA fragmentation, peroxynitrite (PN) formation, and cytoplasmic cytochrome c accumulation. Surprisingly, caspase-3 activity was undetectable by both immunoblotting and fluorogenic activity assays. Therefore, we hypothesized that PN directly inhibits caspases in these neurons. Consistent with this, stretch injury in cultured neurons elicited tyrosine nitration of procaspase-3, but not caspase-9 or Apaf-1, suggesting a direct interaction of PN with caspase-3. In an ex vivo system, PN inhibited the activity of caspase-3, and this inhibition was reversible with the addition of the sulfhydryl reducing agent dithiothreitol, indicating that PN inhibits caspases by cysteinyl oxidation. Moreover, in cultures, the PN donor 3-morpholinosydnonimine (SIN-1) blocked staurosporine-induced caspase-3 activation and its downstream effects including PARP-1 [poly-(ADP-ribose) polymerase-1] cleavage and phosphotidylserine inversion, suggesting that peroxynitrite can inhibit caspase-3-mediated apoptosis. To examine these mechanisms in vivo, rats were exposed to a lateral fluid percussion injury (FPI). FPI caused increased neuronal protein nitration that colocalized with TUNEL staining, indicating that PN was associated with neurodegeneration. Caspase-3 activity was inhibited in brain lysates harvested after FPI and was restored by adding dithiothreitol. Our data show that caspase-mediated apoptosis is inhibited in neurons subjected to stretch in vitro and to TBI in vivo, mostly because of cysteinyl oxidation of caspase-3 by PN. However, this is insufficient to prevent cell death, indicating that the TBI therapy may, at a minimum, require a combination of both anti-apoptotic and anti-oxidant strategies.
Keywords: peroxynitrite, traumatic brain injury, caspase-3, apoptosis, nitric oxide, reactive oxygen species
Introduction
Traumatic brain injuries (TBIs) represent a significant socioeconomic burden (Marshall, 2000). In the United States, TBI incidence is 618 per 100,000 person-years (Sosin et al., 1996), and an estimated 80,000–90,000 individuals sustain long-term disabilities annually (Thurman et al., 1999). TBI can result in neurological impairment because of immediate CNS tissue disruption (primary injury), but, additionally, surviving cells may be secondarily damaged by complex mechanisms triggered by the primary event, leading to further damage and disability (Teasdale and Graham, 1998; Amar and Levy, 1999). Several mechanisms have been implicated, including necrotic and apoptotic pathways (for review, see Beattie et al., 2000; Raghupathi et al., 2000; Zipfel et al., 2000). However, their complex interactions have made it difficult to determine which pathways should be addressed therapeutically in TBI. To date, most approaches to the treatment of TBI that target a single injury mechanism have failed in clinical trials (Ikonomidou and Turski, 2002; Tolias and Bullock, 2004; McKee et al., 2005). Consequently, it has been suggested that “recognition of multiple cell death pathways should lead to new treatment strategies, including both combination drug treatment and drugs that affect multiple components of the secondary injury cascade” (Faden, 2002). However, compelling evidence to indicate which of the many components of secondary injury should be addressed by these means is lacking. This report begins to address this need.
We previously developed and characterized an in vitro model of neuronal stretch injury to study secondary injury mechanisms (Arundine et al., 2003, 2004). In brief, cultured cortical neurons were exposed to sublethal stretch to remove the confounding effects of primary mechanical damage. The neurons maintained cell membrane integrity, viability, and electrophysiological function but exhibited an enhanced vulnerability to subsequent challenges with l-glutamate or NMDA (Arundine et al., 2003). This was attributable to a combination of stretch-induced, mitochondrially derived, reactive oxygen species (ROS) compounded by NMDA-mediated nitric oxide (NO) production. Together, these formed the lethal oxidant peroxynitrite (PN), which killed the cells (Arundine et al., 2004). However, cell death in the stretch plus NMDA paradigm was also associated with nuclear irregularities and DNA fragmentation suggestive of apoptosis. Because apoptotic mechanisms have been implicated in TBI previously (see reviews above), we set out to determine how they interact with PN-dependent neurotoxic mechanisms.
We studied these mechanisms in vitro using a cell stretch paradigm and in vivo using the rodent lateral fluid percussion injury (FPI) model of TBI. We found that neuronal stretch injury induces cytochrome c release into cytoplasm as anticipated by others (Buki et al., 2000; Fiskum, 2000). However, whereas cytochrome c release is expected to trigger classical, caspase-mediated apoptosis (Kluck et al., 1997; P. Li et al., 1997), this process was aborted in traumatized neurons because of the actions of PN. Here, we show that PN inactivates classical apoptosis in vitro and in vivo by directly and preferentially targeting caspase-3, a key effector caspase. This inhibition was primarily attributable to cysteinyl oxidation of caspase-3 by PN, thus aborting caspase-mediated effects and switching cell death away from a classical apoptotic mechanism.
Materials and Methods
Unless otherwise indicated, all chemicals are from Sigma-Aldrich (Oakville, Ontario, Canada). All in vitro experiments were performed at 37°C. All experiments were approved by the University Health Network Animal Resource Center, which is fully accredited by the Canadian Council on Animal Care.
Primary murine cortical cultures.
Mixed cortical cell cultures containing both neurons and glia were prepared from embryonic day 15 (E15) Swiss mice as described previously (Arundine et al., 2004). In brief, cerebral cortices were extracted from embryos and incubated for 10 min in 0.05% trypsin-EDTA. Digested cortices were dissociated by trituration and plated on poly-l-ornithine-coated six-well plates at ∼3.6 × 106 cells per well. Flexible membrane plates (Flexcell International Corporation, Hillsborough, NC) were used for stretch injury experiments, and standard plates (Corning, Corning, NY) were used for all other experiments. All cell culture reagents were obtained from Invitrogen (Mississauga, Ontario, Canada). Plating medium consisted of minimum essential medium supplemented with 10% heat-inactivated horse serum, 10% fetal bovine serum, 1% glutamine, and 25 mm glucose. After 3 d in vitro, growth of non-neuronal cells was halted by exposure to 10 μm FDU-solution (5 μm uridine, 5 μm (+)-5-fluor-2′-deoxyuridine) for 48 h. Previous studies have shown that these cultures are >85% neurons (Sattler et al., 1997). Cultures were maintained at 37°C in a humidified 5% CO2 atmosphere until used at 11 d after plating.
3-Morpholinosydnonimine and staurosporine treatments.
At 11 d after dissection, the growth medium is aspirated and cultures are washed twice with HEPES solution. A 100 mm 3-morpholinosydnonimine (SIN-1) stock was prepared in ddH2O immediately before use, and a 100 μm staurosporine stock solution was dissolved in DMSO. Appropriate volumes of the stock solution (according the experimental protocol) were added directly to the wells. For the cotreatment paradigm, 10 μl of 100 mm SIN-1 and 10 μl of 100 μm staurosporine were added directly to the wells, resulting in a final concentration of 1 mm SIN-1 and 1 μm staurosporine. Accordingly, the vehicle controls for the cotreatment paradigm were treated with 10 μl each of ddH2O and DMSO. Cultures were gently shaken to promote equal distribution. Treatments were left in the wells until appropriate time points in a 37°C humidified incubator for the remainder of the experiments.
In vitro cell stretch model.
This was performed as described previously (Arundine et al., 2003, 2004). In brief, the cultures were washed twice with HEPES solution composed of the following (in mm): 121 NaCl, 10 HEPES acid, 7 HEPES Na, 20 d-glucose, 0.01 glycine, 0.5 KCl, 1 Na pyruvate, 1.8 CaCl2, and 0.3 NaHCO3. Cultures were then stretched to 130% of their original length for 1 s using the Flexercell FX-3000 Strain Unit (Flexcell, Hillsborough, NC). This degree of stretch did not cause the cultures to detach from their underlying substrate and, in itself, is nonlethal to the cells (Arundine et al., 2004). Immediately after stretch, the cells were washed with either HEPES solution alone or in combination with NMDA for 1 h and then returned to HEPES solution. Control cultures were identically manipulated, except that no stretch was applied. All solutions contained nimodipine (2 μm) and CNQX (10 μm) to restrict the actions of applied NMDA to NMDA receptors (NMDARs) by preventing the secondary activation of other pathways (Sattler et al., 1998).
Lateral FPI.
FPI was performed as described by McIntosh et al. (1989) using 350–370 g male Sprague Dawley rats (Charles River, St. Constant, Quebec, Canada). In brief, 24 h before injury, the animal was anesthetized using 2% halothane and 2:1 nitrous oxide:oxygen. A 4.8 mm craniotomy was made with its center between the bregma and lambda and between the sagittal suture and right temporal ridge. A modified Leur-loc fitting was attached over the craniotomy and cemented in place using Acron MC/R dental acrylic (GC America, Alsip, IL). The Leur-loc was filled with saline, and a small piece of gelfoam was inserted. The incision was sutured, and the animal was returned to its cage overnight. The following day, the incision was reopened, cleaned, and the gelfoam was removed. The animal was attached to the fluid percussion injury device (Custom Design and Fabrication, Richmond, VA) and was administered a 2.4 atm injury, which resulted in a mortality rate of ∼6%. Injury severity was recorded using an oscilloscope attached to a transducer. Immediately after injury, the Leur-loc setup was removed en bloc, the wound was cleaned, and the incision was resutured. The animal was then returned to its cage.
2,3,5-Triphenyltetrazolium chloride staining.
Twenty-four hours after fluid percussion injury, animals were reanesthetized as above and decapitated. Whole brains were extracted and chilled at −20°C for 10 min. Coronal brain sections were made and incubated in 2% 2,3,5-triphenyltetrazolium chloride (TTC) in saline solution for 30 min at 37°C (Joshi et al., 2004).
Protein extraction.
After in vitro treatments, cells were washed twice with HEPES solution. Mammalian protein extraction reagent (M-PER; Pierce, Rockford, IL) was added to each well, and the cell lysates were collected and centrifuged at 10,000 rpm for 10 min. Supernatant was collected for analysis in activity assays and Western blots. After FPI, brain tissue samples were extracted from TTC-treated brains and placed into modified radioimmunoprecipitation assay buffer (50 mm Tris-HCl, 150 mm NaCl, 1 mm EDTA, pH 7.4) with protease inhibitors for Western blotting. Protease inhibitors were omitted for caspase activity assays. To extract protein from the injury site, all cortical areas negatively stained with TTC were extracted from 2 mm sections spanning from approximately −2.0 to −9.0 mm (bregma levels) and combined per animal for analysis. Brain sections were triturated using a 25-gauge needle, followed by finer dissociation using a 30-gauge needle. Samples were centrifuged at 2900 × g for 5 min, and pellets were discarded. Samples were stored at −80°C until used. Protein concentrations from both extractions were determined using the Bio-Rad Protein Assay (Bio-Rad, Hercules, CA) in conjunction with bovine serum albumin standards.
SDS-PAGE.
Each lane was loaded with 20 μg of boiled sample suspended in 4× sample buffer consisting of the following: 0.125 m Tris-HCl, pH 6.8, 20% glycerol, 4% SDS, 5% β-mercaptoethanol, and 0.025% bromophenol blue. Gels were run at 70 V. All Western blots were run on 12% Tris/glycine gels except poly-(ADP-ribose) polymerase-1 (PARP-1) gels, which were run with 7.5% Tris/glycine gels. Gels were transferred onto nitrocellulose paper in 4°C in transfer buffer (0.2 m glycine, 0.025 m Tris, 0.04% SDS, 20% methanol) at 100 V for 1 h.
Immunoblotting.
After transfer, gels were blocked in 5% blotting grade blocker nonfat dry milk (Bio-Rad) in TBST (0.01 m Tris, 0.1 m NaCl, 0.05% Tween 20) for 50 min. Immunoblots were performed with either a monoclonal mouse anti-cytochrome c antibody (1:200; PharMingen, San Diego, CA); polyclonal rabbit anti-caspase-3 antibody (1:1000; Stressgen, Victoria, British Columbia, Canada) recognizing both active and inactive (pro-) caspase-3; polyclonal rabbit anti-3-nitrotyrosine antibody (2 μg/ml; Upstate Biotechnology, Lake Placid, NY); monoclonal mouse anti-3-nitrotyrosine (2 μg/ml; Biomol, Plymouth Meeting, PA); polyclonal rabbit anti-caspase-9 (1:500; Stressgen); monoclonal mouse anti-apoptotic protease activating factor-1 (APAF-1) (1:500; Stressgen); monoclonal anti-cytochrome c (1:200, PharMingen); or monoclonal mouse anti-PARP-1 (2 μg/ml; PharMingen) recognizing both cleaved and uncleaved PARP-1. Primary antibodies were incubated overnight at 4°C. After washing, HRP-conjugated goat anti-rabbit IgG (1:5000; Jackson ImmunoResearch, West Grove, PA) or HRP-conjugated goat anti-mouse IgG (1:5000; Pierce Biotechnology) was added at room temperature for 1 h. Immunoreactivity was visualized using an ECL Western blotting detection system (GE Healthcare, Piscataway, NJ). To ensure equal protein loading in all lanes within each immunoblot, the amount of protein pipetted into each lane (usually 40 μg) was quantified using the Bio-Rad Protein Assay Dye Reagent (catalog #500–0006). Also, the blots were then stripped and reprobed with anti-β-actin antibodies (1:5000; Sigma). As a further confirmation, proteins from the same sample lysates were run on parallel gels, transferred onto nitrocellulose, and stained with Ponceau stain. Representative examples are provided in Figure 2A but are generally omitted for clarity.
Figure 2.

Stretch plus 30 mm NMDA injury produces cytochrome c release, but not caspase-3 activation. A, Immunoblot of the cytoplasmic fraction of injured cells using anti-cytochrome c (top) antibodies. Reblotting with anti-β-actin (middle) antibodies and Ponceau staining of parallel samples (bottom) was performed as control for protein loading. Cytochrome c is released into cytoplasm almost immediately following stretch plus 30 μm NMDA and persists for at least 20 h (n = 3). B, Immunoblot of caspase-3 (both pro- and active/cleaved forms) from whole-cell lysates taken 20 h after the indicated insult. The procaspase-3 form (∼32 kDa) was detectable under all conditions, whereas only cultures treated with staurosporine displayed the active caspase-3 band (∼17 kDa). Data are representative of four experiments. C, Cell lysates taken 20 h after the indicated insults and incubated with Ac-DEVD-AMC show no increases in caspase-3-like proteolytic activity compared with controls (n = 3 experiments). Asterisk, Difference from HEPES buffer-treated controls (*p < 0.05; n = 3). Stauro, Staurosporine (1 μm) controls harvested at 24 h posttreatment in B and C. D, Immunoblot of whole-cell lysates taken 20 h after the indicated insults using anti-3-nitrotyrosine antibodies (n = 4 experiments). SIN-1 (1 mm for 24 h) was used as a positive control.
Subcellular fractionation.
HEPES solution in the wells was aspirated and replaced with 1 ml of ice-cold PBS. Cells were collected and samples were centrifuged at 300 × g for 1 min at 4°C. The supernatant was discarded, and the pellet was resuspended in 300 μl of ice-cold PBS. Samples were centrifuged at 300 × g for 1 min at 4°C and supernatant was discarded. The pellet was then resuspended in 500 μl of digitonin release buffer (250 mm sucrose, 17 mm 4-morpholinepropanesulfonic acid, 2.5 mm EDTA, 0.8 mg/ml digitonin) and transferred to a 2 ml Dounce homogenizer. Samples were homogenized with 30 ups and downs. Lysates were spun at 5000 × g for 5 min, the supernatant was discarded, and the lysates were centrifuged again at 21,000 × g for 30 min all at 4°C. The supernatant was collected and designated the cytoplasmic fraction, whereas the pellet was resuspended in 250 μl of M-PER and designated the mitochondrial fraction.
Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling assay.
Nuclear DNA strand breaks were detected by enzymatically labeling free 3′-OH termini with modified nucleotides provided in the Apoptag kit (Intergen, Purchase, NY) using the manufacturer instructions. Visualization of terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL)-stained nuclei was achieved using an anti-digoxigenin antibody conjugated to a fluorescein or peroxidase reporter molecule (ApopTag Kits S7110 and S7100, respectively). In fluorescence experiments, nuclei were counterstained using 5 ng/ml Hoechst 33258 (Invitrogen, Eugene, OR) and visualized using 465–495 nm excitation and 515–555 nm emission for fluorescein and 340 nm excitation and 510–40 nm emission for Hoechst. The number of TUNEL-positive cells was expressed as a fraction of the total cell number in the field.
Phosphatidylserine inversion.
After chemical treatment, the cells were washed twice with HEPES solution. A kit using FITC-conjugated annexin V to label inverted phosphatidylserine (ApopNexin kit; Intergen) was used according to the specifications of the manufacturer. After application of the labeled annexin V, the cells were washed with HEPES solution and observed at 200× magnification and visualized using 465–495 nm excitation and 515–555 nm emission for fluorescein.
Quantification of in vitro cell death.
Cell death was determined by serial quantitative measurements of propidium iodide (PI) fluorescence using a multiwell plate fluorescence scanner (Fluoroskan Ascent FL; ThermoLab Systems, Burlington, Ontario, Canada) as described previously (Sattler et al., 1997, 1998). In brief, the medium in each culture well was replaced with HEPES solution containing 50 μg/ml PI, and a baseline fluorescence reading was taken. Sequential readings were then taken at appropriate intervals over the observation period. The fraction of dead cells in each culture at a given time was calculated as: fraction dead = (Ft − Fo)/FNMDA where Ft is the PI fluorescence at time t, Fo is the initial PI fluorescence at time 0, and FNMDA is the background subtracted PI fluorescence of identical cultures from the same dissection and plating, 20–24 h after a 60 min exposure to 1 mm NMDA at 37°C. Based on manual observations at the time of validation of this technique, this NMDA exposure routinely produced near complete neuronal death in each culture but had no effect on surrounding glia (Bruno et al., 1994; David et al., 1996; Sattler et al., 1997). Adding Triton X-100 (0.1%) to cultures treated in this manner produced an additional 10–15% increase in PI fluorescence resulting from permeabilization of non-neuronal cell membranes, consistent with a 10–15% glial component in the cultures.
Fluorogenic quantification of caspase-3 and caspase-9 activity.
After quantification of protein concentration, cell lysates from in vitro experiments were incubated with 10 μm N-acetyl-Asp-Glu-Val-Asp-7-amino-4-methylcoumarin (EMD Biosciences, San Diego, CA) in M-PER at 37°C for 1 h. Protein lysates from whole animal experiments were treated identically, except 40 μm Ac-DEVD-AMC was used for caspase-3 activity measurements, and 100 μm Ac-N-acetyl-Leu-Glu-His-Asp-7-amido-trifluoromethylcoumarin (EMD Biosciences) was used to measure caspase-9 activity. Plates were placed into a multiwell plate fluorescence scanner, and fluorescence was quantified at 355 nm excitation and 460 nm emission for Ac-DEVD-AMC, or 380 nm excitation and 510 nm emission for Ac-LEHD-AFC. We note here that these fluorogenic caspase substrates have the highest specificity to caspase-3 and caspase-9, respectively, but we do not rule out possible interactions with other effector or initiator caspases, respectively. However, any additional, as yet undemonstrated, interactions with these caspases would not affect the interpretation of the results provided herein.
Ex vivo recombinant caspase-3 activity system.
Recombinant procaspase-3 was resuspended in PBS, pH 7.4, to a concentration of 0.5 μg/ml and incubated with SIN-1 or peroxynitrite (EMD Biosciences) for 1 h at 37°C. In some experiments, 10 mm dithiothreitol (DTT) or ddH2O as a negative control was added to the system. To trigger caspase-3 activity, 0.5 ng (3 U) of recombinant caspase-9 (Calbiochem, La Jolla, CA) was added. Activity was assessed using Ac-DEVD-AMC as above.
Ex vivo recombinant caspase-9 activity system.
Recombinant caspase-9 (Calbiochem; 1 U) was diluted in M-PER to a final concentration of 0.01 U/μl and incubated with peroxynitrite for 1 h at 37°C. DTT (10 mm), or ddH2O as a negative control, was applied immediately after incubation with peroxynitrite. Caspase-9 activity was assessed using Ac-LEHD-AFC as above.
Immunoprecipitation.
Protein lysates from cell cultures were incubated with agarose beads and agarose beads conjugated to nonspecific mouse IgG before incubation with agarose-conjugated anti-3-nitrotyrosine (Upstate Biotechnology). Lysates were incubated overnight at 4°C with agarose-conjugated anti-3-nitrotyrosine, and the supernatant was discarded. The beads were suspended in 2× sample buffer without β-mercaptoethanol and boiled for 10 min before being run with SDS-PAGE. Gels were then transferred for Western blotting.
DNA gel electrophoresis.
At 20 h after the experimental manipulation, cells were collected from each tissue culture well in HEPES solution and pelleted by centrifugation at 1000 rpm. The cells were digested for 2 h at 56°C in DNA lysis buffer containing the following (in mm): 10 Tris, pH 8.0, 2 EDTA, pH 8.0, 400 NaCl, 0.5% SDS, and 1 mg/ml proteinase K. Total DNA was subsequently extracted and precipitated using the phenol/chloroform method. In brief, an equal volume of phenol was added to the cell extract, followed by centrifugation at 13,000 rpm for 10 min. The top layer was removed, and an equal volume of chloroform was added thereafter. This process was repeated twice. After the final chloroform extraction, 100% ethanol was added to the top fraction and placed on ice for 10 min. DNA was pelleted by centrifugation 13,000 rpm for 10 min. The DNA pellet was dried, and 50 μl of ddH2O was added thereafter. The DNA pellet was left to dissolve into the ddH2O overnight at 4°C, gently mixed, and then quantified spectrophotometrically (260/280 absorption). DNA (10 μg) was gently mixed with loading buffer, loaded on a 2% agarose gel, and electrophoretically separated at 100 V. Gels were stained with ethidium bromide and photographed using UV transillumination.
Immunohistochemistry.
For frozen brain sections, animals were anesthetized and transcardially perfused using heparinized saline followed by 4% (w/v) paraformaldehyde in PBS. Whole brains were extracted from the fixed animals and were cryoprotected in 30% sucrose with 4% paraformaldehyde in PBS solution at 4°C overnight. They were sectioned into 25 μm free-floating slices in PBS using a Kryostat 1720 (Leica, Wetzler, Germany). Immunostaining was performed as described previously (Sun et al., 2006). In brief, sections were blocked using a 1% BSA, 3% goat serum, 0.3% Triton X-100 in PBS solution for 90 min and doubled labeled with rabbit anti-3-nitrotyrosine (1:50; Upstate Biotechnology) and mouse anti-neuronal-specific nuclear protein (NeuN; 1:100; Chemicon, Temecula, CA) overnight at 4°C. Sections were subsequently washed and blocked briefly with blocking solution and incubated with goat anti-rabbit Alexa568 (1:100; Invitrogen) and goat anti-mouse FITC (1:200; Sigma) for 1 h at room temperature. Slides were mounted using ProLong Gold antifade reagent (Invitrogen).
For paraffin-embedded tissue, anesthetized animals were transcardially perfused using 10% phosphate-buffered formalin and decapitated, and the heads were placed into 10% phosphate-buffered formalin overnight. Whole brains were extracted the following day, processed, embedded in paraffin wax, and sectioned into 7-μm-thick slices. The slices were kept at room temperature until use, after which they were rehydrated in a reverse order of decreasing xylene and alcohol concentrations. TUNEL staining was performed according to the manufacturer protocol using an Apoptag kit (Intergen). After TUNEL staining, the specimens were washed with PBS and blocked using 1% BSA in PBS for 30 min. A secondary block with 2% goat serum in PBS for 1 h was followed by treatment with 1:100 anti-3-nitrotyrosine (Upstate Biotechnology) overnight at 4°C. The next morning, sections were treated with anti-rabbit FITC (1:1000; Jackson ImmunoResearch) for 1.5 h.
Confocal microscopy.
Frozen sections (25 μm) taken from brains of animals that underwent a 2.4 atm FPI were double stained with anti-NeuN and anti-3-nitrotyrosine. These were visualized with a laser-scanning confocal microscope (Zeiss LSM 510 META) using 25× or 63× microscope lenses (Zeiss). In each preparation, the section was initially scanned to determine a depth halfway between its two surfaces, and the midslice depth was used for imaging. Optical stacks of 5–10 confocal images taken at 0.5 μm intervals were used to generate figures as described previously (Sun et al., 2006).
Statistics.
All data were expressed as mean ± SEM and analyzed using ANOVA or Student’s t test as appropriate (SigmaStat3.0; SPSS, Chicago, IL). Statistical significance was considered at the p < 0.05 level.
Results
Sublethally stretched neurons exhibit enhanced vulnerability to NMDA toxicity and apoptotic-like DNA fragmentation
We previously developed and characterized a model of sublethal neuronal stretch injury to study secondary mechanisms of traumatic damage (see Materials and Methods). In brief, stretched neurons maintain viability, cell membrane integrity, and electrophysiological function but exhibit a heightened vulnerability to subsequent challenges with NMDAR agonists (Arundine et al., 2003). Consistent with this, cultured cortical neurons subjected to 130% stretch for 1 s (hereafter termed “stretch”) were unharmed by this insult as measured by PI uptake (Fig. 1A). However, the addition of 30 μm NMDA induced significantly more cell death in stretched compared with unstretched cultures. The enhanced vulnerability of stretched neurons to NMDA insults is the consequence of a combination of stretch-induced, mitochondrially derived, ROS compounded by NMDA-mediated NO production. Together, these form the lethal oxidant PN, which accounts for the lethality of the stretch plus NMDA paradigm (Arundine et al., 2004).
Figure 1.

Enhanced vulnerability and apoptosis-like features of sublethally stretched neurons in response to NMDA. A, Neuronal death 20 h after the indicated insults as assessed by propidium iodide uptake. Where used, NMDA was applied for 1 h after the stretch. Asterisk, Difference from 30 μm NMDA (*p < 0.05; n = 4). B, Staurosporine and stretch plus 30 μm NMDA, but not stretch alone, induces internucleosomal DNA laddering (arrowheads) after insult. Data are representative of three separate experiments. C, Staurosporine and stretch plus 30 μm NMDA, but not HEPES-treated controls, enhances TUNEL (green) labeling of the cultures. Cultures were costained with Hoescht 33258 to illustrate cell nuclei (red). Images are representative of three separate experiments. Scale bar, 200 μm.
Neurons subjected to stretch plus 30 μm NMDA, but not neurons exposed to control medium or to stretch without NMDA, exhibited internucleosomal DNA fragmentation as demonstrable by DNA gel electrophoresis (Fig. 1B). The “DNA ladder” was similar to that obtainable by treating the cultures with staurosporine, a nonselective protein kinase inhibitor, which induces caspase-3 activation and classical apoptosis in cultured central neurons (Koh et al., 1995). Also, neurons subjected to stretch plus NMDA exhibited increased TUNEL, a feature that was also produced by the staurosporine treatment (Fig. 1C). Thus, stretch plus NMDA injury resulted in cell death exhibiting apoptotic-like features.
Neuronal stretch induces cytoplasmic cytochrome c release but without concomitant caspase-3 activation
In vitro, neuronal stretch produces mitochondrial dysfunction and ROS formation (Arundine et al., 2004). In vivo, neurotrauma is associated with the release of mitochondrial cytochrome c into cytoplasm (Buki et al., 2000; Fiskum, 2000). Because cytochrome c release is recognized as a trigger of classical type II caspase-mediated apoptosis (Kluck et al., 1997; P. Li et al., 1997), we further investigated the cause of DNA degradation in our stretched neurons by measuring cytochrome c release after stretch. Neurons were subjected to the stretch plus NMDA insult, and cytochrome c immunoreactivity in cytoplasmic fractions was determined by Western blots. Stretch plus 30 μm NMDA induced a rapid and robust increase in cytoplasmic cytochrome c, which was measurable immediately after the insult and persisted for at ≥20 h (Fig. 2A).
Cytochrome c release is the critical point in the formation of the apoptosome, a cytoplasmic protein complex that leads to the processing and activation of effector caspases that, in turn, lead to apoptotic neuronal death (for review, see Green and Reed, 1998; Stefanis, 2005). Although apoptosis is not always caspase dependent, it is anticipated that once released, cytochrome c should lead to caspase-3 activation (Dawson and Dawson, 2004). To test this, whole-cell lysates were taken from cultures subjected to stretch plus NMDA and assayed in Western blots using an anti-caspase-3 antibody recognizing both active (cleaved) and inactive (pro-) caspase-3 (molecular weight of ∼17 kDa and ∼32 kDa, respectively; see Materials and Methods). Surprisingly, no active caspase-3 immunoreactivity was observed at the expected ∼17 kDa band (Fig. 2B). To probe caspase-3 proteolytic activity, we incubated the whole-cell lysates taken from cells exposed to stretch plus NMDA with the fluorogenic caspase-3 substrate Ac-DEVD-AMC (10 μm; see Materials and Methods). This yielded no increase in AMC fluorescence 20 h after the insult when compared with controls (Fig. 2C). Staurosporine (1 μm), the positive control that elicits caspase-3-mediated classic apoptosis in neurons (Koh et al., 1995), induced both caspase-3 cleavage (Fig. 2B, right lane) and significantly increased Ac-DEVD-AMC fluorescence (Fig. 2C). Thus, despite robust cytoplasmic cytochrome c release (Fig. 2A), the cleavage and activity of caspase-3, the main effector caspase of apoptosis, was undetectable in stretch plus NMDA injured neurons.
Neuronal stretch injury is associated with increased peroxynitrite formation
Cell death in the in vitro stretch model of neuronal injury was associated with PN formation (Arundine et al., 2004), and PN formation has been reported to trigger cell death in neurons and neuronal cell lines (Estevez et al., 1995, 1998; Spear et al., 1998; Virag et al., 1998; Bao and Liu, 2003). Thus, we next examined PN formation in our model by measuring protein tyrosine nitration. Tyrosine nitration occurs nearly exclusively by PN (Beckman and Koppenol, 1996) and is detectable immunologically using anti-3-nitrotyrosine antibodies (Ye et al., 1996).
Western blots were performed using whole-cell lysates taken from cultures that had undergone stretch in paradigms identical to those in Figure 2, B and C, and then immunostained with an anti-3-nitrotyrosine antibody (see Materials and Methods). Stretch alone failed to elicit nitrotyrosine formation, whereas stretch plus 30 μm NMDA elicited increased tyrosine nitration, confirming that PN was formed under this condition (Fig. 2D). SIN-1 (1 mm), a compound known to release equimolar amounts of superoxide and nitric oxide to form peroxynitrite in cortical cultures (Trackey et al., 2001), was used as a positive control for peroxynitrite production and 3-nitrotyrosine detection.
From these findings, we conclude that in vitro neuronal stretch injury elicits a form of cell death characterized by cytochrome c release, PN formation, and DNA fragmentation but without caspase-3 activation. Although PN is known to produce DNA fragmentation without apoptosis (Bolanos et al., 1997; Szabo and Ohshima, 1997; Hill et al., 2000), cytochrome c release is widely viewed as a step that initiates caspase-mediated apoptotic death pathways (Green and Reed, 1998). Thus, to account for the lack of caspase-3 activation, we hypothesized that, in addition to damaging neurons directly, PN actively inhibits classical apoptosis in mechanically injured neurons.
Procaspase-3 is nitrated after stretch injury in cortical neuronal cultures
We next asked whether, in the stretch injury model, PN may act on caspase-3 directly or whether it acts earlier on steps that intervene between cytochrome c release and caspase-3 activation. After its release, cytochrome c should bind with procaspase-9 and Apaf-1 to form an apoptosome complex (P. Li et al., 1997). Apoptosome formation effectively activates caspase-9 (Zou et al., 1999), leading to the cleavage and activation of caspase-3. However, treatment of cytochrome c with peroxynitrite did not affect apoptosome formation and subsequent caspase-3 activation in previous studies (Ueta et al., 2003). Consequently, we evaluated the interaction of PN with key constituents downstream from cytochrome c by measuring tyrosine nitration of caspase-9, Apaf-1, and caspase-3 from stretched neurons. Nitrated proteins taken from cortical cultures subjected to stretch plus 30 μm NMDA were immunoprecipitated by incubating whole-cell lysates with an anti-3-nitrotyrosine agarose conjugate (see Materials and Methods). The immunoprecipitated lysates were then run on SDS-PAGE and assayed with appropriate antibodies. As controls, we used untreated cultures (negative control) and cultures treated with staurosporine (1 μm; positive control for identification of the immunoreactive bands). Whereas caspase-9 (Fig. 3A) and Apaf-1 (Fig. 3B) were detectable in the nonimmunoprecipitated positive (staurosporine) controls, neither could be immunoprecipitated with anti-3-nitrotyrosine antibodies, arguing against protein nitration of these apoptosome components after stretch injury. However, the same immunoprecipitates exhibited a strong immunoreactive band at ∼32 kDa corresponding to the pro- (inactive) form of caspase-3 (Fig. 3C), indicating that procaspase-3 is directly nitrated by PN after the stretch injury
Figure 3.

Stretch plus 30 μm NMDA nitrates procaspase-3. A–C, Whole-cell lysates from cultures exposed to stretch plus 30 μm NMDA were immunoprecipitated using an agarose-bound monoclonal anti-3-nitrotyrosine antibody and run on immunoblots (middle lanes) along with nonimmunoprecipitated samples from staurosporine-treated cultures as positive controls and from untreated cultures as negative controls. A, Anti-caspase-9 antibody. B, Anti-Apaf-1 antibody. C, Anti-caspase-3 (pro- and active/cleaved forms). n = 2 experiments/blot. The dotted line encompasses the lanes containing the immunoprecipitated sample. The untreated and staurosporine controls were not immunoprecipitated with anti-3-NT antibodies as, in the absence of nitration, these proteins cannot be precipitated with the antibody-agarose conjugate.
Peroxynitrite inhibits caspase activity directly via cysteine oxidation
Tyrosine nitration is an index of PN activity but may not necessarily lead to caspase-3 inactivation. Having shown that PN elicits the tyrosine nitration of procaspase-3 (Fig. 3C), we next used an ex vivo system to measure the effects of PN on the activity of both caspase-3 and -9.
First, to examine the direct effect of PN on caspase-3 activity, recombinant procaspase-3 was reconstituted in PBS, pH 7.4, at 0.5 μg/ml and incubated with either SIN-1 or pure peroxynitrite (Fig. 4A). After 1 h incubation at 37°C, recombinant active caspase-9 (Sigma) was added to the system, and caspase-3 activity was assessed using the fluorogenic Ac-DEVD-AMC assay (see Materials and Methods). Caspase-3 activity was significantly decreased after treatment with as little as 300 μm SIN-1 and was completely abolished with peroxynitrite treatment at a similar concentration.
Figure 4.

Peroxynitrite can inhibit caspase-3 and caspase-9 activity ex vivo via cysteinyl oxidation. A, Effects of the PN donor SIN-1, or of direct PN addition, on caspase-3-like activity. Recombinant procaspase-3 was incubated with the indicated concentrations of SIN-1 or peroxynitrite before activation with recombinant caspase-9 and Ac-DEVD-AMC fluorescence was measured. Both compounds significantly reduced (p < 0.05) caspase-3 activity as shown (n = 3 experiments per compound; asterisks, different from own control). B, C, Recombinant caspase-3 (B) and caspase-9 (C) were incubated for 1 h with the indicated PN concentrations, and proteolytic activity was assessed using Ac-DEVD-AMC and Ac-LEHD-AFC, respectively. The inhibition of activity of both caspases by PN was significantly reversed by DTT (10 mm; p < 0.05; n = 3 experiments per concentration/condition).
PN may affect protein function by protein tyrosine nitration (Ischiropoulos et al., 1992; Beckman and Koppenol, 1996) but is also a potent oxidant that affects cysteine residues and may thus exert its effects in part by oxidizing tissue sulfhydryls (Radi et al., 1991). Although PN elicits the tyrosine nitration of procaspase-3 (Fig. 3C), we have not yet determined whether this posttranslational modification, or cysteine oxidation, is the mechanism of caspase inhibition. Consequently, we next repeated the same ex vivo experiment using a lower range of PN concentrations (0–100 μm). Then, after 1 h of incubation, we added 10 mm DTT, a sulfhydryl reducing reagent. This treatment significantly reduced the inhibition of caspase-3 activity by PN, indicating that an oxidative modification of cysteine residues is primarily responsible for the inhibition of caspase-3 in this recombinant system (Fig. 4B).
Last, to test whether PN can have similar effects on caspase-9, recombinant caspase-9 was incubated with 0–100 μm PN for 1 h at 37°C, and 10 mm DTT was applied immediately thereafter. Caspase-9 activity was assessed using the fluorogenic Ac-LEHD-AFC assay (see Materials and Methods). PN strongly inhibited caspase-9 activity, and this was almost completely reversed by the DTT treatment (Fig. 4C). These data suggest that, at least in a cell-free system, PN inhibits the activity of both caspase-3 and caspase-9 by reversible cysteinyl oxidation.
Next, we sought to determine whether PN can inactivate caspase-3 in cultured cortical neurons otherwise committed to apoptosis. To this end, we developed and optimized two separate paradigms. In the first, PN formation was elicited without caspase-3 activation, and in the second, caspase-3 activation was elicited without PN formation. Combining these should demonstrate whether caspase-3 activation is affected by PN.
Staurosporine treatment causes caspase-3-dependent cell death without protein nitration
First, the cultures were treated with 1 μm staurosporine, a general protein kinase inhibitor that induces caspase-3-mediated death in murine cortical cell cultures (Koh et al., 1995). The cells were harvested at different times (3–24 h) and assayed in Western blots for cleaved (active) caspase-3 (Fig. 5A) and for 3-nitrotyrosine (Fig. 5B). Staurosporine-induced progressive caspase-3 cleavage (∼17 kDa band in Fig. 5A) was observable until 24 h, and this occurred in the absence of 3-nitrotyrosine formation (Fig. 5B). TUNEL staining 24 h after staurosporine treatment revealed a marked increase in DNA fragmentation (Fig. 5C, right panel), and this was concomitant with a progressive reduction in cell viability as assayed with PI at 3 and 24 h (Fig. 5D). Thus, staurosporine induces caspase-3 activation and cell death without protein nitration.
Figure 5.

Dependence of staurosporine-mediated neuronal death on caspase-3 activation but not on protein nitration. A, B, Immunoblots using anti-caspase-3 (pro- and active/cleaved forms) and anti-3-nitrotyrosine antibodies of whole-cell lysates from cultures treated with 1 μm staurosporine for the indicated times. Active caspase-3 immunoreactivity (∼17 kDa band) increases with time (A; n = 4 experiments), whereas anti-NT staining does not (B; n = 3 experiments). SIN-1 (1 mm) was used as a positive control in B. C, Fluorescent TUNEL labeling (green) of staurosporine-treated cultures (right) compared with controls (left). Cells were counterstained with Hoescht (red) to show all nuclei (n = 3 experiments). Scale bar, 100 μm. D, Cell death resulting from staurosporine treatment (Stauro) at the indicated times (n = 4 experiments; asterisks, different from control at p < 0.05). Right panels, Representative PI fluorescence images at 24 h from the indicated conditions. Scale bar, 100 μm.
SIN-1 treatment causes protein nitration without caspase-3 activation
Next, sister cortical cultures were treated with 1 mm SIN-1. This treatment did not induce caspase-3 activation (Fig. 6A), whereas it induced robust protein nitration at all time points between 3 and 24 h (Fig. 6B). This same concentration of SIN-1 did not cause detectable TUNEL staining in the cultures (Fig. 6C, middle panel), indicating that 1 mm SIN-1 can be used to elicit protein nitration without TUNEL-detectable DNA damage. Notably, DNA fragmentation was not seen until the concentration of SIN-1 was raised to 3 mm (Fig. 6C, right panel). Concomitant cell viability assays revealed that 1 mm SIN-1 was not toxic to the cultures, whereas 3–10 mm SIN-1 significantly increased cell death at both 3 and 24 h after treatment (Fig. 6D). Thus, 1 mm SIN-1 elicits protein nitration without inducing caspase-3 activation or cell death.
Figure 6.

Dependence of SIN-1-mediated neuronal death on protein nitration but without caspase-3 activation. A, B, Immunoblots using anti-caspase-3 (pro- and active/cleaved forms) and anti-3-nitrotyrosine antibodies of whole-cell lysates from cultures treated with 1 μm staurosporine for the indicated times. Active caspase-3 immunoreactivity (∼17 kDa band) does not increase at any time (A; n = 2 experiments), whereas anti-NT staining increases at all time points assayed (B; n = 2 experiments). Staurosporine (1 μm) was used as a positive control in A. C, DNA fragmentation observed with TUNEL staining (green) after 1 and 3 mm SIN-1 (middle and right panels, respectively). Cells were counterstained with Hoescht (red) to visualize nuclei (n = 3 experiments). Scale bar, 100 μm. D, Cell death at 3 and 24 h resulting from SIN-1 treatment at the indicated concentrations (n = 4 experiments; asterisks, different from control at p < 0.05). Right panels, Representative PI fluorescence images at the indicated times and conditions. Scale bar, 100 μm.
SIN-1 blocks staurosporine-induced caspase-3 activation and its downstream effects but not cell death
In the context of the above, we next combined the SIN-1 and staurosporine treatments. If PN formation can inhibit caspase-mediated apoptosis, then SIN-1 treatment should inhibit staurosporine-induced caspase-3 activation. Using cultures treated with 1 μm staurosporine, we coapplied SIN-1 at 1 mm, the concentration that nitrated proteins in Figure 6B without impacting cell survival as shown in Figure 6D. Coapplication of SIN-1 to the cultures abolished staurosporine-induced caspase-3 cleavage as detected in immunoblots using cells harvested for up to 24 h after the insult (Fig. 7A).
Figure 7.

SIN-1 attenuates staurosporine-induced caspase-3 activation and apoptotic events, but not cell death. A, Caspase-3 immunoblot of whole-cell lysates taken at the indicated times after treatment with 1 μm staurosporine alone or in combination with 1 mm SIN-1. Active caspase-3 immunoreactivity (∼17 kDa band) induced by staurosporine (lanes 2–5) was abolished after the application of 1 mm SIN-1 (lanes 6–9). B, Caspase-3 activity measurements with Ac-DEVD-AMC using cell lysates taken (24 h) after the indicated insults (n = 4 experiments; asterisk, different from control at p < 0.05). C, Effect of staurosporine alone and in combination with SIN-1 on PARP-1 cleavage (n = 2 experiments; +Ve indicates positive control using camptothecin-treated Jurkat cells). D, Staining of the cultures using FITC-conjugated annexin V, a label of inverted phosphatidylserine, after treatment with staurosporine alone or in combination with SIN- 1 (n = 3 experiments). Scale bar, 50 μm. For quantification, FITC fluorescence was averaged from several images taken using identical settings, normalized to baseline controls, and expressed as a percentage increase over baseline controls. The application of 1 mm SIN-1 significantly reduced FITC staining compared with staurosporine treatment alone (p < 0.05; n = 4 experiments per condition). E, TUNEL (green) and Hoechst (red) nuclear labeling of cultures after treatment with staurosporine alone or the staurosporine/SIN-1 combination (n = 3 experiments per condition). Scale bar, 100 μm. No significant difference (see Results) is observed in cell death after the application of 1 mm SIN-1 compared with staurosporine alone (also compare images with Fig. 5C).
To test whether the suppression of caspase-3 cleavage seen in immunoblots corresponded to a reduction in caspase activity, we next examined caspase-3 activity in the fluorogenic activity assay (see Materials and Methods). Cell lysates were obtained from the cortical cultures 24 h after treatment with either SIN-1 (1 mm), staurosporine (1 μm), 1 mm SIN-1 plus 1 μm staurosporine, or HEPES control solution. The lysates were incubated with the caspase substrate Ac-DEVD-AMC (10 μm), and the fluorescence was quantified. Consistent with the results obtained by immunoblotting, only the staurosporine treatment induced a significant increase in caspase-3 activity (Fig. 7B) (p < 0.05; n = 4 assays). Caspase-3 activity was undetectable above control levels in staurosporine-treated cultures that were cotreated with SIN-1. Thus, a PN donor can inhibit caspase activity in otherwise apoptotic primary cell cultures.
To further test for inhibition of caspase-3, we evaluated whether any of its downstream effects were detectable. A direct downstream effect of activated caspase-3 is the proteolytic cleavage of the ∼116 kDa DNA repair enzyme PARP-1 into a smaller ∼85 kDa inactive fragment (Lazebnik et al., 1994). Because the response of PARP-1 to DNA damage requires ATP, PARP-1 inhibition by caspases may serve to preserve ATP to enable apoptosis (Stefanis, 2005). If PN inactivates caspase-3, then this should prevent PARP-1 cleavage. To check this, cell lysates obtained from the cortical cultures 12 h after treatment with 1 mm SIN-1 in combination with 1 μm staurosporine were assayed for PARP-1 cleavage on immunoblots. These cells exhibited the anticipated PARP-1 band at ∼116 kDA, but no band corresponding to the cleaved form of PARP-1 (∼85 kDa) was observable at any time point assayed (Fig. 7C). As controls for PARP-1 cleavage, we used both cell lysates from staurosporine-treated cortical cultures at 12 h, and from Jurkat T cells treated with camptothecin (PharMingen).
Phosphotidylserine inversion is another hallmark of apoptosis that occurs on the plasma membrane (Fadok et al., 1992). Annexin V is a protein that binds with high affinity to phosphotidylserine and, when conjugated to a visualization agent, can be used to assess apoptotic-like cellular behavior. The application of an annexin V-FITC conjugate to cell cultures treated with 1 μm staurosporine resulted in an expected increase in fluorescence (Fig. 7D). The addition of SIN-1 to staurosporine-treated cells significantly attenuated this increase in fluorescence (Fig. 7D), indicating attenuation of phosphatidylserine inversion.
Despite the inhibition of caspase-3 and its downstream effects, the cotreatment of cells with SIN-1 and staurosporine resulted in a similar extent of TUNEL staining compared with staurosporine treatment alone (Fig. 7E). Quantification of TUNEL staining was then performed by normalizing TUNEL-positive cells to the number of Hoechst-stained cells in each randomly selected field. No significant difference was observed in DNA fragmentation with SIN-1 and staurosporine-cotreated cultures compared with staurosporine treatments alone (86.5 ± 5.1% compared with 82.1 ± 1.9% TUNEL-positive cells; from an average of at least 12 random fields per condition). Thus, we conclude that cell death in the apoptotic paradigm induced by staurosporine could be shifted by PN to a caspase-independent death.
Lateral FPI causes an increase in peroxynitrite production
Our in vitro and ex vivo experiments have shown that PN is formed in our stretch injury model and, while creating DNA damage and causing neurotoxicity, it also nitrates, oxidizes, and inactivates key components of caspase-mediated apoptotic mechanisms. To determine the in vivo relevance of these findings, we next set out to examine them further in an animal model of TBI. We selected the lateral FPI model in the rat, an established and validated animal model of TBI (Dixon et al., 1987; McIntosh et al., 1989). Sprague Dawley rats were subjected to a 2.4 atm FPI (see Materials and Methods), an injury severity that, in pilot studies using our apparatus, was the highest impact injury that caused minimal (∼6%) early (postimpact) mortalities in test animals. Animals that underwent FPI experienced <30 s of apnea.
First, we determined whether FPI resulted in peroxynitrite production as had the stretch injury in the cultured neurons. Coronal histological sections were taken from paraffin-embedded brains removed from animals 24 h after FPI. The injured area could be readily visualized in hematoxylin and eosin (H+E)-stained sections when these were viewed at low-power magnification with bright-field optics (Fig. 8A) or with higher-power magnification using phase contrast optics (Fig. 8B). Coronal sections were also immunostained with anti-3-nitrotyrosine antibodies and visualized with DAB (Fig. 8C). FPI caused increased 3-nitrotyrosine immunoreactivity in a cortical region corresponding to the injured areas as observed using H+E staining (Fig. 8A, black arrows). Animals undergoing sham surgery did exhibit increased 3-nitrotyrosine immunoreactivity in the cortex (data not shown).
Figure 8.

Lateral fluid percussion TBI induces protein nitration in the rat brain. A, H+E-stained coronal section demonstrating gross histological damage (arrow) 7 d after fluid percussion injury. B, Low-power view (phase contrast) of the area of cortical injury (24 h) after lateral fluid percussion injury in the rat. The dotted line outlines the injury area. Scale bar, 500 μm. C, Anti-NT staining of same section, indicating increased NT staining (arrow) in the injured area. Immunoreactivity was visualized using DAB, and sections were counterstained using cresyl violet. A–C are representative of at least six animals per experiment. D, Representative brain sections from sham and injured animals stained with TTC to delineate the injury site (arrow) for protein harvest. E, Immunoblots using anti-3-nitrotyrosine antibodies from injured (I) and contralateral (C) cortex taken 24 h after TBI or control (sham) animals (n = 6 animals/experiment). Notably, this monoclonal antibody (4 μg/ml; BioMol) detects this ∼30 kDa band more selectively than the polyclonal antibody (Upstate Biotechnology) used in the tissue culture experiments (Fig. 2D). F, Immunoblot of caspase-3 (both pro- and active/cleaved forms) from brain lysates taken at 24 h from cortex ipsilateral (I) and contralateral (C) to FPI in four animals subjected to a 2.4 atm injury. Positive control, 20 μg recombinant human caspase-3 (Sigma).
To corroborate our histological findings, 2-mm-thick coronal sections were obtained from injured and uninjured animals. Coronal sections were then stained with the mitochondrial dye TTC. This dye is most commonly used in stroke research to delineate infarct size by staining living tissue red (Bederson et al., 1986). Here, it was used to delineate the damaged cortical area in unfixed tissue (Fig. 8D, left brain), thus enabling the harvesting of cortical tissue samples from the injury area. Samples from both hemispheres were taken from injured and uninjured shams and run on SDS-PAGE. Western blots using an anti-3-nitrotyrosine antibody with these samples revealed increased levels of nitrotyrosine in the ipsilateral, injured cortex at 32 kDa (Fig. 8E). However, immunoblots for caspase-3 (both pro- and active/cleaved forms) from the same lysates failed to reveal any increased caspase-3 activation in the injured cortex (Fig. 8F). These results suggest that cortical histological damage is associated with mitochondrial dysfunction, PN formation, and lack of caspase-3 activation at 24 h after FPI.
Peroxynitrite formation after fluid percussion injury is associated with DNA damage and neurodegeneration
To determine the cellular localization of peroxynitrite formation in vivo, we next doubly immunostained 25 μm coronal sections taken from animals subjected to FPI using antibodies against 3-nitrotyrosine and anti-NeuN (Mullen et al., 1992). There was marked colocalization of both markers, indicating protein nitration by PN in neurons (Fig. 9A, top panels). Notably, anti-3-nitrotyrosine staining was not observed in the contralateral (uninjured) cortex (Fig. 9A, bottom panels). Higher-power confocal images clearly showed that the nitration was preferentially distributed in cytoplasm of injured neurons in the ipsilateral cortex (Fig. 9A). 3-Nitrotyrosine-positive neurons also demonstrated irregular nuclear morphology when compared with neurons in the contralateral cortex, suggestive of ongoing degenerative processes (data not shown).
Figure 9.
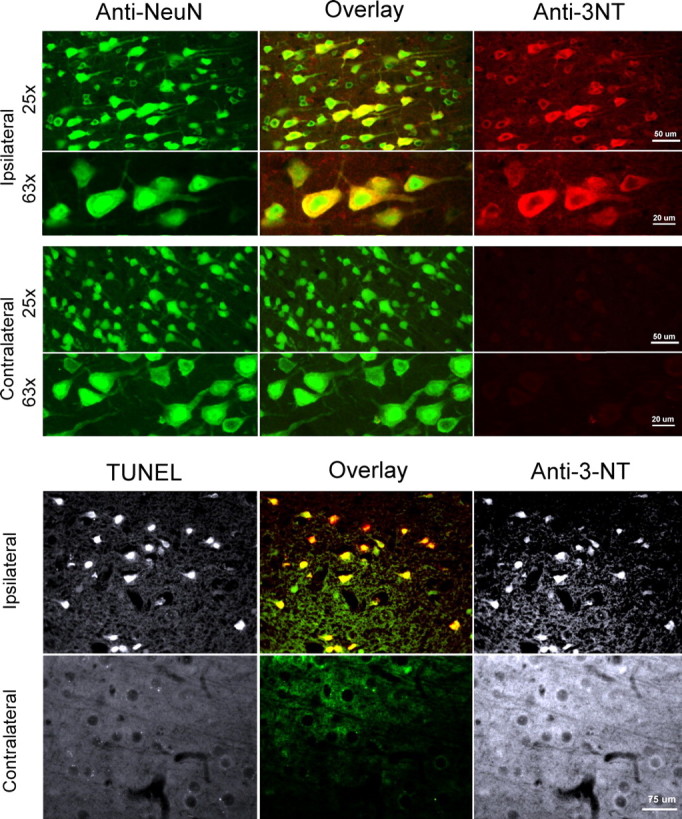
Lateral fluid percussion TBI induces TUNEL and protein nitration in neurons in vivo. Rats underwent a 2.4 atm FPI. The brains were extracted at 24 h and sectioned. A, Confocal imaging of the colocalization of Anti-3-NT staining with the neuronal marker NeuN in 25 μm frozen sections from injured (ipsilateral, top panels) cortex. Only anti-NeuN staining is observed in uninjured cortex (contralateral, bottom panels) (n = 3 experiments). At higher power (63×), the anti-3-nitrotyrosine staining is visible mainly in the cytoplasm. B, TUNEL staining colocalizes with anti-3-NT staining in injured (ipsilateral) cortex. Immunostaining was performed on 7-μm-thick paraffin-embedded coronal sections from injured cortical areas (n = 3 experiments). Uninjured (contralateral) cortex did not stain with either TUNEL or anti-3-NT (bottom panels). Yellow/orange in A indicates colocalization in the overlay panels.
We next examined whether neurons that exhibit peroxynitrite formation after in vivo TBI also exhibit DNA damage as detectable by TUNEL staining. Paraffin-embedded 7-μm-thick coronal sections were doubly immunostained with TUNEL and with anti-3-nitrotyrosine. TUNEL staining indicative of DNA fragmentation was colocalized with anti-3-nitrotyrosine-stained neurons (Fig. 9B, top panels). However, no detectable TUNEL or anti-3-nitrotyrosine staining was observed in the contralateral cortex (Fig. 9B, bottom panels). Together, these results demonstrate a neuronal pathology in rats subjected to FPI that is similar to that observed in the stretch plus 30 μm NMDA cell culture model of TBI in which peroxynitrite formation is associated with DNA degradation and neurodegeneration.
Cysteinyl oxidation inhibits caspase-3 activity after fluid percussion injury
Having shown that PN can inactivate caspases in an ex vivo system (Fig. 4) and in cultured neurons (Figs. 5–7), we next determined whether the PN formed in TBI has similar effects in vivo.
To determine the levels of caspase-3 activity after FPI, injured and uninjured cortical regions were harvested 24 h after injury. Forty micrograms of each sample were then incubated with either recombinant caspase-9 (Sigma), DTT, the caspase inhibitor z-VAD-fmk (EMD Biosciences), or DMSO as a control for 1 h at 37°C. After incubation, 40 μm Ac-DEVD-AMC was added, and the fluorescence was observed. It is expected that the application of z-VAD-fmk should produce activity measurements commensurate with minimum caspase-3 activity levels, whereas caspase-9 treatment should verify that caspase-3 pathway was able to be activated.
Analysis of samples taken from contralateral (uninjured) cortex of animals after TBI revealed that DMSO-treated samples elicited Ac-DEVD-AMC fluorescence values similar to those of samples treated with z-VAD-fmk, suggesting that baseline active caspase-3 level in uninjured cortex is low (Fig. 10A, left panel). The addition of caspase-9 to samples from uninjured cortex increased Ac-DEVD-AMC fluorescence over the ensuing hours, indicating that procaspase-3 was present and available to be activated. The addition of DTT had relatively little effect on caspase-3 activity in the uninjured cortex compared with the ipsilateral cortex, suggesting that in uninjured cortex, caspase-3 is in a relatively nonoxidized state (Fig. 10A, left panel).
Figure 10.

Caspase inhibition via cysteinyl oxidation in FPI affects caspase-3 preferentially to caspase-9. Cortical lysates were harvested at 24 h from FPI injured animals (2.4 atm) and assayed for caspase-3 and caspase-9 activity using Ac-DEVD-AMC and Ac-LEHD-AFC, respectively (n = 6 animals). A, Effects of incubation with DTT, activated caspase-9 (3 U), or z-VAD-fmk on caspase-3 activity in cortex ipsilateral (right) and contralateral (left) to the FPI. DTT-treated samples exhibited significantly higher levels of caspase-3 proteolytic activity in the ipsilateral cortex compared with the contralateral cortex. B, Effects of incubation with DTT or z-VAD-fmk on caspase-9 activity in cortex ipsilateral (right) and contralateral (left) to the FPI. No significant differences in caspase-9 activity between the ipsilateral and contralateral cortex were observed with any treatment assayed.
When samples taken from the ipsilateral (injured) cortex were processed and analyzed identically to the uninjured samples, similar results were obtained for the baseline (DMSO control) and z-VAD-fmk treatment, indicating that in the absence of exogenous, recombinant caspase-9, no significant caspase-3 activity was observable from injured cortex (Fig. 10A, right panel). Also, treatment with exogenous caspase-9 increased Ac-DEVD-AMC fluorescence to levels similar to those measured in the contralateral cortex, suggesting that procaspase-3 is also available for activation in the injured cortex. However, unlike in contralateral cortex, the addition of DTT increased the Ac-DEVD-AMC fluorescence significantly (Fig. 10A, right panel), demonstrating that a substantial amount of caspase-3 must have existed in an inhibited, oxidized state after FPI. As shown in the ex vivo studies (Fig. 4B), restoration of function using DTT suggests that an oxidative modification of cysteine residues is responsible for the inhibition of caspases after FPI.
Our ex vivo experiments suggested that caspase-9 activity was also affected by PN (Fig. 7C). To examine whether the same occurs in FPI, we examined its effects on endogenous caspase-9 activity. Samples taken from uninjured (contralateral) and injured (ipsilateral) brain cortices were incubated with DTT or with z-VAD-fmk. Unlike with caspase-3, no significant differences in endogenous caspase-9 activity were observed between injured and uninjured cortex with or without DTT. This suggests that, in vivo, PN affects caspase-3-like activity preferentially over caspase-9-like activity. This in vivo result is supported by our finding that PN preferentially nitrated procaspase-3 but not caspase-9 in the in vitro stretch model (Fig. 3A,C). It is possible that native caspases might have different susceptibilities to inactivation by PN.
Discussion
Our data, derived from ex vivo assays, in vitro neuronal stretch, and in vivo FPI, demonstrate inactivation of classical apoptosis mediated by PN mostly by targeting caspase-3 in TBI. Furthermore, this inactivation of apoptosis was shown to be primarily dependent on cysteinyl oxidation and inhibition of proteolytic function. Thus, PN acts to switch off caspase-mediated effects and apoptotic mechanisms in TBI.
Our data do not exclude a role for apoptosis in TBI, nor do they exclude a role for other mechanisms of neuronal damage. Indeed, many neurotoxic signaling mechanisms have been implicated after TBI. Caspases (Yakovlev et al., 1997; Clark et al., 1999, 2000; Beer et al., 2000, 2001; Knoblach et al., 2002), calpains (Pike et al., 1998, 2000; Buki et al., 1999; Knoblach et al., 2004), PARP-1 (LaPlaca et al., 2001; Besson et al., 2003; Hortobagyi et al., 2003), reactive nitrogen species (Mesenge et al., 1998; Hall et al., 2004), and reactive oxygen species (Marklund et al., 2001a,b) have all been suggested to play key roles in TBI. In addition, it is recognized that cross talk may exist between signaling pathways. For example, calpains can proteolytically inactivate procaspase-9 in neurons after NO application (Volbracht et al., 2005), NO can inhibit caspase-3 directly in human umbilical vein endothelial cells (Dimmeler et al., 1997), and caspase-3 can proteolytically inactivate PARP-1 (Lazebnik et al., 1994). Our present study contributes to unraveling this complexity by elucidating the interplay between caspase-mediated apoptosis and PN-mediated damage. The results here suggest that treating apoptosis alone may be insufficient, because PN-mediated damage would be unaffected. Similarly, treating PN-mediated effects using ROS scavengers or NO inhibitors may be insufficient, because as PN levels drop, proteolytic apoptotic mechanisms could be disinhibited and come into play. Thus, at a minimum, multimodality therapy in TBI should focus on anti-oxidant and anti-apoptotic strategies.
Other studies have indirectly supported the possibility that apoptosis may be oxidatively inhibited after TBI. For example, a study by Lewen et al. (2001) demonstrated that scavenging reactive oxygen species using α-phenyl-tert-N-butyl nitrone actually increased levels of detectable DNA fragmentation after TBI at 24 h. In this scenario, it is possible that the inhibition of peroxynitrite formation caused a disinhibition of caspase-3 and permitted apoptotic DNA degradation to occur. Moreover, other studies have noted cytochrome c release into the cytoplasm, and yet no protection with the caspase inhibitor z-DEVD-fmk (Sullivan et al., 2002). Together with our results, these studies support a therapeutic approach in which coinhibition of both oxidative and proteolytic mechanisms are necessary.
In vivo, it is possible that different injury mechanisms may dominate in different cell populations or brain regions. For example, Figure 8C illustrates that anti-nitrotyrosine staining, a surrogate measure of PN activity, is not evenly distributed in all brain regions or among all neurons in the injured areas (Fig. 9A,B). Thus, it is possible that, because of this heterogeneous injury pattern, PN-mediated damage may occur in some cells, whereas apoptotic mechanisms could predominate in others (Conti et al., 1998). Supporting evidence is provided by Pike et al. (1998), who demonstrated that, while evidence of caspase-3 proteolysis is absent in the cortex after TBI, significant increases in caspase-3 proteolysis was observed in the hippocampus. Moreover, our data do not rule out the possibility that, whereas PN formation occurs acutely, additional apoptotic triggers could arise in a delayed manner and initiate delayed apoptosis beyond the times studied here. Such explanations could account for previous studies in which caspase-3 activity was reported after TBI in animal studies (Yakovlev et al., 1997; Beer et al., 2000; Clark et al., 2000; Knoblach et al., 2002) and human studies (Clark et al., 1999). However, others suggest that procaspase-3 cleavage is not detectable even at 24 h after FPI (Knoblach et al., 2004). Notably, Knoblach et al. (2002) and Clark et al. (2000) noted that many TUNEL-stained neurons in TBI were not colocalized with active caspase-3, supporting the notion that caspase-3 activity, if it occurs, might arise from only a subset of a larger population of neurons in which DNA degradation occurs. In the end, all of these alternatives further support the prediction that a strategy using a single treatment will be less effective than a treatment strategy that targets both PN and caspase-dependent mechanisms. Although targeting multiple mechanisms is an intuitive approach to improving the success of TBI therapies, our work additionally provides specifics as to which mechanisms should be targeted.
Our findings show that PN formation in neurons subjected to TBI can act as a molecular switch that predisposes neurons to die via oxidative mechanisms by simultaneously inhibiting proteolytic cell death pathways. An implication of these findings is that, if caspases are being inhibited, then other proteolytic pathways of relevance to TBI may likewise be affected. Of particular interest are calpains which, like caspases, are cysteine proteases and have been implicated mediating secondary damage in TBI. Accordingly, an oxidative inhibition of calpains in hippocampi in a model of Alzheimer’s disease is reported (Marcum et al., 2005), although the extent of oxidation-mediated inhibition of calpains in TBI remains to be seen.
Although our work supports a role for PN as a key oxidant molecule in TBI, we cannot exclude contributions from other oxidative mechanisms in caspase inhibition. NO-mediated inhibition of caspases is a possibility. Initially reported by Mannick et al. (1994) in B lymphocytes, J. Li et al. (1997) later demonstrated in an ex vivo system a pan-inhibitory effect of NO on caspases -1, -2, -3, -4, -6, -7, and -8. However, PN inhibits caspases more potently compared with NO (Mohr et al., 1997), and NO produced after TBI may preferentially interact with superoxide and form PN before peptide interaction (Cherian and Robertson, 2003).
The origin of PN in TBI also remains to be determined. In the in vitro stretch model, it is likely produced in the neurons as PN formation is triggered by the NMDA application (Arundine et al., 2004). However, in whole animal models and in human cases of TBI, it has also been demonstrated that inducible nitric oxide synthase (iNOS) production of NO increases after injury (Wada et al., 1998). Recently, inhibition of neuronal NOS and endothelial NOS has been demonstrated to reduce nitrotyrosine staining (Gahm et al., 2005), although it remains to be determined whether inhibition of iNOS will yield similar results. It may be that the location of superoxide radical is the key to the localization of PN formation, because NO is more freely diffusible through plasma membranes. Superoxide forms in stretched neurons (Arundine et al., 2004) but in vivo may arise in other cell types as well.
In conclusion, our finding that PN acts to switch off caspase-mediated apoptosis reveals that there is cross talk among key neurotoxic signaling mechanisms in TBI. Unraveling these signaling pathways may be essential to establishing future rational therapeutic approaches. In TBI, both anti-oxidant and anti-apoptotic strategies may be the minimum necessary to reduce traumatic brain damage. Our findings likely have implications to other acute CNS disorders in which multiple pathways have been implicated as being causative in mediating neuronal damage.
References
- Amar AP, Levy ML. Pathogenesis and pharmacological strategies for mitigating secondary damage in acute spinal cord injury. Neurosurgery. 1999;44:1027–1039. doi: 10.1097/00006123-199905000-00052. [DOI] [PubMed] [Google Scholar]
- Arundine M, Chopra GK, Wrong A, Lei S, Aarts MM, MacDonald JF, Tymianski M. Enhanced vulnerability to NMDA toxicity in sublethal traumatic neuronal injury in vitro. J Neurotrauma. 2003;20:1377–1395. doi: 10.1089/089771503322686166. [DOI] [PubMed] [Google Scholar]
- Arundine M, Aarts M, Lau A, Tymianski M. Vulnerability of central neurons to secondary insults after in vitro mechanical stretch. J Neurosci. 2004;24:8106–8123. doi: 10.1523/JNEUROSCI.1362-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao F, Liu D. Peroxynitrite generated in the rat spinal cord induces apoptotic cell death and activates caspase-3. Neuroscience. 2003;116:59–70. doi: 10.1016/s0306-4522(02)00571-7. [DOI] [PubMed] [Google Scholar]
- Beattie MS, Farooqui AA, Bresnahan JC. Rev of current evidence for apoptosis after spinal cord injury. J Neurotrauma. 2000;17:915–925. doi: 10.1089/neu.2000.17.915. [DOI] [PubMed] [Google Scholar]
- Beckman JS, Koppenol WH. Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. Am J Physiol. 1996;271:C1424–C1437. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- Bederson JB, Pitts LH, Germano SM, Nishimura MC, Davis RL, Bartowski HM. Evaluation of 2,3,5-triphenyltetrazolium chloride as a stain for detection and quantification of experimental cerebral infarction in rats. Stroke. 1986;17:1304–1308. doi: 10.1161/01.str.17.6.1304. [DOI] [PubMed] [Google Scholar]
- Beer R, Franz G, Srinivasan A, Hayes RL, Pike BR, Newcomb JK, Zhao X, Schmutzhard E, Poewe W, Kampfl A. Temporal profile and cell subtype distribution of activated caspase-3 following experimental traumatic brain injury. J Neurochem. 2000;75:1264–1273. doi: 10.1046/j.1471-4159.2000.0751264.x. [DOI] [PubMed] [Google Scholar]
- Beer R, Franz G, Krajewski S, Pike BR, Hayes RL, Reed JC, Wang KK, Klimmer C, Schmutzhard E, Poewe W, Kampfl A. Temporal and spatial profile of caspase 8 expression and proteolysis after experimental traumatic brain injury. J Neurochem. 2001;78:862–873. doi: 10.1046/j.1471-4159.2001.00460.x. [DOI] [PubMed] [Google Scholar]
- Besson VC, Croci N, Boulu RG, Plotkine M, Marchand-Verrecchia C. Deleterious poly(ADP-ribose)polymerase-1 pathway activation in traumatic brain injury in rat. Brain Res. 2003;989:58–66. doi: 10.1016/s0006-8993(03)03362-6. [DOI] [PubMed] [Google Scholar]
- Bolanos JP, Almeida A, Stewart V, Peuchen S, Land JM, Clark JB, Heales SJ. Nitric oxide-mediated mitochondrial damage in the brain: mechanisms and implications for neurodegenerative diseases. J Neurochem. 1997;68:2227–2240. doi: 10.1046/j.1471-4159.1997.68062227.x. [DOI] [PubMed] [Google Scholar]
- Bruno VMG, Goldberg MP, Dugan LL, Giffard RG, Choi DW. Neuroprotective effect of hypothermia in cortical cultures exposed to oxygen-glucose deprivation or excitatory amino acids. J Neurochem. 1994;63:1398–1406. doi: 10.1046/j.1471-4159.1994.63041398.x. [DOI] [PubMed] [Google Scholar]
- Buki A, Siman R, Trojanowski JQ, Povlishock JT. The role of calpain-mediated spectrin proteolysis in traumatically induced axonal injury. J Neuropathol Exp Neurol. 1999;58:365–375. doi: 10.1097/00005072-199904000-00007. [DOI] [PubMed] [Google Scholar]
- Buki A, Okonkwo DO, Wang KK, Povlishock JT. Cytochrome c release and caspase activation in traumatic axonal injury. J Neurosci. 2000;20:2825–2834. doi: 10.1523/JNEUROSCI.20-08-02825.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherian L, Robertson CS. l-arginine and free radical scavengers increase cerebral blood flow and brain tissue nitric oxide concentrations after controlled cortical impact injury in rats. J Neurotrauma. 2003;20:77–85. doi: 10.1089/08977150360517209. [DOI] [PubMed] [Google Scholar]
- Clark RS, Kochanek PM, Chen M, Watkins SC, Marion DW, Chen J, Hamilton RL, Loeffert JE, Graham SH. Increases in Bcl-2 and cleavage of caspase-1 and caspase-3 in human brain after head injury. FASEB J. 1999;13:813–821. doi: 10.1096/fasebj.13.8.813. [DOI] [PubMed] [Google Scholar]
- Clark RS, Kochanek PM, Watkins SC, Chen M, Dixon CE, Seidberg NA, Melick J, Loeffert JE, Nathaniel PD, Jin KL, Graham SH. Caspase-3 mediated neuronal death after traumatic brain injury in rats. J Neurochem. 2000;74:740–753. doi: 10.1046/j.1471-4159.2000.740740.x. [DOI] [PubMed] [Google Scholar]
- Conti AC, Raghupathi R, Trojanowski JQ, McIntosh TK. Experimental brain injury induces regionally distinct apoptosis during the acute and delayed posttraumatic period. J Neurosci. 1998;18:5663–5672. doi: 10.1523/JNEUROSCI.18-15-05663.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David JC, Yamada KA, Bagwe MR, Goldberg MP. AMPA receptor activation is rapidly toxic to cortical astrocytes when desensitization is blocked. J Neurosci. 1996;16:200–209. doi: 10.1523/JNEUROSCI.16-01-00200.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson VL, Dawson TM. Deadly conversations: nuclear-mitochondrial cross-talk. J Bioenerg Biomembr. 2004;36:287–294. doi: 10.1023/B:JOBB.0000041755.22613.8d. [DOI] [PubMed] [Google Scholar]
- Dimmeler S, Haendeler J, Nehls M, Zeiher AM. Suppression of apoptosis by nitric oxide via inhibition of interleukin-1beta-converting enzyme (ICE)-like and cysteine protease protein (CPP)-32-like proteases. J Exp Med. 1997;185:601–607. doi: 10.1084/jem.185.4.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon CE, Lyeth BG, Povlishock JT, Findling RL, Hamm RJ, Marmarou A, Young HF, Hayes RL. A fluid percussion model of experimental brain injury in the rat. J Neurosurg. 1987;67:110–119. doi: 10.3171/jns.1987.67.1.0110. [DOI] [PubMed] [Google Scholar]
- Estevez AG, Radi R, Barbeito L, Shin JT, Thompson JA, Beckman JS. Peroxynitrite-induced cytotoxicity in PC12 cells: evidence for an apoptotic mechanism differentially modulated by neurotrophic factors. J Neurochem. 1995;65:1543–1550. doi: 10.1046/j.1471-4159.1995.65041543.x. [DOI] [PubMed] [Google Scholar]
- Estevez AG, Spear N, Manuel SM, Radi R, Henderson CE, Barbeito L, Beckman JS. Nitric oxide and superoxide contribute to motor neuron apoptosis induced by trophic factor deprivation. J Neurosci. 1998;18:923–931. doi: 10.1523/JNEUROSCI.18-03-00923.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faden AI. Neuroprotection and traumatic brain injury: theoretical option or realistic proposition. Curr Opin Neurol. 2002;15:707–712. doi: 10.1097/01.wco.0000044767.39452.bf. [DOI] [PubMed] [Google Scholar]
- Fadok VA, Voelker DR, Campbell PA, Cohen JJ, Bratton DL, Henson PM. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J Immunol. 1992;148:2207–2216. [PubMed] [Google Scholar]
- Fiskum G. Mitochondrial participation in ischemic and traumatic neural cell death. J Neurotrauma. 2000;17:843–855. doi: 10.1089/neu.2000.17.843. [DOI] [PubMed] [Google Scholar]
- Gahm C, Danilov A, Holmin S, Wiklund PN, Brundin L, Mathiesen T. Reduced neuronal injury after treatment with NG-nitro-L-arginine methyl ester (L-NAME) or 2-sulfo-phenyl-N-tert-butyl nitrone (S-PBN) following experimental brain contusion. Neurosurgery. 2005;57:1272–1281. doi: 10.1227/01.neu.0000187317.32529.06. [DOI] [PubMed] [Google Scholar]
- Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281:1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- Hall ED, Detloff MR, Johnson K, Kupina NC. Peroxynitrite-mediated protein nitration and lipid peroxidation in a mouse model of traumatic brain injury. J Neurotrauma. 2004;21:9–20. doi: 10.1089/089771504772695904. [DOI] [PubMed] [Google Scholar]
- Hill IE, Murray C, Richard J, Rasquinha I, MacManus JP. Despite the internucleosomal cleavage of DNA, reactive oxygen species do not produce other markers of apoptosis in cultured neurons. Exp Neurol. 2000;162:73–88. doi: 10.1006/exnr.2000.7322. [DOI] [PubMed] [Google Scholar]
- Hortobagyi T, Gorlach C, Benyo Z, Lacza Z, Hortobagyi S, Wahl M, Harkany T. Inhibition of neuronal nitric oxide synthase-mediated activation of poly(ADP-ribose) polymerase in traumatic brain injury: neuroprotection by 3-aminobenzamide. Neuroscience. 2003;121:983–990. doi: 10.1016/s0306-4522(03)00482-2. [DOI] [PubMed] [Google Scholar]
- Ikonomidou C, Turski L. Why did NMDA receptor antagonists fail clinical trials for stroke and traumatic brain injury? Lancet Neurol. 2002:383–386. doi: 10.1016/s1474-4422(02)00164-3. [DOI] [PubMed] [Google Scholar]
- Ischiropoulos H, Zhu L, Chen J, Tsai M, Martin JC, Smith CD, Beckman JS. Peroxynitrite-mediated tyrosine nitration catalyzed by superoxide dismutase. Arch Biochem Biophys. 1992;298:431–437. doi: 10.1016/0003-9861(92)90431-u. [DOI] [PubMed] [Google Scholar]
- Joshi CN, Jain SK, Murthy PS. An optimized triphenyltetrazolium chloride method for identification of cerebral infarcts. Brain Res Brain Res Protoc. 2004;13:11–17. doi: 10.1016/j.brainresprot.2003.12.001. [DOI] [PubMed] [Google Scholar]
- Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD. The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science. 1997;275:1132–1136. doi: 10.1126/science.275.5303.1132. [DOI] [PubMed] [Google Scholar]
- Knoblach SM, Nikolaeva M, Huang X, Fan L, Krajewski S, Reed JC, Faden AI. Multiple caspases are activated after traumatic brain injury: evidence for involvement in functional outcome. J Neurotrauma. 2002;19:1155–1170. doi: 10.1089/08977150260337967. [DOI] [PubMed] [Google Scholar]
- Knoblach SM, Alroy DA, Nikolaeva M, Cernak I, Stoica BA, Faden AI. Caspase inhibitor z-DEVD-fmk attenuates calpain and necrotic cell death in vitro and after traumatic brain injury. J Cereb Blood Flow Metab. 2004;24:1119–1132. doi: 10.1097/01.WCB.0000138664.17682.32. [DOI] [PubMed] [Google Scholar]
- Koh JY, Wie MB, Gwag BJ, Sensi SL, Canzoniero LM, Demaro J, Csernansky C, Choi DW. Staurosporine-induced neuronal apoptosis. Exp Neurol. 1995;135:153–159. doi: 10.1006/exnr.1995.1074. [DOI] [PubMed] [Google Scholar]
- LaPlaca MC, Zhang J, Raghupathi R, Li JH, Smith F, Bareyre FM, Snyder SH, Graham DI, McIntosh TK. Pharmacologic inhibition of poly(ADP-ribose) polymerase is neuroprotective following traumatic brain injury in rats. J Neurotrauma. 2001;18:369–376. doi: 10.1089/089771501750170912. [DOI] [PubMed] [Google Scholar]
- Lazebnik YA, Kaufmann SH, Desnoyers S, Poirier GG, Earnshaw WC. Cleavage of poly(ADP-ribose) polymerase by a proteinase with properties like ICE. Nature. 1994;371:346–347. doi: 10.1038/371346a0. [DOI] [PubMed] [Google Scholar]
- Lewen A, Skoglosa Y, Clausen F, Marklund N, Chan PH, Lindholm D, Hillered L. Paradoxical increase in neuronal DNA fragmentation after neuroprotective free radical scavenger treatment in experimental traumatic brain injury. J Cereb Blood Flow Metab. 2001;21:344–350. doi: 10.1097/00004647-200104000-00003. [DOI] [PubMed] [Google Scholar]
- Li J, Billiar TR, Talanian RV, Kim YM. Nitric oxide reversibly inhibits seven members of the caspase family via S-nitrosylation. Biochem Biophys Res Commun. 1997;240:419–424. doi: 10.1006/bbrc.1997.7672. [DOI] [PubMed] [Google Scholar]
- Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- Mannick JB, Asano K, Izumi K, Kieff E, Stamler JS. Nitric oxide produced by human B lymphocytes inhibits apoptosis and Epstein-Barr virus reactivation. Cell. 1994;79:1137–1146. doi: 10.1016/0092-8674(94)90005-1. [DOI] [PubMed] [Google Scholar]
- Marcum JL, Mathenia JK, Chan R, Guttmann RP. Oxidation of thiol-proteases in the hippocampus of Alzheimer’s disease. Biochem Biophys Res Commun. 2005;334:342–348. doi: 10.1016/j.bbrc.2005.06.089. [DOI] [PubMed] [Google Scholar]
- Marklund N, Clausen F, McIntosh TK, Hillered L. Free radical scavenger posttreatment improves functional and morphological outcome after fluid percussion injury in the rat. J Neurotrauma. 2001a;18:821–832. doi: 10.1089/089771501316919184. [DOI] [PubMed] [Google Scholar]
- Marklund N, Clausen F, Lewander T, Hillered L. Monitoring of reactive oxygen species production after traumatic brain injury in rats with microdialysis and the 4-hydroxybenzoic acid trapping method. J Neurotrauma. 2001b;18:1217–1227. doi: 10.1089/089771501317095250. [DOI] [PubMed] [Google Scholar]
- Marshall LF. Epidemiology and cost of central nervous system injury. Clin Neurosurg. 2000;46:105–112. [PubMed] [Google Scholar]
- McIntosh TK, Vink R, Noble L, Yamakami I, Fernyak S, Soares H, Faden AL. Traumatic brain injury in the rat: characterization of a lateral fluid-percussion model. Neuroscience. 1989;28:233–244. doi: 10.1016/0306-4522(89)90247-9. [DOI] [PubMed] [Google Scholar]
- McKee JA, Brewer RP, Macy GE, Borel CO, Reynolds JD, Warner DS. Magnesium neuroprotection is limited in humans with acute brain injury. Neurocrit Care. 2005;2:342–351. doi: 10.1385/NCC:2:3:342. [DOI] [PubMed] [Google Scholar]
- Mesenge C, Charriaut-Marlangue C, Verrecchia C, Allix M, Boulu RR, Plotkine M. Reduction of tyrosine nitration after N(omega)-nitro-l-arginine-methylester treatment of mice with traumatic brain injury. Eur J Pharmacol. 1998;353:53–57. doi: 10.1016/s0014-2999(98)00432-4. [DOI] [PubMed] [Google Scholar]
- Mohr S, Zech B, Lapetina EG, Brune B. Inhibition of caspase-3 by S-nitrosation and oxidation caused by nitric oxide. Biochem Biophys Res Commun. 1997;238:387–391. doi: 10.1006/bbrc.1997.7304. [DOI] [PubMed] [Google Scholar]
- Mullen RJ, Buck CR, Smith AM. NeuN, a neuronal specific nuclear protein in vertebrates. Development. 1992;116:201–211. doi: 10.1242/dev.116.1.201. [DOI] [PubMed] [Google Scholar]
- Pike BR, Zhao X, Newcomb JK, Posmantur RM, Wang KK, Hayes RL. Regional calpain and caspase-3 proteolysis of alpha-spectrin after traumatic brain injury. NeuroReport. 1998;9:2437–2442. doi: 10.1097/00001756-199808030-00002. [DOI] [PubMed] [Google Scholar]
- Pike BR, Zhao X, Newcomb JK, Glenn CC, Anderson DK, Hayes RL. Stretch injury causes calpain and caspase-3 activation and necrotic and apoptotic cell death in septo-hippocampal cell cultures. J Neurotrauma. 2000;17:283–298. doi: 10.1089/neu.2000.17.283. [DOI] [PubMed] [Google Scholar]
- Radi R, Beckman JS, Bush KM, Freeman BA. Peroxynitrite oxidation of sulfhydryls. The cytotoxic potential of superoxide and nitric oxide. J Biol Chem. 1991;266:4244–4250. [PubMed] [Google Scholar]
- Raghupathi R, Graham DI, McIntosh TK. Apoptosis after traumatic brain injury. J Neurotrauma. 2000;17:927–938. doi: 10.1089/neu.2000.17.927. [DOI] [PubMed] [Google Scholar]
- Sattler R, Charlton MP, Hafner M, Tymianski M. Determination of the time-course and extent of neurotoxicity at defined temperatures in cultured neurons using a modified multi-well plate fluorescence scanner. J Cereb Blood Flow Metab. 1997;17:455–463. doi: 10.1097/00004647-199704000-00011. [DOI] [PubMed] [Google Scholar]
- Sattler R, Charlton MP, Hafner M, Tymianski M. Distinct influx pathways, not calcium load, determine neuronal vulnerability to calcium neurotoxicity. J Neurochem. 1998;71:2349–2364. doi: 10.1046/j.1471-4159.1998.71062349.x. [DOI] [PubMed] [Google Scholar]
- Sosin DM, Sniezek JE, Thurman DJ. Incidence of mild and moderate brain injury in the United States, 1991. Brain Inj. 1996;10:47–54. doi: 10.1080/026990596124719. [DOI] [PubMed] [Google Scholar]
- Spear N, Estevez AG, Johnson GV, Bredesen DE, Thompson JA, Beckman JS. Enhancement of peroxynitrite-induced apoptosis in PC12 cells by fibroblast growth factor-1 and nerve growth factor requires p21Ras activation and is suppressed by Bcl-2. Arch Biochem Biophys. 1998;356:41–45. doi: 10.1006/abbi.1998.0741. [DOI] [PubMed] [Google Scholar]
- Stefanis L. Caspase-dependent and -independent neuronal death: two distinct pathways to neuronal injury. Neuroscientist. 2005;11:50–62. doi: 10.1177/1073858404271087. [DOI] [PubMed] [Google Scholar]
- Sullivan PG, Keller JN, Bussen WL, Scheff SW. Cytochrome c release and caspase activation after traumatic brain injury. Brain Res. 2002;949:88–96. doi: 10.1016/s0006-8993(02)02968-2. [DOI] [PubMed] [Google Scholar]
- Sun HS, Feng ZP, Miki T, Seino S, French RJ. Enhanced neuronal damage after ischemic insults in mice lacking Kir6.2-containing ATP-sensitive K+ channels. J Neurophysiol. 2006;95:2590–2601. doi: 10.1152/jn.00970.2005. [DOI] [PubMed] [Google Scholar]
- Szabo C, Ohshima H. DNA damage induced by peroxynitrite: subsequent biological effects. Nitric Oxide. 1997;1:373–385. doi: 10.1006/niox.1997.0143. [DOI] [PubMed] [Google Scholar]
- Teasdale GM, Graham DI. Craniocerebral trauma: protection and retrieval of the neuronal population after injury. Neurosurgery. 1998;43:723–737. doi: 10.1097/00006123-199810000-00001. [DOI] [PubMed] [Google Scholar]
- Thurman DJ, Alverson C, Dunn KA, Guerrero J, Sniezek JE. Traumatic brain injury in the United States: a public health perspective. J Head Trauma Rehabil. 1999;14:602–615. doi: 10.1097/00001199-199912000-00009. [DOI] [PubMed] [Google Scholar]
- Tolias CM, Bullock MR. Critical appraisal of neuroprotection trials in head injury: what have we learned? NeuroRx. 2004;1:71–79. doi: 10.1602/neurorx.1.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trackey JL, Uliasz TF, Hewett SJ. SIN-1-induced cytotoxicity in mixed cortical cell culture: peroxynitrite-dependent and -independent induction of excitotoxic cell death. J Neurochem. 2001;79:445–455. doi: 10.1046/j.1471-4159.2001.00584.x. [DOI] [PubMed] [Google Scholar]
- Ueta E, Kamatani T, Yamamoto T, Osaki T. Tyrosine-nitration of caspase 3 and cytochrome c does not suppress apoptosis induction in squamous cell carcinoma cells. Int J Cancer. 2003;103:717–722. doi: 10.1002/ijc.10832. [DOI] [PubMed] [Google Scholar]
- Virag L, Marmer DJ, Szabo C. Crucial role of apopain in the peroxynitrite-induced apoptotic DNA fragmentation. Free Radic Biol Med. 1998;25:1075–1082. doi: 10.1016/s0891-5849(98)00139-7. [DOI] [PubMed] [Google Scholar]
- Volbracht C, Chua BT, Ng CP, Bahr BA, Hong W, Li P. The critical role of calpain versus caspase activation in excitotoxic injury induced by nitric oxide. J Neurochem. 2005;93:1280–1292. doi: 10.1111/j.1471-4159.2005.03122.x. [DOI] [PubMed] [Google Scholar]
- Wada K, Chatzipanteli K, Kraydieh S, Busto R, Dietrich WD. Inducible nitric oxide synthase expression after traumatic brain injury and neuroprotection with aminoguanidine treatment in rats. Neurosurgery. 1998;43:1427–1436. doi: 10.1097/00006123-199812000-00096. [DOI] [PubMed] [Google Scholar]
- Yakovlev AG, Knoblach SM, Fan L, Fox GB, Goodnight R, Faden AI. Activation of CPP32-like caspases contributes to neuronal apoptosis and neurological dysfunction after traumatic brain injury. J Neurosci. 1997;17:7415–7424. doi: 10.1523/JNEUROSCI.17-19-07415.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye YZ, Strong M, Huang ZQ, Beckman JS. Antibodies that recognize nitrotyrosine. Methods Enzymol. 1996;269:201–209. doi: 10.1016/s0076-6879(96)69022-3. [DOI] [PubMed] [Google Scholar]
- Zipfel GJ, Babcock DJ, Lee JM, Choi DW. Neuronal apoptosis after CNS injury: the roles of glutamate and calcium. J Neurotrauma. 2000;17:857–869. doi: 10.1089/neu.2000.17.857. [DOI] [PubMed] [Google Scholar]
- Zou H, Li Y, Liu X, Wang X. An APAF-1.cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9. J Biol Chem. 1999;274:11549–11556. doi: 10.1074/jbc.274.17.11549. [DOI] [PubMed] [Google Scholar]