

Abstract
Protein kinase A (PKA) modulates several steps of synaptic transmission. However, the identification of the mediators of these effects is as yet incomplete. Synapsins are synaptic vesicle (SV)-associated phosphoproteins that represent the major presynaptic targets of PKA. We show that, in hippocampal neurons, cAMP-dependent pathways affect SV exocytosis and that this effect is primarily brought about through synapsin I phosphorylation. Phosphorylation by PKA, by promoting dissociation of synapsin I from SVs, enhances the rate of SV exocytosis on stimulation. This effect becomes relevant when neurons are challenged with sustained stimulation, because it appears to counteract synaptic depression and accelerate recovery from depression by fostering the supply of SVs from the reserve pool to the readily releasable pool. In contrast, synapsin phosphorylation appears to be dispensable for the effects of cAMP on the frequency and amplitude of spontaneous synaptic currents and on the amplitude of evoked synaptic currents. The modulation of depolarization-evoked SV exocytosis by PKA phosphorylation of synapsin I is primarily caused by calmodulin (CaM)-dependent activation of cAMP pathways rather than by direct activation of CaM kinases. These data define a hierarchical crosstalk between cAMP- and CaM-dependent cascades and point to synapsin as a major effector of PKA in the modulation of activity-dependent SV exocytosis.
Keywords: knock-out mice, lentiviruses, neurotransmitter release, phosphorylation, synapse, trafficking
Introduction
cAMP-dependent signaling cascades modulate several aspects of neuronal physiology, including synaptic transmission. Most of the effects of cAMP are mediated by protein kinase A (PKA). PKA-dependent protein phosphorylation at both presynaptic and postsynaptic sites modulates synaptic activity and is crucially involved in certain forms of plasticity and long-term memory (Nguyen and Woo, 2003). The pleiotrophic effects of cAMP on synapse physiology and its crosstalk and convergence with Ca2+-dependent pathways (Wang and Storm, 2003) have hampered the identification of the possible targets mediating the control of presynaptic functions by PKA. Experimental evidence supports the idea that the cAMP/PKA pathway regulates synaptic activity by modulating the exocytic machinery at the presynaptic terminal (Evans and Morgan, 2003). The PKA-dependent facilitation of synaptic transmission occurs without altering either the number of synapses or the number of docked vesicles (Trudeau et al., 1996). Indeed, PKA was shown to participate in the recruitment of synaptic vesicles (SVs) from the reserve pool to the readily releasable pool of vesicles, a presynaptic mechanism that controls synaptic efficacy and plasticity (Kuromi and Kidokoro, 2000).
The SV-associated phosphoproteins of the synapsin family, which act at the crossroad between cAMP and Ca2+-dependent cascades, play important roles in synaptic transmission (Hilfiker et al., 1999) and are thus optimal candidates to translate changes in intracellular cAMP levels into modulation of SV recycling. Interestingly, although synapsins were discovered because of their prominent phosphorylation by PKA (Johnson et al., 1972; Huttner and Greengard, 1979), the functional relevance of synapsin phosphorylation by PKA still awaits to be completely clarified.
Three synapsin genes have been identified. Transcripts of each of these genes are differentially spliced to give rise to the various isoforms (Sudhof et al., 1989). A single phosphorylation site (site 1) for PKA and Ca2+/calmodulin dependent kinase (CaMK) I/IV is highly conserved among the various isoforms and across vertebrate and invertebrate species. However, it is currently unknown which kinase is predominantly involved in site 1 phosphorylation in vivo. Synapsin I is phosphorylated also by CaMKII at two additional sites (sites 2 and 3). In addition, all synapsin isoforms identified contain multiple consensus sites for the mitogen-activated protein (MAP) kinases extracellular signal-regulated kinase 1/2 and cyclin-dependent kinase 1/5 (sites 4–6). Synapsins have been proposed to maintain a reserve pool of SVs in the proximity of the active zone by tethering them to each other and to the presynaptic actin cytoskeleton, to control the availability of SVs through their ability to dissociate from the SV membrane and actin in a phosphorylation-dependent manner and to play a role in the postdocking steps of exocytosis (Hilfiker et al., 1999; Chi et al., 2001).
In this study, we investigated the functional role of cAMP-dependent pathways in the control of SV recycling. We show that PKA plays a major role in tuning the size of the pool of recycling SVs in response to depolarization and that this effect is strictly dependent on PKA-mediated phosphorylation of synapsin I at site 1.
Materials and Methods
Materials.
The anti-synapsin I/II (clone 19.11) mouse monoclonal antibody, phospho-site 3-specific (RU19) and phospho-site 1-specific (G257) anti-synapsin I polyclonal antibodies, and anti-active CaMKII polyclonal antibodies were prepared and characterized at the Rockefeller University (New York, NY) (Menegon et al., 2000, 2002; Bonanomi et al., 2005). The polyclonal antibody against synaptophysin I (SypI) has been described previously (Valtorta et al., 1988). The monoclonal anti-vesicle-associated membrane protein 2 (VAMP2) antibody was from Synaptic Systems (Göttingen, Germany). The following antibodies were purchased from Jackson ImmunoResearch (West Grove, PA): FITC-conjugated goat anti-mouse, tetramethylrhodamine isothiocyanate-conjugated goat anti-rabbit, and 7-amino-4methylcoumarin-3-acetic acid-conjugated goat anti-mouse. Forskolin, H89 (N-[2-(p-bromo-cinnamylamino)-ethyl]-5-isoquinoline-sulfon-amide 2HCl), KN93 (2-[N-(2-hydroxyethyl)]-N-(4-methoxybenzenesulfonyl)amino-N-(4-chlorocinnamyl)-N-methylbenzylamine), and W7 [N-(6-aminohexyl)-5-chloro-1-naphthalenesulfonamide] were purchased from Calbiochem (La Jolla, CA); CNQX, bicuculline, d-AP-5, CGP55845 [(2S)-3-[[(1S)-1-(3,4-dichlorophenyl)ethyl]amino-2-hydroxypropyl](phenylmethyl)phosphinic acid], and QX-314 [2(triethylamino)-N-(2,6-dimethylphenyl) acetamine] were from Tocris Cookson (Ellisville, MO); FM4-64 [N-(3-triethylammoniumpropyl)-4-(6-(4-diethylamino)phenyl)hexatrienyl)pyridinium dibromide] and N6,2′-O-dibutyryl-cAMP (BT-cAMP) were from Sigma (St. Louis, MO); and tetrodotoxin (TTX) was from Latoxan (Valence, France).
All chemicals were diluted in Krebs'–Ringer's–HEPES (KRH) (in mm: 130 NaCl, 5 KCl, 1.2 MgSO4, 1.2 KH2PO4, 2 CaCl2, 6 glucose, and 25 HEPES/Na, pH 7.4) at the following final concentrations: H89, 5–10 μm, as indicated; W7, 50 μm; KN93, 4 μm (i.e., 5-fold the Ki for CaMKI/II and 1.2-fold the Ki for CaMKIV); forskolin, 50 μm; TTX, 1 μm; CNQX, 10 μm; and BT-cAMP 20 mm.
For the preparation of plasmids and viruses, see Bonanomi et al. (2005).
Cell culture and transduction of neurons.
Primary neuronal cultures were prepared from the hippocampi of Sprague Dawley embryonic day 18 (E18) rat embryos (Charles River, Calco, Italy) or E17 synapsin I knock-out mice (obtained from the laboratory of Paul Greengard, The Rockefeller University) as described previously (Banker and Cowan, 1977). After being cultured for 8–9 d in vitro (DIV), neurons were placed in a clean dish containing glia-conditioned medium [MEM supplemented with 1% N2 supplement (Invitrogen, Carlsbad, CA), 2 mm glutamine (BioWhittaker, Walkersville, MD), 0.1% ovalbumin, 1 mm sodium pyruvate (Sigma), and 4 mm glucose] and incubated for 10–15 h at 37°C in a 5% CO2-humidified atmosphere in the presence of viral supernatant at 0.1–1 multiplicities of infection. After transduction, neurons were returned to the original dishes and maintained in culture until 12–14 DIV in glia-conditioned medium.
Transfections were performed with Effectene (Qiagen, Hilden, Germany) according to the instructions of the manufacturer using 0.4 μg of DNA per Petri dish. Neurons at 10–12 DIV were placed in a clean dish, incubated for 2 h with glia-conditioned medium containing Effectene and DNA, returned to the original dishes, and analyzed at 12–14 DIV.
Electrophysiological experiments were performed on hippocampal neurons prepared from synapsin I mutant mice, as described previously (Baldelli et al., 2005). The isolated hippocampal neurons were plated at low density (100 cells/mm2) and maintained in a culture medium consisting of Neurobasal, B-27 (1:50), 1% glutamine, and 1% penicillin—streptomycin. Reagents were from Invitrogen. Electrophysiological recordings were performed on 10–14 DIV neurons.
Cell labeling protocols.
For standard double-immunofluorescence analysis, cells were rapidly rinsed with KRH/EGTA and fixed for 30 min with 4% paraformaldehyde, 4% sucrose in 120 mm sodium phosphate buffer, pH 7.4, supplemented with 2 mm EGTA, and processed as described previously (Menegon et al., 2002). For detection of synapsin phosphorylation at site 3, incubation with the cyanine 3-conjugated phosphosite-3-specific anti-synapsin I antibody (RU19) (Menegon et al., 2002) was performed in the presence of 10 μg/ml rabbit IgG and was preceded by a 20 min blocking reaction in the same solution.
For FM4-64 uptake experiments, 12–14 DIV hippocampal neurons were stimulated for 1 min (or 30 s, when indicated) with 55 mm KCl, in the presence of FM4-64 (10 μm) and TTX. After FM4-64 loading, neurons were washed by two complete medium substitutions, followed by perfusion for 10 min with warmed KRH (37°C) supplemented with TTX and CNQX.
For FM4-64 release experiments, 10 min after washing, the neurons were exposed to a second pulse of 55 mm KCl in the absence of the dye.
Video microscopy and image analysis.
Specimens were viewed through a PlanApo 63×/1.4 oil immersion objective at 37°C in KRH with a Zeiss (Oberkochen, Germany) Axiovert 135 TV inverted microscope equipped with epifluorescence optics. Images were recorded with a C4742-98 ORCA II cooled charge-coupled device camera (Hamamatsu, Hamamatsu City, Japan) and processed using Image Pro Plus 4.5 (Media Cybernetics, Silver Spring, MD) and Adobe Photoshop 6.0 (Adobe System, San Jose, CA).
To selectively analyze FM4-64 uptake at synaptic boutons, neurons were infected with lentiviruses to express GFP variant-tagged SV proteins. As a minimum, we analyzed three independent coverslips of cultured neurons and acquired 10–15 fields of view per condition in each experiment. All of the synapses that were present in randomly selected subfields were analyzed. Multiple fluorescence images of the same field of view were stacked as a red–green–blue or multiple channel image type. Synapses expressing fluorescent SV proteins [enhanced yellow fluorescent protein (EYFP)-tagged synaptophysin or enhanced cyan fluorescent protein (ECFP)-tagged synapsins] were selected with an adequate color range, and a reference mask was generated. The mask was subsequently used to analyze the FM4-64 fluorescence image of the stack. FM4-64 fluorescence was measured on digital images within an area of 3 × 3 pixels at the center of synapses expressing SypI–EYFP, ECFP–SynI, or ECFP–SynI S9A as indicated. A similar routine was performed to measure the levels of immunoreactivity for synaptophysin shown in supplemental Figure 1 (available at www.jneurosci.org as supplemental material).
In the experiments in which FM4-64 release was measured, the kinetics of FM4-64 destaining elicited from a second depolarizing pulse was evaluated by acquiring a stack of images at 2 s intervals and analyzing the progressive decrease in fluorescence at single synaptic boutons.
Electrophysiology.
Patch-clamp recordings were performed in whole-cell configuration (Vh of −70 mV) using an EPC-10 amplifier (HEKA Elektronik, Lambrecht, Germany). Cultured neurons were perfused with Tyrode's solution containing the following (in mm): 2 CaCl2, 150 NaCl, 1 MgCl2, 10 HEPES, 4 KCl, and 10 glucose, pH 7.4. Bicuculline (20 μm), d-AP-5 (50 μm), and CGP58845 (5 μm) (Tocris Cookson, Bristol, UK) was included in the bathing solution to isolate evoked EPSCs (eEPSCs). The patch pipette (3–4 MΩ) solution contained the following (in mm): 90 CsCl, 20 tetraethylammonium-Cl, 10 EGTA, 10 glucose, 1 MgCl2, 4 ATP, 0.5 GTP, and 15 phosphocreatine, pH 7.4. QX-314 (10 mm) was added to block Na+ currents activated during eEPSCs. Presynaptic stimuli were delivered through a glass pipette of 1 μm tip diameter filled with Tyrode's solution placed in contact with the soma of the glutamatergic neuron in a loose-seal configuration. Current pulses of 0.1/0.3 ms and variable amplitude (5–25 μA) delivered by an insulated pulse stimulator (model 2100; A-M Systems, Carlsborg, WA) were required to induce monosynaptic eEPSCs with short latency (1–2.5 ms). To ensure that only synaptic contacts of the selected presynaptic neuron were stimulated by the extracellular stimulating pipette, we included in the analysis only those eEPSCs that were completely lost after a few micrometer displacement from the soma of the presynaptic neuron and responded in an all-or-none manner to stimuli of graded intensity. The stimulation intensity was set at 1.5-fold the threshold for all experiments. Recordings with leak currents >100 pA or series resistance >20 MΩ were discarded. Data acquisition was performed using PATCHMASTER programs (HEKA Elektronik). All of the experiments were performed at room temperature (23–25°C).
Statistical analysis.
Statistical analysis was performed using ANOVA. Two-way ANOVA was used to assess between-experiment variability, which in all cases accounted for <1.25% of the total variance. Post hoc Bonferroni's or Duncan's test was then performed to analyze the effects of the various treatments. Correlation analysis was performed using Pearson's test. The 95% confidence interval and two-tailed analysis were used as test parameters. Statistical analysis was performed using the program Prism from GraphPad Software (San Diego, CA).
Results
PKA modulates SV recycling evoked by depolarization
To accurately monitor SV exo-endocytosis at single synapses in living neurons, we generated the fluorescent synaptic marker SypI–EYFP by tagging the integral SV protein SypI with EYFP. Neurons were infected with lentiviruses engineered to express SypI–EYFP under the control of a constitutive cellular promoter, the phosphoglycerate kinase (PGK) promoter (Bonanomi et al., 2005). The use of a lentiviral-based expression system allowed high-infection efficiency in postmitotic cells, whereas expression under the PGK promoter ensured expression of the protein at quasi-physiological level. The fluorescent styryl dye FM4-64 (Betz et al., 1996) was loaded into infected neurons using high K+ depolarization, in a well established protocol that induces multiple rounds of exo-endocytosis, leading to labeling of the entire pool of recycling SVs (Klingauf et al., 1998; Pyle et al., 2000; Sara et al., 2002), and FM4-64 uptake was measured exclusively at synapses, identified by the presence of SypI–EYFP. An additional depolarizing stimulus released virtually all the dye from SypI–EYFP-expressing synapses, confirming the correct targeting of the chimera at functional synaptic boutons and the loading of FM4-64 in the releasable pool of vesicles (Fig. 1A).
Figure 1.
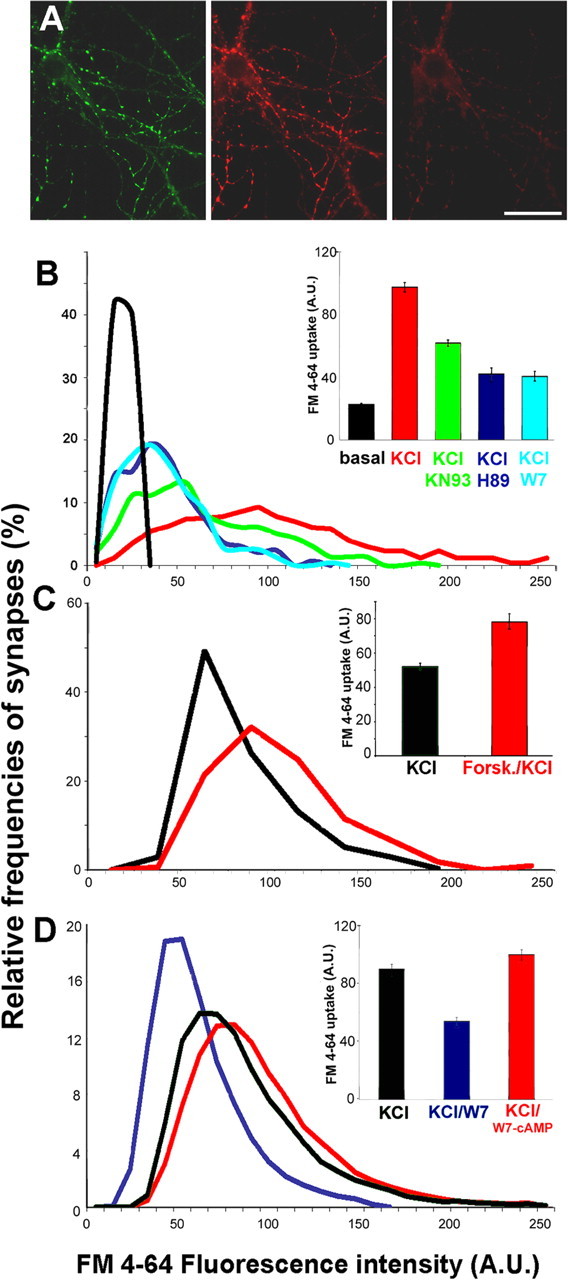
PKA modulates SV recycling. A, Rat hippocampal neurons expressing SypI–EYFP (left) were loaded with FM4-64 by exposure for 1 min to KRH–55 mm KCl (depolarizing solution) (middle). Neurons were subsequently exposed to a second depolarizing stimulus to unload the dye (right). Scale bar, 20 μm. B–D, Quantitative analysis of FM4-64 uptake in SypI–EYFP-positive synapses. The intensity of FM4-64 uptake was measured at single synaptic boutons. The distributions of classes of FM4-64 fluorescence are shown as smoothened histograms, and the mean ± SE fluorescence values (n = 3 independent experiments) expressed in arbitrary units (A.U.) are shown in the inset with the corresponding color coding. B, Neurons were incubated for 30 min in KRH in the presence or absence of H89, KN93, or W7 and were then loaded with FM4-64 during a 1 min incubation in depolarizing solution; a 1 min incubation in KRH was used as control (black). p < 0.001 for KCl versus KCl/W7, KCl/H89, or KCl/KN93, Bonferroni's multiple comparison test (n = 250 synapses from 3 independent experiments). C, Quantitative analysis of FM4-64 uptake at SypI-EYFP-positive synapses elicited by a depolarizing stimulus applied for 30 s after a 10 min incubation in KRH in either the absence (black) or presence (red) of forskolin. p < 0.001, Bonferroni's multiple comparison test (n = 250 synapses from 3 independent experiments). D, Quantitative analysis of FM4-64 uptake at SypI–EYFP-positive synapses elicited by a depolarizing stimulus applied for 1 min after a 30 min incubation in the absence (black) or presence of either W7 (blue) or W7/dibutyryl-cAMP (red). p < 0.001, Bonferroni's multiple comparison test (n = 350 synapses from 3 independent experiments).
Quantitation of FM4-64 uptake induced by exposure to 55 mm KCl for 1 min was performed by measuring fluorescence intensity at single synaptic boutons. Fluorescence intensity values displayed a broad distribution, indicating variability among individual synapses. However, in >95% of the synapses analyzed, depolarization induced an increase in FM4-64 labeling with respect to synapses belonging to resting neurons (Fig. 1B). FM4-64 uptake levels were not influenced by expression of SypI–EYFP, indicating that the chimera did not impair synaptic activity (data not shown). Incubation of the neurons with either the PKA inhibitor H89 or the CaM antagonist W7, which inhibits all CaM-dependent enzymes, greatly reduced depolarization-induced SV recycling. Selective inhibition of CaMKs with KN93 (Hook and Means, 2001) was less effective in reducing FM4-64 uptake compared with H89 and W7. The decrease in SV recycling induced by PKA inhibition was reversible (Fig. 2), because a depolarizing pulse delivered 30 min after removal of H89 induced FM4-64 uptake at levels corresponding to 88 ± 4% of those observed before exposure to the drug. The increase in FM4-64 uptake after recovery from PKA inhibition was not attributable to synaptic potentiation, because it was not observed when two 30-min-spaced depolarizing stimuli were applied in the absence of the drug (data not shown).
Figure 2.

The effects of PKA inhibition on SV recycling are reversible. A, B, Rat hippocampal neurons were incubated for 30 min with H89 and subsequently loaded with FM4-64 by exposure to 55 mm KCl for 1 min (left column). After a 30 min incubation in the absence of H89 to recover from PKA inhibition and bleaching of the residual fluorescence, a second pulse of FM4-64 uptake was induced with the same depolarizing protocol (right column). Note the variability among individual synapses in the SV recycling activity reached on recovery from PKA inhibition (arrowheads in B). Scale bar: A, 15 μm; B, 5.5 μm.
To reveal whether SV recycling could be modulated by PKA activation, we treated neurons with the adenylate cyclase activator forskolin. Because in our model system 55 mm KCl for 1 min produced a maximal FM4-64 uptake (data not shown), the depolarizing stimulus was applied for 30 s to achieve a submaximal stimulation. Under these conditions, forskolin treatment fostered SV recycling induced by depolarization (Fig. 1C), indicating that PKA-dependent pathways bidirectionally modulate SV recycling at mature synapses. Importantly, the strong reduction in FM4-64 uptake observed under inhibition of CaM-dependent activities with W7 was effectively overcome by the application of the cell-permeant cAMP analog BT-cAMP (Fig. 1D). Thus, elevation of intracellular cAMP levels, presumably leading to PKA activation, appears to be necessary and sufficient to sustain SV recycling at normal rates.
PKA-mediated synapsin phosphorylation occurs in response to depolarization and leads to synapsin dissociation from SVs
To characterize the PKA-dependent phosphorylation of synapsin at mature synapses, we exploited a phospho-specific antibody directed against synapsin phosphorylated on the PKA/CaMKI site (Ser 9, site 1) (Fig. 3) (see Fig. 5). At rest, a basal level of synapsin phosphorylation on site 1 could be detected in a small population of terminals (Fig. 3A). In neurons treated with TTX, immunolabeling with the phosphospecific anti-site 1 antibody was barely detectable in virtually all terminals, suggesting that the variability observed in untreated samples is linked to the spontaneous activity of the neuronal network (Fig. 3B). Interestingly, in the same cultures, TTX did not affect the phosphorylation of synapsin in axonal growth cones (Fig. 3, compare A′, B′). Inhibition of PKA by treatment with H89 virtually abolished and elevation of intracellular cAMP by treatment with either forskolin or BT-cAMP greatly increased synapsin phosphorylation on site 1 (Fig. 3C,D and data not shown). The basal level of synapsin phosphorylation at site 1 was virtually abolished on inhibition of CaM-dependent enzymes with W7 and effectively restored by the concomitant application of BT-cAMP. After cAMP increase, synapsin phosphorylation at site 1 was no longer restricted to a small percentage of terminals but was instead associated with virtually all synapses (Fig. 4A,B).
Figure 3.

PKA activation induces synapsin phosphorylation and diffusion along the axon. A–E, Hippocampal neurons were processed for double immunofluorescence with a phosphosite-1-specific anti-synapsin antibody (red) and an antibody that recognizes all synapsin isoforms (green). F, Neurons were stained with an anti-VAMP2 antibody (red) and a phosphosite-1-specific anti-synapsin antibody (green). Neurons were fixed in unstimulated conditions (A, A′) or after incubation with TTX (B, B′), H89 (C), forskolin (D, F), or forskolin plus KN93 (E). Under basal conditions, synapsin phosphorylation at site 1 can be detected in a subpopulation of synaptic boutons (arrowheads in A) as well as in growth cones (A′). Treatment of the neurons with TTX strongly reduces phosphorylation in nerve terminals (B) but not in growth cones (B′). Inhibition of PKA abolishes (C) whereas inhibition of CaM kinases only modestly decreases (E) synapsin phosphorylation. PKA activation induces site 1 phosphorylation and synapsin dispersion out of the terminals (D, F), whereas VAMP2 immunoreactivity remains confined to the synapses (F). G, Time-lapse experiment showing the dispersion of ECFP–SynI out of the synapses induced by forskolin (Forsk) and the recovery (Rec.) after removal of the drug. Scale bar: A, B, 10 μm; A′, B′, 15 μm; C–F, 20 μm; G, 5 μm.
Figure 5.
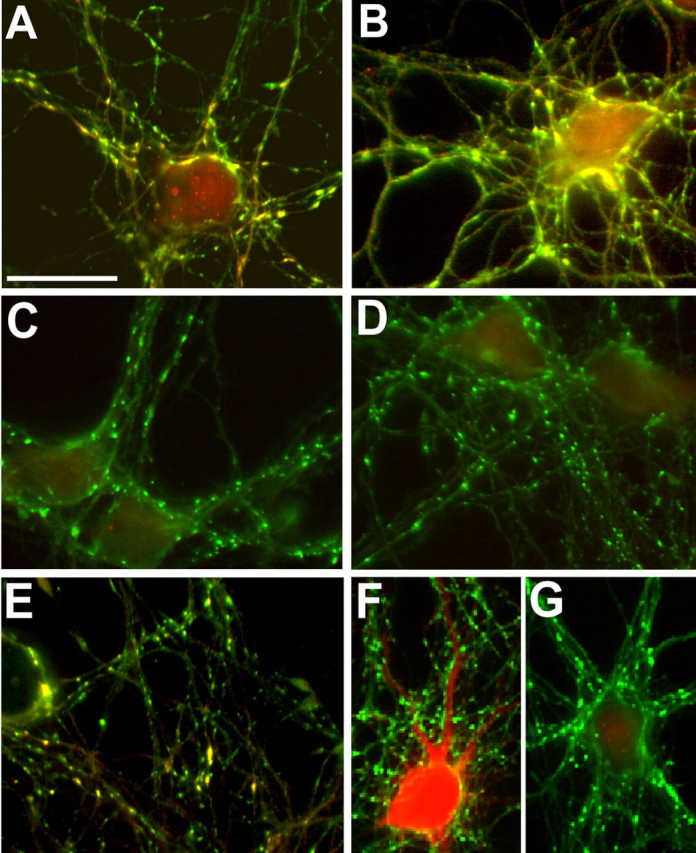
Depolarization-induced phosphorylation of synapsin at site 1 requires PKA activation. A–G, After incubation in the absence (A, B, F) or presence of H89 (C), W7 (D), or KN93 (E, G), rat hippocampal neurons were exposed to 55 mm KCl for 1 min, except for the sample shown in B, in which the depolarizing stimulus was applied for 2 min. Neurons were fixed and processed for double immunofluorescence with an antibody that recognizes all synapsin isoforms (A–G, green) and either a phosphosite-1-specific anti-synapsin antibody (A–E, red) or an antibody against autophosphorylated CaMKII (F, G, red). Note that depolarization-induced phosphorylation of synapsin at site 1 is inhibited by both H89 and W7 but not by KN93, although KN93 prevents CaMKII autophosphorylation. Scale bar, 15 μm.
Figure 4.
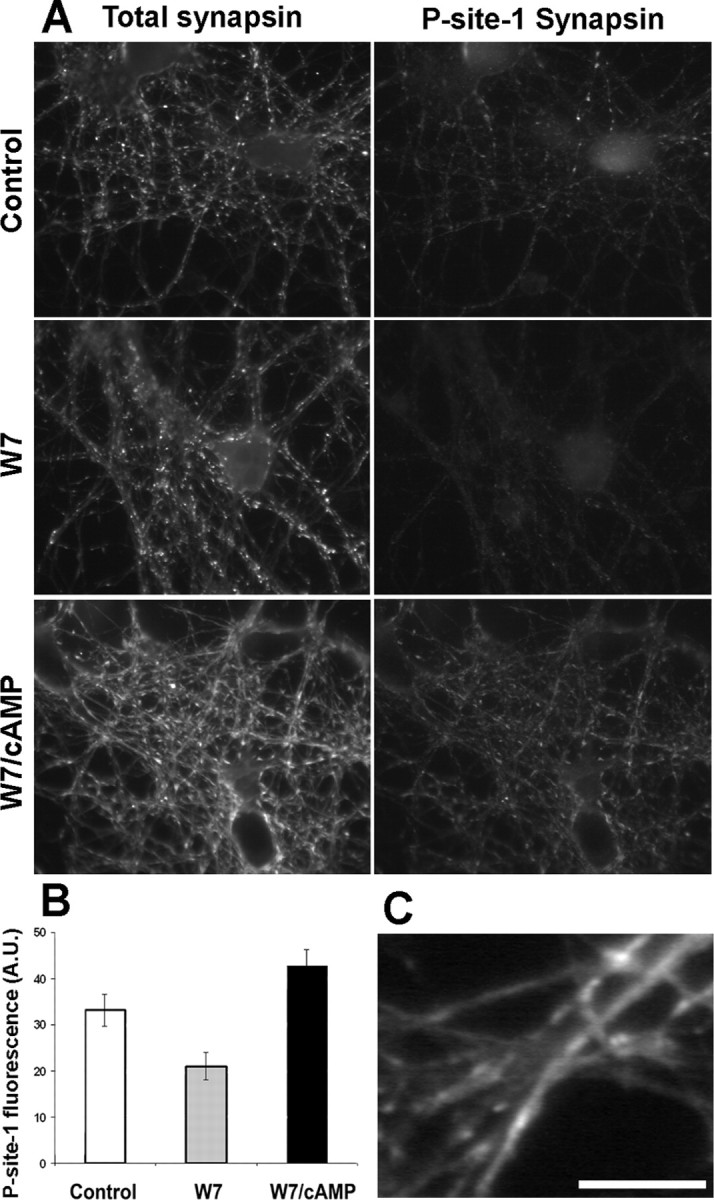
Phosphorylation of synapsin at site 1 and synapsin dispersion occur independently of CaM-dependent enzymatic activities. A, Rat hippocampal neurons were fixed either at rest (control) or after a 30 min treatment with W7 in the presence or absence of dibutyryl-cAMP. Neurons were processed for double immunofluorescence with an antibody that recognizes all synapsin isoforms (left column) and a phosphosite-1-specific anti-synapsin antibody (right column). B, Histograms of the mean ± SE fluorescence intensities of site1-phosphorylated synapsin of the samples shown in A. A.U., Arbitrary units. C, High-magnification image of a sample treated with W7 and cAMP and processed as described in A. Note the diffusion of synapsin along the axons. Scale bar: A, 20 μm; C, 10 μm.
Remarkably, cAMP-dependent phosphorylation of synapsin at site 1 induced dispersion of the protein along the axons but left SVs concentrated at synapses (Figs. 3D,F, 4C). In vivo imaging of a fusion protein of ECFP and wild-type synapsin Ia (ECFP–SynI) expressed by lentiviral vectors under the control of the PGK promoter (Bonanomi et al., 2005) (supplemental Fig. 1A,B, available at www.jneurosci.org as supplemental material) showed that dispersion of the chimera out of the terminals was complete within 10 min of treatment with forskolin, and its localization at the synapse was restored 10 min after removal of the drug (Fig. 3G). CaMK inhibition by KN93 did not noticeably prevent the effects of forskolin on either site 1 phosphorylation or synapsin dispersion (Fig. 3E).
Synapsin phosphorylation on site 1 was also elicited by applying a depolarizing stimulus. Synapsin dispersion along the axons was barely detectable after a 1 min depolarization (Fig. 5A) and greatly increased when depolarization was applied for 2 min (Fig. 5B). Interestingly, both depolarization-induced synapsin phosphorylation on site 1 and dispersion were prevented by either H89 (Fig. 5C) or W7 (Fig. 5D). At the concentrations used in these experiments, H89 did not inhibit CaM kinases (data not shown). Thus, these data indicate that an activation of the PKA pathway is implicated in the effects of depolarization on synapsin I. In contrast, exposure to the CaMK inhibitor KN93 did not completely prevent site 1 phosphorylation induced by depolarization (Fig. 5E), although it effectively inhibited CaMKII autophosphorylation (Fig. 5F,G).
Synapsin phosphorylation and SV recycling are closely correlated
To evaluate the existence of a relationship between site 1 phosphorylation and SV exo-endocytosis, we correlated the extent of depolarization-induced FM4-64 uptake with the state of site 1 phosphorylation, retrospectively determined using phosphorylation state-specific antibodies.
In the absence of a secretagogue stimulus, most synapses remained essentially inactive in terms of SV recycling, although a small proportion of them showed an appreciable level of site 1 phosphorylation (Fig. 6A,A′). After a depolarizing stimulus, we found a higher level of phosphorylation in those terminals in which FM4-64 uptake had been more intense (Fig. 6B,B′). Quantitative analysis confirmed the existence of a significant correlation between the ratio of site 1-phosphorylated to total synapsin at a given terminal and the ability of that terminal to sustain SV recycling (Fig. 6C, top). In contrast, the intensity of SV recycling did not correlate with the absolute content of either site 1-phosphorylated (data not shown) or total synapsin (Fig. 6C, bottom).
Figure 6.
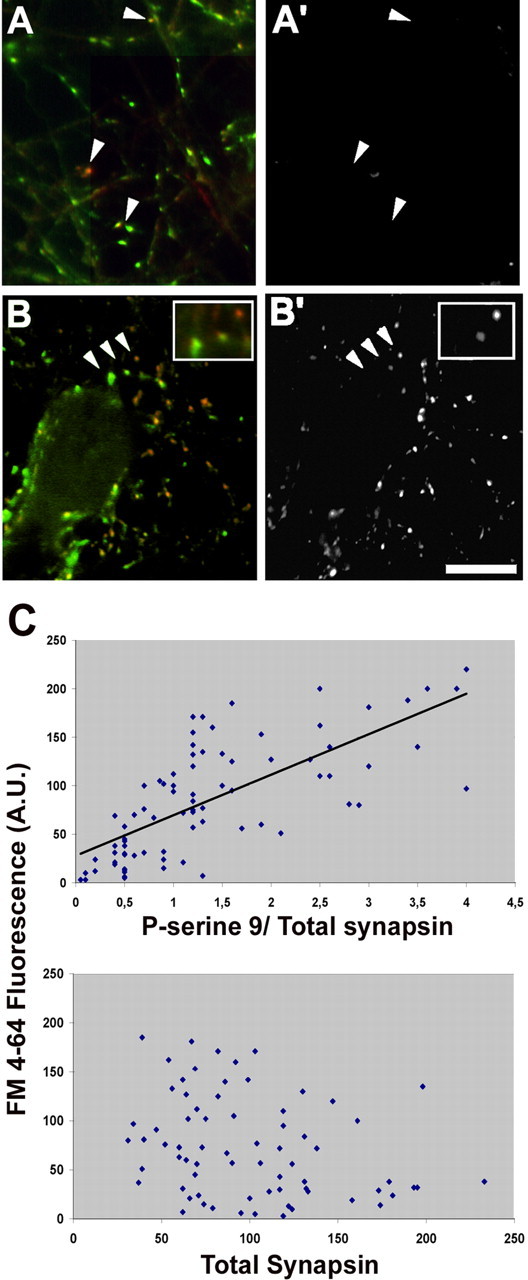
Depolarization-induced SV recycling correlates with the intensity of synapsin phosphorylation at site 1. A–B′, Rat hippocampal neurons were loaded for 1 min with FM4-64 (A′, B′) in either KRH (A′) or depolarizing solution (B′), fixed, and processed for retrospective double immunofluorescence (A, B) with an antibody that recognizes all synapsin isoforms (green) and with a phosphosite-1-specific anti-synapsin antibody (red). Arrowheads in A and A′ point to unstimulated synapses showing a high level of site-1 phosphorylation not accompanied by detectable FM4-64 internalization. Arrowheads in B and B′ point to stimulated synapses (shown at higher magnification in the insets) in which the intensity of staining for site 1-phosphorylated synapsin correlates with the intensity of FM4-64 labeling. Scale bar, 10 μm. C, Quantitative correlation between apparent stoichiometry of synapsin phosphorylation at site 1 (top) (r = 0.71; p < 0.001, Pearson's test) or total synapsin immunoreactivity (bottom) (r = −0.28; p > 0.1) and the level of FM4-64 uptake induced by depolarization. Each point corresponds to one synapse. The intensity of the labeling for phosphorylated synapsin is normalized for the total synapsin immunoreactivity in that terminal. A.U., Arbitrary units.
Synapsin phosphorylation at site 1 accounts for PKA-dependent modulation of SV exocytosis
To investigate whether synapsin phosphorylation at site 1 is essential for the physiological regulation of SV exo-endocytosis that occurs during synaptic activity, neurons were infected with lentiviruses engineered to express either wild-type synapsin or a synapsin mutant lacking phosphorylation site 1 fused to ECFP (ECFP–SynI S9A). Expression was driven by the cytomegalovirus (CMV) promoter, which allows the production of the large amount of exogenous protein needed to interfere with endogenous synapsins. Semiquantitative immunocytochemistry confirmed that both chimeras were produced in similar amounts. The synapsin S9A chimera maintained the specific targeting to nerve terminals and could be phosphorylated on the CaMKII sites. However, in contrast to wild-type synapsin, after forskolin treatment, Syn S9A was not phosphorylated and retained a synaptic distribution (supplemental Fig. 1A,B, available at www.jneurosci.org as supplemental material).
A marked reduction in the intensity of FM4-64 loading induced by depolarization was observed in neurons expressing ECFP–SynI S9A. In contrast, the extent of FM4-64 uptake in synapses expressing ECFP–SynI was similar to that observed in untreated neurons, in agreement with previous results (Chi et al., 2001) (compare Figs. 7A, 1B; and data not shown).
Figure 7.

PKA-mediated synapsin phosphorylation at site 1 modulates SV recycling. A, Rat hippocampal neurons transfected to express either ECFP–SynI or ECFP–SynI S9A under the control of the CMV promoter were loaded with FM4-64 by exposure to 55 mm KCl for 1 min. The distribution of classes of FM4-64 fluorescence in ECFP fluorescent synapses is shown in the histograms with the corresponding color coding. p < 0.001 for ECFP–SynI versus ECFP–SynIS9A, Bonferroni's multiple comparison test (n = 300 synapses from 3 independent experiments). Mean ± SE values of FM4-64 uptake (n = 3 independent experiments) are shown in the insets. A.U., Arbitrary units. B, Rat hippocampal neurons transfected to express either ECFP–SynI or ECFP–SynI S9A under the CMV promoter were loaded with FM4-64 by exposure to 55 mm KCl for 1 min after a 30 min incubation with H89. A second epoch of FM4-64 uptake was induced by applying the same depolarizing stimulus 30 min after the washing out of H89 and bleaching of the residual fluorescence. FM4-64 was visualized exclusively in the synaptic boutons of the transfected neurons, selected using a digital masking system. The increase in FM4-64 uptake after removal of the PKA inhibition is prominent in synapses expressing ECFP–SynI (arrowheads), whereas it is partially inhibited in synapses expressing ECFP–SynI S9A (arrows). Scale bar, 4 μm. C, Quantitative analysis of FM4-64 uptake. p < 0.001 for KCl/H89 versus KCl for both chimeras and for ECFP–Syn I KCl versus ECFP–Syn I S9A KCl, Bonferroni's multiple comparison test (n = 200 synapses from 3 independent experiments).
To evaluate whether synapsin I phosphorylation at site 1 is necessary and sufficient for the PKA-induced modulation of SV recycling, depolarization-induced FM4-64 loading was measured in synapses expressing either ECFP–SynI or ECFP–SynI S9A in the presence or absence of modulators of PKA activity. Remarkably, when PKA was inhibited by H89, SV recycling was low, with no significant differences between synapses expressing ECFP–SynI and synapses expressing ECFP–SynI S9A. Only after removal of the PKA inhibition, both SynI- and SynI S9A-expressing synapses reset to their standard recycling rates, revealing a significant difference between the two populations of terminals (Fig. 7B,C). Indeed, the distribution of classes of FM4-64 uptake induced by depolarization was clearly left shifted in synapses expressing the mutant form of the synapsin I chimera (Fig. 7A).
Activation of PKA by forskolin enhanced depolarization-evoked SV recycling (Fig. 1). To determine whether this effect required synapsin phosphorylation at site 1, we measured FM4-64 uptake induced by a 30 s depolarization either before or after incubation with forskolin in synapses expressing either ECFP–SynI or ECFP–SynI S9A (Fig. 8). After forskolin treatment, the intensity of FM4-64 uptake increased in both kinds of synapses. However, in synapses expressing ECFP–SynI S9A, the increase in FM4-64 internalization was limited compared with that observed in synapses expressing ECFP–synapsin I. Thus, the ability of PKA to modulate the efficiency of SV recycling mostly depends on synapsin phosphorylation at site 1.
Figure 8.

Phosphorylation of synapsin at site 1 mostly accounts for the increase in SV recycling induced by PKA activation. Rat hippocampal neurons infected to express either ECFP–SynI or ECFP–SynI S9A under the CMV promoter were loaded with FM4-64 by exposure to 55 mm KCl for 30 s. A second epoch of FM4-64 uptake was induced by applying the same depolarizing stimulus after 20 min of recovery, followed by a 10 min incubation with forskolin (Forsk.) and bleaching of the residual fluorescence. The distribution of classes of FM4-64 fluorescence are shown (light gray, KCl; black, forskolin/KCl). p < 0.001 for neurons infected with vector before and after forskolin and for neurons expressing ECFP–SynI versus neurons expressing ECFP–SynI S9A both before and after forskolin; Bonferroni's multiple comparison test (n = 500 synapses from 3 independent experiments). Mean ± SE values of FM4-64 uptake (n = 3 independent experiments) are shown in the bottom. A.U., Arbitrary units.
To establish whether the effects produced by the expression of exogenous synapsin on SV recycling were confounded by the presence of endogenous synapsin I, we studied depolarization-induced FM4-64 uptake in neurons from synapsin I knock-out mice expressing either ECFP–SynI or ECFP–SynI S9A under the control of the PGK promoter. In synapses expressing SynI S9A, SV recycling was significantly reduced compared with synapses expressing the wild-type protein (Fig. 9A). In addition, the reintroduction of ECFP–SynI in neurons derived from synapsin I knock-out mice led to a much higher potentiation of SV recycling after recovery from PKA inhibition compared with either uninfected or ECFP–SynI S9A-expressing synapses (Fig. 9B).
Figure 9.
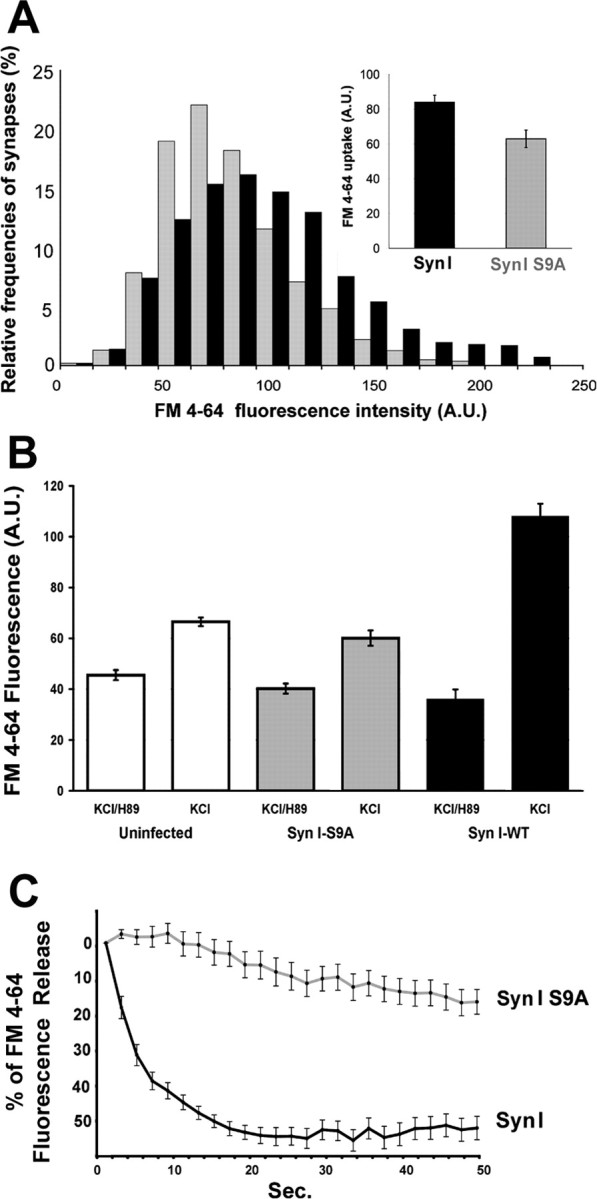
Expression of SynI S9A impairs SV exocytosis in synapsin I knock-out neurons. Hippocampal neurons in culture from synapsin I knock-out mice infected to express either ECFP–SynI or ECFP–SynI S9A under the PGK promoter were loaded with FM4-64 using a depolarizing stimulus for 1 min. A, Distribution of classes of FM4-64 fluorescence are shown in the histograms with the corresponding color coding. p < 0.001 for ECFP–SynI versus ECFP–SynI S9A, Bonferroni's multiple comparison test (n = 300 synapses from 3 independent experiments). Mean ± SE values of FM4-64 uptake (n = 3 independent experiments) are shown in the inset. B, Before the depolarizing pulse, neurons were incubated for 30 min with 10 μm H89. A second epoch of FM4-64 uptake was induced by applying the same depolarizing stimulus 30 min after the washing out of H89 and bleaching of the residual fluorescence. The increase in FM4-64 uptake after removal of the PKA inhibition is prominent in synapses expressing ECFP–SynI, whereas it is partially inhibited in synapses either uninfected or expressing ECFP–SynI S9A. p < 0.001 for neurons treated with H89 (either uninfected or infected with either vector) versus the same neurons after recovery from PKA inhibition and for neurons infected with ECFP–SynI versus either uninfected or ECFP–SynI S9A-infected neurons, after recovery from PKA inhibition; Bonferroni's multiple comparison test (n = 300 synapses from 2 independent experiments). C, The kinetics of FM4-64 destaining elicited from a second depolarizing pulse was evaluated by acquiring a stack of images at 2 s intervals and analyzing the progressive decrease in fluorescence at single synaptic boutons. Values have been corrected for bleaching and normalized. The average ± SE fluorescence intensities of 32 synaptic boutons per sample from three independent experiments are shown. A.U., Arbitrary units.
Synapsin is known to modulate multiple steps in the exocytotic limb of the SV recycling process, although it has not been reported to affect endocytosis (for review, see Hilfiker et al., 1999). Thus, all of the effects on FM4-64 uptake described in this paper are likely to reflect modifications in SV exocytosis. However, to confirm that synapsin phosphorylation selectively affects exocytosis, synapses from synapsin I knock-out neurons expressing either SynI or SynI S9A were loaded with FM4-64 during a 1 min depolarization and the kinetics of dye unloading induced by a second depolarizing stimulus were analyzed. Given the short interval elapsed between the two depolarizing pulses, both samples released only a fraction of the loaded dye. However, a noticeable difference in the kinetics of FM4-64 release was detected between knocked out neurons infected with viruses expressing wild-type and those infected with viruses expressing mutant synapsin I, indicating that site 1 phosphorylation of synapsin I impinges on SV exocytosis (Fig. 9C).
Because synapsins have been described to promote synaptic maturation in a PKA-dependent manner (Fiumara et al., 2004) and mediate SV clustering at the synaptic terminal (Hilfiker et al., 1999), we ascertained that, under our conditions, the density of synaptic contacts and/or the number of SVs present in the terminals (indirectly evaluated by measuring the levels of synaptophysin I immunoreactivity) were unchanged by expression of exogenous synapsin I in synapsin I knock-out neurons (supplemental Fig. 1C, available at www.jneurosci.org as supplemental material).
Mutation of phosphorylation site 1 in synapsin affects the kinetics of synaptic depression and recovery in response to sustained high-frequency stimulation and impairs the effect of forskolin
To better evaluate the functional role of PKA-mediated phosphorylation of synapsin, we performed electrophysiological recordings on hippocampal neurons from synapsin I knock-out mice infected with lentiviruses engineered to express either wild-type ECFP–Syn I or ECFP–SynI S9A.
No significant difference was detected in the frequency or amplitude of miniature EPSCs (mEPSCs) between the two types of neurons, in either the absence or presence of forskolin. However, in both cases, forskolin increased the mean frequency of mEPSCs to a similar extent (supplemental Fig. 2, available at www.jneurosci.org as supplemental material), consistent with the previously reported effect of forskolin on release probability (Nguyen and Woo, 2003). Similarly, the amplitude of eEPSCs after single stimuli was similar for the two groups of neurons and was equally increased by forskolin (Fig. 10A,B).
Figure 10.

Mutation of phosphorylation site 1 in synapsin affects the kinetics of synaptic depression and recovery in response to sustained high-frequency stimulation and impairs the response to activation or inhibition of the PKA pathway. Recordings were performed on hippocampal neurons (9–15 DIV) from synapsin I knock-out mice expressing either ECFP–SynI or ECFP–SynI S9A, in the absence (control conditions) or presence of either forskolin (50 μm) or H89 (5 μm). Black bars, ECFP–SynI under control conditions; dark gray bars, ECFP–SynI S9A under control conditions; light gray bars, ECFP–SynI after forskolin; white bars, ECFP–SynI S9A after forskolin; dotted bars, ECFP–SynI after H89; dashed bars, ECFP–SynI S9A after H89. A, Representative traces of eEPSC amplitude versus time recorded during repeated presynaptic stimulation at 0.1 Hz in neurons expressing either wild-type or mutated synapsin I. Addition of 50 μm forskolin causes a clear increase in current amplitude in both neurons. Traces of single eEPSCs taken at 50 and 350 s are shown in the inset. B, Mean ± SE eEPSC amplitude of samples treated as in A (9 < n <13 cells for the various experimental groups). C, Plot of the 15 first eEPSCs evoked by a stimulation train at 10 Hz. The postsynaptic response is expressed as percentage of the first eEPSC of the train. A representative postsynaptic response evoked by the train is shown in the inset. Depression is increased by forskolin, but no significant differences are detected between the two groups of neurons (9 < n <11 cells for the various experimental groups). D, Plot of the normalized mean ± SE eEPSC amplitude (14 < n < 16 cells for the various experimental groups) recorded before, during, and after a 1 min pulse of stimulation at 16 Hz. The basal frequency of stimulation was 0.1 Hz. A clear difference in the rate of recovery from depression can be detected between neurons expressing wild-type or mutated synapsin I. In E, the time course of depression of the samples shown in D is shown on an expanded scale and fitted with a double-exponential decay function. For clarity, only every 15th point is shown. F, Mean ± SE values of eEPSC amplitude at steady state (Iss) and of the time constants of the fast (τ1) and slow (τ2) decay components of the samples in E. The slow component is strongly accelerated in neurons expressing synapsin S9A and accounts for 23 ± 0.5% of the total decay (vs 11 ± 0.2% in neurons expressing wild-type synapsin I). G, H, Effects of forskolin and H89 on the time courses of recovery from depression induced by the protocol described in D. Recovery curves were fitted according to a monoexponential function. I, The mean ± SE values (7 < n < 16 cells for the various experimental groups) of the time constants of recovery under basal conditions are shown in the left. In the right, the percentage ± SE changes in the time constants observed after treatment with forskolin or H89 with respect to the control values are shown. Forskolin markedly accelerates and H89 markedly slows down recovery in neurons expressing wild-type synapsin I. Both treatments had smaller effects in neurons expressing the mutated protein. Statistical analysis was performed by using two-way ANOVA, followed by the Duncan's multiple comparison test (*p < 0.05 and **p < 0.01 forskolin or H89 vs respective control; Δp < 0.05 and ΔΔp < 0.01 forskolin vs H89; °°p < 0.01 SynI S9A vs SynI).
Because the synapsins are known to affect trafficking of SVs between the reserve and the releasable pools, we explored the effect of the S9A mutation and of forskolin treatment on the response to trains of stimuli. When short trains of stimuli were applied, no significant difference in the kinetics of synaptic depression could be detected between the two groups of neurons. Consistent with its effects on release probability, forskolin increased depression, particularly at early time points, to a similar extent in wild-type and S9A synapsin-expressing neurons (Fig. 10C).
In contrast, after the application of long trains (1 min at 16 Hz) of stimuli, ECFP–SynI S9A-expressing neurons exhibited a more intense depression and an impaired recovery (Fig. 10D). A detailed analysis of the kinetics of depression revealed that the absence of phosphorylation site 1 markedly decreased the steady-state level of depression and mainly affected the slower decay component of eEPSC amplitude, whose time constant became considerably shorter, with a concomitant increase (from 11 ± 0.2% in ECFP–SynI neurons to 23 ± 0.5% in ECFP–SynI S9A neurons) in the magnitude of its contribution to depression (Fig. 10E,F). With this protocol, recovery from depression in ECFP–SynI S9A neurons was markedly slower than that observed in ECFP–SynI neurons (Fig. 10D,G–I). Acute treatments affecting the PKA pathway markedly affected the recovery kinetics in neurons expressing ECFP–SynI, with a 40% decrease and an 80% increase in the time constants of recovery after forskolin and H89 treatments, respectively. When the same treatments were applied to ECFP–SynI S9A-expressing neurons, the effects were significantly smaller than those observed in neurons expressing the wild-type protein (Fig. 10G–I).
Discussion
The synapsins are a family of SV-associated phosphoproteins that have been shown to play important roles in synapse development and neurotransmitter release (Valtorta et al., 1992) (for review, see Hilfiker et al., 1999). The conservation of the PKA/CaMKI site in all synapsin isoforms and throughout evolution (Kao et al., 1999) is suggestive of an essential role for this site in the regulation of synapsin function. Indeed, recent evidence supports a role of site 1 phosphorylation, mediated by PKA, in axonal elongation as well as in synapse development (Kao et al., 2002; Fiumara et al., 2004; Bonanomi et al., 2005). However, thus far, the possibility that PKA-mediated synapsin phosphorylation plays a role in mature nerve terminals has been essentially overlooked.
In this study, we show that, in hippocampal neurons, PKA potentiates activity-dependent SV exocytosis and that site 1 phosphorylation of synapsin I mostly accounts for this modulatory effect.
The extent of SV exo-endocytosis induced by depolarization with high K+, as measured by FM4-64 loading of the recycling pool of vesicles, was decreased by treatment with the PKA inhibitor H89 and increased by the adenylyl cyclase activator forskolin, indicating that, in resting terminals, a basal level of PKA activity is present and that increase in this activity leads to enhancement of neurotransmitter release elicited by Ca2+ influx. Consistently, basal phosphorylation of synapsin I at site 1, which could be detected in a minority of untreated nerve terminals, was virtually abolished after treatment with H89 and was strongly increased by forskolin. The massive increase in synapsin phosphorylation induced by forskolin was accompanied by diffusion of the protein into the axonal shaft, an effect that was readily reversible on removal of the drug. Because the localization of SV integral membrane proteins at nerve terminals was not affected, these observations are consistent with the idea that the phosphorylation-dependent detachment of synapsin I from SVs observed in vitro (Schiebler et al., 1986; Benfenati et al., 1989; Hosaka and Sudhof, 1998) also occurs in intact neurons.
It has been reported previously that, in neurons from synapsin I knock-out mice, the recycling pool of SVs is reduced (Ryan et al., 1996). We observed that, in these neurons, reintroduction of site 1-mutated synapsin I reduces the normal secretory response to depolarization with respect to wild-type synapsin I, in the absence of variations in the total numbers of SVs per terminal. The levels of depolarization-induced SV exocytosis were directly related to the apparent stoichiometry of site 1-synapsin phosphorylation.
The existence of a causal relationship between synapsin I phosphorylation and enhancement of the neurosecretory response is supported by several evidences. In synapses expressing mutated synapsin I, the size of the recycling SV pool and the rate of SV exocytosis induced by depolarization were reduced with respect to those observed in synapses expressing the wild-type form of the protein. Expression of nonphosphorylatable synapsin I strongly reduced the enhancement of depolarization-evoked SV exocytosis detected after treatment with forskolin or after recovery from PKA inhibition, suggesting that synapsin I phosphorylation accounts for most of the effects of PKA activity on neurosecretion.
The functional importance of synapsin I phosphorylation on site 1 is also reflected by the more intense depression and much slower recovery rate observed after long trains of action potentials in neurons expressing synapsin S9A. These observations suggest a role for site 1 phosphorylation in the supply of SVs from the reserve pool, thus affecting both the steady-state level of activity and the reconstitution of the readily releasable pool after intense stimulation. This hypothesis is further strengthened by the observation that acute perturbations of the PKA pathway with either forskolin or H89 markedly affected the rate of recovery from depression in neurons expressing wild-type synapsin I and had a much smaller effect in those expressing the mutated protein. Together, the effects of the S9A mutation on FM4-64 uptake and recovery from depression indicate that phosphorylation of synapsin site 1 is the major, albeit not the exclusive, mechanism by which PKA modulates recovery from depression at the presynaptic level. Our data point to a major role of synapsin phosphorylation in the PKA-dependent regulation of SV exocytosis and are in agreement with the current models envisioning synapsin as a negative regulator of SV turnover: synapsin I, in its dephosphorylated state, links SVs to the nerve terminal cytoskeleton. Activity-dependent synapsin I phosphorylation causes its dissociation from SVs and increases the availability of vesicles for release (Valtorta et al., 1992; Chi et al., 2001).
Previous reports considered site 1 as a site phosphorylated in response to [Ca2+]i increases, similarly to sites 2 and 3, which are phosphorylated by CaMKII (Chi et al., 2001). Indeed, site 1 is a conserved site for PKA and CaMKI/IV. Our data indicate that in vivo synapsin I phosphorylation at site 1 is primarily mediated by PKA. In fact, depolarization-stimulated phosphorylation of site 1 was abolished by H89 but was essentially insensitive to CaMK inhibition by KN93, although depolarization strongly enhances CaMKI activation (Matsushita and Nairn, 1999). In addition, elevation of intracellular cAMP levels was sufficient to reverse the inhibitory effect of W7, which affects all CaM-dependent activities. Most important, although in untreated neurons depolarization-induced SV exocytosis was impaired by expression of synapsin S9A, under conditions of PKA inhibition the FM4-64 uptake did not differ significantly between synapses expressing wild-type synapsin I and synapses expressing the synapsin mutant.
Under all conditions tested, the rate of recovery from depression in S9A synapsin I-expressing neurons was always slower than that measured in neurons expressing wild-type synapsin I. Although the effects of an acute perturbation of PKA phosphorylation cannot be fully compared with those of a long-term expression of a mutated protein, this observation may suggest the possibility that S9A synapsin I, expressed in synapsin I knock-out neurons, inhibits SV recycling independently of its inability to be phosphorylated. Although this possibility cannot be excluded, several considerations indicate that the effects of the point mutation are specific.
Site 1 mutation leaves intact the targeting of synapsin I to nerve terminals and its ability to associate with SVs and to cause their aggregation (Bonanomi et al., 2005), although it selectively prevents the PKA-induced dissociation of synapsin I from SVs. The differential ability of nerve terminals expressing wild-type or mutant synapsin I to sustain SV recycling is concealed by PKA inhibition, supporting the specific dependency of the effect of the S9A mutation on PKA activity. Moreover, the effects of both forskolin and H89 on recovery from depression become markedly weaker in nerve terminals expressing mutant synapsin I, although other physiological parameters are fully preserved.
The intense effects of PKA inhibition on SV recycling may be explained by the involvement of PKA in both the direct regulation of components of the exocytic machinery and the indirect control of Ca2+-dependent pathways known to play a relevant role in synaptic transmission. However, the present data indicate that, despite its pleiotrophic effects on synaptic physiology, the PKA-dependent control of SV recycling is underlain to a considerable extent by phosphorylation of synapsin I at site 1.
It is noteworthy that inhibition of all CaM-dependent pathways by W7 hampered site 1 phosphorylation on depolarization. This was most likely attributable to blockade of the CaM-dependent adenylate cyclase (type 1), which is expressed in hippocampal neurons (Wong et al., 1999). The effective reversal of the W7 block on SV recycling after elevation of cAMP levels indicates that site 1 phosphorylation is sensitive to intracellular Ca2+ levels, but this regulation appears to predominantly depend on PKA rather than CaMKI activation.
Work from other laboratories indicates that, in the presynaptic compartment, PKA substrates other than synapsin are present and may thus contribute to the effects of cAMP on the regulation of SV exocytosis (Hell et al., 1995; Surmeier et al., 1995; Hirling and Scheller, 1996; Graham and Burgoyne, 2000; Lonart et al., 2003; Nagy et al., 2004; Thakur et al., 2004; Baba et al., 2005). Effects of cAMP variations on the activity of these proteins might explain the limited, but statistically significant, increase in SV exocytosis observed in our experiments on recovery from PKA inhibition in synapses expressing synapsin S9A, in either wild-type or synapsin I-deficient nerve terminals.
The data presented in this study add complexity to the current model of synapsin regulation and function at the synapse. Indeed, it appears that, beside stimulating the CaMKII and mitogen-activated protein kinase-dependent pathways (Jovanovic et al., 1996; Chi et al., 2003), Ca2+ influx also regulates SV recycling by activating the PKA/synapsin machinery. Together, our results support a model in which, under resting conditions, tuning of the level of synapsin phosphorylation by modulators of PKA activity may set the efficiency of SV exocytosis in response to depolarization and Ca2+ influx.
Footnotes
This work was supported by grants from the Italian Ministry of University (Cofin 2004 and 2005, University Excellence Center on Physiopathology of Cell Differentiation, and Fondo per gli Investimenti della Ricerca di Base), Telethon (F.B., F.V., G.F.), and the Cariplo Foundation. We thank Dr. Paul Greengard (The Rockefeller University, New York, NY) for providing antibodies and synapsin I knock-out mice.
References
- Baba T, Sakisaka T, Mochida S, Takai Y. PKA-catalyzed phosphorylation of tomosyn and its implication in Ca2+-dependent exocytosis of neurotransmitter. J Cell Biol. 2005;170:1113–1125. doi: 10.1083/jcb.200504055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldelli P, Hernandez-Guijo JM, Carabelli V, Carbone E. Brain-derived neurotrophic factor enhances GABA release probability and nonuniform distribution of N- and P/Q-type channels on release site of hippocampal inhibitory synapses. J Neurosci. 2005;25:3358–3368. doi: 10.1523/JNEUROSCI.4227-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banker GA, Cowan WM. Rat hippocampal neurons in dispersed cell culture. Brain Res. 1977;126:397–442. doi: 10.1016/0006-8993(77)90594-7. [DOI] [PubMed] [Google Scholar]
- Benfenati F, Bahler M, Jahn R, Greengard P. Interactions of synapsin I with small synaptic vesicles: distinct sites in synapsin I bind to vesicle phospholipids and vesicle proteins. J Cell Biol. 1989;108:1863–1872. doi: 10.1083/jcb.108.5.1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betz WJ, Mao F, Smith CB. Imaging exocytosis and endocytosis. Curr Opin Neurobiol. 1996;6:365–371. doi: 10.1016/s0959-4388(96)80121-8. [DOI] [PubMed] [Google Scholar]
- Bonanomi D, Menegon A, Miccio A, Ferrari G, Corradi A, Kao HT, Benfenati F, Valtorta F. Phosphorylation of synapsin I by cAMP-dependent protein kinase controls synaptic vesicle dynamics in developing neurons. J Neurosci. 2005;25:7299–7308. doi: 10.1523/JNEUROSCI.1573-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi P, Greengard P, Ryan TA. Synapsin dispersion and reclustering during synaptic activity. Nat Neurosci. 2001;4:1187–1193. doi: 10.1038/nn756. [DOI] [PubMed] [Google Scholar]
- Chi P, Greengard P, Ryan TA. Synaptic vesicle mobilization is regulated by distinct synapsin I phosphorylation pathways at different frequencies. Neuron. 2003;38:69–78. doi: 10.1016/s0896-6273(03)00151-x. [DOI] [PubMed] [Google Scholar]
- Evans GJ, Morgan A. Regulation of the exocytotic machinery by cAMP-dependent protein kinase: implications for presynaptic plasticity. Biochem Soc Trans. 2003;31:824–827. doi: 10.1042/bst0310824. [DOI] [PubMed] [Google Scholar]
- Fiumara F, Giovedi S, Menegon A, Milanese C, Merlo D, Montarolo PG, Valtorta F, Benfenati F, Ghirardi M. Phosphorylation by cAMP-dependent protein kinase is essential for synapsin-induced enhancement of neurotransmitter release in invertebrate neurons. J Cell Sci. 2004;117:5145–5154. doi: 10.1242/jcs.01388. [DOI] [PubMed] [Google Scholar]
- Graham ME, Burgoyne RD. Comparison of cysteine string protein (Csp) and mutant α-SNAP overexpression reveals a role for csp in late steps of membrane fusion in dense-core granule exocytosis in adrenal chromaffin cells. J Neurosci. 2000;20:1281–1289. doi: 10.1523/JNEUROSCI.20-04-01281.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hell JW, Yokoyama CT, Breeze LJ, Chavkin C, Catterall WA. Phosphorylation of presynaptic and postsynaptic calcium channels by cAMP-dependent protein kinase in hippocampal neurons. EMBO J. 1995;14:3036–3044. doi: 10.1002/j.1460-2075.1995.tb07306.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilfiker S, Pieribone VA, Czernik AJ, Kao HT, Augustine GJ, Greengard P. Synapsins as regulators of neurotransmitter release. Philos Trans R Soc Lond B Biol Sci. 1999;354:269–279. doi: 10.1098/rstb.1999.0378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirling H, Scheller RH. Phosphorylation of synaptic vesicle proteins: modulation of the alpha SNAP interaction with the core complex. Proc Natl Acad Sci USA. 1996;93:11945–11949. doi: 10.1073/pnas.93.21.11945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hook SS, Means AR. Ca2+/CaM-dependent kinases: from activation to function. Annu Rev Pharmacol Toxicol. 2001;41:471–505. doi: 10.1146/annurev.pharmtox.41.1.471. [DOI] [PubMed] [Google Scholar]
- Hosaka M, Sudhof TC. Synapsin III, a novel synapsin with an unusual regulation by Ca2+ J Biol Chem. 1998;273:13371–13374. doi: 10.1074/jbc.273.22.13371. [DOI] [PubMed] [Google Scholar]
- Huttner WB, Greengard P. Multiple phosphorylation sites in protein I and their differential regulation by cyclic AMP and calcium. Proc Natl Acad Sci USA. 1979;76:5402–5406. doi: 10.1073/pnas.76.10.5402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson EM, Ueda T, Maeno H, Greengard P. Adenosine 3′,5-monophosphate-dependent phosphorylation of a specific protein in synaptic membrane fractions from rat cerebrum. J Biol Chem. 1972;247:5650–5652. [PubMed] [Google Scholar]
- Jovanovic JN, Benfenati F, Siow YL, Sihra TS, Sanghera JS, Pelech SL, Greengard P, Czernik AJ. Neurotrophins stimulate phosphorylation of synapsin I by MAP kinase and regulate synapsin I-actin interactions. Proc Natl Acad Sci USA. 1996;93:3679–3683. doi: 10.1073/pnas.93.8.3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao HT, Porton B, Hilfiker S, Stefani G, Pieribone VA, DeSalle R, Greengard P. Molecular evolution of the synapsin gene family. J Exp Zool. 1999;285:360–377. [PubMed] [Google Scholar]
- Kao HT, Song HJ, Porton B, Ming GL, Hoh J, Abraham M, Czernik AJ, Pieribone VA, Poo MM, Greengard P. A protein kinase A-dependent molecular switch in synapsins regulates neurite outgrowth. Nat Neurosci. 2002;5:431–437. doi: 10.1038/nn840. [DOI] [PubMed] [Google Scholar]
- Klingauf J, Kavalali ET, Tsien RW. Kinetics and regulation of fast endocytosis at hippocampal synapses. Nature. 1998;394:581–585. doi: 10.1038/29079. [DOI] [PubMed] [Google Scholar]
- Kuromi H, Kidokoro Y. Tetanic stimulation recruits vesicles from reserve pool via a cAMP-mediated process in Drosophila synapses. Neuron. 2000;27:133–143. doi: 10.1016/s0896-6273(00)00015-5. [DOI] [PubMed] [Google Scholar]
- Lonart G, Schoch S, Kaeser PS, Larkin CJ, Sudhof TC, Linden DJ. Phosphorylation of RIM1alpha by PKA triggers presynaptic long-term potentiation at cerebellar parallel fiber synapses. Cell. 2003;115:49–60. doi: 10.1016/s0092-8674(03)00727-x. [DOI] [PubMed] [Google Scholar]
- Matsushita M, Nairn AC. Inhibition of the Ca2+/calmodulin-dependent protein kinase I cascade by cAMP-dependent protein kinase. J Biol Chem. 1999;274:10086–10093. doi: 10.1074/jbc.274.15.10086. [DOI] [PubMed] [Google Scholar]
- Menegon A, Dunlap DD, Castano F, Benfenati F, Czernik AJ, Greengard P, Valtorta F. Use of phosphosynapsin I-specific antibodies for image analysis of signal transduction in single nerve terminals. J Cell Sci. 2000;113:3573–3582. doi: 10.1242/jcs.113.20.3573. [DOI] [PubMed] [Google Scholar]
- Menegon A, Verderio C, Leoni C, Benfenati F, Czernik AJ, Greengard P, Matteoli M, Valtorta F. Spatial and temporal regulation of Ca2+/calmodulin-dependent protein kinase II activity in developing neurons. J Neurosci. 2002;22:7016–7026. doi: 10.1523/JNEUROSCI.22-16-07016.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy G, Reim K, Matti U, Brose N, Binz T, Rettig J, Neher E, Sorensen JB. Regulation of releasable vesicle pool sizes by protein kinase A-dependent phosphorylation of SNAP-25. Neuron. 2004;41:417–429. doi: 10.1016/s0896-6273(04)00038-8. [DOI] [PubMed] [Google Scholar]
- Nguyen PV, Woo NH. Regulation of hippocampal synaptic plasticity by cyclic AMP-dependent protein kinases. Prog Neurobiol. 2003;71:401–437. doi: 10.1016/j.pneurobio.2003.12.003. [DOI] [PubMed] [Google Scholar]
- Pyle JL, Kavalali ET, Piedras-Renteria ES, Tsien RW. Rapid reuse of readily releasable pool vesicles at hippocampal synapses. Neuron. 2000;28:221–231. doi: 10.1016/s0896-6273(00)00098-2. [DOI] [PubMed] [Google Scholar]
- Ryan TA, Li L, Chin LS, Greengard P, Smith SJ. Synaptic vesicle recycling in synapsin I knock-out mice. J Cell Biol. 1996;134:1219–1227. doi: 10.1083/jcb.134.5.1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sara Y, Mozhayeva MG, Liu X, Kavalali ET. Fast vesicle recycling supports neurotransmission during sustained stimulation at hippocampal synapses. J Neurosci. 2002;22:1608–1617. doi: 10.1523/JNEUROSCI.22-05-01608.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiebler W, Jahn R, Doucet JP, Rothlein J, Greengard P. Characterization of synapsin I binding to small synaptic vesicles. J Biol Chem. 1986;261:8383–8390. [PubMed] [Google Scholar]
- Sudhof TC, Czernik AJ, Kao HT, Takei K, Johnston PA, Horiuchi A, Kanazir SD, Wagner MA, Perin MS, De Camilli P, Greengard P. Synapsins: mosaics of shared and individual domains in a family of synaptic vesicle phosphoproteins. Science. 1989;245:1474–1480. doi: 10.1126/science.2506642. [DOI] [PubMed] [Google Scholar]
- Surmeier DJ, Bargas J, Hemmings HC, Jr, Nairn AC, Greengard P. Modulation of calcium currents by a D1 dopaminergic protein kinase/phosphatase cascade in rat neostriatal neurons. Neuron. 1995;14:385–397. doi: 10.1016/0896-6273(95)90294-5. [DOI] [PubMed] [Google Scholar]
- Thakur P, Stevens DR, Sheng ZH, Rettig J. Effects of PKA-mediated phosphorylation of Snapin on synaptic transmission in cultured hippocampal neurons. J Neurosci. 2004;24:6476–6481. doi: 10.1523/JNEUROSCI.0590-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trudeau LE, Emery DG, Haydon PG. Direct modulation of the secretory machinery underlies PKA-dependent synaptic facilitation in hippocampal neurons. Neuron. 1996;17:789–797. doi: 10.1016/s0896-6273(00)80210-x. [DOI] [PubMed] [Google Scholar]
- Valtorta F, Jahn R, Fesce R, Greengard P, Ceccarelli B. Synaptophysin (p38) at the frog neuromuscular junction: its incorporation into the axolemma and recycling after intense quantal secretion. J Cell Biol. 1988;107:2717–2727. doi: 10.1083/jcb.107.6.2717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valtorta F, Benfenati F, Greengard P. Structure and function of the synapsins. J Biol Chem. 1992;267:7195–7198. [PubMed] [Google Scholar]
- Wang H, Storm DR. Calmodulin-regulated adenylyl cyclases: cross-talk and plasticity in the central nervous system. Mol Pharmacol. 2003;63:463–468. doi: 10.1124/mol.63.3.463. [DOI] [PubMed] [Google Scholar]
- Wong ST, Athos J, Figueroa XA, Pineda VV, Schaefer ML Chavkin CC, Muglia LJ, Storm DR. Calcium-stimulated adenylyl cyclase activity is critical for hippocampus-dependent long-term memory and late phase LTP. Neuron. 1999;23:787–798. doi: 10.1016/s0896-6273(01)80036-2. [DOI] [PubMed] [Google Scholar]