

Abstract
Missense mutations in the human PLP1 gene lead to dysmyelinating diseases with a broad range of clinical severity, ranging from severe Pelizaeus–Merzbacher disease (PMD) to milder spastic paraplegia type 2 (SPG-2). The molecular pathology has been generally attributed to endoplasmic reticulum (ER) retention of misfolded proteolipid protein (PLP) (and its splice isoform DM20) and induction of the unfolded protein response. As opposed to previous studies of heterologous expression systems, we have analyzed PLP/DM20 trafficking in oligodendroglial cells, thereby revealing differences between PMD and SPG-2-associated PLP/DM20 isoforms. PLPA242V and DM20A242V (jimpy-msd in mice), associated with severe PMD-like phenotype in vivo, were not only retained in the ER but also interfered with oligodendroglial process formation. In contrast, glial cells expressing SPG-2-associated PLPI186T or DM20I186T (rumpshaker in mice) developed processes, and mutant PLP/DM20 reached a late endosomal/lysosomal compartment. Unexpectedly, PLP/DM20 with either substitution exhibited impaired cholesterol binding, and the association with lipid raft microdomains was strongly reduced. Turnover analysis demonstrated that mutant PLP was rapidly degraded in oligodendroglial cells, with half-lives for PLP > PLPI186T > PLPA242V. Protein degradation was specifically sensitive to proteasome inhibition, although PLP/DM20I186T degradation was also affected by inhibition of lysosomal enzymes. We conclude that, in addition to ER retention and unfolded protein response (UPR) induction, impaired cholesterol binding and lipid raft association are characteristic cellular defects of PLP1-missense mutations. Mutant protein is rapidly cleared and does not accumulate in oligodendroglial cells. Whereas UPR-induced cell death governs the PMD phenotype of the msd mutation, we propose that impaired cholesterol and lipid raft interaction of the rsh protein may contribute to the dysmyelination observed in SPG-2.
Keywords: myelin, PLP-related disorders, trafficking, lipid rafts, cholesterol, turnover
Introduction
The myelin proteolipid protein (PLP) is the major protein of mammalian CNS myelin with proposed functions in myelin membrane stability and maintenance (Griffiths et al., 1998a; Hudson, 2004). Mutations in the human PLP1 gene lead to dysmyelinating diseases of a broad clinical spectrum, classified as connatal and classic Pelizaeus–Merzbacher disease (PMD) and spastic paraplegia type 2 (SPG-2) (Koeppen and Robitaille, 2002; Inoue 2005). Whereas a null mutation of the gene results in a mild and late-onset phenotype with morphologically normal myelin (Griffiths et al., 1998b; Garbern et al., 2002), missense mutations are characterized by dysmyelination, suggesting that they act through gain-of-function mechanisms. Point mutations resulting in single amino acid substitutions have been identified throughout the coding sequence (Garbern, 2005). They contribute to ∼20% of all cases and cover the whole spectrum of the disease from the severest forms of connatal PMD over classical PMD to the milder SPG-2 form. How the different mutations in the PLP gene result in divergent phenotypes is still a matter of debate.
Mouse models of all forms of the disease are available and facilitate the analysis of PLP1-related pathogenesis (Werner et al., 1998; Duncan, 2005). The natural mouse mutations jimpy-msd (msd, A242V) and rumpshaker (rsh, I186T) have also been identified in humans and model severe conatal PMD and mild SPG-2, respectively (Kobayashi et al., 1994; Komaki et al., 1999). Msd mice develop progressive tremors and seizures, causing death between the third and fourth week of life. Morphologically, myelin is almost absent in the CNS, most likely because of oligodendroglial apoptosis (Knapp et al., 1986; Gow et al., 1998). In contrast, rsh mice on the C3H/101 genetic background develop ataxia in the third postnatal week but rarely exhibit seizures and have a normal life span. Remarkably, enhanced oligodendroglial apoptosis is not a feature of these mice (Schneider et al., 1992), demonstrating that the observed dysmyelination is not the result of oligodendroglial degeneration.
The molecular pathogenesis underlying PLP1 missense mutations has been generally attributed to retention and accumulation of misfolded PLP protein in the endoplasmic reticulum (ER), accompanied by induction of the unfolded protein response (UPR) (Gow and Lazzarini, 1996; Jung et al., 1996; Gow et al., 1998; Southwood et al., 2002). So far, all studies of cellular and molecular pathology have been performed in heterologous expression systems using high-level expression in fibroblastoid cells. However, differences in the level of ER retention and cell surface trafficking of mutant PLP have been observed in different cell lines (Thomson et al., 1997) suggesting cell type-specific influences.
Here, we compared the behavior of PLP/DM20 with the severe msd mutation and the mild rsh mutation in oligodendroglia. rsh-PLP/DM20 overcame ER retention and trafficked to late endosomes/lysosomes. However, cholesterol binding and lipid raft association of both msd- and rsh-PLP/DM20 was impaired. Mutant protein had a higher turnover and was rapidly degraded by the proteasome, implicating that accumulation and deposition of misfolded protein is not characteristic for PMD and SPG-2. We suggest that impaired cholesterol and lipid raft interaction contributes to the dysmyelination observed in SPG-2.
Materials and Methods
Reagents.
The l-[35S]methionine/cysteine in vitro labeling mix and ECL reagents were from Amersham-Buchler (Braunschweig, Germany); 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS), filipin, N-acetyl-l-leucyl-l-leucyl-l-norleucinal (ALLN), leupeptin, methyl-β-cyclodextrin (MβCD), dbcAMP (di-butyryl-adenosine-3′,5′-cyclic monophosphate), and Optiprep were from Sigma (Deisenhofen, Germany). Protein A-Sepharose CL4B was from Pharmacia (Freiburg, Germany). Lipoluma and Lumasolve were from PerkinElmer (Rodgau, Germany). The following rabbit polyclonal antibodies were used: antibodies A431 against the C-terminal peptide of PLP (Jung et al., 1996) and anti-calnexin (StressGen, Victoria, British Columbia, Canada). The following monoclonal antibodies were used: rat AA3 against the C terminus of PLP (a kind gift from Dr. M. B. Lees, E. K. Shriver Center, Waltham, MA), mouse IgM O10 against a cell surface epitope of PLP (Jung et al., 1996), anti-lysosome associated membrane protein 1 (LAMP1) and anti-BiP/Grp78 (BD Pharmingen, Erembodegem, Belgium). Fluorochrome-conjugated secondary antibodies were from Dianova (Hamburg, Germany).
Cell culture and transfection.
Primary cultures of oligodendrocytes were prepared and maintained as described previously (Krämer et al., 1999). The cell line Oli-neu was cultured in Sato medium containing 1% horse serum (HS) (Jung et al., 1995). Transfection was performed by electroporation. The expression vectors used for expression of wild-type and mutant PLP and DM20 were described previously (Jung et al., 1996). Plasmid-DNA (20 μg) was added to 3 × 106 cells in 600 μl of Sato medium and cells were electroporated at 220 V and 950 μF using the Bio-Rad Gene Pulser Xcell (exponential decay program). Di-butyryl-cAMP (1 mm) and sodium butyrate (2 mm) were added 2 and 4 h after plating, respectively. Cells were analyzed 20–48 h after the transfection.
COS7 cells were maintained in DMEM containing 10% fetal calf serum. Transfection was performed using the jetPEI transfection reagent according to the instructions of the manufacturer (QBiogene, Illkirch, France).
Immunofluorescence staining.
For cell surface staining of PLP and DM20, primary oligodendrocytes and Oli-neu cells were live stained. Cells were blocked for 10 min in BME medium containing 10% HS and incubated for 30 min with O10 antibody diluted in blocking medium. Subsequently, cells were fixed in 4% paraformaldehyde in PBS, and permeabilized for 2 min with 0.1% Triton X-100. After blocking, cells were incubated for 30 min with polyclonal A431 antibody recognizing the intracellular C terminus of PLP, followed by dye-conjugated secondary antibodies, and were finally mounted. For simultaneous detection of two intracellular epitopes, cells were first fixed, and then permeabilized and blocked as above, and primary as well as secondary anti-species antibodies were applied as a mixture. Cells were analyzed by fluorescence light microscopy (DMLB microscope connected to a DFC 350F digital camera; Leica, Bensheim, Germany). To obtain focal planes, images of cells were taken in stacks and deconvolved using the Openlab 2.3 software (Improvision, Coventry, UK).
Lipid raft isolation and immunoblotting.
Lipid raft isolation was performed as described by (Simons et al. 2000). Briefly, transfected Oli-neu cells were washed in TBS, scraped into 300 μl of extraction buffer (50 mm Tris/HCl, pH 7.4, 5 mm EDTA, 20 mm CHAPS) supplemented with protease inhibitors and extracted for 30 min at 4°C. The extract was adjusted to 30% Optiprep (600 μl), overlaid with 20% Optiprep in extraction buffer (1.4 ml) and with extraction buffer only (200 μl), followed by ultracentrifugation for 2 h at 55,000 rpm in a TLS-55 rotor (Beckman, Fullerton, CA). Six fractions of 360 μl were collected from the top and processed for SDS-PAGE.
Proteins were blotted onto a polyvinylidene difluoride membrane (Amersham Biosciences, Freiburg, Germany), which was blocked with 4% milk powder/0.1% Tween in PBS. Proteins were detected by sequential incubation of the membrane with primary antibodies overnight at 4°C and second anti-species antibodies conjugated with HRP for 30–60 min at room temperature. The blots were developed with ECL reagents (Amersham Biosciences) according to the manufacturer's instructions.
Photocholesterol labeling.
The synthesis of 6,6-[3α-3H]azocholestan-3β-ol (referred to as photocholesterol) has been described previously (Thiele et al., 2000). For preparation of the [3H]photocholesterol/MβCD inclusion complex, the steroid (final concentration, 0.3 mm) was added to an aqueous solution of MβCD (40 mg/ml). The mixture was overlaid with N2, and was continuously vortexed under light protection at 30°C for 24 h in a thermomixer. Labeling of living Oli-neu cells was performed as described previously (Simons et al., 2000). Cells were washed with Sato medium supplemented with 1% HS, which had been depleted of lipids (dlHS), and incubated with 100 μl (50 μCi) of [3H]photocholesterol–MβCD inclusion complex in 2 ml of Sato/1% dlHS for 16 h. Cells were irradiated for 20 min with UV light at 4°C and subsequently disrupted in lysis buffer (25 mm Tris/HCl, pH 7.4, 150 mm NaCl, 1% Triton X-100) supplemented with protease inhibitors for 15 min on ice, followed by a brief spin (10 min; 200 × g) to deplete nuclei. A small sample of the lysate was directly processed for SDS-PAGE and fluorography, although the rest was subjected to PLP immunoprecipitation as described.
For labeling of isolated membranes, a total membrane fraction of Oli-neu cells was prepared according to (Krämer et al. 1997). Briefly, a postnuclear supernatant was prepared by shearing cells through a 22 gauge needle followed by centrifugation for 10 min at 200 × g. Membranes were pelleted by ultracentrifugation for 30 min at 100,000 × g and resuspended in 50 μl of PBS (500 μg/ml protein) followed by addition of 2 μl of [3H]photocholesterol/MβCD (0.7 μCi). The mixture was continuously vortexed in a thermomixer at 30°C for 10 min in the dark. Thereafter, the sample was irradiated at 30°C for 5 min using a mercury-arc lamp (200 W; λ > 335 nm). Proteins were separated by SDS-tube gel electrophoresis (12%) according to (Laemmli 1970). After electrophoresis, gels were sliced for liquid scintillation counting. Gel slices were incubated in 4 ml of a scintillator mixture [Lipoluma/Lumasolve/H2O 893:89:18 (v/v)] at room temperature for 72 h. Radioactivity was measured in a LKB 1215 Rackbeta liquid scintillation counter.
Pulse-chase experiments.
Oli-neu cells maintained in 3 cm dishes 20 h after transfection were starved for 1 h at 37°C in methionine/cysteine-free DMEM and labeled for 30 min at 37°C with [35S]methionine/cysteine. Cells were washed and chased in prewarmed Sato/1% HS supplemented with 0.5 mm methionine and 0.5 mm cysteine. In some experiments, ALLN (100 μm) or leupetin (100 μg/ml) were included in the chase medium, to inhibit proteasomal or lysosomal degradation, respectively. After the appropriate length of the chase, cells were washed twice in ice-cold TBS, lysed in 0.5 ml of lysis buffer (25 mm Tris/HCl, pH 7.4, 150 mm NaCl, 5 mm EDTA, 1 mm PMSF, 1% NP-40) for 10 min on ice and centrifuged for 10 min at 2000 × g. The supernatant was further processed for immunoprecipitation.
Immunoprecipitation.
Cell monolayers were washed twice in cold TBS and were scraped in 0.5–1 ml of lysis buffer (25 mm Tris/HCl, pH 7.4, 150 mm NaCl, 5 mm EDTA, 1 mm PMSF, 1% NP-40). Lysates were precleared by incubation with protein A-Sepharose for 2 h at 4°C. After the protein A-Sepharose had been removed by centrifugation, the lysates were incubated with anti-PLP antibodies (polyclonal A431, 1:500) overnight at 4°C and for 2 h with protein A-Sepharose on a head-over-tail rotator. Sepharose beads were washed twice in 50 mm Tris/HCl, pH 7.4, 150 mm NaCl, 5 mm EDTA, 1% NP-40, 0.1% SDS, and twice under high salt conditions in the same buffer but containing 400 mm NaCl. Immunoprecipitates were analyzed by SDS-PAGE and phosphoimaging (Fuji Bas 1000 and MacBas Software; Fuji Medical Systems, Stanford, CA). Densitometric analysis was performed using Fuji Film Science Lab 99 software.
Results
Previous reports on the cellular pathology of PLP1 mutations using fibroblastoid cell lines revealed differences in the cell surface transport of mutant PLP between different cell lines, suggesting that transport and retention mechanisms vary between different cell types. To analyze PLP1 missense mutations in a more physiological environment, we used the oligodendroglial cell line Oli-neu (Jung et al., 1995), which shares many features of O4-positive immature oligodendroglial cells but survives when expressing misfolded proteins. A small subpopulation of Oli-neu cells weakly transcribes the endogenous Plp1 gene, expressing the DM20 isoform, and a substantial fraction of the cells expresses the myelin-associated glycoprotein, indicating the initiation of myelin gene expression.
Distinct subcellular localization of PLP/DM20 in oligodendroglial cells and COS7 cells
To verify normal trafficking of PLP in Oli-neu cells, we compared the subcellular localization of wild-type PLP and DM20 after transfection to that of endogenous PLP/DM20 in primary cultured oligodendrocytes. As expected for a major myelin protein, PLP/DM20 is efficiently integrated in the plasma membrane of primary oligodendrocytes and Oli-neu cells (see supplemental Fig. 1, available at www.jneurosci.org as supplemental material). In permeabilized cells, a substantial fraction of the PLP/DM20 protein was found in intracellular compartments. In both primary cells and Oli-neu cells, PLP/DM20 did not obviously colocalize with ER markers (BiP or calnexin) and only transiently colocalized with Golgi markers (58K or GM130) (Krämer et al., 2001). Confocal or deconvolution analysis demonstrated that intracellular PLP/DM20 in these cells mostly overlapped with the marker LAMP1 (Fig. 1A–C), revealing that these compartments are of late endosomal/lysosomal origin. In contrast, PLP and DM20 overexpressed in COS7 cells showed a striking degree of overlap with the ER marker calnexin (Fig. 1D). Only in a small fraction of PLP/DM20-expressing COS7 cells of <5% was the protein additionally present in an endosomal/lysosomal compartment (see supplemental Fig. 2, available at www.jneurosci.org as supplemental material).
Figure 1.

Localization of PLP and DM20 in oligodendroglial and COS7 cells. Primary cultured oligodendrocytes (A), wild-type PLP and DM20 transfected Oli-neu cells (B, C), and wild-type PLP transfected COS7 cells (D) were costained with antibodies AA3 recognizing PLP (green) and LAMP1 (red; A–C) or calnexin (red; D). In A and C, confocal images are shown, and B is a deconvolved image. Insets show enlarged areas. In primary oligodendrocytes and in Oli-neu cells, PLP/DM20 is efficiently incorporated in the plasma membrane and localized in endosomes/lysosomes [plasma membrane and endosomes/lysosomes are primarily detected in distinct focal planes; plasma membrane localization of PLP/DM20 is shown in supplemental Fig. 2 (available at www.jneurosci.org as supplemental material)]. In COS7 cells, a large proportion of the protein is found in the ER. The dashed lines outline the position of the nuclei. The minor endosomal/lysosomal localization of PLP in COS7 is shown in supplemental Figure 2 (available at www.jneurosci.org as supplemental material). Scale bars, 5 μm. pOL, Primary oligodendrocyte; wt, wild type; Cnx, calnexin.
We determined the cell surface transport efficacy of PLP/DM20 in transfected Oli-neu and COS7 cells by live-cell staining of transfected Oli-neu cells using the monoclonal antibody O10, which defines a conformation-sensitive extracellular epitope (Jung et al., 1996). Costaining with the AA3 antibody recognizing the cytoplasmic C terminus of PLP/DM20 identified the total cellular pool of PLP/DM20 (Fig. 2A). Cells expressing PLP/DM20, which is not reaching the plasma membrane, were thus visualized in the staining with the AA3 antibody but did not stain with the O10 antibody. In Oli-neu cells, the plasma membrane of 82.7 ± 1.9% of the PLP and 77 ± 7% of the DM20-expressing cells was brightly stained with the O10 antibody, whereas only 26.2 ± 1.4% of the PLP and 22.3 ± 0.8% of the DM20-expressing COS7 cells displayed surface expression (Fig. 2B). Thus, trafficking of PLP and DM20 to the cell surface is more efficient in Oli-neu cells than in COS7 cells.
Figure 2.
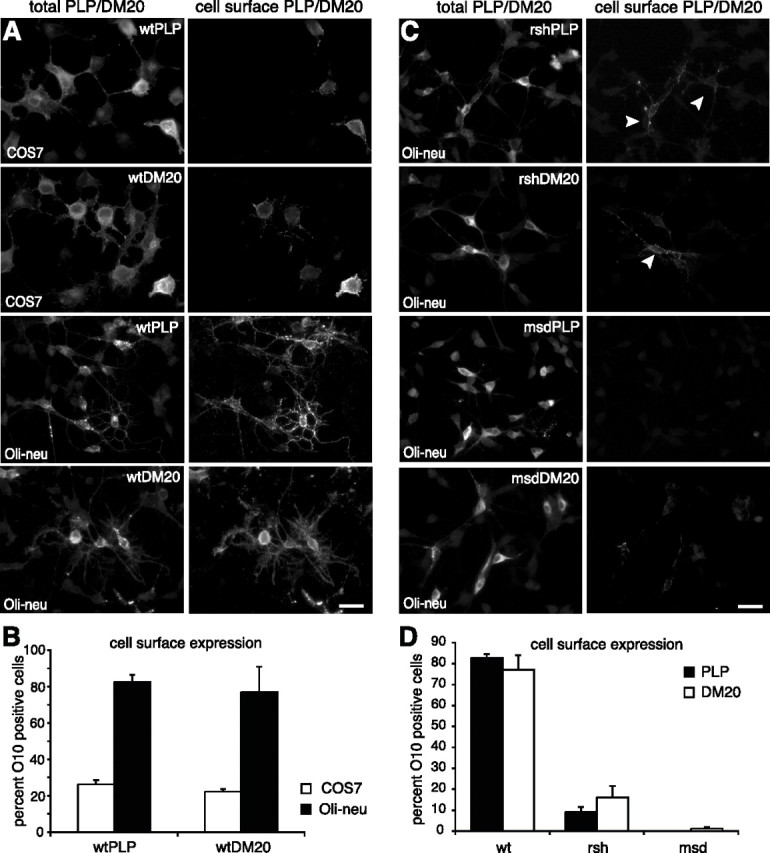
Cell surface transport efficiency of wild-type and mutant PLP and DM20. PLP and DM20 transfected cells were stained live with the O10 antibody, which recognizes an extracellular epitope, and thus selectively marks cells, where PLP/DM20 is localized to the cell surface (right picture columns in A and C, “cell surface PLP/DM20”). After O10 surface staining, cells were fixed, permeabilized, and costained with the AA3 antibody, which binds to the cytoplasmic C terminus of PLP/DM20 and thus identifies all cells expressing PLP/DM20 (left picture columns in A and C, “total PLP/DM20”). Cells expressing PLP/DM20 that has not reached the plasma membrane are visualized in the staining with the AA3 antibody but do not stain with the O10 antibody. The O10-positive cells (“cell surface PLP/DM20”) were counted and expressed as a percentage of all PLP/DM20-expressing cells (“total PLP/DM20”), evaluating the surface transport efficiency of PLP/DM20 (B, D). A, B, Cell surface transport efficiency of wild-type PLP and DM20 expressed in COS7 cells and Oli-neu cells. In Oli-neu cells, wild-type PLP and DM20 are efficiently transported to the plasma membrane, whereas in a large fraction of COS7 cells PLP/DM20 is retained intracellularly. A total of 400–550 PLP/DM20-expressing cells was counted in each of three independent experiments. C, D, Cell surface expression of rsh- and msd-mutant PLP and DM20 in Oli-neu cells. A total of 350–600 PLP/DM20-expressing cells was counted in each of four independent experiments. Error bars represent SEM. Note that rsh-PLP and rsh-DM20 retain some capacity for cell surface transport (arrowheads). Scale bars, 30 μm.
PLP/DM20 trafficking in COS7 cells thus differs from that of oligodendroglial cells in that its localization is shifted toward the ER, whereas in oligodendroglial cells the protein seems to quickly progress through the early secretory pathway and is incorporated into the plasma membrane. The subcellular localization and cell surface expression of ectopically expressed PLP and DM20 in Oli-neu cells is identical with that of the endogenous protein in primary cultured oligodendroglial cells. Hence, this expression system is a superior tool to study PLP-related cellular pathomechanisms.
Glial cell morphology and O10 expression as indicators of disease severity
Wild-type PLP- or DM20-expressing Oli-neu cells exhibited a complex morphology, typical of oligodendrocytes with many branched processes (Fig. 2A). With expression of the mutant PLP alleles msd (A242V) and rsh (I186T) in Oli-neu cells, we observed that expression of the msd allele, associated with a severe disease phenotype in vivo, frequently led to spindle-shaped cells and cells with short processes (see Figs. 2C, 3, 4A, 8). In contrast, the more benign rsh mutation was expressed in cells with several processes, demonstrating that the severity of dysmyelination caused by these mutations is reflected in deviations from normal cellular morphology.
Figure 3.

Distinct subcellular localization of rsh- and msd-PLP. Oli-neu cells were transfected with wild-type (wt), rsh-, and msd-PLP and colabeled using antibodies that recognize PLP (green) and the ER marker protein BiP (A; red) or LAMP1 (B; red). Deconvolved images are shown. Wild-type as well as rsh protein colocalize primarily with the lysosomal marker, whereas msd protein colocalizes with the ER marker. Insets show enlargements. Scale bars, 5 μm. Note dramatic differences in cellular phenotype induced by msd protein.
Figure 4.
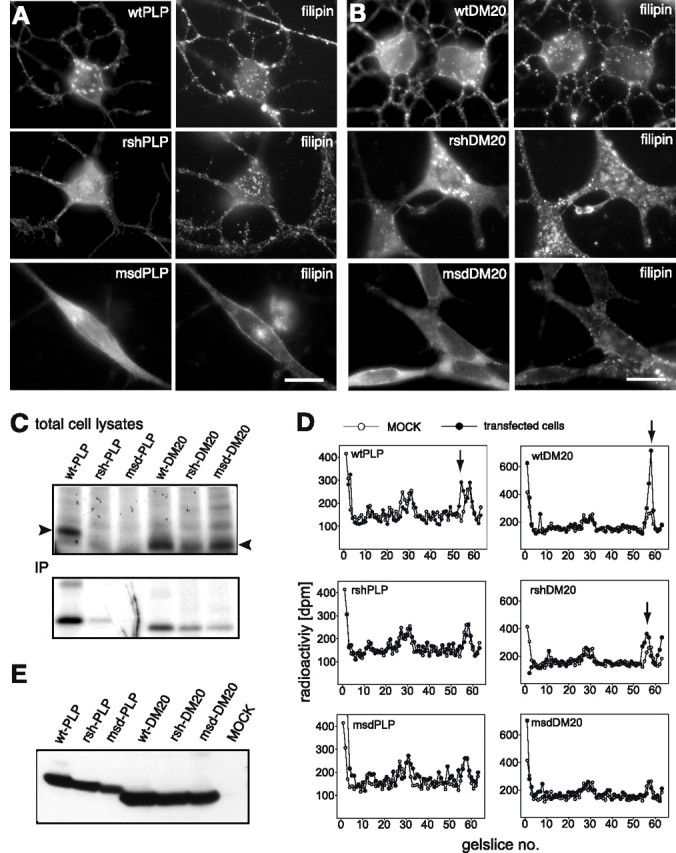
Cholesterol interaction of mutant PLP/DM20. Oli-neu cells transfected with wild-type, rsh and msd mutant PLP (A), or DM20 (B) were stained with antibodies AA3 to PLP/DM20 (left pictures) and with the cholesterol interacting agent filipin, which is autofluorescent (right pictures). Expression of mutant PLP/DM20 does not induce redistribution of cholesterol. Scale bars, 10 μm. C, Transfected Oli-neu cells were labeled overnight with [3H]photocholesterol. Cells were UV-irradiated to covalently cross-link cholesterol binding proteins, and total cell lysates were directly analyzed by SDS-PAGE (top panel) or subjected to PLP/DM20 immunoprecipitation using AA3 antibodies (IP; bottom panel). [3H]Photocholesterol-modified proteins were visualized by autoradiography. The arrowheads mark the position of PLP and DM20 in the gel. D, [3H]Photocholesterol labeling and UV cross-linking of total membranes isolated from transfected Oli-neu cells. Proteins were separated by SDS-tube gel electrophoresis and slices were analyzed for associated radioactivity by scintillation counting. Results obtained from PLP/DM20 transfected (closed circles) are plotted over those from control cells (open circles). Peaks of radioactivity resulting from [3H]photocholesterol modification of transfected PLP or DM20 are marked by arrows. E, A Western blot of the same membrane fractions analyzed in Figure 4D with antibodies AA3 recognizing PLP/DM20 demonstrates that equal amounts of total PLP/DM20 were loaded.
Figure 8.

Shift of the subcellular PLP localization by ALLN and leupeptin treatment. Wild-type, rsh- and msd-PLP transfected cells were treated with ALLN (100 μm) (A) or leupeptin (100 μg/ml) (B). After 8 h of treatment, cells were fixed and stained with antibodies to PLP/DM20 and calnexin (A) or LAMP1 (B). Note that ALLN or leupeptin treatment induces a shift in the subcellular localization of wild-type and rsh-PLP. Scale bars, 20 μm.
It has been reported previously that, in transfected COS7 cells, mutant PLP fails to be transported to the cell surface (Gow and Lazzarini, 1996; Jung et al., 1996). Because surface trafficking of PLP is generally more efficient in Oli-neu cells, we reinvestigated this issue in Oli-neu cells (expressing rsh- and msd-PLP/DM20) by surface staining of live cells with the O10 antibody and costaining with the AA3 antibody recognizing the cytoplasmic C terminus of both wild-type and mutant PLP/DM20, thus labeling all PLP/DM20-expressing cells, independent of cell surface localization of the protein (Fig. 2C). Remarkably, 9 ± 2.5% of rsh-PLP- and 16.1 ± 5.4% of rsh-DM20-expressing cells exhibited correctly folded protein at the cell surface, although at reduced levels compared with wild-type PLP/DM20-expressing cells (Fig. 2D). In msd-PLP-expressing cells, the O10 epitope was totally absent, only a small fraction (∼1%) of msd-DM20-expressing cells showed a weak O10 signal. Interestingly, coexpression of either rsh- or msd-PLP/DM20 with wild-type protein revealed surface expression levels similar to the expression of wild-type PLP/DM20 alone (data not shown), demonstrating that mutant PLP/DM20 does not inhibit surface transport of wild-type protein in a dominant-negative manner.
Distinct subcellular localization of rsh- and msd-PLP/DM20
To define possible sites of subcellular pathology, we costained wild-type, rsh and msd-expressing Oli-neu cells with intracellular compartment markers and analyzed focal planes by deconvolution. Deconvolved images demonstrated intense colocalization of msd-PLP with the ER marker BiP, whereas colocalization of rsh-PLP with BiP was not prominent (Fig. 3A). Surprisingly, a substantial fraction of rsh-PLP was found in LAMP-1-positive endocytic compartments (Fig. 3B), whereas msd-PLP showed no overlap with this late endosomal/lysosomal marker. None of the mutant isoforms colocalized with Golgi markers. The subcellular distribution of DM20 mutant isoproteins was qualitatively identical with the respective PLP isoform (data not shown). Thus, the major fraction of rsh protein is not retained in the ER, but progressing through the secretory pathway and finally targeted to late endosomes/lysosomes (although processing kinetics in the ER may be slower compared with wild type).
Impaired cholesterol interaction of mutant PLP/DM20
It had been previously demonstrated that PLP and DM20 bind cholesterol and associate with lipid rafts, suggesting a role in sorting and trafficking of the protein to compact myelin (Simons et al., 2000). We tested to what extent mutant and mislocalized PLP/DM20 sequesters cholesterol, which could result in an imbalance of the cellular cholesterol homeostasis, as shown for overexpression of PLP (Simons et al., 2002). We made use of the fluorescent fungal antibiotic filipin, which interacts with cholesterol in membranes and stains cholesterol when applied to cells. Filipin staining of Oli-neu cells transfected with wild-type and mutant PLP or DM20 revealed that cholesterol did not colocalize with mutant PLP and DM20, and no major redistribution of cholesterol with mutant protein was observed (Fig. 4A,B).
To monitor the interaction of mutant PLP with cholesterol more directly, the ability of mutant PLP and DM20 to bind [3H]photocholesterol, which is modified by an azo group that forms a free radical in response to irradiation with UV light, was tested (Thiele et al., 2000). Briefly, we labeled transfected Oli-neu cells with [3H]photocholesterol using methyl-β-cyclodextrin as a carrier and triggered covalent modification of interacting proteins by UV irradiation. The transfection efficiency was checked by immunofluorescence staining to confirm equal expression levels of wild-type and mutant PLP/DM20 in the different populations of transfected cells (transfection efficiency of 61 ± 2.3%) (see supplemental Fig. 3, available at www.jneurosci.org as supplemental material). Autoradiograms of total cell lysates from transfected cells showed [3H]photocholesterol modified proteins over a wide range of molecular weights. In the lysates of wild-type PLP and DM20 transfected cells, a prominent band appeared at 23 and 20 kDa, respectively, which was absent or weaker in lysates from rsh and msd transfected cells (Fig. 4C, arrowheads). Immunoprecipitation of PLP and DM20 from the same lysates confirmed the reduced yield of [3H]photocholesterol modified mutant PLP and DM20 compared with wild-type protein. This decrease of the [3H]photocholesterol signal potentially indicates a reduced interaction of mutant PLP/DM20 with cholesterol but, in contrast, could be attributable to the reduced yield of total mutant PLP/DM20 [however, supplemental Fig. 3B (available at www.jneurosci.org as supplemental material) shows an example of an average immunoprecipitation of PLP/DM20 from transfected cells, in which only the yield of msd-PLP is slightly reduced]. In addition, the [3H]photocholesterol labeling procedure of living cells could potentially favor labeling of plasma membrane over intracellular pools of PLP/DM20, if the labeling reagent is not efficiently transported to the intracellular compartments.
To finally exclude these concerns, we performed the [3H]photocholesterol labeling on isolated crude membranes from transfected Oli-neu cells to ensure that plasma membrane and intracellular membranes are equally accessible to the labeling probe. After UV-induced cross-linking, samples were separated by SDS tube gel electrophoresis and gel slices were analyzed for associated [3H]photocholesterol by scintillation counting. Radioactivity profiles of membranes from wild-type PLP and DM20 transfected cells superimposed on the profile of MOCK transfected cells demonstrated a clear extra peak of 3H radioactivity, which corresponded to the predicted mobility of PLP and DM20, respectively (Fig. 4D, arrows). In radioactivity profiles from rsh-PLP, msd-PLP, and msd-DM20 transfected cells, this peak was absent, whereas the profile of rsh-DM20 transfected cells displayed a peak of reduced size. Equal loading of wild-type and mutant PLP/DM20 was tested by Western blotting of the same membrane preparations subjected to tube gel analysis (Fig. 4E). In summary, these experiments suggest that cholesterol interaction of mutant PLP and DM20 is impaired. Only rsh-PLP/DM20 appears to retain some capacity to bind cholesterol. This observation could explain its presence at low abundance in purified myelin from rsh mice (Schneider et al., 1992; McLaughlin et al., 2006).
Impaired association of mutant PLP/DM20 with lipid rafts
The interaction with cholesterol favors the association of PLP and DM20 with lipid rafts, which are defined by their resistance to the detergent CHAPS at 4°C and can be isolated after density gradient centrifugation (Simons et al., 2000). We examined lipid rafts from transfected Oli-neu cells to test whether association of mutant PLP/DM20 was maintained. Western blot analysis of density gradient fractions showed that lipid raft association of rsh- and msd-mutant PLP/DM20 was drastically reduced (Fig. 5). This was not surprising in the case of msd-PLP and msd-DM20, because both proteins are retained in the ER, whereas the association of PLP with lipid rafts occurs at later stage in the secretory pathway in the Golgi compartment (Simons et al., 2000). Intriguingly, although rsh-PLP and rsh-DM20 are able to traffic through the secretory pathway, they are not efficiently incorporated into lipid rafts.
Figure 5.
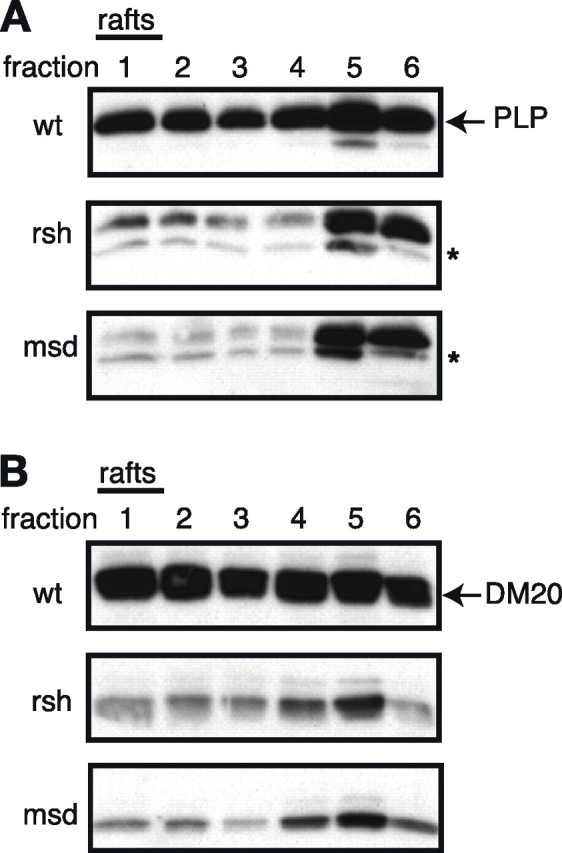
Impaired association of mutant PLP/DM20 with lipid rafts. Wild-type, rsh-, and msd-PLP (A) and DM20 (B) transfected Oli-neu cells were subjected to lipid raft analysis, by cell extraction in a CHAPS-containing buffer at 4°C followed by density gradient centrifugation. Fractions 1 (light fraction) to 6 (dense fraction) were collected and analyzed by Western blotting using antibodies recognizing PLP/DM20. Because of their low density, lipid rafts float in the light fractions of the gradient and are collected from fractions 1 and 2. A high proportion of wild-type PLP and DM20 is present in the lipid raft fraction, whereas rsh- and msd-PLP/DM20 are almost absent from this fraction. The asterisk (*) marks an endogenous DM20 band, which is occasionally seen in Western blots.
We then tested whether rafts are generally affected in their organization or trafficking by mutant PLP/DM20 expression, resulting in possible mislocalization and malfunction of lipid raft components such as signaling proteins. Analysis of other lipid raft-associated molecules, such as the cell adhesion molecule NCAM-120 or Fyn kinase (Krämer et al., 1999), showed that these are present in lipid raft fractions and in the plasma membrane of msd- or rsh-PLP/DM20-expressing cells. In addition, coexpression of mutant PLP or DM20 did not influence the distribution and trafficking of GPI-EGFP (glycosylphosphatidylinositol-enhanced green fluorescent protein), a protein widely used as a lipid raft marker for imaging studies (data not shown).
Thus, the absence of mutant PLP and DM20 from lipid rafts does not have a major effect on lipid raft trafficking and function. The absence of rsh-PLP/DM20 from lipid rafts is most likely attributable to the failure of the protein to interact with cholesterol and may indeed result in the reduced localization of rsh protein in the plasma membrane.
Mutant PLP is rapidly degraded
It is generally believed that unfolded and mislocalized mutant proteins accumulate in cells because of an overload of the cellular protein degradation machinery (Forman et al., 2003). To compare the turnover of wild-type and mutant PLP/DM20, we performed pulse-chase experiments. Transfected glial cells were radiolabeled for 30 min with [35S]methionine/cysteine (“pulse”), followed by increasing periods of incubation in cold medium (“chase”). After the chase period, cells were lysed and PLP/DM20 was immunoprecipitated. Labeled PLP/DM20 was displayed by phosphoimaging (Fig. 6A) and quantified by densitometric analysis (Fig. 6B). The percentage of PLP/DM20 recovery after the different chase periods was calculated in relation to the total amount of labeled PLP/DM20 precipitated directly after the 30 min labeling procedure (0 h). In some experiments, a slight shift in PLP molecular weight was observed during the chase, most likely because of posttranslational modification (e.g., acylation) (Weimbs and Stoffel, 1992). Interestingly, the degradation of rsh- and msd-PLP progressed significantly faster than wild-type PLP (Fig. 6B, top graph), with msd proceeding even faster than rsh (with factors of 1.5 for rsh and 2.0 for msd). Wild-type PLP had a medium half-life of 6.5 h, rsh-PLP of 3.5 h, and msd-PLP of 3 h. Mutant PLP-expressing cells were not affected by apoptosis during the chase period, because the number of PLP/DM20-expressing cells did not change (data not shown).
Figure 6.

Pulse-chase turnover analysis of wild-type and mutant PLP/DM20. Wild-type, rsh-, and msd-PLP or DM20 transfected Oli-neu cells were pulse labeled for 30 min with [35S]methionine/cysteine followed by incubation in normal growth medium for indicated times (chase). Labeled PLP and DM20 were recovered by immunoprecipitation. A, Immunoprecipitates were analyzed by SDS-PAGE, and 35S-labeled PLP/DM20 was detected by phosphoimaging. B, C, The recovery of 35S-labeled PLP/DM20 was densitometrically quantified. The amount of PLP/DM20 recovered immediately after the pulse (0 h chase) was equalized to 100%, and the percentage of recovery after the different chase times was calculated. Error bars represent SEM. Statistical significance was determined using the t test (*p < 0.05; **p < 0.01; ***p < 0.001). rsh- and msd-PLP are significantly faster degraded than wild-type PLP, whereas degradation of mutant DM20 versus wild-type DM20 is not significantly enhanced (B). C illustrates the turnover of wild-type and mutant PLP compared with the respective DM20 isoform. n = 12 for PLP 4 h plus 8 h, n = 8 for DM20 4 h plus 8 h, n = 3 for PLP plus DM20 24 h.
Moreover, wild-type DM20 had an inherently faster turnover compared with PLP (Fig. 6C) (half-life, 3.5 h). Consequently, the turnover of mutant DM20 was not significantly increased compared with wild-type DM20 (Fig. 6B, bottom graph). If the turnover of the respective PLP and DM20 mutations is compared (Fig. 6C), rsh-DM20 is still degraded faster than rsh-PLP, whereas this difference in turnover between PLP and DM20 is absent in the msd mutation.
These results demonstrate that mutant PLP/DM20 is rapidly cleared from the cells. Misfolded PLP/DM20 is not accumulating in intracellular deposits in oligodendroglial cells.
The proteasome is the major degradation site of wild-type and mutant PLP/DM20
Normal turnover of membrane proteins includes their degradation in lysosomes. In contrast, misfolded membrane proteins that fail to pass the ER quality control are retranslocated from the ER to the cytoplasm and degraded by the proteasome (Trombetta and Parodi, 2003). To identify the degradation site of mutant PLP/DM20, we inhibited these degradation pathways by performing similar pulse-chase experiments as described above in the presence of the proteasomal inhibitor ALLN or the lysosomal inhibitor leupeptin (Fig. 7A,B). Degradation of wild-type PLP and DM20 was inhibited by both ALLN and leupeptin, demonstrating that both proteasomal and lysosomal pathways are involved in the turnover of the wild-type proteins. However, the degradation of all mutant PLP and DM20 isoforms was most strongly inhibited by ALLN, demonstrating the major involvement of the proteasome. A statistically significant inhibition of lysosomal degradation by leupeptin was only detected for rsh-DM20, although both rsh-PLP and rsh-DM20 were located in the lysosome. In some experiments, we could observe effects of leupeptin on rsh-PLP degradation and also on msd-DM20 degradation (see Fig. 7A).
Figure 7.
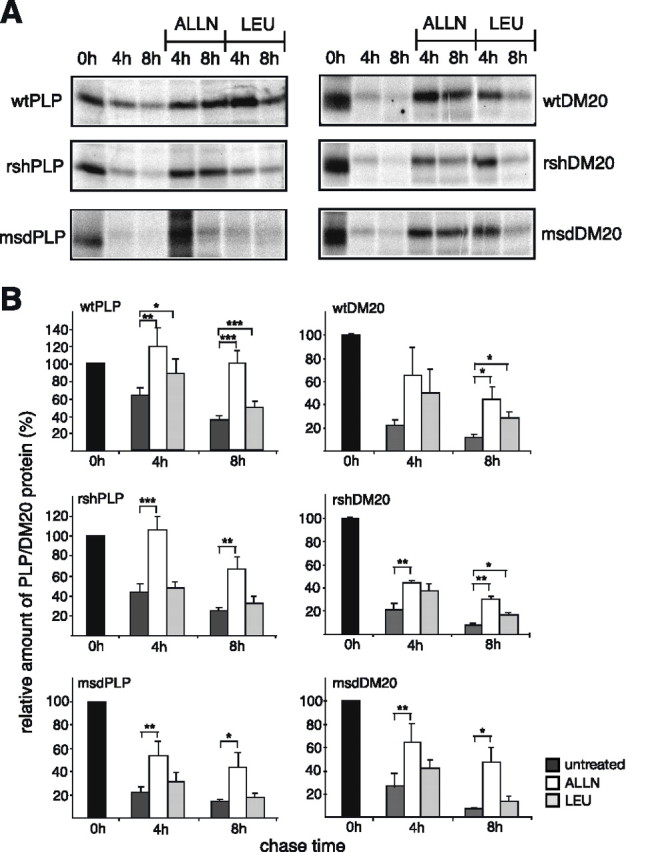
Proteasomal and lysosomal degradation of PLP and DM20. Wild-type and mutant PLP- or DM20-expressing Oli-neu cells were subjected to pulse-chase analysis as described in Figure 6. During the chase, the cells were either left untreated, or treated with the proteasome inhibiting peptide ALLN (100 μm), or the lysosomal enzyme inhibitor leupeptin (LEU) (100 μg/ml). A, Immunoprecipitation of wild-type and mutant PLP (left panels) and DM20 (right panels) analyzed by SDS-PAGE and phosphoimaging of 35S-labeled PLP/DM20. B, Densitometric quantification of recovered PLP/DM20. Statistical significance was determined by applying the paired t test (*p < 0.05; **p < 0.01; ***p < 0.001). Wild-type PLP and DM20 are degraded by both the proteasome and the lysosome. Mutant PLP and DM20 are predominantly degraded by the proteasome. Significant lysosomal degradation of rsh-DM20 is observed. n = 8 for wild-type and rsh-PLP; n = 6 for msd-PLP; n = 3 for wild-type and rsh-DM20; n = 4 for msd-DM20. Error bars indicate SEM.
Does the inhibition of proteasomal or lysosomal functions induce the accumulation of mutant PLP/DM20 or influence its subcellular distribution? Inhibition of degradation could favor aggresome formation (Johnston et al., 1998). We therefore costained ALLN- and leupeptin-treated cells with antibodies recognizing PLP/DM20 and calnexin (ER marker) or LAMP1 (late endosomal/lysosomal marker). After 8 h of ALLN treatment, ER staining of rsh-PLP was pronounced and cells adopted a rounded morphology, reminiscent of msd-expressing cells (Fig. 8A). In contrast, with leupeptin treatment, the lysosomal localization of rsh-PLP was strongly increased and lysosomes appeared swollen, demonstrating that rsh-PLP accumulates in late endosomes/lysosomes with inhibition of lysosomal function (Fig. 8B). Treatment with either of the inhibitors did not induce a change in distribution of msd-PLP, which remained strictly localized to the ER. Formation of aggresomes in response to the inhibition of protein degradation was not observed.
We conclude that the turnover of wild-type PLP and DM20 includes proteasomal as well as lysosomal degradation. Mutant PLP/DM20 is degraded predominantly by the proteasome, reflecting the degradation of misfolded protein. Lysosomal degradation is additionally involved in degradation of rsh-DM20, which behaves in many respects like a wild-type protein.
Discussion
Based on studies in heterologous expression systems, a common pathomechanism has been suggested for missense mutations of the PLP1 gene, involving accumulation of misfolded mutant protein in the ER and induction of the UPR (Gow and Lazzarini, 1996; Jung et al., 1996; Gow et al., 1998; Southwood et al., 2002). However, the choice of cell line has an unexpected influence on the distribution of PLP and DM20, making the analysis of PLP1 mutations in glial cells essential. We have used the oligodendroglial cell line Oli-neu to examine the msd (A242V) and rsh (I186T) mutations, which lead to severe connatal PMD and mild SPG-2, respectively. We found that distinct trafficking defects are associated with these mutations: Whereas msd-derived gene products are strictly retained in the ER, both rsh-DM20 and rsh-PLP overcome ER retention and localize to late endosomes/lysosomes. However, rsh-PLP and rsh-DM20 exhibit reduced cholesterol binding and impaired lipid raft association. This strongly suggests that cholesterol binding is essential for proteolipids to reach the myelin compartment. Because cholesterol is by itself essential and rate-limiting for myelin growth (Saher et al., 2005), our studies provide a plausible explanation for the hypomyelinated phenotype of rsh mutant mice and humans suffering from SPG-2. Finally, we demonstrate that, in oligodendroglial cells, mutant PLP/DM20 is rapidly degraded, predominantly by the proteasome.
High surface transport efficiency of PLP/DM20 in oligodendroglial cells
The subcellular distribution of wild-type PLP/DM20 overexpressed in Oli-neu cells mimics that of endogenous expression in primary oligodendrocytes. When wild-type PLP or DM20 are expressed in fibroblast cell lines, a considerable fraction of the protein is localized to the ER, and only a subpopulation of cells manage to transport the correctly folded protein to the cell surface. In contrast, oligodendroglial cells exhibit a high level of surface expression, and ER localization of wild-type PLP/DM20 is barely detectable.
Similarly, folding and transport of mutant PLP/DM20 proceeds with higher efficiency in oligodendroglial cells compared with COS7 cells. Oli-neu cells incorporate correctly folded rsh-PLP also in the plasma membrane, albeit at reduced levels compared with wild-type protein. Thus, the folding of PLP/DM20 and progression through the secretory pathway appears inherently more efficient in oligodendroglial cells than in fibroblasts, possibly because of the action of cell type specific chaperones and/or sorting signals. In COS7 cells, ER retention of msd-PLP is characterized by a strong interaction with the ER chaperones BiP and calnexin (Swanton et al., 2003) and is accompanied by premature oligomerization of mutant proteins (Swanton et al., 2005). Chaperones are also good candidates for modifier genes, which have been postulated to influence the severity of the rsh pathology (Al-Saktawi et al., 2003).
Mistrafficking and cellular pathology
Previous studies suggested that ER retention is the common feature of all PLP missense mutations (Gow and Lazzarini, 1996; Gow et al., 1998). The milder phenotype of some mutations was explained by limited export of mutant DM20. Here, we show, using oligodendroglial cells, that rsh- and msd-PLP/DM20 adopt distinct subcellular localizations, and that loss of cholesterol binding as well as lipid raft association is a common molecular signature of mutant PLP isoforms. Whereas msd-PLP/DM20 was completely retained in the ER, a substantial fraction of rsh-PLP and rsh-DM20 was localized in late endosomes/lysosomes, similar to wild-type PLP/DM20. A similar endosomal trafficking has been observed for disease-associated missense mutations within the second extracellular loop (A. Dhaunchak and K. A. Nave, unpublished observations).
Msd mice and humans affected with connatal PMD suffer from hypomyelination caused by oligodendroglial apoptosis as a result of UPR induction (Gow et al., 1998). Because oligodendrocyte degeneration and cell death is not observed in rsh mice (Schneider et al., 1992), the mechanism of hypomyelination in these mice (and likewise in SPG-2) requires a different explanation. Our study revealed that both rsh-PLP and rsh-DM20 escape ER retention in Oli-neu cells, similar to rsh oligodendrocytes in vivo (Thomson et al., 1997; McLaughlin et al., 2006). Moreover, we demonstrated impaired cholesterol binding and inefficient association of rsh-PLP/DM20 with lipid rafts, which have been proposed to mediate efficient sorting of PLP/DM20 to myelin (Simons et al., 2000). Most likely, cholesterol modification of PLP/DM20 promotes raft association. Rsh-PLP/DM20 may additionally reach the plasma membrane independent of lipid rafts by bulk flow. However, absence of rsh-PLP/DM20 from lipid rafts (and thus partitioning into the liquid-disordered lipid phase of the membrane) could potentially reduce its half-life and impair its transport into the myelin compartment.
Transgenic overexpression of PLP/DM20, which causes dysmyelination (Readhead et al., 1994), has been associated with abnormal endosomal/lysosomal accumulation of PLP and cholesterol in oligodendrocytes (Simons et al., 2002). Moreover, it has been suggested that this sequestration of PLP-associated cholesterol in lysosomes (possibly with other lipid raft components) underlies the dysmyelination, both in mice and in PMD patients with a PLP1 genomic duplication. Indeed, lipid rafts are implicated in the sorting of myelin components to the myelin sheath (Simons et al., 2000; Lee, 2001), and cholesterol availability is a rate-limiting factor for myelination in cholesterol synthesis-deficient conditional mouse mutants (Saher et al., 2005). Thus, impaired cholesterol binding and lipid raft association of the rsh proteolipids, as determined in the present study, may be an important factor contributing to dysmyelination in rsh mice.
Turnover and proteasomal degradation of mutant PLP/DM20
We found that in Oli-neu cells, the turnover rate of mutant PLP is increased compared with wild-type protein. Interestingly, the more aggressive msd-PLP was degraded even faster than rsh-PLP. This is in agreement with in vivo studies, which suggest limited accumulation of mutant PLP/DM20 in the cell bodies of oligodendrocytes (Schiffmann and Boespflug-Tanguy, 2001). Increased turnover of rsh-PLP/DM20 has also been observed recently in primary oligodendrocytes isolated from rsh mice (McLaughlin et al., 2006). Apparently, accumulation of misfolded PLP/DM20 and deposition of insoluble aggregates is not a key feature of the pathology of PLP1 missense mutations. Rather, ER quality control and induction of the UPR may increase disposal of the abnormal deleterious proteins and contribute to the survival of rsh-PLP/DM20-expressing cells. However, the gateway to cell toxicity and death in the life cycle of msd-PLP/DM20 remains obscure.
The degradation of mutant PLP/DM20 in Oli-neu cells was dependent on proteasomal activity. A fraction of wild-type PLP/DM20 was also degraded by the proteasome, most likely reflecting incompletely folded wild-type PLP/DM20 molecules that are removed by ER-associated degradation. Conformational maturation of PLP/DM20 appears to be a slow and inefficient event, which is characteristic of other polytopic and oligomeric membrane proteins involved in diseases such as the CFTR (cystic fibrosis transmembrane conductance regulator) and connexin-32 (Ward et al., 1995; VanSlyke et al., 2000). Lysosomal degradation most likely is responsible for the turnover of PLP/DM20 molecules, which initially passed ER quality control and were inserted in the plasma membrane. Turnover of rsh-PLP/DM20 was also dependent on lysosomal function, reflecting the population of molecules that had overcome ER retention.
Interestingly, with proteasomal inhibition, mutant PLP did not induce aggresome formation, which indicates an overloading of the cellular degradation machinery and has been shown to play a role in other neurodegenerative disorders, including peripheral neuropathies caused by PMP-22 mutations (Johnston et al., 1998; Kopito, 2000; Ryan et al., 2002). Proteasome or lysosome inhibition shifted the subcellular distribution of rsh protein toward ER and lysosome, respectively, whereas the distribution of msd protein was unaffected. Increased ER retention of rsh protein frequently resulted in morphologically abnormal cells reminiscent of msd-expressing cells, indicating that indeed the level of ER retention of mutant protein is a factor determining disease severity. A low level of ER retention of rsh-PLP/DM20 under normal conditions may thus be linked to the milder phenotype of the disease.
Footnotes
This work was supported by Deutsche Forschungsgemeinschaft Grant SFB523 (K.-A.N.) and a grant from the European Leukodystrophy Association (E.-M.K.-A.). We thank Ulrike Stapf and Lilja Niedens for excellent technical assistance, Christine Winterstein for providing images, and Gesine Saher for critical comments on this manuscript.
References
- Al-Saktawi K, McLaughlin M, Klugmann M, Schneider A, Barrie JA, McCulloch MC, Montague P, Kirkham D, Nave KA, Griffiths IR. Genetic background determines phenotypic severity of the PLP-rumpshaker mutation. J Neurosci Res. 2003;72:12–24. doi: 10.1002/jnr.10561. [DOI] [PubMed] [Google Scholar]
- Duncan ID. The PLP mutants from mouse to man. J Neurol Sci. 2005;228:204–205. doi: 10.1016/j.jns.2004.10.011. [DOI] [PubMed] [Google Scholar]
- Forman MS, Lee VY, Trojanowski JQ. “Unfolding” pathways in neurodegenerative disease. Trends Neurosci. 2003;26:407–410. doi: 10.1016/S0166-2236(03)00197-8. [DOI] [PubMed] [Google Scholar]
- Garbern JY. Pelizaeus-Merzbacher disease: pathogenic mechanisms and insights into the roles of proteolipid protein 1 in the nervous system. J Neurol Sci. 2005;228:201–203. doi: 10.1016/j.jns.2004.10.010. [DOI] [PubMed] [Google Scholar]
- Garbern JY, Yool DA, Moore GJ, Wilds IB, Faulk MW, Klugmann M, Nave KA, Sistermans EA, van der Knaap MS, Bird TD, Shy ME, Kamholz JA, Griffiths IR. Patients lacking the major CNS myelin protein, proteolipid protein 1, develop length-dependent axonal degeneration in the absence of demyelination and inflammation. Brain. 2002;125:551–561. doi: 10.1093/brain/awf043. [DOI] [PubMed] [Google Scholar]
- Gow A, Lazzarini RA. A cellular mechanism governing the severity of Pelizaeus-Merzbacher disease. Nat Genet. 1996;13:422–428. doi: 10.1038/ng0896-422. [DOI] [PubMed] [Google Scholar]
- Gow A, Southwood CM, Lazzarini RA. Disrupted proteolipid trafficking results in oligodendrocyte apoptosis in an animal model of Pelizaeus-Merzbacher disease. J Cell Biol. 1998;140:925–934. doi: 10.1083/jcb.140.4.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths I, Klugmann M, Anderson TJ, Thomson C, Vouyiouklis D, Nave KA. Current concepts of PLP and its role in the nervous system. Microsc Res Tech. 1998a;41:344–358. doi: 10.1002/(SICI)1097-0029(19980601)41:5<344::AID-JEMT2>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Griffiths I, Klugmann M, Anderson T, Yool D, Thomson C, Schwab MH, Schneider A, Zimmermann F, McCulloch M, Nadon N, Nave KA. Axonal swellings and degeneration in mice lacking the major proteolipid of myelin. Science. 1998b;280:1610–1613. doi: 10.1126/science.280.5369.1610. [DOI] [PubMed] [Google Scholar]
- Hudson LD. Proteolipid protein gene. In: Lazzarini RA, editor. Myelin biology and disorders. London: Elsevier AP; 2004. pp. 401–420. [Google Scholar]
- Inoue K. PLP1-related inherited dysmyelinating disorders: Pelizaeus-Merzbacher disease and spastic paraplegia type 2. Neurogenetics. 2005;6:1–16. doi: 10.1007/s10048-004-0207-y. [DOI] [PubMed] [Google Scholar]
- Johnston JA, Ward CL, Kopito RR. Aggresomes: a cellular response to misfolded proteins. J Cell Biol. 1998;143:1883–1898. doi: 10.1083/jcb.143.7.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung M, Krämer EM, Grzenkowski M, Tang K, Blakemore W, Aguzzi A, Khazaie K, Chlichlia K, von Blankenfeld G, Kettenmann H, Trotter J. Lines of murine oligodendroglial precursor cells immortalized by an activated neu tyrosine kinase show distinct degrees of interaction with axons in vitro and in vivo. Eur J Neurosci. 1995;7:1245–1265. doi: 10.1111/j.1460-9568.1995.tb01115.x. [DOI] [PubMed] [Google Scholar]
- Jung M, Sommer I, Schachner M, Nave KA. Monoclonal antibody O10 defines a conformationally sensitive cell-surface epitope of proteolipid protein (PLP): evidence that PLP misfolding underlies dysmyelination in mutant mice. J Neurosci. 1996;16:7920–7929. doi: 10.1523/JNEUROSCI.16-24-07920.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knapp PE, Skoff RP, Redstone DW. Oligodendroglial cell death in jimpy mice: an explanation for the myelin deficit. J Neurosci. 1986;6:2813–2822. doi: 10.1523/JNEUROSCI.06-10-02813.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi H, Hoffman EP, Marks HG. The rumpshaker mutation in spastic paraplegia. Nat Genet. 1994;7:351–352. doi: 10.1038/ng0794-351. [DOI] [PubMed] [Google Scholar]
- Koeppen AH, Robitaille Y. Pelizaeus-Merzbacher disease. J Neuropathol Exp Neurol. 2002;61:747–759. doi: 10.1093/jnen/61.9.747. [DOI] [PubMed] [Google Scholar]
- Komaki H, Sasaki M, Yamamoto T, Iai M, Takashima S. Connatal Pelizaeus-Merzbacher disease associated with the jimpy(msd) mice mutation. Pediatr Neurol. 1999;20:309–311. doi: 10.1016/s0887-8994(98)00160-x. [DOI] [PubMed] [Google Scholar]
- Kopito RR. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 2000;10:524–530. doi: 10.1016/s0962-8924(00)01852-3. [DOI] [PubMed] [Google Scholar]
- Krämer EM, Koch T, Niehaus A, Trotter J. Oligodendrocytes direct glucosylphosphatidylinositol-anchored proteins to the myelin sheath in glycosphingolipid-rich complexes. J Biol Chem. 1997;272:8937–8945. doi: 10.1074/jbc.272.14.8937. [DOI] [PubMed] [Google Scholar]
- Krämer EM, Klein C, Koch T, Boytinck M, Trotter J. Compartmentation of Fyn kinase with glycosylphosphatidylinositol-anchored molecules in oligodendrocytes facilitates kinase activation during myelination. J Biol Chem. 1999;274:29042–29049. doi: 10.1074/jbc.274.41.29042. [DOI] [PubMed] [Google Scholar]
- Krämer EM, Schardt A, Nave KA. Membrane traffic in myelinating oligodendrocytes. Microsc Res Tech. 2001;52:656–671. doi: 10.1002/jemt.1050. [DOI] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Lee AG. Myelin: delivery by raft. Curr Biol. 2001;11:R60–R62. doi: 10.1016/s0960-9822(01)00008-2. [DOI] [PubMed] [Google Scholar]
- McLaughlin M, Barrie JA, Karim S, Montague P, Edgar JM, Kirkham D, Thomson CE, Griffiths IR. Processing of PLP in a model of Pelizaeus-Merzbacher disease/SPG-2 due to the rumpshaker mutation. Glia. 2006;53:715–722. doi: 10.1002/glia.20325. [DOI] [PubMed] [Google Scholar]
- Readhead C, Schneider A, Griffiths I, Nave KA. Premature arrest of myelination in transgenic mice with increased proteolipid protein gene dosage. Neuron. 1994;12:583–595. doi: 10.1016/0896-6273(94)90214-3. [DOI] [PubMed] [Google Scholar]
- Ryan MC, Shooter EM, Notterpek L. Aggresome formation in neuropathy models based on peripheral myelin protein 22 mutations. Neurobiol Dis. 2002;10:109–118. doi: 10.1006/nbdi.2002.0500. [DOI] [PubMed] [Google Scholar]
- Saher G, Brugger B, Lappe-Siefke C, Mobius W, Tozawa R, Wehr MC, Wieland F, Ishibashi S, Nave KA. High cholesterol level is essential for myelin membrane growth. Nat Neurosci. 2005;8:468–475. doi: 10.1038/nn1426. [DOI] [PubMed] [Google Scholar]
- Schiffmann R, Boespflug-Tanguy O. An update on the leukodsytrophies. Curr Opin Neurol. 2001;14:789–794. doi: 10.1097/00019052-200112000-00018. [DOI] [PubMed] [Google Scholar]
- Schneider A, Montague P, Griffiths I, Fanarraga M, Kennedy P, Brophy P, Nave KA. Uncoupling of hypomyelination and glial cell death by a mutation in the proteolipid protein gene. Nature. 1992;358:758–761. doi: 10.1038/358758a0. [DOI] [PubMed] [Google Scholar]
- Simons M, Krämer EM, Thiele C, Stoffel W, Trotter J. Assembly of myelin by association of proteolipid protein with cholesterol- and galactosylceramide-rich membrane domains. J Cell Biol. 2000;151:143–154. doi: 10.1083/jcb.151.1.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons M, Krämer EM, Macchi P, Rathke-Hartlieb S, Trotter J, Nave KA, Schulz JB. Overexpression of the myelin proteolipid protein leads to accumulation of cholesterol and proteolipid protein in endosomes/lysosomes: implications for Pelizaeus-Merzbacher disease. J Cell Biol. 2002;157:327–336. doi: 10.1083/jcb.200110138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southwood CM, Garbern J, Jiang W, Gow A. The unfolded protein response modulates disease severity in Pelizaeus-Merzbacher disease. Neuron. 2002;36:585–596. doi: 10.1016/s0896-6273(02)01045-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanton E, High S, Woodman P. Role of calnexin in the glycan-independent quality control of proteolipid protein. EMBO J. 2003;22:2948–2958. doi: 10.1093/emboj/cdg300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanton E, Holland A, High S, Woodman P. Disease-associated mutations cause premature oligomerization of myelin proteolipid protein in the endoplasmic reticulum. Proc Natl Acad Sci USA. 2005;102:4342–4347. doi: 10.1073/pnas.0407287102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiele C, Hannah MJ, Fahrenholz F, Huttner WB. Cholesterol binds to synaptophysin and is required for biogenesis of synaptic vesicles. Nat Cell Biol. 2000;2:42–49. doi: 10.1038/71366. [DOI] [PubMed] [Google Scholar]
- Thomson CE, Montague P, Jung M, Nave KA, Griffiths IR. Phenotypic severity of murine Plp mutants reflects in vivo and in vitro variations in transport of PLP isoproteins. Glia. 1997;20:322–332. doi: 10.1002/(sici)1098-1136(199708)20:4<322::aid-glia5>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- Trombetta ES, Parodi AJ. Quality control and protein folding in the secretory pathway. Annu Rev Cell Dev Biol. 2003;19:649–676. doi: 10.1146/annurev.cellbio.19.110701.153949. [DOI] [PubMed] [Google Scholar]
- VanSlyke JK, Deschenes SM, Musil LS. Intracellular transport, assembly, and degradation of wild-type and disease-linked mutant gap junction proteins. Mol Biol Cell. 2000;11:1933–1946. doi: 10.1091/mbc.11.6.1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward CL, Omura S, Kopito RR. Degradation of CFTR by the ubiquitin-proteasome pathway. Cell. 1995;83:121–127. doi: 10.1016/0092-8674(95)90240-6. [DOI] [PubMed] [Google Scholar]
- Weimbs T, Stoffel W. Proteolipid protein (PLP) of CNS myelin: positions of free, disulfide-bonded, and fatty acid thioester-linked cysteine residues and implications for the membrane topology of PLP. Biochemistry. 1992;31:12289–12296. doi: 10.1021/bi00164a002. [DOI] [PubMed] [Google Scholar]
- Werner H, Jung M, Klugmann M, Sereda M, Griffiths IR, Nave KA. Mouse models of myelin diseases. Brain Pathol. 1998;8:771–793. doi: 10.1111/j.1750-3639.1998.tb00200.x. [DOI] [PMC free article] [PubMed] [Google Scholar]