

Abstract
Amyloid-β peptide (Aβ)-induced death in cerebral endothelial cells (CECs) is preceded by mitochondrial dysfunction and signaling events characteristic of apoptosis. Mitochondria-dependent apoptosis engages Bcl-2 family proteins, especially the BH3-only homologues, which play a key role in initiating the apoptotic cascade. Here, we report that the expression of bim, but not other BH3-only members, was selectively increased in cerebral microvessels isolated from 18-month-old APPsw (Tg2576) mice, a model of cerebral amyloid angiopathy (CAA), suggesting a pivotal role for Bim in Aβ-induced cerebrovascular degeneration in vivo. A similar expression profile was observed in Aβ-treated CECs. Furthermore, Aβ induction of bim expression involved a pro-apoptotic transcription factor, FKHRL1. FKHRL1 bound to a consensus sequence in the bim promoter region and was activated by Aβ before bim expression. FKHRL1 activity was negatively regulated by phosphorylation catalyzed by Akt, an anti-apoptotic kinase. Akt upregulation by adenoviral gene transfer inhibited Aβ-induced FKHRL1 activation and bim induction. In addition, Aβ increased the activity of protein phosphatase 2A (PP2A), a ceramide-activated protein phosphatase. Suppression of PP2A activity by RNA interference or a specific inhibitor, okadaic acid, effectively suppressed Aβ-induced Akt inactivation and FKHRL1 activation, leading to an attenuation of bim expression and cell death in CECs. Coimmunoprecipitation experiments revealed that Aβ enhanced the binding of the PP2A regulatory subunit PP2ACαβ to Akt. These results implicate PP2A as an early regulator of Aβ-induced bim expression and CEC apoptosis via the Akt/FKHRL1 signaling pathway. We raise the possibility that this pathway may play a role in cerebrovascular degeneration in CAA.
Keywords: amyloid β, peptide, apoptosis, bim, ceramide-activated protein phosphatases, cerebral endothelial cells, FKHRL1
Introduction
Amyloid-β peptide (Aβ) has been implicated in the pathogenesis of Alzheimer's disease (AD) (Wisniewski and Wegiel, 1995; Yankner, 1996). In addition to parenchymal accumulation and neuronal degeneration in AD brains (Yankner et al., 1989; Behl et al., 1994), Aβ also accumulates in the cerebrovascular wall of elderly individuals with or without AD, leading to cerebral amyloid angiopathy (CAA) (Perlmutter et al., 1994; Wisniewski et al., 2000). Aβ induces death of cerebral endothelial cells (CECs) (Price et al., 1997; Preston et al., 1998) with features suggestive of apoptosis (Blanc et al., 1997; Suo et al., 1997; Xu et al., 2001; Yin et al., 2002b). We have recently reported that Aβ-induced CEC apoptosis was associated with the induction of Bim, an apoptotic initiator, to release pro-apoptotic mitochondrial molecules into the cytosol (Yin et al., 2002b). Bim belongs to the “BH3-only proteins,” a subgroup of Bcl-2 apoptotic regulators that contain only one of the bcl-2 homology regions (BH3). In response to apoptotic stimuli, BH3-only proteins translocate to the mitochondrial membrane from other cellular compartments to interfere with the function of antiapoptotic Bcl-2 family members to initiate apoptosis (Huang and Strasser, 2000). Although some BH3-only proteins may be post-translationally modified by upstream death signaling events to trigger translocation (Dijkers et al., 2000; Huang and Strasser, 2000; Whitfield et al., 2001), others such as Bim are transcriptionally regulated to initiate apoptosis (Bouillet et al., 2001). Bim expression is regulated by transcription factors of the forkhead in rhabdomyosarcoma (FKHR) family, which includes FKHR (also known as FOXO1) and FKHRL1 (FOXO3a) (Burgering and Kops, 2002; Gilley et al., 2003). Forkhead protein activity, in turn, is negatively regulated via phosphorylation by the survival promoting kinase Akt (Brunet et al., 1999; Tang et al., 1999).
Akt maintains cell viability by phosphorylating pro-apoptotic mediators including forkhead proteins (Brunet et al., 1999; Zheng et al., 2000). Dephosphorylation of Akt by protein phosphatases disinhibits pro-apoptotic proteins to initiate apoptotic processes (Ugi et al., 2004; Gao et al., 2005). Protein phosphatase 2A (PP2A), a member of the ceramide-activated protein phosphatases (CAPPs) family, is a serine/threonine-specific protein phosphatase that regulates the activities of several major protein kinase families, including Akt to drive apoptotic processes (Millward et al., 1999; Silverstein et al., 2002).
We have shown previously that Aβ cytotoxicity is mediated by ceramide (Xu et al., 1998; Lee et al., 2004; Yang et al., 2004), a pro-apoptotic lipid mediator (Obeid et al., 1993; Brugg et al., 1996; Mangoura and Dawson, 1998). This Aβ-ceramide death pathway involves the activation of neutral sphingomyelinase (nSMase) (Lee et al., 2004; Yang et al., 2004), a ceramide-synthesizing enzyme. We also demonstrated that Aβ-induced CEC death is associated with the rapid induction of Bim and the activation of subsequent apoptotic pathways (Yin et al., 2002b). The link between these two cell death pathways remains to be established. In the present study, we report that activation of PP2A is the intermediate step between the Aβ-ceramide cascade and the subsequent inactivation of Akt, activation of FKHRL1, and upregulation of bim.
Materials and Methods
Mouse CEC primary cultures.
Mouse CECs were prepared as described previously (Xu et al., 1992). Briefly, mouse CECs migrating from isolated microvessel preparations were pooled to form a proliferating cell culture that was maintained in DMEM, with high glucose and l-glutamine supplemented with 10% FBS, 0.5 mg/ml heparin, and 75 μg/ml endothelial cell growth supplements. Mouse CECs (between passages 4 and 15) uniformly positive for factor VIII, vimentin, and characteristic bradykinin receptors (>95% endothelial cell purity) were grown to 85–95% confluence before use (Xu et al., 1992).
Treatment with Aβ and other chemicals.
Aβ1–40 is the major component of amyloid deposits in cerebrovascular vessels in CAA. However, in most experimental systems, the biological effects of fragment Aβ25–35 are comparable (Loo et al., 1993; Behl et al., 1994). We have shown previously that Aβ25–35 has approximately the same potency as Aβ1–40 in inducing cell death in CECs (Xu et al., 2001; Yin et al., 2002b). Based on these data, we used Aβ25–35 in most experiments in this study and confirmed key findings with Aβ1–40. Several protocols for Aβ aggregation were used in the present studies. For Aβ25–35 (Sigma, St. Louis, MO), a 1 mm peptide stock solution was prepared in sterile double-distilled H2O and maintained for 3 d at room temperature to allow polymerization. For Aβ1–40 (Global Peptides, Fort Collins, CO), the peptide was first dissolved in dimethyl sulfoxide to a concentration of 5 mm and then diluted into PBS to a final concentration of 500 μm. The diluted peptide solution was allowed to aggregate at 37°C for 5 d in a 5% CO2-supplemented atmosphere. CECs were treated with 25 μm Aβ25–35, 25 μm Aβ1–40, or 25 μm C2 ceramide (Biomol, Plymouth Meeting, PA) in serum-free growth medium for various time intervals. In some experiments, CECs were treated with okadaic acid (OA), a selective PP2A inhibitor, or 1,2-dioleoyl-sn-glycero-3-phosphate (PA), a selective PP1 inhibitor (Upstate Biotechnology, Lake Placid, NY) 1 h before Aβ exposure. For nSMase inhibitor studies, CECs were treated with 1 μm 3-O-methyl-sphingomyelin (3-OMe-SM), a specific nSMase inhibitor, for 2 h before 25 μm Aβ25–35 exposure for 4 h.
APPsw transgenic mice and cerebral microvessel isolation.
APPsw mice overexpress human APP695 with the familial Swedish AD mutations at positions 670/671 under control of the prion promoter and were a generous gift from Dr. K. Hsiao-Ashe (Minneapolis, MN). The production, genotyping, and background strain (B6/SJL) of APPsw (Tg2576) mice used in this study have been described previously (Hsiao et al., 1996). Cerebral microvessel isolation used previously described methods with modifications (Pardridge et al., 1985; Zlokovic et al., 1993). Briefly, mice were killed by decapitation under anesthesia. The brains were immediately removed from the skull and immersed in ice-cold buffer A (in mm: 103 NaCl, 4.7 KC1, 2.5 CaC12, 1.2 KH2PO4, 1.2 MgSO4, and 15 HEPES, pH 7.4). The brain was homogenized in a fivefold volume excess of buffer B (103 mm NaCl, 4.7 mm KC1, 2.5 mm CaC12, 1.2 mm KH2PO4, 1.2 mm MgSO4, 15 mm HEPES, 25 mm HCO3, 10 mm glucose, 1 mm sodium pyruvate, and 1 g/100 ml dextran, pH 7.4) with a Teflon homogenizer. The homogenate was suspended in an equal volume of 25% BSA and centrifuged at 5800 × g at 4°C for 30 min. The pellet was resuspended in 10 ml of buffer B and passed over an 85 μm nylon mesh (Tetko, Depu, NY). This filtrate was then passed over a 3 × 4 cm glass bead column (0.45 mm glass beads) with a 44 μm nylon mesh at the bottom and washed with 400 ml of buffer B. Microvessels adhere to the glass beads, whereas contaminants pass unimpeded. Microvessels were recovered by repeated gentle agitation of the glass beads in buffer B. The supernatant containing microvessels was decanted and spun at 500 × g for 5 min, and the final pellet was stored at −80°C until RNA extraction.
Real-time PCR and reverse transcription-PCR.
Real-time PCR was used to measure mRNA expression of BH3-only proteins. Total RNA from the mouse brain microvessel fraction or cultured mouse CECs were isolated with the RNeasy Mini kit (Qiagen, Valencia, CA). Equal amounts of total RNA (600 ng) were reverse transcribed with 1 μmol/L oligo (dT) and 0.5 mmol/L dNTPs (Qiagen), 10 U of Rnasin (Promega, Madison, WI), and 4 U of reverse transcriptase (Qiagen) for 1 h at 37°C. Ten nanograms of cDNA were subjected to PCR using the ABI Prism 7000 Sequence Detection System and SYBR Green PCR Master Mix (both from Applied Biosystems, Foster City, CA). The primers of the BH3-only genes were designed using Primer Express software (Applied Biosystems) as follows: bim (forward, 5′-CGGATCGGAGACGAGTTCA-3′; reverse, 5′-TTCAGCCTCGCGGTAATCA-3′); bid (forward, 5′- GAAGACGAGCTGCAGACAGATG-3′; reverse, 5′- TGGCTCTATTCTTCCTTGGTTGA-3′); bad (forward, 5′- GCAGGCACTGCAACACAGAT-3′; reverse, 5′- CTCCTTTGCCCAAGTTTCGAT-3′); bmf (forward, 5′- TACGCAACAACACCAGCAGAA-3′; reverse, 5′- CGAGGTTTTGAAGGAAGAGGAA-3′); dp5 (forward, 5′- TGGAGGAAGCTGGTTCCTGTT-3′; reverse, 5′- CAGCTCTTTACAATTCTGCTTCCTT-3′); cyclophilin (forward, 5′-CGCTTCCCAGATGAGAACTTCA-3′; reverse, 5- ACTGTGGTTATGAAGAACTGTGA-3′). A standard amplification program was used (1 cycle of 50°C for 2 min, 1 cycle of 95°C for 10 min, 45 cycles of 95°C for 15 s, and 60°C for 1 min). All quantifications were normalized to cyclophilin to account for variability in the initial concentration and quality of total RNA and in the conversion efficiency of the reverse transcription (RT) reaction. The expression of bim mRNA was also detected by conventional RT-PCR (Yin et al., 2002a). cDNA was amplified in 0.2 mmol/L dNTPs, 1 μmol/L of each primer, 1.5 mmol/L MgCl2, and 2.5 U of Taq polymerase (Roche, Indianapolis, IN). PCR was performed for 26 cycles alternating between 95°C for 20 s, 53°C for 30 s, followed by extension at 72°C for 1 min. Primers were designed based on the mouse bim sequences (forward, 5′-ctgagtgtgacagagaaggtg-3′; reverse, 5′-gtggtcttcagcctcgcggt-3′). The amplified products were analyzed on a 1% agarose gel. The relative mRNA level of bim was normalized to endogenous cyclophilin mRNA for each sample. The RT-PCR was conducted within the linear ranges of PCR cycles and RNA input. The PCR experiments were repeated at least three times.
Protein phosphatase activity assay.
An immunoprecipitation phosphatase assay kit (Upstate Biotechnology) was used to measure phosphate release as an index of phosphatase activity. Total cellular proteins from Aβ-treated mouse CECs or cerebral microvessels in APPsw transgenic mice were extracted in radioimmunoprecipitation assay buffer. Protein concentrations were determined using a Bio-Rad (Hercules, CA) bicinchoninic acid assay. Assays were run in 96-well plates according to the manufacturer's instructions. Briefly, 50–100 μg of cellular proteins was immunoprecipitated with a mouse anti-PP2A catalytic subunit antibody (2 μg/ml; Upstate Biotechnology) or a mouse anti-PP1 antibody (2 μg/ml; Santa Cruz Biotechnology, Santa Cruz, CA) and incubated with 4.5 μl of the substrate, phosphoprotein (amino acid sequence: KRpTIRR) at a concentration of 175 μm, and protein phosphatase assay buffer (20 mm 4-morpholinepropanesulfonic acid, pH 7.5, 60 mm 2-mercaptoethanol, 0.1 m NaCl, and 0.1 mg/ml serum albumin) in a final volume of 25 μl. Reactions were started with the addition of the phosphoprotein substrate and conducted for 10 min at room temperature. Reactions were terminated by the addition of 100 μl of malachite green solution, and color was developed for 15 min before reading the plate at 650 nm.
Electrophoretic mobility shift assay.
Electrophoretic mobility shift assay (EMSA) to assess DNA binding activity has been described in detail previously (Yin et al., 2002a,b). The following FKHRL1 consensus oligonucleotides were used, 5′-AATAGATCTTAAATAAATAGATCTTTA-3′ and 5′- TAAAGATCTATTTATTTAAGATCTATT-3′ (Brunet et al., 1999), and labeled with γ-32P-ATP. The binding reaction was performed in a final volume of 20 μl of binding buffer containing 0.0175 pmol of labeled probe (>10,000 cpm), 20 μg of nuclear proteins, and 1 μg of poly dIdC. After incubation for 20 min at room temperature, the mixture was subjected to electrophoresis on a nondenaturing 6% polyacrylamide gel at 180 V for 2 h under low ionic strength conditions. The gel was dried and subjected to autoradiography. For supershift assays, samples were incubated with anti-FKHRL1 (1:100; Upstate Biotechnology) or anti-Bad (1:100; Cell Signaling, Beverly, MA) antibody for 1 h before the addition of the γ-32P-ATP FKHRL1 oligonucleotide probe. The specificity of FKHRL1 DNA binding activity was also demonstrated by the complete inhibition of FKHRL1 binding in the presence of a 100-fold molar excess of the cold FKHRL1 oligonucleotide.
Western blot analysis.
Cytoplasmic and nuclear proteins were isolated from CECs after treatment as reported previously (Xu et al., 2001; Yin et al., 2002b). Twenty to 40 μg of proteins from each sample were analyzed by 10–15% SDS-PAGE gel and transferred to polyvinylidene difluoride (PVDF) membranes. The blot was incubated with various primary antibodies including rabbit anti-phospho-Akt (Thr308) (1;1000; Cell Signaling), rabbit anti-phospho-Akt (Ser473) (1:1000; Cell Signaling), rabbit anti-phospho-FKHRL1 (Ser253) (1;500; Upstate Biotechnology), rabbit anti-phospho-FKHRL1 (Thr32) (1:1000; Upstate Biotechnology), rabbit anti-phospho-ERK1/2 (1:1000; Cell Signaling), mouse anti-phospho-JNK (1:1000; Cell Signaling), mouse anti-PP2A catalytic subunit (1:1000; Upstate Biotechnology), and mouse anti-actin (1:200; Santa Cruz Biotechnology). The membranes were then incubated with the appropriate secondary antibody (1:5000; anti-rabbit or anti-mouse IgG conjugated with alkaline phosphatase; Promega) at room temperature for 1 h. The color reaction was developed using the AP Blot System (Promega) according to the manufacturer's instructions.
Construction of retrovirus-mediated RNA interference of PP2A in CECs.
A retrovirus vector carrying small interfering RNAs (siRNAs) targeting the mouse PP2A gene was prepared by the BD Knockout RNAi System (BD Biosciences Clontech, Palo Alto, CA). The PAGE-purified complementary oligonucleotide pair for the hairpin siRNA was synthesized to target the coding region of mouse PP2A catalytic subunit mRNA as follows: 5′-gatccGTGTGTGACTTGCTGTGGTCTTCAAGAGAGA-CCACAGCAAGTCACACATTTTTTg-3′, 5′-aattcAAAAAATGTG-TGACTTGCTGTGGTCTCTCTTGAAGACCACAGCAAGTCACA-CACg-3′. A complementary pair of oligonucleotides for a sense-only control RNA was also designed using the following sequences: 5′-gatccGTGTGTGACTTGCTGTGGTCTTCAAGAGATTTTTTg-3′, 5′-aattcAAAAAATCTCTTGAAGACCACAGCAAGTCACACACg-3′. These oligonucleotide pairs containing BamHI and EcoRI overhangs were annealed and ligated to a linearized RNAi-Ready pSIREN-RetroQ-ZsGreen vector digested with BamHI and EcoRI (BD Biosciences Clontech). The RNAi-Ready pSIREN-RetroQ-ZsGreen vector is a self-inactivating retroviral expression vector designed to express a small hairpin RNA using the human U6 promoter. The resultant constructs were amplified, purified, and sequenced. For production of high-titer retrovirus vectors expressing PP2A small hairpin RNA, EcoPack-293 cell lines (BD Biosciences Clontech) were plated at ∼50–75% confluence, and the recombinant vectors were transfected into EcoPack-293 cells by a modified calcium phosphate method. After incubation at 32°C for 2 d, the supernatant fraction containing the viral vector was collected and passed through a 0.45 μm filter. CECs were infected with the retroviral vector in the presence of Polybrene (5–10 μg/ml) and incubated for 3 h. The supernatant fraction containing the retroviral vector was removed and replaced with normal growth medium. Cells grown for 48–72 h were assessed by fluorescence microscopy. The ZsGreen fluorescent marker yields a bright green fluorescence, permitting direct monitoring of the delivery efficiency in CECs. Infected populations exhibiting between 60 and 75% green fluorescent cells were used for additional experimentation.
Construction of adenovirus-mediated overexpression of Akt in CECs.
Akt cDNAs, including a nonfunctional mutant and an overexpressing construct, were obtained from pCMV-HA-K179m-Akt (mutant Akt) and pCMV-myr-HA-Akt by PCR amplification (Franke et al., 1995). The PCR products were cloned into a pGEM–T Easy vector (Promega), isolated by NotI-NotI digestion, and subcloned into a p-Shuttle vector (BD Biosciences Clontech). The p-Shuttle vectors were subcloned into the Adeno-X expression system (BD Biosciences Clontech) using PI-SceI and I-CeuI. The resultant Adeno-X-containing Akt and mutant Akt were transformed into Escherichia coli for amplification, purified, and analyzed by PCR. Recombinant Adeno-X viral DNAs were confirmed and amplified. The recombinant Adeno-X DNAs were linearized by digestion with PacI for transfection. For production of high-titer adenoviral vector expressing Akt, 10 μg of recombinant Adeno-X DNA vector was transfected into HEK 293 cells. The adenoviral vector was collected from the cells 10–14 d after transfection and amplified for another 10–14 d to generate high-titer stocks. To concentrate adenovirus, we used a BD Adeno-X virus purification kit (BD Biosciences Clontech), and titers were determined with Adeno-X rapid titer kits (BD Biosciences Clontech). Normally, we obtained titers in the range of 1010 to 1012 ifu/ml. CECs were infected with the adenoviral vectors at a titer of 1010 ifu/ml for 48 h, and the culture medium was changed. Infected populations exhibiting between 85 and 95% green fluorescent cells were used for additional experimentation.
Coimmunoprecipitation.
Coimmunoprecipitation was conducted as described previously (Yin et al., 2002b). Aβ-treated CECs were homogenized in a lysis buffer (1% Triton X-100, 150 mm NaCl, 10 mm Tris, pH 7.4, 1 mm EDTA, 1 mm EGTA, pH 8.0, 0.2 mm sodium ortho-vanadate, 0.5% NP-40, 0.2 mm PMSF, 4 μg/ml pepstatin, 4 μg/ml leupeptin, and 5 μg/ml aprotinin), and the supernatant was incubated with a rabbit anti-Akt antibody (2 μg/ml; Cell Signaling) as described above. Protein G-Sepharose 4 fast flow (Amersham Biosciences, Uppsala, Sweden) was added to the antigen–antibody mixture and incubated with gentle agitation for 1–2 h. The immunoprecipitate was washed with the same lysis buffer, resuspended in 6× SDS loading buffer, separated in a SDS-PAGE gel, transferred to the PVDF membrane, and analyzed by Western blotting with a goat anti-PP2A-Aα, goat anti-PP2A-Bα/β/γ, goat anti-PP2A-B56α, or goat anti-PP2A-Cα/β antibody (1:200; Santa Cruz Biotechnology).
Assessment of CEC death.
CEC survival or death was assessed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) and lactate dehydrogenase (LDH) assays as described previously (Yin et al., 2002b)
Quantitative and statistical analysis.
Quantitative analysis of selected bands in Western blots, PCR images, and EMSA gels was performed using the NIH Image Analysis System.
Quantitative data were expressed as mean ± SD based on at least three separate experiments of triplicate samples. Differences among groups were statistically analyzed by one-way ANOVA, followed by Bonferroni's post hoc t test. Comparison between two experimental groups was based on the two-tailed t test. p < 0.05 was considered significant.
Results
bim mRNA expression was upregulated in amyloid-laden vessels in vivo and in Aβ-treated CECs in vitro
In a previous study, we have shown that Aβ induced Bim expression, leading to Smac release from mitochondria into cytosol to bind XIAP, a member of the IAP (inhibitor of apoptosis protein) family. This series of events resulted in CEC apoptosis (Yin et al., 2002b). Bim belongs to the BH3-only subgroup of the Bcl-2 family of apoptosis regulators (Kelekar and Thompson, 1998; Huang and Strasser, 2000). To examine the potential role of BH3-only regulators in cerebrovascular degeneration in CAA, we measured the mRNA from various BH3-only genes including bim, bid, bad, bmf, and dp5 in cerebral microvessels isolated from APPsw (Tg2576) mice at age 18 months when CAA is fully manifested (Fryer et al., 2003; Lee et al., 2003). Compared with age-matched wild-type controls, bim mRNA was selectively elevated based on real-time PCR (Fig. 1A), suggesting that the induction of bim may play an important role in cerebrovascular degeneration in this animal model of CAA. We also examined the expression of BH3-only genes in CECs treated with Aβ for 4 h. bim mRNA was also selectively induced (Fig. 1B). A time course study revealed that Aβ rapidly induced all three isoforms of bim (bimEL, bimL, bimS) in CECs, as early as 1 h after exposure; this induction was transient, because very low levels of the bim isoforms were detected beyond 24 h (Fig. 1C,D).
Figure 1.
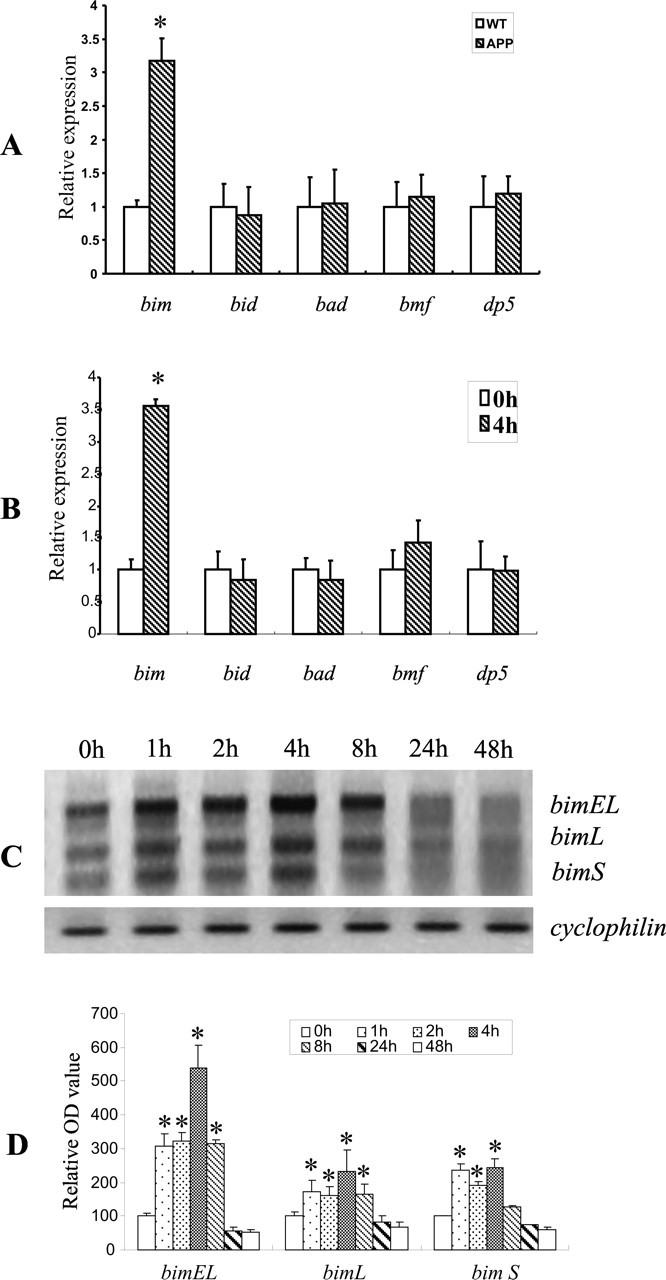
bim expression was selectively increased in amyloid-laden vessels in vivo and in Aβ-treated CECs in vitro. A, Total RNA from the cerebral microvessel fraction of 18-month-old APPsw transgenic or age-matched wild-type (WT) mice was analyzed by quantitative real-time PCR to assess expression of BH3-only genes. Only the mRNA level of bim was increased in amyloid-laden vessels of APPsw transgenic mice compared with the wild-type controls (n = 3 per group). Data are expressed as mean ± SD. *p < 0.05, significant difference from wild type. B, CECs treated with 25 μm Aβ25–35 at 0 or 4 h were isolated for extraction of total RNA for quantitative real-time PCR. Aβ exposure for 4 h significantly increased the mRNA level of bim but not other BH3-only genes. Data are expressed as mean ± SD *p < 0.05, significant difference from 0 h. C, Expression of the BH3-only genes in CECs treated with 25 μm Aβ25–35 for the indicated times were analyzed by RT-PCR. Aβ increased the expression of three isoforms of bim (bimEL, bimL, bimS) as early as 1 h after exposure. Upregulation of these three isoforms was sustained for 8 h before declining to basal levels. Levels of cyclophilin mRNA were used as an internal control. The data shown are representative of three separate experiments with similar results. D, Quantitative analysis of three PCR images (normalized to cyclophilin) using the NIH Image Analysis System graphically illustrates a time-dependent increase in the three isoforms of bim after Aβ treatment. Data (relative optical density) are expressed as mean ± SD. *p < 0.05 versus the 0 h group.
Aβ dephosphorylated Akt and increased FKHRL1 binding activity
FKHRL1, a member of the forkhead family of transcription factors (Burgering and Kops, 2002; Gilley et al., 2003), was recently shown to transactivate bim in apoptotic lymphocytes in response to cytokine withdrawal (Dijkers et al., 2000). Furthermore, overexpression of a constitutively active mutant of FKHRL1 was sufficient to increase bim expression in B-cells (Dijkers et al., 2000). The action of forkhead family members is negatively regulated by the phosphorylation of the serine and threonine residues. FKHRL1 has been identified as a principal substrate of Akt in neurons (Brunet et al., 1999; Zheng et al., 2000). Therefore, we first examined the phosphorylation status of Akt and FKHRL1 in mouse CECs after Aβ treatment using immunoblotting with phospho-site-specific antibodies. Exposure of CECs to Aβ (25 μm) resulted in a substantial decrease in the levels of phospho-Akt at both Ser 473 and Thr 308, as early as 1 h after treatment, and persisted throughout the 48 h period of exposure (Fig. 2A). In parallel with this decreased phosphorylation of Akt, a marked decline in phospho-FKHRL1 levels on Ser 253 and Thr32 in the cytoplasmic fraction was observed (Fig. 2A,B), suggesting that Aβ exposure resulted in a net dephosphorylation of Akt and the subsequent reduction in FKHRL1 phosphorylation.
Figure 2.

Aβ dephosphorylated Akt resulting in an increase in FKHRL1 DNA binding activity. A, B, CECs treated with 25 μm Aβ25–35 for the indicated times were analyzed by immunoblotting using anti-p-Akt (Thr308 and Ser473) and anti-p-FKHRL1 (Thr32 and Ser253) antibodies. A decrease in p-Akt and p-FKHRL1 levels was evident at both sites within 1 h and persisted for at least 48 h after exposure. C, D, EMSA demonstrated a time-dependent increase in FKHRL1 binding activity after Aβ25–35 treatment in CECs, increasing 1 h after exposure and returning to the basal level by 24 h. Note the low basal level of FKHRL1 binding activity in cultures unexposed to Aβ25–35 (0 h). The specificity of FKHRL1 DNA binding activity was confirmed by the complete inhibition of FKHRL1 binding in the presence of a 100-fold molar excess of cold FKHRL1 oligonucleotide (SC) or in the absence of nuclear protein (FP). Suppression of FKHRL1 binding activity by an anti-FKHRL1 antibody, but not a nonspecific anti-Bad antibody (compare with the 2 h lane), also supports the specificity of the FKHRL1 EMSA. The data shown in A and C are representative from three separate experiments with similar results. The relative optical density in Figure B and D is expressed as mean ± SD. *p < 0.05 versus the 0 h group.
Phosphorylation of FKHRL1 by Akt leads to its sequestration by the 14-3-3 protein, preventing its translocation into the nucleus to act as a pro-apoptotic transcription factor (Brunet et al., 1999). We next examined whether FKHRL1 dephosphorylation enhanced its specific binding to the cognate sequences in the promoter region of bim in Aβ-treated CECs using EMSA. As illustrated in Figure 2, C and D, Aβ treatment increased FKHRL1 DNA binding activity in a time-dependent manner, starting as early as 1 h and persisting for at least 8 h after Aβ exposure. A low basal level of FKHRL1 binding activity was noted in CECs without Aβ treatment. Preincubation of the nuclear extract with anti-FKHRL1 antibody, but not anti-Bad antibody, reduced FKHRL1 DNA binding activity (Fig. 2C). The addition of excess unlabeled FKHRL1 oligonucleotides abolished the observed DNA–protein complex. These results demonstrated the specificity of FKHRL1 DNA binding activity in the present study.
Viral vector-mediated gene transfer to overexpress Akt attenuated Aβ-induced dephosphorylation of FKHRL1 and bim induction
To assess the direct role of Akt in the regulation of FKHRL1 activity and bim expression in CECs, we infected cells with an adenoviral vector to overexpress Akt in an attempt to increase phospho-Akt levels in CECs. CECs exhibited an infection efficiency of 70–80%, and no significant cell death or proliferation was observed in control cultures (data not shown).
As shown in Figure 3, A and B, adenovirus-mediated gene transfer overexpressing Akt reduced Aβ-induced FKHRL1 binding activity and subsequent expression of bim mRNA, whereas a nonfunctional mutant (Aktm) had no effect. Results from these experiments support the causal role of Akt in regulating Aβ-induced FKHRL1 transactivation of bim expression.
Figure 3.
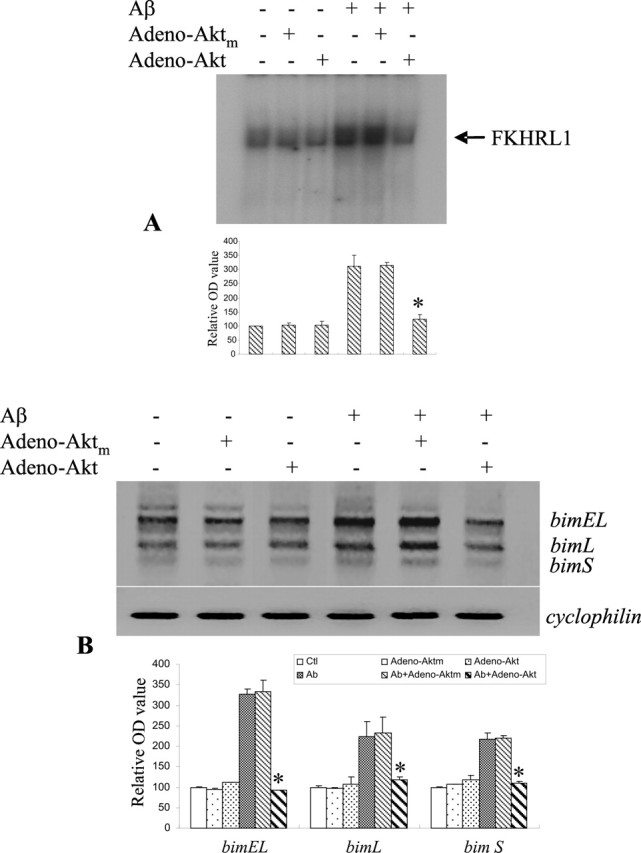
Adenoviral gene transfer overexpressing Akt reduced Aβ activation of FKHRL1 and subsequent bim transactivation. A, Adenoviral gene transfer to overexpress Akt in CECs reduced FKHRL1 binding activity, whereas an inactive mutant (Aktm) had no effect. B, Adeno-Akt-infected CECs showed a decrease in Aβ-induced upregulation of the three isoforms of bim. Adeno-Aktm-infected CECs had approximately the same level of FKHRL1 binding activity as the noninfectced cells. Quantitative analysis of three sets of data is shown in the bottom panel in A and B, respectively, with mean ± SD. *p < 0.05 versus the Aβ or Aβ/Adeno-Aktm group.
Aβ increased protein phosphatase activity in cultured CECs via nSMase activation and ceramide synthesis
The regulation of phosphorylated protein is balanced by the activity of kinases and phosphatases. Aβ dephosphorylation of Akt and FKHRL1 raises the possibility that phosphatases may be activated in Aβ-induced CEC apoptosis. In light of our previous finding that Aβ promoted the generation of ceramide via nSMase activation (Lee et al., 2004; Yang et al., 2004), we examined the activities of two protein phosphatases, PP1 and PP2A, known to be activated by ceramide (Dobrowsky and Hannun, 1993; Dobrowsky et al., 1993; Law and Rossie, 1995; Chalfant et al., 1999). These two serine phosphatases regulate the activities of several major protein kinase signaling pathways in a variety of cell systems, including the phosphatidylinositol 3′-kinase/Akt signaling pathway (Millward et al., 1999; Silverstein et al., 2002). CECs treated with both Aβ25–35 (Fig. 4A) and Aβ1–40 (Fig. 4B) significantly increased PP2A activity as early as 1 h after exposure and persisted for 24 h. Aβ peptides also induced a modest but nonsignificant increase in PP1 activity in a similar manner (Fig. 4A,B). We further examined the effect of the ceramide analog C2 ceramide on the activities of PP2A and PP1 in CECs. Similar to Aβ treatment, 25 μm C2 ceramide increased PP2A activity as early as 0.5 h after exposure but had little effect on PP1 (Fig. 4C).
Figure 4.

Aβ increased PP2A activity via nSMase activation and ceramide generation. A, B, CECs were treated with 25 μm Aβ25–35 (A) or Aβ1–40 (B) for the indicated times, and PP2A activity was determined. Aβ peptides increased PP2A activity in CECs in a time-dependent manner. C, C2 ceramide treatment at a concentration of 25 μm also increased PP2A activity in a similar manner. A–C, However, Aβ and C2 ceramide increased PP1 activity to a much lesser extent. D, Pretreatment with 3-OMe-SM, a specific nSMase inhibitor at a concentration of 1 μm, for 2 h before 25 μm Aβ25–35 exposure for 4 h prevented Aβ activation of PP2A. E, Total proteins from the cerebral microvessel in 18-month-old APPsw transgenic or age-matched wild-type (WT) mice were analyzed to assess protein phosphatase activity. PP2A but not PP1 activity was increased in amyloid-laden vessels of APPsw transgenic mice in compared with the wild-type controls (n = 3 per group). The data shown are mean ± SD of three separate experiments in triplicate. *p < 0.05, significant difference from the 0 h (A–C), Aβ-treated (D), or WT (E) group.
To determine whether Aβ-induced activation of PP2A was mediated via nSMase, we blocked the activity of this ceramide-synthesizing enzyme using a selective nSMase inhibitor, 3-OMe-SM. 3-OMe-SM has been shown previously to block Aβ activation of nSMase and Aβ stimulation of ceramide synthesis (Lee et al., 2004; Yang et al., 2004). 3-OMe-SM blocked Aβ-induced increase in PP2A activity (Fig. 4D), suggesting that Aβ activation of nSMase is causally related to ceramide generation and PP2A activation.
To further explore the potential involvement of CAPPs in cerebrovascular degeneration in CAA, we measured protein phosphatase activity in cerebral microvessels isolated from 18-month-old APPsw mice. PP2A, but not PP1, activity was significantly increased compared with age-matched wild-type animals (Fig. 4E), implying that the induction of PP2A may also play a regulatory role in cerebrovascular degeneration in CAA.
PP2A, but not PP1, dephosphorylated Akt, resulting in subsequent bim induction and CEC death
To determine whether PP2A regulates Akt activity after Aβ treatment in CECs, we blocked PP2A activity applying both pharmacological and genetic strategies. OA is a selective inhibitor of PP2A at low concentrations (0.1–10 nm) but inhibits PP1 at much higher concentrations (Cohen, 1989; Cohen and Cohen, 1989; Dobrowsky and Hannun, 1992). Treatment with OA at the concentration of 0.1–100 nm resulted in a dose-dependent increase in the levels of phospho-Akt at both Ser 473 and Thr 308 in CECs after Aβ exposure as demonstrated by Western blot analysis (Fig. 5A). In parallel with this increase, a marked increase in phospho-FKHRL1 level was observed (Fig. 5A). This OA-induced increase in phospho-Akt/FKHRL1 level was associated with a dose-dependent inhibition of Aβ-induced activation of bim mRNA expression (Fig. 5B) and in turn resulted in an attenuation of Aβ-induced CEC death (Fig. 5C). Because OA at higher doses (≥100 nm) may have inhibitory effects on both PP2A and PP1, we used PA, a selective inhibitor of PP1 (Kishikawa et al., 1999), to confirm that PP2A was the pivotal regulator of bim mRNA expression. Treatment with PA at two doses (100 and 500 nm) resulted in no change in the levels of phospho-Akt and phospho-FKHRL1, as assessed by immunoblotting (Fig. 5D). This is in contrast to the findings with OA, a selective inhibitor of PP2A, at low concentrations (Fig. 5A). Furthermore, PA treatment did not have any effect on Aβ-induced bim mRNA expression (Fig. 5E) and cell death in CECs (Fig. 5F).
Figure 5.
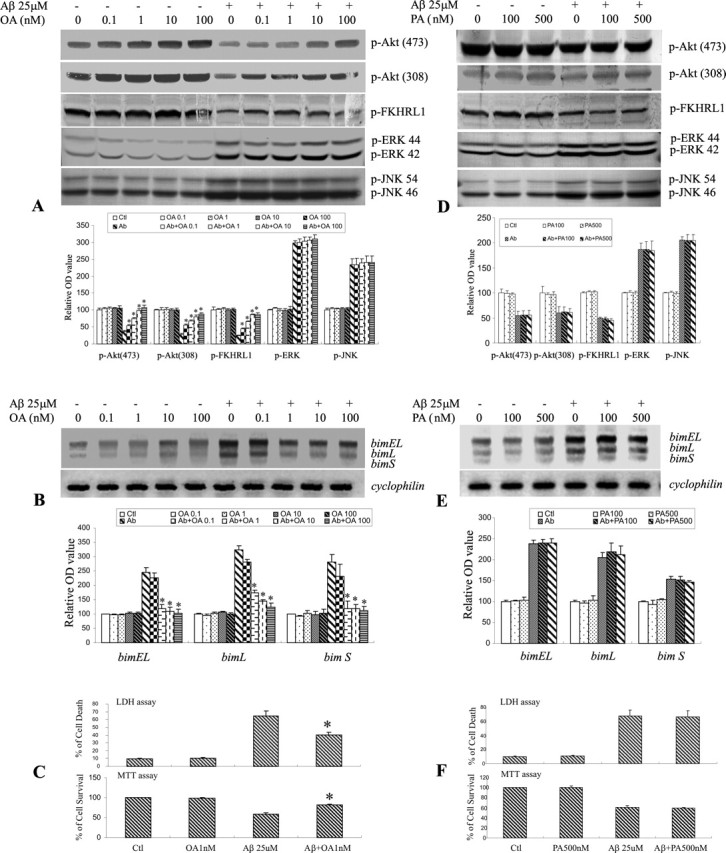
PP2A inhibition reduced dephosphorylation of Akt and FKHRL1, subsequent induction of bim, and CEC death. CECs, treated with 25 μm Aβ25–35 in the presence or absence of OA, a PP2A inhibitor, or PA, a PP1 inhibitor, were fractionated and analyzed by immunoblotting with the indicated antibodies. A, OA decreased Aβ-induced dephosphorylation of Akt and FKHRL1 in a dose-dependent manner. However, OA had no effect on dephosphorylation of ERK and JNK. B, OA also decreased subsequent induction of bim, including bimEL and bimL in CECs (determined by RT-PCR) after Aβ exposure. C, Moreover, treatment with OA at a concentration of 1 nm effectively reduced CEC death (LDH assay) or increased CEC survival (MTT assay) after Aβ exposure. D–F, In contrast to OA, PA, at concentrations up to 500 nm, did not affect the levels of p-Akt, p-FKHRL1, p-ERK, p-JNK (D), bim expression (E), or Aβ-induced CEC death (F). The quantitative analysis of data with representative blots shown in A, B, D, and E is in the respective bottom panels. Data are expressed as mean ± SD. *p < 0.05 versus the Aβ-treated group.
In previous studies, we found that the JNK/AP1/Bim signaling pathway is activated in CECs after Aβ exposure (Yin et al., 2002b). To determine whether the effect of PP2A was mediated via the mitogen-activated protein kinase (MAPK) pathways, we examined the effect of PP2A inhibition on the expression of extracellular signal-related kinase (ERK) and c-Jun N-terminal kinase (JNK), in CECs after Aβ exposure. OA had no effect on the levels of phospho-ERK and phospho-JNK after Aβ treatment (Fig. 5A). Furthermore, inhibition of PP1 with PA also had no effect on phospho-ERK or phospho-JNK in CECs treated with Aβ (Fig. 5D). These results suggest that the effect of PP2A on Akt/FKHRL1/Bim cascade and subsequent CEC death is independent of the JNK/Bim signaling pathway.
To further confirm the involvement of PP2A in regulating Aβ-induced transactivation of bim mRNA, we used a retroviral system for delivery of siRNA into CECs. We generated retroviral vectors expressing hairpin siRNA of the PP2A catalytic unit (pS-PP2A) or the corresponding nonfunctional sense siRNA control (pS-PP2Ac). Successful delivery of both recombinant retroviral vectors into CECs was confirmed by fluorescence microscopy, which revealed abundant cytoplasmic staining of the virally expressed ZsGreen (Fig. 6A). CECs were infected with the retroviral vectors for 48 h before Aβ exposure. PP2A mRNA levels (Fig. 6B), protein expression (Fig. 6C), and activity (Fig. 6D) were significantly repressed by treatment with pS-PP2A for 48 h, whereas the nonfunctional sense siRNA control (pS-PP2Ac) had no effect (Fig. 6B–D). Retroviral siRNA vectors did not alter levels of cyclophilin mRNA and actin protein, suggesting specificity of the knock-down strategy. PP2A siRNA gene transfer effectively attenuated Aβ-induced Akt/FKHRL1 dephosphorylation (Fig. 6E), bim mRNA expression (Fig. 6F), and subsequent CEC death (Fig. 6G,H), whereas pS-PP2Ac had none of these effects. These results provide further support for the contention that PP2A can actively regulate Aβ-induced bim expression and CEC apoptosis via the Akt/FKHRL1 signaling pathway.
Figure 6.
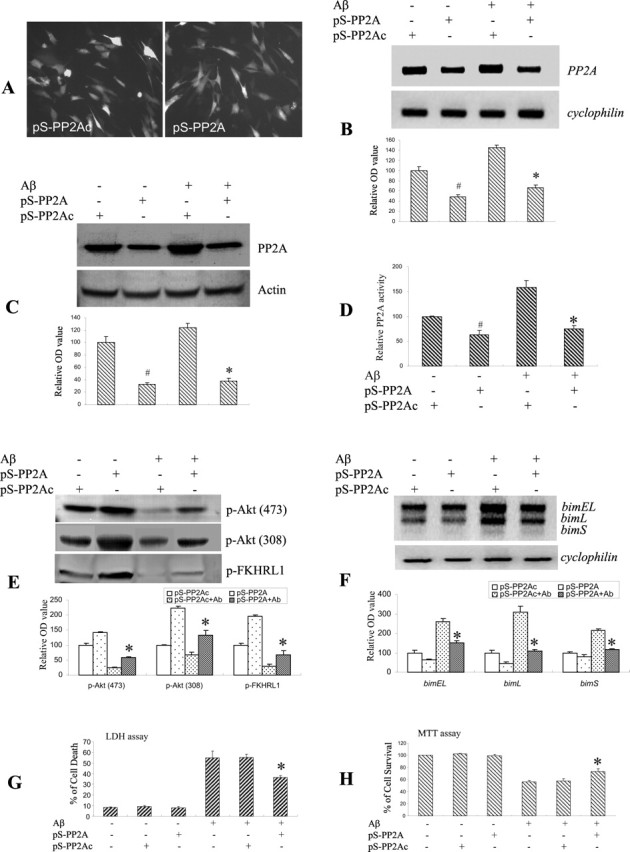
PP2A knockdown with siRNA reduced dephosphorylation of Akt and FKHRL1, upregulation of bim, and subsequent cell death. A, Effective gene transfer with retroviral vectors carrying PP2A siRNA (pS-PP2A) or sense siRNA control (pS-PP2Ac) was confirmed by ZsGreen fluorescence. Magnification, 200×. B, Total RNA from CECs infected with retroviral vectors carrying PP2A siRNA (pS-PP2A) or sense siRNA control (pS-PP2Ac) was analyzed by RT-PCR 48 h after infection. PP2A siRNA reduced the mRNA level of PP2A. The sense siRNA control was without effects. Note that PP2A siRNA did not alter cyclophilin mRNA levels, suggesting the specificity of this knockdown strategy. C, D, Total protein from CECs infected with retroviral vectors carrying PP2A siRNA (pS-PP2A) or sense siRNA control (pS-PP2Ac) was analyzed by immunoblotting (C) and PP2A activity assay (D) 48 h after infection. PP2A siRNA reduced the level of PP2A protein and PP2A activity. The sense siRNA control had no effect. E, CECs infected with retroviral vector containing PP2A siRNA (pS-PP2A) for 48 h before Aβ treatment were fractionated and analyzed by immunoblotting with the indicated antibodies. PP2A siRNA (pS-PP2A) effectively reduced dephosphorylation of Akt and FKHRL1 in CECs at 4 h after Aβ treatment. The sense siRNA control (pS-PP2Ac) was without effects. F, PP2A knockdown also reduced bim EL, bim L, and bim S mRNA induction after Aβ exposure. G, H, PP2A downregulation by RNAi also protected CECs from cell death after Aβ exposure by LDH (G) and MTT (H) assays. Data shown in the top panels of B, C, E, and F are representative of three separate experiments with similar results. The quantitative analysis of data in D, G, and H and in the bottom panels of B, C, E, and F is expressed as mean ± SD. #p < 0.05, significant differences compared with the sense siRNA control group (B–D); *p < 0.05 versus Aβ cotreatment with the sense siRNA control group.
Aβ induced interaction between PP2A and Akt
If PP2A directly dephosphorylates Akt, one might expect a physical interaction between these two enzymes after Aβ exposure. Indeed, a similar interaction was observed between PP2A and the stress-related kinase, JNK (Shanley et al., 2001). To test this hypothesis, lysates from CECs with and without Aβ treatment were immunoprecipitated with anti-Akt antibodies and examined for the presence of a structural (PP2A Aα) or regulatory (PP2A Bαβγ, B56α, Cαβ) subunits of PP2A by immunoblotting. Levels of PP2A subunits were generally unchanged by Aβ treatment (Fig. 7A,B). As illustrated in Figure 7, C and D, the structural subunit PP2A Aα and the regulatory subunit PP2A Cαβ exhibited a specific interaction in the anti-Akt-immunoprecipitated product (Fig. 7C,D), suggesting that these 2 PP2A subunits had increased physical association with Akt after Aβ exposure.
Figure 7.

PP2A interacted with Akt after Aβ exposure. A, B, Immunoblotting shows the presence of various PP2A subunits in CECs. Structural (PP2A Aα) and regulatory (PP2A Bαβγ, B56α, Cαβ) subunits were immunoblotted in whole-cell lysates at the times indicated after Aβ exposure. Little change in protein levels of these PP2A subunits was noted up to 48 h of Aβ treatment. C, D, Selected PP2A subunits coimmunoprecipitated with Akt. Whole-cell lysates were immunoprecipitated (IP) with anti-Akt antibodies and probed with the indicated anti-PP2A subunit antibodies. Unstimulated cells (0 h) demonstrated a weak association between Akt and the four PP2A subunits. After Aβ exposure for 4 h, anti-Akt immunoprecipitate demonstrated increased density in the PP2A Aα and PP2A Cαβ bands, suggesting that Aβ increased the physical association between Akt and these two subunits of PP2A. Data shown in A and C are representative of three separate experiments. Quantitative analysis of the data are shown in B and D with data expressed as mean ± SD. *p < 0.05 versus the 0 h group.
Discussion
Aβ has been implicated in neuronal and vascular degeneration in patients with AD and CAA. Increasing evidence suggests that Aβ induces CEC death via apoptosis (Blanc et al., 1997; Suo et al., 1997; Xu et al., 2001; Yin et al., 2002b). We have reported that Aβ-induced CEC apoptosis was associated with the induction of Bim (Yin et al., 2002b). Bim is an apoptotic initiator belonging to the BH3-only proteins. In this study, we note that bim mRNA expression was selectively elevated in cerebral microvessels isolated from 18-month-old APPsw mutant transgenic mice with phenotypic presentation of CAA compared with the age-matched wild-type animals. No increased expression of other genes in the BH3-only family (bid, bad, bmf, and dp5) was noted. APPsw mice develop age-dependent accumulation in parenchymal and vascular amyloid, reproducing many of the major pathological features of AD and CAA (Hsiao et al., 1996). At 18 months of age, APPsw mice demonstrate prominent amyloid accumulation surrounding cortical arterioles, and microvascular hemorrhage is evident throughout the cortex (Fryer et al., 2003; Lee et al., 2003), suggesting extensive cerebrovascular degeneration. Therefore, the selective induction of bim suggests that this apoptotic initiator of the BH3-only family may play a role in cerebrovascular degeneration. To investigate the regulatory mechanism of bim expression in CAA, we studied the effects of Aβ on mouse CECs. Paralleling the finding in amyloid-laden vessels in APPsw mice, selective bim induction was observed in CECs treated with Aβ; all three isoforms of bim were induced in a time-dependent manner, peaking at 4 h.
FKHRL1, a member of the forkhead transcription factor family, transactivates bim expression after its dephosphorylation (Burgering and Kops, 2002; Gilley et al., 2003). In viable cells, forkhead protein activity is kept in phosphorylated and inactive state by the survival-promoting kinase Akt (Brunet et al., 1999; Tang et al., 1999). Dephosphorylation and the consequent inactivation of Akt by protein phosphatases releases pro-apoptotic proteins such as forkhead proteins from the phosphorylated and inactive state to initiate apoptotic processes (Ugi et al., 2004; Gao et al., 2005). Aβ induction of bim expression was associated with the dephosphorylation of Akt (at Ser 473 and Thr 308). This was accompanied by dephosphorylation and activation of FKHRL1, leading to an increase in DNA binding activity of FKHRL1. Consensus binding sites for FKHRL1 are present in the promoter region of bim. FKHRL1 activation (via dephosphorylation) has been shown to increase bim expression after cytokine withdrawal in lymphocytes (Dijkers et al., 2000) and IGF-I withdrawal in cerebellar granule neurons (Linseman et al., 2002). We also found that increasing Akt activity by viral gene transfer attenuated Aβ-induced increase in FKHRL1 DNA binding activity and bim induction in CECs, suggesting that FKHRL1 activity is negatively regulated via phosphorylation by Akt. These results are consistent with previous reports that phosphorylation of FKHRL1 by Akt results in FKHRL1 sequestration in cytosol by 14-3-3 (Brunet et al., 1999; Tang et al., 1999). The phorphorylation state of any protein is the result of a net balance between kinase activity and phosphatase activity.
In a previous study, we have demonstrated that Aβ activated nSMase to generate ceramide (Lee et al., 2004; Yang et al., 2004). Ceramide, a lipid mediator of apoptosis, mediates Aβ-induced CEC death (Yang et al., 2004). Here, we report that Aβ induced a rapid activation of the CAPP PP2A. Inhibition of PP2A with a selective PP2A inhibitor, OA, or PP2A gene knockdown by an RNA interference (RNAi) strategy attenuated Aβ-induced dephosphorylation of Akt and FKHRL1. Furthermore, PP2A inhibition prevented bim activation and subsequent CEC death induced by Aβ. PA, a specific inhibitor of PP1, did not alter the phosphorylation of Akt and FKHRL1, induction of bim, or cell death induced by Aβ. Collectively, these results demonstrate for the first time that PP2A, but not PP1, is selectively involved (at least in part) in the induction of bim-mediated apoptosis via the Akt/FKHRL1 pathway. We also found that inhibition of ceramide synthesis by a selective nSMase inhibitor (3-OMe-SM) prevented PP2A activation by Aβ. Thus, we have established the following pathway for Aβ-induced CEC apoptosis: Aβ → nSMase → ceramide → PP2A activation → Akt dephosphorylation → FKHRL1 dephosphorylation → bim induction → apoptotic cascade → apoptosis.
Recent studies have suggested that PP2A may play a critical role in the regulation of the MAPK cascades by dephosphorylation of selective substrates, including ERKs and the JNKs. ERKs and JNKs may be activated by cellular stress or apoptotic signals (Millward et al., 1999; Shanley et al., 2001; Silverstein et al., 2002; Kins et al., 2003). We and other investigators have also reported that activation of the JNK signaling pathway may contribute to Bim-mediated apoptosis in CECs in vitro (Yin et al., 2002b) and neuronal apoptosis after transient focal cerebral ischemia (Okuno et al., 2004). To determine whether MAPK signaling is actively associated with PP2A regulation of bim-mediated apoptosis in CECs after Aβ exposure, we examined the effects of PP2A inhibition on dephosphorylation of ERK and JNK. Treatment of CEC with a PP2A inhibitor (OA) or a PP1 inhibitor (PA) at different doses failed to lead to the alteration of phospho-ERK/JNK levels (Fig. 5A,D), implying that these pathways may not be involved in PP2A-modulated transactivation of bim in CECs. Additional studies are needed to determine the relative importance of PP2A-dependent and -independent mechanisms in Bim-mediated CEC death induced by Aβ.
In the present study, we found that PP2A activity was elevated in cultured CECs after Aβ exposure as well as in cerebral microvessels isolated from 18-month-old APPsw transgenic mice. PP2A, a serine/threonine phosphatase, is involved in a number of signaling pathways in mammalian cells (Mumby and Walter, 1993). It is a heterotrimer containing a catalytic subunit (C), a structural scaffolding subunit (A), and a regulatory subunit (B). The catalytic and structural subunits, forming the catalytic complex of the phosphatase, are evolutionary conserved (Stone et al., 1987; Hemmings et al., 1990; Mumby and Walter, 1993); only two closely related isoforms (α and β) of both subunits have been identified (Aα and Cα are the most abundant). However, there are four known families of diverse B-subunits (B, B′, B″, or B‴ family) that vary in expression temporally and by tissue types (Ruediger et al., 1991; Mumby and Walter, 1993; McCright and Virshup, 1995; Csortos et al., 1996). Substrate specificity seems to be determined by the B-subunits (Cegielska et al., 1994). Moreover, there are multiple isoforms (α, β, γ, δ, etc.) and splice variants for each family, allowing for a wide variety of possible PP2A holoenzymes. The B-subunits may also target the PP2A catalytic complex to intracellular sites such as microtubules (Sontag et al., 1995) or the nucleus (McCright et al., 1996a,b). These features of the PP2A regulatory B-subunits suggest that it is the B-subunit that defines PP2A isoforms and their physiological roles. In this study, we found that mouse CECs express the structural subunit (PP2A Aα) and many of the regulatory B-subunit families: PP2A Bαβγ (B), B56α (B′), Cαβ (B″). Protein levels of all of the subunits remained unchanged after Aβ treatment for up to 48 h. To determine which subunit might be involved in the regulation of Akt, we performed coimmunoprecipitation experiments with subunit-selective antibodies in CECs grown in the presence and absence of Aβ. The structural subunit PP2A Aα coimmunoprecipitated with anti-Akt antibodies in CECs treated with Aβ but not in control cultures. Furthermore, regulatory subunits from the B (Bαβγ), B′ (B56α), and B″ family (PP2A Cαβ) coimmunoprecipitated with anti-Akt antibodies in Aβ-treated CECs. However, only the B″ subunit showed an increase in coimmunoprecipitation after Aβ treatment, suggesting that this subunit is selectively involved in regulating Akt phosphorylation, subsequent bim induction, and apoptotic cell death resulting from Aβ toxicity.
In conclusion, the present study suggests a novel role for PP2A, a ceramide- activated protein phosphatase, in mediating Aβ-induced apoptosis in CECs. Our data suggest that PP2A may mediate Aβ-induced bim expression and subsequent apoptotic cascades via the Akt/FKHRL1 signaling pathway. That importance of bim upregulation by PP2A in cerebrovascular degeneration is suggested by increased PP2A activity and bim expression in microvascular preparations isolated from APPsw mice. The elucidation of the signaling events involved in Aβ-induced CEC death may be important for understanding molecular mechanisms of cerebrovascular degeneration to design effective strategies to attenuate the pathogenesis of CAA.
Footnotes
This work was supported by National Institutes of Health Grants K08 NS002190 and NS40525 (J.-M.L.), American Heart Association Grants 0365378Z (K.-J.Y.) and 0460066Z (J.-M.L.), and National Science Council Grant 93-2321-B-038-003 (C.Y.H.).
References
- Behl C, Davis JB, Lesley R, Schubert D (1994). Hydrogen peroxide mediates amyloid beta protein toxicity. Cell 77:817–827. [DOI] [PubMed] [Google Scholar]
- Blanc EM, Toborek M, Mark RJ, Hennig B, Mattson MP (1997). Amyloid beta-peptide induces cell monolayer albumin permeability, impairs glucose transport, and induces apoptosis in vascular endothelial cells. J Neurochem 68:1870–1881. [DOI] [PubMed] [Google Scholar]
- Bouillet P, Zhang LC, Huang DC, Webb GC, Bottema CD, Shore P, Eyre HJ, Sutherland GR, Adams JM (2001). Gene structure alternative splicing, and chromosomal localization of pro-apoptotic Bcl-2 relative Bim. Mamm Genome 12:163–168. [DOI] [PubMed] [Google Scholar]
- Brugg B, Michel PP, Agid Y, Ruberg M (1996). Ceramide induces apoptosis in cultured mesencephalic neurons. J Neurochem 66:733–739. [DOI] [PubMed] [Google Scholar]
- Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME (1999). Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96:857–868. [DOI] [PubMed] [Google Scholar]
- Burgering BM, Kops GJ (2002). Cell cycle and death control: long live Forkheads. Trends Biochem Sci 27:352–360. [DOI] [PubMed] [Google Scholar]
- Cegielska A, Shaffer S, Derua R, Goris J, Virshup DM (1994). Different oligomeric forms of protein phosphatase 2A activate and inhibit simian virus 40 DNA replication. Mol Cell Biol 14:4616–4623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalfant CE, Kishikawa K, Mumby MC, Kamibayashi C, Bielawska A, Hannun YA (1999). Long chain ceramides activate protein phosphatase-1 and protein phosphatase-2A. Activation is stereospecific and regulated by phosphatidic acid. J Biol Chem 274:20313–20317. [DOI] [PubMed] [Google Scholar]
- Cohen P (1989). The structure and regulation of protein phosphatases. Annu Rev Biochem 58:453–508. [DOI] [PubMed] [Google Scholar]
- Cohen P, Cohen PT (1989). Protein phosphatases come of age. J Biol Chem 264:21435–21438. [PubMed] [Google Scholar]
- Csortos C, Zolnierowicz S, Bako E, Durbin SD, DePaoli-Roach AA (1996). High complexity in the expression of the B′ subunit of protein phosphatase 2A0. Evidence for the existence of at least seven novel isoforms. J Biol Chem 271:2578–2588. [DOI] [PubMed] [Google Scholar]
- Dijkers PF, Medema RH, Lammers JW, Koenderman L, Coffer PJ (2000). Expression of the pro-apoptotic Bcl-2 family member Bim is regulated by the forkhead transcription factor FKHR-L1. Curr Biol 10:1201–1204. [DOI] [PubMed] [Google Scholar]
- Dobrowsky RT, Hannun YA (1992). Ceramide stimulates a cytosolic protein phosphatase. J Biol Chem 267:5048–5051. [PubMed] [Google Scholar]
- Dobrowsky RT, Hannun YA (1993). Ceramide-activated protein phosphatase: partial purification and relationship to protein phosphatase 2A. Adv Lipid Res 25:91–104. [PubMed] [Google Scholar]
- Dobrowsky RT, Kamibayashi C, Mumby MC, Hannun YA (1993). Ceramide activates heterotrimeric protein phosphatase 2A. J Biol Chem 268:15523–15530. [PubMed] [Google Scholar]
- Franke TF, Yang SI, Chan TO, Datta K, Kazlauskas A, Morrison DK, Kaplan DR, Tsichlis PN (1995). The protein kinase encoded by the Akt proto-oncogene is a target of the PDGF-activated phosphatidylinositol 3-kinase. Cell 81:727–736. [DOI] [PubMed] [Google Scholar]
- Fryer JD, Taylor JW, DeMattos RB, Bales KR, Paul SM, Parsadanian M, Holtzman DM (2003). Apolipoprotein E markedly facilitates age-dependent cerebral amyloid angiopathy and spontaneous hemorrhage in amyloid precursor protein transgenic mice. J Neurosci 23:7889–7896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao T, Furnari F, Newton AC (2005). PHLPP: a phosphatase that directly dephosphorylates Akt, promotes apoptosis, and suppresses tumor growth. Mol Cell 18:13–24. [DOI] [PubMed] [Google Scholar]
- Gilley J, Coffer PJ, Ham J (2003). FOXO transcription factors directly activate bim gene expression and promote apoptosis in sympathetic neurons. J Cell Biol 162:613–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemmings BA, Adams-Pearson C, Maurer F, Muller P, Goris J, Merlevede W, Hofsteenge J, Stone SR (1990). Alpha- and beta-forms of the 65-kDa subunit of protein phosphatase 2A have a similar 39 amino acid repeating structure. Biochemistry 29:3166–3173. [DOI] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G (1996). Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 274:99–102. [DOI] [PubMed] [Google Scholar]
- Huang DC, Strasser A (2000). BH3-only proteins-essential initiators of apoptotic cell death. Cell 103:839–842. [DOI] [PubMed] [Google Scholar]
- Kelekar A, Thompson CB (1998). Bcl-2-family proteins: the role of the BH3 domain in apoptosis. Trends Cell Biol 8:324–330. [DOI] [PubMed] [Google Scholar]
- Kins S, Kurosinski P, Nitsch RM, Gotz J (2003). Activation of the ERK and JNK signaling pathways caused by neuron-specific inhibition of PP2A in transgenic mice. Am J Pathol 163:833–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishikawa K, Chalfant CE, Perry DK, Bielawska A, Hannun YA (1999). Phosphatidic acid is a potent and selective inhibitor of protein phosphatase 1 and an inhibitor of ceramide-mediated responses. J Biol Chem 274:21335–21341. [DOI] [PubMed] [Google Scholar]
- Law B, Rossie S (1995). The dimeric and catalytic subunit forms of protein phosphatase 2A from rat brain are stimulated by C2-ceramide. J Biol Chem 270:12808–12813. [DOI] [PubMed] [Google Scholar]
- Lee JM, Yin KJ, Hsin I, Chen S, Fryer JD, Holtzman DM, Hsu CY, Xu J (2003). Matrix metalloproteinase-9 and spontaneous hemorrhage in an animal model of cerebral amyloid angiopathy. Ann Neurol 54:379–382. [DOI] [PubMed] [Google Scholar]
- Lee JT, Xu J, Lee JM, Ku G, Han X, Yang DI, Chen S, Hsu CY (2004). Amyloid-beta peptide induces oligodendrocyte death by activating the neutral sphingomyelinase-ceramide pathway. J Cell Biol 164:123–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linseman DA, Phelps RA, Bouchard RJ, Le SS, Laessig TA, McClure ML, Heidenreich KA (2002). Insulin-like growth factor-I blocks Bcl-2 interacting mediator of cell death (Bim) induction and intrinsic death signaling in cerebellar granule neurons. J Neurosci 22:9287–9297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo DT, Copani A, Pike CJ, Whittemore ER, Walencewicz AJ, Cotman CW (1993). Apoptosis is induced by beta-amyloid in cultured central nervous system neurons. Proc Natl Acad Sci USA 90:7951–7955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangoura D, Dawson G (1998). Programmed cell death in cortical chick embryo astrocytes is associated with activation of protein kinase PK60 and ceramide formation. J Neurochem 70:130–138. [DOI] [PubMed] [Google Scholar]
- McCright B, Virshup DM (1995). Identification of a new family of protein phosphatase 2A regulatory subunits. J Biol Chem 270:26123–26128. [DOI] [PubMed] [Google Scholar]
- McCright B, Brothman AR, Virshup DM (1996a). Assignment of human protein phosphatase 2A regulatory subunit genes b56alpha, b56beta, b56gamma, b56delta, and b56epsilon (PPP2R5A-PPP2R5E), highly expressed in muscle and brain, to chromosome regions 1q41, 11q12, 3p21, 6p21.1, and 7p11.2 → p12. Genomics 36:168–170. [DOI] [PubMed] [Google Scholar]
- McCright B, Rivers AM, Audlin S, Virshup DM (1996b). The B56 family of protein phosphatase 2A (PP2A) regulatory subunits encodes differentiation-induced phosphoproteins that target PP2A to both nucleus and cytoplasm. J Biol Chem 271:22081–22089. [DOI] [PubMed] [Google Scholar]
- Millward TA, Zolnierowicz S, Hemmings BA (1999). Regulation of protein kinase cascades by protein phosphatase 2A. Trends Biochem Sci 24:186–191. [DOI] [PubMed] [Google Scholar]
- Mumby MC, Walter G (1993). Protein serine/threonine phosphatases: structure, regulation, and functions in cell growth. Physiol Rev 73:673–699. [DOI] [PubMed] [Google Scholar]
- Obeid LM, Linardic CM, Karolak LA, Hannun YA (1993). Programmed cell death induced by ceramide. Science 259:1769–1771. [DOI] [PubMed] [Google Scholar]
- Okuno S, Saito A, Hayashi T, Chan PH (2004). The c-Jun N-terminal protein kinase signaling pathway mediates Bax activation and subsequent neuronal apoptosis through interaction with Bim after transient focal cerebral ischemia. J Neurosci 24:7879–7887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardridge WM, Eisenberg J, Yamada T (1985). Rapid sequestration and degradation of somatostatin analogues by isolated brain microvessels. J Neurochem 44:1178–1184. [DOI] [PubMed] [Google Scholar]
- Perlmutter LS, Myers MA, Barron E (1994). Vascular basement membrane components and the lesions of Alzheimer's disease: light and electron microscopic analyses. Microsc Res Tech 28:204–215. [DOI] [PubMed] [Google Scholar]
- Preston JE, Hipkiss AR, Himsworth DT, Romero IA, Abbott JN (1998). Toxic effects of beta-amyloid(25–35) on immortalised rat brain endothelial cell: protection by carnosine, homocarnosine and beta- alanine. Neurosci Lett 242:105–108. [DOI] [PubMed] [Google Scholar]
- Price JM, Sutton ET, Hellermann A, Thomas T (1997). Beta-amyloid induces cerebrovascular endothelial dysfunction in the rat brain. Neurol Res 19:534–538. [DOI] [PubMed] [Google Scholar]
- Ruediger R, Van Wart Hood JE, Mumby M, Walter G (1991). Constant expression and activity of protein phosphatase 2A in synchronized cells. Mol Cell Biol 11:4282–4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanley TP, Vasi N, Denenberg A, Wong HR (2001). The serine/threonine phosphatase, PP2A: endogenous regulator of inflammatory cell signaling. J Immunol 166:966–972. [DOI] [PubMed] [Google Scholar]
- Silverstein AM, Barrow CA, Davis AJ, Mumby MC (2002). Actions of PP2A on the MAP kinase pathway and apoptosis are mediated by distinct regulatory subunits. Proc Natl Acad Sci USA 99:4221–4226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sontag E, Nunbhakdi-Craig V, Bloom GS, Mumby MC (1995). A novel pool of protein phosphatase 2A is associated with microtubules and is regulated during the cell cycle. J Cell Biol 128:1131–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone SR, Hofsteenge J, Hemmings BA (1987). Molecular cloning of cDNAs encoding two isoforms of the catalytic subunit of protein phosphatase 2A. Biochemistry 26:7215–7220. [DOI] [PubMed] [Google Scholar]
- Suo Z, Fang C, Crawford F, Mullan M (1997). Superoxide free radical and intracellular calcium mediate A beta(1–42) induced endothelial toxicity. Brain Res 762:144–152. [DOI] [PubMed] [Google Scholar]
- Tang ED, Nunez G, Barr FG, Guan KL (1999). Negative regulation of the forkhead transcription factor FKHR by Akt. J Biol Chem 274:16741–16746. [DOI] [PubMed] [Google Scholar]
- Ugi S, Imamura T, Maegawa H, Egawa K, Yoshizaki T, Shi K, Obata T, Ebina Y, Kashiwagi A, Olefsky JM (2004). Protein phosphatase 2A negatively regulates insulin's metabolic signaling pathway by inhibiting Akt (protein kinase B) activity in 3T3–L1 adipocytes. Mol Cell Biol 24:8778–8789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitfield J, Neame SJ, Paquet L, Bernard O, Ham J (2001). Dominant-negative c-Jun promotes neuronal survival by reducing BIM expression and inhibiting mitochondrial cytochrome c release. Neuron 29:629–643. [DOI] [PubMed] [Google Scholar]
- Wisniewski HM, Wegiel J (1995). The neuropathology of Alzheimer's disease. Neuroimaging Clin N Am 5:45–57. [PubMed] [Google Scholar]
- Wisniewski HM, Wegiel J, Vorbrodt AW, Mazur-Kolecka B, Frackowiak J (2000). Role of perivascular cells and myocytes in vascular amyloidosis. Ann NY Acad Sci 903:6–18. [DOI] [PubMed] [Google Scholar]
- Xu J, Qu ZX, Moore SA, Hsu CY, Hogan EL (1992). Receptor-linked hydrolysis of phosphoinositides and production of prostacyclin in cerebral endothelial cells. J Neurochem 58:1930–1935. [DOI] [PubMed] [Google Scholar]
- Xu J, Yeh CH, Chen S, He L, Sensi SL, Canzoniero LM, Choi DW, Hsu CY (1998). Involvement of de novo ceramide biosynthesis in tumor necrosis factor-α/cycloheximide-induced cerebral endothelial cell death. J Biol Chem 273:16521–16526. [DOI] [PubMed] [Google Scholar]
- Xu J, Chen S, Ku G, Ahmed SH, Chen H, Hsu CY (2001). Amyloid beta peptide-induced cerebral endothelial cell death involves mitochondrial dysfunction and caspase activation. J Cereb Blood Flow Metab 21:702–710. [DOI] [PubMed] [Google Scholar]
- Yang DI, Yeh CH, Chen S, Xu J, Hsu CY (2004). Neutral sphingomyelinase activation in endothelial and glial cell death induced by amyloid beta-peptide. Neurobiol Dis 17:99–107. [DOI] [PubMed] [Google Scholar]
- Yankner BA (1996). Mechanisms of neuronal degeneration in Alzheimer's disease. Neuron 16:921–932. [DOI] [PubMed] [Google Scholar]
- Yankner BA, Dawes LR, Fisher S, Villa-Komaroff L, Oster-Granite ML, Neve RL (1989). Neurotoxicity of a fragment of the amyloid precursor associated with Alzheimer's disease. Science 245:417–420. [DOI] [PubMed] [Google Scholar]
- Yin KJ, Chen SD, Lee JM, Xu J, Hsu CY (2002a). ATM gene regulates oxygen-glucose deprivation-induced nuclear factor-kappaB DNA-binding activity and downstream apoptotic cascade in mouse cerebrovascular endothelial cells. Stroke 33:2471–2477. [DOI] [PubMed] [Google Scholar]
- Yin KJ, Lee JM, Chen SD, Xu J, Hsu CY (2002b). Amyloid-β induces Smac release via AP-1/Bim activation in cerebral endothelial cells. J Neurosci 22:9764–9770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng WH, Kar S, Quirion R (2000). Insulin-like growth factor-1-induced phosphorylation of the forkhead family transcription factor FKHRL1 is mediated by Akt kinase in PC12 cells. J Biol Chem 275:39152–39158. [DOI] [PubMed] [Google Scholar]
- Zlokovic BV, Mackic JB, Wang L, McComb JG, McDonough A (1993). Differential expression of Na,K-ATPase alpha and beta subunit isoforms at the blood-brain barrier and the choroid plexus. J Biol Chem 268:8019–8025. [PubMed] [Google Scholar]