

Abstract
Evidence that cerebellar learning involves more than one site of plasticity comes from, in part, pavlovian eyelid conditioning, where disconnecting the cerebellar cortex abolishes one component of learning, response timing, but spares the expression of abnormally timed short-latency responses (SLRs). Here, we provide evidence that SLRs unmasked by cerebellar cortex lesions are mediated by an associative form of learning-induced plasticity in the anterior interpositus nucleus (AIN) of the cerebellum. We used pharmacological inactivation and/or electrical microstimulation of various sites afferent and efferent to the AIN to systematically eliminate alternative candidate sites of plasticity upstream or downstream from this structure. Collectively, the results suggest that cerebellar learning is mediated in part by plasticity in target nuclei downstream of the cerebellar cortex. These data demonstrate an instance in which an aspect of associative learning, SLRs, can be used as an index of plasticity at a specific site in the brain.
Keywords: cerebellum, deep cerebellar nucleus, eyelid conditioning, learning, plasticity, timing
Introduction
Understanding the brain mechanisms of learning requires, in part, establishing causal relationships between the processes of neural plasticity and changes in behavior. Identifying the sites of plasticity involved is an important step in this process, generally requiring a combination of approaches such as reversible lesions, stimulation, and recordings in instances where the underlying pathways are reasonably well worked out (Davis et al., 1982). One such instance is eyelid conditioning, in which repeatedly presenting a tone conditioned stimulus (CS) together with mild electrical stimulation around the eye [the unconditioned stimulus (US)] promotes learning of a well-timed closure of the eyelid in response to the CS. The CS and US are conveyed to the cerebellum, respectively, by mossy and climbing fiber afferents (Mauk et al., 1986; Steinmetz et al., 1989; Hesslow et al., 1999), and the learned eyelid closure is driven by activity in the anterior interpositus nucleus (AIN) of the cerebellum (McCormick and Thompson, 1984; Choi and Moore, 2003) (see Fig. 1). This relatively direct relationship between eyelid conditioning and the cerebellum greatly facilitates the analysis of sites of plasticity and their relative contributions to learning.
Figure 1.

A schematic showing how eyelid conditioning engages the cerebellum. The tone-CS activates mossy fiber inputs from the pontine nuclei, mild electrical stimulation around the eye (US) activates climbing fiber inputs (green), and output from the AIN of the cerebellum drives CR expression (blue trace). Removing or pharmacologically disconnecting the cerebellar cortex via GABAA antagonists in the AIN or by electrolytic lesions of Purkinje cells (dotted blue) unmasks SLRs (red trace). The expression pathway underlying SLRs and the essential site of plasticity was localized using a variety of treatments, including glutamate receptor antagonists [experiment 1 (E1) and E2], the GABAA agonist muscimol (E1–4), and microstimulation of mossy fibers (E3, 4).
The cerebellar cortex and/or AIN have been proposed as candidate sites of plasticity, because the inputs from the CS and US converge at these sites (see Fig. 1). One line of evidence for multiple plasticity sites comes from studies of posttraining lesions of the cerebellar cortex. Early results in rabbits ranged from complete abolition of the learned responses pointing to the cerebellar cortex as the key site (Yeo, 1991) to nominal effects that suggested a more fundamental role for the AIN (McCormick and Thompson, 1984). Numerous subsequent studies measuring closure of the external eyelid or nictitating membrane have reported responses with relatively fixed and short latencies to onset (∼80–150 ms) after direct lesions (McCormick and Thompson, 1984; Perrett et al., 1993; Perrett and Mauk, 1995; Garcia et al., 1999; Medina et al., 2000) or infusing GABAA antagonists into the AIN (Garcia and Mauk, 1998; Medina et al., 2001; Ohyama and Mauk, 2001; Bao et al., 2002; Ohyama et al., 2003; Aksenov et al., 2004). We previously hypothesized that these short-latency responses (SLRs) (see Fig. 1) are mediated by plasticity in the AIN (Raymond et al., 1996; Mauk and Donegan, 1997).
Here, we use microstimulation and reversible lesions to test this hypothesis. Our results eliminate alternative candidate sites of plasticity that are either upstream or downstream from the AIN, suggesting that SLRs reflect a form of plasticity at the excitatory synapses of mossy fibers onto AIN neurons (Racine et al., 1986; Kleim et al., 2002; Pugh and Raman, 2006; Zhang and Linden, 2006). They provide new support for the view that cerebellar learning in general involves plasticity in the cerebellar cortex and in its downstream target neurons in the deep cerebellar and vestibular nuclei. The link between SLRs and plasticity in the AIN also suggests the potential for using SLRs to measure plasticity induced at a particular cell and/or synapse in vivo.
Materials and Methods
Subjects and surgery.
Data were obtained from 24 naive New Zealand albino rabbits (Oryctolagus cuniculus) treated according to National Institutes of Health guidelines and protocols approved by the Animal Welfare Committee at the University of Texas-Houston Health Science Center. Subjects were preanesthetized with acepromazine (1.5 mg/kg) and maintained under isoflurane anesthesia (∼1–2% mixed in oxygen). The head was immobilized in a stereotaxic restrainer with lamba 1.5 mm ventral to bregma. Four craniotomies surrounding bregma were made with a dremel drill, and screws were inserted in each to support the headstage. In all experiments, a cannula (Plastics One, Roanoke, VA) was stereotaxically placed in the ipsilateral AIN [+0.7 to ∼1.0 mm anteroposterior (AP), −5.0 mm mediolateral (ML), −14.5 mm dorsoventral (DV) from lamda]. Additionally, the following instruments were implanted in experiments 1–4, respectively: an array of stainless-steel electrodes (exposed ∼0.5–1 mm at tip; catalog #571000, A-M Systems, Carlsborg, WA) oriented coronally (n = 2; two spaced 1 mm apart) or sagittally (n = 4; four spaced 0.75 mm apart) in the cerebellar cortex (+2.5 to ∼5 mm AP, −4.5 to ∼5.5 mm ML, −14 to ∼14.5 mm DV from lambda; experiment 1), a cannula in the red nucleus (9.0 mm AP, +1.2 mm ML, −14.8 mm DV from bregma; experiment 2), or a tungsten electrode (tip exposed to obtain impedance of ∼100–200 kΩ; catalog #575500, A-M Systems) in the middle cerebellar peduncle (+3.0 mm AP, −5.5 mm ML, −16 mm DV from lambda; experiments 3 and 4). Implants were secured with dental acrylic together with a headbolt screw placed between the four skullscrews, and any openings were closed with sutures. Stimulating electrodes were attached ∼1 cm rostral and caudal to the left eye. Training began at least 1 week after recovery.
Drugs and infusions.
Chemicals (Sigma, St. Louis, MO) were dissolved in artificial CSF (ACSF) (in mm: 119 NaCl, 2.5 KCl, 1.2 NaH2PO4, 2 MgCl2, 2 CaCl2, 26 NaHCO3, 10 d-glucose, 20 HEPES, pH ∼7.35–7.4). Each of the five experiments used the GABAA antagonists picrotoxin (200 μm) or SR 95531 (gabazine; 20 μm), and/or the GABAA agonist muscimol (1 mm). The glutamate antagonists d-(−)-APV (1 mm) and CNQX (.5 mm), or kynurenic acid (2 mm), respectively, were used in experiments 2 and 3. Drugs were delivered through a 50 μl Hamilton syringe coupled to a 33 gauge internal cannula protruding 1.2 mm beyond the guide cannula. The syringe was mounted on an automated injector system (model MD-1001; Bioanalytical Systems, West Lafayette, IN) and driven by an electronic pump (model MD-1020; Bioanalytical Systems).
Training and testing.
Subjects were trained in custom-designed chambers equipped with a speaker connected to an audio source module (model V85–05; Coulbourn Instruments, Allentown, PA) to generate tones and isolated pulse stimulators (model 2100; A-M Systems) to deliver electrical pulses through the periorbital electrodes. For experiments involving electrical microstimulation, stimulus isolators (model A360; World Precision Instruments, Sarasota, FL) coupled to an additional stimulator were used to provide a constant current through lead wires connected to the implanted electrodes via gold pins. An infrared emitter/detector attached to the headstage of each rabbit was used to record movements of the left external eyelid by detecting changes in the amount of reflected light. Maximum eyelid closure was calibrated daily before each session by manually applying an electric pulse through the periorbital electrode to elicit a full eyelid closure, and the voltage-to-millimeter relationship was then determined assuming full closure to be 6 mm.
For all experiments, the 550 ms CS was either a 1 kHz tone (85 dB, ∼70 dB background) or cathodal stimulation (100 Hz, 40 μs pulse width, 100 or 150 μA) of the middle cerebellar peduncle. The US was a 50 ms pulse train (100 Hz, 1 ms pulse width, ∼1–2.5 mA). Paired sessions involved 12 nine-trial blocks [one CS-only plus eight paired (CS coterminating with US)]. Extinction involved 108 CS-only trials. For both paired and extinction sessions, the average time between trials was 30 s (range, 20–40). In unpaired training, the CS or US occurred every 15 s (range, 5–25) for 216 trials. Stimulus presentation was controlled by custom-designed software. Data were sampled at a rate of 1 kHz, and individual sweeps consisting of 2500 data points (200 ms before and 2300 ms after CS onset) were collected for each trial and stored for subsequent off-line analysis.
In each experiment, we trained subjects for at least five sessions to establish robust responding to the CS and then (with the exception of experiment 5) tested for appropriate placement of the cannula in the AIN by confirming the abolition of learned responses with muscimol (Garcia and Mauk, 1998). Subsequently, to unmask SLRs (see definition below) we either passed anodal current (2 mA, ∼1–1.5 min) through one or two stainless steel electrodes at a time with the intent of creating electrolytic lesions of the anterior lobe (Garcia et al., 1999) (n = 6; experiment 1) or infused a GABAA antagonist into the AIN (Garcia and Mauk, 1998) (n = 22, experiments 1–5).
Unless otherwise noted, for fast drug infusions (experiments 2 and 5, and four animals from experiment 1), 1 μl was infused at 0.5 μl/min 30 and 5 min before initiating or resuming testing. Testing involved 24 or 108 CS-only trials postinfusion or a paired session in which infusions preceded or began 18 trials after session initiation. [For four animals in experiment 1 with sagittal arrays in which repeated electrolytic lesions were ineffective, we examined the effects of infusing d-(−)-APV and CNQX (2 μl at 0.5 μl/min) into the AIN on SLRs unmasked by presession gabazine infusions through the same cannula. In this case, the infusion began after trial 21 of the paired session and testing resumed immediately.] For slower infusions (experiments 1, 3, and 4), 4 μl of muscimol or 2 μl of gabazine, respectively, were infused at 0.2 or 0.1 μl/min after trial 36 of a paired session and testing began immediately after initiating infusions. We used slower GABAA antagonist infusions (experiments 3 and 4) with the intent of eventually titrating volumes for each animal. As a result, SLRs occurred less frequently than during fast GABAA antagonist infusions (experiments 1, 2, and 5) owing to the greater variability in their time of emergence.
The mossy fiber stimulation current was 100 μA (experiment 3, n = 4; increased to 150 μA in one animal after three sessions) or 150 μA (experiment 4, n = 4). Animals in experiment 3 received a second gabazine test after at least four sessions of extinction. Animals in experiment 4 received a second gabazine test with a white noise stimulus (85 dB) presented on trial 7 of each nine-trial block.
Histology.
Infusion sites were marked by passing direct anodal current (200 μA, ∼30 s) through a stainless-steel wire cut to the length of the internal cannula. Animals were killed with sodium pentobarbital and perfused intracardially with 2 L of 10% formalin. Brains were embedded and sectioned (80 μm) with a freezing microtome. Slices were mounted and stained with cresyl violet.
Data analysis.
For each trial, we obtained the latency to a criterion amplitude of 0.3 mm eyelid closure. This criterion value is >100 SDs above the noise level of the recording system (SD, ∼0.002 mm) and detects fairly robust SLRs visible to the naked eye without inflating the contribution of very small SLRs. A conditioned response (CR) was defined as a response attaining the criterion within 500 ms of CS onset. An SLR was defined as a CR having a latency to criterion <200 ms. Cannula placements in the AIN were judged appropriate if the percentage of SLRs during picrotoxin or gabazine in the AIN exceeded 8%, and histological examination confirmed the placements in or near the AIN.
Tests for differences in mean percentage CRs and mean percentage SLRs were conducted with repeated-measures ANOVA (RM-ANOVA) and paired-sample t tests, with the level of α set at 0.05. For the sake of simplicity, we report only these measures, because maximum CR and SLR amplitudes also showed similar patterns of results. For experiment 1, a two-way RM-ANOVA (n = 4) was performed on percentage of SLRs during test sessions with treatment (glutamate antagonists or ACSF) and 21-trial blocks as within-subject factors. For experiment 2 (n = 3), a paired sample t test was performed on the first 24 postpicrotoxin/gabazine trials before and after training with the red nucleus inactivated. For experiment 3 (n = 4), paired-sample t tests were performed on averaged percentage SLRs before and after infusions during the posttraining gabazine test. For experiment 4 (n = 4), paired sample t tests were performed on trials during postgabazine test trials with white noise and mossy fiber stimulation. For experiment 5 (n = 7), a one-way RM-ANOVA and follow-up paired-sample t tests were performed on the first 24 postpicrotoxin trials before training, after unpaired training, and after paired training. In addition, for experiments 1 (n = 6), 3 (n = 4), and 4 (n = 4), one-way RM-ANOVAs were performed on percentage CRs as a function of 36-trial blocks during a posttraining test session with muscimol.
Results
Experiment 1: glutamatergic transmission in the AIN is necessary for SLR expression
To test the hypothesis that activity in the AIN is necessary for expressing SLRs, we first examined the effects of pharmacologically manipulating AIN activity. If the AIN is part of the necessary expression pathway, then inactivating this nucleus should abolish the expression of SLRs (Fig. 1). We initially gave pairings of a tone-CS and US to six rabbits to produce robust learning. As an independent control to confirm proper cannula placement, we then infused the GABAA agonist muscimol to test the effects of silencing the AIN on the expression of normal conditioned responses. Consistent with previous studies, this abolished normal conditioned responses to the tone-CS (Fig. 2, top left and bottom, cyan) (n = 6; F(2,10) = 49.7; p < 0.001) (Krupa et al., 1993; Garcia and Mauk, 1998). Subsequently, we made electrolytic lesions of the cerebellar cortex to unmask SLRs (Fig. 2, top right, dark gray) (Garcia et al., 1999). In two animals with successful lesions, muscimol infusions into the AIN also abolished the expression of SLRs (Fig. 2, top right, cyan). These data show that like normal conditioned responses, SLRs require proper AIN activity to be expressed.
Figure 2.
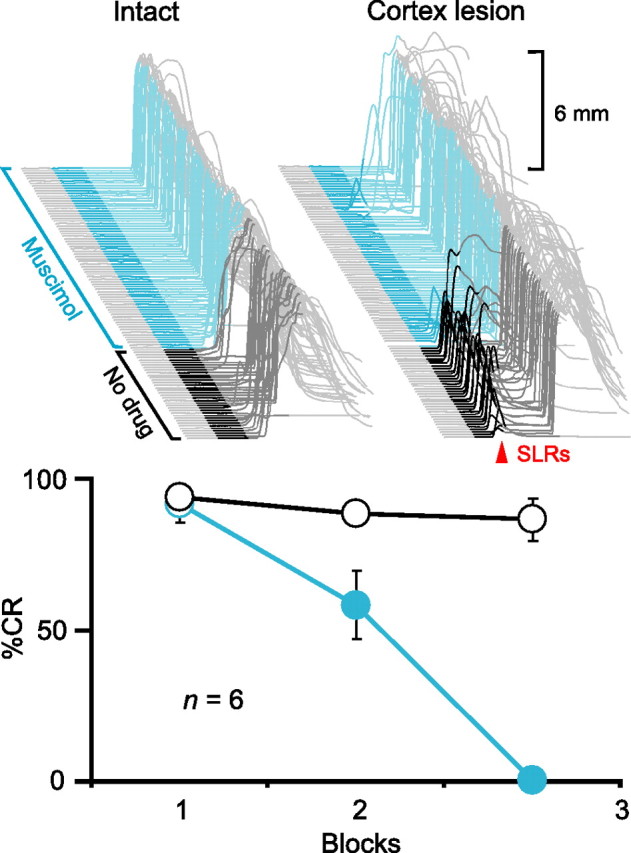
Expression of CRs and SLRs requires activity in the AIN. Top left, Sample traces from an animal during a test session in which the GABAA agonist muscimol infused into the AIN abolished CRs. Top right, Sample traces from the same animal in which muscimol abolished SLRs (red arrowhead) unmasked by electrolytic lesions of the cerebellar cortex. In this and all subsequent figures, sample eyelid traces are arranged chronologically from front to back with the time of CS presentation highlighted (dark gray, no drug; color, after start of infusion) and the first 200 ms of the CS darkened to emphasize SLRs (upward deflection, eyelid closure). Scale bars denote the magnitude of full eyelid closure (6 mm). Bottom, Summary data showing percentage CRs as a function of 36-trial blocks during test sessions with muscimol (cyan) or a standard training session (black and white) before the cerebellar cortex lesion. For summary data in this and all subsequent figures, circles and bars (open, no drug; filled, after start of infusion) indicate CRs and SLRs, respectively. Error bars show SEM.
To examine more precisely the necessary conditions for SLR expression, we next tested the effects of blocking excitatory transmission at mossy fiber-to-AIN synapses by infusing ionotropic glutamate receptor antagonists into the AIN (Fig. 1, E1, dotted black line). If SLRs are mediated by an expression pathway involving the AIN, this treatment should also abolish SLRs, because the mossy fiber-to-AIN synapses are the only route through which the tone-CS could drive their expression. In four animals where the electrolytic lesion fell outside of the cerebellar cortex, we first reestablished robust baseline responding to overcome nonspecific temporary decrements in performance. Then, in a subsequent test session, we preinfused the GABAA antagonist SR 95531 (gabazine) into the AIN to unmask SLRs from the start of the session (Fig. 3, top left, red). [In later experiments, we also used picrotoxin (Garcia and Mauk, 1998). These pharmacologically induced SLRs are selective to GABAA antagonists and resemble those revealed by physical cerebellar cortex lesions and lidocaine infusions into the cerebellar cortex (J. Kreider, T. Ohyama, W. L. Nores, and M. D. Mauk, unpublished observation). Thus, in all likelihood, infusing GABAA antagonists into the AIN causes SLRs by reversibly blocking inhibitory input from Purkinje cells.]
Figure 3.
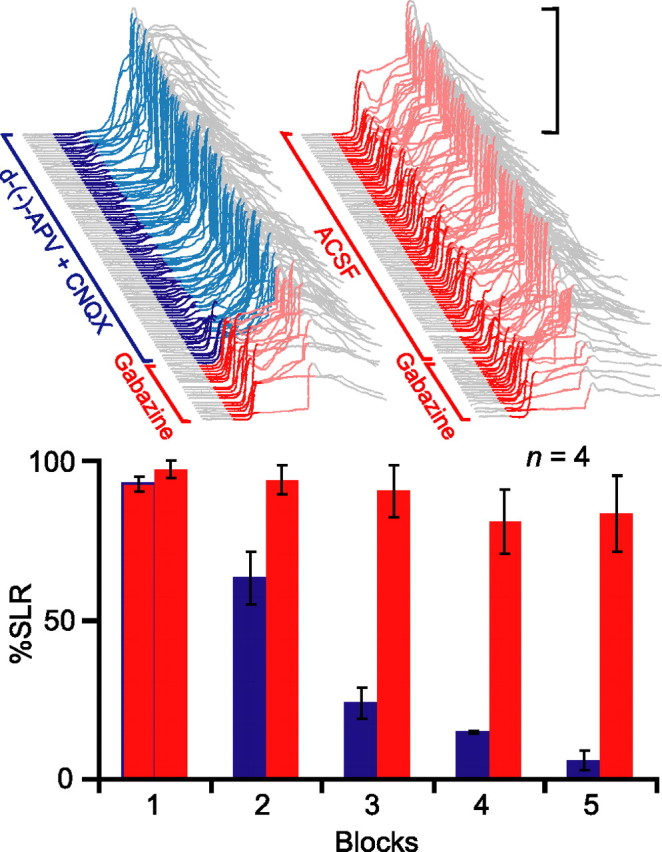
Expression of SLRs requires AIN activity mediated by glutamatergic synaptic transmission through mossy fiber synapses. Top, Sample traces from an individual subject during two test sessions in which presession infusions of the GABAA antagonist gabazine unmasked SLRs from the start of testing (red traces). Infusing the ionotropic glutamate receptor antagonists d-(−)-APV and CNQX into the AIN after trial 21 suppressed SLRs (left), whereas infusing ACSF did not (right). Bottom, Summary data of percentage SLRs as a function of 21-trial blocks during the two test sessions (glutamate antagonist session, red to blue; ACSF session, all red).
Once the gabazine infusion unmasked stable SLRs, we infused the NMDA receptor antagonist d-(−)-APV and the AMPA receptor antagonist CNQX through the same cannula. This led to a gradual suppression of SLRs over the course of the next few blocks of trials (Fig. 3, top left and bottom, red to blue). The suppression was not a result of washout of gabazine, because a subsequent control session in which the vehicle (ACSF) was infused after gabazine did not affect SLR expression (Fig. 3, top right and bottom, all red) (n = 4; F(4,12) = 44.1; p < 0.001). The nearly complete abolition of SLRs indicates that AIN activity mediated by AMPA and NMDA receptor-mediated synaptic transmission is required for their expression.
Experiment 2: the site of plasticity underlying SLRs is upstream of the red nucleus
The previous experiment demonstrated that the AIN is required for the expression of SLRs but did not provide sufficient evidence that the site of plasticity underlying SLRs is in the AIN. Excitatory transmission in the AIN may only be necessary either to induce plasticity at the AIN or at sites further downstream, or it may simply relay neural changes induced upstream from the AIN down to premotor/motor neurons. To distinguish these possibilities, we first tested whether the site of plasticity is downstream from the AIN. Specifically, we asked whether SLRs could be learned when excitatory synaptic transmission is blocked in the red nucleus, the first downstream target of the AIN (Fig. 1, E2, dotted black line). If the site is in the AIN or upstream from it, acquisition of SLRs should occur normally, because the activity required for plasticity should be unaffected by inactivating the red nucleus. In contrast, SLRs should not be acquired if the site is downstream of the AIN, because blocking excitatory transmission in the red nucleus should prevent any activity at or further downstream from it.
We trained rabbits for five sessions while infusing kynurenic acid, a broad-spectrum glutamate receptor antagonist, into the red nucleus. The effectiveness of the infusion was assessed by its ability to prevent expression of normal conditioned eyelid responses (Chapman et al., 1990; Krupa et al., 1993). Therefore, animals were included in the analysis only if they expressed <10% conditioned responses across the five sessions of training (n = 3) (Fig. 4, bottom, blue). Despite this blockade of response expression, robust SLRs were unmasked during the first posttraining test session in which we infused either picrotoxin (n = 2) or gabazine (n = 1) into the AIN (Fig. 4, top right) (n = 3; t(2) = 32.01; p < 0.001; T1 vs T2). In one animal, we were also able to test the effects of silencing the red nucleus on the SLR expression; the SLRs unmasked by infusing gabazine into the AIN were reversibly abolished by simultaneous presession infusions of muscimol into the red nucleus (data not shown). Thus, like normal conditioned responses, blocking activity in the red nucleus prevents the expression of SLRs but not the induction of the plasticity mediating them (Krupa et al., 1993). These data indicate that the site of plasticity underlying SLRs is upstream of the red nucleus, either at or upstream from the AIN.
Figure 4.
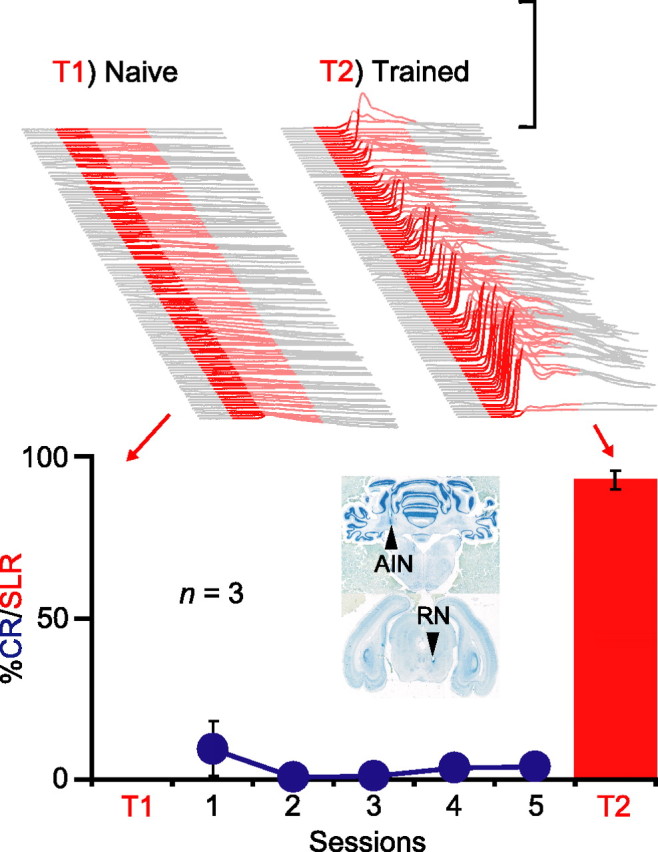
Plasticity underlying SLRs is upstream from the red nucleus. Top, Sample traces from one animal during two separate sessions involving 108 CS-only trials after presession infusions of gabazine into the AIN before (left, T1) and after (right, T2) training with the red nucleus blocked. Bottom, Summary data showing that animals not exhibiting CRs during training (blue) nonetheless acquired SLRs (red bars, T2). The inset shows coronal sections from an individual subject showing the placement of the internal cannulas in the AIN and red nucleus (RN), respectively.
Experiment 3: the site of plasticity underlying SLRs is downstream of mossy fibers
Next, to address whether the site of plasticity underlying SLRs is in the AIN or upstream from it, we asked whether substituting microstimulation of mossy fibers for the tone-CS (Fig. 1, E3) would support the acquisition of SLRs. Mossy fiber stimulation should support SLRs only if the site of plasticity underlying them is at or downstream of the AIN, and not if the site of plasticity is upstream from the mossy fiber input to the AIN.
We first trained four animals using direct microstimulation of the middle cerebellar peduncle, a tract containing mossy fibers arising from the pontine nuclei, as the CS. Consistent with previous studies, mossy fiber stimulation supported robust learning (Fig. 5, top) (n = 4; F(4,12) = 18.2; p < 0.001) (Steinmetz et al., 1989; Hesslow et al., 1999). In an initial test session to confirm proper cannula placement, we infused muscimol to silence the AIN. Like conditioned responses to a tone-CS, this abolished the learned responses to the mossy fiber stimulation CS (Fig. 5, bottom left) (n = 4; F(2, 6) = 39.9; p < 0.001). We then pharmacologically disconnected the cerebellar cortex during a subsequent test session by infusing gabazine into the AIN. As with a tone-CS, this unmasked SLRs to the mossy fiber stimulation CS (Fig. 5, bottom center, training) (n = 4; t(3) = 4.47; p < 0.05; percentage SLRs preinfusion and postinfusion).
Figure 5.
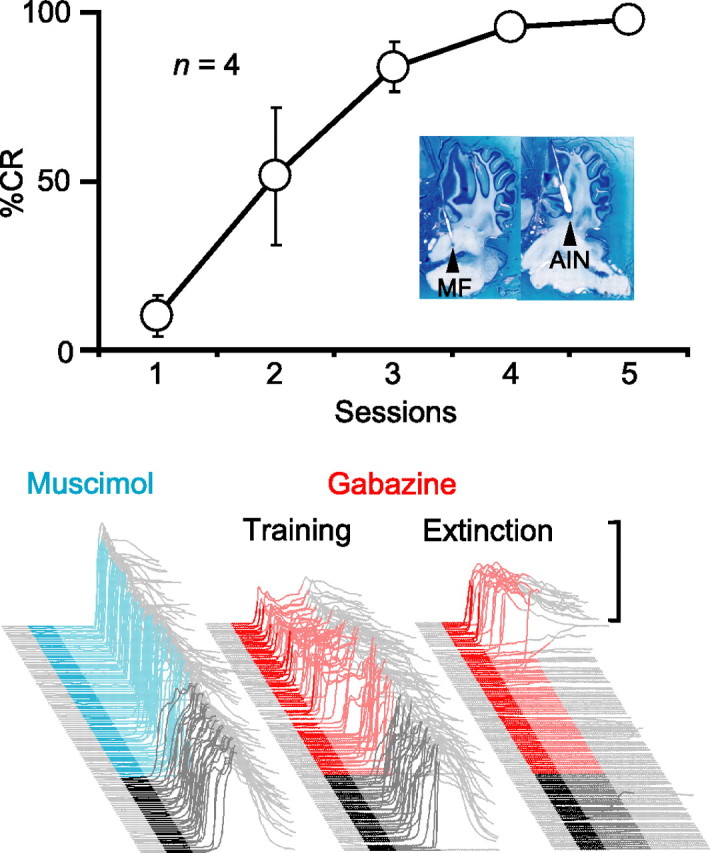
Plasticity underlying SLRs is downstream of the mossy fibers. Top, Microstimulation of mossy fibers supported the acquisition of normal conditioned responses. The inset shows parasagittal sections from one animal, with the approximate locations of the tips of the stimulating electrode (MF) and internal cannula (AIN). Bottom, Sample traces from a subject during test sessions in which either muscimol (cyan) or gabazine (red) were infused into the AIN during paired sessions (left and center) or on the fifth session of extinction (right). Muscimol abolishes CRs (left), whereas gabazine unmasks SLRs (center) even after extinction (right).
A previous study showed that SLRs are unmasked by disconnection of the cerebellar cortex even when conditioned responses have been fully extinguished by repeated presentations of the tone-CS alone (Medina et al., 2001). To ensure that SLRs supported by mossy fiber stimulation display properties similar to those supported by a tone-CS, we performed a second gabazine infusion after at least four sessions of extinction. In two rabbits, we were able to unmask SLRs during a final extinction session, despite a complete absence of responding before the infusion (Fig. 5, bottom right, extinction). This reinforces the parallels between SLRs mediated by mossy fiber stimulation and those by a standard tone-CS. Together, these data suggest that, like normal conditioned responses, the essential plasticity underlying SLRs is downstream from mossy fiber axons entering the cerebellum.
Experiment 4: the plasticity underlying SLRs is input specific
The combined results of the previous two experiments suggest that the site of plasticity is in the AIN, because it is upstream of the red nucleus and downstream of mossy fibers. One of two general mechanisms, synaptic plasticity at mossy fiber-to-AIN synapses (Racine et al., 1986; Kleim et al., 2002; Pugh and Raman, 2006; Zhang and Linden, 2006) or a cell-wide increase in the intrinsic excitability of AIN neurons (Aizenman and Linden, 2000; Zhang et al., 2004), could mediate SLRs. To test these alternatives, we examined the stimulus specificity of SLRs established with a mossy fiber stimulation CS (Fig. 1, E4). If SLRs reflect activity-dependent plasticity and/or the formation of new mossy fiber-to-AIN synapses, they should be evoked by the training CS but not by untrained stimuli that nonetheless have (or with training eventually gain) access to the AIN. In contrast, if SLRs are mediated solely by a cell-wide increase in the excitability of AIN neurons, they should generalize broadly to these stimuli.
As in the previous experiment, we trained four animals with a mossy fiber stimulation CS and then confirmed proper placement of the cannula by showing that muscimol infusions abolished learned responses (data not shown). In a subsequent test session, we infused gabazine into the AIN to unmask SLRs. During this session, white noise trials were interspersed among standard stimulation trials to activate as many auditory mossy fibers as possible and increase the likelihood of detecting generalization. Nonetheless, SLRs completely failed to generalize to the white noise (Fig. 6A) (n = 4; t(3) = 3.72; p < 0.05; percentage SLRs postinfusion, mossy fiber vs white noise). To assess whether this specificity was attributable to a failure of the white noise to support SLRs, we trained two of the animals for three sessions with the white noise paired with the US and then infused gabazine into the AIN a second time. The white noise supported robust learning and SLRs (data not shown). Thus, the stimulus specificity cannot be attributed to an inability of the white noise to gain access to the AIN. The data suggest that a highly input-specific form of plasticity in the AIN underlies SLRs (Perrett and Mauk, 1995; Ohyama et al., 2003).
Figure 6.
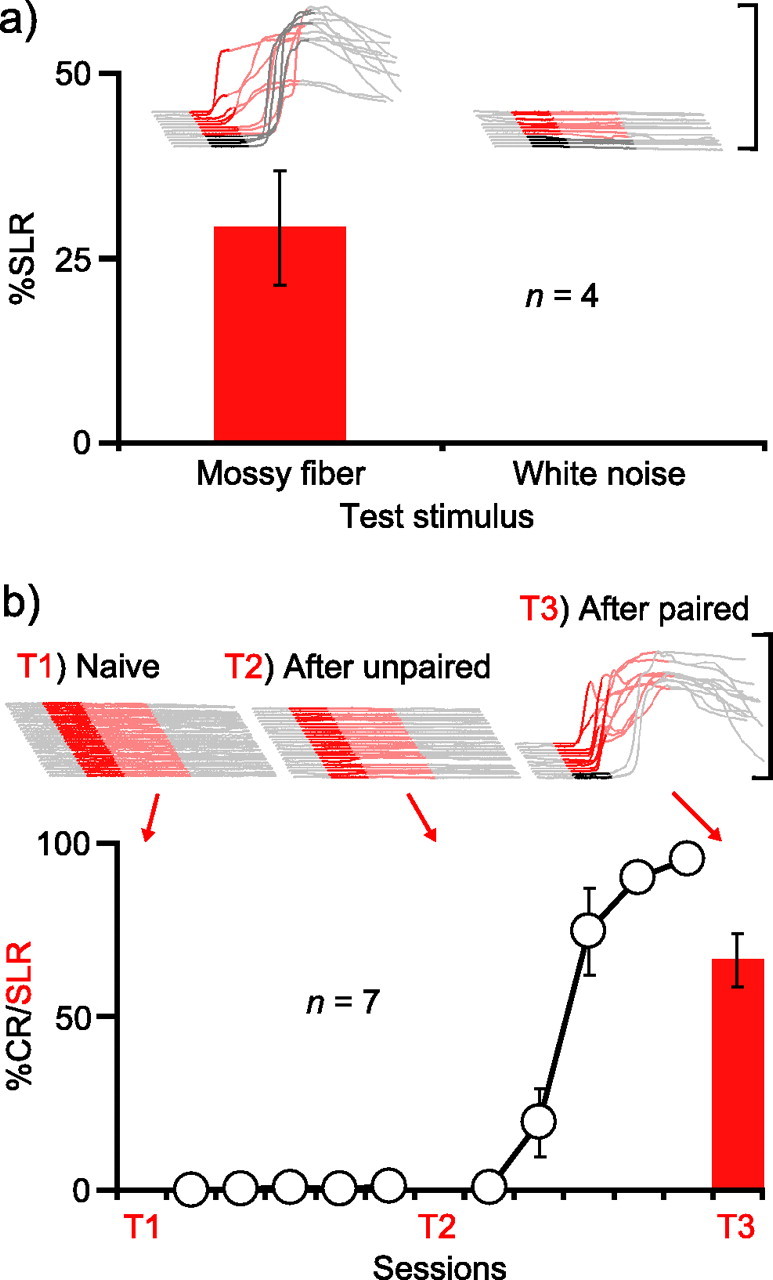
SLRs are stimulus specific and associative. A, SLRs supported by mossy fiber stimulation are stimulus specific. Sample traces and summary data showing that SLRs established to mossy fiber stimulation do not generalize to a white noise. Because subsequent training to the white noise enabled gabazine to unmask SLRs, this stimulus specificity of SLRs is not a result of the inability of the white noise to gain access to the AIN. B, SLRs are induced only after paired training. All traces in this figure are from CS-only trials. Infusing the GABAA antagonist picrotoxin into the AIN was ineffective before (T1) or after (T2) unpaired training, but unmasked SLRs after subsequent paired training (T3).
Experiment 5: SLRs are learned and associative
It has been noted previously that SLRs may simply reflect a nonassociative change resulting as a consequence of disinhibiting the AIN (Attwell et al., 2002). Although the result of the previous experiment (Fig. 6A) suggests that they are associative, in a final experiment we examined this further by asking whether SLRs would fail to be induced by unpaired presentations of the CS and US (Gormezano et al., 1983). If SLRs are mediated by an associative form of plasticity in the AIN, then they should be induced only after specifically pairing the CS with the US.
We tested for the presence of SLRs at three different time points: before training, after unpaired training in which a tone-CS and US were explicitly not paired, and finally after paired training during which conditioned eyelid responses were acquired to the tone-CS. During each test, we infused picrotoxin into the AIN (Garcia and Mauk, 1998). Consistent with previous studies, we observed no SLRs before training (Fig. 6B, T1) (Medina et al., 2001; Ohyama et al., 2003). Five sessions of unpaired training failed to promote learning and, importantly, also failed to support the acquisition of SLRs (Fig. 6B, T2). This failure was not a result of improper cannula placement, because SLRs were unmasked in a test session conducted after five sessions of paired training (Fig. 2, T3) (n = 7; F(2,12) = 75.0; p < 0.001; ts(6) = 8.66; p < 0.001; T1 or T2 vs T3). Therefore, SLRs are associative.
To summarize, the expression of SLRs requires activity in the AIN, mediated specifically by glutamatergic synaptic transmission (Figs. 2, 3). Thus, the AIN is part of the necessary pathway for expressing SLRs (Fig. 7). The site of plasticity within this expression pathway must be upstream of the red nucleus (Fig. 7, dotted arrow), because its inactivation prevents the expression but not the induction of SLRs (Fig. 4). The site must also be downstream of the pontine nuclei (Fig. 7, dashed arrow), because microstimulation of their mossy fiber axons supports SLRs (Fig. 5). Therefore, both SLRs and normal conditioned responses are learned during red nucleus inactivation (Krupa et al., 1993) and with mossy fiber stimulation as the CS (Steinmetz et al., 1989; Hesslow et al., 1999). [It is unlikely that plasticity in a pathway downstream of the AIN that initially bypasses the red nucleus (e.g., via thalamus, cortex, and back to red nucleus) or presynaptic plasticity (e.g., increased transmitter release) in the red nucleus mediates SLRs, because the threshold for evoking eyelid closure by stimulating the AIN does not change with training (Tracy et al., 1998).] Therefore, because SLRs are seen only when input from the cerebellar cortex is blocked, the most likely explanation is that the underlying site of plasticity is in the AIN. Finally, SLRs are highly stimulus-specific and induced only after pairing the CS and US (Fig. 6). These observations are incompatible with notions that SLRs reflect nonassociative changes resulting from disinhibiting the AIN (Attwell et al., 2002), increases in tonic AIN activity levels (Aksenov et al., 2005), or cell-wide increases in the intrinsic excitability of AIN neurons (Aizenman and Linden, 2000; Zhang et al., 2004). Rather, they suggest that SLRs reflect an associative and input-specific form of AIN plasticity.
Figure 7.

The expression pathway and site of plasticity underlying SLRs. Because proper AIN activity is necessary for SLR expression (E1), the AIN must be part of the expression pathway. The site of plasticity must be upstream of the red nucleus, because inactivating this structure does not prevent the acquisition of SLRs (E2, dotted line), and downstream of the pontine nuclei, because direct stimulation of their mossy fibers supports SLRs (E3, 4, dashed line). The underlying plasticity in the AIN must be input specific and associative, because SLRs are strongly stimulus specific (E4) and induced only by paired training (E5). Thus, SLRs are probably mediated by synaptic plasticity at mossy fiber-to-AIN synapses (green asterisk) or possibly a dendrite-specific increase in the intrinsic excitability of AIN neurons (blue asterisk).
Discussion
Cerebellar learning induces plasticity in target nuclei downstream of cerebellar cortex
Our results add to the evidence that in addition to the classical site of plasticity in the cerebellar cortex (Marr, 1969; Albus, 1971), eyelid conditioning induces plasticity in the AIN (Kleim et al., 2002). The existence of an analogous form of plasticity in the vestibular nucleus and the notion that its induction is controlled by inputs from Purkinje cells was first suggested by Miles and Lisberger (1981) for another form of cerebellar motor learning known as vestibulo-ocular reflex (VOR) adaptation, and evidence in support of this view has been obtained in subsequent studies (Lisberger, 1994; du Lac et al., 1995). The present findings underscore the similarities between eyelid conditioning and adaptation of the VOR, in that they are both forms of cerebellar motor learning that induce plasticity at two sites, one in the cerebellar cortex and another in their target neurons in the deep cerebellar/vestibular nuclei. As such, our results suggest the generality of two sites of plasticity in cerebellar learning (Raymond et al., 1996; Mauk, 1997; Mauk and Donegan, 1997).
Candidate plasticity mechanisms underlying SLRs
A number of plasticity mechanisms in the AIN could underlie SLRs. A pairing-specific increase in the number of excitatory synapses in the AIN (Kleim et al., 2002) and/or long-term potentiation (LTP) at the mossy fiber-to-AIN synapse (Racine et al., 1986; Pugh and Raman, 2006) have been reported. Persistent increases in the intrinsic excitability of deep cerebellar nucleus neurons (Aizenman and Linden, 2000; Nelson et al., 2003, 2005; Zhang et al., 2004) could also contribute, but such changes must be region specific (Zhang and Linden, 2003) to explain the stimulus specificity of SLRs (Perrett and Mauk, 1995; Ohyama et al., 2003).
Understanding the role of the input-specific plasticity in the AIN will require elucidation of the signals that control its induction. In principle, the plasticity could be driven by the activity in any of the inputs to the AIN or by activity in the AIN itself (Medina and Mauk, 1999). Miles and Lisberger (1981) first proposed that Purkinje cells could control the induction of synaptic changes in the deep cerebellar/vestibular nuclei, and subsequent computational studies were in agreement (Medina and Mauk, 1999). Recent studies have shown that rebound excitation of AIN neurons following burst-pause modulation of Purkinje cell activity is sufficient to increase both the intrinsic excitability of AIN neurons (Zhang et al., 2004) and the strength of mossy fiber synapses (Pugh and Raman, 2006). Because such a modulation occurs during eyelid conditioning (Hesslow and Ivarsson, 1994), it seems likely that both types of change are induced in the AIN during learning.
Functional contributions of plasticity in the AIN
We propose the following hypothesis for how plasticity in the AIN cooperates with plasticity in the cerebellar cortex in eyelid conditioning. Initially, CS–US pairings induce plasticity in the cerebellar cortex in the form of long-term depression of granule cell–Purkinje cell synapses coactive with climbing fiber activity (Ito, 2001). This leads to the establishment of a transient, well-timed suppression of Purkinje cell activity to the previously neutral CS (Hesslow and Ivarsson, 1994). Additional training induces plasticity in the AIN at mossy fiber synapses coactive with this Purkinje cell pause (Medina and Mauk, 1999; Pugh and Raman, 2006). For instances of learning such as eyelid conditioning in which the CS-activated mossy fiber input has not previously modulated the target response, mossy fiber-to-AIN synapses may not yet exist, and thus their formation (Kleim et al., 2002) may be necessary before they can undergo LTP.
Current evidence is consistent with the hypothesis that plasticity in the AIN is necessary for the expression of temporally specific learning initially induced in the cerebellar cortex. First, infusing picrotoxin into the AIN at different time points during initial acquisition reveals that the probability of observing SLRs parallels that of normal conditioned responses, consistent with the notion that plasticity induction in the AIN is the rate limiting factor in acquisition (Medina et al., 2001). Second, the observation of initial learning in the cerebellar cortex that is later revealed by inducing AIN plasticity suggests that AIN plasticity is necessary for the expression of conditioned responses and requires initial plasticity in the cerebellar cortex for its induction (Ohyama and Mauk, 2001). Third, the hypothesis explains a form of savings, the rapid reacquisition of learned eyelid responses after extinction (Kehoe, 1988). Consistent with a detailed simulation of the cerebellum, SLRs unmasked by picrotoxin in the AIN are more resistant to extinction than normal conditioned responses, and their frequency predicts the rate of subsequent relearning (Medina et al., 2001). This suggests that initial acquisition of eyelid responses is slow, because it requires establishing plasticity at two sites, whereas relearning after extinction is faster, because plasticity is already induced in the AIN. Interestingly, a more natural form of cerebellar learning such as VOR adaptation does not display robust savings (Miles and Lisberger, 1981), perhaps because mossy fiber synapses onto the vestibular nuclei already exist. Nonetheless, recent studies of VOR adaptation showing that the effects of reversible cerebellar cortex lesions depend on the amount of training are generally consistent with these findings, further underscoring the parallels between eyelid conditioning and VOR adaptation (Kassardjian et al., 2005; Shutoh et al., 2006).
The residual responses observed after infusions of glutamate antagonists into the AIN (Fig. 3, top left, blue), as well as the failure of similar infusions to abolish the expression of normal conditioned responses (Attwell et al., 2002; Aksenov et al., 2005), appear to contradict the proposed hypothesis. However, glutamate blockade may be incomplete because of either low-drug concentrations or a wider area of inactivation required compared with muscimol. Consistent with a partial glutamate block, simulations in which a fraction of the mossy fiber-to-AIN synapses is inactivated can reproduce the selective abolition of SLRs and the sparing of residual timed responses (data not shown). Alternatively, other excitatory mossy fiber inputs through metabotropic (Zhang and Linden, 2006) or cholinergic (Jaarsma et al., 1997) receptors, and/or increased intrinsic excitability of AIN neurons (Aizenman and Linden, 2000; Zhang et al., 2004), may contribute to the expression of residual responses. Additional work will be necessary to test these alternative hypotheses in detail.
The nature of SLRs in other species
In rabbits, SLRs are seen only after treatments that remove the cerebellar cortex or disconnect it from the AIN. In other species, however, SLRs have been observed even with the cerebellar cortex intact. An early short-latency component to learned responses in cats survives inactivation of the posterior interpositus nucleus (the relevant deep cerebellar nucleus in cats) (Delgado-Garcia and Gruart, 2005), and similar responses in mice survive lesions of the AIN (Koekkoek et al., 2003, 2005). Turtles without a cerebellum also acquire SLRs (Anderson and Keifer, 1997). In each of these cases, an alternative site of plasticity has been suggested: the amygdala in mice (Koekkoek et al., 2005), the motor cortex in cats (Delgado-Garcia and Gruart, 2006), or the brainstem in turtles (Keifer, 2003).
Nevertheless, these findings do not necessarily indicate that SLRs unmasked by cerebellar cortex lesions, and the associated plasticity in the deep cerebellar nuclei, are specific to rabbits. Rather, species may differ on the stimulus parameters required for engaging noncerebellar systems and thus differ on whether the conditioned responses are well timed or instead display an untimed noncerebellar component superimposed on a well-timed component driven by cerebellar learning. It is an entirely separate question as to whether for each species there is an untimed (that is, short-latency) response that is revealed by cerebellar cortex lesions. Resolving this issue will require systematically varying stimulus parameters to identify a set of parameters for each species that confines learning to the cerebellum (Bitterman, 1965). Then, it will be possible to ask whether the relative contributions of plasticity in the cerebellar cortex and deep cerebellar nucleus are similar across different species.
Conclusions
We show in a series of experiments that the SLRs unmasked by posttraining cerebellar cortex lesions in the rabbit are mediated by plasticity in the AIN of the cerebellum. SLRs are expressed via glutamatergic transmission in the AIN, induced only when afferents to the AIN are stimulated or when the nucleus immediately downstream from the AIN is inactivated, and are associative and stimulus specific. Together, our results suggest that SLRs reflect a synaptic and/or regional cellular change in the AIN and represent a rare instance in which a site of plasticity underlying an aspect of a well-studied form of associative learning has been identified. This implies that SLRs can be used as a tool for analyzing the cellular and molecular bases of learning-induced plasticity in vivo. Given the relative ease of obtaining SLRs and the rich behavioral properties of eyelid conditioning (Gormezano et al., 1983), such analyses could contribute significantly to establishing firm links between the molecular, cellular, and behavioral levels of analysis.
Footnotes
This work was supported by National Institute of Mental Health Grants MH46904 and MH57051 (M.D.M.). We thank N. Taylor for histology; P. Bek, A. Polozeck, and S. Woltmann for assistance with surgeries; and T. Blasdel for veterinary support.
References
- Aizenman and Linden, 2000.Aizenman CD, Linden DJ. Rapid, synaptically driven increases in the intrinsic excitability of cerebellar deep nuclear neurons. Nat Neurosci. 2000;3:109–111. doi: 10.1038/72049. [DOI] [PubMed] [Google Scholar]
- Aksenov et al., 2004.Aksenov D, Serdyukova N, Irwin K, Bracha V. GABA neurotransmission in the cerebellar interposed nuclei: involvement in classically conditioned eyeblinks and neuronal activity. J Neurophysiol. 2004;91:719–727. doi: 10.1152/jn.00859.2003. [DOI] [PubMed] [Google Scholar]
- Aksenov et al., 2005.Aksenov DP, Serdyukova NA, Bloedel JR, Bracha V. Glutamate neurotransmission in the cerebellar interposed nuclei: involvement in classically conditioned eyeblinks and neuronal activity. J Neurophysiol. 2005;93:44–52. doi: 10.1152/jn.00586.2004. [DOI] [PubMed] [Google Scholar]
- Albus, 1971.Albus JS. A theory of cerebellar function. Math Biosci. 1971;10:25–61. [Google Scholar]
- Anderson and Keifer, 1997.Anderson CW, Keifer J. The cerebellum and red nucleus are not required for in vitro classical conditioning of the turtle abducens nerve response. J Neurosci. 1997;17:9736–9745. doi: 10.1523/JNEUROSCI.17-24-09736.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attwell et al., 2002.Attwell PJ, Ivarsson M, Millar L, Yeo CH. Cerebellar mechanisms in eyeblink conditioning. Ann NY Acad Sci. 2002;978:79–92. doi: 10.1111/j.1749-6632.2002.tb07557.x. [DOI] [PubMed] [Google Scholar]
- Bao et al., 2002.Bao S, Chen L, Kim JJ, Thompson RF. Cerebellar cortical inhibition and classical eyeblink conditioning. Proc Natl Acad Sci USA. 2002;99:1592–1597. doi: 10.1073/pnas.032655399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitterman, 1965.Bitterman ME. Phyletic differences in learning. Am Psychol. 1965;20:396–410. doi: 10.1037/h0022328. [DOI] [PubMed] [Google Scholar]
- Chapman et al., 1990.Chapman PF, Steinmetz JE, Sears LL, Thompson RF. Effects of lidocaine injection in the interpositus nucleus and red nucleus on conditioned behavioral and neuronal responses. Brain Res. 1990;537:149–156. doi: 10.1016/0006-8993(90)90351-b. [DOI] [PubMed] [Google Scholar]
- Choi and Moore, 2003.Choi JS, Moore JW. Cerebellar neuronal activity expresses the complex topography of conditioned eyeblink responses. Behav Neurosci. 2003;117:1211–1219. doi: 10.1037/0735-7044.117.6.1211. [DOI] [PubMed] [Google Scholar]
- Davis et al., 1982.Davis M, Gendelman DS, Tischler MD, Gendelman PM. A primary acoustic startle circuit: lesion and stimulation studies. J Neurosci. 1982;2:791–805. doi: 10.1523/JNEUROSCI.02-06-00791.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado-Garcia and Gruart, 2005.Delgado-Garcia JM, Gruart A. Firing activities of identified posterior interpositus nucleus neurons during associative learning in behaving cats. Brain Res Brain Res Rev. 2005;49:367–376. doi: 10.1016/j.brainresrev.2004.10.006. [DOI] [PubMed] [Google Scholar]
- Delgado-Garcia and Gruart, 2006.Delgado-Garcia JM, Gruart A. Building new motor responses: eyelid conditioning revisited. Trends Neurosci. 2006;29:330–338. doi: 10.1016/j.tins.2006.05.003. [DOI] [PubMed] [Google Scholar]
- du Lac et al., 1995.du Lac S, Raymond JL, Sejnowski TJ, Lisberger SG. Learning and memory in the vestibulo-ocular reflex. Annu Rev Neurosci. 1995;18:409–441. doi: 10.1146/annurev.ne.18.030195.002205. [DOI] [PubMed] [Google Scholar]
- Garcia and Mauk, 1998.Garcia KS, Mauk MD. Pharmacological analysis of cerebellar contributions to the timing and expression of conditioned eyelid responses. Neuropharmacology. 1998;37:471–480. doi: 10.1016/s0028-3908(98)00055-0. [DOI] [PubMed] [Google Scholar]
- Garcia et al., 1999.Garcia KS, Steele PM, Mauk MD. Cerebellar cortex lesions prevent acquisition of conditioned eyelid responses. J Neurosci. 1999;19:10940–10947. doi: 10.1523/JNEUROSCI.19-24-10940.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gormezano et al., 1983.Gormezano I, Kehoe EJ, Marshall MS. Twenty years of classical conditioning research with the rabbit. In: Sprague JM, Epstein AN, editors. Progress in psychobiology and physiological psychology. New York: Academic; 1983. pp. 197–275. [Google Scholar]
- Hesslow and Ivarsson, 1994.Hesslow G, Ivarsson M. Suppression of cerebellar Purkinje cells during conditioned responses in ferrets. NeuroReport. 1994;5:649–652. doi: 10.1097/00001756-199401000-00030. [DOI] [PubMed] [Google Scholar]
- Hesslow et al., 1999.Hesslow G, Svensson P, Ivarsson M. Learned movements elicited by direct stimulation of cerebellar mossy fiber afferents. Neuron. 1999;24:179–185. doi: 10.1016/s0896-6273(00)80831-4. [DOI] [PubMed] [Google Scholar]
- Ito, 2001.Ito M. Cerebellar long-term depression: characterization, signal transduction, and functional roles. Physiol Rev. 2001;81:1143–1195. doi: 10.1152/physrev.2001.81.3.1143. [DOI] [PubMed] [Google Scholar]
- Jaarsma et al., 1997.Jaarsma D, Ruigrok TJ, Caffe R, Cozzari C, Levey AI, Mugnaini E, Voogd J. Cholinergic innervation and receptors in the cerebellum. Prog Brain Res. 1997;114:67–96. doi: 10.1016/s0079-6123(08)63359-2. [DOI] [PubMed] [Google Scholar]
- Kassardjian et al., 2005.Kassardjian CD, Tan YF, Chung JY, Heskin R, Peterson MJ, Broussard DM. The site of a motor memory shifts with consolidation. J Neurosci. 2005;25:7979–7985. doi: 10.1523/JNEUROSCI.2215-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kehoe, 1988.Kehoe EJ. A layered network model of associative learning: learning to learn and configuration. Psychol Rev. 1988;95:411–433. doi: 10.1037/0033-295x.95.4.411. [DOI] [PubMed] [Google Scholar]
- Keifer, 2003.Keifer J. In vitro classical conditioning of the turtle eyeblink reflex: approaching cellular mechanisms of acquisition. Cerebellum. 2003;2:55–61. doi: 10.1080/14734220310015610. [DOI] [PubMed] [Google Scholar]
- Kleim et al., 2002.Kleim JA, Freeman JHJ, Bruneau R, Nolan BC, Cooper NR, Zook A, Walters D. Synapse formation is associated with memory storage in the cerebellum. Proc Natl Acad Sci USA. 2002;99:13228–13231. doi: 10.1073/pnas.202483399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koekkoek et al., 2003.Koekkoek SK, Hulscher HC, Dortland BR, Hensbroek RA, Elgersma Y, Ruigrok TJ, De Zeeuw CI. Cerebellar LTD and learning-dependent timing of conditioned eyelid responses. Science. 2003;301:1736–1739. doi: 10.1126/science.1088383. [DOI] [PubMed] [Google Scholar]
- Koekkoek et al., 2005.Koekkoek SK, Yamaguchi K, Milojkovic BA, Dortland BR, Ruigrok TJ, Maex R, De Graaf W, Smit AE, Vanderwerf F, Bakker CE, Willemsen R, Ikeda T, Kakizawa S, Onodera K, Nelson DL, Mientjes E, Joosten M, De Schutter E, Oostra BA, Ito M, et al. Deletion of FMR1 in Purkinje cells enhances parallel fiber LTD, enlarges spines, and attenuates cerebellar eyelid conditioning in fragile X syndrome. Neuron. 2005;47:339–352. doi: 10.1016/j.neuron.2005.07.005. [DOI] [PubMed] [Google Scholar]
- Krupa et al., 1993.Krupa DJ, Thompson JK, Thompson RF. Localization of a memory trace in the mammalian brain. Science. 1993;260:989–991. doi: 10.1126/science.8493536. [DOI] [PubMed] [Google Scholar]
- Lisberger, 1994.Lisberger SG. Neural basis for motor learning in the vestibuloocular reflex of primates. III. Computational and behavioral analysis of the sites of learning. J Neurophysiol. 1994;72:974–998. doi: 10.1152/jn.1994.72.2.974. [DOI] [PubMed] [Google Scholar]
- Marr, 1969.Marr D. A theory of cerebellar cortex. J Physiol (Lond) 1969;202:437–470. doi: 10.1113/jphysiol.1969.sp008820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauk, 1997.Mauk MD. Roles of cerebellar cortex and nuclei in motor learning: contradictions or clues? Neuron. 1997;18:343–346. doi: 10.1016/s0896-6273(00)81235-0. [DOI] [PubMed] [Google Scholar]
- Mauk and Donegan, 1997.Mauk MD, Donegan NH. A model of pavlovian eyelid conditioning based on the synaptic organization of the cerebellum. Learn Mem. 1997;4:130–158. doi: 10.1101/lm.4.1.130. [DOI] [PubMed] [Google Scholar]
- Mauk et al., 1986.Mauk MD, Steinmetz JE, Thompson RF. Classical conditioning using stimulation of the inferior olive as the unconditioned stimulus. Proc Natl Acad Sci USA. 1986;83:5349–5353. doi: 10.1073/pnas.83.14.5349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick and Thompson, 1984.McCormick DA, Thompson RF. Cerebellum: essential involvement in the classically conditioned eyelid response. Science. 1984;223:296–299. doi: 10.1126/science.6701513. [DOI] [PubMed] [Google Scholar]
- Medina and Mauk, 1999.Medina JF, Mauk MD. Simulations of cerebellar motor learning: computational analysis of plasticity at the mossy fiber to deep nucleus synapse. J Neurosci. 1999;19:7140–7151. doi: 10.1523/JNEUROSCI.19-16-07140.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina et al., 2000.Medina JF, Garcia KS, Nores WL, Taylor NM, Mauk MD. Timing mechanisms in the cerebellum: testing predictions of a large-scale computer simulation. J Neurosci. 2000;20:5516–5525. doi: 10.1523/JNEUROSCI.20-14-05516.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina et al., 2001.Medina JF, Garcia KS, Mauk MD. A mechanism for savings in the cerebellum. J Neurosci. 2001;21:4081–4089. doi: 10.1523/JNEUROSCI.21-11-04081.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles and Lisberger, 1981.Miles FA, Lisberger SG. Plasticity in the vestibulo-ocular reflex: a new hypothesis. Annu Rev Neurosci. 1981;4:273–299. doi: 10.1146/annurev.ne.04.030181.001421. [DOI] [PubMed] [Google Scholar]
- Nelson et al., 2003.Nelson AB, Krispel CM, Sekirnjak C, du Lac S. Long-lasting increases in intrinsic excitability triggered by inhibition. Neuron. 2003;40:609–620. doi: 10.1016/s0896-6273(03)00641-x. [DOI] [PubMed] [Google Scholar]
- Nelson et al., 2005.Nelson AB, Gittis AH, du Lac S. Decreases in CaMKII activity trigger persistent potentiation of intrinsic excitability in spontaneously firing vestibular nucleus neurons. Neuron. 2005;46:623–631. doi: 10.1016/j.neuron.2005.04.009. [DOI] [PubMed] [Google Scholar]
- Ohyama and Mauk, 2001.Ohyama T, Mauk M. Latent acquisition of timed responses in cerebellar cortex. J Neurosci. 2001;21:682–690. doi: 10.1523/JNEUROSCI.21-02-00682.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohyama et al., 2003.Ohyama T, Nores WL, Mauk MD. Stimulus generalization of conditioned eyelid responses produced without cerebellar cortex: implications for plasticity in the cerebellar nuclei. Learn Mem. 2003;10:346–354. doi: 10.1101/lm.67103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrett and Mauk, 1995.Perrett SP, Mauk MD. Extinction of conditioned eyelid responses requires the anterior lobe of cerebellar cortex. J Neurosci. 1995;15:2074–2080. doi: 10.1523/JNEUROSCI.15-03-02074.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrett et al., 1993.Perrett SP, Ruiz BP, Mauk MD. Cerebellar cortex lesions disrupt learning-dependent timing of conditioned eyelid responses. J Neurosci. 1993;13:1708–1718. doi: 10.1523/JNEUROSCI.13-04-01708.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugh and Raman, 2006.Pugh JR, Raman IM. Potentiation of mossy fiber EPSCs in the cerebellar nuclei by NMDA receptor activation followed by postinhibitory rebound current. Neuron. 2006;51:113–123. doi: 10.1016/j.neuron.2006.05.021. [DOI] [PubMed] [Google Scholar]
- Racine et al., 1986.Racine RJ, Wilson DA, Gingell R, Sunderland D. Long-term potentiation in the interpositus and vestibular nuclei in the rat. Exp Brain Res. 1986;63:158–162. doi: 10.1007/BF00235658. [DOI] [PubMed] [Google Scholar]
- Raymond et al., 1996.Raymond JL, Lisberger SG, Mauk MD. The cerebellum: a neuronal learning machine? Science. 1996;272:1126–1131. doi: 10.1126/science.272.5265.1126. [DOI] [PubMed] [Google Scholar]
- Shutoh et al., 2006.Shutoh F, Ohki M, Kitazawa H, Itohara S, Nagao S. Memory trace of motor learning shifts transsynaptically from cerebellar cortex to nuclei for consolidation. Neuroscience. 2006;139:767–777. doi: 10.1016/j.neuroscience.2005.12.035. [DOI] [PubMed] [Google Scholar]
- Steinmetz et al., 1989.Steinmetz JE, Lavond DG, Thompson RF. Classical conditioning in rabbits using pontine nucleus stimulation as a conditioned stimulus and inferior olive stimulation as an unconditioned stimulus. Synapse. 1989;3:225–233. doi: 10.1002/syn.890030308. [DOI] [PubMed] [Google Scholar]
- Tracy et al., 1998.Tracy JA, Thompson JK, Krupa DJ, Thompson RF. Evidence of plasticity in the pontocerebellar conditioned stimulus pathway during classical conditioning of the eyeblink response in the rabbit. Behav Neurosci. 1998;112:267–285. doi: 10.1037//0735-7044.112.2.267. [DOI] [PubMed] [Google Scholar]
- Yeo, 1991.Yeo CH. Cerebellum and classical conditioning of motor responses. Ann NY Acad Sci. 1991;627:292–304. doi: 10.1111/j.1749-6632.1991.tb25933.x. [DOI] [PubMed] [Google Scholar]
- Zhang and Linden, 2003.Zhang W, Linden DJ. The other side of the engram: experience-driven changes in neuronal intrinsic excitability. Nat Rev Neurosci. 2003;4:885–900. doi: 10.1038/nrn1248. [DOI] [PubMed] [Google Scholar]
- Zhang and Linden, 2006.Zhang W, Linden DJ. Long-term depression at the mossy fiber-deep cerebellar nucleus synapse. J Neurosci. 2006;26:6935–6944. doi: 10.1523/JNEUROSCI.0784-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang et al., 2004.Zhang W, Shin JH, Linden DJ. Persistent changes in the intrinsic excitability of rat deep cerebellar nuclear neurones induced by EPSP or IPSP bursts. J Physiol (Lond) 2004;561:703–719. doi: 10.1113/jphysiol.2004.071696. [DOI] [PMC free article] [PubMed] [Google Scholar]