

Abstract
Disease processes such as infection, leukemia, and autoimmune disorders are often associated with nausea, emesis, and anorexia. A common denominator of these rather disparate states is the production of the early, proinflammatory cytokine tumor necrosis factor-α (TNF) in significant quantities. Recent studies have shown that TNF may act as a neuromodulator in the hindbrain to produce malaise by potentiating visceral afferent signaling at the central processes of the vagus nerve. However, the mechanism by which TNF produces this signal amplification is not known. Our time-lapse calcium imaging studies of individual central vagal afferent varicosities in the caudal brainstem slice preparation show that, although TNF has minimal direct effects to elevate terminal intracellular calcium levels, TNF does potentiate the terminal afferent responses to other stimuli through a ryanodine-based, calcium-induced calcium release mechanism. Such a scheme may explain how TNF sensitizes visceral as well as somatosensory primary afferents.
Keywords: calcium imaging, cytokine, CD38, c-ADPR, brainstem, visceral afferents
Introduction
Activation of the immune system in response to infection, injury, or chronic disease is associated with high levels of cytokine release. The primary function of cytokine production is elimination of the source of the “challenge.” However, a constellation of debilitating physiological and behavioral events is often associated with immune activation. These include fever, fatigue, loss of appetite, gastric stasis, nausea, and hypersensitivity to touch or pain (Hermann et al., 2005a).
Tumor necrosis factor-α (TNF) is one of the early, proinflammatory cytokines. Cancer or exposure to radiation evokes production of TNF by non-immune cell types as well (Vassalli, 1992). Systemic injection of TNF can mimic many of the autonomic signs associated with illness, such as increased body temperature, gastric hypomotility, suppressed appetite, as well as nausea and vomiting (Hermann et al., 2005a).
The dorsal vagal complex (DVC) contains the basic neural circuitry for the vago-vagal reflex control of gastrointestinal function (e.g., gastric motility, tone, and acid secretion). This medullary complex is composed of the sensory nuclei of the solitary tract (NST) and the area postrema and their interconnections with the dorsal motor nucleus of the vagus (Rogers et al., 1995). Most vago-vagal control of gastric motility and tone is inhibitory, i.e., activation of vagal afferents by distension of the stomach, intestine, or esophagus results in a marked reduction in gastric motility and tone by initiating gastric vago-vagal reflexes (Rogers et al., 2005). Gastroinhibition may be perceived as nausea and serves as an essential prodrome to emesis (Syed et al., 2005). Indeed, this region of the caudal brainstem is also essential for the generation of the emetic motor program (Blessing, 1997).
The DVC possesses vascular characteristics of a circumventricular organ with fenestrated capillaries and enlarged perivascular spaces (Gross et al., 1990). Additionally, dendrites from neurons of the DVC penetrate the ependymal layer and enter the floor of the fourth ventricle (Rogers and McCann, 1993). Therefore, this complex is in position to monitor blood- and CSF-borne chemicals, hormones, peptides, and cytokines (Hermann et al., 2005a) and evoke appropriate physiological responses such as gastric stasis (Rogers et al., 1995).
Peripherally generated TNF causes gastric stasis by acting at the DVC (Hermann et al., 2002; Hermann et al., 2005a). TNF injected directly into the NST also causes a substantial reduction in gastric motility. This effect may be attributable to the TNF-mediated activation of vagal afferent fibers synapsing on NST neurons that form part of the vagal gastroinhibitory reflex (Rogers et al., 2005). This hypothesis is supported by immunohistochemical studies showing that the TNF receptor type 1 is highly concentrated on vagal afferent fibers and terminals in the NST (Hermann et al., 2004). Indeed, our neurophysiological studies suggested that TNF amplifies the responsiveness of vagal afferent pathways to otherwise innocuous sensory signals (Emch et al., 2000). To test this hypothesis, we developed a live-cell calcium imaging method designed to address the mechanism by which TNF activates or amplifies vagal afferent signaling at the level of the terminal varicosity.
Materials and Methods
Long–Evans rats of either gender (100–200 g body weight; Simonsen Laboratories, Gilroy, CA) were maintained on ad libitum food and water in a 12 h light/dark cycled room with constant temperature and humidity. All experimental protocols were performed according to the guidelines set forth by the National Institutes of Health and were approved by the Institutional Animal Care and Use Committees at the Pennington Biomedical Research Center.
Vagal afferent labeling for in vitro calcium imaging.
A complete description of the in vitro vagal afferent labeling procedure was published previously (Rogers et al., 2006). Briefly, a glass microinjection pipette was filled with a mixture of 20% CalciumGreen 1-dextran 3000 molecular weight conjugate (CG) (Invitrogen, Carlsbad, CA), 1% Triton X-100, and distilled water for calcium imaging experiments (n = 30 rats). A similar mixture with 5% rhodamine dextran 3000 molecular weight [FluoroRuby (FR); Invitrogen] was used to identify vagal afferent fibers and terminals in the NST (n = 5); this prelabeled tissue was exposed to immunohistochemical demonstrations of the distribution of ryanodine receptors (RyRs) and IP3 receptors (IP3Rs) in these vagal processes.
Rats were anesthetized with pentobarbital (30 mg/kg, i.p.; Nembutal; Abbott Labs, Chicago, IL). Using aseptic technique, the nodose ganglion was exposed at the jugular foramen. The micropipette containing either CG or FR was connected to a source of pulsed air pressure. The tip of the pipette was guided by hand through the sheath of the nodose ganglion. Pressure pulses (2–10 psi) were applied to the pipette and dye was injected; total injected volume was ∼100 nl. The cervical wound was closed with 4-0 nylon suture, and animals were returned to their home cage for 2–5 d to allow anterograde transport of dextran-conjugated constructs.
Brainstem slice preparation.
Animals were anesthetized with ethyl carbamate (urethane, 1.5 g/kg body weight, i.p.; Sigma, St. Louis, MO); this anesthesia readily washes out of tissue and does not have long-term effects on the activity of neurons in these slices (Hara and Harris, 2002). After decapitation and swift removal of the brainstem to cold (∼4°C), carbogenated (95% O2/5% CO2) cutting solution, 300-μm-thick slices were cut coronally with a sapphire knife on a vibratome (model 1500; Vibratome, St. Louis, MO). Slices were placed in a scintillation vial containing carbogenated normal Krebs' solution (29°C) for ∼1 h before conducting experiments.
Slices are transferred to a temperature-regulated recording chamber maintained at 33°C with a solution flow rate of 3 ml/min. Stability of the slice preparation is of paramount importance to the imaging of small fibers and terminals. The confocal depth of field of 1–2 μm is just sufficient to image terminal varicosities and fibers that are, themselves, no more than 3–4 μm in diameter (Rogers et al., 2006). No drift can be tolerated. To solve this problem, we opted to support the recording chamber on a large fixed stage (Gibralter platform; Burley, Victor, NY); this fixed stage supports and moves the microscope in the x–y plane.
In vitro drugs and solutions.
The cutting solution contained the following (in mm): 110 choline chloride, 25 NaHCO3, 2.5 KCl, 7 MgSO4-7H2O, 1.25 NaH2PO4, 10 glucose, and 0.5 CaCl2-2H2O (bubbled with 95% O2/5% CO2 during the entire cutting process). Normal Krebs' solution contained the following (in mm): 124 NaCl, 25 NaHCO3, 3.0 KCl, 1 MgSO4-7H2O, 1.5 NaH2PO4, 10 glucose, and 1.5 CaCl2-2H2O, pH 7.3 (bubbled with 95% O2/5% CO2, continuously; osmolarity was 300 ± 10 mOsm).
When specified below (see Experimental design), one of the following drugs was added to the normal Krebs' solution (note that the specified concentrations of reagents are based on references cited for similar in vitro studies): 100 μm ATP was used to activate P2X3 ligand-gated cation channels on vagal afferent terminals (Jin et al., 2004; Shigetomi and Kato, 2004); 1 nm TNF [comparable with the circulating concentration after a significant immune challenge (Hermann et al., 2002)]; 10 μm ryanodine [ryanodine endoplasmic reticular calcium channel antagonist at this concentration (Lancaster et al., 2002)]; 10 μm U73122 (1-[6[[(17β)-3-methoxyestra-1,3,5(10)-trien-17-yl]amino]hexyl]-1H-pyrrole-2,5-dione) [selective inositol 1,4,5 tris-phosphate synthesis inhibitor (Hermann et al., 2005b)]; and 100 μm 8-bromo-cADP ribose (8-BR-cADPR) [an antagonist of intracellular cADPR action (Deshpande et al., 2003)].
Imaging instrumentation.
Time-lapse laser confocal calcium imaging was performed with an Ultraview Nipkow disk imager (model CSU21; PerkinElmer, Boston, MA) combined with a digital camera (Orca-ER; Hamamatsu, Shizouka, Japan) on a Nikon (Tokyo, Japan) E600FN fixed-stage upright microscope equipped with 10× and 40× water immersion objectives. CalciumGreen preloaded fibers were visualized using the 488 nm excitation/509 nm emission filter arrangement. Data were collected at a rate of three frames per second. At 400× total magnification, full frames were ∼75 × 100 μm of brainstem slice area.
In vitro calcium signal analysis.
Relative changes in cytoplasmic calcium were expressed as changes in fluorescence: (ΔF/F)%, where F is the fluorescence intensity within an area of interest (e.g., the outline of a vagal afferent fibers or varicosities) before stimulation, and ΔF is the change from this value during neuronal activity (Helmchen, 2000). Background fluorescence (i.e., non-involved areas adjacent to the area of interest) was subtracted from both ΔF and F. A responsive component was classified as one in which an agonist substance (e.g., TNF or ATP) produced a calcium signal elevation (ΔF/F) of 5% or greater.
Drug challenges and statistical analysis.
Initial studies focused on the direct actions of TNF to generate calcium signals within vagal afferent varicosities in the NST. Here, TNF was applied to the perfusion chamber for 1–10 min while fluorescence intensity of CG-labeled varicosities was monitored.
In subsequent experiments, ATP was applied to the bath for 30 s to activate a ligand-gated calcium release mechanism known to operate on vagal afferents (Jin et al., 2004; Shigetomi and Kato, 2004). The exposure to ATP was repeated 10 min later to provide a “time control.” Thus, the experimental design of these studies was such that each varicosity served as its own control. Next, we determined whether TNF could modulate the primary ATP effect. In this case, 30 s exposure to ATP occurred first, followed by 10 min perfusion with TNF, followed by a second 30 s exposure to ATP.
Last, to evaluate the likely basis for any TNF modulatory effects on ATP-evoked activity, other slices were continuously perfused with normal Krebs' solution that also contained one of the antagonist drugs ryanodine or U73122 or 8-Br-cADPR, reagents known to influence the control over intracellularly stored calcium. The ATP–TNF–ATP challenge was repeated against a background of one of these antagonist drugs. Thus, these studies were performed to account for any direct effects of the antagonists on the direct ability of ATP to alter relative calcium levels in afferent terminals.
Given that each varicosity was exposed to ATP twice (e.g., before and after exposure to TNF), each varicosity acted as its own control. This pattern was followed even when we were evaluating the actions of specific antagonists (see above). As such, paired Student's t tests were used for comparisons; values of p < 0.05 were considered statistically significant.
RyR and IP3R immunohistochemistry on identified vagal afferent fibers.
Rats that had received FR injections into the nodose ganglion were anesthetized with urethane and transcardially perfused with PBS and then PBS containing 4% paraformaldehyde. Brainstems were removed and postfixed overnight in a solution of 4% paraformaldehyde in 20% sucrose and PBS. The caudal brainstem containing the NST region was sectioned (40 μm) on a freezing microtome. Tissue sections were reacted with a primary antibody directed against either RyR [ascites; mouse monoclonal; MA3-925 (Affinity BioReagents, Golden, CO); 1:500, which reacts specifically with the 565 kDa protein identified as the ryanodine receptor in rat as determined by Western blot] or IP3R [1 mg/ml rabbit polyclonal; AB1622 (Chemicon, Temecula, CA); 1:50 raised against the C-terminal epitope (KDSTEYTGPESYV) of the human IP3 type 1 receptor as determined by ELISA]. Both antibodies have been used together to examine the distribution of these two intracellular calcium regulatory channels in previous studies (Clark and Petty, 2005). After rinsing, sections reacted with the primary antibodies (as well as controls with no exposure to primary antibody) were treated with donkey anti-mouse or rabbit secondary antibody (1:500) conjugated with an Alexa 488 fluorophore (Invitrogen). Sections were air dried and coverslipped with Vectashield Hard Set (Vector Laboratories, Burlingame, CA). The medial NST at the level of the area postrema was then examined with the Ultraview confocal microscope.
Results
Forty brainstem slices from 10 rats were exposed to TNF alone. Only five terminal varicosities were observed to be responsive in these preparations (Fig. 1). We next examined the effects of TNF to modulate cytoplasmic calcium signals generated by ATP. For time control purposes, slices were exposed to two applications of ATP with an intervening 10 min interval of normal Krebs' solution alone. Of the 80 responsive varicosities recorded, the first ATP application caused a relative increase in CG fluorescence (ΔF/F)% of 36 ± 2.1%. The second application of ATP 10 min later (i.e., time control) produced a diminished change in CG fluorescence (ΔF/F)% of 27 ± 2.7% (Figs. 2, 3). This decline in calcium response to the second application of ATP, relative to the first, was statistically significant (paired t test; t = 4.044; df = 79; p < 0.0001). In contrast, perfusion of slices for 10 min with TNF between the first and second application of ATP (n = 128 varicosities) resulted in an enhanced ATP-generated calcium signal from (ΔF/F)% of 34 ± 2.2% (first ATP) to 48 ± 3.2% (second ATP). This increase in calcium signal is significant (paired t test; t = 4.569 df = 127; p < 0.0001), particularly in light of the fact that, normally, the magnitude of the second response is smaller than the first (Figs. 2, 3).
Figure 1.
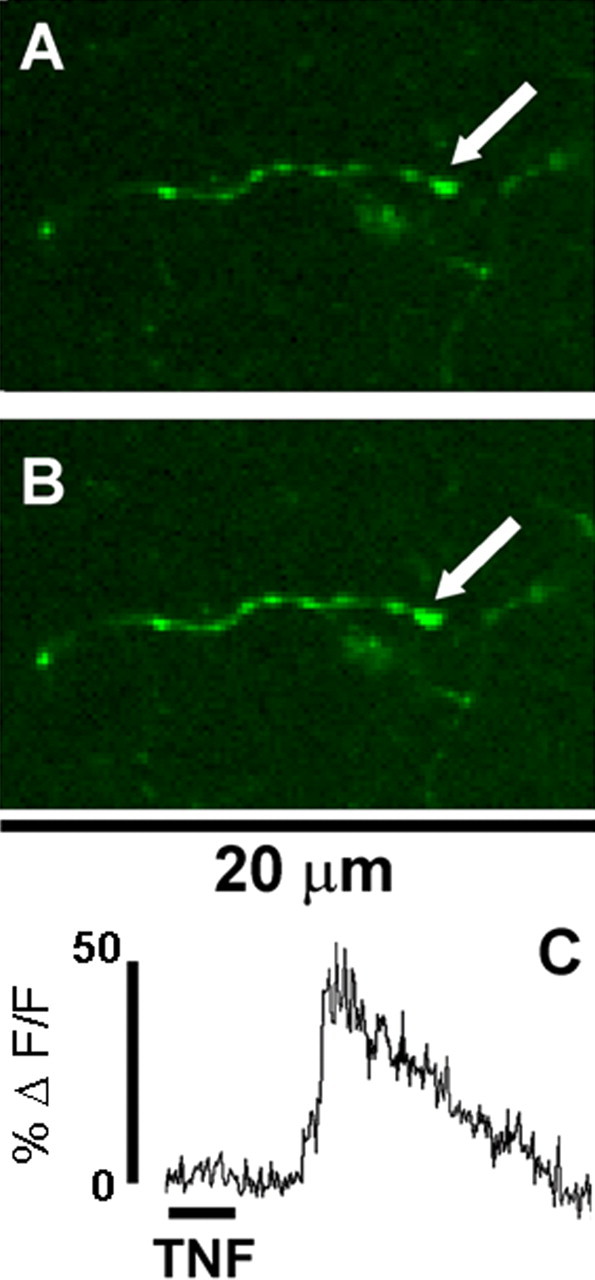
Direct effect of TNF on vagal varicosities. One of the few central vagal afferent processes observed that responded directly to TNF (1 nm for 1 min) alone: A, before TNF stimulation; B, during TNF stimulation; C, relative fluorescence plot generated by area of interest (i.e., area encompassed by the varicosity indicated by the arrow).
Figure 2.

Modulation of vagal afferent terminals responses to ATP by exposure to TNF. A low-power field containing numerous calcium green-labeled central vagal afferent terminals in the NST is shown. A and B were taken before exposure of the slice to 1 nm TNF. A, Varicosities at rest just before perfusion of the slice with ATP (100 μm). B, Varicosities in the same field as A showing the peak effect of ATP. C and D were taken just after 10 min exposure to TNF. C, Varicosities at rest. D, Varicosities showing peak effect of ATP. E, Relative fluorescence plot of the initial response of ATP to elevate calcium signal in the varicosity indicated by the arrow (refer to B) before TNF exposure. F, Relative fluorescence plot of the response of ATP to elevate calcium signal in the same varicosity indicated by the arrow (refer to D) after TNF exposure. This TNF amplification of the calcium signal is sensitive to ryanodine (see Fig. 3).
Figure 3.

TNF-mediated amplification of presynaptic calcium signal. A, Time control, effects of two separate exposures of the brainstem slice to 100 μm ATP (i.e., no exposure to TNF). Data are presented as mean ± SE. *p < 0.0001; n = 80 varicosities. B, Exposing slices to 1 nm TNF amplified the second response to ATP. *p < 0.0001; n = 128 varicosities. C, Exposing the slice to 10 μm ryanodine plus 100 μm TNF effectively blocked the effect of TNF to amplify terminal calcium signals produced by ATP (n = 63 varicosities). D, Similarly, blockade of receptors for cADP ribose by 8-Br-cADPR suppressed TNF effect to amplify the ATP response (n = 29 varicosities). E, Blockade of IP3 production by U73122 had no effect on the ability of TNF to amplify the ATP response. *p < 0.0001; n = 82 varicosities. F, TNF can amplify terminal calcium signaling by activating ryanodine channels. ATP acts at terminal ligand-gated cation channels to produce an increase in intra-terminal calcium that activates CICR through ryanodine channels. TNF apparently amplifies this effect based on the generation of cADP ribose, a known ryanodine agonist molecule. CD38 generates cADP ribose; TNF drives CD38 transcription (Iqbal et al., 2006) and may enhance CD38 trafficking.
When the intracellular calcium release antagonist ryanodine was applied, the TNF-induced augmentation of the ATP effect was blocked. Under these conditions, the first ATP application produced (ΔF/F)% of 23.3 ± 1.9%, and the post-TNF ATP stimulation produced the same result, i.e., (ΔF/F)% of 22.8 ± 1.9% (n = 63 varicosities). A similar result was obtained with a cADP ribose antagonist (8-Br-cADPR); the pre-TNF ATP application yielded (ΔF/F)% of 15.5 ± 1.4%, whereas the post-TNF ATP stimulus produced (ΔF/F)% of 12.3 ± 1.3% (n = 29 varicosities). However, U73122 was not able to block the TNF augmentation of the ATP-induced increase in vagal afferent terminal calcium; TNF produced a significant elevation in vagal afferent calcium signals (ΔF/F)% of 32 ± 2.2% (first ATP) versus 50 ± 4.1% (second ATP) (t = 5.8; df = 81; p < 0.0001).
Immunohistochemical studies revealed a substantial difference in the presence of RyRs versus IP3Rs in vagal afferent varicosities (Fig. 4). A total of 23 histological sections taken from five animals were examined: 12 were exposed to primary antibodies for RyR, 11 to antibodies for IP3R. Sixty-three percent of the 6090 FR-labeled vagal varicosities (i.e., 3893 of 6090, 63%) were immunoreactive positive to RyR staining, whereas only 20% of the 6316 FR-labeled vagal varicosities (i.e., 1469 of 6316) were IP3R immunoreactive positive.
Figure 4.
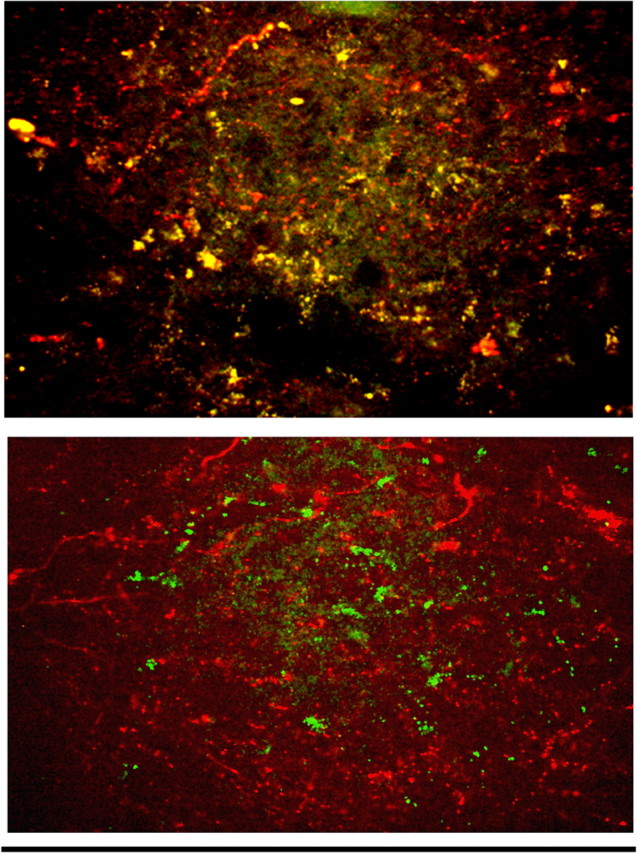
Immunohistochemical demonstration of receptors in vagal afferents. Top, Ryanodine receptors (green dots) are highly expressed in the central processes of vagal afferents prelabeled with rhodamine dextran (red fibers). Colocalization of these components is evident by the yellow areas. Bottom, IP3 receptor (green dots) immunoreactivity, although present, is significantly less common in identified vagal afferent terminal processes (red fibers). Scale bar, 100 μm.
Discussion
TNF has been implicated as an activator or amplifier of glutamatergic neurotransmission in somatosensory and visceral afferents as well as in the intrinsic innervation of the hippocampus and spinal cord (Pickering et al., 2005). This feature of TNF action may explain the multifunctional aspects of the effects of cytokine on the CNS. Disease processes provoking elevated TNF release cause allodynia and nausea as a consequence of effects on spinal and vagal afferents, respectively (Hermann et al., 2005a; Pickering et al., 2005). Furthermore, TNF release secondary to neurotrauma or neuroinfection may accelerate neurodegeneration and contribute to dementia by augmenting glutamate neurotransmission (Pickering et al., 2005). Previous studies have suggested that TNF may cause some of the augmentation of glutamate transmission by increasing the membrane trafficking of the AMPA receptor postsynaptic to glutamatergic inputs (Stellwagen et al., 2005). Other work implicated a TNF amplification effect at presynaptic sites (Emch et al., 2000; Beattie et al., 2002; Hermann et al., 2005a; Rogers et al., 2005; Stellwagen et al., 2005), but a physiological mechanism explaining this action had not been advanced.
By applying calcium imaging methods to vagal afferent terminal varicosities in vitro, we find that TNF has a weak effect, at best, to directly increase the presynaptic calcium signal, hence, transmitter release. However, TNF strongly amplifies the terminal calcium signal produced by ATP. ATP activates a transmembrane calcium flux by causing P2X3 ligand-gated cation channels to open. As a consequence, ATP causes glutamate release from vagal afferents onto second-order NST neurons (Jin et al., 2004). Although the direct entry of calcium into the terminal region certainly activates the transmitter release mechanism, entering calcium can also serve as a trigger for the release of endoplasmic reticulum (ER)-stored calcium through calcium-induced calcium release (CICR) mechanism (Bardo et al., 2006). Two parallel processes regulate this release of stored calcium: ryanodine channels and the IP3–calmodulin release mechanism (Hoesch et al., 2002). Calcium entering the terminal interacts with binding sites on the ryanodine channel to provoke release of ER calcium stores. Our results suggest that TNF operates predominantly on the ryanodine stores release mechanism as ryanodine blocks the TNF effect on ATP-evoked calcium release, whereas U73122, an IP3 synthesis inhibitor, has no effect. Although our immunohistochemistry studies demonstrated that both receptor types are expressed on central vagal afferent fibers, our pharmacological studies indicated that the IP3 receptor did not play a role in TNF modulation of the ATP-evoked calcium mobilization. However, there is no a priori reason to believe that TNF might not modulate other afferent responses through the IP3 (or some other) transduction mechanisms that impact stored calcium release.
The mechanisms by which agonist molecules affect the function of ryanodine receptors, hence, presynaptic potentiation and depression, are incompletely understood. However, studies of the relationship between TNF and ryanodine receptor function in non-neural tissues strongly suggest that TNF can increase the expression of CD38 (Yusufi et al., 2001; Deshpande et al., 2003). CD38 (the transmembrane ectoenzyme ADP-ribosyl-cyclase) generates cADP ribose. This product is a potent activator of the ryanodine channel. cADPR, acting on presynaptic ryanodine channels, may serve as a potent positive modulator of CNS neurotransmission (Barata et al., 2004; Collin et al., 2005). Our studies show that 8-Br-cADPR, a selective antagonist of cADPR, completely blocks the action of TNF to augment calcium signaling in vagal afferents. Although this result suggests a connection between TNF receptor activation and upregulation of CD38-mediated cADPR production, the time course of the effect is problematic. Although CD38 transcription can be upregulated by exposure to TNF in as little as 3 h (Iqbal et al., 2006), the effects we observe here develop much more rapidly.
It has been suggested that the effectiveness of CD38 in regulating cytoplasmic calcium is a function of the delivery of substrate (oxidized nicotinamide-adenine dinucleotide) and the destination of the cADPR product. Connexin hemichannels or nucleoside transporters (Zocchi et al., 1999; De Flora et al., 2004) may perform these functions. Trafficking of the CD38 molecule itself has also been suggested as a mechanism for controlling the production of cADPR, hence, the activity of the ryanodine channel (De Flora et al., 2004). Although the connection between TNF receptor activation and the rapid regulation of CD38 is not presently clear, convincing evidence suggests that TNF can potently affect neurotransmission through rapid control of receptor trafficking (Stellwagen et al., 2005).
The present results provide a physiological explanation for the mechanism by which TNF sensitizes afferent nerve pathways to the CNS. TNF induces the production of cADPR, which in turn, sensitizes the ryanodine ER calcium release mechanism to augment the calcium-induced calcium release. This, in turn, sensitizes presynaptic neurotransmission mechanisms. Because TNF also induces the trafficking of postsynaptic AMPA glutamate receptors (Stellwagen et al., 2005), TNF emerges as a powerful positive, pleiotropic modulator of glutamate neurotransmission. This effect may be the basis for many of the neuropathological events associated with the release of this cytokine.
Footnotes
This work was supported by National Institutes of Health Grants DK52142, DK56373, and HD47643.
References
- Barata et al., 2004.Barata H, Thompson M, Zielinska W, Han YS, Mantilla CB, Prakash YS, Feitoza S, Sieck G, Chini EN. The role of cyclic-ADP-ribose-signaling pathway in oxytocin-induced Ca2+ transients in human myometrium cells. Endocrinology. 2004;145:881–889. doi: 10.1210/en.2003-0774. [DOI] [PubMed] [Google Scholar]
- Bardo et al., 2006.Bardo S, Cavazzini MG, Emptage N. The role of the endoplasmic reticulum Ca2+ store in the plasticity of central neurons. Trends Pharmacol Sci. 2006;27:78–84. doi: 10.1016/j.tips.2005.12.008. [DOI] [PubMed] [Google Scholar]
- Beattie et al., 2002.Beattie EC, Stellwagen D, Morishita W, Bresnahan JC, Ha BK, Von Zastrow M, Beattie MS, Malenka RC. Control of synaptic strength by glial TNFalpha. Science. 2002;295:2282–2285. doi: 10.1126/science.1067859. [DOI] [PubMed] [Google Scholar]
- Blessing, 1997.Blessing WW. The lower brainstem and bodily homeostasis. Oxford: Oxford UP; 1997. Eating and metabolism; pp. 323–372. [Google Scholar]
- Clark and Petty, 2005.Clark AJ, Petty HR. Differential intracellular distributions of inositol trisphosphate and ryanodine receptors within and among hematopoietic cells. J Histochem Cytochem. 2005;53:913–916. doi: 10.1369/jhc.4B6610.2005. [DOI] [PubMed] [Google Scholar]
- Collin et al., 2005.Collin T, Marty A, Llano I. Presynaptic calcium stores and synaptic transmission. Curr Opin Neurobiol. 2005;15:275–281. doi: 10.1016/j.conb.2005.05.003. [DOI] [PubMed] [Google Scholar]
- De Flora et al., 2004.De Flora A, Zocchi E, Guida L, Franco L, Bruzzone S. Autocrine and paracrine calcium signaling by the CD38/NAD+/cyclic ADP-ribose system. Ann NY Acad Sci. 2004;1028:176–191. doi: 10.1196/annals.1322.021. [DOI] [PubMed] [Google Scholar]
- Deshpande et al., 2003.Deshpande DA, Walseth TF, Panettieri RA, Kannan MS. CD38/cyclic ADP-ribose-mediated Ca2+ signaling contributes to airway smooth muscle hyper-responsiveness. FASEB J. 2003;17:452–454. doi: 10.1096/fj.02-0450fje. [DOI] [PubMed] [Google Scholar]
- Emch et al., 2000.Emch GS, Hermann GE, Rogers RC. TNF-alpha activates solitary nucleus neurons responsive to gastric distension. Am J Physiol Gastrointest Liver Physiol. 2000;279:G582–G586. doi: 10.1152/ajpgi.2000.279.3.G582. [DOI] [PubMed] [Google Scholar]
- Gross et al., 1990.Gross PM, Wall KM, Pang JJ, Shaver SW, Wainman DS. Microvascular specializations promoting rapid interstitial solute dispersion in nucleus tractus solitarius. Am J Physiol Regul Integr Comp Physiol. 1990;259:R1131–R1138. doi: 10.1152/ajpregu.1990.259.6.R1131. [DOI] [PubMed] [Google Scholar]
- Hara and Harris, 2002.Hara K, Harris RA. The anesthetic mechanism of urethane: the effects on neurotransmitter-gated ion channels. Anesth Analg. 2002;94:313–318. doi: 10.1097/00000539-200202000-00015. table of contents. [DOI] [PubMed] [Google Scholar]
- Helmchen, 2000.Helmchen F. Calibration of fluorescent calcium indicators. In: Yuste R, Lanni F, Konnerth A, editors. Imaging neurons: a laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 2000. pp. 31–39. [Google Scholar]
- Hermann et al., 2002.Hermann GE, Tovar CA, Rogers RC. LPS-induced suppression of gastric motility relieved by TNFR:Fc construct in dorsal vagal complex. Am J Physiol Gastrointest Liver Physiol. 2002;283:G634–G639. doi: 10.1152/ajpgi.00412.2001. [DOI] [PubMed] [Google Scholar]
- Hermann et al., 2004.Hermann GE, Hebert SL, Van Meter MJ, Holmes GM, Rogers RC. TNF(alpha)-p55 receptors: medullary brainstem immunocytochemical localization in normal and vagus nerve-transected rats. Brain Res. 2004;1004:156–166. doi: 10.1016/j.brainres.2003.11.078. [DOI] [PubMed] [Google Scholar]
- Hermann et al., 2005a.Hermann GE, Holmes GM, Rogers RC. TNF(alpha) modulation of visceral and spinal sensory processing. Curr Pharm Des. 2005a;11:1391–1409. doi: 10.2174/1381612053507828. [DOI] [PubMed] [Google Scholar]
- Hermann et al., 2005b.Hermann GE, Nasse JS, Rogers RC. Alpha-1 adrenergic input to solitary nucleus neurones: calcium oscillations, excitation and gastric reflex control. J Physiol (Lond) 2005b;562:553–568. doi: 10.1113/jphysiol.2004.076919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoesch et al., 2002.Hoesch RE, Yienger K, Weinreich D, Kao JP. Coexistence of functional IP(3) and ryanodine receptors in vagal sensory neurons and their activation by ATP. J Neurophysiol. 2002;88:1212–1219. doi: 10.1152/jn.2002.88.3.1212. [DOI] [PubMed] [Google Scholar]
- Iqbal et al., 2006.Iqbal J, Kumar K, Sun L, Zaidi M. Selective upregulation of the ADPribosyl-cyclases CD38 and CD157 by TNF but not by RANK-L reveals differences in downstream signaling. Am J Physiol Renal Physiol. 2006;291:F557–F566. doi: 10.1152/ajprenal.00066.2006. [DOI] [PubMed] [Google Scholar]
- Jin et al., 2004.Jin YH, Bailey TW, Li BY, Schild JH, Andresen MC. Purinergic and vanilloid receptor activation releases glutamate from separate cranial afferent terminals in nucleus tractus solitarius. J Neurosci. 2004;24:4709–4717. doi: 10.1523/JNEUROSCI.0753-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster et al., 2002.Lancaster E, Oh EJ, Gover T, Weinreich D. Calcium and calcium-activated currents in vagotomized rat primary vagal afferent neurons. J Physiol (Lond) 2002;540:543–556. doi: 10.1113/jphysiol.2001.013121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickering et al., 2005.Pickering M, Cumiskey D, O'Connor JJ. Actions of TNF-alpha on glutamatergic synaptic transmission in the central nervous system. Exp Physiol. 2005;90:663–670. doi: 10.1113/expphysiol.2005.030734. [DOI] [PubMed] [Google Scholar]
- Rogers and McCann, 1993.Rogers RC, McCann MJ. Intramedullary connections of the gastric region in the solitary nucleus: a biocytin histochemical tracing study in the rat. J Auton Nerv Syst. 1993;42:119–130. doi: 10.1016/0165-1838(93)90043-t. [DOI] [PubMed] [Google Scholar]
- Rogers et al., 1995.Rogers RC, McTigue DM, Hermann GE. Vagovagal reflex control of digestion: afferent modulation by neural and “endoneurocrine” factors. Am J Physiol Gastrointest Liver Physiol. 1995;268:G1–G10. doi: 10.1152/ajpgi.1995.268.1.G1. [DOI] [PubMed] [Google Scholar]
- Rogers et al., 2005.Rogers RC, Hermann GE, Travagli RA. Brainstem control of gastric function. In: Johnson LR, editor. Physiology of the gastrointestinal tract. Ed 4. New York: Elsevier; 2005. pp. 851–875. [Google Scholar]
- Rogers et al., 2006.Rogers RC, Nasse JS, Hermann GE. Live-cell imaging methods for the study of vagal afferents within the nucleus of the solitary tract. J Neurosci Methods. 2006;150:47–58. doi: 10.1016/j.jneumeth.2005.05.020. [DOI] [PubMed] [Google Scholar]
- Shigetomi and Kato, 2004.Shigetomi E, Kato F. Action potential-independent release of glutamate by Ca2+ entry through presynaptic P2X receptors elicits postsynaptic firing in the brainstem autonomic network. J Neurosci. 2004;24:3125–3135. doi: 10.1523/JNEUROSCI.0090-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stellwagen et al., 2005.Stellwagen D, Beattie EC, Seo JY, Malenka RC. Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-alpha. J Neurosci. 2005;25:3219–3228. doi: 10.1523/JNEUROSCI.4486-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syed et al., 2005.Syed AA, Rattansingh A, Furtado SD. Current perspectives on the management of gastroparesis. J Postgrad Med. 2005;51:54–60. [PubMed] [Google Scholar]
- Vassalli, 1992.Vassalli P. The pathophysiology of tumor necrosis factors. Annu Rev Immunol. 1992;10:411–452. doi: 10.1146/annurev.iy.10.040192.002211. [DOI] [PubMed] [Google Scholar]
- Yusufi et al., 2001.Yusufi AN, Cheng J, Thompson MA, Dousa TP, Warner GM, Walker HJ, Grande JP. cADP-ribose/ryanodine channel/Ca2+-release signal transduction pathway in mesangial cells. Am J Physiol Renal Physiol. 2001;281:F91–F102. doi: 10.1152/ajprenal.2001.281.1.F91. [DOI] [PubMed] [Google Scholar]
- Zocchi et al., 1999.Zocchi E, Usai C, Guida L, Franco L, Bruzzone S, Passalacqua M, De Flora A. Ligand-induced internalization of CD38 results in intracellular Ca2+ mobilization: role of NAD+ transport across cell membranes. FASEB J. 1999;13:273–283. doi: 10.1096/fasebj.13.2.273. [DOI] [PubMed] [Google Scholar]