

Abstract
Brain-derived neurotrophic factor (BDNF) polymorphism is associated with the pathophysiology of several neurodegenerative disorders, including Huntington's disease. In view of these data and the involvement of huntingtin in intracellular trafficking, we examined the intracellular transport and release of Val66Val BDNF (Val-BDNF) and Val66Met BDNF (Met-BDNF) in transfected striatal knock-in cells expressing wild-type or mutant full-length huntingtin. Colocalization studies with specific markers for endoplasmic reticulum showed no differences between the Val-BDNF and Met-BDNF and were not modified by mutant huntingtin. However, post-Golgi trafficking was altered by mutant huntingtin dependent on the BDNF form. Thus, fluorescence recovery after photobleaching (FRAP) and inverse FRAP analysis showed retention of Met-BDNF in the Golgi apparatus with respect to Val-BDNF in wild-type cells. Strikingly, mutant huntingtin diminished post-Golgi trafficking of Val-BDNF, whereas Met-BDNF was not modified. Accordingly, a reduction in the number of transport vesicles was only observed in mutant huntingtin cells transfected with Val-BDNF but not Met-BDNF. Moreover, mutant huntingtin severely affected the KCl-evoked release of Val-BDNF, although it had little effect on Met-BDNF regulated release. The constitutive release of Val-BDNF or Met-BDNF in mutant cells was only slightly reduced. Interestingly, mutant huntingtin only perturbed post-Golgi trafficking of proteins that follow the regulated secretory pathway (epidermal growth factor receptor or atrial natriuretic factor), whereas it did not change those that follow the constitutive pathway (p75NTR).
We conclude that mutant huntingtin differently affects intracellular transport and release of Val-BDNF and Met-BDNF. In addition, our findings reveal a new role for huntingtin in the regulation of the post-Golgi trafficking of the regulated secretory pathway.
Keywords: Huntington's disease, neurotrophin, vesicular transport, trans-Golgi network, p75, EGFR
Introduction
Brain-derived neurotrophic factor (BDNF) plays a critical role in the pathophysiology of different neurodegenerative diseases, such as Huntington's disease (HD) (Zuccato et al., 2001; Canals et al., 2004). HD is a neurodegenerative disorder caused by a multiple CAG expansion in exon1 of huntingtin (htt) gene (Kremer et al., 1994), producing a significant dysfunction and neural death, especially in the medium spiny neurons of the striatum. Htt is a ubiquitously expressed large protein that participates in a plethora of functions, including clathrin-mediated endocytosis, vesicle transport, transcriptional regulation, and cell survival (for review, see Harjes and Wanker, 2003; Cattaneo et al., 2005). In relation to BDNF, htt enhances the transcription of BDNF by the inhibition of the neuron restrictive silencer element (Zuccato et al., 2003) and promotes the transport of BDNF-containing vesicles along microtubules (Gauthier et al., 2004). Thus, the mutation of htt produces a reduction in BDNF expression (Ferrer et al., 2000; Zuccato et al., 2001) and release (Gauthier et al., 2004), which in turn affect the survival action of BDNF on striatal neurons (Canals et al., 2004).
As in all neurotrophins, BDNF is synthesized as a protein precursor composed by two domains, the prodomain and the BDNF domain (Seidah et al., 1996). The BDNF prodomain has an important role in the intracellular traffic of BDNF, promoting the sorting of proBDNF to the regulated secretory pathway because of its interaction with sortilin at the trans-Golgi network (TGN) (Chen et al., 2005). The sorting of BDNF to the regulated secretory pathway is impaired by a BDNF polymorphism consisting in a valine to methionine substitution at codon 66 in the prodomain (Val66Met BDNF) (Chen et al., 2004), reducing the amount of secreted BDNF after neural cell depolarization (Egan et al., 2003; Chen et al., 2004).
The characterization of this BDNF polymorphism has also highlighted the importance of this neurotrophin in different neuropsychiatric disorders. In fact, several studies have associated this polymorphism with increased susceptibility to anorexia nervosa (Ribases et al., 2003), obsessive-compulsive disorders (Hall et al., 2003), eating disorders (Friedel et al., 2005), depression (Sen et al., 2003), schizophrenia (Skibinska et al., 2004), Alzheimer's disease (Ventriglia et al., 2002), and Parkinson's disease (Momose et al., 2002).
We reported recently that heterozygous patients with HD carrying Val66Met BDNF (Met-BDNF) allele have a later age of onset compared with homozygous Val66Val BDNF (Val-BDNF) patients (Alberch et al., 2005). These data and the emerging properties of htt in intracellular trafficking prompted us to study the role of mutant htt (mhtt) in the intracellular transport of Val-BDNF and Met-BDNF in striatal knock-in cells that express wild-type (wt) full-length htt (with 7Q, wt cells) or full-length mhtt (with 111Q, mhtt cells). Here, we also report that mhtt impairs post-Golgi trafficking of Val-BDNF but not of Met-BDNF, which in turn produces an impairment of Val-BDNF-containing vesicles and a reduction of the KCl-evoked release of Val-BDNF. In addition, the results show that htt participates in the post-Golgi transport of proteins that follow the regulated secretory pathway.
Materials and Methods
Reagents and antibodies.
Anti-giantin antibody was kindly supplied by H. P. Hauri (Basel, Switzerland), anti-calnexin peptide 4 (residues 555–573) was from the Bergeron Laboratory (Montreal, Quebec, Canada) (Wada et al., 1991), and anti-secretogranin II was from Biodesign (Saco, ME). Anti-BDNF antibody was from Alomone Labs (Jerusalem, Israel), anti-epidermal growth factor receptor (EGFR) was from Santa Cruz Biotechnology (Santa Cruz, CA), and anti-htt MAB 2166 was from Chemicon (Temecula, CA). Fluorescent cyanine 3 (Cy3) (anti-rabbit) and Alexa 647 (anti-mouse and anti-rabbit) secondary antibodies were from Invitrogen (Carlsbad, CA) and rhodamine-conjugated anti-mouse antibody was from Jackson ImmunoResearch (West Grove, PA). Lipofectamine 2000 was from Invitrogen.
DNA constructs.
The human BDNF and its Val66Met polymorphism subcloned into pCDNA3.1hygro expression vector (Invitrogen) with hemagglutinin epitope tag at 3′ and FLAG epitope tag at 5′ was kindly supplied by Dr. Francis S. Lee (Weill Medical College of Cornell University, New York, NY) (Chen et al., 2004). BDNF and its variant Val66Met polymorphism subcloned into enhanced green fluorescent protein (eGFP)–N1 plasmid (Clontech, Palo Alto, CA) was described by Egan et al. (2003). The p75NTR receptor fused to GFP was kindly supplied by Dr. Roman Polishchuck (Consorzio Mario Negri Sud, Santa Maria Imbaro, Italy). EGFR fused to GFP was generously supplied by Dr. Francesc Tebar (Universitat de Barcelona, Barcelona, Spain) (Carter and Sorkin, 1998), atrial natriuretic factor (ANF) fused to GFP was a gift from Dr. Edwin S. Levitan (University of Pittsburgh, Pittsburgh, PA) (Burke et al., 1997), and galactosyltransferase fused to yellow fluorescent protein (GT–YFP) was kindly supplied by Dr. Jennifer Lippincott-Schwartz (National Institutes of Health, Bethesda, MD). Full-length 17Q htt (FL-17Q-htt) and full-length 75Q htt (FL-75Q-htt) was a gift from Drs. Fréderic Saudou and Sandrine Humbert (Institut Curie, Orsay, France) (Gauthier et al., 2004). Htt exon1 with 25Q (exon1–25Q-htt) and 103Q (exon1–103Q-htt) was generously provided by Dr. George M. Lawless (Cure HD Initiative, Reagent Resource Bank of the Hereditary Disease Foundation, New York, NY).
Striatal knock-in wt and mhtt cells and M213 and C17.2 cell culture and transfection.
To study the effect of htt in the intracellular transport of BDNF, we used the following cell lines: striatal knock-in cells stably expressing full-length htt (7Q/7Q) or full-length mhtt (111Q/111Q) established from HdhQ111 knock-in mice (Trettel et al., 2000); the M213 cells, conditionally immortalized striatal derived neural stem cells (Giordano et al., 1993); and the C17.2 cells, a multipotent neural stem cell line generated by retrovirus-mediated v-myc transfer into murine cerebellar progenitor cells (Ryder et al., 1990). All cell lines were transfected using Lipofectamine 2000 as instructed by the manufacturer.
Immunocytochemical staining and confocal microscopy analysis.
Twenty-four hours after transfection, cells were fixed in 4% paraformaldehyde for 10 min and 0.2 m glycine for 20 min and permeabilized in 0.1% saponin for 10 min. Blocking was done in 1% BSA in PBS for 1 h. Specimens were incubated with primary antibodies: anti-BDNF (1:50), anti-giantin (1:500), anti-calnexin (1:100), anti-secretogranin II (1:100), anti-EGFR (1:100), and anti-htt (1:100). Afterward, specimens were incubated with subtype-specific fluorescent secondary antibodies: Cy3 and rhodamine-conjugated anti-mouse (1:300) and Alexa 647 (1:100). Double-stained cells were examined by confocal microscopy using Leica (Mannheim, Germany) TCS SL laser scanning confocal spectral microscope with argon and helium–neon lasers attached to a Leica DMIRE2 inverted microscope. Images were taken using 63× numerical aperture (NA) objective with 4× digital zoom and standard (one Airy disc) pinhole. For each cell, the entire three-dimensional stack of images was obtained by the use of the Z drive present in the Leica TCS-SL microscope, and the size of the optical image was 0.5 μm. The colocalization was measured using the freeware NIH ImageJ version 1.33 by Wayne Rasband (National Institutes of Health, Bethesda, MD). Briefly, for each cell stack, the cell area was delineated, and the total number of double-positive pixels for BDNF/EGFR and a specific subcellular region staining for each slice were summed. This value was divided by the number of total positive pixels for BDNF/EGFR in the stack and multiplied by 100. For BDNF–eGFP colocalization studies, 70–80 cells were randomly selected, whereas for endogenous BDNF and EGFR colocalization, 20–25 cells were randomly selected.
Fluorescence recovery after photobleaching and inverse fluorescence recovery after photobleaching experiments.
Fluorescence recovery after photobleaching (FRAP) and inverse FRAP (iFRAP) experiments were performed using the Leica confocal microscope mentioned above equipped with an incubation system with temperature and CO2 control. Cells, 4 × 105, were seeded to 35 mm plate (Nunc, Roskilde, Denmark) containing a glass coverslip of 22 mm (Micro cover glass; Electron Microscopy Sciences, Fort Washington, PA). After 24 h, cells were transfected with 4 μg of either Val-BDNF–eGFP or Met-BDNF–eGFP constructs. For FRAP experiments 24 h after transfection, the glass coverslip was mounted in the video confocal chamber, keeping the cells at 33°C, whereas for iFRAP experiments, cells were before incubated for 2 h in media at 20°C (last hour in the presence of 0.1 mg/ml cycloheximide).
For visualization of eGFP, images were acquired using 63× oil immersion objective lens (numerical aperture, 1.32), 488 nm laser line, excitation beam splitter RSP 500, 500–600 nm emission range detection, and the confocal pinhole set at 2–3 Airy units to minimize changes in fluorescence efficiency attributable to eGFP proteins moving away from the plane of focus.
For FRAP experiments, the whole Golgi area was photobleached using 40 scans with the 488 nm laser line at full power. To correctly detect the fast component of the recovery, the first 30 images were taken during each half of a second, and the rest were taken each 2 s during 10 min. For iFRAP experiments, the whole cytoplasm area of the transfected cell was photobleached using 80 scans with the 488 nm laser line at full power. Afterward, images were collected in stream mode each 5 s during 20 min.
All FRAP and iFRAP experiments were corrected and normalized using the equation described by Phair and Misteli (2000) and Dundr et al. (2002), respectively (supplemental data, available at www.jneurosci.org as supplemental material). FRAP curves were fitted assuming one-phase exponential association equation, whereas iFRAP curves were modeled equally well with a two-phase exponential decay equation (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Fifteen cells per condition were randomly selected for FRAP experiments, whereas six to nine cells per condition were randomly selected for iFRAP experiments.
Video microscopy experiments and analysis.
Video microscopy experiments were done 24 h after transfection. Cells were transfected with either Val-BDNF–eGFP or Met-BDNF–eGFP as described for FRAP experiments. One day after, the glass coverslip was mounted in the video confocal chamber of the confocal microscope, keeping the cells at 33°C. Images were collected in stream mode using the 63× oil immersion objective with 4× of digital zoom, each second during 1 min. The pinhole was set to 350 μm, and the laser power was to 10%. All vesicles in each cell were selected and tracked using the particle-tracking software Diatrack version 3.0 (Semasopht, Chavannes, Switzerland) measuring the mean velocity of each vesicle. The mean velocity of each vesicle was calculated over the 1 min of acquisition time. In this way, the length of a vesicle trajectory, computed as the sum of the lengths of displacement vectors composing it, was divided by the lifetime of the vesicle. A displacement vector is the distance covered by one vesicle between two successive images of the sequence. The Cartesian coordinates of the center of the vesicles were used to calculate dynamic parameters as described by Gauthier et al. (2004). To calculate the diffusion pattern of Val-BDNF and Met-BDNF vesicles in wt and mhtt cells, the squared distance of each vesicle from its position at time 0–10 s was averaged and reported in a graph as a function of time. Ten to 15 cells per condition were randomly selected for video microscopy experiments.
ELISA assay.
Fifty thousand cells per well were seeded to 24-well plates. After 24 h, cells were transfected with 0.8 μg of dual-epitope-tagged Val-BDNF and Met-BDNF to avoid GFP interference with the ELISA method. At 24 h later, cells were washed with PBS, and 500 μl/well N2 medium was added. The conditioned media was collected after 24 h of incubation with N2 medium and used as a measure of constitutive secretion. To determine regulated secretion, cells were washed twice with PBS, followed by 15 min incubation at 33°C with modified Krebs–Ringer–Henseleit buffer with the following composition (in mm): 75 NaCl, 56 KCl, 2.6 CaCl2, 25 HEPES, 1.2 MgSO4, 5.6 glucose, 1 sodium ascorbate, and 1.2 KH2PO4, pH 7.4, as described previously (Chen et al., 2004). The BDNF protein concentrations in the respective media samples were measured using the BDNF Emax immunoassay system (Promega, Madison, WI). Standards and samples were performed in duplicates, and each group contained six independent samples.
Statistical analysis.
For statistical analysis, Prism version 4.0 (GraphPad Software, San Diego, CA) software was used, performing nonparametric one-way ANOVA and Bonferroni's post hoc test. For iFRAP experiments, with two conditions, the nonparametric t test followed by the Mann–Whitney test was used.
Results
Full-length but not exon1 mhtt accumulates endogenous BDNF in the Golgi apparatus
We first analyzed whether full-length or exon1 mhtt affects BDNF intracellular trafficking in different cell lines: striatal knock-in wt cells and M213 and C17.2 cells. In all three lines, colocalization studies revealed an increased colocalization of endogenous BDNF with the Golgi apparatus in the presence of FL-75Q-htt but not in the presence of exon1–103Q-htt (Fig. 1) (supplemental Figs. 2–4, available at www.jneurosci.org as supplemental material). For exon1 colocalization analysis, cells were transfected with exon1–25Q-htt, exon1–103Q-htt fused to GFP, or with GFP alone (control condition). After 24 h of transfection, cells were fixed and immunostained against BDNF and giantin. No differences in the colocalization of BDNF with giantin were observed in all cell lines. In the case of full-length htt, because there is not any tag in the vector, we cotransfected FL-17Q-htt and FL-75Q-htt with GT–YFP (which selectively marks the Golgi apparatus, ratio 8:1, respectively) or we transfected with GT–YFP alone as a control condition. After 24 h of transfection, cells were fixed and immunostained against BDNF and htt. Cells transfected with FL-17Q-htt or FL-75Q-htt were selected because of an increased immunoreactivity for anti-htt antibody. An enhanced colocalization of BDNF with GT–YFP was observed in the presence of FL-75Q-htt relative to FL-17Q-htt and the control condition. Moreover, we also performed the same colocalization studies in striatal knock-in wt or mhtt cells with 111Q (Trettel et al., 2000). mhtt cells showed increased colocalization of BDNF with giantin with respect to wt cells (Fig. 1) (supplemental Fig. 5, available at www.jneurosci.org as supplemental material).
Figure 1.

Full-length, but not exon1, mhtt increases BDNF accumulation in Golgi. Wt cells and M213 and C17.2 cells were transfected with GFP, exon1–25Q-htt of exon1–103Q-htt fused to GFP (Exon1 section), or with GT–YFP or GT–YFP plus FL-17Q-htt or FL-75Q-htt (Full-length htt overexpression section). Colocalization studies of endogenous BDNF with giantin showed no difference between GFP-expressing cells and exon1-Q25-htt or exon1–103Q-htt. In contrast, colocalization of endogenous BDNF with GT–YFP showed an increased accumulation of BDNF in the Golgi apparatus in cells transfected with FL-75Q-htt with respect to FL-17Q-htt- or GT–YFP-transfected cells. mhtt cells showed an increased BDNF accumulation in the Golgi apparatus compared with wt cells (Full-length htt endogenous section). *p < 0.05; **p < 0.01.
Val-BDNF but not Met-BDNF is retained at the Golgi apparatus in mhtt cells
We next studied whether full-length htt differentially affects Val-BDNF or Met-BDNF trafficking because expression of FL-75Q-htt increased accumulation of endogenous BDNF in Golgi apparatus. Thus, we performed several colocalization studies between Val-BDNF or Met-BDNF fused to eGFP with a variety of endomembrane markers in striatal knock-in wt or mhtt cells. These cell lines express full-length htt or full-length mhtt at endogenous levels, reflecting the closest situation to HD patients. After 24 h of transfection, cells were fixed and immunostained with calnexin or giantin, which are well established markers of the endoplasmatic reticulum (ER) and the Golgi apparatus, respectively. The colocalization between Val-BDNF or Met-BDNF with calnexin in either wt or mhtt cells showed no differences (Fig. 2A), giving an averaged colocalization in all cases of ∼25% (Fig. 2B). Conversely, when the colocalization of Val-BDNF or Met-BDNF with giantin was analyzed (Fig. 2C), we found an increased colocalization of Val-BDNF in mhtt cells compared with wt cells up to 15%. No significant differences in the Golgi colocalization were found between wt and mhtt cells expressing Met-BDNF, but both were significantly higher with respect to Val-BDNF in wt cells (Fig. 2D).
Figure 2.

In mhtt cells, Val-BDNF but not Met-BDNF is retained at the Golgi apparatus. A, B, Colocalization of Val-BDNF or Met-BDNF with calnexin in wt and mhtt cells did not show any difference between all conditions. C, D, Colocalization of Val-BDNF or Met-BDNF with giantin, a Golgi marker, revealed a higher colocalization of Val-BDNF in mhtt cells compared with wt cells, although these effects were not observed in mhtt cells expressing Met-BDNF compared with wt cells. Results are represented as a mean ± SEM determined from analysis of three independent experiments [not significant (n.s.), p > 0.05; ***p < 0.001 vs Val-BDNF in wt cells]. Scale bar, 8 μm.
mhtt differentially impairs post-Golgi trafficking of Val-BDNF and Met-BDNF
To verify whether the different accumulation of Val-BDNF in the Golgi area was caused by an impaired post-Golgi transport, we performed FRAP studies. For FRAP studies, the Golgi area was marked and photobleached, and the recovery of Golgi fluorescence was monitored by time lapse until a plateau was reached (Fig. 3A). A fast increase in the Golgi fluorescence recovery was detected during the first 60 s, reflecting the transport of new fluorescent proteins from the ER to the Golgi apparatus (Fig. 3B). No differences were observed in the transport of Val-BDNF or Met-BDNF from the ER to the Golgi apparatus in wt or mhtt cells, showing similar kinetics values in all conditions (Table 1A). Nonetheless, the analysis of the total fluorescence recovery showed a 25% reduction of Val-BDNF post-Golgi trafficking in mhtt cells compared with wt cells (Fig. 3A), which is indicative of a reduced mobile fraction (Table 1A). In addition, the effects of mhtt in Met-BDNF trafficking were different from those obtained for Val-BDNF. Interestingly, no differences were observed between wt or mhtt cells expressing Met-BDNF. Moreover, wt cells expressing Met-BDNF showed a reduced Golgi fluorescence recovery compared with wt cells expressing Val-BDNF. Altogether, these results are indicative of an impaired post-Golgi trafficking of Met-BDNF compared with Val-BDNF in wt cells.
Figure 3.

mhtt differentially impairs post-Golgi trafficking of Val-BDNF and Met-BDNF. A, FRAP recovery curves, in which the rectangle shows the rapid diffusion between ER-to-Golgi (enhanced region shown in B) and the dotted rectangle shows the plateau reached in its condition showing a reduced Golgi fluorescence recovery of Val-BDNF in mhtt and Met-BDNF in either wt or mhtt cells compared with Val-BDNF in wt cells. B, Graph showing no differences in the earlier transport from ER-to-Golgi between all conditions. C, iFRAP curves showing protein exit as Golgi fluorescence decay. Val-BDNF expressed in mhtt cells as well as Met-BDNF expressed in both cells showed a reduced Golgi exit compared with wt cells expressing Val-BDNF. Results are represented as a mean ± SEM determined from analysis of three independent experiments. ***p < 0.001.
Table 1.
Kinetic values and mobility fractions calculated for FRAP and iFRAP experiments
| Experimental condition | ER → Golgi |
Golgi → Plasma membrane |
|||
|---|---|---|---|---|---|
| K constant | T1/2 (0.69/K) (s) | Mobile fraction % | K constant | T1/2 (0.69/K) (s) | |
| Val | |||||
| wt | 0.043 | 16.04 | 88.36 ± 1.74 | 0.007 | 97.47 |
| mhtt | 0.049 | 14.08 | 68.05 ± 3.48* | 0.016 | 43.12 |
| Met | |||||
| wt | 0.045 | 15 | 69.77 ± 6.32* | 0.013 | 52.55 |
| mhtt | 0.048 | 14.37 | 72.37 ± 4.27* | 0.011 | 60.26 |
| Experimental condition | Golgi → Plasma membrane |
||||
|---|---|---|---|---|---|
| Mobile fraction % | K1 constant | K2 constant | T1/2 (s) | ||
| Val | |||||
| wt | 64.74 ± 3.1 | 0.0016 | 0.0023 | 236.8 | |
| mhtt | 42.65 ± 6.83* | 0.001 | 0.005 | 245.9 | |
| Met | |||||
| wt | 42.83 ± 4.59* | 1.35e-5 | 0.0025 | 279.4 | |
| mhtt | 39.63 ± 4.09* | 1.65e-5 | 0.0028 | 253.1 | |
| p75 | |||||
| wt | 64.97 ± 5.75 | 0.0018 | 0.029 | 265.2 | |
| mhtt | 63.16 ± 4.65 | 0.0014 | 0.0011 | 200.1 | |
| EGFR | |||||
| wt | 63.82 ± 2.51 | 0.0013 | 0.0155 | 210.5 | |
| mhtt | 35.02 ± 5.75# | 0.0014 | 0.0135 | 185.3 | |
| ANF | |||||
| wt | 54.46 ± 6.65 | 0.0013 | 0.0026 | 398.8 | |
| mhtt | 22.46 ± 8.7## | 1.27e-5 | 0.0042 | 170.3 | |
Top, FRAP studies showing no differences in the transport between the ER to the Golgi apparatus in all experimental conditions (Fig. 3B), whereas total fluorescence values indicated a reduced mobile fraction of Val-BDNF in mhtt cells and Met-BDNF in both wt and mhtt cells
(*p < 0.05) (Fig. 3A). Bottom, iFRAP studies showing a reduced mobile fraction of Val-BDNF in mhtt cells and Met-BDNF in both cells compared with Val-BDNF in wt cells (*p < 0.05 vs Val-BDNF in wt cells). In contrast to p75NTR, EGFR and ANF expressed in mhtt cells showed a reduced mobile fraction compared with wt cells (Fig. 4)
(#p < 0.05 vs EGFR in wt cells;
##p < 0.05 vs ANF in wt cells).
Next, we performed iFRAP studies to verify the alteration of post-Golgi trafficking of Val-BDNF in mhtt cells (supplemental Movie 1, available at www.jneurosci.org as supplemental material). For iFRAP studies, Val-BDNF and Met-BDNF proteins were accumulated in the Golgi apparatus as described in Materials and Methods. Afterward, the entire cytoplasmic area, except the Golgi region, was photobleached, making possible the determination of the protein fraction leaving the Golgi region as the percentage of Golgi fluorescence decay. Val-BDNF expressed in mhtt cells showed a 33% reduction in the Golgi exit compared with wt cells (Fig. 3C). No differences were observed between wt and mhtt cells expressing Met-BDNF, although both showed an altered post-Golgi trafficking when compared with Val-BDNF-expressing wt cells (Fig. 3C, Table 1B). These results strongly suggest that mhtt impairs post-Golgi trafficking of Val-BDNF.
To find out whether mhtt-induced post-Golgi transport impairment is restricted to Val-BDNF or whether it is also involved in post-Golgi transport in general, we examined a variety of cargo proteins exiting from the TGN in route to the plasma membrane. Thus, p75NTR receptor, which exits TGN via constitutive pathway (De Lisle and Bansal, 1996), showed the same post-Golgi traffic kinetics in both wt and mhtt cells (Fig. 4A, Table 1B) (supplemental Movie 2, available at www.jneurosci.org as supplemental material). Conversely, the EGFR, which follows the constitutive and clathrin-dependent post-Golgi pathways (Willingham and Pastan, 1982; Sorkina et al., 1999), showed a 44% reduction in its post-Golgi trafficking in mhtt cells (Fig. 4B) (supplemental Movie 3, available at www.jneurosci.org as supplemental material). Accordingly, colocalization data of endogenous EGFR and giantin showed an accumulation of EGFR in the Golgi of mhtt cells (7.93 ± 0.9%) compared with wt cells (2.81 ± 0.38%) (supplemental Fig. 6, available at www.jneurosci.org as supplemental material). The ANF, which exits TGN by the regulated pathway (Bloch et al., 1986; De Young et al., 1994), showed a higher decrease (60%) in its post-Golgi trafficking in mhtt cells compared with wt cells (Fig. 4C) (supplemental Movie 4, available at www.jneurosci.org as supplemental material).
Figure 4.
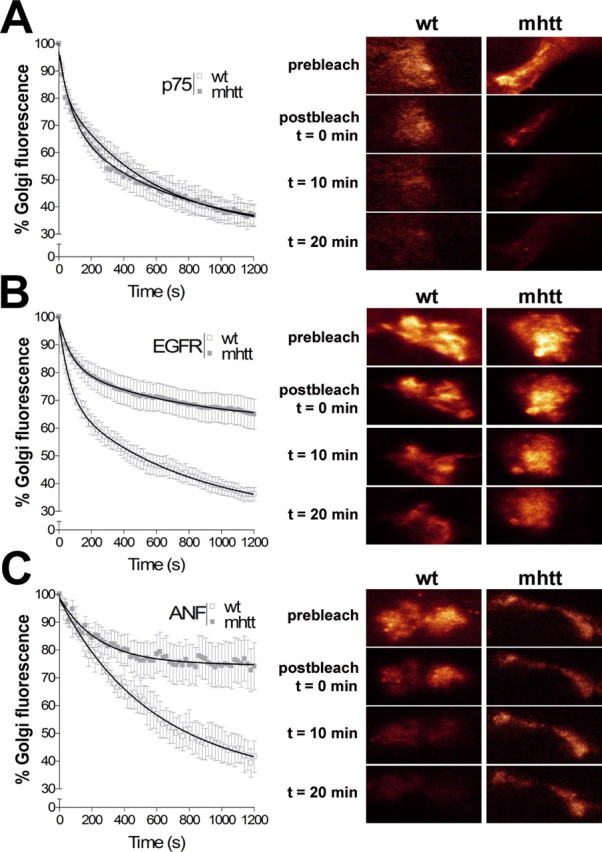
mhtt impairs the post-Golgi transport to the regulated pathway. A, No differences were observed in wt and mhtt cells expressing p75NTR. EGFR (B) and ANF (C) showed an impaired post-Golgi transport in mhtt cells compared with wt cells. Representative Golgi apparatus fluorescence for each condition is shown on the right side. Results are represented as a mean ± SEM determined from analysis of three independent experiments. p < 0.001 for ANF and EGFR in mhtt versus wt cells.
mhtt reduces the number of Val-BDNF- but not Met-BDNF-containing vesicles
To analyze the impaired post-Golgi traffic of Val-BDNF in mhtt cells, we next measured the total number of BDNF-containing vesicles produced per cell 24 h after transfection. Because in our images one pixel represents 54 nm, we considered a vesicle those fluorescent spots comprising between 2 and 10 pixels (both included, 108–540 nm) (Fig. 5A). We found a 50% reduction in the number of Val-BDNF-containing vesicles in mhtt cells. In contrast, no significant differences in the number of Met-BDNF-containing vesicles were found between wt and mhtt cells. However, in both cells, the number of Met-BDNF vesicles was lower than Val-BDNF in wt cells (Fig. 5B). In addition, the colocalization of Val-BDNF with secretogranin II, which is a specific marker of secretory vesicles, was higher in wt cells than in mhtt cells (Fig. 5C), whereas no differences were observed between wt and mhtt cells expressing Met-BDNF (Fig. 5D), although they showed a reduced colocalization with secretogranin II compared with wt cells expressing Val-BDNF.
Figure 5.

mhtt reduces the number of Val-BDNF but not Met-BDNF vesicles. A, For counting BDNF-containing vesicles, each stack of images was thresholded, and BDNF staining was marked in red. B, All of the marked spots comprising 2–10 pixels were counted, showing a reduction in Val-BDNF-containing vesicles in mhtt cells compared with wt cells. C, Colocalization between Val-BDNF or Met-BDNF and secretogranin II (SecII). D, Val-BDNF in mhtt cells showed a reduced colocalization with secretogranin II compared with wt cells. Results are represented as a mean ± SEM determined from analysis of three independent experiments [not significant (n.s.), p > 0.05; **p < 0.01; ***p < 0.001 vs Val-BDNF in wt cells). Scale bar, 8 μm.
Val-BDNF and Met-BDNF-containing vesicles show similar transport dynamics in knock-in striatal cells
To determine a possible differential effect of mhtt in the transport of Val-BDNF or Met-BDNF vesicles, we next examined the dynamics of BDNF-containing vesicles using confocal video microscopy. All Val-BDNF- or Met-BDNF-containing transport vesicles in either wt or mhtt cells were selected and tracked (Fig. 6A). We found an increase up to 16% for Val-BDNF and 24% for Met-BDNF in the number of vesicles with velocities between 0 and 30 μm/min in mhtt cells compared with wt cells, followed by a drastic decrease in the number of vesicles with velocities higher than 30 μm/min (Fig. 6B). Consequently, the overall mean velocity of Val-BDNF and Met-BDNF vesicles in mhtt cells was significantly lower (18.1 ± 2.54 and 20.4 ± 0.74 μm/min, respectively) compared with wt cells (24.53 ± 1.1 and 28.20 ± 3.1 μm/min, respectively). Moreover, these overall mean velocities are in agreement with previous studies (Kohara et al., 2001; Gauthier et al., 2004; Adachi et al., 2005). The fact that mhtt reduces the velocity of each vesicle was also observed when the diffusion pattern was calculated (Fig. 6C) (see Materials and Methods). Val-BDNF- and Met-BDNF-containing vesicles showed the same pattern of diffusion in wt cells, and the diffusion of both Val-BDNF and Met-BDNF vesicles was similarly reduced in mhtt cells.
Figure 6.
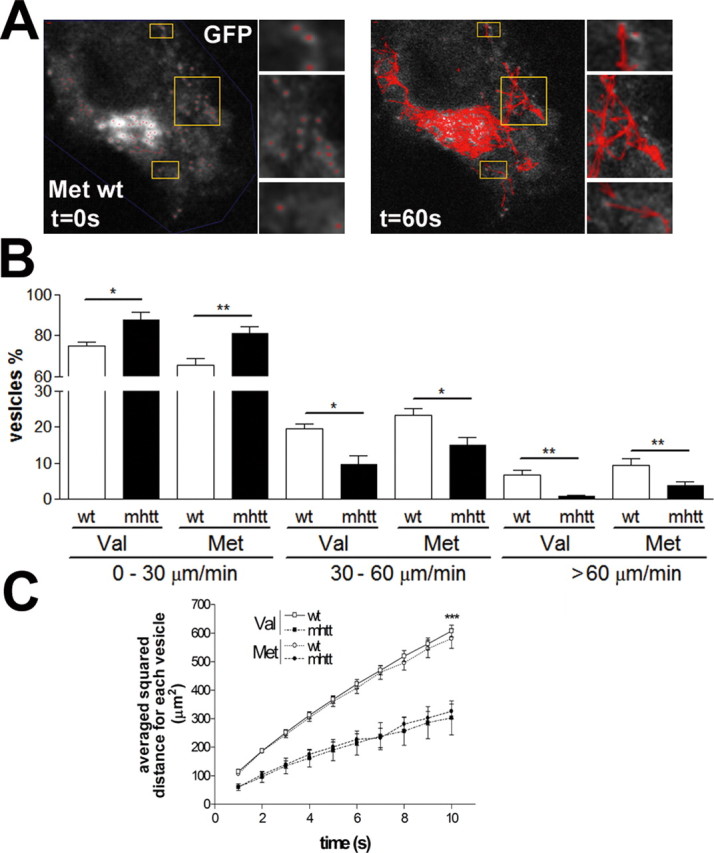
Val-BDNF- and Met-BDNF-containing vesicles have similar transport dynamics. A, All vesicles in each cell were selected and tracked during 60 s. B, The mean velocity of all tracked vesicles showed no differences between Val-BDNF and Met-BDNF vesicles. C, Val-BDNF- and Met-BDNF-containing vesicles showed the same pattern of diffusion in wt cells, which was similarly reduced in mhtt cells. Val-BDNF in wt cells, 4795 measures; Met-BDNF in wt cells, 3627 measures; Val-BDNF in mhtt cells, 3761 measures; Met-BDNF in mhtt cells, 5177 measures. Results are represented as a mean ± SEM determined from analysis of three independent experiments (*p < 0.05; **p < 0.01; ***p < 0.001).
mhtt preferentially reduces regulated secretion of Val-BDNF compared with Met-BDNF
Next, we assessed in striatal knock-in cells the constitutive and regulated secretion of Val-BDNF and Met-BDNF. There were no differences in the amount of BDNF released by constitutive secretion between Val-BDNF or Met-BDNF in either wt and mhtt cells. However, mhtt cells secreted ∼10% less Val-BDNF and Met-BDNF compared with wt cells (Fig. 7A).
Figure 7.
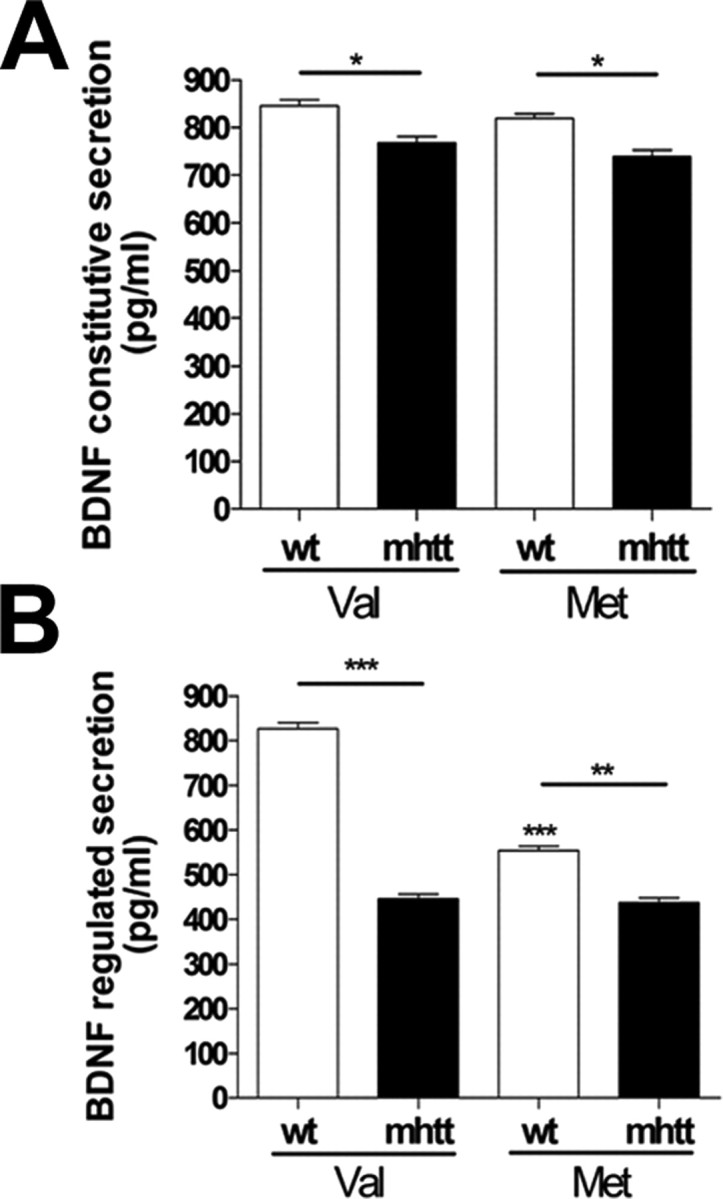
mhtt preferentially reduces regulated secretion of Val-BDNF compared with Met-BDNF. Knock-in cells were transfected with dual-epitope-tagged BDNF constructs. After 24 h, media was collected under constitutive (A) and depolarization (B) secretion conditions and analyzed by ELISA. Results are presented as a mean ± SEM determined from analysis of three independent experiments (*p < 0.05; **p < 0.01; ***p < 0.001 vs Val-BDNF in wt cells).
Nevertheless, during cell depolarization (56 mm KCl), we found a drastic reduction of a 48% in the release of Val-BDNF expressed in mhtt cells with respect to wt cells expressing Val-BDNF. In contrast, mhtt only reduced 20% of the evoked secretion of Met-BDNF (Fig. 7B).
Discussion
Although htt has been related to diverse processes, its biological function has not been completely elucidated (Harjes and Wanker, 2003). Here, we show that full-length, but not exon1, mhtt increases the accumulation of endogenous BDNF in the Golgi apparatus. Moreover, we also demonstrate that mhtt affects the trafficking to the regulated secretory pathway of Val-BDNF with low effect on Met-BDNF, suggesting that htt has an important role in the post-Golgi transport of BDNF. These data provide new insights into the intracellular transport defect of BDNF associated with HD.
Our experiments showed that full-length, but not exon1, mhtt is required to induce an effect on BDNF post-Golgi trafficking. This effect could be explained by several protein interactions that are lost in the case of exon1 htt. In fact, previous studies have shown that exon1 htt does not modify BDNF transport because it cannot bind HAP1 (Gauthier et al., 2004).
Next we studied whether mhtt affects differentially Val-BDNF or Met-BDNF forms. It has been shown that Met-BDNF has an impaired sorting to the regulated secretory pathway with respect to Val-BDNF because of a reduced interaction with sortilin at the TGN (Egan et al., 2003; Chen et al., 2004). Nonetheless, the reduced sorting of Met-BDNF to the regulated secretory pathway was not accompanied by an increase in the constitutive secretion (Chen et al., 2004, 2005). According to these results, we show that Met-BDNF is retained in the Golgi apparatus compared with Val-BDNF in wt cells. However, we observed no differences in the trafficking from the ER to the Golgi apparatus. Colocalization studies using calnexin and FRAP experiments show that the presence of mhtt does not impair the transport of either Val-BDNF or Met-BDNF from ER to Golgi apparatus. This result is in agreement with a previous report suggesting that the trafficking between the ER and the Golgi in mhtt cells might not be affected compared with wt cells (Trettel et al., 2000). In contrast, we found that the post-Golgi trafficking of Val-BDNF was significantly blocked in mhtt cells, whereas mhtt did not produce a major retention of Met-BDNF in the Golgi apparatus. These findings, corroborated by colocalization studies with giantin and FRAP/iFRAP analyses, suggest that the effect of mhtt on the post-Golgi trafficking might be subsequent to the BDNF sorting. In addition, the results are indicative that htt seems to be involved in the post-Golgi-regulated pathway. To study this hypothesis, we analyzed the effect of mhtt on the trafficking of P75NTR, a protein that exits the TGN via the constitutive pathway (De Lisle and Bansal, 1996), EGFR, which follows both constitutive and clathrin-dependent pathways (Willingham and Pastan, 1982; Sorkina et al., 1999), and ANF, which is transported through regulated pathway (Bloch et al., 1986; De Young et al., 1994). iFRAP studies showed that mhtt does not affect the trafficking of P75NTR, but, in contrast, it impaired to a different extent the post-Golgi trafficking of EGFR and ANF. All of these data support a new role for htt in modulating the sorting and/or post-Golgi trafficking of different proteins. In accordance with this, mhtt only affects trafficking of proteins by the regulated pathway (Fig. 8). Another possibility, which is currently examined, is that mhtt may also disrupt the regulation of routing the EGFR to the lysosomal degradation pathway.
Figure 8.

Schematic diagram of the effects of mhtt in post-Golgi trafficking of proteins depending on the secretory pathway. mhtt impairs trafficking of proteins secreted by the regulated clathrin-dependent pathway (Val-BDNF, EGFR, and ANF). In contrast, p75 and Met-BDNF were not severely affected. PM, Plasma membrane.
According to the results obtained about changes in post-Golgi trafficking, we observed that mhtt reduces the number of Val-BDNF but not of Met-BDNF transport vesicles. The fact that wt cells expressing Met-BDNF produced less vesicles compared with wt cells expressing Val-BDNF is in agreement with previous studies reporting an altered intracellular trafficking of Met-BDNF producing less BDNF vesicles and therefore a reduced amount of secreted BDNF after neural cell depolarization (Chen et al., 2004). However, mhtt equally impairs the transport dynamic properties of both Val-BDNF and Met-BDNF vesicles. These findings are in agreement with the role of htt in vesicle transport reported in previous studies by Gauthier et al. (2004). Thus, the effect of mhtt on the post-Golgi trafficking has a different functional repercussion on Val-BDNF or Met-BDNF release. mhtt cells showed a dramatic decrease in the KCl-evoked release of Val-BDNF, whereas the impairment of Met-BDNF release during cell depolarization was very little. In contrast, the constitutive release of Val-BDNF and Met-BDNF was similarly affected by mhtt; this effect could be explained by a reduced transport of BDNF vesicles in their way to the plasma membrane, in accordance with previous studies (Gauthier et al., 2004).
All of these results localize the effect of mhtt on BDNF polymorphism at the Golgi apparatus level. Previous studies already linked htt with Golgi apparatus. Hence, the size of this perinuclear organelle is reduced by htt deficiency (Hilditch-Maguire et al., 2000). Furthermore, some htt-interacting proteins, such as HIP-14 (Singaraja et al., 2002) and HIP1R (Carreno et al., 2004), are located in Golgi apparatus. Therefore, it is reasonable to postulate that mhtt could induce aberrant interactions with proteins involved in post-Golgi sorting and trafficking (Egea et al., 2006). Our results also suggest that an incorrect folding attributable to the mutation of the prodomain of BDNF produces an accumulation of Met-BDNF in the Golgi apparatus. In fact, modeling of the prodomain on Val-BDNF and Met-BDNF shows that the substitution of valine to methionine at codon 66 might increase the prodomain stability, which leads to an altered interaction with sortilin (supplemental data and supplemental Fig. 7, available at www.jneurosci.org as supplemental material). However, additional experiments are required to confirm this hypothesis.
The findings of the present work and those reported previously (Chen et al., 2004, 2005) demonstrate that Val-BDNF and Met-BDNF differ in their post-Golgi trafficking in nonpathological cells. It has been estimated that ∼20–30% of the healthy human population is heterozygous for Met-BDNF (Egan et al., 2003; Sen et al., 2003; Alberch et al., 2005), and these people only exhibit a relatively low diminished memory performance (Hariri et al., 2003) and hippocampal volume (Szeszko et al., 2005), probably attributable to a reduced regulated secretion of Met-BDNF. Nevertheless, this mutation in the BDNF prodomain is not associated with any neurological alteration per se.
A previous study showed that patients with HD carrying Met-BDNF allele have a later age of onset compared with those carrying Val-BDNF (Alberch et al., 2005). However, it has been reported recently that there is no evidence of association between Met-BDNF and the age at onset of HD (Di Maria et al., 2006; Metzger et al., 2006), which could be related to the different origin population and/or size sample. In fact, differences in the influence of Met-BDNF has been described in different neurological disorders depending on the population analyzed (Ribases et al., 2004; Tochigi et al., 2006; Tsai et al., 2004; Green et al., 2006), suggesting that other factors can be involved (Rubinsztein et al., 1997; Wexler et al., 2004).
In conclusion, our findings involve htt in post-Golgi transport regulation and that mhtt differently affects the post-Golgi transport of Val-BDNF and Met-BDNF, affecting more severely the regulated release of Val-BDNF compared with Met-BDNF. Thus, the Val66Met BDNF polymorphism supply different traffic support that could modulate the neurodegenerative process observed in HD.
References
- Adachi et al., 2005.Adachi N, Kohara K, Tsumoto T. Difference in trafficking of brain-derived neurotrophic factor between axons and dendrites of cortical neurons, revealed by live-cell imaging. BMC Neurosci. 2005;6:42. doi: 10.1186/1471-2202-6-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberch et al., 2005.Alberch J, Lopez M, Badenas C, Carrasco JL, Mila M, Munoz E, Canals JM. Association between BDNF Val66Met polymorphism and age at onset in Huntington disease. Neurology. 2005;65:964–965. doi: 10.1212/01.wnl.0000175977.57661.b1. [DOI] [PubMed] [Google Scholar]
- Bloch et al., 1986.Bloch KD, Seidman JG, Naftilan JD, Fallon JT, Seidman CE. Neonatal atria and ventricles secrete atrial natriuretic factor via tissue-specific secretory pathways. Cell. 1986;47:695–702. doi: 10.1016/0092-8674(86)90512-x. [DOI] [PubMed] [Google Scholar]
- Burke et al., 1997.Burke NV, Han W, Li D, Takimoto K, Watkins SC, Levitan ES. Neuronal peptide release is limited by secretory granule mobility. Neuron. 1997;19:1095–1102. doi: 10.1016/s0896-6273(00)80400-6. [DOI] [PubMed] [Google Scholar]
- Canals et al., 2004.Canals JM, Pineda JR, Torres-Peraza JF, Bosch M, Martin-Ibanez R, Munoz MT, Mengod G, Ernfors P, Alberch J. Brain-derived neurotrophic factor regulates the onset and severity of motor dysfunction associated with enkephalinergic neuronal degeneration in Huntington's disease. J Neurosci. 2004;24:7727–7739. doi: 10.1523/JNEUROSCI.1197-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carreno et al., 2004.Carreno S, Engqvist-Goldstein AE, Zhang CX, McDonald KL, Drubin DG. Actin dynamics coupled to clathrin-coated vesicle formation at the trans-Golgi network. J Cell Biol. 2004;165:781–788. doi: 10.1083/jcb.200403120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter and Sorkin, 1998.Carter RE, Sorkin A. Endocytosis of functional epidermal growth factor receptor-green fluorescent protein chimera. J Biol Chem. 1998;273:35000–35007. doi: 10.1074/jbc.273.52.35000. [DOI] [PubMed] [Google Scholar]
- Cattaneo et al., 2005.Cattaneo E, Zuccato C, Tartari M. Normal huntingtin function: an alternative approach to Huntington's disease. Nat Rev Neurosci. 2005;6:919–930. doi: 10.1038/nrn1806. [DOI] [PubMed] [Google Scholar]
- Chen et al., 2004.Chen ZY, Patel PD, Sant G, Meng CX, Teng KK, Hempstead BL, Lee FS. Variant brain-derived neurotrophic factor (BDNF) (Met66) alters the intracellular trafficking and activity-dependent secretion of wild-type BDNF in neurosecretory cells and cortical neurons. J Neurosci. 2004;24:4401–4411. doi: 10.1523/JNEUROSCI.0348-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen et al., 2005.Chen ZY, Ieraci A, Teng H, Dall H, Meng CX, Herrera DG, Nykjaer A, Hempstead BL, Lee FS. Sortilin controls intracellular sorting of brain-derived neurotrophic factor to the regulated secretory pathway. J Neurosci. 2005;25:6156–6166. doi: 10.1523/JNEUROSCI.1017-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Lisle and Bansal, 1996.De Lisle RC, Bansal R. Brefeldin A inhibits the constitutive-like secretion of a sulfated protein in pancreatic acinar cells. Eur J Cell Biol. 1996;71:62–71. [PubMed] [Google Scholar]
- De Young et al., 1994.De Young MB, Keller JC, Graham RM, Wildey GM. Brefeldin A defines distinct pathways for atrial natriuretic factor secretion in neonatal rat atrial and ventricular myocytes. Circ Res. 1994;74:33–40. doi: 10.1161/01.res.74.1.33. [DOI] [PubMed] [Google Scholar]
- Di Maria et al., 2006.Di Maria E, Marasco A, Tartari M, Ciotti P, Abbruzzese G, Novelli G, Bellone E, Cattaneo E, Mandich P. No evidence of association between BDNF gene variants and age-at-onset of Huntington's disease. Neurobiol Dis. 2006;24:274–279. doi: 10.1016/j.nbd.2006.07.002. [DOI] [PubMed] [Google Scholar]
- Dundr et al., 2002.Dundr M, Hoffmann-Rohrer U, Hu Q, Grummt I, Rothblum LI, Phair RD, Misteli T. A kinetic framework for a mammalian RNA polymerase in vivo. Science. 2002;298:1623–1626. doi: 10.1126/science.1076164. [DOI] [PubMed] [Google Scholar]
- Egan et al., 2003.Egan MF, Kojima M, Callicott JH, Goldberg TE, Kolachana BS, Bertolino A, Zaitsev E, Gold B, Goldman D, Dean M, Lu B, Weinberger DR. The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell. 2003;112:257–269. doi: 10.1016/s0092-8674(03)00035-7. [DOI] [PubMed] [Google Scholar]
- Egea et al., 2006.Egea G, Lazaro-Dieguez F, Vilella M. Actin dynamics at the Golgi complex in mammalian cells. Curr Opin Cell Biol. 2006;18:168–178. doi: 10.1016/j.ceb.2006.02.007. [DOI] [PubMed] [Google Scholar]
- Ferrer et al., 2000.Ferrer I, Goutan E, Marin C, Rey MJ, Ribalta T. Brain-derived neurotrophic factor in Huntington disease. Brain Res. 2000;866:257–261. doi: 10.1016/s0006-8993(00)02237-x. [DOI] [PubMed] [Google Scholar]
- Friedel et al., 2005.Friedel S, Horro FF, Wermter AK, Geller F, Dempfle A, Reichwald K, Smidt J, Bronner G, Konrad K, Herpertz-Dahlmann B, Warnke A, Hemminger U, Linder M, Kiefl H, Goldschmidt HP, Siegfried W, Remschmidt H, Hinney A, Hebebrand J. Mutation screen of the brain derived neurotrophic factor gene (BDNF): identification of several genetic variants and association studies in patients with obesity, eating disorders, and attention-deficit/hyperactivity disorder. Am J Med Genet B Neuropsychiatr Genet. 2005;132:96–99. doi: 10.1002/ajmg.b.30090. [DOI] [PubMed] [Google Scholar]
- Gauthier et al., 2004.Gauthier LR, Charrin BC, Borrell-Pages M, Dompierre JP, Rangone H, Cordelieres FP, De MJ, MacDonald ME, Lessmann V, Humbert S, Saudou F. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell. 2004;118:127–138. doi: 10.1016/j.cell.2004.06.018. [DOI] [PubMed] [Google Scholar]
- Giordano et al., 1993.Giordano M, Takashima H, Herranz A, Poltorak M, Geller HM, Marone M, Freed WJ. Immortalized GABAergic cell lines derived from rat striatum using a temperature-sensitive allele of the SV40 large T antigen. Exp Neurol. 1993;124:395–400. doi: 10.1006/exnr.1993.1213. [DOI] [PubMed] [Google Scholar]
- Green et al., 2006.Green EK, Raybould R, Macgregor S, Hyde S, Young AH, O'Donovan MC, Owen MJ, Kirov G, Jones L, Jones I, Craddock N. Genetic variation of brain-derived neurotrophic factor (BDNF) in bipolar disorder: case-control study of over 3000 individuals from the UK. Br J Psychiatry. 2006;188:21–25. doi: 10.1192/bjp.bp.105.009969. [DOI] [PubMed] [Google Scholar]
- Hall et al., 2003.Hall D, Dhilla A, Charalambous A, Gogos JA, Karayiorgou M. Sequence variants of the brain-derived neurotrophic factor (BDNF) gene are strongly associated with obsessive-compulsive disorder. Am J Hum Genet. 2003;73:370–376. doi: 10.1086/377003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hariri et al., 2003.Hariri AR, Goldberg TE, Mattay VS, Kolachana BS, Callicott JH, Egan MF, Weinberger DR. Brain-derived neurotrophic factor val66met polymorphism affects human memory-related hippocampal activity and predicts memory performance. J Neurosci. 2003;23:6690–6694. doi: 10.1523/JNEUROSCI.23-17-06690.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harjes and Wanker, 2003.Harjes P, Wanker EE. The hunt for huntingtin function: interaction partners tell many different stories. Trends Biochem Sci. 2003;28:425–433. doi: 10.1016/S0968-0004(03)00168-3. [DOI] [PubMed] [Google Scholar]
- Hilditch-Maguire et al., 2000.Hilditch-Maguire P, Trettel F, Passani LA, Auerbach A, Persichetti F, MacDonald ME. Huntingtin: an iron-regulated protein essential for normal nuclear and perinuclear organelles. Hum Mol Genet. 2000;9:2789–2797. doi: 10.1093/hmg/9.19.2789. [DOI] [PubMed] [Google Scholar]
- Kohara et al., 2001.Kohara K, Kitamura A, Morishima M, Tsumoto T. Activity-dependent transfer of brain-derived neurotrophic factor to postsynaptic neurons. Science. 2001;291:2419–2423. doi: 10.1126/science.1057415. [DOI] [PubMed] [Google Scholar]
- Kremer et al., 1994.Kremer B, Goldberg P, Andrew SE, Theilmann J, Telenius H, Zeisler J, Squitieri F, Lin B, Bassett A, Almqvist E. A worldwide study of the Huntington's disease mutation. The sensitivity and specificity of measuring CAG repeats. N Engl J Med. 1994;330:1401–1406. doi: 10.1056/NEJM199405193302001. [DOI] [PubMed] [Google Scholar]
- Metzger et al., 2006.Metzger S, Bauer P, Tomiuk J, Laccone F, Didonato S, Gellera C, Mariotti C, Lange HW, Weirich-Schwaiger H, Wenning GK, Seppi K, Melegh B, Havasi V, Baliko, Wieczorek S, Zaremba J, Hoffman-Zacharska D, Sulek A, Basak AN, Soydan E, et al. Genetic analysis of candidate genes modifying the age-at-onset in Huntington's disease. Hum Genet. 2006;120:285–292. doi: 10.1007/s00439-006-0221-2. [DOI] [PubMed] [Google Scholar]
- Momose et al., 2002.Momose Y, Murata M, Kobayashi K, Tachikawa M, Nakabayashi Y, Kanazawa I, Toda T. Association studies of multiple candidate genes for Parkinson's disease using single nucleotide polymorphisms. Ann Neurol. 2002;51:133–136. doi: 10.1002/ana.10079. [DOI] [PubMed] [Google Scholar]
- Phair and Misteli, 2000.Phair RD, Misteli T. High mobility of proteins in the mammalian cell nucleus. Nature. 2000;404:604–609. doi: 10.1038/35007077. [DOI] [PubMed] [Google Scholar]
- Ribases et al., 2003.Ribases M, Gratacos M, Armengol L, de CR, Badia A, Jimenez L, Solano R, Vallejo J, Fernandez F, Estivill X. Met66 in the brain-derived neurotrophic factor (BDNF) precursor is associated with anorexia nervosa restrictive type. Mol Psychiatry. 2003;8:745–751. doi: 10.1038/sj.mp.4001281. [DOI] [PubMed] [Google Scholar]
- Ribases et al., 2004.Ribases M, Gratacos M, Fernandez-Aranda F, Bellodi L, Boni C, Anderluh M, Cristina Cavallini M, Cellini E, Di Bella D, Erzegovesi S, Foulon C, Gabrovsek M, Gorwood P, Hebebrand J, Hinney A, Holliday J, Hu X, Karwautz A, Kipman A, Komel R. Association of BDNF with anorexia, bulimia and age of onset of weight loss in six European populations. Hum Mol Genet. 2004;13:1205–1212. doi: 10.1093/hmg/ddh137. [DOI] [PubMed] [Google Scholar]
- Rubinsztein et al., 1997.Rubinsztein DC, Leggo J, Chiano M, Dodge A, Norbury G, Rosser E, Craufurd D. Genotypes at the GluR6 kainate receptor locus are associated with variation in the age of onset of Huntington disease. Proc Natl Acad Sci USA. 1997;94:3872–3876. doi: 10.1073/pnas.94.8.3872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryder et al., 1990.Ryder EF, Snyder EY, Cepko CL. Establishment and characterization of multipotent neural cell lines using retrovirus vector-mediated oncogene transfer. J Neurobiol. 1990;21:356–375. doi: 10.1002/neu.480210209. [DOI] [PubMed] [Google Scholar]
- Seidah et al., 1996.Seidah NG, Benjannet S, Pareek S, Savaria D, Hamelin J, Goulet B, Laliberte J, Lazure C, Chretien M, Murphy RA. Cellular processing of the nerve growth factor precursor by the mammalian pro-protein convertases. Biochem J. 1996;314:951–960. doi: 10.1042/bj3140951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen et al., 2003.Sen S, Nesse RM, Stoltenberg SF, Li S, Gleiberman L, Chakravarti A, Weder AB, Burmeister M. A BDNF coding variant is associated with the NEO personality inventory domain neuroticism, a risk factor for depression. Neuropsychopharmacology. 2003;28:397–401. doi: 10.1038/sj.npp.1300053. [DOI] [PubMed] [Google Scholar]
- Singaraja et al., 2002.Singaraja RR, Hadano S, Metzler M, Givan S, Wellington CL, Warby S, Yanai A, Gutekunst CA, Leavitt BR, Yi H, Fichter K, Gan L, McCutcheon K, Chopra V, Michel J, Hersch SM, Ikeda JE, Hayden MR. HIP14, a novel ankyrin domain-containing protein, links huntingtin to intracellular trafficking and endocytosis. Hum Mol Genet. 2002;11:2815–2828. doi: 10.1093/hmg/11.23.2815. [DOI] [PubMed] [Google Scholar]
- Skibinska et al., 2004.Skibinska M, Hauser J, Czerski PM, Leszczynska-Rodziewicz A, Kosmowska M, Kapelski P, Slopien A, Zakrzewska M, Rybakowski JK. Association analysis of brain-derived neurotrophic factor (BDNF) gene Val66Met polymorphism in schizophrenia and bipolar affective disorder. World J Biol Psychiatry. 2004;5:215–220. doi: 10.1080/15622970410029936. [DOI] [PubMed] [Google Scholar]
- Sorkina et al., 1999.Sorkina T, Bild A, Tebar F, Sorkin A. Clathrin, adaptors and eps15 in endosomes containing activated epidermal growth factor receptors. J Cell Sci. 1999;112:317–327. doi: 10.1242/jcs.112.3.317. [DOI] [PubMed] [Google Scholar]
- Szeszko et al., 2005.Szeszko PR, Lipsky R, Mentschel C, Robinson D, Gunduz-Bruce H, Sevy S, Ashtari M, Napolitano B, Bilder RM, Kane JM, Goldman D, Malhotra AK. Brain-derived neurotrophic factor val66met polymorphism and volume of the hippocampal formation. Mol Psychiatry. 2005;10:631–636. doi: 10.1038/sj.mp.4001656. [DOI] [PubMed] [Google Scholar]
- Tochigi et al., 2006.Tochigi M, Otowa T, Suga M, Rogers M, Minato T, Yamasue H, Kasai K, Kato N, Sasaki T. No evidence for an association between the BDNF Val66Met polymorphism and schizophrenia or personality traits. Schizophr Res. 2006;87:45–47. doi: 10.1016/j.schres.2006.06.029. [DOI] [PubMed] [Google Scholar]
- Trettel et al., 2000.Trettel F, Rigamonti D, Hilditch-Maguire P, Wheeler VC, Sharp AH, Persichetti F, Cattaneo E, MacDonald ME. Dominant phenotypes produced by the HD mutation in STHdh(Q111) striatal cells. Hum Mol Genet. 2000;9:2799–2809. doi: 10.1093/hmg/9.19.2799. [DOI] [PubMed] [Google Scholar]
- Tsai et al., 2004.Tsai SJ, Hong CJ, Liu HC, Liu TY, Hsu LE, Lin CH. Association analysis of brain-derived neurotrophic factor Val66Met polymorphisms with Alzheimer's disease and age of onset. Neuropsychobiology. 2004;49:10–12. doi: 10.1159/000075332. [DOI] [PubMed] [Google Scholar]
- Ventriglia et al., 2002.Ventriglia M, Bocchio CL, Benussi L, Binetti G, Zanetti O, Riva MA, Gennarelli M. Association between the BDNF 196 A/G polymorphism and sporadic Alzheimer's disease. Mol Psychiatry. 2002;7:136–137. doi: 10.1038/sj.mp.4000952. [DOI] [PubMed] [Google Scholar]
- Wada et al., 1991.Wada I, Rindress D, Cameron PH, Ou WJ, Doherty JJ, Louvard D, Bell AW, Dignard D, Thomas DY, Bergeron JJ. SSR alpha and associated calnexin are major calcium binding proteins of the endoplasmic reticulum membrane. J Biol Chem. 1991;266:19599–19610. [PubMed] [Google Scholar]
- Wexler et al., 2004.Wexler NS, Lorimer J, Porter J, Gomez F, Moskowitz C, Shackell E, Marder K, Penchaszadeh G, Roberts SA, Gayan J, Brocklebank D, Cherny SS, Cardon LR, Gray J, Dlouhy SR, Wiktorski S, Hodes ME, Conneally PM, Penney JB, Gusella J, et al. Venezuelan kindreds reveal that genetic and environmental factors modulate Huntington's disease age of onset. Proc Natl Acad Sci USA. 2004;101:3498–3503. doi: 10.1073/pnas.0308679101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willingham and Pastan, 1982.Willingham MC, Pastan IH. Transit of epidermal growth factor through coated pits of the Golgi system. J Cell Biol. 1982;94:207–212. doi: 10.1083/jcb.94.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuccato et al., 2001.Zuccato C, Ciammola A, Rigamonti D, Leavitt BR, Goffredo D, Conti L, MacDonald ME, Friedlander RM, Silani V, Hayden MR, Timmusk T, Sipione S, Cattaneo E. Loss of huntingtin-mediated BDNF gene transcription in Huntington's disease. Science. 2001;293:493–498. doi: 10.1126/science.1059581. [DOI] [PubMed] [Google Scholar]
- Zuccato et al., 2003.Zuccato C, Tartari M, Crotti A, Goffredo D, Valenza M, Conti L, Cataudella T, Leavitt BR, Hayden MR, Timmusk T, Rigamonti D, Cattaneo E. Huntingtin interacts with REST/NRSF to modulate the transcription of NRSE-controlled neuronal genes. Nat Genet. 2003;35:76–83. doi: 10.1038/ng1219. [DOI] [PubMed] [Google Scholar]