

Abstract
Uptake of l-glutamate into synaptic vesicles is mediated by vesicular glutamate transporters (VGLUTs). Three transporters (VGLUT1–VGLUT3) are expressed in the mammalian CNS, with partial overlapping expression patterns, and VGLUT2 is the most abundantly expressed paralog in the thalamus, midbrain, and brainstem. Previous studies have shown that VGLUT1 is necessary for glutamatergic transmission in the hippocampus, but the role of VGLUT2 in excitatory transmission is unexplored in glutamatergic neurons and in vivo. We examined the electrophysiological and behavioral consequences of loss of either one or both alleles of VGLUT2. We show that targeted deletion of VGLUT2 in mice causes perinatal lethality and a 95% reduction in evoked glutamatergic responses in thalamic neurons, although hippocampal synapses function normally. Behavioral analysis of heterozygous VGLUT2 mice showed unchanged motor function, learning and memory, acute nociception, and inflammatory pain, but acquisition of neuropathic pain, maintenance of conditioned taste aversion, and defensive marble burying were all impaired. Reduction or loss of VGLUT2 in heterozygous and homozygous VGLUT2 knock-outs led to a graded reduction in the amplitude of the postsynaptic response to single-vesicle fusion in thalamic neurons, indicating that the vesicular VGLUT content is critically important for quantal size and demonstrating that VGLUT2-mediated reduction of excitatory drive affects specific forms of sensory processing.
Keywords: synaptic vesicles, glutamate, neurotransmitter transporter, thalamus, chronic pain, quantal size
Introduction
Before release of l-glutamate from excitatory synapses, glutamate is transported into synaptic vesicles by vesicular glutamate transporters (VGLUTs). Three distinct VGLUT genes have been identified in mammals (Ni et al., 1994; Aihara et al., 2000; Bellocchio et al., 2000; Bai et al., 2001; Fremeau et al., 2001; Herzog et al., 2001; Takamori et al., 2001, 2002; Schafer et al., 2002). The transporters use a membrane potential gradient set by the vesicular H+-ATPase for glutamate uptake (Maycox et al., 1988).
It is not known whether the three VGLUTs perform distinct functions in the nervous system. Uptake studies did not reveal significant differences in transport rates and substrate affinity for VGLUT1–VGLUT3, and overexpression of VGLUT1 or VGLUT2 in nonglutamatergic neurons showed similar spontaneous release, indicating that both gene products function with similar efficiency and are functionally redundant (Bellocchio et al., 2000; Takamori et al., 2000, 2001, 2002; Herzog et al., 2001). However, VGLUT1–VGLUT3 have distinct temporal and spatial expression profiles (Fujiyama et al., 2001; Herzog et al., 2001; Gras et al., 2002; Kaneko et al., 2002; Fremeau et al., 2004a). Whereas VGLUT1 is present in neurons of the cerebral cortex, hippocampus, and cerebellar cortex and has a late onset of expression, VGLUT2 is expressed in early development and is most abundant in the thalamus and lower brainstem regions of adult rodents. Spinal cord and primary sensory neurons primarily express both VGLUT1 and VGLUT2 (Landry et al., 2004). Genetic deletion of VGLUT1 has been shown by two laboratories to disrupt synaptic glutamate release in hippocampus (Fremeau et al., 2004b; Wojcik et al., 2004), but the role of VGLUT2 in glutamatergic transmission in the hippocampus or elsewhere is not well characterized.
In addition to different roles for each VGLUT isoform, transport numbers may also be functionally important. Increasing the amount of VGLUT1 at the synapse has been shown to increase quantal size (Wojcik et al., 2004; Wilson et al., 2005). Alterations in VGLUT expression levels have been measured in response to pharmacologic manipulations (De Gois et al., 2005; Wilson et al., 2005) and in brains of schizophrenic and Parkinson's patients (Eastwood and Harrison, 2005; Kashani et al., 2006). The effects of these alterations in fast excitatory transmission on behavior is unknown, but alterations in the levels of the vesicular monoamine transporter VMAT2 have been shown to affect both quantal size and behavior (Takahashi et al., 1997; Pothos et al., 2000).
Here we deleted the Slc17a6 gene encoding VGLUT2 in mice and show that homozygous null mice die at birth. Although heterozygous mice express 50% of wild-type levels of VGLUT2 protein, they show discrete behavioral phenotypes that suggest a deficit in thalamic processing. Electrophysiological analysis shows that VGLUT2 is necessary for glutamatergic signaling in the thalamus but not the hippocampus. In addition, we observed graded reduction in miniature EPSC (mEPSC) amplitude in VGLUT2 +/− and VGLUT2 −/− thalamic neurons compared with VGLUT2 +/+, demonstrating that the overall VGLUT content controls the amount of glutamate molecules loaded into single vesicles.
Materials and Methods
Generation of VGLUT2 knock-out mice
VGLUT2 knock-out mice were developed in collaboration with Lexicon Genetics (The Woodlands, TX) using a PCR probe derived from the second coding exon of the murine VGLUT2 gene. Clones were isolated by screening of the 129SvEvBrd-derived lambda pKOS genomic library (Wattler et al., 1999). A 9.8 kb genomic clone spanning the first three coding exons was used to generate the targeting vector. An IRESBgeo/PGKpuro selection cassette was inserted as an SfiI fragment to replace a 287 bp VGLUT2 genomic fragment spanning exon 2 after yeast-mediated homologous recombination. The NotI-linearized vector was electroporated into 129 Sv/Evbrd(LEX1) embryonic stem (ES) cells, and puromycine-resistant ES cell clones were isolated and analyzed for homologous recombination by Southern blot analysis. Targeted ES cell clones were injected into C57BL/6(albino) blastocysts, and the resulting chimeras were mated to C57BL/6(albino) females to generate animals heterozygous for the mutation. These were subsequently crossed to generate all three genotypes used in these studies. PCR was used to screen genotypes by using DNA isolated from mouse-tail biopsy samples. Primers 5′-GGTGCTGGAGAAGAAGCAG-3′ and 5′-CAAGCCTTCCT-CTTTCAGTGG-3′ amplified a 333 bp band from the wild-type allele, whereas primers 5′-CAAGCCTTCCTCTTTCAGTGG-3′ and 5′-CGCAAGCCCGGTGCCTGA-3′ amplified a 503 bp band from the knock-out allele.
Quantitative reverse transcription-PCR analysis
Quantitative reverse transcription (RT)-PCR analysis was used to show expression of the VGLUT2 transcript. Total RNA was isolated from different tissues derived from embryonic day 18 (E18) embryos, using Trizol (Invitrogen, Carlsbad, CA), and first-strand cDNA synthesis was performed on 0.5 μg of total RNA using random hexamer primers and SuperscriptII RT (Invitrogen). Quantitative PCR was performed on an ABI Prism 7700 cycler (Applied Biosystems, Foster City, CA) using a Taqman PCR kit. Serial dilutions of cDNA were used to generate standard curves of threshold cycles versus the logarithms of concentration for β-actin VGLUT1 and VGLUT2. A linear regression line calculated from the standard curves allowed the determination of transcript levels in RNA samples from mice. The primer–probe pair of the respective genes of interest (see below) relative to actin was used to assess expression levels: VGLUT2, primer 5′-TGCTACCTCACAGGAGAATGGA-3′; primer 5′-GCGCACCTTCTTGCACAAAT-3′; probe, 5′-CCTTTTTCT-CCCAGCCGTTAGGCCA-3′ [5′, 6-carboxyfluorescein (FAM); 3′, 6-carboxytetramethylrhodamine (TAMRA)]; actin, primer 5′-CATCTTGGCCTCACTGTCCAC-3′; primer 5′-GGGCCGGACTCATCGTA-CT-3′; probe 5′-TGCTTGCTGATCCACATCTGCTGGA-3′ (5′, FAM; 3′, TAMRA).
Western blot analysis
Brains from E18.5 wild-type, heterozygous, and homozygous embryos were used to prepare a vesicular enriched fraction with the MITO-ISO1 kit from Sigma (St. Louis, MO) following the technical bulletin of the manufacturer. Pellets were resuspended in 75 μl of 1× storage buffer, followed by a BCA protein determination (Pierce, Rockford, IL). VGLUT 2 aggregates after boiling, so SDS-PAGE was performed only with 25 μg/lane nonboiled samples on 7.5% Tris-glycine PAGEr Duramide precast gels (Cambrex, East Rutherford, NJ) and transferred by semi-dry blotting (1 mA/cm2 for 1 h) to Invitrolon polyvinylidene difluoride membranes 0.45 μm (Invitrogen). After blocking in 5% nonfat dry milk in Tris-buffered saline plus 0.1% Tween 20, blots were incubated with anti-VGLUT2 affinity-purified rabbit polyclonal antibody with an identified epitope located between amino acid residue 510 and 582 (Synaptic Systems, Göttingen, Germany) at 1:5000, followed by reaction with secondary anti-rabbit horseradish peroxidase and Supersignal West-Dura detection (Pierce). The signal intensity was measured using the Lumi-Imager from Roche Applied Science (Basel, Switzerland).
Mouse care
Animals were maintained in an specific pathogen-free facility that meets all Belgian and European Union requirements for animal care. Mice were group housed in a climate-controlled animal colony with a 12 h light/dark cycle (light on 6:00 A.M.) with access to food and water ad libitum, except during some of the behavioral testing. Adequate measures were taken to minimize pain or discomfort. All experiments were performed in accordance with the European Communities Council Directives (86/609/EEC) and Baylor College of Medicine guidelines and were approved by the local ethical committee.
Behavioral analysis
In the different behavioral tests, VGLUT2 +/+ and VGLUT2 +/− littermates (n = 12 for each group), aged 3–4 months, were used. General assessment of neuromotor performance and fear-related behavior were done using standard behavior protocols (supplemental data, available at www.jneurosci.org as supplemental material).
Defensive burying test.
Mice were placed individually in transparent plastic cages (15 × 26 × 42 cm) containing 5 cm of sawdust and 24 identical glass marbles (∼1.5 cm diameter) evenly spaced 2 cm from the cage wall (Njung'e and Handley, 1991; Millan et al., 2000). The cages were placed on a platform 80 cm above the floor and under bright illumination. After 30 min, the mice were returned to their home cage, and the number of marbles buried at least two-thirds by sawdust was counted.
Morris water maze.
Spatial memory abilities were examined in the standard hidden-platform acquisition and retention and reversal learning versions of the Morris maze. A 150 cm circular pool was filled with water, made opaque with nontoxic white paint, and kept at 26°C as described previously (Morris, 1989; D'Hooge et al., 2005). A 15 cm round platform was hidden 1 cm beneath the surface of the water at a fixed position. Each daily trial block consisted of four swimming trials (10 min intertrial interval) starting randomly from each of four starting positions. Mice that failed to find the platform within 2 min were guided to the platform, where they remained for 15 s before being returned to their cages. At the end of acquisition training, a probe trial was performed during which the platform was removed from the pool, and the search pattern of the mice was recorded for 100 s, followed by a reversal trial in which the platform was moved to the opposite quadrant (supplemental data, available at www.jneurosci.org as supplemental material). Swimming paths of the animals were recorded using EthoVision video tracking equipment and software (Noldus, Wageningen, The Netherlands).
Context-dependent fear conditioning.
Cue- and context-dependent fear conditioning were tested in a previously described protocol (Paradee et al., 1999). On the first day, the animals were placed in the testing chamber (22.5 × 32.5 × 33.3 cm; Plexiglas cage with a grid floor) and were allowed to acclimate for 5 min. On day 2, they were first allowed to explore the testing chamber for 2 min [pre-unconditioned stimulus (US) score]. A 30 s tone [conditioned stimulus (CS)] was delivered (frequency, 2150 ± 200 Hz; Star Micronics, Piscataway, NJ), which coterminated with a 2 s, 0.35 mA footshock (the US). Two minutes later, a second pairing of the CS and US was presented, followed by another 30 s exploration (post-US score). Twenty-four hours later, the animals were returned to the testing chamber for 5 min exploration in the same context as the previous day (context score). Ninety minutes later, the animals were returned to the test chamber, but now the grid floor was hidden with a Plexiglas plate and odorized sawdust to alter the context of the testing chamber. The animals were observed for 6 min. During the first 3 min, no stimulus was delivered (pre-CS score). During the next 3 min phase, the auditory cue was delivered (CS score). Under the different conditions, animals were scored for freezing every 10 s, leading to a maximum score of 12 bouts of freezing during baseline trials, 21 during the shock trials, 30 during the context trials, and 18 for both the pre-CS and CS trials. A freezing score was expressed as the percentage of freezing bouts versus the total number of bouts in each of the five trial blocks.
Conditioned taste aversion.
Mice were trained to drink water from modified 15 ml Falcon tubes two times a day for 30 min. On conditioning day, mice were presented saccharin-flavored water (0.5% saccharin sodium in water) to drink, and, 30 min later, they were injected intraperitoneally with lithium chloride in saline solution (4.5 mEq/kg) to induce nausea. Six, 24, 48, 72, and 96 h later, mice were presented simultaneously water and saccharin solution from two drinking tubes, allowing free choice. The aversion index (AI) was calculated as AI = water drunk/(water intake + saccharin intake).
Hot plate test.
Mice were placed on a metal surface maintained at 50 ± 0.3, 52 ± 0.3, 54 ± 0.3, or 56 ± 0.3°C. The temperature of the plate was monitored at all times. Locomotion of the mouse on the plate was constrained by a colorless acrylic cylinder (outer diameter, 20 cm). To remove traces of urine and feces, the plate was wiped with a cloth. Mice were placed on the hot plate one at a time, and the response latency to the heat stimulus was measured to the nearest 0.1 s. To prevent tissue damage, a cutoff time was used (30 s). Mice remained on the plate until they performed one of the behaviors regarded as indicative of nociception: hindpaw lick, hindpaw shake/flutter, and hindpaw lift, or until the cutoff time has elapsed. Only hindpaw responses were used, because forepaw licking and lifting are components of normal grooming behavior. Each mouse was tested two times for each temperature with at least 15 min between each measurement. An interval of at least 30 min was used between testing the mice for a different temperature.
Tail withdrawal test.
Mice were slightly restrained in a cloth “pocket” into which they entered voluntarily. The distal half of the tail was then dipped into a bath of warm water, which was thermostatically controlled (47 ± 0.5, 49 ± 0.5, 51 ± 0.5, or 53 ± 0.5°C). Latency to respond to the heat stimulus with vigorous flexion of the tail was measured to the nearest 0.1 s using a manual stopwatch. Animals were removed immediately after responding and the tail was wiped off with a cloth. A cutoff time of 25 s was adopted. Two separate withdrawal latency determinations (separated by ∼15 min) for each animal were averaged.
Hargreaves' test.
Before testing, mice were acclimatized in the clear plastic testing chambers (outer diameter, 14 cm) until exploratory activity has ceased. Thermal pain thresholds of the hindpaws were determined by applying radiant heat to the plantar surface of the hindpaws from underneath. The radiant heat produced a continuous rise in temperature of the paw until the pain threshold of the animal was reached and the animal withdrew the paw from the stimulus. Paw withdrawal latencies were recorded three times on each hindpaw with an interval of at least 2 min. Results were expressed as the mean withdrawal latencies (seconds).
Formalin test.
To assess the paw flinch behavior after injection of formalin, the automated nociception analyzer (University of California, San Diego, La Jolla, CA) was used. A soft metal band (“C” shaped) was placed on the hindpaw, and mice were placed in the plastic testing chambers (outer diameter, 8 cm). After 2 h of habituation in the cylinder, mice were injected with 25 μl of a 1% formalin solution in the subplantar surface of the banded paw. Each animal's flinch count value over a 1 min interval is measured over the duration of the study (60 min). For statistical comparison, the total number of flinches observed during the selected time periods (phase I, 0–9 min; phase II, 10–60 min; phase IIA, 10–40 min; phase IIB, 41–60 min) is calculated by accumulating each individual animal's flinches over that time period and averaging the group.
Carrageenan subplantar injection.
Peripheral inflammation was produced in the mice by a 25 μl subcutaneous injection of a 2% carrageenan solution (Sigma, Steinheim, Germany) into the subplantar side of the left hindpaw using a syringe with a 30 gauge needle. Control animals received an injection of saline (25 μl). Mice were tested for thermal sensitivity on the heat test of Hargreaves' as described above. Before the injection, baseline latencies were measured twice with an interval of 15 min. Postinjection latencies were measured after 3, 4, 5, and 6 h.
Spared nerve injury.
Baseline thresholds were recorded from VGLUT2 +/+ and VGLUT2 +/− littermates performing the automated von Frey system and the acetone spray test. Surgical procedures as described by Shields et al. (2003) were performed under gas anesthesia with a mixture of isoflurane (5% for induction and 3% for maintenance), oxygen, and N2O. After skin and muscle incision of the left hindleg, two of the three terminal branches of the sciatic nerve (sural and common peroneal nerve) were tightly ligated with 9-0 virgin silk suture (Ethicon, Somerville, NJ). The ligated branches were then transected distal to the ligation, and a 2–4 mm section of the nerve was removed to prevent rejoining. Caution was taken to minimize any contact with the spared nerve (tibial nerve). The muscle and skin layer were closed separately by using 6-0 Vicryl suture (Ethicon) and 6-0 Mersilk suture (Ethicon), respectively. Sham mice were made by following the same surgical procedure without touching any nerve. Responses to mechanical and thermal thresholds were measured 2, 6, 9, 14, and 21 d after injury.
von Frey test.
To assess mechanical allodynia, mice were placed in individual wire-mesh floored cages to allow insertion of the mechanical probe against the plantar surface of the hindpaw. The animals were allowed to habituate to the testing chambers for at least 1 h before the first measurement. By using a mechanical force transducer and metal probe, the mean force required to evoke a withdrawal response was calculated from three measurements for both hindpaws.
Acetone test.
This test was used for the determination of the reactivity to a cold, chemical, and possibly mechanical stimulation. The mice were housed in a similar manner as in the von Frey test. From below the grid, 50 μl of acetone was sprayed onto the plantar surface of the left hindpaw with an Eppendorf multistepper pipette. The time spent licking, flinching, and lifting during 1 min after being exposed to the acetone was recorded.
Statistics.
In all behavior studies, results are expressed as mean ± SEM. To identify significant differences between VGLUT2 +/+ and VGLUT2 +/− mice, statistical comparisons were done with ANOVA. When appropriate, pairwise multiple comparison procedures were performed using the Holm–Sidak method. In all cases, significant levels were set at p < 0.05. Correlations between variables were assessed using Pearson's product-moment correlation coefficient.
Primary neuronal cell culture and electrophysiology
Standard procedures were taken to culture both thalamic and hippocampal neurons from E18.5 mouse embryos. Both cultures were plated on astrocytes derived from neonatal cortex tissue. Neurons were plated on island cultures at a density of 2000–3000 neurons per 35 mm dish. Recordings were performed from 14- to 18-d-old neurons. Standard extracellular solution contained the following (in mm): 140 NaCl, 2.4 KCl, 10 HEPES, 10 glucose, 4 MgCl2, and 4 CaCl2, pH 7.3 (305 mOsm). Internal solution contained the following: 135 mm KCl, 18 mm HEPES, 1 mm EGTA, 4.6 mm MgCl2, 4 mm ATP, 0.3 mm GTP, 15 mm creatine phosphate, and 20 U/ml phosphocreatine kinase. Patch pipettes were fabricated from thick-walled borosilicate glass and had resistances of 2.5–3.5 MΩ. Series resistance was compensated by 75% and was usually below 12 MΩ before compensation. The readily releasable vesicle pool (RRP) was estimated using published procedures (Rosenmund and Stevens, 1996).
Miniature EPSC analysis
mEPSCs were detected for 60–80 s with 3 mm kynurenic acid applied for 2 s of every 10 s for background noise subtraction. For each cell, data were filtered at 1 kHz and analyzed using template-based mEPSC detection algorithms implemented in analysis software Axograph 4.9 (Molecular Devices, Union City, CA). Threshold for detection was set at three times the baseline SD from a template of 0.5 ms rise time and 3 ms decay. mEPSCs were captured from the time during the trace without kynurenic acid, and those outside allowable ranges for amplitude, rise time, and half-width were rejected. The remaining number of events were counted and averaged to determine frequency and mean amplitude. The same procedure was then repeated for the time during the trace when kynurenic acid was applied. The frequency and amplitude of these captured events was considered the rate and amplitudes of false positives. It was subtracted from the original frequency, and the average amplitude was adjusted accordingly.
Results
Generation of the VGLUT2 knock-out mice
In mouse, the VGLUT2 gene contains 12 exons. We performed homologous recombination resulting in deletion of exon 2 and disruption of the open reading frame (Fig. 1 A,B). Correct targeting in ES cells was confirmed by Southern blot analysis (results not shown), and loss of the wild-type VGLUT2 allele was confirmed by PCR analysis. Expression of the VGLUT2 transcript was strongly reduced in VGLUT2 −/− embryos, whereas intermediate levels were obtained in the VGLUT2 +/− embryos (Fig. 1 C). A residual transcript was still present in the VGLUT2 −/− embryo (Fig. 1 C), but no VGLUT2 protein was detectable in VGLUT2 −/− animals (Fig. 1 D,E). The VGLUT2 protein level in brain from VGLUT2 +/− animals was found to be reduced by 54 ± 5% (mean ± SEM) compared with VGLUT2 +/+ animals (Fig. 1 E), suggesting gene dosage dependency of VGLUT2 expression. To look for potential compensatory mechanisms, VGLUT1 and VGLUT3 mRNA levels were analyzed in brain, spinal cord, and DRG of adult mice, but no changes in expression level could be detected between VGLUT2 +/+ and VGLUT2 −/− mice (data not shown). Western analysis showed that VGLUT1 protein levels were not affected in VGLUT2−/− mice (Fig. 1 F).
Figure 1.
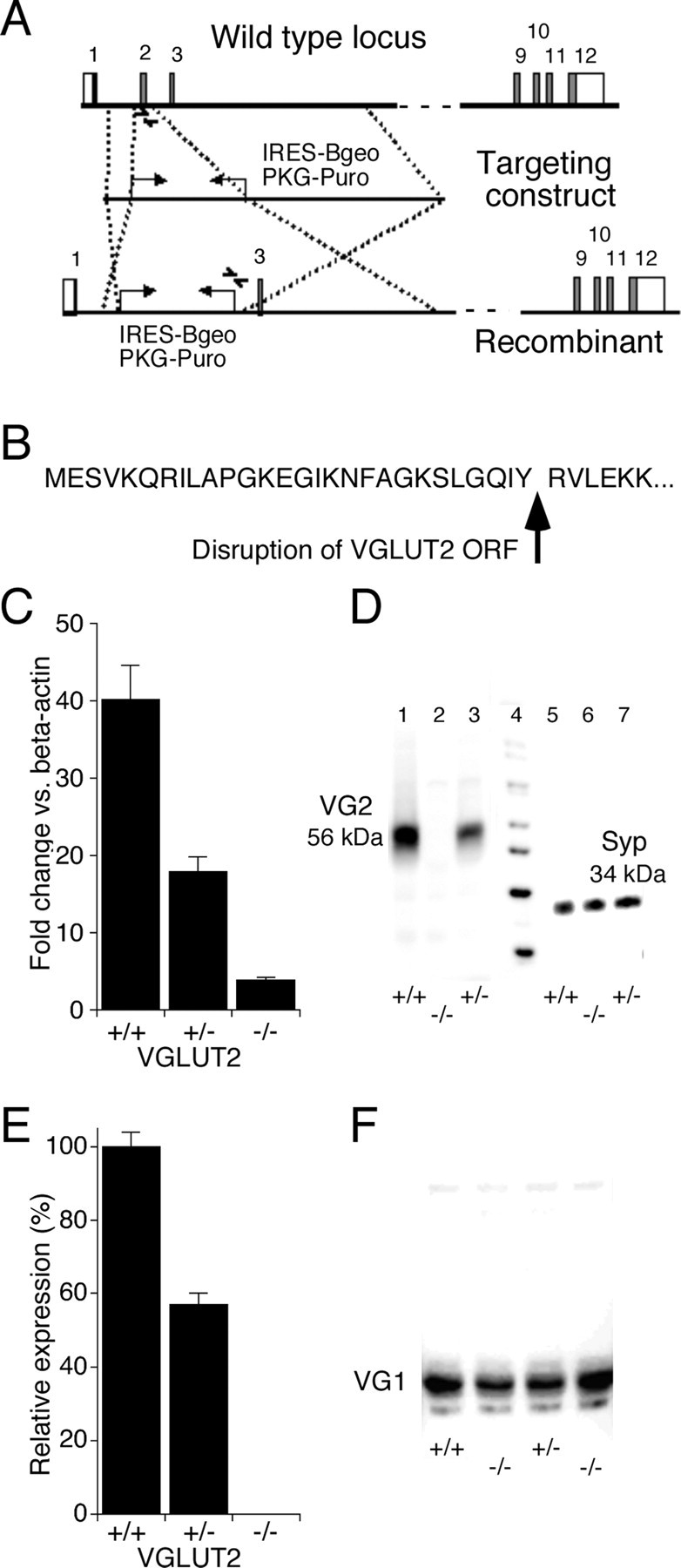
Targeted disruption of the VGLUT2 gene. A, Structure of the wild-type locus, targeting vector, and recombinant locus. Boxes represent the known exons; in white and gray are indicated the noncoding and coding regions. A lambda pKOS-based targeting construct was generated by replacing exon 2 by the IRESBgeo/PGK-Puro selection cassette, disrupting the open reading frame (ORF; site of interruption shown in B). Arrows indicate positions of the PCR primers used for genotyping the wild-type and targeted allele. C, Expression of the VGLUT2 transcript in brain derived from 18.5-d-old embryos was strongly reduced in the VGLUT2 −/− mouse as determined by quantitative RT-PCR. Values expressed are mean ± SD (n = 3) relative expression levels after normalization to β-actin. D, Expression of the VGLUT2 protein (56 kDa) in brain derived from 18.5-d-old VGLUT2 +/+ embryos (lane 1) was absent in the VGLUT2 −/− embryos, whereas intermediate levels were observed in VGLUT2 +/− embryos (lane 2) as determined by Western blot analysis. Detection of synaptophysin (Syp; 34 kDa) was used as a positive control for the presence of synaptic vesicles in the vesicular preparation. E, Expression of the VGLUT2 protein in brain derived from 18.5-d-old embryos was absent in the VGLUT2 −/− mouse as determined by Western blot analysis. Values expressed are mean ± SD (n = 6). F, Western blot analysis of VGLUT1 in VGLUT2 +/+ and VGLUT2 −/− brains showed no difference in expression levels between genotypes.
Lethality of VGLUT2 −/− newborns
VGLUT2-deficient animals were born at the expected Mendelian frequency, but VGLUT2 −/− mice died at or immediately after birth. E18.5 embryos, derived from VGLUT2 +/− × VGLUT2 +/− breeding, were collected to investigate whether the lack of VGLUT2 protein caused any developmental abnormalities. After cesarean delivery, mutant embryos did not react to forceps stimuli, remained in a fetal position, and did not exhibit a breathing reflex. They became rapidly cyanotic and died a few minutes after delivery as judged from cessation of heartbeats. Full necropsy and histological examination did not reveal any apparent developmental abnormalities. Serial histological sections through the brain did not show differences in cytoarchitecture (supplemental data, available at www.jneurosci.org as supplemental material). In the spinal cord region, histology of the motor neurons and dorsal root ganglia along the VGLUT2 −/− spinal cords was normal (supplemental data, available at www.jneurosci.org as supplemental material).
Although the complete loss of VGLUT2 is lethal, VGLUT2 +/− mice were not obviously different from their wild-type littermates. However, the loss of 50% of VGLUT2 protein may affect excitatory drive, at least in some parts of the brain in which VGLUT2 is present, because the level of expression of vesicular transporters has been shown to affect the postsynaptic response to single-vesicle fusion (Wojcik et al., 2004; Wilson et al., 2005). Because a gene dosage effect for the vesicular monoamine transporter has not only been linked to changes in quantal responses but also to behavioral phenotypes (Takahashi et al., 1997), we pursued a series of behavioral tests on VGLUT2 +/− mice and compared them with their wild-type littermates.
Behavioral analysis of the VGLUT2 +/− mice
VGLUT2 +/+ and VGLUT2 +/− mice were tested in measurements of spontaneous activity in the home cage, open field test, rotarod, elevated plus maze, social interaction, passive avoidance learning, Morris water maze, and context-dependent fear conditioning. None of these tests showed significant differences between the genotypes, indicating that the VGLUT2 +/− mice have normal neuromotor performance, innate fear-related behavior, and learning and memory performance (supplemental data, available at www.jneurosci.org as supplemental material). In the water maze test (Fig. 2 A), genotype had no effect on ability to improve performance as a result of training or escape latency (F (1,56) > 1; p = 0.3), and VGLUT2 +/− mice reached the escape platform even faster than VGLUT2 +/+ mice during trial block 2 (p = 0.04). Also, both genotypes showed similar cue- and context-dependent fear response learning (Fig. 2 B). Both groups responded with increasing freezing after two light footshocks (F (1,22) = 23.76; p < 0.001), and both showed robust fear response to the tone–shock pairing, with comparable freezing scores. Freezing score in both genotypes was significantly reduced when the context was changed (F (1,22) = 37.24; p < 0.001), but tone exposure still elicited a significant response in both genotypes (F (1,22) = 58.299; p < 0.001).
Figure 2.
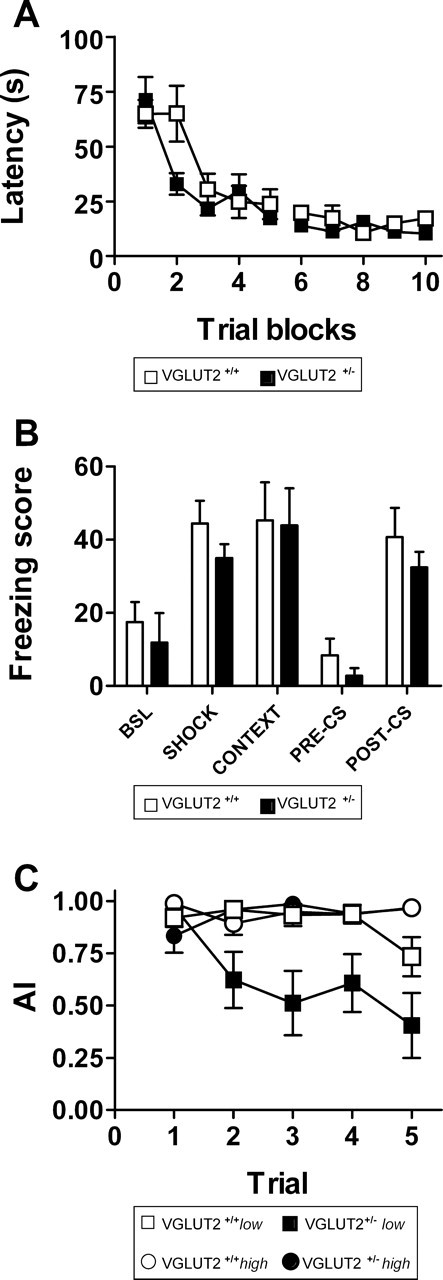
Behavioral performance of male VGLUT2 +/− mice and VGLUT2 +/+ littermates (n = 12 in each group) in water maze learning, cue- and context-dependent fear conditioning, and conditioned taste aversion. A, Acquisition training of the water maze task shows comparable learning performance in both groups. Mean escape latency during the 10 trial blocks does not differ significantly between VGLUT2 +/+ (open symbols) and VGLUT2 +/− (filled symbols) mice. B, Cue- and context-dependent fear conditioning also showed no differences between VGLUT2 +/− (filled bars) and VGLUT2 +/+ (open bars) littermates during different fear conditioning trials. C, Conditioned taste aversion during five extinction trials revealed a difference between VGLUT2 +/+ (open symbols) and VGLUT2 +/− (filled symbols) mice when the group was split into high (circles) and low (squares) responders.WT, VGLUT2 +/+ mice; HZ, VGLUT2 +/− mice. Data are presented as mean ± SEM.
Acute nociception is normal in VGLUT2 +/− mice
To test for sensory performance, we used acute nociceptive perception tests, including the hot plate test, tail withdrawal, and Hargreaves' test (Table 1), and did not observe any differences between the genotypes. In the hot plate test, licking or jumping responses are considered to be the result of supra-spinal sensory integration. Higher temperatures led to lower response latencies in both genders (temperature effect, F (3,92) = 110.3, p < 0.001; F (3,92) = 137.0, p < 0.001 in males and females, respectively), and the response latency was not different between genotypes in females (genotype effect, F (1,94) = 0.1; p = NS) at any temperature (Table 1). In males, a significant difference between genotypes was observed (genotype effect, F (1,94) = 5.7; p = 0.019), but post hoc analysis attributed this entirely to a small difference at the lowest temperature only (Table 1). Similar results were obtained in the tail withdrawal test, in which the response is considered to be a spinal reflex. Again a clear temperature effect was observed in both genders (temperature effect, F (3,92) = 16.3, p < 0.001; F (3,92) = 115.8, p < 0.001 in males and females, respectively), and the response latency was not different between genotypes (genotype effect, F (1,94) = 0.01, p = NS; F (1,94) = 0.01, p = NS in males and females, respectively) across the range of temperatures (Table 1). Applying a radiant heat stimulus to the subplantar side of the hindpaw using Hargreaves' test also showed no significant differences in withdrawal latencies between the two genotypes (genotype effect, F (1,46) = 0.8, p = NS; F (1,46) = 0.5, p = NS in males and females, respectively), on either the left or the right side (Table 1). The lack of significant effects in acute pain tests indicate that acute nociception is intact, on the peripheral, spinal, and supra-spinal level.
Table 1.
Nociceptive responses in acute tests
| Thermal nociception |
||||
|---|---|---|---|---|
| Males | Females | |||
| Hot plate | VGLUT2 +/+ | VGLUT2 +/− | VGLUT2 +/+ | VGLUT2 +/− |
| 50°C | 20.7 ± 1.0 | 23.3 ± 1.3* | 24.8 ± 1.3 | 24.3 ± 1.2 |
| 52°C | 12.7 ± 1.0 | 15.0 ± 1.1 | 16.1 ± 1.1 | 15.5 ± 1.3 |
| 54°C | 8.7 ± 0.8 | 10.0 ± 0.7 | 9.7 ± 0.5 | 10.3 ± 1.0 |
| 56°C | 6.5 ± 0.5 | 6.4 ± 0.5 | 6.3 ± 0.4 | 5.9 ± 0.4 |
| Males | Females | |||
| Tail withdrawal | VGLUT2 +/+ | VGLUT2 +/− | VGLUT2 +/+ | VGLUT2 +/− |
| 47°C | 16.4 ± 1.4 | 15.9 ± 1.7 | 17.2 ± 1.5 | 17.0 ± 1.5 |
| 49°C | 8.0 ± 2.3 | 7.1 ± 1.7 | 6.4 ± 0.5 | 6.4 ± 0.6 |
| 51°C | 6.4 ± 2.5 | 5.7 ± 2.1 | 4.4 ± 0.4 | 4.5 ± 0.4 |
| 53°C | 2.6 ± 0.4 | 5.1 ± 2.1 | 2.8 ± 0.2 | 3.1 ± 0.3 |
| Males | Females | |||
| Hargreaves' test | VGLUT2 +/+ | VGLUT2 +/− | VGLUT2 +/+ | VGLUT2 +/− |
| Left | 8.0 ± 0.9 | 7.6 ± 0.5 | 6.6 ± 0.4 | 6.9 ± 0.6 |
| Right | 7.7 ± 1.0 | 6.7 ± 0.6 | 6.7 ± 0.4 | 7.1 ± 0.8 |
Response latencies (seconds) in the hot plate test, the tail withdrawal test, and the Hargreaves' test. Data are presented as mean ± SEM; n = 12. No significant difference were obtained between VGLUT2 +/+ and VGLUT2 +/− mice in the Hargreaves' test or between VGLUT2 +/+ and VGLUT2 +/− mice within a temperature group in the tail withdrawal test and the hot plate test apart from the males at 50°C;
*p < 0.05.
Inflammatory and neuropathic pain models in VGLUT2 +/− mice
We next examined whether the absence of VGLUT2 affected the behavioral responses of mice in inflammatory and neuropathic pain models. Inflammatory pain and neuropathic pain involve an activity-dependent facilitation in excitability of both peripheral neurons (peripheral sensitization) and spinal and supra-spinal neurons (central sensitization), including different brain regions such as the thalamus and cortex (Bridges et al., 2001; Kidd and Urban, 2001). To examine the contribution of VGLUT2 to inflammatory pain signaling, we used two models: peripheral injection of formalin and peripheral injection of carrageenan. Formalin was injected into the plantar side of the hindpaw. Mice responded by flinching of the injected paw, and this behavior was typically concentrated in two distinct phases (Fig. 3 A), with peak flinch rates during phases I and II being in the order of 60 and 40 flinches/min (phase effect, F (3,88) = 92.0; p < 0.001). However, no difference in behavioral response was observed between VGLUT2 +/+ and VGLUT2 +/− mice (F (1,90) = 0.007; p = NS) (Fig. 3 B). VGLUT2 +/− and VGLUT2 +/+ littermates were also compared in a second test of inflammatory pain, the carrageenan model (Fig. 3 C). Intraplantar injection of carrageenan induced a significant decrease of paw withdrawal latencies in the VGLUT2 +/+ mice (treatment effect, F (1,70) = 15.7, p < 0.001; F (1,70) = 59.2, p < 0.001 for males and females, respectively), as well as in the VGLUT2 +/− mice (treatment effect, F (1,70) = 11.936, p = 0.001; F (1,70) = 20.3, p < 0.001 for males and females, respectively), demonstrating the development of hyperalgesia as a consequence of sensitization in both genotypes.
Figure 3.
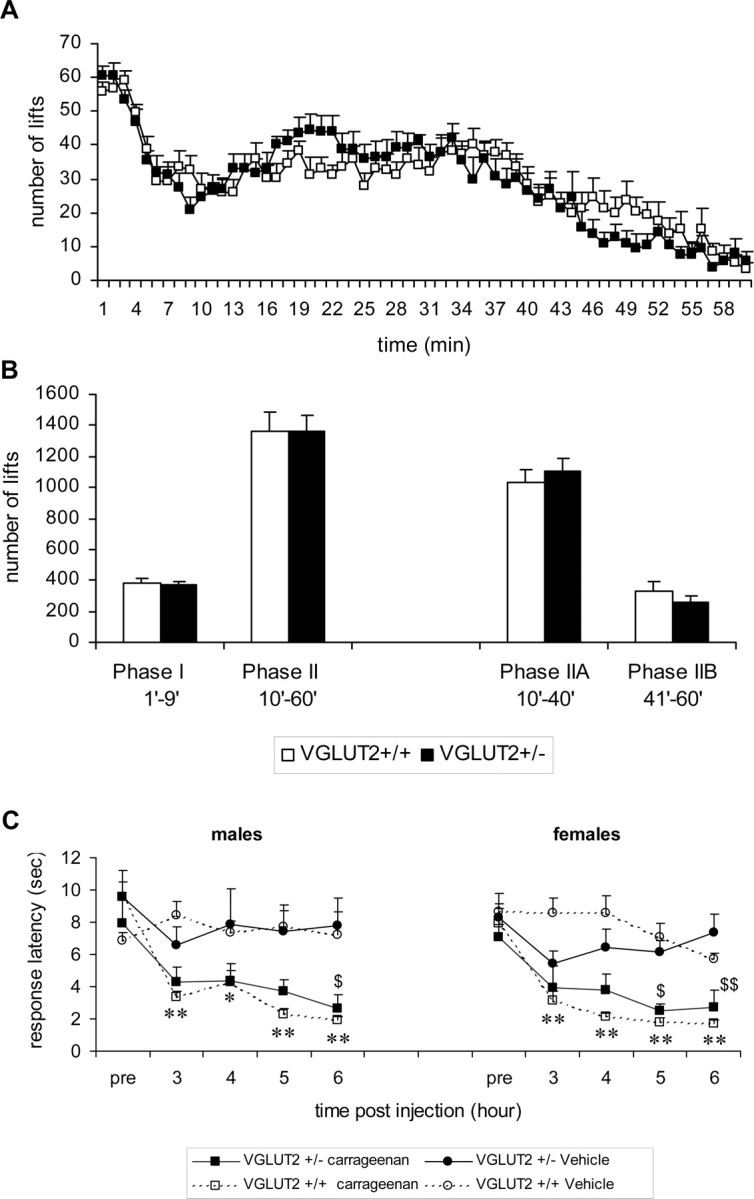
Pain response in inflammation models. Effect of formalin (1%) subplantar injection on the flinching behavior of VGLUT2 +/+ and VGLUT2 +/− male littermates. A, Frequency of flinching per minute is presented over a 60 min period. B, Mean number of flinches in each phase of the automated formalin test. No significant differences between genotypes were found. Data are presented as mean ± SEM; n VGLUT+/− = 11; n VGLUT+/+ = 12. C, Effect of carrageenan (2%) injection on thermal sensitivity in VGLUT2 +/+ and VGLUT2 +/− littermates (Hargreaves' test). Paw withdrawal latencies are presented for the carrageenan/vehicle-injected paw. For VGLUT2 +/+ animals, differences between carrageenan- (open squares) and vehicle- (open circles) injected mice were statistically significant on all time points after injection for both sexes. For VGLUT2 +/− mice, a significant effect of treatment was observed, and post hoc analysis (Holm–Sidak method) showed significant differences at one (males) or two (females) time points after injection. Data are presented as mean ± SEM; n = 6. $ p < 0.05, $$ p < 0.001 within VGLUT2 +/− groups; *p < 0.05, **p < 0.001 within VGLUT2 +/+ groups.
To evaluate neuropathic pain, we evaluated the behavioral responses of mice in the spared nerve injury (SNI) model. In SNI, two of the three terminal branches of the sciatic nerve are sectioned. Two tests were performed to measure the degree of neuropathic pain: mechanical allodynia (von Frey test) and cold allodynia (acetone test). Responses to mechanical and cold stimuli were measured at the plantar surface of the foot before and 2, 6, 9, 14, and 21 d after SNI surgery. Before surgery, no significant difference in mechanical threshold was observed between VGLUT2 +/+ and VGLUT2 +/− mice (7.66 ± 0.25 and 8.07 ± 0.30 g, respectively, F (1,46) = 1.03, p = NS in males; 6.48 ± 0.27 and 6.33 ± 0.24 g, respectively, F (1,46) = 0.43, p = NS in females) (Fig. 4 A). In the VGLUT2 +/+ mice, an SNI operation induced a significant 30–40% decrease in the mechanical threshold for ipsilateral hindpaw withdrawal that was apparent at the first test day (day 2 after injury) and lasted for >21 d after injury (treatment effect, F (1,142) = 69.3, p < 0.001; F (1,142) = 123.5, p < 0.001 in males and females, respectively). However, mechanical allodynia was completely absent in the male VGLUT2 +/− SNI-operated mice (treatment effect, F (1,142) = 3.4; p = NS). A weak allodynia was observed in the female VGLUT2 +/− SNI-operated mice (treatment effect, F (1,142) = 15.4; p < 0.01), with a significant difference in threshold between SNI- and sham-operated female VGLUT2 +/− mice only at days 9 and 21 after injury (Fig. 4 A). As a control, the mechanical thresholds of the right (contralateral) non-operated hindpaw of all groups did not show a decrease after surgery (data not shown).
Figure 4.

Pain response in neuropathic pain model. A, Effect of the SNI on mechanical thresholds in VGLUT2 +/− and VGLUT2 +/+ littermates (von Frey test). The mean force required to evoke a withdrawal response is presented for the ipsilateral hindpaw. For the VGLUT2 +/+ animals, differences between SNI-operated (open squares) and sham-operated (open circles) mice were statistically significant on all days for both sexes. No such significant differences were found between the VGLUT2 +/− male groups (filled symbols), whereas for VGLUT2 +/− females, a significant difference in mechanical allodynia was only found 9 and 21 d after surgery. Data are presented as mean ± SEM; n = 12. $ p < 0.05 within VGLUT2 +/− groups; *p < 0.05, **p < 0.001 within VGLUT2 +/+ groups. B, Effect of the spared nerve injury on the acetone test in both genotypes. The average ± SEM response time (in seconds) of the ipsilateral hindpaw during a 1 min observation period is plotted against the time. Similar to the results of mechanical allodynia, we observed highly significant differences in cold allodynia within the VGLUT2 +/+ groups (*p < 0.05; **p < 0.001) but not in VGLUT2 +/− mice (only significant difference at day 2 in females; $ p < 0.05). Pre, Preoperative control value. Data are presented as mean ± SEM; n = 12.
Cold allodynia tests showed no significant difference in reaction time to acetone between VGLUT2 +/+ and VGLUT2 +/− mice before the operation (1.31 ± 0.29 and 1.01 ± 0.27 s, respectively, F (1,46) = 0.59, p = NS in males; 1.68 ± 0.39 and 1.86 ± 0.38 s, respectively, F (1,46) = 0.34, p = NS in females) (Fig. 4 B). In the VGLUT2 +/+ mice, the SNI operation induced a hypersensitivity of the operated hindpaw, as measured by the increased response time of paw withdrawal after the acetone spray. This effect was seen already on the first test day (day 2 after injury) and lasted for >21 d after injury (treatment effect, F (1,142) = 68.2, p < 0.001; F (1,142) = 29.0, p < 0.001 in males and females, respectively). In contrast to VGLUT2 +/+ mice, hypersensitivity was completely absent in the female VGLUT2 +/− SNI-operated mice (treatment effect, F (1,142) = 3.2; p = NS), and only a relatively weak increased sensitivity was observed in the male VGLUT2 +/− SNI-operated mice (treatment effect, F (1,142) = 10.0; p < 0.01), with no significant difference in response time between SNI and sham-operated male VGLUT2 +/− mice observed at any specific day after injury (Fig. 4 B).
Abnormal marble burying and conditioned taste aversion in VGLUT2 +/− mice
Both the marble burying test, a measure of defensive behavior, and the conditioned taste aversion test showed significant differences between VGLUT2 +/+ and VGLUT2 +/− mice. In the defensive marble burying test, VGLUT2 +/− mice buried significantly fewer marbles than their VGLUT2 +/+ littermates (7.5 ± 0.92 and 13 ± 1.82 for VGLUT2 +/− and VGLUT2 +/+, respectively; F (1,22) = 7.273; p = 0.013). Genotype effects were also observed in the conditioned taste aversion test (Fig. 2 C). After intraperitoneal injection of lithium, both VGLUT2 +/− mice and VGLUT2 +/+ littermates displayed a clear aversion toward saccharin-flavored water. Repeated exposure to saccharin solutions gradually extinguished the initial taste aversion with marginally more pronounced extinction in the VGLUT2 +/− group (F (1,22) = 4.128; p = 0.054). When the groups were split in high (AI >0.9) and low (AI <0.9) responders, much more pronounced extinction was observed in the low-responder VGLUT2 +/− (Fig. 2 C). In the VGLUT2 +/+ group, this subdivision resulted in four low and eight high responders that only showed a difference on extinction trial 5. The VGLUT2 +/− group was divided into five low and seven high responders, with a significant effect of group (F (1,40) = 16.123; p = 0.002) and trial × group interaction (F (1,40) = 6.952; p < 0.001). Notably, marble burying and taste aversion (on trial 5) were significantly correlated in the VGLUT2 +/− mice (r = 0.615; p < 0.05).
Given these discrete effects of the loss of one allele of VGLUT2 on behavior, we wanted to provide physiological evidence for the role of VGLUT2 in central excitatory synaptic transmission and also try to identify a functional correlate for the phenotype observed in the heterozygous mice.
Synaptic transmission in VGLUT2 −/− and VGLUT2 +/− mice
To study synaptic transmission, we relied on autaptic cultured neurons obtained from VGLUT2 +/+, VGLUT2 +/−, and VGLUT2 −/− mice, because this system is ideally suited to quantify changes in basic synaptic properties. We made autaptic cultures of both hippocampal and thalamic neurons, because the other major VGLUT isoform, VGLUT1, is predominantly expressed in neurons of the forebrain and hippocampus and shows weak expression in thalamus, whereas VGLUT2 is strongly expressed in thalamus and weakly expressed in hippocampus (Hisano et al., 2000; Fremeau et al., 2001; Herzog et al., 2001). Autaptic cultures of thalamus have, to our knowledge, not been examined previously. In our thalamic cultures, ∼60–70% of the neurons were excitatory and 30–40% inhibitory. Both cell types were distinguished by the time course of the synaptic current in response to a brief depolarization and by the sensitivity of this current to the AMPA receptor antagonist kynurenic acid (3 mm). The mean EPSC peak amplitude in VGLUT2 +/+ and VGLUT2 +/− thalamic neurons was 2.44 ± 0.33 nA (n = 37) and 2.41 ± 0.32 nA (n = 32), respectively (Fig. 5 A). In contrast, EPSC amplitudes from VGLUT2 −/− neurons averaged only 91 ± 16 pA (n = 33; p < 0.001), which corresponds to >95% reduction in synaptic response. An additional 21% of non-GABAergic VGLUT2 −/− neurons showed no evoked response at all, although this was never observed in the VGLUT2 +/+ or VGLUT2 +/− cultures. To examine whether a change in glutamate receptors was responsible for the observed decrease in the evoked response, we applied 10 μm kainate, a glutamate receptor agonist that elicits a nondesensitizing response from AMPA receptors, and measured the steady-state current. Kainate responses were slightly larger in VGLUT2 −/− (890 ± 115 pA; n = 17) and VGLUT2 +/− (802 ± 77 pA; n = 27) neurons than in wild type (782 ± 114 pA; n = 24), but the difference was not significant.
Figure 5.
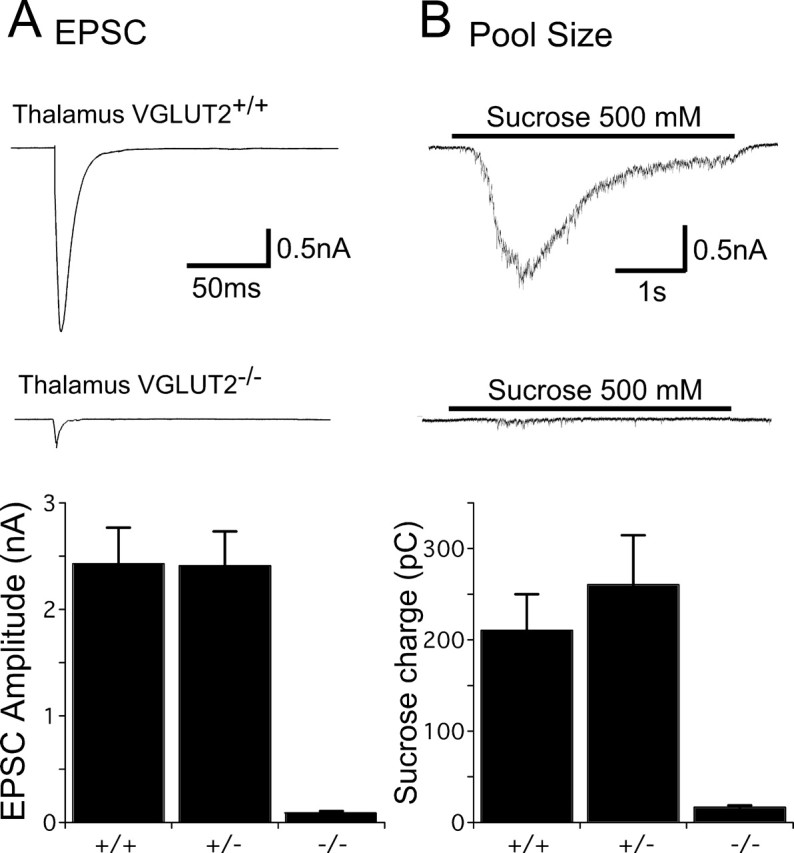
EPSC amplitudes and vesicle pool sizes in thalamic neurons. A, Top, Representative traces showing EPSCs in response to 2 ms depolarization to 0 mV in VGLUT2 +/+ and VGLUT2 −/− neurons. The somatic current is blanked for display. Bottom, Bar graph showing amplitudes (mean ± SEM) of EPSCs from VGLUT2 +/+, VGLUT2 +/−, and VGLUT2 −/− thalamic neurons. B, Top, Representative traces showing responses of VGLUT2 +/+ and VGLUT2 −/− thalamic neurons to 4 s hypertonic sucrose. Bottom, Bar graph showing the charge transferred during sucrose application in VGLUT2 +/+, VGLUT2 +/−, and VGLUT2 −/− thalamic neurons (mean ± SEM).
To identify what caused the severe reduction of the evoked response in the VGLUT2 −/− neurons, we first determined the RRP size of VGLUT2 +/+, VGLUT2 +/−, and VGLUT2 −/− neurons by applying hypertonic sucrose (Rosenmund and Stevens, 1996). Whereas VGLUT2 +/+ and VGLUT2 +/− neurons had similar RRP charges of 211 ± 39 pC (n = 37) and 260 ± 55 pC (n = 32), the charge in the VGLUT2 −/− neurons was reduced by >90% to 16.1 ± 3.5 pC (n = 33; p < 0.001) (Fig. 5 B). This suggests that the reduction of the evoked response in the VGLUT2 −/− neurons is mainly attributable to a reduction of release-ready, glutamate-containing vesicles.
In the mature hippocampus, glutamatergic transmission is almost entirely dependent on VGLUT1 (Fremeau et al., 2004b; Wojcik et al., 2004), but VGLUT2 is expressed transiently during development and at low levels in adulthood (Fremeau et al., 2001, 2004b; Herzog et al., 2001; Boulland et al., 2004). In contrast to thalamic neurons, however, 15–18 d in vitro hippocampal neurons showed no significant effect from the removal of VGLUT2 (Fig. 6). Evoked EPSC amplitudes (VGLUT2 +/+, 10.4 ± 1.9 nA, n = 16; VGLUT2 −/−, 9.6 ± 2.0 nA, n = 20) and the RRP size (VGLUT2 +/+, 1115 ± 128 pC, n = 16; VGLUT2 −/−, 1325 ± 1314 pC, n = 20) were similar in both genotypes.
Figure 6.

EPSC amplitudes and readily releasable vesicle pool size in hippocampal neurons. A, Representative traces showing EPSCs in response to 2 ms depolarization to 0 mV in VGLUT2 +/+ and VGLUT2 −/− hippocampal neurons. The somatic current is blanked for display. B, Representative traces showing responses of VGLUT2 +/+ and VGLUT2 −/− hippocampal neurons to 4 s hypertonic sucrose.
Spontaneous release in VGLUT2 −/− and VGLUT2 +/− mice
Although the analysis of evoked responses and the readily releasable pool revealed a clear phenotype in the VGLUT2 −/− thalamic neurons, the neurons from heterozygous mice showed no significant effects. This could be attributable to the intrinsically large variability of the quantity of synapses formed in culture or to a possible compensatory effect. A reduction in the number of VGLUTs may, however, also lead to a reduced content of glutamate in the vesicle and consequently to a reduced mEPSC amplitude (Wojcik et al., 2004), and this property will be insensitive to synapse number variability. We therefore performed an analysis of spontaneous release activity (Fig. 7). In thalamic VGLUT2 −/− neurons, mEPSC frequency was reduced by almost 90% (VGLUT2 +/+, 6.7 ± 1.2 Hz, n = 34; VGLUT2 −/−, 0.82 ± 0.2 Hz, n = 29; p < 0.001), a finding consistent with the reduced RRP size (Fig. 5 B). The mEPSC frequency in VGLUT2 +/− neurons was 5.6 ± 0.8 Hz, which was slightly lower but not significantly different from VGLUT2 +/+ neurons. The mean amplitude of mEPSCs, however, was significantly different in all three groups (Fig. 7 B). VGLUT2 +/+ neurons had the largest mEPSC amplitudes (20.9 ± 1.5 pA; n = 34), whereas the amplitude of VGLUT2 −/− mEPSCs was just more than half of that value (11.3 ± 1.0 pA; n = 29; p < 0.001). mEPSC amplitude measured from VGLUT2 +/− neurons was between the wild-type and knock-out value at 15.9 ± 0.9 pA (n = 34; p < 0.01 vs VGLUT2 +/+; p < 0.05 vs VGLUT2 −/−), suggesting that total glutamate content per vesicle is extremely sensitive to VGLUT protein levels. Hippocampal neurons in contrast showed no significant change in spontaneous release rate or mEPSC amplitude. Thus, the physiological analysis not only demonstrates the absolute importance of VGLUT2 for thalamic function, it also provided evidence for a significant reduction in quantal size when VGLUT2 levels are reduced to 50%.
Figure 7.

Miniature EPSC frequency and amplitude. A, Bar diagram showing frequency (mean ± SEM) of mEPSCs recorded from thalamic VGLUT2 +/+ (n = 34), VGLUT2 +/− (n = 34), and VGLUT2 −/− (n = 29) and hippocampal VGLUT2 +/+ (n = 16) and VGLUT2 −/− (n = 18) neurons. B, Cumulative distribution of mean mEPSC amplitudes from VGLUT2 +/+, VGLUT2 +/−, and VGLUT2 −/− thalamic neurons. C, Representative traces of mEPSC activity. Black bar indicates application of kynurenic acid (3 mm). Bottom trace is a magnification of a trace from a VGLUT2 −/− thalamic neuron. HC, Hippocampus; Thal, thalamus; Kyn, kynurenic acid.
Discussion
Findings from the VGLUT2 −/− mouse now confirm hypotheses suggested by the localization of VGLUT mRNA and electrophysiological results from the VGLUT1 −/− mouse but also demonstrate an essential role for expression levels of vesicular glutamate transporters in regulating the strength of individual vesicle release events (Hisano et al., 2000; Fremeau et al., 2001, 2004b; Herzog et al., 2001; Wojcik et al., 2004). In addition, we demonstrate this subtle reduction in excitatory drive is correlated with changes in behavioral responses to neuropathic lesions and defensive behavior.
VGLUT2 −/− mice die at birth, whereas VGLUT1 −/− mice survive for ∼3 weeks, a contrast that demonstrates the importance of VGLUT2 in early development and its predominance in the brainstem and pons, in which the neurons controlling the basic functions of respiration and the autonomic nervous system reside. The collapsed lung alveoli of the VGLUT2-deficient embryos indicate that they are dying as a result of respiratory failure. Because many neurons of the respiratory network contain VGLUT2 mRNA, such as the neurokinin-1-expressing cells of the medullary ventral respiratory group that are required for normal respiratory rhythmogenesis (Stornetta et al., 2003a, b; Weston et al., 2004), it is likely that the loss of VGLUT2 in these neurons disrupts glutamate release in a similar manner to the thalamic neurons we tested and underlies the respiratory failure.
Our results also functionally demonstrate what is suggested by the localization of the two isoforms, that VGLUT2 is necessary for glutamatergic transmission in the diencephalon but not the telencephalon. Together with previous studies, we can now state that the presence or absence of VGLUT1 and/or VGLUT2 determines whether a neuron releases glutamate or not at the majority of CNS synapses (Bellocchio et al., 2000; Takamori et al., 2000, 2001; Fremeau et al., 2004b; Wojcik et al., 2004). Whether VGLUT isoforms also regulate how a neuron releases glutamate remains controversial. Comparing results from autaptic VGLUT2 −/− neurons from the thalamus with those from hippocampal VGLUT1 −/− neurons shows a similar physiological phenotype, indicating that the differences between the two isoforms may be limited to where and when they are expressed. Both isoforms play an essential role in vesicle filling, because their removal leads to a dramatic reduction in evoked responses, pools of glutamate containing and readily releasing vesicles, and spontaneous release. The sparse release activity that remains in each case is most likely mediated by sporadic expression of the other isoform.
A 50% reduction in VGLUT2 protein levels resulted in a surprisingly specific abolishment of neuropathic pain in the spared nerve injury model, a previously described robust model with substantial and prolonged changes in mechanical sensitivity and thermal responsiveness in both rat and mouse (Decosterd and Woolf, 2000; Shields et al., 2003). In contrast, several general behavioral tests, as well as behavior associated with sensory processing, acute nociception, or inflammatory pain, were not affected. Where does this specificity in behavior come from?
Changes in glutamatergic signaling at the peripheral, spinal, and supra-spinal levels have been reported to impact neuropathic pain (Chiechio et al., 2004; Zieglgansberger et al., 2005) Although our experiments do not address the likely involvement of peripheral and spinal circuitry in neuropathic pain, our data provide strong support of the involvement of VGLUT2-dependent thalamic signaling in neuropathic pain. First, the finding that acute nociception is normal indicates that baseline glutamatergic signaling in nociceptive pathways at the peripheral, spinal, and supra-spinal levels is unchanged in the VGLUT2 +/− mice. Second, inflammatory pain and, more specifically, the behavioral response in the second phase of the formalin test, which involves glutamate-mediated sensitization at the level of the periphery and the spinal cord (Tjolsen et al., 1992; Taylor et al., 1995; Wei et al., 2001), is normal in the VGLUT2 +/−. This suggests that VGLUT2 reduction in these areas is not sufficient to impair mechanisms of pain sensitization. A simple explanation is that, on the peripheral and spinal level, a partial loss of VGLUT2 is compensated by VGLUT1 function, because VGLUT1 is abundantly expressed in most parts of the spinal cord (Li et al., 2003; Alvarez et al., 2004; Landry et al., 2004) Third, forebrain synapses use VGLUT1 as their main vesicular glutamate transporter and are unlikely to be affected by reduction of VGLUT2 content (Fremeau et al., 2001, 2004b; Herzog et al., 2001; Wojcik et al., 2004) (Fig. 6).
VGLUT2 has its most exclusive expression patterns in brainstem and the thalamus. Among those regions, the importance of the thalamus in the development and maintenance of neuropathic pain is well documented. The neuropathic pain model has been shown to lead to a functional increase in the strength of lateral connections in the somatosensory thalamus (Bruggemann et al., 2001). Studies in rat neuropathic pain models, as well as physiological studies in humans with chronic pain, have demonstrated abnormal discharge patterns in the thalamus (Guilbaud et al., 1990; Lenz et al., 1994, 1995). In addition, NMDA receptor antagonists have been shown to reduce nociceptive-induced activity in the thalamus, whereas NMDA antagonism and modulation of metabotropic glutamate receptor signaling at the level of the thalamus block the development of hyperalgesia in both rat and primate neuropathic pain models (Dougherty et al., 1996; Kolhekar et al., 1997; Bordi and Quartaroli, 2000; Neugebauer, 2002; Chiechio et al., 2004).
Additional behavioral data suggest that partial loss of VGLUT2 function affects thalamic processing. The faster conditioned taste aversion extinction in these mice mimics results from rats in which the thalamus was chemically lesioned (Yamamoto et al., 1995). This does not reflect a mere anxiolytic effect of VGLUT2 deficiency, because VGLUT2 +/− mice did display reduced marble burying, but the prototypical anxiety test, the elevated plus maze, was unaltered in these animals. De Boer and Koolhaas (2003) demonstrated that marble burying and elevated plus maze performance are in fact not correlated and suggest that these tests assess active and passive emotional coping strategies, respectively, that are known to depend on different neuroanatomical circuits. Older works has shown that such active, defensive behaviors are indeed impaired in rats with thalamic lesions (Alexinsky et al., 1971; Kanki and Adams, 1978), and more recently, Bhatnagar et al. (2003) described distinct changes in defensive burying in rats with paraventricular thalamic lesions. However, defensive behaviors seem to depend especially on midline thalamic nuclei that relay information between prefrontal cortex and limbic structures (Phelps and LeDoux, 2005; Vertes, 2006). Kolb and Whishaw (1981) have shown that neonatal lesions that destroy frontal corticothalamic pathways abolish defensive burying in rats. Interestingly, marble burying has also been suggested as a murine model for obsessive-compulsive disorder (OCD) (Njung'e and Handley, 1991; Ichimaru et al., 1995), and a group II metabotropic glutamate receptor antagonist that reduced marble-burying in mice was suggested for clinical use in OCD (Shimazaki et al., 2004).
The possible involvement of thalamus in the VGLUT +/−-associated behavior was supported by the electrophysiological analysis of thalamic neuronal cultures, in which loss of both VGLUT2 alleles almost completely abolished excitatory synaptic transmission, whereas the heterozygous neurons showed a discrete but significant 25% reduction in the postsynaptic response to single vesicle fusion (quantal size). In our previous study on VGLUT1 −/− neurons in hippocampus, spontaneous release events were ∼50% reduced in amplitude (similar to the magnitude of reduction seen in VGLUT2 −/− thalamic neurons) (Fig. 7 B), indicating that the number of vesicular transporters can dynamically influence the glutamate filling state of synaptic vesicles and hence the mEPSC amplitude (Wojcik et al., 2004). Other studies using brain slices did not detect a reduction in mEPSC amplitude with deletion of VGLUT1 (Fremeau et al., 2004b) and have found that one transporter may suffice to fill a synaptic vesicle to normal levels at the Drosophila neuromuscular junction (Daniels et al., 2006). Here we show that loss of both VGLUT2 alleles in thalamic neurons leads to a 50% reduction in mEPSC amplitude and that a reduction in VGLUT2 levels by 50% leads to a 25% reduction in mEPSC amplitude, a result that is most likely attributable to the fact that, in central mammalian synapses, individual vesicles contain multiple glutamate transporters per vesicle. A significant reduction of transporters then leads to a reduced vesicle filling state and thus a reduced quantal response size. Because overexpression studies have found that increased VGLUT levels can increase quantal size (Daniels et al., 2004; Wojcik et al., 2004; Wilson et al., 2005), both increasing and decreasing levels of VGLUTs seems to have a significant effect on the amount of glutamate in a synaptic vesicle. Because altered levels of VGLUTs have been demonstrated in response to pharmacological treatment and in disease states (De Gois et al., 2005; Eastwood and Harrison, 2005; Wilson et al., 2005; Kashani et al., 2006), bidirectional regulation of VGLUT levels is likely an endogenous mechanism for controlling synaptic strength in glutamatergic synapses.
Although all of these data are in good agreement with a thalamic location of the neuropathic pain defect and the other behavioral deficits, future behavioral analysis using region specific knockdown methodologies for VGLUT2 (conditional mouse knock-out, region-specific in vivo RNA interference, etc.) will allow to specifically test the involvement of the thalamus and other morphological substrates such as the spinal cord. At the spinal level, NMDA- and mGluR-mediated glutamatergic signaling have been shown to play a key role in the sensitization of dorsal horn neurons and the establishment of hyperalgesia and allodynia, the typical features of neuropathic pain (Zieglgansberger et al., 2005). Regardless of the exact circuitry that is impaired in the VGLUT2 +/− mice, we have identified VGLUT2 as a molecular player in the development and/or maintenance of neuropathic pain, and these animals will be valuable tools in studying the underlying disease mechanisms. Furthermore, VGLUT2 antagonism could present an attractive therapeutic strategy in treatment of neuropathic pain, because current therapeutic strategies for neuropathic pain have relatively poor overall efficacy, and tolerability or side effects are often limiting, particularly when therapeutic strategies target glutamatergic neurotransmission. Longer-term treatment with NMDA antagonists has been associated with serious adverse effects, including memory loss (Harris et al., 1975; Collins and Chessell, 2005). Importantly, no impairment in memory and learning, neuromotor performance, or anxiety-related behavior was observed in the VGLUT2 +/− mice, suggesting that such unwanted side effects could be avoided in this therapeutic approach. The observed decrease in defensive behavior in the VGLUT2 +/− mice suggests that antagonism of VGLUT2-mediated glutamatergic neurotransmission could also be applied for clinical use in anxiety-related disorders, including OCD.
Footnotes
This work was supported by National Institutes of Health Grant R01 NS051262 (C.R.), the KUL2004 Impulse Programme for Biological Psychology, and Fonds voor Wetenschappelijk Onderzoek Grant G.0271.06 (R.D.). Z.C.-V. is a Katholieke Universiteit Leuven Postdoctoral Fellow. We thank Hui Deng, Hongmei Chen, and Mingshan Xue for their help with the cell culture and genotyping and Leen Van Aerschot for help with the behavioral assessment.
References
- Aihara Y, Mashima H, Onda H, Hisano S, Kasuya H, Hori T, Yamada S, Tomura H, Yamada Y, Inoue I, Kojima I, Takeda J. Molecular cloning of a novel brain-type Na+-dependent inorganic phosphate cotransporter. J Neurochem. 2000;74:2622–2625. doi: 10.1046/j.1471-4159.2000.0742622.x. [DOI] [PubMed] [Google Scholar]
- Alexinsky T, Delacour J, Libouban S. Thalamus and defensive conditioning: relative importance of the three medial structures. Physiol Behav. 1971;6:439–446. doi: 10.1016/0031-9384(71)90180-6. [DOI] [PubMed] [Google Scholar]
- Alvarez FJ, Villalba RM, Zerda R, Schneider SP. Vesicular glutamate transporters in the spinal cord, with special reference to sensory primary afferent synapses. J Comp Neurol. 2004;472:257–280. doi: 10.1002/cne.20012. [DOI] [PubMed] [Google Scholar]
- Bai L, Xu H, Collins JF, Ghishan FK. Molecular and functional analysis of a novel neuronal vesicular glutamate transporter. J Biol Chem. 2001;276:36764–36769. doi: 10.1074/jbc.M104578200. [DOI] [PubMed] [Google Scholar]
- Bellocchio EE, Reimer RJ, Fremeau RT, Jr, Edwards RH. Uptake of glutamate into synaptic vesicles by an inorganic phosphate transporter. Science. 2000;289:957–960. doi: 10.1126/science.289.5481.957. [DOI] [PubMed] [Google Scholar]
- Bhatnagar S, Huber R, Lazar E, Pych L, Vining C. Chronic stress alters behavior in the conditioned defensive burying test: role of the posterior paraventricular thalamus. Pharmacol Biochem Behav. 2003;76:343–349. doi: 10.1016/j.pbb.2003.08.005. [DOI] [PubMed] [Google Scholar]
- Bordi F, Quartaroli M. Modulation of nociceptive transmission by NMDA/glycine site receptor in the ventroposterolateral nucleus of the thalamus. Pain. 2000;84:213–224. doi: 10.1016/s0304-3959(99)00205-5. [DOI] [PubMed] [Google Scholar]
- Boulland JL, Qureshi T, Seal RP, Rafiki A, Gundersen V, Bergersen LH, Fremeau RT, Jr, Edwards RH, Storm-Mathisen J, Chaudhry FA. Expression of the vesicular glutamate transporters during development indicates the widespread corelease of multiple neurotransmitters. J Comp Neurol. 2004;480:264–280. doi: 10.1002/cne.20354. [DOI] [PubMed] [Google Scholar]
- Bridges D, Thompson S, Rice R. Mechanisms of neuropathic pain. Br J Anaesth. 2001;87:12–26. doi: 10.1093/bja/87.1.12. [DOI] [PubMed] [Google Scholar]
- Bruggemann J, Galhardo V, Apkarian AV. Immediate reorganization of the rat somatosensory thalamus after partial ligation of sciatic nerve. J Pain. 2001;2:220–228. doi: 10.1054/jpai.2001.22792. [DOI] [PubMed] [Google Scholar]
- Chiechio S, Copani A, Melchiorri D, Canudas AM, Storto M, Calvani M, Nicolai R, Nicoletti F. Metabotropic receptors as targets for drugs of potential use in the treatment of neuropathic pain. J Endocrinol Invest. 2004;27:171–176. [PubMed] [Google Scholar]
- Collins SD, Chessell IP. Emerging therapies for neuropathic pain. Expert Opin Emerg Drugs. 2005;10:95–108. doi: 10.1517/14728214.10.1.95. [DOI] [PubMed] [Google Scholar]
- Daniels RW, Collins CA, Gelfand MV, Dant J, Brooks ES, Krantz DE, DiAntonio A. Increased expression of the Drosophila vesicular glutamate transporter leads to excess glutamate release and a compensatory decrease in quantal content. J Neurosci. 2004;24:10466–10474. doi: 10.1523/JNEUROSCI.3001-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels RW, Collins CA, Chen K, Gelfand MV, Featherstone DE, DiAntonio A. A single vesicular glutamate transporter is sufficient to fill a synaptic vesicle. Neuron. 2006;49:11–16. doi: 10.1016/j.neuron.2005.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Boer SF, Koolhaas JM. Defensive burying in rodents: ethology, neurobiology and psychopharmacology. Eur J Pharmacol. 2003;463:145–161. doi: 10.1016/s0014-2999(03)01278-0. [DOI] [PubMed] [Google Scholar]
- Decosterd I, Woolf CJ. Spared nerve injury: an animal model of persistent peripheral neuropathic pain. Pain. 2000;87:149–158. doi: 10.1016/S0304-3959(00)00276-1. [DOI] [PubMed] [Google Scholar]
- De Gois S, Schafer MK, Defamie N, Chen C, Ricci A, Weihe E, Varoqui H, Erickson JD. Homeostatic scaling of vesicular glutamate and GABA transporter expression in rat neocortical circuits. J Neurosci. 2005;25:7121–7133. doi: 10.1523/JNEUROSCI.5221-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Hooge R, Lullmann-Rauch R, Beckers T, Balschun D, Schwake M, Reiss K, von Figura K, Saftig P. Neurocognitive and psychotiform behavioral alterations and enhanced hippocampal long-term potentiation in transgenic mice displaying neuropathological features of human alpha-mannosidosis. J Neurosci. 2005;25:6539–6549. doi: 10.1523/JNEUROSCI.0283-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty PM, Li YJ, Lenz FA, Rowland L, Mittman S. Evidence that excitatory amino acids mediate afferent input to the primate somatosensory thalamus. Brain Res. 1996;728:267–273. doi: 10.1016/0006-8993(96)00550-1. [DOI] [PubMed] [Google Scholar]
- Eastwood SL, Harrison PJ. Decreased expression of vesicular glutamate transporter 1 and complexin II mRNAs in schizophrenia: further evidence for a synaptic pathology affecting glutamate neurons. Schizophr Res. 2005;73:159–172. doi: 10.1016/j.schres.2004.05.010. [DOI] [PubMed] [Google Scholar]
- Fremeau RT, Jr, Troyer MD, Pahner I, Nygaard GO, Tran CH, Reimer RJ, Bellocchio EE, Fortin D, Storm-Mathisen J, Edwards RH. The expression of vesicular glutamate transporters defines two classes of excitatory synapse. Neuron. 2001;31:247–260. doi: 10.1016/s0896-6273(01)00344-0. [DOI] [PubMed] [Google Scholar]
- Fremeau RT, Jr, Voglmaier S, Seal RP, Edwards RH. VGLUTs define subsets of excitatory neurons and suggest novel roles for glutamate. Trends Neurosci. 2004a;27:98–103. doi: 10.1016/j.tins.2003.11.005. [DOI] [PubMed] [Google Scholar]
- Fremeau RT, Jr, Kam K, Qureshi T, Johnson J, Copenhagen DR, Storm-Mathisen J, Chaudhry FA, Nicoll RA, Edwards RH. Vesicular glutamate transporters 1 and 2 target to functionally distinct synaptic release sites. Science. 2004b;304:1815–1819. doi: 10.1126/science.1097468. [DOI] [PubMed] [Google Scholar]
- Fujiyama F, Furuta T, Kaneko T. Immunocytochemical localization of candidates for vesicular glutamate transporters in the rat cerebral cortex. J Comp Neurol. 2001;435:379–387. doi: 10.1002/cne.1037. [DOI] [PubMed] [Google Scholar]
- Gras C, Herzog E, Bellenchi GC, Bernard V, Ravassard P, Pohl M, Gasnier B, Giros B, El Mestikawy S. A third vesicular glutamate transporter expressed by cholinergic and serotoninergic neurons. J Neurosci. 2002;22:5442–5451. doi: 10.1523/JNEUROSCI.22-13-05442.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilbaud G, Benoist JM, Jazat F, Gautron M. Neuronal responsiveness in the ventrobasal thalamic complex of rats with an experimental peripheral mononeuropathy. J Neurophysiol. 1990;64:1537–1554. doi: 10.1152/jn.1990.64.5.1537. [DOI] [PubMed] [Google Scholar]
- Harris JA, Biersner RJ, Edwards D, Bailey LW. Attention, learning, and personality during ketamine emergence: a pilot study. Anesth Analg. 1975;54:169–172. [PubMed] [Google Scholar]
- Herzog E, Bellenchi GC, Gras C, Bernard V, Ravassard P, Bedet C, Gasnier B, Giros B, El Mestikawy S. The existence of a second vesicular glutamate transporter specifies subpopulations of glutamatergic neurons. J Neurosci. 2001;21(RC181):1–6. doi: 10.1523/JNEUROSCI.21-22-j0001.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hisano S, Hoshi K, Ikeda Y, Maruyama D, Kanemoto M, Ichijo H, Kojima I, Takeda J, Nogami H. Regional expression of a gene encoding a neuron-specific Na+-dependent inorganic phosphate cotransporter (DNPI) in the rat forebrain. Brain Res Mol Brain Res. 2000;83:34–43. doi: 10.1016/s0169-328x(00)00194-7. [DOI] [PubMed] [Google Scholar]
- Ichimaru Y, Egawa T, Sawa A. 5-HT1A-receptor subtype mediates the effect of fluvoxamine, a selective serotonin reuptake inhibitor, on marble-burying behavior in mice. Jpn J Pharmacol. 1995;68:65–70. doi: 10.1254/jjp.68.65. [DOI] [PubMed] [Google Scholar]
- Kaneko T, Fujiyama F, Hioki H. Immunohistochemical localization of candidates for vesicular glutamate transporters in the rat brain. J Comp Neurol. 2002;444:39–62. doi: 10.1002/cne.10129. [DOI] [PubMed] [Google Scholar]
- Kanki JP, Adams DB. Ventrobasal thalamus necessary for visually-released defensive boxing of rat. Physiol Behav. 1978;21:7–12. doi: 10.1016/0031-9384(78)90268-8. [DOI] [PubMed] [Google Scholar]
- Kashani A, Betancur C, Giros B, Hirsch E, Mestikawy SE. Altered expression of vesicular glutamate transporters VGLUT1 and VGLUT2 in Parkinson disease. Neurobiol Aging. 2006 doi: 10.1016/j.neurobiolaging.2006.02.010. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolhekar R, Murphy S, Gebhart GF. Thalamic NMDA receptors modulate inflammation-produced hyperalgesia in the rat. Pain. 1997;71:31–40. doi: 10.1016/s0304-3959(97)03334-4. [DOI] [PubMed] [Google Scholar]
- Kidd B, Urban L. Mechanisms of inflammatory pain. Br J Anaesth. 2001;87:3–11. doi: 10.1093/bja/87.1.3. [DOI] [PubMed] [Google Scholar]
- Kolb B, Whishaw IQ. Neonatal frontal lesions in the rat: sparing of learned but not species-typical behavior in the presence of reduced brain weight and cortical thickness. J Comp Physiol Psychol. 1981;95:863–879. doi: 10.1037/h0077849. [DOI] [PubMed] [Google Scholar]
- Landry M, Bouali-Benazzouz R, El Mestikawy S, Ravassard P, Nagy F. Expression of vesicular glutamate transporters in rat lumbar spinal cord, with a note on dorsal root ganglia. J Comp Neurol. 2004;468:380–394. doi: 10.1002/cne.10988. [DOI] [PubMed] [Google Scholar]
- Lenz FA, Gracely RH, Hope EJ, Baker FH, Rowland LH, Dougherty PM, Richardson RT. The sensation of angina can be evoked by stimulation of the human thalamus. Pain. 1994;59:119–125. doi: 10.1016/0304-3959(94)90055-8. [DOI] [PubMed] [Google Scholar]
- Lenz FA, Gracely RH, Romanoski AJ, Hope EJ, Rowland LH, Dougherty PM. Stimulation in the human somatosensory thalamus can reproduce both the affective and sensory dimensions of previously experienced pain. Nat Med. 1995;1:910–913. doi: 10.1038/nm0995-910. [DOI] [PubMed] [Google Scholar]
- Li JL, Fujiyama F, Kaneko T, Mizuno N. Expression of vesicular glutamate transporters, VGLUT1 and VGLUT2, in axon terminals of nociceptive primary afferent fibers in the superficial layers of the medullary and spinal dorsal horns of the rat. J Comp Neurol. 2003;457:236–249. doi: 10.1002/cne.10556. [DOI] [PubMed] [Google Scholar]
- Maycox PR, Deckwerth T, Hell JW, Jahn R. Glutamate uptake by brain synaptic vesicles. Energy dependence of transport and functional reconstitution in proteoliposomes. J Biol Chem. 1988;263:15423–15428. [PubMed] [Google Scholar]
- Millan MJ, Lejeune F, Gobert A, Brocco M, Auclair A, Bosc C, Rivet JM, Lacoste JM, Cordi A, Dekeyne A. S18616, a highly potent spiroimidazoline agonist at alpha(2)-adrenoceptors. II. Influence on monoaminergic transmission, motor function, and anxiety in comparison with dexmedetomidine and clonidine. J Pharmacol Exp Ther. 2000;295:1206–1222. [PubMed] [Google Scholar]
- Morris RG. Synaptic plasticity and learning: selective impairment of learning rats and blockade of long-term potentiation in vivo by the N-methyl-d-aspartate receptor antagonist AP5. J Neurosci. 1989;9:3040–3057. doi: 10.1523/JNEUROSCI.09-09-03040.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neugebauer V. Metabotropic glutamate receptors–important modulators of nociception and pain behavior. Pain. 2002;98:1–8. doi: 10.1016/s0304-3959(02)00140-9. [DOI] [PubMed] [Google Scholar]
- Ni B, Rosteck PR, Jr, Nadi NS, Paul SM. Cloning and expression of a cDNA encoding a brain-specific Na+-dependent inorganic phosphate cotransporter. Proc Natl Acad Sci USA. 1994;91:5607–5611. doi: 10.1073/pnas.91.12.5607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Njung'e K, Handley SL. Evaluation of marble-burying behavior as a model of anxiety. Pharmacol Biochem Behav. 1991;38:63–67. doi: 10.1016/0091-3057(91)90590-x. [DOI] [PubMed] [Google Scholar]
- Paradee W, Melikian HE, Rasmussen DL, Kenneson A, Conn PJ, Warren ST. Fragile X mouse: strain effects of knockout phenotype and evidence suggesting deficient amygdala function. Neuroscience. 1999;94:185–192. doi: 10.1016/s0306-4522(99)00285-7. [DOI] [PubMed] [Google Scholar]
- Phelps EA, LeDoux JE. Contributions of the amygdala to emotion processing: from animal models to human behavior. Neuron. 2005;48:175–187. doi: 10.1016/j.neuron.2005.09.025. [DOI] [PubMed] [Google Scholar]
- Pothos EN, Larsen KE, Krantz DE, Liu Y, Haycock JW, Setlik W, Gershon MD, Edwards RH, Sulzer D. Synaptic vesicle transporter expression regulates vesicle phenotype and quantal size. J Neurosci. 2000;20:7297–7306. doi: 10.1523/JNEUROSCI.20-19-07297.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenmund C, Stevens CF. Definition of the readily releasable pool of vesicles at hippocampal synapses. Neuron. 1996;16:1197–1207. doi: 10.1016/s0896-6273(00)80146-4. [DOI] [PubMed] [Google Scholar]
- Schafer MK, Varoqui H, Defamie N, Weihe E, Erickson JD. Molecular cloning and functional identification of mouse vesicular glutamate transporter 3 and its expression in subsets of novel excitatory neurons. J Biol Chem. 2002;277:50734–50748. doi: 10.1074/jbc.M206738200. [DOI] [PubMed] [Google Scholar]
- Shields SD, Eckert WA, III, Basbaum AL. Spared nerve injury model of neuropathic pain in the mouse: a behavioral and anatomic analysis. J Pain. 2003;4:465–470. doi: 10.1067/s1526-5900(03)00781-8. [DOI] [PubMed] [Google Scholar]
- Shimazaki T, Iijima M, Chaki S. Anxiolytic-like activity of MGS0039, a potent group II metabotropic glutamate receptor antagonist, in a marble-burying behavior test. Eur J Pharmacol. 2004;501:121–125. doi: 10.1016/j.ejphar.2004.08.016. [DOI] [PubMed] [Google Scholar]
- Stornetta RL, Sevigny CP, Guyenet PG. Inspiratory augmenting bulbospinal neurons express both glutamatergic and enkephalinergic phenotypes. J Comp Neurol. 2003a;455:113–124. doi: 10.1002/cne.10486. [DOI] [PubMed] [Google Scholar]
- Stornetta RL, Rosin DL, Wang H, Sevigny CP, Weston MC, Guyenet PG. A group of glutamatergic interneurons expressing high levels of both neurokinin-1 receptors and somatostatin identifies the region of the pre-Botzinger complex. J Comp Neurol. 2003b;455:499–512. doi: 10.1002/cne.10504. [DOI] [PubMed] [Google Scholar]
- Takahashi N, Miner LL, Sora I, Ujike H, Revay RS, Kostic V, Jackson-Lewis V, Przedborski S, Uhl GR. VMAT2 knockout mice: heterozygotes display reduced amphetamine-conditioned reward, enhanced amphetamine locomotion, and enhanced MPTP toxicity. Proc Natl Acad Sci USA. 1997;94:9938–9943. doi: 10.1073/pnas.94.18.9938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamori S, Rhee JS, Rosenmund C, Jahn R. Identification of a vesicular glutamate transporter that defines a glutamatergic phenotype in neurons. Nature. 2000;407:189–194. doi: 10.1038/35025070. [DOI] [PubMed] [Google Scholar]
- Takamori S, Rhee JS, Rosenmund C, Jahn R. Identification of differentiation-associated brain-specific phosphate transporter as a second vesicular glutamate transporter (VGLUT2) J Neurosci. 2001;21(RC182):1–6. doi: 10.1523/JNEUROSCI.21-22-j0002.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamori S, Malherbe P, Broger C, Jahn R. Molecular cloning and functional characterization of human vesicular glutamate transporter 3. EMBO Rep. 2002;3:798–803. doi: 10.1093/embo-reports/kvf159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor BK, Peterson MA, Basbaum AI. Persistent cardiovascular and behavioral nociceptive responses to subcutaneous formalin require peripheral nerve input. J Neurosci. 1995;15:7575–7584. doi: 10.1523/JNEUROSCI.15-11-07575.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tjolsen A, Berge OG, Hunskaar S, Rosland JH, Hole K. The formalin test: an evaluation of the method. Pain. 1992;51:5–17. doi: 10.1016/0304-3959(92)90003-T. [DOI] [PubMed] [Google Scholar]
- Vertes RP. Interactions among the medial prefrontal cortex, hippocampus and midline thalamus in emotional and cognitive processing in the rat. Neuroscience. 2006;142:1–20. doi: 10.1016/j.neuroscience.2006.06.027. [DOI] [PubMed] [Google Scholar]
- Wattler S, Kelly M, Nehls M. Construction of gene targeting vectors from lambda KOS genomic libraries. Biotechniques. 1999;26:1150–1156. doi: 10.2144/99266rr02. 1158, 1160. [DOI] [PubMed] [Google Scholar]
- Wei F, Wang GD, Kerchner GA, Kim SJ, Xu HM, Chen ZF, Zhuo M. Genetic enhancement of inflammatory pain by forebrain NR2B overexpression. Nat Neurosci. 2001;4:164–169. doi: 10.1038/83993. [DOI] [PubMed] [Google Scholar]
- Weston MC, Stornetta RL, Guyenet PG. Glutamatergic neuronal projections from the marginal layer of the rostral ventral medulla to the respiratory centers in rats. J Comp Neurol. 2004;473:73–85. doi: 10.1002/cne.20076. [DOI] [PubMed] [Google Scholar]
- Wilson NR, Kang J, Hueske EV, Leung T, Varoqui H, Murnick JG, Erickson JD, Liu G. Presynaptic regulation of quantal size by the vesicular glutamate transporter VGLUT1. J Neurosci. 2005;25:6221–6234. doi: 10.1523/JNEUROSCI.3003-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojcik SM, Rhee JS, Herzog E, Sigler A, Jahn R, Takamori S, Brose N, Rosenmund C. An essential role for vesicular glutamate transporter 1 (VGLUT1) in postnatal development and control of quantal size. Proc Natl Acad Sci USA. 2004;101:7158–7163. doi: 10.1073/pnas.0401764101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto T, Fujimoto Y, Shimura T, Sakai N. Conditioned taste aversion in rats with excitotoxic brain lesions. Neurosci Res. 1995;22:31–49. doi: 10.1016/0168-0102(95)00875-t. [DOI] [PubMed] [Google Scholar]
- Zieglgansberger W, Berthele A, Tolle TR. Understanding neuropathic pain. CNS Spectr. 2005;10:298–308. doi: 10.1017/s1092852900022628. [DOI] [PubMed] [Google Scholar]