

Abstract
The p75 neurotrophin receptor (p75NTR) has been implicated in diverse neuronal responses, including survival, cell death, myelination, and inhibition of regeneration. However, the role of p75NTR in neuropathic pain, for which there is currently no effective therapy, has not been explored. Here, we report that the pharmacological blockade of p75NTR in primary sensory neurons reversed neuropathic pain after nerve injury. Nerve injury increased the expression and axonal transport of p75NTR and phosphorylation of TrkA in the uninjured primary afferents. Functional inhibition of p75NTR suppressed injury-induced neuropathic pain and decreased the phosphorylation of TrkA and p38 mitogen-activated protein kinase, and the induction of transient receptor potential channels in dorsal root ganglion (DRG) neurons. Our results show that p75NTR induced in undamaged DRG neurons facilitates TrkA signaling and contributes to heat and cold hyperalgesia.
Keywords: p75 neurotrophin receptor, nerve growth factor, p38 mitogen-activated protein kinase, transient receptor potential channels, primary afferent neurons, neuropathic pain
Introduction
The neurotrophins are a family of secreted growth factors that include nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), neurotrophin 3 (NT-3), and NT-4. They bind to two types of receptors: a common receptor, the p75 neurotrophin receptor (p75NTR), which binds all neurotrophins with a similar affinity; and members of the Trk family of receptor tyrosine kinases, TrkA, TrkB, and TrkC, which bind with different neurotrophins. NGF was identified originally as a survival factor for sensory and sympathetic neurons in the developing nervous system. In adults, NGF is not required for survival, but it has a crucial role in the generation of acute and chronic pain (McMahon et al., 1995; Sah et al., 2003; Hefti et al., 2006). Levels of NGF increase dramatically in injured tissues, and the enhanced transport of NGF activate and sensitize primary afferent neurons that express the NGF receptors, TrkA and/or p75NTR. However, it is not clear whether it is the interaction of NGF with TrkA, p75NTR, or both, that is important for pain after nerve damage.
p75NTR, a member of the tumor necrosis factor (TNF) receptor superfamily, has attracted attention because of its many neuronal functions (Chao, 2003; Barker, 2004; Lu et al., 2005). p75NTR enhances the ability of Trk receptors to respond to neurotrophins and the discrimination of Trk receptors for their preferred neurotrophin ligands (Hempstead et al., 1991; Benedetti et al., 1993; Bibel et al., 1999). Furthermore, under conditions of either reduced or absent Trk signaling, neurotrophin binding to p75NTR promotes apoptosis. p75NTR also binds and transduces the effects of potentially toxic, amyloid β and prion protein peptides. Importantly, it is also a coreceptor (with the Nogo receptor and LINGO-1) for the myelin-derived proteins Nogo, myelin-associated glycoprotein (MAG), and oligodendrocyte myelin glycoprotein (OmgP), which inhibit axon growth (Yamashita and Tohyama, 2003; Mi et al., 2004).
Neuropathic pain models are created by partial nerve injury in which some primary afferents are axotomized and others are spared. There is compelling evidence indicating that not only injured primary afferents, but also their spared neighbors, show an alteration of excitability and gene expression and that these changes have functional roles in neuropathic pain (Gold, 2000; Fukuoka and Noguchi, 2002; Ringkamp and Meyer, 2005). We now show that nerve injury increases the expression and axonal transport of p75NTR, and enhances TrkA signaling in uninjured dorsal root ganglion (DRG) neurons. This augmentation of NGF/TrkA signaling contributes to heat and cold hyperalgesia by regulating transient receptor potential (TRP) channel expression.
Materials and Methods
Animals.
Male Sprague Dawley rats weighing 200–250 g were used. All procedures were approved by the Hyogo College of Medicine Committee on Animal Research and were performed in accordance with National Institutes of Health guidelines on animal care. Rats that did not receive surgery (n = 12) were used as naive controls for in situ hybridization histochemistry, reverse transcription (RT)-PCR, immunohistochemistry, and Western blotting.
Surgical procedures.
All procedures were performed with the rats under pentobarbital anesthesia (50 mg/kg, i.p.). Additional doses of the anesthetics were given as needed. In all rats, no surgery was performed on the right side. Special care was taken to prevent infection and to minimize the influence of inflammation. The hair of the rat's lower back was shaved and the skin was sterilized with 0.5% chlorhexidine and covered with clean paper. Sterile operating instruments were used.
To produce an L5 spinal nerve ligation (SNL), a skin incision (3–4 cm) was made in the midline lumbar region (L4–S1) (Kim and Chung, 1992). The L6 transverse process was identified, freed of muscular attachments, and partially removed with the help of bone ronguers. This exposed the L5 spinal nerve. The L5 ventral ramus was isolated, freed from the adjacent nerves, and then the L5 spinal nerve was tightly ligated with silk suture and transected distal to the ligature. After surgery, the wound was washed with saline and closed in layers (fascia and skin) with 3-0 silk thread. Animals were allowed to survive for 3, 5, 7, or 14 d after surgery. In sham-operated rats, the nerve was exposed without ligation.
The intrathecal delivery of 10 μl of NGF (1 or 10 μg; R & D Systems, Minneapolis, MN), BDNF (10 μg; R & D Systems), NT-3 (10 μg; R & D Systems), and glial cell line-derived neurotrophic factor (GDNF) (10 μg; R & D Systems) was performed as described previously (Fukuoka et al., 2001). A laminectomy of the L5 vertebra was performed under adequate anesthesia with sodium pentobarbital. The dura was cut, and a soft tube (Silascon; outer diameter, 0.64 mm; Kaneka Medix, Osaka, Japan) was inserted into the subarachnoid space of the spinal cord at the L4/5 DRG level. To obtain a sustained drug infusion, an ALZET osmotic pump (7 d pump; 1 μl/h; Durect, Cupertino, CA) was filled with anti-NGF antibody (1 μg/μl; Chemicon, Temecula, CA) in saline, anti-p75NTR antibody (1 μg/μl; Sigma, St. Louis, MO) in saline, or K252a (0.1 μg/μl; Calbiochem, La Jolla, CA) in 50% DMSO, and the associated catheter was implanted intrathecally 12 h before L5 SNL. Normal saline was used as the vehicle control. The lack of effect of DMSO was determined in preliminary experiments, which was consistent with previous reports (Ji et al., 2002a; Obata et al., 2003). Antisense oligodeoxynucleotide (AS-ODN) (5′-CAGGGCGGCTAAAAG-3′), mismatch ODN (MM-ODN) (5′-ACTACTACACTAGACTAC-3′), and FITC-labeled ODN directed at the p75NTR were designed and manufactured by Biognostik (Göttingen, Germany). Although it has frequently been questioned whether ODN can reach the DRG in a sufficient concentration by intrathecal delivery, several reports have demonstrated that intrathecal ODN accumulates in DRG cells (Barclay et al., 2002; Lai et al., 2002). Therefore, AS-ODN (0.5 nmol · μl−1 · h−1) and MM-ODN (0.5 nmol · μl−1 · h−1) were delivered intrathecally by an osmotic pump 12 h before L5 SNL.
The local application of anti-NGF was performed basically as described previously (Fukuoka et al., 2001; Obata et al., 2004). A soft tube (Silascon; outer diameter, 0.64 mm; Kaneka Medix) was placed on the surface of the L4 spinal nerve. To obtain a sustained drug infusion, an ALZET osmotic pump (7 d pump; 1 μl/h; Durect) was filled with anti-NGF antibody (1 μg/μl; Chemicon) in saline. The catheter of the pump was implanted at least 12 h before the L5 SNL. Normal saline was used as the vehicle control.
Behavioral tests.
All rats were tested for heat and cold hyperalgesia and mechanical allodynia of the plantar surface of the hindpaw 1 d before surgery and 3, 7, or 14 d after surgery. Room temperature and humidity remained stable for all experiments. On each testing day, the rats were brought into the behavior room 1 h before the test session to allow them to habituate to the environment.
Heat hyperalgesia was tested using the Hargreaves radiant heat apparatus (7370; Ugo Basile, Comerio, Italy). A radiant heat source beneath a glass floor was aimed at the plantar surface of the hindpaw. Three measurements of latency were taken for each hindpaw in each test session. The hindpaws were tested alternately, with intervals between consecutive tests of >5 min. The three measurements of latency per side were averaged.
To estimate cold sensitivity of the paw, the cold plate test was performed as previously described (Choi et al., 1994; Jasmin et al., 1998). Each rat was placed in a plastic cage with a Peltier cooled cold plate (Neuroscience, Osaka, Japan) kept at a cold temperature (5 ± 0.5°C). After 5 min of adaptation, the numbers of paw lifts within the next 5 min were recorded. Foot lifts associated with locomotion or reposition of the body were not counted. An assistant, who was unaware of the treatment group, performed all of the behavioral experiments.
Mechanical allodynia was assessed with a “dynamic plantar aesthesiometer” (Ugo Basile), which is an automated von Frey type system (Kalmar et al., 2003; Lever et al., 2003). To measure rat hindpaw mechanical thresholds, rats were placed in plastic cages with a wire mesh floor and allowed to acclimate for 15 min before each test session. A paw-flick response was elicited by applying an increasing force (measured in grams) using a plastic filament (0.5 mm diameter) focused on the middle of the plantar surface of the ipsilateral hindpaw. The force applied was initially below detection threshold and then increased from 1 to 50 g in 1 g steps over 20 s, and then held at 50 g for an additional 10 s. The rate of force increase was 2.5 g/s. The force applied to elicit a reflex removal of the ipsilateral hindpaw was monitored. This was defined as the mean of three measurements at 1 min intervals. The variability between trials was ∼2 g.
Data are expressed as mean ± SEM. Differences in changes of values over time of each group were tested using one-way ANOVA, followed by individual post hoc comparisons (Fisher's exact test). One-way ANOVA, followed by individual post hoc comparisons (Fisher's exact test) or pairwise comparisons (t test), were used to assess differences of values between the intrathecal groups. A difference was accepted as significant if p < 0.05.
Immunohistochemistry.
The rats were deeply anesthetized with sodium pentobarbital and perfused transcardially with 1% paraformaldehyde in 0.1 m phosphate buffer (PB), pH 7.4, followed by 4% paraformaldehyde in 0.1 m PB at 7 d after surgery. After the perfusion, the L5 spinal cord segments, L4 spinal nerve, and L4/5 DRGs were dissected out and postfixed in the same fixative for 12 h, and then replaced with 20% sucrose overnight. Transverse spinal sections (free-floating; 25 μm) and DRG sections (16 μm) were cut in a cryostat and processed for p75NTR, TrkA, glial fibrillary acid protein (GFAP), phosphorylated-Trk (p-Trk), p-TrkA, and p-p38 mitogen-activated protein kinases (MAPK) immunohistochemistry according to previously described methods (Noguchi et al., 1995). The polyclonal primary antibody for TrkA (1:400; Chemicon), GFAP (1:400; DakoCytomation, Glostrup, Denmark), p-Trk (1:400; Santa Cruz Biotechnology, Santa Cruz, CA), and p-p38 (1:400; Cell Signaling Technology, Beverly, MA) and the monoclonal primary antibody for p75NTR (1:400; Roche Molecular Biochemicals, Mannheim, Germany) and p-TrkA (1:200; Santa Cruz Biotechnology) were used for DAB staining.
The number of p75NTR-, TrkA-, p-Trk-, p-TrkA-, and p-p38-immunoreactive (IR) neurons per section was counted in the left DRG. The proportion of p75NTR-, TrkA-, p-Trk-, p-TrkA-, and p-p38-expressing DRG neurons was determined by counting the neuronal profiles that showed distinctive labeling in the DRG sections. In each rat, four to six sections of the L4/5 DRG at each time point were selected randomly and 2000–3000 profiles were counted. An average percentage of p75NTR-, TrkA-, p-Trk-, p-TrkA-, and p-p38-IR neurons, relative to the total number of neurons, was obtained for each animal across the different tissue sections; then the mean ± SD across animals was determined. Because a stereological approach was not used in this study, quantification of the data may represent a biased estimate of the actual number of cells and neurons. An assistant, who was unaware of the treatment group of the tissue sections, performed all counting.
Differences in changes of values over time were tested using one-way ANOVA, followed by individual post hoc comparisons (Fisher's exact test). Pairwise comparisons (t test) were used to assess differences of values between the intrathecal groups. A difference was accepted as significant if p < 0.05.
Western blotting.
Tissue samples from the L5 spinal cord segments, L4 spinal nerve, and L4/5 DRGs were lysed by homogenizing in 200 μl of lysis buffer containing 20 mm Tris-HCl, pH 8.0, 150 mm NaCl, 1 mm EDTA, 1% Igepal CA-630, 2 mm Na3VO4, 0.5 mm DTT, 1 mm PMSF, 1 μg/ml pepstatin, 5 μg/ml leupeptin, 9 μg/ml aprotinin, and 10% glycerol. Lysates were centrifuged at 14,400 × g for 60 min, and the concentration of protein in each sample (supernatant) was determined using the Bio-Rad (Hercules, CA) dye binding. Samples with equal amounts of protein were then separated by 10–20% polyacrylamide gel electrophoresis, and the resolved proteins were electrotransferred to Hybond-P Nitrocellulose (Amersham Biosciences, Little Chalfont, UK). Membranes were incubated with 5% nonfat milk in Tris buffer containing Tween 20 (TBST) (10 mm Tris-HCl, pH 8.0, 150 mm NaCl, and 0.2% Tween 20) for at least 10 min at room temperature and incubated with the polyclonal primary antibody for TrkA (1:1000; Chemicon) and the monoclonal primary antibody for p75NTR (1:500; Roche Molecular Biochemicals), p-TrkA (1:500; Santa Cruz Biotechnology), and β-actin (1:1000; Sigma) at 4°C overnight. Membranes were then washed twice with TBST and probed with goat anti-rabbit IgG conjugated with horseradish peroxidase (Vector Laboratories, Burlingame, CA) at room temperature for 2 h. Membranes were finally washed several times with TBST to remove unbound secondary antibodies and visualized using enhanced chemiluminescence (Boehringer Mannheim, Indianapolis, IN). Each experiment was repeated at least twice, and in all cases the same results were obtained. The density of specific bands was measured with a computer-assisted imaging analysis system (Densitograph, version 4.02; ATTO, Tokyo, Japan) and normalized against a loading control (β-actin). The protein level was expressed as a percentage of the protein level in the naive control.
Data are expressed as mean ± SD. Differences in changes of values over time of each group were tested using one-way ANOVA, followed by individual post hoc comparisons (Fisher's exact test). One-way ANOVA, followed by individual post hoc comparisons (Fisher's exact test) or pairwise comparisons (t test), were used to assess differences of values between the intrathecal groups. A difference was accepted as significant if p < 0.05.
In situ hybridization histochemistry.
For the in situ hybridization histochemistry (ISHH), the tissue was sectioned (16 μm thick) with a cryostat, thaw-mounted onto Vectabond (Vector Laboratories)-coated slides and stored at −80°C until ready for use. The procedure for ISHH was performed according to previously described methods (Yamanaka et al., 1999; Hashimoto et al., 2001). Briefly, the rat TrkA, TrkB, TrkC, p75NTR, TRPV1, and TRPA1 cRNA probes corresponding to nucleotides 560–979, 277–585, 867–1380, 157–600, 149–505, and 302–788, respectively, were prepared. The sections were treated with 10 μg/ml proteinase K in 50 mm Tris-HCl and 5 mm EDTA for 3 min and acetylated with 0.25% acetic anhydride in 0.1 m triethanolamine; then 35S-labeled RNA probe (5 × 106 cpm/ml) was placed on these sections overnight at 55°C. Hybridized sections were rinsed in 5× SSC, 5 mm DTT for 30 min at 65°C; washed in high-stringency buffer for 30 min at 65°C; and treated with 2 μg/ml RNase A for 30 min at 37°C. Sections were rinsed, dehydrated in an ascending ethanol series, and air dried. For autoradiography, the sections were coated with NTB-3 emulsion (Eastman Kodak, Rochester, NY), diluted 6:4 with distilled water at 45°C, and exposed for 2 weeks in light-tight boxes at 4°C. After development in D19 (Eastman Kodak) and fixation in 24% sodium thiosulfate, the sections were rinsed in distilled water, stained with hematoxylin–eosin, dehydrated in a graded ethanol series, cleared in xylene, and coverslipped. For the double labeling of TrkB, TrkC, TRPV1 or TRPA1 mRNA and p75NTR or p-Trk protein, immunohistochemical staining of p75NTR or p-Trk was performed using an ABC (avidin–biotinylated peroxidase complex) kit (Vector Laboratories). Afterward, TrkB, TrkC, TRPV1 or TRPA1 mRNA were detected using ISHH with a radioisotope-labeled probe as described previously (Kobayashi et al., 2005).
Measurements of the density of silver grains over randomly selected tissue profiles were performed using the NIH Image program by a blinded assistant, where only neuronal profiles that contained nuclei were used for quantification. At a magnification of 200× and with bright-field illumination, upper and lower thresholds of gray level density were set such that only silver grains were accurately discriminated from the background in the outlined cell or tissue profile and read by the computer pixel-by-pixel. Subsequently, the area of discriminated pixels was measured and divided by the area of the outlined profile, giving a grain density for each cell or tissue profile. To reduce the risk of biased sampling of the data because of varying emulsion thickness, we used a signal/noise (S/N) ratio for each cell in each tissue section. The S/N ratio of an individual neuron and its cross-sectioned area, which was computed from the outlined profile, was plotted. Based on this scattergram, neurons with a grain density of 10-fold the background level or higher (10 ≤ S/N ratio) were considered positively labeled for p75NTR, TRPA1, and TRPV1 mRNA. At least 1200 neurons from four rats were quantified for each antisense probe in the ISHH study according to previously described methods (Kobayashi et al., 2005). The number of positively labeled DRG neurons was divided by the number of neuronal profiles counted in each DRG.
Data are expressed as mean ± SD. One-way ANOVA, followed by individual post hoc comparisons (Fisher's exact test), was used to assess differences of values between the intrathecal groups. A difference was accepted as significant if p < 0.05.
RT-PCR.
For the RT-PCR, the rats were killed by decapitation under deep anesthesia at 7 d after surgery and the left L4 DRG was removed and rapidly frozen with powdered dry ice and stored at −80°C until ready for use. The procedure of extraction of total RNA using a RNA extraction regent ISOGEN (Nippon Gene, Tokyo, Japan) was described in our previous study (Fukuoka et al., 2001). PCR primers for TrkA, TrkB, TrkC, p75NTR, and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) cDNA were designed corresponding to the coding region of the genes as follows: TrkA primers, sense, 5′-GTGCCCAGAGGGCACATACT-3′, and antisense, 5′-CGGCTGTTGGCGCCTTGTTT-3′; TrkB primers, sense, 5′-GCCCACGTCGTAGCAAACCA-3′, and antisense, 5′-GGGCTCATACCAGGGCTTG-3′; TrkC primers, sense, 5′-GGTGGGCATGAGCAATGCCA-3′, and antisense, 5′-GGACCGTCGACCGTACTTGT-3′; p75NTR primers, sense, 5′-GCCTGCTGCTGCTGCTGATT-3′, and antisense, 5′-CCACGTGGTTGGCTTCGTCT-3′; GAPDH primers, sense, 5′-TGCTGGTGCTGAGTATGTCG-3′, and antisense, 5′-GCATGTCAGATCCACAACGG-3′. Subsequent PCR was performed in a 50 μl solution of 1× PCR buffer (PerkinElmer, Boston, MA), 0.2 mm dNTP, and 1.25 U of AmpliTaq (PerkinElmer) with a pair of 20 pmol TrkA primers, 20 pmol TrkB primers, 10 pmol TrkC primers, 20 pmol p75NTR primers, or 20 pmol GAPDH primers on Perkin DNA Thermal Cycler (PerkinElmer), and the PCR program was 15 s at 94°C, 15 s at 57°C, and 45 s at 72°C. The intensity of stained bands was measured with a computer-assisted imaging analysis system (ATTO Densitograph, version 4.02). The density of PCR product bands of TrkA, TrkB, TrkC, p75NTR, and GAPDH mRNAs was increased between 25 and 35 PCR cycles, depending on the number of cycles; therefore, the number of PCR cycles of 30 was used. The ratio of TrkA, TrkB, TrkC, or p75NTR to GAPDH mRNAs was considered to indicate the level of each transcript. The mRNA level was expressed as a percentage of the mRNA level in the normal control ganglia. Samples without the addition of reverse transcriptase or without the addition of RNA (negative controls) revealed no detectable product.
Data are expressed as mean ± SD. Differences in changes of values between the intrathecal groups were tested using one-way ANOVA, followed by individual post hoc comparisons (Fisher's exact test) or pairwise comparisons (t test). A difference was accepted as significant if p < 0.05.
Results
p75NTR, but not Trks, mRNA increases in intact DRG neurons after nerve injury
The L5 SNL model (Kim and Chung, 1992), which is one of the most popular models of neuropathic pain, is unique because the L4 DRG neurons are clearly separated from the axotomized L5 neurons (Obata et al., 2004). Therefore, we investigated the role of p75NTR in nerve injury-induced heat and cold hyperalgesia using the L5 SNL model. Rats with L5 nerve injury displayed marked heat and cold hyperalgesia: the paw withdrawal latency to a noxious heat stimulus decreased from 10.4 ± 0.7 s before the injury to 7.0 ± 0.5 s at day 3 and 6.9 ± 0.5 s at day 14 after surgery. In addition, the number of paw lifts on a cold plate for the 5 min testing period increased from 2.3 ± 0.4 before the injury to 12.8 ± 2.6 at day 3 and 11.8 ± 1.9 at day 14 after surgery. The contralateral side of the nerve-ligated rats and both sides of sham-operated rats did not show pain behaviors (data not shown).
ISHH revealed that TrkA and p75NTR were expressed by DRG neurons of all sizes, whereas TrkB and TrkC were seen mainly in large-size neurons (Fig. 1 A,B), consistent with previous studies (Verge et al., 1992; Wright and Snider, 1995). Using a computerized image analysis, we found that 28.4 ± 1.7% of DRG neurons were positively labeled for p75NTR mRNA in naive control rats (Fig. 1C,D). The percentages of p75NTR mRNA-positive neurons in the present study are much lower than those previously reported (50–60%) (Zhou et al., 1996; Bergman et al., 1999). This discrepancy may be attributable to the differences in the criterion used to distinguish between positively and negatively labeled neurons.
Figure 1.
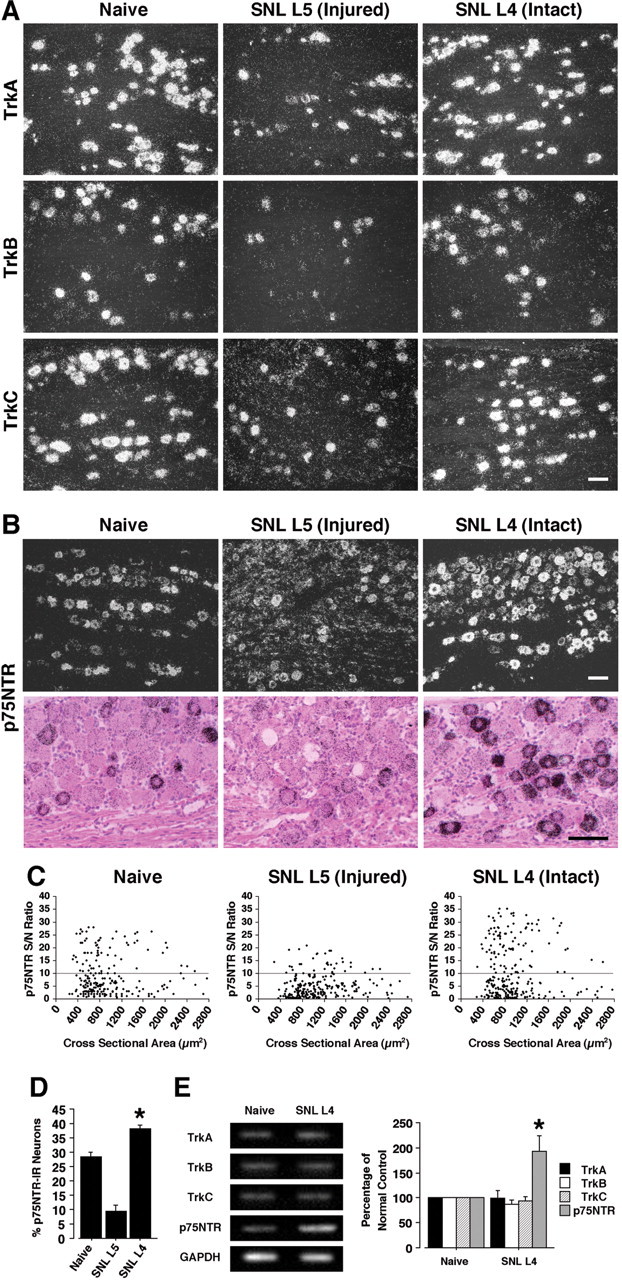
L5 SNL induces an increase of p75NTR, but not Trks, mRNA in spared L4 DRG neurons. A, Dark-field photomicrograph of ISHH showing the expression of Trks mRNA in the naive DRG, the injured L5 DRG, and the intact L4 DRG at day 7 after L5 SNL. B, Bright- and dark-field photomicrographs of ISHH showing the expression of p75NTR mRNA in the naive DRG, the injured L5 DRG, and the intact L4 DRG at day 7 after L5 SNL. C, Scatterplot diagrams made by plotting the individual cell profiles at day 7 after L5 SNL. The gray lines represent the borderlines between the negatively and positively labeled neurons (S/N ratio, 10). D, Quantification of the percentage of p75NTR mRNA-positive neurons at day 7 after surgery. E, mRNA expression of Trks and p75NTR in the L4 DRG at day 7 after nerve injury, as detected by RT-PCR. Quantification of the RT-PCR data is shown at the right. Data represent mean ± SD; n = 4 per group. *p < 0.05 compared with the naive control. Scale bars, 100 μm.
Seven days after surgery, the L5 SNL decreased p75NTR mRNA expression in the injured L5 DRG neurons and 9.5 ± 2.0% of DRG neurons were positively labeled for p75NTR mRNA. There was no change in p75NTR mRNA expression on the contralateral side (data not shown); however, the percentage of p75NTR-positive neurons significantly increased in the L4 DRG at day 7 after surgery and 38.1 ± 1.3% of DRG neurons were positively labeled (Fig. 1C,D). The increase in p75NTR was seen mainly in small- to medium-diameter neurons, and thus they were presumed to be nociceptive neurons. In contrast, there was no change in the expression of TrkA, TrkB, and TrkC in the undamaged L4 DRG, although the L5 SNL decreased Trks mRNA expression in the damaged L5 DRG. These changes in p75NTR and Trks mRNA in the spared L4 DRG were confirmed by RT-PCR (Fig. 1E).
p75NTR protein increases in TrkA-expressing DRG neurons after nerve injury
We then examined the changes of p75NTR and TrkA expression at the protein level using immunohistochemistry (Fig. 2A). Seven days after surgery, both p75NTR and TrkA protein expression decreased in the L5 DRG neurons, consistent with our data obtained from ISHH and previous reports (Zhou et al., 1996; Bergman et al., 1999), whereas the number of p75NTR-IR glial cells was greatly increased, particularly around large-diameter neurons (Fig. 2A). In contrast, the L5 SNL induced a significant increase in the percentage of p75NTR-IR neurons in the L4 DRG at day 7 (Fig. 2B). However, there was no difference in the percentages of TrkA-IR neurons (Fig. 2C). These changes in p75NTR and TrkA protein in the intact L4 DRG were also confirmed by Western blotting (Fig. 2D). Double labeling showed that, in the L5 DRG, p75NTR was present in ring structures and was colocalized with GFAP, indicating that p75NTR was localized with satellite glial cells, but not neurons, in the damaged DRG (Fig. 2E). In the L4 DRG, in contrast, the increase in p75NTR was seen mainly in small- and medium-size neurons (Fig. 1C). Cutaneous nociceptors can be divided into two broad groups: NGF-responsive/TrkA-expressing neurons and GDNF-responsive/c-ret-expressing neurons (Snider and McMahon, 1998). We found that p75NTR heavily colocalized with TrkA in the L4 DRG at day 7 after surgery (Fig. 2F). However, p75NTR-positive small neurons coexpressed neither TrkB nor TrkC (Fig. 2G). These findings indicate that injury increases p75NTR in NGF-responsive neurons in the uninjured DRG.
Figure 2.

Marked upregulation of p75NTR protein and overlap between p75NTR- and TrkA-expressing neurons in the spared L4 DRG after L5 SNL. A, Immunohistochemistry showing the expression of p75NTR and TrkA protein in the naive DRG, the injured L5 DRG, and the intact L4 DRG at day 7 after L5 SNL. B, C, Quantification of the percentage of p75NTR- and TrkA-IR neurons at day 7 after surgery. D, Protein expression of p75NTR and TrkA in the L4 DRG at day 7 after surgery, as detected by Western blotting. Quantification of the Western blot data are shown at the right. E, Immunohistochemical colocalization of green reaction product for GFAP and red product for p75NTR in the injured L5 DRG at day 7 after surgery. F, Double staining for p75NTR and TrkA revealed heavy colocalization in the intact L4 DRG at day 7 after surgery. The open arrows indicate double-labeled neurons. G, Double labeling by a combined method of ISHH and immunohistochemistry for TrkB or TrkC mRNA and p75NTR-IR in the L4 DRG. The arrows indicate single-labeled neurons for p75NTR protein, whereas the arrowheads indicate single-labeled neurons for TrkB or TrkC mRNA. Data represent mean ± SD; n = 4 per group. *p < 0.05 compared with the naive control. Scale bars, 50 μm.
NGF increases p75NTR expression in intact DRG neurons after nerve injury
NGF is known to increase the expression of p75NTR in sensory neurons after peripheral inflammation (Verge et al., 1992; Pezet et al., 2001). To determine whether alterations of endogenous NGF might be involved in p75NTR upregulation in the uninjured L4 DRG after nerve injury, anti-NGF was delivered intrathecally before L5 SNL and maintained for 7 d via a catheter whose tip was positioned close to the L4 DRG. The L5 SNL induced a substantial increase in the percentage of p75NTR-IR neurons in the vehicle group at day 7 after surgery, but anti-NGF application prevented this increase (Fig. 3A,B). However, there was no apparent change in TrkA expression (Fig. 3C).
Figure 3.

NGF, but not BDNF, NT-3, or GDNF, regulates p75NTR expression in DRG neurons. A, Effects of intrathecal infusion of anti-NGF on the L5 SNL-induced increase in p75NTR expression in the L4 DRG. Immunohistochemistry showing the expression of p75NTR protein in the intact L4 DRG at day 7 after L5 SNL is shown. B, C, Quantification of the percentage of p75NTR- and TrkA-IR neurons at day 7 after surgery. D, E, Effects of local application of anti-NGF on the L5 SNL-induced increase in p75NTR expression in the L4 DRG. Quantification of the percentage of p75NTR- and TrkA-IR neurons at day 7 after surgery is shown. F, Immunohistochemistry showing the expression of p75NTR in the DRG at day 3 after injection. G,H, Effects of NGF, BDNF, NT-3, or GDNF on p75NTR expression in the DRG neurons. Quantification of the percentage of p75NTR- and TrkA-IR neurons at day 3 after injection is shown. Data represent mean ± SD; n = 4 per group. *p < 0.05 compared with the naive control; #p < 0.05 compared with the vehicle group. Scale bar, 50 μm.
NGF is a neurotrophic factor that is retrogradely transported to the DRG from the periphery. Therefore, we aimed to block the NGF being transported to the L4 DRG using local application of anti-NGF antibody on the surface of the L4 spinal nerve. We found that local anti-NGF application significantly reduced the L5 SNL-induced increase in p75NTR protein in the L4 DRG at day 7 (Fig. 3D,E). Consistent with our previous reports (Obata et al., 2004, 2005), intrathecal or local anti-NGF application reversed the development of heat and cold hyperalgesia at day 7 after surgery (data not shown).
To further investigate the effects of NGF on p75NTR expression in the DRG neurons, we injected NGF (1 or 10 μg in 10 μl of saline) intrathecally into naive rats. Three days after injection, the percentage of p75NTR-IR neurons in the DRG was significantly higher in the 10 μg NGF group than in the saline group (Fig. 3F,G). However, p75NTR levels did not change after the 10 μg BDNF, NT-3, or GDNF injections. In contrast, NGF, as well as other neurotrophins, did not significantly alter the expression of TrkA (Fig. 3H). Thus, these findings suggest that nerve injury increases p75NTR in the intact DRG through alterations in target-derived NGF.
Nerve injury elevates the transport of p75NTR in the peripheral nerve terminals
p75NTR and TrkA are anterogradely transported from the DRG, along the spinal nerve, to peripheral nerve terminals, and via the dorsal root to central terminals in the spinal dorsal horn (Delcroix et al., 2003; Zweifel et al., 2005). We examined the presence of p75NTR and TrkA protein in the spinal cord and L4 spinal nerve after nerve injury. Seven days after surgery, both p75NTR and TrkA protein levels in the dorsal horn did not change significantly (Fig. 4A). In contrast, immunohistochemical staining revealed a dramatic increase in p75NTR, but not TrkA, protein levels in the spinal nerve (Fig. 4B). Consistent with this immunohistochemical evidence, Western blot analysis showed a significant increase in p75NTR labeling in the spinal nerve, but not the spinal cord (Fig. 4C,D). Furthermore, we performed double immunofluorescence with p75NTR and TrkA in the L4 spinal nerve (Fig. 4E). p75NTR was heavily colocalized with TrkA, implying that p75NTR synthesized in the spared DRG is transported selectively to peripheral nerve terminals with TrkA.
Figure 4.

Marked upregulation of p75NTR protein and overlap between p75NTR- and TrkA-labeled nerve fibers in the L4 spinal nerve after L5 SNL. A, Immunohistochemistry showing the expression of p75NTR and TrkA protein in the spinal cord at day 7 after L5 SNL. Scale bar, 200 μm. B, Immunohistochemistry showing the expression of p75NTR and TrkA protein in the L4 spinal nerve at day 7 after L5 SNL. Scale bar, 100 μm. C, D, Protein expression of p75NTR and TrkA in the spinal cord and in the L4 spinal nerve at day 7 after surgery, as detected by Western blotting. Quantification of the Western blot data is shown at the right. E, Double staining for p75NTR and TrkA revealed heavy colocalization in the spinal nerve at day 7 after surgery. Scale bar, 100 μm. Data represent mean ± SD; n = 4 per group. *p < 0.05 compared with the naive control.
Nerve injury induces the phosphorylation of TrkA in intact DRG neurons
p75NTR acts as a Trk coreceptor that increases the binding specificity and affinity of Trk receptors for neurotrophins (Hempstead et al., 1991; Benedetti et al., 1993; Bibel et al., 1999). Therefore, we first analyzed the phosphorylation levels of TrkA by immunohistochemistry and Western blotting using antibodies specific to the phosphoproteins. Seven days after surgery, the L5 SNL induced a significant increase in p-TrkA in the L4 DRG; the increase in p-TrkA was seen mainly in small- to medium-diameter neurons (Fig. 5A). The percentage of p-TrkA-IR neurons in the L4 DRG increased to 27.0 ± 4.0% compared with that in the naive control ganglia (19.0 ± 1.8%). Western blot analysis also showed a marked increase in TrkA phosphorylation (Fig. 5B). To examine the state of other Trks signaling, we then examined p-Trk immunoreactivity in the L4 DRG at day 7 after nerve injury (Fig. 5C). The number of p-Trk-IR neurons in the L4 DRG significantly increased to 20.3 ± 5.4% compared with that in the naive control ganglia (10.7 ± 1.4%), mainly in small- and medium-size neurons. Double labeling showed that the majority of the p-Trk-labeled neurons also expressed both TrkA and p75NTR (Fig. 5D,E). However, p-Trk-positive neurons coexpressed neither TrkB nor TrkC (Fig. 5F), indicating that, although there is no change in TrkA expression, nerve injury enhances TrkA signaling in the spared DRG.
Figure 5.

L5 SNL enhances NGF/TrkA signaling in the undamaged L4 DRG neurons. A, Immunohistochemistry showing the phosphorylation of TrkA in the spared L4 DRG at day 7 after L5 SNL. B, Protein expression of p-TrkA in the L4 DRG at day 7 after surgery, as detected by Western blotting. C, Immunohistochemistry showing the phosphorylation of Trk in the spared L4 DRG at day 7 after L5 SNL. D, Immunohistochemical colocalization of green reaction product for p-Trk and red product for TrkA in the L4 DRG at day 7 after surgery. The open arrows indicate double-labeled neurons. E, Double staining for p-Trk and p75NTR revealed heavy colocalization in the L4 DRG at day 7 after surgery. F, Double labeling by a combined method of ISHH and immunohistochemistry for TrkB or TrkC mRNA and p-Trk-IR in the L4 DRG. The arrows indicate single-labeled neurons for p-Trk protein, whereas the arrowheads indicate single-labeled neurons for TrkB or TrkC mRNA. Data represent mean ± SD; n = 4 per group. *p < 0.05 compared with the naive control. Scale bars, 50 μm.
Knock-down of the p75NTR gene reverses nerve injury-induced heat and cold hyperalgesia and mechanical allodynia, and decreases the phosphorylation of TrkA and p38 MAPK
To elucidate whether p75NTR induced in spared DRG neurons might contribute to the development of heat and cold hyperalgesia after nerve injury, rats with nerve injury were intrathecally treated with either an AS-ODN targeting p75NTR or an MM-ODN beginning 12 h before L5 SNL. We found that p75NTR AS-ODN (0.5 nmol · μl−1 · h−1) significantly inhibited nerve injury-induced heat and cold hyperalgesia and mechanical allodynia at days 3 and 7 after surgery (Fig. 6A–C). Both latencies and the number of paw lifts in the MM-ODN group (0.5 nmol · μl−1 · h−1) were not different from those of the vehicle control rats. We then confirmed that the level of p75NTR protein in the DRG neurons of the AS-ODN-treated rats was significantly lower than that in the MM-ODN-treated rats (Fig. 6D,E), whereas there was no difference in TrkA expression between p75NTR AS-ODN-treated rats and control rats at day 7 after surgery (Fig. 6F). Consistent with a previous study (Obata et al., 2005), the distribution of FITC-labeled ODN showed a progressive increase in fluorescence associated with DRG cell bodies, indicating that the uptake of the ODN occurred in a time-dependent manner (data not shown). Intrathecal administration of AS-ODN or MM-ODN into naive animals produced no significant changes in basal pain sensitivity.
Figure 6.

Reversal of heat and cold hyperalgesia, mechanical allodynia, and enhanced NGF/TrkA signaling in the spared L4 DRG by a selective blockade of p75NTR expression. A–C, Heat and cold hyperalgesia were tested using the plantar test and the cold-plate test, respectively, at days 3 and 7 after L5 SNL. Mechanical allodynia was determined with a dynamic plantar aesthesiometer. Data represent mean ± SEM; n = 8 per group. D, Immunohistochemistry showing the expression of p75NTR in the L4 DRG at day 7 after L5 SNL. E, F, Quantification of the percentage of p75NTR- and TrkA-IR neurons in the L4 DRG at day 7 after surgery. G, Immunohistochemistry showing the expression of p-TrkA and p-p38 MAPK in the L4 DRG at day 7 after L5 SNL. H, I, Quantification of the percentage of p-TrkA- and p-p38 MAPK-IR neurons in the L4 DRG at day 7 after surgery. J, Dark-field photomicrograph of ISHH showing the expression of TRPV1 and TRPA1 mRNA in the L4 DRG at day 7 after surgery. K,L, Quantification of the percentage of TRPV1 and TRPA1 mRNA-positive neurons in the L4 DRG at day 7 after surgery. Data represent mean ± SD; n = 4 per group. *p < 0.05 compared with the naive control; #p < 0.05 compared with the MM-ODN group. Scale bar, 100 μm.
NGF induces autophosphorylation of TrkA followed by phosphorylation of signaling molecules such as MAPK (Huang and Reichardt, 2003). Furthermore, it has been demonstrated that activation of p38 MAPK occurs in uninjured DRG neurons after nerve injury (Obata et al., 2004). We therefore investigated whether p75NTR affects the phosphorylation of TrkA and p38 MAPK in the L4 DRG. Phosphorylation of TrkA and p38 MAPK in L4 DRG neurons increased strikingly at day 7 after surgery, but pretreatment with AS-ODN blocked this increase (Fig. 6G–I). These results suggest that, in intact DRG neurons, p75NTR facilitates NGF/TrkA signaling through sustained phosphorylation of TrkA and the resultant p38 MAPK.
Increased p75NTR and enhanced TrkA signaling regulates TRPV1 and TRPA1 expression after nerve injury
NGF-induced p38 activation in DRG neurons participates in the development of heat and cold hyperalgesia by regulating TRPV1 and TRPA1 expression, respectively (Ji et al., 2002b; Obata et al., 2004, 2005). We therefore hypothesized that p75NTR induced in the intact sensory neurons increases the expression of TRPV1 and TRPA1 by augmentation of NGF signaling through TrkA. The L5 SNL induced a substantial increase in the percentage of TRPV1- and TRPA1-positive neurons in the L4 DRG in the vehicle group at day 7 after surgery, which was seen mainly in small- and medium-size neurons (Fig. 6J–L). In addition, we found that pretreatment with p75NTR AS-ODN significantly diminished nerve injury-induced elevation of both TRPV1 and TRPA1 expression.
To further ascertain whether increased p75NTR and enhanced TrkA signaling in the uninjured DRG regulates TRPV1 and TRPA1 expression, and contributes to heat and cold hyperalgesia, we administered anti-p75NTR or the NGF/TrkA receptor antagonist K252a into the intrathecal space. Intrathecal administration of anti-p75NTR or K252a into naive animals produced no significant changes in basal pain sensitivity. The treatment of anti-p75NTR and K252a diminished nerve injury-induced heat and cold hyperalgesia and mechanical allodynia at days 3 and 7 after surgery (Fig. 7A–C). Furthermore, both anti-p75NTR and K252a blocked the nerve injury-induced increase in p-TrkA, p-p38 MAPK, TRPV1, and TRPA1 in the L4 DRG at day 7 after surgery (Fig. 7D–G). Double labeling showed that p-p38 MAPK was predominantly expressed in p-TrkA-positive neurons in the L4 DRG (Fig. 7H). We also found that the majority of the p75NTR-labeled neurons also expressed TRPV1 and TRPA1 (Fig. 7I).
Figure 7.

Anti-p75NTR and an NGF/TrkA receptor antagonist attenuate nerve injury-induced heat and cold hyperalgesia and mechanical allodynia, and suppress the increase in p-TrkA, p-p38 MAPK, TRPV1, and TRPA1 expression in the spared L4 DRG neurons. A–C, Heat and cold hyperalgesia were tested using the plantar test and the cold-plate test, respectively, at days 3 and 7 after L5 SNL. Mechanical allodynia was determined with a dynamic plantar aesthesiometer. Data represent mean ± SEM; n = 8 per group. D,E, Quantification of the percentage of p-TrkA- and p-p38 MAPK-IR neurons in the L4 DRG at day 7 after surgery. F,G, Quantification of the percentage of TRPV1 and TRPA1 mRNA-positive neurons in the L4 DRG at day 7 after surgery. H, Double staining for p-TrkA and p-p38 MAPK revealed heavy colocalization in the intact L4 DRG at day 7 after surgery. The open arrows indicate double-labeled neurons. I, Double labeling by a combined method of ISHH and immunohistochemistry for TRPV1 or TRPA1 mRNA and p75NTR-IR in the uninjured L4 DRG at day 7 after surgery. Data represent mean ± SD; n = 4 per group. #p < 0.05 compared with the vehicle group. Scale bar, 50 μm.
Discussion
p75NTR upregulation in the adjacent intact DRG neurons and Wallerian degeneration
In previous studies, much attention was focused on the directly damaged primary afferents and their influence on the activity of the dorsal horn neurons (Woolf and Salter, 2000; Scholz and Woolf, 2002). However, it is evident that the spared DRG neurons and their fibers are also functionally important in the maintenance of neuropathic pain (Gold, 2000; Fukuoka and Noguchi, 2002; Ringkamp and Meyer, 2005). For example, L5 SNL increases the expression of growth factors, channels, neurotransmitters, and receptors in the uninjured L4 DRG, which indicates that the molecular phenotypic changes in the spared DRG are just like those produced by inflammation (Fukuoka et al., 2001; Obata et al., 2004, 2005). In the present study, SNL induced p75NTR upregulation in the intact DRG neurons, but anti-NGF blocked this increase. Furthermore, NGF injection increased p75NTR in DRG neurons. Wallerian degeneration after nerve lesion leads to an increase in cytokines and growth factors (Sommer and Schafers, 1998; Cui et al., 2000; Shamash et al., 2002). NGF content increases as early as 1 d after L5 SNL in the sciatic nerve, which contains both axotomized L5 nerves and spared L4 nerves (Fukuoka et al., 2001). Together, our observations suggest that, after nerve injury, NGF is synthesized and released in the degenerative nerve fibers and acts on nearby sensory fibers, inducing p75NTR upregulation in the spared DRG. However, considering that local application of anti-NGF did not completely block the L5 SNL-induced increase in p75NTR expression, we cannot deny the possibility that anti-NGF did not reach the DRG in a sufficient concentration. Alternatively, other cytokines and growth factors, such as TNF-α, also may be involved in p75NTR upregulation in the L4 DRG (Schafers et al., 2003).
The intrathecal injection of MAPK inhibitors has been shown to affect DRG neurons (Jin et al., 2003; Zhuang et al., 2005). Svensson et al. (2005) demonstrated that intrathecal injection of recombinant TNF receptor–Fc fusion protein etanercept reduced L5 SNL-induced increase in p38 MAPK phosphorylation in the DRG. Recently, Zhuang et al. (2006) reported an uptake of the peptide inhibitor by DRG neurons after intrathecal injection, using Tat peptide. However, it is not clear how intrathecal anti-NGF is able to block what is assumed to be the influence of transported NGF, although the intrathecal route has been used to deliver molecules directly to DRG neurons including anti-NGF (Christensen and Hulsebosch, 1997; Amaya et al., 2004; Obata et al., 2004). Indeed, Reichardt and colleagues have reported that p38 MAPK activation occurs at the site of NGF binding to TrkA in the periphery (Huang and Reichardt, 2003).
Interactions between TrkA and p75NTR in the uninjured primary afferents
In the present study, increased p75NTR in DRG neurons was transported selectively to peripheral nerve terminals. Furthermore, the population of fibers transporting p75NTR appeared to be larger than that shifting TrkA. Delcroix et al. (1998) reported that NGF treatment increased axonal transport of p75NTR without modifying TrkA expression or axonal transport.
The role of the p75NTR axonal transport is unclear, because studies in p75NTR knock-out mice indicate that the transport of NGF depends mostly on the presence of TrkA (Curtis et al., 1995). Furthermore, NGF-induced hyperalgesia occurs in p75NTR knock-out mice, indicating that the TrkA receptor is sufficient to mediate the acute, noxious actions of NGF (Bergmann et al., 1998). However, we found that p75NTR was heavily colocalized with TrkA, not only in the intact DRG neurons but also in the spinal nerve. Therefore, it is likely that p75NTR and TrkA receptors cooperate to increase the capacity of peripheral nerve terminals of primary afferents to capture and transport NGF. Indeed, many previous studies have shown that, when these two receptors are coexpressed, p75NTR enhances the ability of TrkA receptors to bind and respond to NGF and discriminates between preferred and nonpreferred neurotrophin ligands (Hempstead et al., 1991; Benedetti et al., 1993; Bibel et al., 1999). We also found that, although there was no change in TrkA expression, nerve injury increased the phosphorylation of TrkA in p75NTR-expressing neurons in the spared DRG. Furthermore, p75NTR AS-ODN reduced nerve injury-induced TrkA phosphorylation. These findings support the idea that p75NTR facilitates NGF binding to TrkA and enhances TrkA activity through sustained phosphorylation of TrkA (Fig. 8). Intriguingly, a recent report showed that necdin and its homologous MAGE (melanoma antigen gene) family proteins modulate NGF signaling for neuronal survival by regulating the association between p75NTR and TrkA (Kuwako et al., 2005).
Figure 8.

Diagram of the intracellular signaling pathways downstream of p75NTR and TrkA in sensory neurons. p75NTR plays an essential role in supporting NGF-triggered TrkA signaling pathways mediating neuropathic pain. After nerve injury, NGF is produced in damaged tissue and increases p75NTR expression in spared sensory neurons. p75NTR then interacts with the TrkA receptor and facilitates the ability of TrkA to respond to NGF. This enhanced TrkA signaling in sensory neurons increases the expression of TRPV1 and TRPA1 through the p38 MAPK pathway, increasing heat and cold hyperalgesia. Previous studies have demonstrated that both anti-NGF and p38 MAPK inhibitor SB203580 not only reverse heat and cold hyperalgesia, but also diminish the induction of TRPV1 and TRPA1 in the spared DRG (Ji et al., 2002b; Obata et al., 2004, 2005). The present study demonstrated that blockade of p75NTR also attenuated injury-induced hyperalgesia and upregulation of TRPV1 and TRPA1.
TRP channel upregulation in adjacent intact DRG neurons and heat and cold hyperalgesia
NGF injection in a peripheral target induces p38 MAPK activation in TrkA-expressing DRG neurons; activation of p38 MAPK in DRG neurons contributes to persistent pain via the transcriptional regulation of key gene products (Ji et al., 2002b). A subset of TRP channels mediate thermosensation and are expressed in sensory tissues, such as nociceptors and skin (Patapoutian et al., 2003; Moran et al., 2004). TRPV1 is activated by noxious heat, whereas TRPA1 is activated at ∼17°C, a temperature that is reported as painfully cold by humans (Story et al., 2003; Bandell et al., 2004). Therefore, we believe that p75NTR induced in spared DRG neurons facilitates NGF/TrkA signaling through phosphorylation of TrkA and p38 MAPK and regulates TRP channels, and thus increasing hyperalgesia (Fig. 8). However, the role of TRPA1 in cold transduction is still controversial (Bautista et al., 2006; Kwan et al., 2006), and additional study is needed to reach a consensus on the role of TRPA1 in cold transduction under normal physiological condition.
An unexpected finding in the present study was that blockade of p75NTR significantly reduced nerve injury-induced mechanical allodynia. Many studies suggest that large-diameter A-fibers mediate mechanical allodynia, whereas nociceptive C-fibers mediate heat and cold hyperalgesia (Shir and Seltzer, 1990; Hao et al., 1996; Ossipov et al., 1999, 2002; Hama, 2002; Pan et al., 2003). Considering that L5 SNL increased the expression of p75NTR in the L4 DRG, mainly small- to medium-diameter neurons, and thus presumed to be nociceptive, p75NTR induced in undamaged DRG neurons might not be involved in the development of mechanical allodynia. Rather, the effects of p75NTR AS-ODN on mechanical allodynia could be mediated by the blockade of p75NTR in spinal cord and the higher CNS. Goettl et al. (2004) have demonstrated that an SNL-induced increase in p75NTR in the nucleus gracilus might mediate expression of mechanical allodynia.
p75NTR intrinsic signaling and neuropathic pain
p75NTR mediates some of the effects of its neurotrophin ligand through intrinsic signaling, which is independent of the Trk receptors (Chao, 2003; Barker, 2004; Lu et al., 2005). The proform of NGF (proNGF) is a high-affinity ligand for p75NTR and induces p75NTR-dependent neuronal apoptosis, which requires sortilin as a coreceptor (Lee et al., 2001; Nykjaer et al., 2004). Many studies suggest that the p75NTR-induced apoptotic cascade involves Rac1 and c-Jun N-terminal kinase/stress-activated protein kinase (JNK/SAPK) activation (Harrington et al., 2002; Barker, 2004). Although p75NTR is coexpressed with TrkA in primary afferents and p75NTR AS-ODN reduced nerve injury-induced TrkA phosphorylation, we cannot rule out the possibility that p75NTR intrinsic signaling pathway is also involved in the development of neuropathic pain. Indeed, Nicol and colleagues have described a mechanism mediating intrinsic signaling via p75NTR that could still account for at least some of the sensitization of sensory neurons in vitro (Zhang and Nicol, 2004). Nonetheless, nerve injury did not induce the activation of JNK/SAPK in the uninjured DRG (Obata et al., 2004; Zhuang et al., 2006). Furthermore, proNGF was markedly reduced in skin extracts from patients with subclinical diabetic neuropathy (Yiangou et al., 2002). Therefore, these findings suggest that, in uninjured sensory neurons, augmentation of NGF/TrkA signaling, rather than p75NTR intrinsic signaling, probably has a crucial role in the pathogenesis of neuropathic pain.
Conclusion
Collectively, our findings provide evidence that p75NTR induced in the spared primary afferents regulates TRP channels by enhancing NGF/TrkA signaling and contributes to neuropathic pain. Alternatively, p75NTR may form a tripartite complex with the Nogo receptor and with LINGO-1, and mediate RhoA activation via a direct interaction with Rho GDP dissociation inhibitor α, that results in growth inhibitory signals to be transduced in response to Nogo, MAG, or OmgP (Yamashita and Tohyama, 2003; Mi et al., 2004). Intriguingly, recent reports have shown that Y-27632 [(+)-(R)-trans-4-(1-aminoethyl)-(4-pyridyl)cyclohexanecarboxamide dihydrochloride], an antagonist of ROCK (Rho kinase), attenuates neuropathic pain (Inoue et al., 2004; Ramer et al., 2004). The participation of p75NTR in the nociceptive pathway may provide additional insight into the function of these receptors.
Footnotes
This work was supported in part by Grants-in-Aid for Scientific Research and the Open Research Center grant, Hyogo College of Medicine, both from the Japanese Ministry of Education, Science, and Culture. This work was also supported by a grant from the Japan Health Sciences Foundation. We thank Yuki Obata, Yuu Wadazumi, and Noriko Kusumoto for technical assistance. We thank D. A. Thomas for correcting the English usage.
References
- Amaya F, Shimosato G, Nagano M, Ueda M, Hashimoto S, Tanaka Y, Suzuki H, Tanaka M. NGF and GDNF differentially regulate TRPV1 expression that contributes to development of inflammatory thermal hyperalgesia. Eur J Neurosci. 2004;20:2303–2310. doi: 10.1111/j.1460-9568.2004.03701.x. [DOI] [PubMed] [Google Scholar]
- Bandell M, Story GM, Hwang SW, Viswanath V, Eid SR, Petrus MJ, Earley TJ, Patapoutian A. Noxious cold ion channel TRPA1 is activated by pungent compounds and bradykinin. Neuron. 2004;41:849–857. doi: 10.1016/s0896-6273(04)00150-3. [DOI] [PubMed] [Google Scholar]
- Barclay J, Patel S, Dorn G, Wotherspoon G, Moffatt S, Eunson L, Abdel'al S, Natt F, Hall J, Winter J, Bevan S, Wishart W, Fox A, Ganju P. Functional downregulation of P2X3 receptor subunit in rat sensory neurons reveals a significant role in chronic neuropathic and inflammatory pain. J Neurosci. 2002;22:8139–8147. doi: 10.1523/JNEUROSCI.22-18-08139.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker PA. p75NTR is positively promiscuous: novel partners and new insights. Neuron. 2004;42:529–533. doi: 10.1016/j.neuron.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Bautista DM, Jordt SE, Nikai T, Tsuruda PR, Read AJ, Poblete J, Yamoah EN, Basbaum AI, Julius D. TRPA1 mediates the inflammatory actions of environmental irritants and proalgesic agents. Cell. 2006;124:1269–1282. doi: 10.1016/j.cell.2006.02.023. [DOI] [PubMed] [Google Scholar]
- Benedetti M, Levi A, Chao MV. Differential expression of nerve growth factor receptors leads to altered binding affinity and neurotrophin responsiveness. Proc Natl Acad Sci USA. 1993;90:7859–7863. doi: 10.1073/pnas.90.16.7859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergman E, Fundin BT, Ulfhake B. Effects of aging and axotomy on the expression of neurotrophin receptors in primary sensory neurons. J Comp Neurol. 1999;410:368–386. doi: 10.1002/(sici)1096-9861(19990802)410:3<368::aid-cne2>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- Bergmann I, Reiter R, Toyka KV, Koltzenburg M. Nerve growth factor evokes hyperalgesia in mice lacking the low-affinity neurotrophin receptor p75. Neurosci Lett. 1998;255:87–90. doi: 10.1016/s0304-3940(98)00713-7. [DOI] [PubMed] [Google Scholar]
- Bibel M, Hoppe E, Barde Y. Biochemical and functional interactions between the neurotrophin receptors trk and p75NTR. EMBO J. 1999;18:616–622. doi: 10.1093/emboj/18.3.616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao MV. Neurotrophins and their receptors: a convergence point for many signalling pathways. Nat Rev Neurosci. 2003;4:299–309. doi: 10.1038/nrn1078. [DOI] [PubMed] [Google Scholar]
- Choi Y, Yoon YW, Na HS, Kim SH, Chung JM. Behavioral signs of ongoing pain and cold allodynia in a rat model of neuropathic pain. Pain. 1994;59:369–376. doi: 10.1016/0304-3959(94)90023-X. [DOI] [PubMed] [Google Scholar]
- Christensen MD, Hulsebosch CE. Spinal cord injury and anti-NGF treatment results in changes in CGRP density and distribution in the dorsal horn in the rat. Exp Neurol. 1997;147:463–475. doi: 10.1006/exnr.1997.6608. [DOI] [PubMed] [Google Scholar]
- Cui JG, Holmin S, Mathiesen T, Meyerson BA, Linderoth B. Possible role of inflammatory mediators in tactile hypersensitivity in rat models of mononeuropathy. Pain. 2000;88:239–248. doi: 10.1016/S0304-3959(00)00331-6. [DOI] [PubMed] [Google Scholar]
- Curtis R, Adryan KM, Stark JL, Park JS, Compton DL, Weskamp G, Huber LJ, Chao MV, Jaenisch R, Lee KF, Lindsay RM, DiStefano PS. Differential role of the low affinity neurotrophin receptor (p75) in retrograde axonal transport of the neurotrophins. Neuron. 1995;14:1201–1211. doi: 10.1016/0896-6273(95)90267-8. [DOI] [PubMed] [Google Scholar]
- Delcroix JD, Michael GJ, Priestley JV, Tomlinson DR, Fernyhough P. Effect of nerve growth factor treatment on p75NTR gene expression in lumbar dorsal root ganglia of streptozocin-induced diabetic rats. Diabetes. 1998;47:1779–1785. doi: 10.2337/diabetes.47.11.1779. [DOI] [PubMed] [Google Scholar]
- Delcroix JD, Patel J, Averill S, Tomlinson DR, Priestley JV, Fernyhough P. Peripheral axon crush elevates transport of p75NTR in the central projection of sensory neurones of rats. Neurosci Lett. 2003;351:181–185. doi: 10.1016/s0304-3940(03)00749-3. [DOI] [PubMed] [Google Scholar]
- Fukuoka T, Noguchi K. Contribution of the spared primary afferent neurons to the pathomechanisms of neuropathic pain. Mol Neurobiol. 2002;26:57–67. doi: 10.1385/MN:26:1:057. [DOI] [PubMed] [Google Scholar]
- Fukuoka T, Kondo E, Dai Y, Hashimoto N, Noguchi K. Brain-derived neurotrophic factor increases in the uninjured dorsal root ganglion neurons in selective spinal nerve ligation model. J Neurosci. 2001;21:4891–4900. doi: 10.1523/JNEUROSCI.21-13-04891.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goettl VM, Hussain SR, Alzate O, Wirtz DJ, Stephens RL, Jr, Hackshaw KV. Differential change in mRNA expression of p75 and Trk neurotrophin receptors in nucleus gracilis after spinal nerve ligation in the rat. Exp Neurol. 2004;187:533–536. doi: 10.1016/j.expneurol.2004.03.011. [DOI] [PubMed] [Google Scholar]
- Gold MS. Spinal nerve ligation: what to blame for the pain and why. Pain. 2000;84:117–120. doi: 10.1016/s0304-3959(99)00309-7. [DOI] [PubMed] [Google Scholar]
- Hama AT. Capsaicin-sensitive primary afferents mediate responses to cold in rats with a peripheral mononeuropathy. NeuroReport. 2002;13:461–464. doi: 10.1097/00001756-200203250-00020. [DOI] [PubMed] [Google Scholar]
- Hao JX, Yu W, Xu XJ, Wiesenfeld-Hallin Z. Capsaicin-sensitive afferents mediate chronic cold, but not mechanical, allodynia-like behavior in spinally injured rats. Brain Res. 1996;722:177–180. doi: 10.1016/0006-8993(96)00216-8. [DOI] [PubMed] [Google Scholar]
- Harrington AW, Kim JY, Yoon SO. Activation of Rac GTPase by p75 is necessary for c-jun N-terminal kinase-mediated apoptosis. J Neurosci. 2002;22:156–166. doi: 10.1523/JNEUROSCI.22-01-00156.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto N, Yamanaka H, Fukuoka T, Dai Y, Obata K, Mashimo T, Noguchi K. Expression of HGF and cMet in the peripheral nervous system of adult rats following sciatic nerve injury. NeuroReport. 2001;12:1403–1407. doi: 10.1097/00001756-200105250-00022. [DOI] [PubMed] [Google Scholar]
- Hefti FF, Rosenthal A, Walicke PA, Wyatt S, Vergara G, Shelton DL, Davies AM. Novel class of pain drugs based on antagonism of NGF. Trends Pharmacol Sci. 2006;27:85–91. doi: 10.1016/j.tips.2005.12.001. [DOI] [PubMed] [Google Scholar]
- Hempstead BL, Martin-Zanca D, Kaplan DR, Parada LF, Chao MV. High-affinity NGF binding requires co-expression of the trk proto-oncogene and the low-affinity NGF receptor. Nature. 1991;350:678–683. doi: 10.1038/350678a0. [DOI] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF. Trk receptors: roles in neuronal signal transduction. Annu Rev Biochem. 2003;72:609–642. doi: 10.1146/annurev.biochem.72.121801.161629. [DOI] [PubMed] [Google Scholar]
- Inoue M, Rashid MH, Fujita R, Contos JJ, Chun J, Ueda H. Initiation of neuropathic pain requires lysophosphatidic acid receptor signaling. Nat Med. 2004;10:712–718. doi: 10.1038/nm1060. [DOI] [PubMed] [Google Scholar]
- Jasmin L, Kohan L, Franssen M, Janni G, Goff JR. The cold plate as a test of nociceptive behaviors: description and application to the study of chronic neuropathic and inflammatory pain models. Pain. 1998;75:367–382. doi: 10.1016/s0304-3959(98)00017-7. [DOI] [PubMed] [Google Scholar]
- Ji RR, Befort K, Brenner GJ, Woolf CJ. ERK MAP kinase activation in superficial spinal cord neurons induces prodynorphin and NK-1 upregulation and contributes to persistent inflammatory pain hypersensitivity. J Neurosci. 2002a;22:478–485. doi: 10.1523/JNEUROSCI.22-02-00478.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Samad TA, Jin SX, Schmoll R, Woolf CJ. p38 MAPK activation by NGF in primary sensory neurons after inflammation increases TRPV1 levels and maintains heat hyperalgesia. Neuron. 2002b;36:57–68. doi: 10.1016/s0896-6273(02)00908-x. [DOI] [PubMed] [Google Scholar]
- Jin SX, Zhuang ZY, Woolf CJ, Ji RR. p38 mitogen-activated protein kinase is activated after a spinal nerve ligation in spinal cord microglia and dorsal root ganglion neurons and contributes to the generation of neuropathic pain. J Neurosci. 2003;23:4017–4022. doi: 10.1523/JNEUROSCI.23-10-04017.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalmar B, Greensmith L, Malcangio M, McMahon SB, Csermely P, Burnstock G. The effect of treatment with BRX-220, a co-inducer of heat shock proteins, on sensory fibers of the rat following peripheral nerve injury. Exp Neurol. 2003;184:636–647. doi: 10.1016/S0014-4886(03)00343-1. [DOI] [PubMed] [Google Scholar]
- Kim SH, Chung JM. An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain. 1992;50:355–363. doi: 10.1016/0304-3959(92)90041-9. [DOI] [PubMed] [Google Scholar]
- Kobayashi K, Fukuoka T, Yamanaka H, Dai Y, Obata K, Tokunaga A, Noguchi K. Differential expression patterns of mRNAs for P2X receptor subunits in neurochemically characterized dorsal root ganglion neurons in the rat. J Comp Neurol. 2005;481:377–390. doi: 10.1002/cne.20393. [DOI] [PubMed] [Google Scholar]
- Kuwako K, Hosokawa A, Nishimura I, Uetsuki T, Yamada M, Nada S, Okada M, Yoshikawa K. Disruption of the paternal necdin gene diminishes TrkA signaling for sensory neuron survival. J Neurosci. 2005;25:7090–7099. doi: 10.1523/JNEUROSCI.2083-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan KY, Allchorne AJ, Vollrath MA, Christensen AP, Zhang DS, Woolf CJ, Corey DP. TRPA1 contributes to cold, mechanical, and chemical nociception but is not essential for hair-cell transduction. Neuron. 2006;50:277–289. doi: 10.1016/j.neuron.2006.03.042. [DOI] [PubMed] [Google Scholar]
- Lai J, Gold MS, Kim CS, Bian D, Ossipov MH, Hunter JC, Porreca F. Inhibition of neuropathic pain by decreased expression of the tetrodotoxin-resistant sodium channel, NaV1.8. Pain. 2002;95:143–152. doi: 10.1016/s0304-3959(01)00391-8. [DOI] [PubMed] [Google Scholar]
- Lee R, Kermani P, Teng KK, Hempstead BL. Regulation of cell survival by secreted proneurotrophins. Science. 2001;294:1945–1948. doi: 10.1126/science.1065057. [DOI] [PubMed] [Google Scholar]
- Lever I, Cunningham J, Grist J, Yip PK, Malcangio M. Release of BDNF and GABA in the dorsal horn of neuropathic rats. Eur J Neurosci. 2003;18:1169–1174. doi: 10.1046/j.1460-9568.2003.02848.x. [DOI] [PubMed] [Google Scholar]
- Lu B, Pang PT, Woo NH. The yin and yang of neurotrophin action. Nat Rev Neurosci. 2005;6:603–614. doi: 10.1038/nrn1726. [DOI] [PubMed] [Google Scholar]
- McMahon SB, Bennett DL, Priestley JV, Shelton DL. The biological effects of endogenous nerve growth factor on adult sensory neurons revealed by a trkA-IgG fusion molecule. Nat Med. 1995;1:774–780. doi: 10.1038/nm0895-774. [DOI] [PubMed] [Google Scholar]
- Mi S, Lee X, Shao Z, Thill G, Ji B, Relton J, Levesque M, Allaire N, Perrin S, Sands B, Crowell T, Cate RL, McCoy JM, Pepinsky RB. LINGO-1 is a component of the Nogo-66 receptor/p75 signaling complex. Nat Neurosci. 2004;7:221–228. doi: 10.1038/nn1188. [DOI] [PubMed] [Google Scholar]
- Moran MM, Xu H, Clapham DE. TRP ion channels in the nervous system. Curr Opin Neurobiol. 2004;14:362–369. doi: 10.1016/j.conb.2004.05.003. [DOI] [PubMed] [Google Scholar]
- Noguchi K, Kawai Y, Fukuoka T, Senba E, Miki K. Substance P induced by peripheral nerve injury in primary afferent sensory neurons and its effect on dorsal column nucleus neurons. J Neurosci. 1995;15:7633–7643. doi: 10.1523/JNEUROSCI.15-11-07633.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nykjaer A, Lee R, Teng KK, Jansen P, Madsen P, Nielsen MS, Jacobsen C, Kliemannel M, Schwarz E, Willnow TE, Hempstead BL, Petersen CM. Sortilin is essential for proNGF-induced neuronal cell death. Nature. 2004;427:843–848. doi: 10.1038/nature02319. [DOI] [PubMed] [Google Scholar]
- Obata K, Yamanaka H, Dai Y, Tachibana T, Fukuoka T, Tokunaga A, Yoshikawa H, Noguchi K. Differential activation of extracellular signal-regulated protein kinase in primary afferent neurons regulates brain-derived neurotrophic factor expression after peripheral inflammation and nerve injury. J Neurosci. 2003;23:4117–4126. doi: 10.1523/JNEUROSCI.23-10-04117.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obata K, Yamanaka H, Kobayashi K, Dai Y, Mizushima T, Katsura H, Fukuoka T, Tokunaga A, Noguchi K. Role of mitogen-activated protein kinase activation in injured and intact primary afferent neurons for mechanical and heat hypersensitivity after spinal nerve ligation. J Neurosci. 2004;24:10211–10222. doi: 10.1523/JNEUROSCI.3388-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obata K, Katsura H, Mizushima T, Yamanaka H, Kobayashi K, Dai Y, Fukuoka T, Tokunaga A, Tominaga M, Noguchi K. TRPA1 induced in sensory neurons contributes to cold hyperalgesia after inflammation and nerve injury. J Clin Invest. 2005;115:2393–2401. doi: 10.1172/JCI25437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossipov MH, Bian D, Malan TP, Jr, Lai J, Porreca F. Lack of involvement of capsaicin-sensitive primary afferents in nerve-ligation injury induced tactile allodynia in rats. Pain. 1999;79:127–133. doi: 10.1016/s0304-3959(98)00187-0. [DOI] [PubMed] [Google Scholar]
- Ossipov MH, Zhang ET, Carvajal C, Gardell L, Quirion R, Dumont Y, Lai J, Porreca F. Selective mediation of nerve injury-induced tactile hypersensitivity by neuropeptide Y. J Neurosci. 2002;22:9858–9867. doi: 10.1523/JNEUROSCI.22-22-09858.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan HL, Khan GM, Alloway KD, Chen SR. Resiniferatoxin induces paradoxical changes in thermal and mechanical sensitivities in rats: mechanism of action. J Neurosci. 2003;23:2911–2919. doi: 10.1523/JNEUROSCI.23-07-02911.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patapoutian A, Peier AM, Story GM, Viswanath V. ThermoTRP channels and beyond: mechanisms of temperature sensation. Nat Rev Neurosci. 2003;4:529–539. doi: 10.1038/nrn1141. [DOI] [PubMed] [Google Scholar]
- Pezet S, Onteniente B, Jullien J, Junier MP, Grannec G, Rudkin BB, Calvino B. Differential regulation of NGF receptors in primary sensory neurons by adjuvant-induced arthritis in the rat. Pain. 2001;90:113–125. doi: 10.1016/s0304-3959(00)00393-6. [DOI] [PubMed] [Google Scholar]
- Ramer LM, Borisoff JF, Ramer MS. Rho-kinase inhibition enhances axonal plasticity and attenuates cold hyperalgesia after dorsal rhizotomy. J Neurosci. 2004;24:10796–10805. doi: 10.1523/JNEUROSCI.3337-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringkamp M, Meyer RA. Injured versus uninjured afferents: who is to blame for neuropathic pain? Anesthesiology. 2005;103:221–223. doi: 10.1097/00000542-200508000-00002. [DOI] [PubMed] [Google Scholar]
- Sah DW, Ossipov MH, Porreca F. Neurotrophic factors as novel therapeutics for neuropathic pain. Nat Rev Drug Discov. 2003;2:460–472. doi: 10.1038/nrd1107. [DOI] [PubMed] [Google Scholar]
- Schafers M, Geis C, Svensson CI, Luo ZD, Sommer C. Selective increase of tumour necrosis factor-alpha in injured and spared myelinated primary afferents after chronic constrictive injury of rat sciatic nerve. Eur J Neurosci. 2003;17:791–804. doi: 10.1046/j.1460-9568.2003.02504.x. [DOI] [PubMed] [Google Scholar]
- Scholz J, Woolf CJ. Can we conquer pain? Nat Neurosci. 2002;5(Suppl):1062–1067. doi: 10.1038/nn942. [DOI] [PubMed] [Google Scholar]
- Shamash S, Reichert F, Rotshenker S. The cytokine network of Wallerian degeneration: tumor necrosis factor-α, interleukin-1α, and interleukin-1β. J Neurosci. 2002;22:3052–3060. doi: 10.1523/JNEUROSCI.22-08-03052.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shir Y, Seltzer Z. A-fibers mediate mechanical hyperesthesia and allodynia and C-fibers mediate thermal hyperalgesia in a new model of causalgiform pain disorders in rats. Neurosci Lett. 1990;115:62–67. doi: 10.1016/0304-3940(90)90518-e. [DOI] [PubMed] [Google Scholar]
- Snider WD, McMahon SB. Tackling pain at the source: new ideas about nociceptors. Neuron. 1998;20:629–632. doi: 10.1016/s0896-6273(00)81003-x. [DOI] [PubMed] [Google Scholar]
- Sommer C, Schafers M. Painful mononeuropathy in C57BL/Wld mice with delayed Wallerian degeneration: differential effects of cytokine production and nerve regeneration on thermal and mechanical hypersensitivity. Brain Res. 1998;784:154–162. doi: 10.1016/s0006-8993(97)01327-9. [DOI] [PubMed] [Google Scholar]
- Story GM, Peier AM, Reeve AJ, Eid SR, Mosbacher J, Hricik TR, Earley TJ, Hergarden AC, Andersson DA, Hwang SW, McIntyre P, Jegla T, Bevan S, Patapoutian A. ANKTM1, a TRP-like channel expressed in nociceptive neurons, is activated by cold temperatures. Cell. 2003;112:819–829. doi: 10.1016/s0092-8674(03)00158-2. [DOI] [PubMed] [Google Scholar]
- Svensson CI, Schafers M, Jones TL, Powell H, Sorkin LS. Spinal blockade of TNF blocks spinal nerve ligation-induced increases in spinal P-p38. Neurosci Lett. 2005;379:209–213. doi: 10.1016/j.neulet.2004.12.064. [DOI] [PubMed] [Google Scholar]
- Verge VM, Merlio JP, Grondin J, Ernfors P, Persson H, Riopelle RJ, Hokfelt T, Richardson PM. Colocalization of NGF binding sites, trk mRNA, and low-affinity NGF receptor mRNA in primary sensory neurons: responses to injury and infusion of NGF. J Neurosci. 1992;12:4011–4022. doi: 10.1523/JNEUROSCI.12-10-04011.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf CJ, Salter MW. Neuronal plasticity: increasing the gain in pain. Science. 2000;288:1765–1769. doi: 10.1126/science.288.5472.1765. [DOI] [PubMed] [Google Scholar]
- Wright DE, Snider WD. Neurotrophin receptor mRNA expression defines distinct populations of neurons in rat dorsal root ganglia. J Comp Neurol. 1995;351:329–338. doi: 10.1002/cne.903510302. [DOI] [PubMed] [Google Scholar]
- Yamanaka H, He X, Matsumoto K, Shiosaka S, Yoshida S. Protease M/neurosin mRNA is expressed in mature oligodendrocytes. Brain Res Mol Brain Res. 1999;71:217–224. doi: 10.1016/s0169-328x(99)00187-4. [DOI] [PubMed] [Google Scholar]
- Yamashita T, Tohyama M. The p75 receptor acts as a displacement factor that releases Rho from Rho-GDI. Nat Neurosci. 2003;6:461–467. doi: 10.1038/nn1045. [DOI] [PubMed] [Google Scholar]
- Yiangou Y, Facer P, Sinicropi DV, Boucher TJ, Bennett DL, McMahon SB, Anand P. Molecular forms of NGF in human and rat neuropathic tissues: decreased NGF precursor-like immunoreactivity in human diabetic skin. J Peripher Nerv Syst. 2002;7:190–197. doi: 10.1046/j.1529-8027.2002.02024.x. [DOI] [PubMed] [Google Scholar]
- Zhang YH, Nicol GD. NGF-mediated sensitization of the excitability of rat sensory neurons is prevented by a blocking antibody to the p75 neurotrophin receptor. Neurosci Lett. 2004;366:187–192. doi: 10.1016/j.neulet.2004.05.042. [DOI] [PubMed] [Google Scholar]
- Zhou XF, Rush RA, McLachlan EM. Differential expression of the p75 nerve growth factor receptor in glia and neurons of the rat dorsal root ganglia after peripheral nerve transection. J Neurosci. 1996;16:2901–2911. doi: 10.1523/JNEUROSCI.16-09-02901.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang ZY, Gerner P, Woolf CJ, Ji RR. ERK is sequentially activated in neurons, microglia, and astrocytes by spinal nerve ligation and contributes to mechanical allodynia in this neuropathic pain model. Pain. 2005;114:149–159. doi: 10.1016/j.pain.2004.12.022. [DOI] [PubMed] [Google Scholar]
- Zhuang ZY, Wen YR, Zhang DR, Borsello T, Bonny C, Strichartz GR, Decosterd I, Ji RR. A peptide c-Jun N-terminal kinase (JNK) inhibitor blocks mechanical allodynia after spinal nerve ligation: respective roles of JNK activation in primary sensory neurons and spinal astrocytes for neuropathic pain development and maintenance. J Neurosci. 2006;26:3551–3560. doi: 10.1523/JNEUROSCI.5290-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zweifel LS, Kuruvilla R, Ginty DD. Functions and mechanisms of retrograde neurotrophin signalling. Nat Rev Neurosci. 2005;6:615–625. doi: 10.1038/nrn1727. [DOI] [PubMed] [Google Scholar]