

Abstract
The brain controls fertility through release of gonadotropin-releasing hormone (GnRH), but the mechanisms underlying action potential patterning and GnRH release are not understood. We investigated whether GnRH neurons exhibit afterdepolarizing potentials (ADPs) and whether these are modified by reproductive state. Whole-cell current-clamp recordings of GnRH neurons in brain slices from ovariectomized mice revealed a slow ADP (sADP) after action potentials generated by brief current injection. Generating two or four spikes enhanced sADP amplitude and duration. sADP amplitude was not affected by blocking selected neurotransmitter/neuromodulator receptors, delayed-rectifier potassium channels, calcium-dependent cation channels, or hyperpolarization-activated cation channels but was halved by the calcium channel blocker cadmium and abolished by tetrodotoxin. Cadmium also reduced peak latency. Intrinsic mechanisms underlying the sADP were investigated using voltage-clamp protocols simulating action potential waveforms. A single action potential produced an inward current, which increased after double and quadruple stimulation. Cadmium did not affect current amplitude but reduced peak latency. Pretreatment with blockers of calcium-activated potassium currents (IKCa) reproduced this shift and blocked subsequent cadmium-induced changes, suggesting cadmium changes latency indirectly by blocking IKCa. Tetrodotoxin abolished the inward current, suggesting that it is carried by sodium. In contrast, IKCa blockers increased the inward current, indicating that IKCa may oppose generation of the sADP. Strong sADPs were suprathreshold, generating repetitive spontaneous firing. IADP, sADP, and excitability were enhanced by in vivo estradiol, which triggers a preovulatory surge of GnRH release. Physiological feedback modification of this inward current and resulting sADP may modulate action potential firing and subsequent GnRH release.
Keywords: GnRH, intrinsic activity, sodium current, excitability, neuroendocrine, afterdepolarizing potential, feedback regulation
Introduction
Gonadotropin-releasing hormone (GnRH) neurons form the final common pathway for the central regulation of fertility. In males, and during most of the female reproductive cycle, GnRH is released in pulses of varying frequencies, altering downstream function (Clarke and Cummins, 1982; Levine and Duffy, 1988; Moenter et al., 1992). Pulse generation appears intrinsic to GnRH neuronal networks, but the mechanisms are not well understood (Moenter et al., 2003). Immortalized GnRH neurons release GnRH in pulses (Krsmanovic et al., 1992; Martinez de la Escalera et al., 1992; Pitts et al., 2001). Green fluorescent protein (GFP)-identified GnRH neurons in brain slices fire spontaneously (Spergel et al., 1999; Suter et al., 2000b; Han et al., 2004) even when fast synaptic transmission is blocked (Nunemaker et al., 2002), and spontaneous firing persists in isolated GnRH neurons (Kuehl-Kovarik et al., 2002). In females, a surge mode of continuous GnRH release also exists; this is induced by estradiol-positive feedback and provides the central signal initiating ovulation (Docke and Dorner, 1965; Moenter et al., 1991). Firing activity of GnRH neurons in brain slices during surge mode is increased relative to that observed during negative feedback in a animal model in which constant estradiol administration induces daily switching between these feedback modes (Christian et al., 2005). This altered activity may be attributable to changes in synaptic transmission to GnRH neurons and/or enhanced intrinsic excitability to increase both response to synaptic changes and spontaneous endogenous activity.
Action potentials, or extracellularly recorded action currents, of GnRH neurons often occur in groupings of two to several (Suter et al., 2000b; Kuehl-Kovarik et al., 2002; Nunemaker et al., 2003b; Abe and Terasawa, 2005). Such burst firing increases hormone release from magnocellular neuroendocrine neurons (Dutton and Dyball, 1979; Poulain and Wakerley, 1982). The mechanisms underlying burst firing in GnRH neurons are unknown. In magnocellular neuroendocrine neurons and neurons of the hippocampus and cortex, action potential firing can be followed by an afterdepolarization potential (ADP) or depolarizing afterpotential (DAP) (Kandel and Spencer, 1961; Fujita, 1975; Connors et al., 1982; Andrew and Dudek, 1984; Azouz et al., 1996; Hestrin and Armstrong, 1996; Li and Hatton, 1997a,b; Armstrong and Stern, 1998; Chu et al., 2003; Metz et al., 2005). ADPs can contribute to intrinsically repetitive burst firing (Andrew and Dudek, 1983, 1984; Bourque, 1986; Jensen et al., 1996; Bourque et al., 1998; Metz et al., 2005). Several ionic mechanisms may underlie the ADP, including low- and high-voltage-activated calcium currents (Li and Hatton, 1997a; Metz et al., 2005), hyperpolarization-activated currents (Ih) (Erickson et al., 1993; Luthi et al., 1998), persistent sodium current (Azouz et al., 1996; Su et al., 2001; Yue et al., 2005), calcium-activated nonselective cation currents (Greene et al., 1994), or reduced outward potassium currents (Li and Hatton, 1997b; Yue and Yaari, 2004).
GnRH neurons exhibit action potential-dependent ADPs of unknown mechanism (Spergel et al., 1999; Sim et al., 2001; Kuehl-Kovarik et al., 2002, 2005). We investigated mechanisms underlying ADP generation, function, and estradiol regulation. The data suggest that ADPs contribute to repetitive firing in GnRH neurons and are steroid sensitive.
Materials and Methods
Animals.
Adult (2–3 months of age) female mice expressing enhanced GFP (Clontech, Palo Alto, CA) under the control of the GnRH promoter were used to facilitate identification of GnRH neurons (Suter et al., 2000a). Mice were maintained under a 14/10 h light/dark photoperiod with Harlan (Indianapolis, IN) 2916 chow and water available ad libitum. To avoid the confounding effects of ovarian hormone changes during the estrous cycle, mice were ovariectomized (OVX) under Metofane (Janssen Pharmaceuticals, Toronto, Ontario, Canada) or isoflurane (Burns Veterinary Supply, Westbury, NY) anesthesia 5–9 d before experimentation; time after gonadectomy did not affect results. Estradiol feedback was studied using a previously characterized model in which treating OVX females with a constant-release estradiol implant produces daily switches between negative and positive feedback that persist in the brain slice preparation (Christian et al., 2005). The long-acting local anesthetic bupivicaine (0.25%; Abbot Labs, North Chicago, IL) was applied to surgical sites to minimize postoperative pain and distress. All procedures were approved by the Animal Care and Use Committee of the University of Virginia.
Brain slice preparation.
All chemicals were purchased from Sigma (St. Louis, MO) unless noted. Brain slices were prepared as described previously (Nunemaker et al., 2002, 2003a; Chu and Moenter, 2005). All solutions were bubbled with 95% O2 and 5% CO2 to maintain pH and oxygenization for at least 15 min before use and throughout experiments. In brief, mice were decapitated, and brains were quickly removed and immersed immediately for 30–60 s in ice-cold sucrose buffer containing the following (in mm): 250 sucrose, 3.5 KCl, 26 NaHCO3, 10 glucose, 1.25 NaHPO4, 1.2 MgSO4, and 3.8 MgCl2. Sagittal brain slices (300 μm) through the preoptic area (POA) and hypothalamus were cut using a Vibratome 1000 or Vibratome 3000 (Technical Products International, St. Louis, MO). Slices were immediately transferred into a holding chamber and incubated at 31–33°C for a 30 min recovery period in a mixture of 50% sucrose saline and 50% normal saline, containing the following (in mm): 135 NaCl, 3.5 KCl, 26 NaHCO3, 1,25NaH2PO4, 2.5 CaCl2, 1.2 MgSO4, 10 d-glucose, pH 7.4. Slices were then transferred to 100% normal saline and maintained at room temperature (∼21–23°C) until study (30 min to 8 h).
Data acquisition.
Slices were transferred to a recording chamber mounted on the stage of an upright microscope (Olympus Optical BX50WI; Opelco, Dulles, VA) and stabilized in the chamber at least 5 min before recording. The chamber was continuously perfused with normal saline at a rate of 4–5 ml/min at 32°C. Pipettes (3–4 MΩ) were pulled from borosilicate glass capillaries (1.65 mm outer diameter; 1.12 mm inner diameter; World Precision Instruments, Sarasota, FL) using a Narishige model pp-830 (Pacer Scientific, Los Angeles, CA) or Flaming/Brown P-97 (Sutter Instruments, Novato, CA). Pipettes were filled with a solution containing the following (in mm): 125 K-gluconate, 20 KCl, 10 HEPES, 5 EGTA, 4.0 MgATP, 0.4 NaGTP, and 0.1 CaCl2, pH 7.2 (300 mOsm). GnRH–GFP neurons from POA and ventral hypothalamus were identified by brief illumination at 470 nm. Data were acquired and digitized using either an EPC-8 (HEKA Elektronik, Mahone Bay, Nova Scotia, Canada) with an ITC-18 interface (InstruTech, Port Washington, NY) controlled by the PulseControl XOP (InstruTech) running in IgorPro (WaveMetrics, Lake Oswego, OR) or using one head stage of an EPC-10 dual amplifier (HEKA Elektronik) controlled by PatchMaster (HEKA Elektronik). There were no differences attributable to acquisition system. Signals were low-pass filtered at 10 kHz. During whole-cell recordings, input resistance (Rin), series resistance (Rs), and membrane capacitance (Cm) were continually measured. Only recordings with stable Rin >500 MΩ and Rs <20 MΩ and stable Cm were used for analysis. Data were further examined to make sure changes in Rin or Rs within acceptable limits did not influence results. Calculated liquid junction potential error, estimated to be −13 mV, was not corrected (Barry, 1994). Neurobiotin or biocytin (0.5%) was included in some cells for morphological study; no differences were observed between cells with and without these compounds.
Current-clamp recordings.
Whole-cell current-clamp recordings were made using bridge balance and capacitance compensation. All cells had an initial membrane potential negative to −55 mV without current injection and action potential amplitude of >90 mV; direct current (DC) injection (<20 pA) was used to normalize membrane potential to the range of −55 to −65 mV to facilitate comparison. Action potentials were elicited with short-duration (3–5 ms) current pulses (200–300 pA). When multiple pulses were given, they were separated by 50 ms (20 Hz). Each protocol was run 10 times at 2 s intervals, and all 10 traces were averaged for analysis. Amplitude of the slow afterdepolarization (sADP) that followed the action potential was measured from the prespike baseline. Latency to peak of the sADP was measured from the action potential threshold (defined as 1 V/s); threshold of the final spike in a train was used if multiple spikes were initiated. sADP duration was the length of time the membrane voltage exceeded prestimulus baseline levels.
Voltage-clamp recordings.
Whole-cell voltage-clamp was used to study currents underlying the sADP. Cells were held at −60 mV. Current response to single, double, or quadruple 2 ms square voltage pulses, or simulated action potentials, separated by 50 ms was evaluated. Stimuli were repeated 50 times, and traces were averaged for analysis. To assess changes in conductance during the sADP, small hyperpolarizing voltage steps (10 mV, 50 ms duration) were given before, during, and after the peak of the inward current; input resistance was the quotient of the magnitude of the voltage step divided by the steady-state current. Amplitude of the inward current (IADP) was measured from the precommand baseline. Latency to peak of IADP was measured from the onset of the final depolarizing step of the voltage command.
Drug treatments.
The following treatments were bath applied. Ionotropic glutamate receptors (iGluRs) were blocked with 20 μm 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) and 20 μm d(−)2-amino-5-phosphonovaleric acid (d-APV). GABAA receptors were blocked with 20 μm bicuculline methiodide. Sodium channels were blocked with tetrodotoxin (TTX) (0.5 μm; Calbiochem, San Diego, CA). GnRH receptors were blocked with d-pGlu1, d-Phe2, d-Trp3,6 (d-P-Glu) (20–40 nm; Bachem, Torrance, CA). Galanin receptors were blocked with galantide (100 nm; Bachem). Metabotropic glutamate receptors (mGluRs) were blocked with (RS)-α-methyl-4-carboxyphenylglycine (1 mm) and (RS)-α-cyclopropyl-4-phosphonophenylglycine (200 μm). P2 purinergic receptors were blocked with 100 μm suramin. Calcium channels were blocked with 200 μm Cd2+. Hyperpolarization-activated cation currents (Ih) were blocked with 40 μm ZD7288 (4-ethylphenylamino-1,2-dimethyl-6-methylaminopyrimidinium chloride). Delayed rectifier potassium currents (IK) were blocked with 10 mm tetraethylammonium (TEA). Small-conductance calcium-activated potassium currents (IKCa) were blocked with 200 nm apamin, large-conductance IKCa were blocked with 100 nm iberiotoxin, persistent sodium current (INaP) was blocked with 5–10 μm riluzole, and calcium-dependent nonselective cation channels were blocked with Cs+ (3 μm) or flufenamic acid (200 μm).
Statistical analyses.
The values are expressed as mean ± SEM. Statistical comparisons were made using paired parametric or nonparametric tests, as dictated by data distribution. All tests were two tailed, and p < 0.05 was considered significant.
Results
GnRH neurons exhibit homogeneous firing properties
Whole-cell current clamp was used to investigate the firing properties of GnRH neurons identified by expression of green fluorescent protein (Fig. 1A). All recorded cells were located in the preoptic area and anterior hypothalamus. Small amounts of positive current consistently evoked repeated action potentials (Fig. 1B). The small amount of current required to initiate action potential firing is consistent with the high input resistance of these cells (Kusano et al., 1995; Spergel et al., 1999; Suter et al., 2000b; DeFazio and Moenter, 2002; Kuehl-Kovarik et al., 2002). There was little evidence of spike frequency adaptation over the 500 ms injection, although a slight decrement in spike amplitude was routinely observed. Similarly, the prominent afterhyperpolarization (AHP) was reduced in amplitude as an evoked firing train proceeded. GnRH neurons were very homogeneous with regard to these firing properties.
Figure 1.
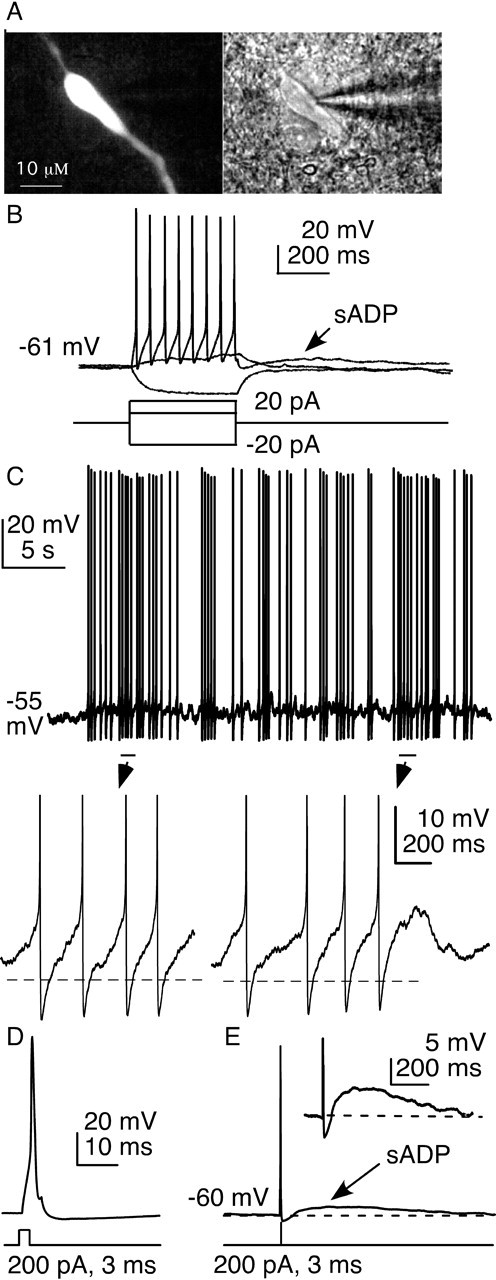
Firing properties and afterdepolarization in GnRH neurons. A, GFP signal (left) and infrared differential interference contrast image (right) of a GFP-identified GnRH neuron. B, Stereotypical response of a GnRH neuron to current injection between pulse and surge modes of secretion that is critical to reproductive success. C, Spontaneous action potentials are followed by ADPs (below, spikes truncated). Dashed line indicates prespike baseline. D, Brief current injections are terminated before the evoked spike begins. Bottom, Current injection protocol; top, membrane potential response. E, Brief current injection triggers action potentials followed by an sADP; inset shows expanded scale (spikes truncated in inset).
GnRH neurons exhibit a prominent sADP
Action potential generation entails regenerative depolarization followed by a restorative hyperpolarization. These two phases are mediated by fast voltage-dependent sodium current and delayed voltage-dependent potassium current, respectively (Hodgkin and Huxley, 1952; Connors et al., 1982). In examining the membrane potential of GnRH neurons in current clamp (Fig. 1C), we noticed that spontaneous action potentials were often followed by ADPs. To study ADPs under consistent conditions, action potentials were induced in current-clamp mode by injection of a brief depolarization current pulse (200–300 pA, 3–5 ms duration). Brief injections were used because they are completed before much of the action potential waveform occurs (Fig. 1D). The changes in membrane potential observed are thus more likely attributable to native conductances, not injected current.
GnRH neurons typically responded to this current injection with a single spike, followed immediately by an AHP, and then an ADP that was slow to develop and decay, hence termed a sADP (Fig. 1E). The sADP is the prominent and prolonged period during which the membrane potential of the cell exceeds the prespike potential. Most GnRH neurons (87%, 114 of 131 cells) in the preoptic area and ventral hypothalamus exhibited sADPs after action potential generation. sADPs were also evident after more prolonged, lower-amplitude current injections that generated action potential firing (Fig. 1B), but the remainder of the characterization of this phenomenon was performed with the above-described brief current injections. Six unidentified neurons in the vicinity of GnRH neurons failed to show sADPs after brief current injections to induce action potential firing (data not shown).
sADP amplitude depends on the number of spikes and membrane potential
When GnRH neurons are recorded extracellularly, bursts of action currents reflecting action potential firing are observed from most cells (Kuehl-Kovarik et al., 2002; Nunemaker et al., 2002, 2003b; Abe and Terasawa, 2005; Chu and Moenter, 2005; Pielecka and Moenter, 2006; Pielecka et al., 2006). These are often grouped so that approximately two to six or more action currents occur over a few hundred milliseconds. We thus examined the effects of multiple induced spikes on the sADP. One, two, four, or eight spikes were induced by 3 ms, 200–300 pA current injection at 50 ms intervals (Fig. 2A,B). sADP amplitude increased (p < 0.01, ANOVA with Bonferroni's post hoc test) from 2.9 ± 0.1 to 3.7 ± 0.1 to 4.5 ± 0.1 mV from one (n = 44) to two (n = 44) to four (n = 44) spikes, respectively, but was not further increased by eight spikes (4.5 ± 0.3 mV; n = 17) (Fig. 2C). Likewise, duration of the sADP was increased from 1.08 ± 0.2 s with one spike (n = 30) to 1.38 ± 0.03 s with two spikes (n = 30; p < 0.001) to 1.71 ± 0.03 s with four spikes (n = 30; p < 0.001) but was not further increased by eight spikes (1.86 ± 0.04 s; n = 10; Kruskal–Wallis with Dunn's post hoc test) (Fig. 2D). Latency to peak of the sADP from the threshold of the final spike was not affected by spike number (191 ± 7 ms, n = 30; 194 ± 7 ms, n = 30; 199 ± 7 ms, n = 30; and 185 ± 7 ms, n = 17, respectively) (Fig. 2E). This suggests that the magnitude, but not the onset, of sADP is dependent on the preceding stimulus up to a point at which amplitude is maximized. Of interest, many extracellularly recorded bursts consist of fewer than eight action currents, suggesting that the regulation of sADP parameters occurs over the physiological range of short-term firing patterns of GnRH neurons.
Figure 2.
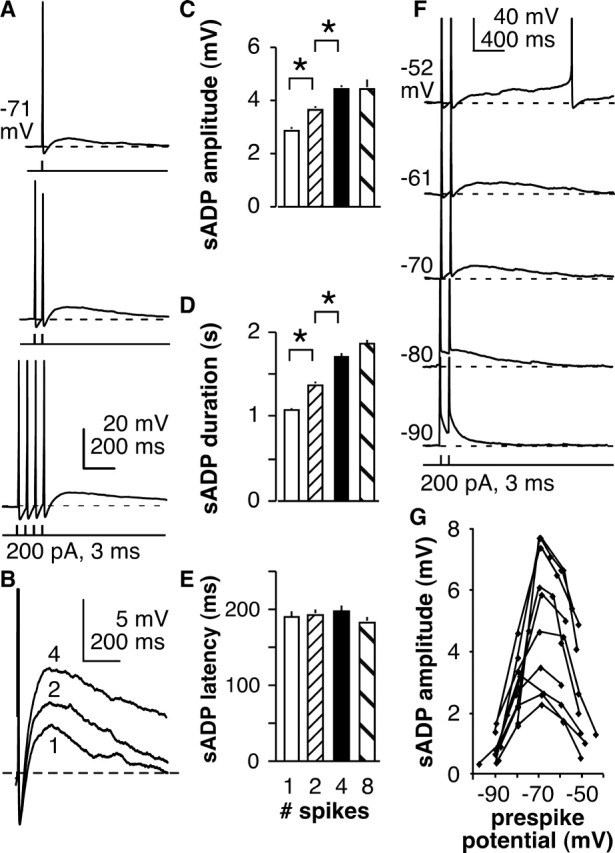
sADPs are dependent on the number of preceding spikes and membrane potential. A, Representative example of sADP after one (top), two (middle), or four (bottom) preceding spikes. Data are aligned by last induced spike. Dashed line shows prespike baseline. B, Expansion of sADP from A; spikes are truncated. The number near the trace indicates number of preceding spikes. C–E, Mean ± SEM sADP amplitude, duration, and latency, respectively, as a function of spike number. *p < 0.05. F, Representative example of sADP after two spikes in an individual cell in which membrane potential was adjusted to the values shown on the left by current injection. Spikes are truncated; dashed lines show prespike baseline. G, Plot of sADP amplitude versus membrane potential for all 10 cells tested.
The voltage dependence of the sADP was investigated next. The membrane potential of GnRH neurons was maintained from −90 to −50 mV at ∼10 mV intervals by injecting negative or positive DC. At each potential, a pair of spikes was initiated (200–350 pA, 3 ms, 50 ms interval), and the sADP amplitude was determined 200 ms after the termination of the final spike. The amplitude of current injection was varied to the minimum needed to induce spikes to avoid overstimulation. A representative example of sADPs from a GnRH neuron at different membrane potentials is illustrated in Figure 2F; data from all 10 cells are plotted in Figure 2G. Amplitude exhibited clear voltage dependence, peaking between −65 and −70 mV. The sADP is less evident at very hyperpolarized potentials near EK. Amplitude of the sADP was also reduced at more depolarized potentials (e.g., near −50 mV). With regard to the latter, it should be noted that spontaneous firing sometimes precluded measurement of the sADP at more depolarized potentials. The peak is near to slightly negative to the membrane potentials typical of quiescent GnRH neurons, suggesting an early net inward postspike current as a possible mechanism for the sADP. The voltage dependence of the sADP indicates that it is poised to alter the function of these cells under physiological conditions.
sADP is not altered by blockade of neurotransmitter/neuromodulator receptors
A net inward current might arise from intrinsic properties of the neuron or from synaptic inputs. We first examined synaptic mechanisms by testing the effects of blocking the receptors for a variety of neurotransmitters and neuromodulators thought to be key inputs to GnRH neurons. GnRH neurons are spiny (Witkin, 1989; Campbell et al., 2005) and express functional iGluRs (Brann and Mahesh, 1994; Eyigor and Jennes, 1997; Adams et al., 1999; Suter, 2004). Recent evidence indicates that they express vesicular glutamate transporter 2 (Hrabovszky et al., 2004) and are functionally glutamatergic (Chu and Moenter, 2005). mGluRs modulate the ADP in CA3 hippocampal neurons (Young et al., 2004). Blockade of either iGluRs (n = 5) or mGluRs (n = 8), however, had no effect on the sADP (Fig. 3A, iGluR raw data, D, summary). A main input to GnRH neurons is GABAergic via the GABAA receptor (Sim et al., 2000; Jansen et al., 2003; Pompolo et al., 2003; Sullivan et al., 2003; Sullivan and Moenter, 2004a,b, 2005), but blocking GABAA receptors did not alter the sADP (n = 7) (Fig. 3B,D). GnRH peptide has been reported to have both stimulatory and inhibitory effects on GnRH neurons and on the anterior pituitary (Tse et al., 1994; Xu et al., 2004), but blocking receptors for this hormone did not alter the sADP (n = 5) (Fig. 3C,D). A high percentage of GnRH neurons coexpress galanin (Merchenthaler et al., 1990; Hohmann et al., 1998) and galanin receptor is also expressed in these neurons (Mitchell et al., 1999), but blocking galanin receptors was also without effect on the sADP (n = 8) (Fig. 3D). ATP can mediate fast synaptic transmission (Edwards et al., 1992; Evans et al., 1992), and a role for purinergic receptors in GnRH release and in synchronizing calcium oscillations in GnRH neurons has been suggested recently (Terasawa et al., 2005). The broadspectrum purinergic receptor antagonist suramin had no effect on the sADP in GnRH neurons (n = 6) (Fig. 3D). Although this list is not exhaustive, the lack of effect of these receptor blockers suggests that the sADP may be intrinsically generated. Further supporting this notion, the sADP persists in isolated GnRH neurons (Kuehl-Kovarik et al., 2005).
Figure 3.
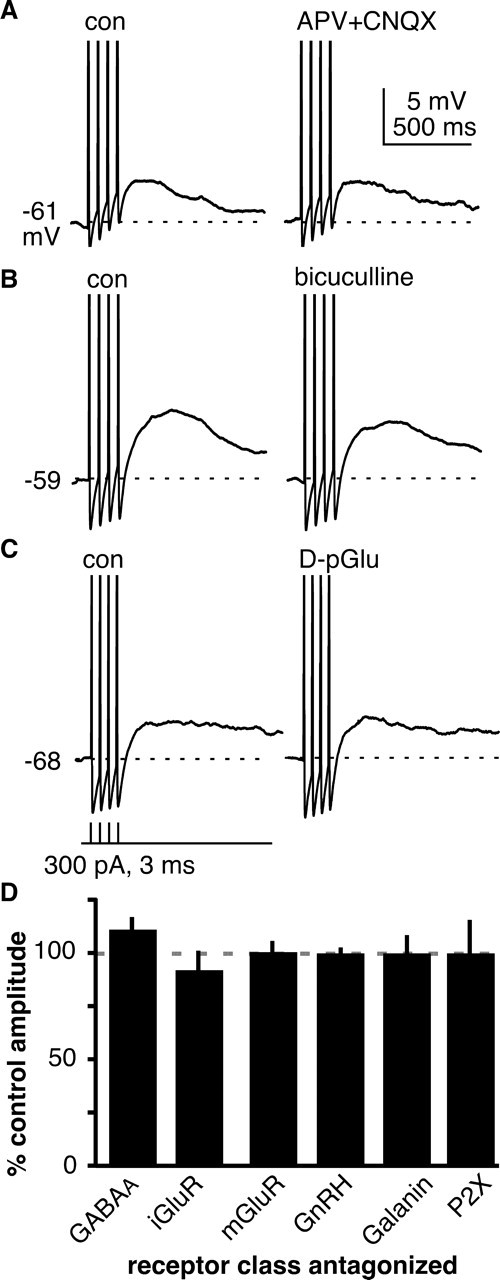
sADPS are not altered by selected neurotransmitter and neuromodulator receptor blockers. A–C, Representative examples of sADP before and after treatment with receptor blockers (A, 20 μm APV plus 20 μm CNQX; B, 20 μm bicuculline; C, 20–40 nm d-P-Glu). Spikes are truncated; dashed lines show prespike baseline. D, Mean ± SEM change in sADP amplitude in response to receptor blockade. Con, Control.
Blocking specific conductances can block the sADP
To explore possible intrinsic mechanisms underlying the sADP, we individually blocked candidate ion channels. Hyperpolarization-activated currents (Ih) are involved in pacemaking and recovery from hyperpolarization in some neurons (Robinson and Siegelbaum, 2003). GnRH neurons have been reported to express Ih in guinea pigs and mice (Kelly and Wagner, 2002) (Z. Chu, H. Takagi, and S. M. Moenter, unpublished observation). ZD7288, a fairly specific inhibitor of the HCN channels through which Ih is conducted, did not significantly alter sADP amplitude in GnRH neurons (n = 6) (Figs. 4A, 5F). An sADP with similar kinetics is observed in magnocellular neuroendocrine neurons (Andrew and Dudek, 1983, 1984; Li and Hatton, 1996); it has been proposed that a reduction in outward TEA-sensitive IK generates this sADP (Li and Hatton, 1997b). The sADP of GnRH neurons, however, was not affected by TEA (n = 7) (Figs. 4B, 5F). Similarly, blockade of calcium-dependent cation channels with either Cs+ (which also blocks Ih and some IK) or flufenamic acid had no effect on the sADP (Fig. 5F) (Ghamari-Langroudi and Bourque, 1998, 2002).
Figure 4.

Effect of channel blockers on the sADP. A, sADP recorded before (black) and after (gray) blocking Ih with ZD7288 (40 μm). B, sADP recorded before (black) and after (gray) blocking IK with TEA (10 μm). C, D, Absolute amplitude of the sADP is decreased by application of cadmium (Cd2+, 200 μm) to block calcium channels. E, F, Cadmium reduces ADP amplitude (E) and latency (F); each individual value is shown with horizontal line indicating mean). G, Cadmium also increases amplitude of the AHP relative to prespike baseline. Spikes are truncated; dashed lines show prespike baseline. *p < 0.05; **p < 0.001. con, Control.
Figure 5.
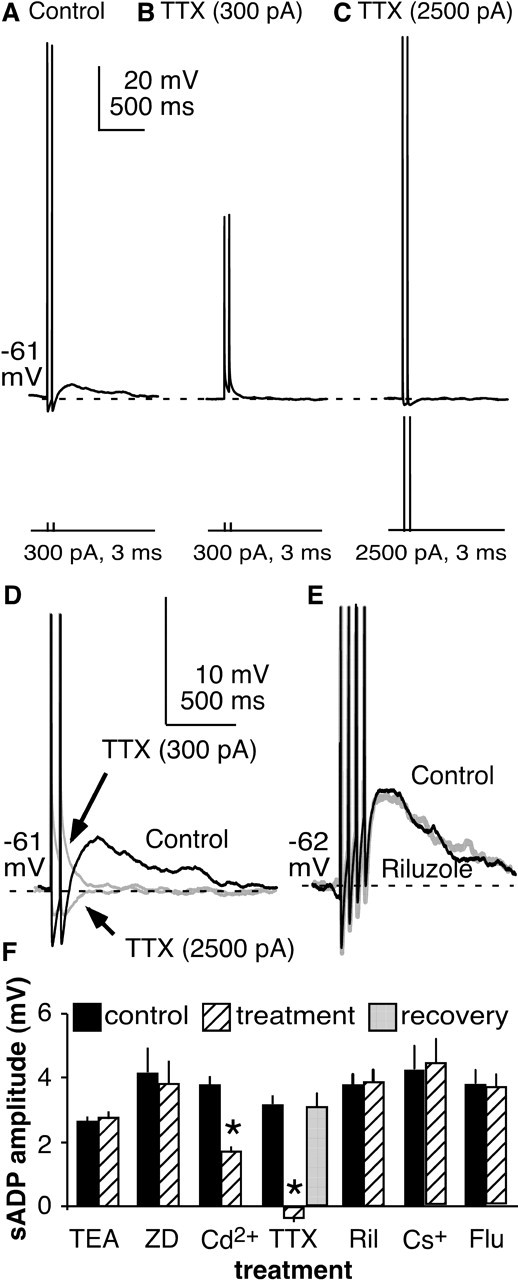
Blocking specific sodium conductances eliminates sADP. A, Two brief depolarizing current pulses (300 pA, 3 ms) evokes two spikes followed by an sADP. B, TTX (0.5 μm) eliminates the sADP. C, Higher-amplitude current pulses (2500 pA, 3 ms) restore membrane potential changes but not sADP. D, Overlap of expanded and truncated traces from A–C. E, The persistent sodium current blocker riluzole (5–10 μm) had no effect on sADP amplitude. F, Summary of pretreatment (black) and posttreatment (hatched) sADP amplitudes by treatment. Dashed lines show prespike baseline. ZD, ZD7288; Ril, riluzole; Flu, flufenamic acid (200 μm). *p < 0.001.
Calcium channels can mediate depolarization either near the resting potential (low-voltage-activated) or as a consequence of activation during action potentials (high-voltage-activated) (Perez-Reyes, 2003; Lacinova, 2005). Elevations in intracellular calcium from a variety of sources are critical in neurotransmitter and hormone release. To test for a role of calcium in mediating the sADP, the broadspectrum calcium channel blocker cadmium (200 μm) was used. Cadmium reduced the amplitude of the sADP by ∼60% (p < 0.001; n = 7) (Figs. 4C–E, 5F). Similarly, removal of calcium from the external solution also reduced the amplitude of the sADP (p < 0.05; n = 4; data not shown). Cadmium also reduced the latency to peak of the sADP (p < 0.002) (Fig. 4F) and increased the amplitude of the AHP (p < 0.05) (Fig. 4G). Overall magnitude of the sADP measured from the most hyperpolarized potential of the AHP to the peak of the sADP was not altered by cadmium (9.3 ± 0.7 mV; control, 9.6 ± 0.6 mV). Together, these studies suggest that blocking calcium channels alters the time course of the sADP, perhaps by blocking the influence of calcium-activated potassium currents, but do not contribute to the depolarization of the sADP itself.
Voltage-dependent sodium channels play a major role in determining neuronal activity. Persistent and resurgent sodium currents can contribute to in regenerative spiking (Crill, 1996; Raman and Bean, 1997; Agrawal et al., 2001). TTX (n = 6; 0.5 μm) reversibly abolished the sADP in GnRH neurons after induced spikes (p < 0.0001) (Fig. 5A,B,F). An obvious caveat of this experiment is that, in the presence of TTX, the membrane does not go through the same changes in potential as during an action potential. A loss of activation of voltage-gated conductances that occurs secondary to loss of the action potential could thus explain the lack of sADP after blocking sodium channels. To correct for this, we increased the amplitude of the current injection so that the capacitive changes in membrane potential were of similar magnitude to the action potential (Swensen and Bean, 2003). Under these conditions, TTX still eliminated the sADP (n = 4) (Fig. 5C,D). The slow kinetics of the sADP suggest that it may be mediated by a persistent sodium current (INaP). Riluzole is reported to block INaP, but it failed to block the sADP, even at high concentrations (n = 7) (Fig. 5E). As a control, we showed that these concentrations of riluzole blocked a majority of the inward persistent sodium current generated by a voltage ramp (−80 to −20 mV at 50 mV/s) in CA1 pyramidal neurons as reported previously (Yue et al., 2005) (data not shown). These data suggest that current through TTX-sensitive voltage-dependent sodium channels is a key mechanism underlying the sADP.
Intrinsic mechanisms underlying the sADP
Voltage clamp was used to explore the current that underlies the sADP (IADP). Two types of voltage commands were used. Short square-wave depolarizing steps (2 ms, −60 to +20 mV) (Fig. 6A, left) approximate the depolarization of an action potential. Because a square wave does not include the afterhyperpolarization, which is typically pronounced in GnRH neurons, a voltage command more closely replicating an action potential was also used (Fig. 6A, middle, right). Both regimens yielded a similar inward current (IADP); data from these two commands were pooled. Latency to the peak of IADP after a single simulated action potential was 95.5 ± 4.3 ms (n = 24), which is shorter than the latency to the peak of the sADP. This advancement in the peak of the current relative to the sADP would be expected by the time required to charge the membrane capacitance in current clamp.
Figure 6.
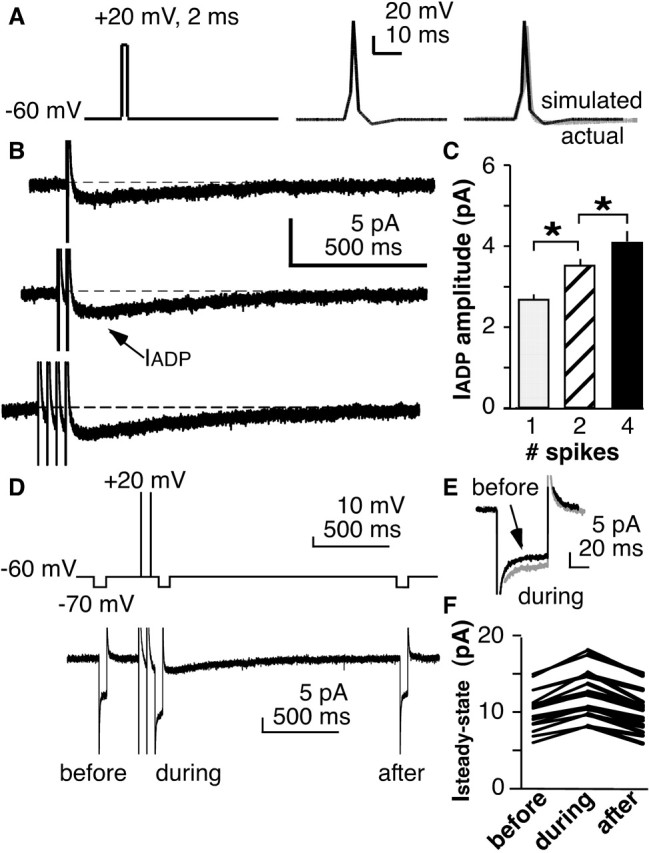
An inward current underlies the sADP. A, Square pulses (left) or simulated action potentials (middle) were used as voltage commands to elicit the current underlying the sADP. Right shows comparison of simulated action potential (black) versus actual action potential (gray). B, Inward current (IADP) is elicited by voltage commands shown in A. B, C, Increasing the number of simulated spikes increases the current. D, Top, Voltage protocol to estimate input resistance before, during, and after induced IADP. Bottom, Corresponding current trace. E, Overlap of current trace from before and during IADP showing increased steady-state conductance during IADP. F, Changes in Isteady-state in all cells tested illustrating the increase in current and return to precommand levels. *p < 0.001.
In current-clamp experiments, inducing multiple spikes increased the amplitude of the sADP (Fig. 2). To test whether IADP had a similar dependence, one, two, or four action potential clamp commands were given at 50 ms intervals (Fig. 6B). A single command produced an inward current of 2.7 ± 0.2 pA; this current increases to 3.5 ± 0.2 pA (p < 0.001) after double stimulation and to 4.1 ± 0.2 pA after quadruple stimulation (p < 0.001; n = 24). Both the timing of the inward current and its dependence on the number of preceding stimuli are consistent with the hypothesis that this inward current mediates the sADP.
Previous studies of DAPs in magnocellular neuroendocrine neurons had proposed a reduction in potassium conductance via delayed rectifier channels as the mediator of the inward current (Li and Hatton, 1997b). The voltage dependence of the sADP in GnRH neurons is perhaps consistent with this notion because the sADP was minimal near the reversal potential for potassium (Fig. 2). We thus explored the change in overall membrane conductance during the IADP by measuring input resistance before and during the inward current (n = 17). A small but consistent reduction in input resistance was observed during the inward current (Fig. 6D–F), suggesting that it is mediated by activation rather than inhibition of a conductance.
Specific blockers were next used to explore the ionic nature of IADP. Consistent with the observed increase in conductance during IADP, TEA had no effect on amplitude of the inward current (n = 10) (Fig. 7A,G,H). Likewise, Cs+ and flufenamic acid had no effect on IADP (Fig. 7H). Cadmium did not alter the amplitude of the IADP (n = 15) (Fig. 7B,C,G,H). Cadmium did, however, reduce the latency to peak of IADP (p < 0.01, n = 14) (Fig. 7D), similar to the reduction it induced in latency to peak of the sADP (Fig. 4). This raised the question of whether or not the latency reduction may represent a secondary effect of cadmium on calcium-sensitive potassium channels. Treatment with blockers (iberiotoxin, 100 nm; apamin, 200 nm; n = 11) of calcium-activated potassium currents (IKCa) reduced latency to peak of the IADP (p < 0.02) and, furthermore, blocked subsequent reductions in latency by cadmium (Fig. 7G,H). These data are consistent with the hypothesis that cadmium reduces latency by blocking IKCa. Interestingly, blocking IKCa tended to increase current amplitude (single spike, 3.0 ± 0.3 vs 3.2 ± 0.4 pA, p < 0.02; double spike, 3.7 ± 0.4 vs 3.9 ± 0.4 pA, p < 0.08; quadruple spike, 4.4 ± 0.5 vs 4.6 ± 0.5 pA, p < 0.05; n = 11) (double spike data shown in Fig. 7H; because this effect varied in significance with spike number, no significance marker is shown in Fig. 7). This suggests that IKCa activated during action potentials may oppose IADP and limit the magnitude of the sADP. In contrast to the lack of effect of other ion channel blockers on IADP, TTX eliminated IADP as it had the sADP (n = 12) (Fig. 7E,F,H). Together, these data indicate the sADP is mediated by a slow inward TTX-sensitive sodium current.
Figure 7.
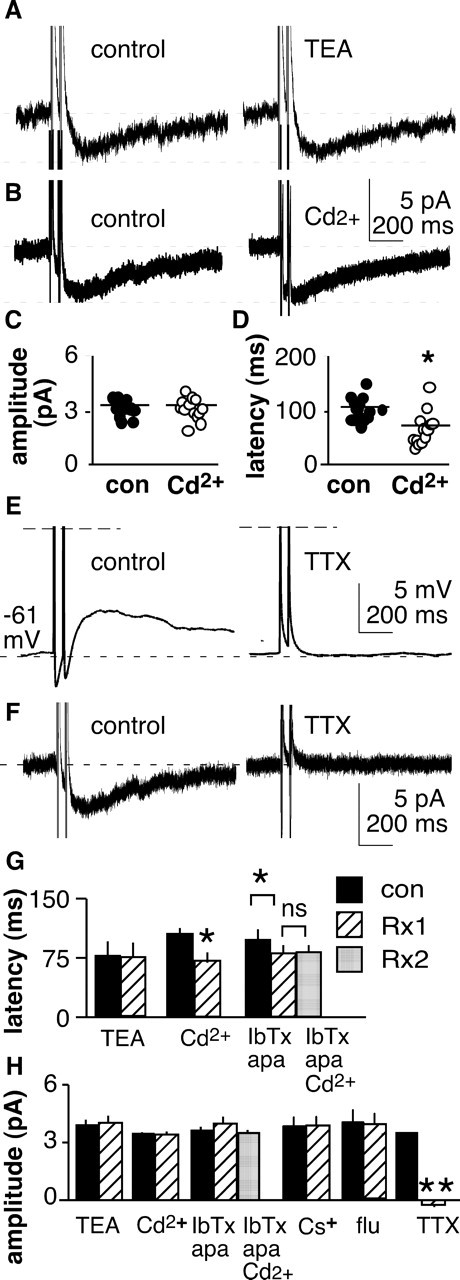
Effect of channel blockers on IADP. A, TEA (10 μm) does not alter IADP. B–D, Cadmium (200 μm) does not affect amplitude of IADP (B, C) but does reduce latency. C and D show each individual cell with horizontal line indicating the mean. *p < 0.05. sADP and IADP are TTX sensitive. con, Control. E, F, Current-clamp (E) and voltage-clamp (F) recordings of the same cell before (left) and after (right) application of TTX. G, H, Summary of effect of channel blockers on IADP latency (G) and amplitude (H). IbTx, 100 μm iberiotoxin; apa, 200 μm apamin; Rx, treatment. *p < 0.05; **p < 0.001.
Function of sADPs in GnRH neuron excitability and regulation by estradiol
The amplitude and duration of the afterpotential, including hyperpolarizing and depolarizing components, can influence neuronal excitability and spike firing patterns. To examine the role of sADPs in controlling GnRH neuron activity, current-clamp recordings were made of GnRH neurons, and the effect on pattern of firing was examined. Strong sADPs were suprathreshold, with spontaneous action potentials riding on the depolarizing envelope (Fig. 8A). This suggests that sADPs can alter the pattern of GnRH neuron firing.
Figure 8.
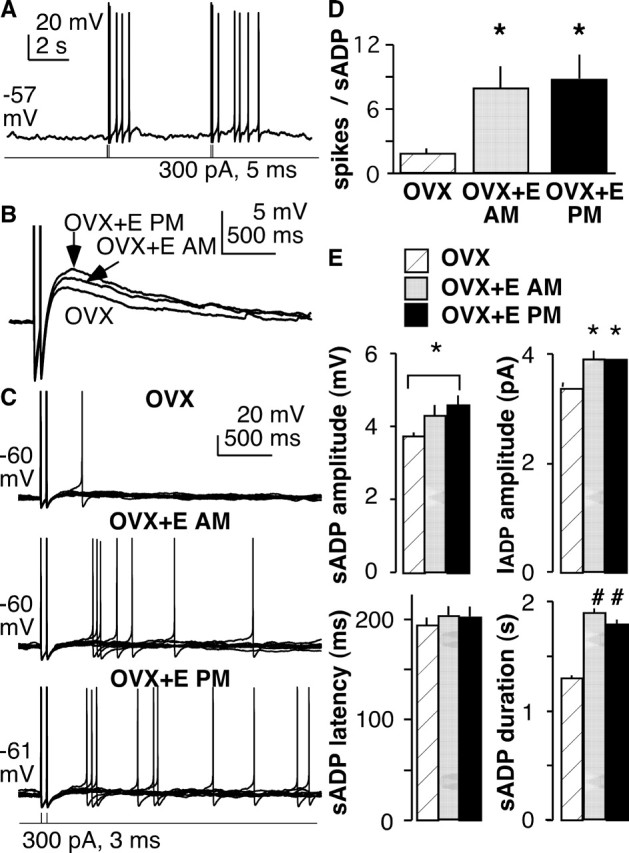
sADPs contribute to repeat firing in GnRH neurons, and both sADPs and the resulting firing are enhanced by estradiol. A, Spontaneous action potentials can occur during the sADP. B, Superimposition of sADP in representative cells (Vm of −58 to −62 mV) from OVX and OVX plus estradiol mice showing the increased magnitude induced by estradiol (measured at 200 ms). C, Current-clamp recordings showing that induction of a pair of spikes by brief current injection elicits spontaneous spikes during the subsequent sADP and that this is enhanced by estradiol treatment in both the morning and evening. Ten traces from each cell are superimposed. D, Effect of estradiol on spikes generated during the sADP. E, Summary of estradiol effects on sADP amplitude, latency, and duration and IADP amplitude. *p < 0.05; #p < 0.01, unpaired analyses. E, Estradiol; AM, morning; PM, evening.
To test whether the sADP is subject to physiological regulation, we chose an established model of estradiol feedback, treating some animals in vivo with estradiol implants (Christian et al., 2005). Recordings from the latter animals were made in both the morning and evening because estradiol feedback is regulated by the circadian clock (Chappell, 2005). Specifically, estradiol exerts negative feedback in the morning and positive feedback in the evening; in ovary-intact nocturnal rodents, this times the ovulatory signal to the onset of night. The mechanisms for this switch in feedback action are unknown but likely involve both synaptic and intrinsic mechanisms. Estradiol-induced feedback persists in GnRH neurons in brain slices, with reduced GnRH neuron activity in the morning and increased activity in the evening; in contrast, there are no diurnal changes in GnRH neuron activity in OVX mice (Christian et al., 2005).
To facilitate comparison between cells, DC (≤10 pA) was injected to maintain membrane potential between −58 and −62 mV. There was no difference in either resting membrane potential before this injection (OVX, n = 20, −64.3 ± 1.1 mV; OVX plus estradiol in the morning, n = 5, 61.6 ± 2.4 mV; OVX plus estradiol in the evening, n = 14, 62.7 ± 2.2 mV) or in input resistance (OVX, 794 ± 57 MΩ; OVX plus estradiol in the morning, 849 ± 69 MΩ; OVX plus estradiol in the evening, 820 ± 61 MΩ) among the cells studied. Two brief current injections were used to trigger action potentials as described above. Estradiol increased sADP duration and amplitude (Fig. 8B,E). Estradiol increased the frequency of spontaneous action potentials during the depolarizing envelope of the sADP by an average of fivefold (Fig. 8C,D). This observation is consistent with a recent report showing that estradiol acutely increases firing activity when applied in the bath solution (Abe and Terasawa, 2005) (note that all estradiol treatments in the present study were in vivo). Estradiol also increased IADP amplitude (Fig. 8E). Most estradiol effects on the sADP were independent of time of day. This suggests that effects on the sADP and resulting changes in GnRH neuron excitability are a response of this system to estradiol alone and that time-of-day information is conveyed via alternative mechanisms such as changes in synaptic transmission. This estradiol-induced increase in sADP and excitability may be an important component of the increased firing rate of GnRH neurons that occurs at the time of the GnRH surge.
Discussion
GnRH neurons express a variety of intrinsic conductances that shape their firing patterns in an incompletely understood manner (Bosma, 1993; Kusano et al., 1995; DeFazio and Moenter, 2002; Kelly and Wagner, 2002; Moenter et al., 2003) (Chu, Takagi, and Moenter, unpublished observation). Here, we show that a TTX-sensitive sodium conductance mediates an intrinsically generated sADP in GnRH neurons. This sADP is modulated by estradiol and can contribute to repetitive firing in these cells and potentially to hormone release. Although these studies were conduced on sADPs after induced spikes, spontaneous spikes are also followed by sADPs, indicating that sADPs are a component of nonstimulated GnRH neuron function.
ADPs in neuroendocrine and non-neuroendocrine neurons can facilitate burst firing. The ADP in GnRH neurons more closely resembles that in other neuroendocrine neurons in amplitude, delayed onset after an AHP, and longer duration (Andrew and Dudek, 1984; Li and Hatton, 1997a,b; Kuehl-Kovarik et al., 2005) than that observed in neurons of the hippocampus and cortex (Metz et al., 2005; Yue et al., 2005). The different characteristics of ADPs may relate to different functions of these cell types. The long duration of neuroendocrine ADPs provides an extended period of increased excitability. Neuroendocrine neurons studied thus far are spontaneously active but tend to have low firing rates compared with other neurons. The activity of a network of neuroendocrine neurons often must be synchronized to produce the discrete pulses of hormone release required for function of downstream targets. The extended period of increased excitability after a GnRH neuron fires an action potential may help increase response to excitatory feedforward and feedback mechanisms required to recruit the network to activity.
Similar to magnocellular neurons (Andrew and Dudek, 1984; Li and Hatton, 1997a), GnRH neuron sADP amplitude was dependent on the number of preceding spikes, but only to a point. sADPs in these cells appear to be self-limiting. By producing a finite increase in excitability, sADPs may dictate a pattern of phasic burst firing, with groupings of action potentials separated by quiescence, rather than a tonic increase in firing rate. Burst firing causes increased hormone release (Dutton and Dyball, 1979), suggesting that sADPs may play a role in this primary function of neuroendocrine neurons. Interestingly, qualitative examination of GnRH neuron firing patterns indicates that many fire groups of action potentials the same size as the number of spikes needed to maximize the sADP.
Mechanisms that underlie ADPs can be broadly classified as intrinsic and network. Blockade of selected transmitter and neuromodulator receptors failed to alter the sADP in GnRH neurons, suggesting that it is intrinsically generated. Consistent with this, sADPs persist in isolated GnRH neurons (Kuehl-Kovarik et al., 2005). Blockade of specific ion channels suggests that calcium, calcium-activated potassium, and sodium conductances play roles in shaping the postspike response in GnRH neurons. Cadmium blockade of calcium channels reduced the absolute amplitude of the sADP, but this appears secondary to reducing latency and increasing the amplitude of the AHP because the trough-to-peak amplitude was similar. This is different from magnocellular neuroendocrine neurons, in which the sADP was completely eliminated by the calcium removal or blockade of release from intracellular stores. In contrast to the altered waveform with cadmium, TTX eliminated the sADP even when the current injection was increased to produce excursions of membrane potential similar to those during an action potential. The voltage dependence of the sADP of GnRH neurons also differs from other neuroendocrine neurons. Specifically, the sADP of GnRH neurons was a bell-shaped curve peaking at approximately −75 mV, whereas that in magnocellular neurons was minimal at −70 mV and not evident at more hyperpolarized potentials. Together, these data suggest different mechanisms.
Voltage clamp was used to explore the mechanism under controlled conditions. In response to a voltage command mimicking an action potential or a square depolarizing pulse of similar duration, GnRH neurons generate an inward current. Several characteristics of this current suggest that it may underlie the sADP. First, it is inward and would thus be depolarizing. Second, its peak is advanced relative to the change in membrane potential of the sADP after a spike; this is expected because of the time required to charge the membrane capacitor. Third, increasing the number of spikes in the voltage command increased the amplitude of the current, just as increasing the number of spikes in current clamp increased the amplitude of the sADP. Finally, the product of this current amplitude and input resistance is similar to the change in membrane potential during the sADP.
Membrane conductance was increased during the inward current of the sADP, suggesting that it is conducted via sodium, calcium, or mixed cation channels rather than reduced outward current. Consistent with this, blocking delayed-rectifier potassium channels did not affect the current. Rather, it was abolished by TTX, suggesting that it is conducted via sodium channels. Cadmium, which reduced the sADP by half, had no effect on the amplitude of the inward current but did reduce the latency to current peak. We hypothesized that this was secondary to blockade of calcium-activated potassium currents in GnRH neurons (Bosch et al., 2002). In support of this, IKCa blockade reduced latency and prevented a subsequent reduction in latency by cadmium. IKCa blockade had a strong tendency to increase amplitude of the inward current, suggesting that IKCa opposes and/or times generation of the sADP.
Two of the remaining questions are the nature of the sodium conductance and the subcellular location of the channels. With regard to the former, two possibilities are persistent sodium current (INaP) and resurgent sodium currents. (INaR). INaP can generate the progressive depolarization of a pacemaker that brings neurons to the threshold for firing (Alonso and Llinas, 1989; Pape et al., 1998; Bennett et al., 2000; Taddese and Bean, 2002; Del Negro et al., 2005), as well as amplify dendritic depolarizations and facilitate repetitive firing (Schwindt and Crill, 1995; Stuart and Sakmann, 1995; Parri and Crunelli, 1998). INaR facilitates conglomerate firing in cerebellar Purkinje neurons (Raman and Bean, 1997). The onset of IADP in GnRH neurons appears slower than reported for INaR, and the time course is prolonged. Although INaR flows through the same pore as the transient sodium current of the action potential spike (INaT) (Grieco et al., 2005), there is ongoing debate about whether INaT and INaP are conducted through the same pore. In the present study, TTX but not riluzole, which blocks INaP, abolished the sADP. These data suggest either that different pores conduct these currents or that the pharmacological sensitivity of a single pore is state dependent, and that the state which conducts IADP is sensitive to TTX but not riluzole.
With regard to subcellular location, dendritic sodium channels can promote retrograde conduction of a spike from soma to dendrite or amplify the weight of synaptic transmission by initiating a spike at the dendritic level (Benardo et al., 1982; Huguenard et al., 1989; Kim and Connors, 1993; Stuart and Sakmann, 1994; Hausser et al., 2000, 2004). Although dendritic sodium currents can modulate the excitability of soma (Fernandez et al., 2005), the persistence of the sADP in isolated GnRH neurons lacking visible dendrites argues that the operative sodium channels are proximal to the soma (Kuehl-Kovarik et al., 2005).
Regardless of the precise mechanism generating sADPs, they clearly modulate the firing properties of GnRH neurons. Spontaneous excitatory discharges rode on top of induced sADPs, changing the firing pattern of GnRH neurons. This response is modulated by an important physiological regulator. Specifically, magnitude of the sADP, IADP, and frequency of spontaneous discharges were all increased by estradiol. The apparent discrepancy between a lack of time of day effect for estradiol enhancement of the sADP and previous work in this animal model indicating firing rate of GnRH neurons is elevated only during positive feedback (in the evening) (Christian et al., 2005) can be resolved by invoking different mechanisms estradiol may use to induce the positive feedback response: changes in synaptic transmission to GnRH neurons and changes in the intrinsic properties of GnRH neurons. The present work indicates that sADPs are intrinsically generated and that estradiol enhancement of sADPs increases firing after initiation of an action potential. Although GnRH neurons fire spontaneously, firing rate is likely modulated by neurotransmission. The response of GnRH neurons to excitatory inputs would be amplified during the sADP, and this amplification would be increased by estradiol. Ongoing studies indicate that estradiol increases depolarizing transmission during positive feedback but decreases transmission during negative feedback (C. A. Christian and S. M. Moenter, unpublished observation). Estradiol enhancement of the intrinsic sADP and perhaps changes in other conductances (DeFazio and Moenter, 2002) may facilitate firing in response to increased transmission in the evening. In ovary-intact cycling animals, such estradiol-induced changes could combine to produce the surge mode of GnRH release and ovulation.
We hypothesize that intrinsic generation of sADPs in GnRH neurons not only integrates with other intrinsic conductances to generate burst firing but also provides, through prolonged depolarization, an altered responsiveness to network influences. This may help increase the robustness of GnRH neuron rhythms, alter the frequency of GnRH pulses, help synchronize the network to produce the overall pattern of hormone release, and participate in the switch.
Footnotes
This work was supported by National Institutes of Health Grant HD34860. We thank Susan Xu and Debra Fisher for excellent technical assistance and Catherine Christian, Manoj Patel, Justyna Pielecka, Alison Roland, and Pei-San Tsai for useful editorial comments. Parts of this work have been presented previously in abstract form at the 2004 and 2005 Society for Neuroscience Meetings.
References
- Abe H, Terasawa E. Firing pattern and rapid modulation of activity by estrogen in primate luteinizing hormone releasing hormone-1 neurons. Endocrinology. 2005;146:4312–4320. doi: 10.1210/en.2005-0435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams MM, Flagg RA, Gore AC. Perinatal changes in hypothalamic N-methyl-d-aspartate receptors and their relationship to gonadotropin-releasing hormone neurons. Endocrinology. 1999;140:2288–2296. doi: 10.1210/endo.140.5.6749. [DOI] [PubMed] [Google Scholar]
- Agrawal N, Hamam BN, Magistretti J, Alonso A, Ragsdale DS. Persistent sodium channel activity mediates subthreshold membrane potential oscillations and low-threshold spikes in rat entorhinal cortex layer V neurons. Neuroscience. 2001;102:53–64. doi: 10.1016/s0306-4522(00)00455-3. [DOI] [PubMed] [Google Scholar]
- Alonso A, Llinas RR. Subtreshold Na+-dependent theta-like rhythmicity in stellate cells of entorhinal cortex layer II. Nature. 1989;342:175–177. doi: 10.1038/342175a0. [DOI] [PubMed] [Google Scholar]
- Andrew RD, Dudek FE. Burst discharge in mammalian neuroendocrine cells involves an intrinsic regenerative mechanism. Science. 1983;221:1050–1052. doi: 10.1126/science.6879204. [DOI] [PubMed] [Google Scholar]
- Andrew RD, Dudek FE. Analysis of intracellularly recorded phasic bursting by mammalian neuroendocrine cells. J Neurophysiol. 1984;51:552–566. doi: 10.1152/jn.1984.51.3.552. [DOI] [PubMed] [Google Scholar]
- Armstrong WE, Stern JE. Electrophysiological distinctions between oxytocin and vasopressin neurons in the supraoptic nucleus. Adv Exp Med Biol. 1998;449:67–77. doi: 10.1007/978-1-4615-4871-3_7. [DOI] [PubMed] [Google Scholar]
- Azouz R, Jensen MS, Yaari Y. Ionic basis of spike after-depolarization and burst generation in adult rat hippocampal CA1 pyramidal cells. J Physiol (Lond) 1996;492:211–223. doi: 10.1113/jphysiol.1996.sp021302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry PH. JPCalc, a software package for calculating liquid junction potential corrections in patch-clamp, intracellular, epithelial and bilayer measurements and for correcting junction potential measurements. J Neurosci Methods. 1994;51:107–116. doi: 10.1016/0165-0270(94)90031-0. [DOI] [PubMed] [Google Scholar]
- Benardo LS, Masukawa LM, Prince DA. Electrophysiology of isolated hippocampal pyramidal dendrites. J Neurosci. 1982;2:1614–1622. doi: 10.1523/JNEUROSCI.02-11-01614.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett BD, Callaway JC, Wilson CJ. Intrinsic membrane properties underlying spontaneous tonic firing in neostriatal cholinergic interneurons. J Neurosci. 2000;20:8493–8503. doi: 10.1523/JNEUROSCI.20-22-08493.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch MA, Kelly MJ, Rønnekleiv OK. Distribution, neuronal colocalization, and 17β-E2 modulation of small conductance calcium-activated K+ channel (SK3) mRNA in guinea pig brain. Endocrinology. 2002;143:1097–1107. doi: 10.1210/endo.143.3.8708. [DOI] [PubMed] [Google Scholar]
- Bosma MM. Ion channel properties and episodic activity in isolated immortalized gonadotropin-releasing hormone (GnRH) neurons. J Membr Biol. 1993;136:85–96. doi: 10.1007/BF00241492. [DOI] [PubMed] [Google Scholar]
- Bourque CW. Calcium-dependent spike after-current induces burst firing in magnocellular neurosecretory cells. J Neurophysiol. 1986;70:204–209. doi: 10.1016/0304-3940(86)90464-7. [DOI] [PubMed] [Google Scholar]
- Bourque CW, Kirkpatrick K, Jarvis CR. Extrinsic modulation of spike afterpotentials in rat hypothalamoneurohypophysial neurons. Cell Mol Neurobiol. 1998;18:3–12. doi: 10.1023/A:1022566924921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brann DW, Mahesh VB. Excitatory amino acids: function and significance in reproduction and neuroendocrine regulation. Front Neuroendocrinol. 1994;15:3–49. doi: 10.1006/frne.1994.1002. [DOI] [PubMed] [Google Scholar]
- Campbell RE, Han SK, Herbison AE. Biocytin filling of adult gonadotropin-releasing hormone neurons in situ reveals extensive, spiny, dendritic processes. Endocrinology. 2005;146:1163–1169. doi: 10.1210/en.2004-1369. [DOI] [PubMed] [Google Scholar]
- Chappell PE. Clocks and the black box: circadian influences on gonadotropin-releasing hormone secretion. J Neuroendocrinol. 2005;17:119–130. doi: 10.1111/j.1365-2826.2005.01270.x. [DOI] [PubMed] [Google Scholar]
- Christian CA, Mobley JL, Moenter SM. Diurnal and estradiol-dependent changes in gonadotropin-releasing hormone neuron firing activity. Proc Natl Acad Sci USA. 2005;102:15682–15687. doi: 10.1073/pnas.0504270102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu Z, Moenter SM. Endogenous activation of metabotropic glutamate receptors modulates GABAergic transmission to gonadotropin-releasing hormone neurons and alters their firing rate: a possible local feedback circuit. J Neurosci. 2005;25:5740–5749. doi: 10.1523/JNEUROSCI.0913-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu Z, Galarreta M, Hestrin S. Synaptic interactions of late-spiking neocortical neurons in layer 1. J Neuorsci. 2003;23:96–102. doi: 10.1523/JNEUROSCI.23-01-00096.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke IJ, Cummins JT. The temporal relationship between gonadotropin releasing hormone (GnRH) and luteinizing hormone (LH) secretion in ovariectomized ewes. Endocrinology. 1982;111:1737–1739. doi: 10.1210/endo-111-5-1737. [DOI] [PubMed] [Google Scholar]
- Connors BW, Gutnick MJ, Prince DA. Electrophysiological properties of neocortical neurons in vitro. J Neurophysiol. 1982;48:1302–1320. doi: 10.1152/jn.1982.48.6.1302. [DOI] [PubMed] [Google Scholar]
- Crill WE. Persistent sodium current in mammalian central neurons. Annu Rev Physiol. 1996;58:349–362. doi: 10.1146/annurev.ph.58.030196.002025. [DOI] [PubMed] [Google Scholar]
- DeFazio RA, Moenter SM. Estradiol feedback alters potassium currents and firing properties of gonadotropin-releasing hormone neurons. Mol Endocrinol. 2002;16:2255–2265. doi: 10.1210/me.2002-0155. [DOI] [PubMed] [Google Scholar]
- Del Negro CA, Morgado-Valle C, Hayes JA, Mackay DD, Pace RW, Crowder EA, Feldman JL. Sodium and calcium current-mediated pacemaker neurons and respiratory rhythm generation. J Neurosci. 2005;25:446–453. doi: 10.1523/JNEUROSCI.2237-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Docke F, Dorner G. The mechanism of the induction of ovulation by oestrogens. J Endocrinol. 1965;33:491–499. doi: 10.1677/joe.0.0330491. [DOI] [PubMed] [Google Scholar]
- Dutton A, Dyball RE. Phasic firing enhances vasopressin release from the rat neurohypophysis. J Physiol (Lond) 1979;290:433–440. doi: 10.1113/jphysiol.1979.sp012781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards FA, Gibb AJ, Colquhoun D. ATP receptor-mediated synaptic currents in the central nervous system. Nature. 1992;359:144–147. doi: 10.1038/359144a0. [DOI] [PubMed] [Google Scholar]
- Erickson KR, Ronnekleiv OK, Kelly MJ. Electrophysiology of guinea-pig supraoptic neurones: role of a hyperpolarization-activated cation current in phasic firing. J Physiol (Lond) 1993;460:407–425. doi: 10.1113/jphysiol.1993.sp019478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans RJ, Derkach V, Suprenant A. ATP mediates fast synaptic transmission in mammalian neurons. Nature. 1992;357:503–505. doi: 10.1038/357503a0. [DOI] [PubMed] [Google Scholar]
- Eyigor O, Jennes L. Expression of glutamate receptor subunit mRNAs in gonadotropin-releasing hormone neurons during the sexual maturation of the female rat. Neuroendocrinology. 1997;66:122–129. doi: 10.1159/000127229. [DOI] [PubMed] [Google Scholar]
- Fernandez FR, Mehaffey WH, Turner RW. Dendritic Na+ current inactivation can increase cell excitability by delaying a somatic depolarizing afterpotential. J Neurophysiol. 2005;94:3836–3848. doi: 10.1152/jn.00653.2005. [DOI] [PubMed] [Google Scholar]
- Fujita Y. Two types of depolarizing after-potentials in hippocampal pyramidal cells of rabbits. Brain Res. 1975;94:435–446. doi: 10.1016/0006-8993(75)90227-9. [DOI] [PubMed] [Google Scholar]
- Ghamari-Langroudi M, Bourque CW. Caesium blocks depolarizing after-potentials and phasic firing in rat supra optic neurones. J Physiol (Lond) 1998;510:165–175. doi: 10.1111/j.1469-7793.1998.165bz.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghamari-Langroudi M, Bourque CW. Flufenamic acid blocks depolarizing afterpotentials and phasic firing in rat supraoptic neurones. J Physiol (Lond) 2002;545:537–542. doi: 10.1113/jphysiol.2002.033589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene CC, Schwindt PC, Crill WE. Properties and ionic mechanisms of a metabotropic glutamate receptor-mediated slow afterdepolarization in neocortical neurons. J Neurophysiol. 1994;72:693–704. doi: 10.1152/jn.1994.72.2.693. [DOI] [PubMed] [Google Scholar]
- Grieco TM, Malhotra JD, Chen C, Isom LL, Raman IM. Open-channel block by the cytoplasmic tail of sodium channel beta4 as a mechanism for resurgent sodium current. Neuron. 2005;45:233–244. doi: 10.1016/j.neuron.2004.12.035. [DOI] [PubMed] [Google Scholar]
- Han SK, Todman MG, Herbison AE. Endogenous GABA release inhibits the firing of adult gonadotropin-releasing hormone neurons. Endocrinology. 2004;145:495–499. doi: 10.1210/en.2003-1333. [DOI] [PubMed] [Google Scholar]
- Hausser M, Spruston N, Stuart GJ. Diversity and dynamics of dendritic signaling. Science. 2000;290:739–744. doi: 10.1126/science.290.5492.739. [DOI] [PubMed] [Google Scholar]
- Hausser M, Raman IM, Otis T, Smith SL, Nelson A, du Lac S, Loewenstein Y, Mahon S, Pennartz C, Cohen I, Yarom Y. The beat goes on: spontaneous firing in mammalian neuronal microcircuits. J Neurosci. 2004;24:9215–9219. doi: 10.1523/JNEUROSCI.3375-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hestrin S, Armstrong WE. Morphology and physiology of cortical neurons in layer I. J Neurosci. 1996;16:5290–5300. doi: 10.1523/JNEUROSCI.16-17-05290.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkin AL, Huxley AF. A quantitative description of membrane current and its application to conduction and excitation in nerve. J Physiol (Lond) 1952;117:500–544. doi: 10.1113/jphysiol.1952.sp004764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohmann JG, Clifton DK, Steiner RA. Galanin: analysis of its coexpression in gonadotropin-releasing hormone and growth hormone-releasing hormone neurons. Ann NY Acad Sci. 1998;863:221–235. doi: 10.1111/j.1749-6632.1998.tb10697.x. [DOI] [PubMed] [Google Scholar]
- Hrabovszky E, Turi GF, Kallo I, Liposits Z. Expression of vesicular glutamate transporter-2 in gonadotropin-releasing hormone neurons of the adult male rat. Endocrinology. 2004;145:4018–4021. doi: 10.1210/en.2004-0589. [DOI] [PubMed] [Google Scholar]
- Huguenard JR, Hamill OP, Prince DA. Sodium channels in dendrites of rat cortical pyramidal neurons. Proc Natl Acad Sci USA. 1989;86:2473–2477. doi: 10.1073/pnas.86.7.2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen HT, Cutter C, Hardy S, Lehman MN, Goodman RL. Seasonal plasticity within the GnRH system of the ewe: changes in identified GnRH inputs and in glial association. Endocrinology. 2003;144:3663–3676. doi: 10.1210/en.2002-0188. [DOI] [PubMed] [Google Scholar]
- Jensen MS, Azouz R, Yaari Y. Spike after-depolarization and burst generation in adult rat hippocampal CA1 pyramidal cells. J Physiol (Lond) 1996;492:199–210. doi: 10.1113/jphysiol.1996.sp021301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandel ER, Spencer WA. Electrophysiology of hippocampal neurons. II. After-potentials and repetitive firing. J Neurophysiol. 1961;24:243–259. doi: 10.1152/jn.1961.24.3.243. [DOI] [PubMed] [Google Scholar]
- Kelly MJ, Wagner EJ. GnRH neurons and episodic bursting activity. Trends Endocrinol Metab. 2002;13:409–410. doi: 10.1016/s1043-2760(02)00698-7. [DOI] [PubMed] [Google Scholar]
- Kim HG, Connors BW. Apical dendrites of the neocortex: correlation between sodium- and calcium-dependent spiking and pyramidal cell morphology. J Neurosci. 1993;13:5301–5311. doi: 10.1523/JNEUROSCI.13-12-05301.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krsmanovic LZ, Stojilkovic SS, Merelli F, Dufour SM, Virmani MA, Catt KJ. Calcium signaling and episodic secretion of gonadotropin-releasing hormone in hypothalamic neurons. Proc Natl Acad Sci USA. 1992;89:8462–8466. doi: 10.1073/pnas.89.18.8462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuehl-Kovarik MC, Pouliot WA, Halterman GL, Handa RJ, Dudek FE, Partin KM. Episodic bursting activity and response to excitatory amino acids in acutely dissociated gonadotropin-releasing hormone neurons genetically targeted with green fluorescent protein. J Neurosci. 2002;22:2313–2322. doi: 10.1523/JNEUROSCI.22-06-02313.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuehl-Kovarik MC, Partin KM, Handa RJ, Dudek FE. Spike-dependent depolarizing afterpotentials contribute to endogenous bursting in gonadotropin releasing hormone neurons. Neuroscience. 2005;134:295–300. doi: 10.1016/j.neuroscience.2005.03.047. [DOI] [PubMed] [Google Scholar]
- Kusano K, Fueshko S, Gainer H, Wray S. Electrical and synaptic properties of embryonic luteinizing hormone-releasing hormone neurons in explant cultures. Proc Natl Acad Sci USA. 1995;92:3918–3922. doi: 10.1073/pnas.92.9.3918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacinova L. Voltage-dependent calcium channels. Gen Physiol Biophys. 2005;24:1–78. [PubMed] [Google Scholar]
- Levine JE, Duffy MT. Simultaneous measurement of luteinizing hormone (LH)-releasing hormone, LH, and follicle-stimulating hormone release in intact and short-term castrate rats. Endocrinology. 1988;122:2211–2221. doi: 10.1210/endo-122-5-2211. [DOI] [PubMed] [Google Scholar]
- Li Z, Hatton GI. Oscillatory bursting of phasically firing rat supraoptic neurones in low-Ca2+ medium: Na+ influx, cytosolic Ca2+ and gap junctions. J Physiol (Lond) 1996;496:379–394. doi: 10.1113/jphysiol.1996.sp021692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Hatton GI. Ca2+ release from internal stores: role in generating depolarizing after-potentials in rat supraoptic neurones. J Physiol (Lond) 1997a;498:339–350. doi: 10.1113/jphysiol.1997.sp021862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Hatton GI. Reduced outward K+ conductances generate depolarizing after-potentials in rat supraoptic nucleus neurones. J Physiol (Lond) 1997b;505:95–106. doi: 10.1111/j.1469-7793.1997.095bc.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luthi A, Bal T, McCormick DA. Periodicity of thalamic spindle waves is abolished by ZD7288, a blocker of Ih. J Neurophysiol. 1998;79:3284–3289. doi: 10.1152/jn.1998.79.6.3284. [DOI] [PubMed] [Google Scholar]
- Martinez de la Escalera G, Choi AL, Weiner RI. Generation and synchronization of gonadotropin-releasing hormone (GnRH) pulses: intrinsic properties of the GT1–1 GnRH neuronal cell line. Proc Natl Acad Sci USA. 1992;89:1852–1855. doi: 10.1073/pnas.89.5.1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merchenthaler I, Lopez FJ, Negro-Vilar A. Colocalization of galanin and luteinizing hormone-releasing hormone in a subset of preoptic hypothalamic neurons: anatomical and functional correlates. Proc Natl Acad Sci USA. 1990;87:6326–6330. doi: 10.1073/pnas.87.16.6326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metz AE, Jarsky T, Martina M, Spruston N. R-type calcium channels contribute to afterdepolarization and bursting in hippocampal CA1 pyramidal neurons. J Neurosci. 2005;25:5763–5773. doi: 10.1523/JNEUROSCI.0624-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell V, Bouret S, Prevot V, Jennes L, Beauvillain JC. Evidence for expression of galanin receptor Gal-R1 mRNA in certain gonadotropin releasing hormone neurones of the rostral preoptic area. J Neuroendocrinol. 1999;11:805–812. doi: 10.1046/j.1365-2826.1999.00399.x. [DOI] [PubMed] [Google Scholar]
- Moenter SM, Caraty A, Locatelli A, Karsch FJ. Pattern of gonadotropin-releasing hormone (GnRH) secretion leading up to ovulation in the ewe: existence of a preovulatory GnRH surge. Endocrinology. 1991;129:1175–1182. doi: 10.1210/endo-129-3-1175. [DOI] [PubMed] [Google Scholar]
- Moenter SM, Brand RM, Midgley AR, Karsch FJ. Dynamics of gonadotropin-releasing hormone release during a pulse. Endocrinology. 1992;130:503–510. doi: 10.1210/endo.130.1.1727719. [DOI] [PubMed] [Google Scholar]
- Moenter SM, DeFazio AR, Pitts GR, Nunemaker CS. Mechanisms underlying episodic gonadotropin-releasing hormone secretion. Front Neuroendocrinol. 2003;24:79–93. doi: 10.1016/s0091-3022(03)00013-x. [DOI] [PubMed] [Google Scholar]
- Nunemaker CS, DeFazio RA, Moenter SM. Estradiol-sensitive afferents modulate long-term episodic firing patterns of GnRH neurons. Endocrinology. 2002;143:2284–2292. doi: 10.1210/endo.143.6.8869. [DOI] [PubMed] [Google Scholar]
- Nunemaker CS, DeFazio RA, Moenter SM. A targeted extracellular approach for recording long-term firing patterns of excitable cells: a practical guide. Biol Proced Online. 2003a;5:53–62. doi: 10.1251/bpo46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunemaker CS, Straume M, DeFazio RA, Moenter SM. Gonadotropin-releasing hormone neurons generate interacting rhythms in multiple time domains. Endocrinology. 2003b;144:823–831. doi: 10.1210/en.2002-220585. [DOI] [PubMed] [Google Scholar]
- Pape HC, Pare D, Driesang RB. Two types of intrinsic oscillations in neurons of the lateral and basolateral nuclei of the amygdala. J Neurophysiol. 1998;79:205–216. doi: 10.1152/jn.1998.79.1.205. [DOI] [PubMed] [Google Scholar]
- Parri HR, Crunelli V. Sodium current in rat and cat thalamocortical neurons: role of a non-inactivating component in tonic and burst firing. J Neurosci. 1998;18:854–867. doi: 10.1523/JNEUROSCI.18-03-00854.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Reyes E. Molecular physiology of low-voltage-activated T-type calcium channels. Physiol Rev. 2003;83:117–161. doi: 10.1152/physrev.00018.2002. [DOI] [PubMed] [Google Scholar]
- Pielecka J, Moenter SM. Steroid negative feedback in males: effect of steroid milieu on GnRH neuron firing pattern and LH levels. Biol Reprod. 2006;74:931–937. doi: 10.1095/biolreprod.105.049619. [DOI] [PubMed] [Google Scholar]
- Pielecka J, Quaynor SD, Moenter SM. Androgens activate GnRH neuron firing in females and interfere with the ability of progesterone to reduce activity of these cells. Endocrinology. 2006;147:1474–1479. doi: 10.1210/en.2005-1029. [DOI] [PubMed] [Google Scholar]
- Pitts GR, Nunemaker CS, Moenter SM. Cycles of transcription and translation do not comprise the gonadotropin-releasing hormone pulse generator in GT1 cells. Endocrinology. 2001;142:1858–1864. doi: 10.1210/endo.142.5.8137. [DOI] [PubMed] [Google Scholar]
- Pompolo S, Pereira A, Kaneko T, Clarke IJ. Seasonal changes in the inputs to gonadotropin-releasing hormone neurones in the ewe brain: an assessment by conventional fluorescence and confocal microscopy. J Neuroendocrinol. 2003;15:538–545. doi: 10.1046/j.1365-2826.2003.01030.x. [DOI] [PubMed] [Google Scholar]
- Poulain DA, Wakerley JB. Electrophysiology of hypothalamic magnocellular neurones secreting oxytocin and vasopressin. Neuroscience. 1982;7:773–808. doi: 10.1016/0306-4522(82)90044-6. [DOI] [PubMed] [Google Scholar]
- Raman IM, Bean BP. Resurgent sodium current and action potential formation in dissociated cerebellar Purkinje neurons. J Neurosci. 1997;17:4517–4526. doi: 10.1523/JNEUROSCI.17-12-04517.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson RB, Siegelbaum SA. Hyperpolarization-activated cation currents: from molecules to physiological function. Annu Rev Physiol. 2003;65:453–480. doi: 10.1146/annurev.physiol.65.092101.142734. [DOI] [PubMed] [Google Scholar]
- Schwindt PC, Crill WE. Amplification of synaptic current by persistent sodium conductance in apical dendrite of neocortical neurons. J Neurophysiol. 1995;74:2220–2224. doi: 10.1152/jn.1995.74.5.2220. [DOI] [PubMed] [Google Scholar]
- Sim JA, Skynner MJ, Pape JR, Herbison AE. Late postnatal reorganization of GABA(A) receptor signalling in native GnRH neurons. Eur J Neurosci. 2000;12:3497–3504. doi: 10.1046/j.1460-9568.2000.00261.x. [DOI] [PubMed] [Google Scholar]
- Sim JA, Skynner MJ, Herbison AE. Heterogeneity in the basic membrane properties of postnatal gonadotropin-releasing hormone neurons in the mouse. J Neurosci. 2001;21:1067–1075. doi: 10.1523/JNEUROSCI.21-03-01067.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spergel DJ, Ulrich D, Hanley DF, Sprengel R, Seeburg PH. GABA- and glutamate-activated channels in green fluorescent protein-tagged gonadotropin-releasing hormone neurons in transgenic mice. J Neurosci. 1999;19:2037–2050. doi: 10.1523/JNEUROSCI.19-06-02037.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart G, Sakmann B. Amplification of EPSPs by axosomatic sodium channels in neocortical pyramidal neurons. Neuron. 1995;15:1065–1076. doi: 10.1016/0896-6273(95)90095-0. [DOI] [PubMed] [Google Scholar]
- Stuart GJ, Sakmann B. Active propagation of somatic action potentials into neocortical pyramidal cell dendrites. Nature. 1994;367:69–72. doi: 10.1038/367069a0. [DOI] [PubMed] [Google Scholar]
- Su H, Alroy G, Kirson ED, Yaari Y. Extracellular calcium modulates persistent sodium current-dependent burst-firing in hippocampal pyramidal neurons. J Neurosci. 2001;21:4173–4182. doi: 10.1523/JNEUROSCI.21-12-04173.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan SD, Moenter SM. γ-Aminobutyric acid neurons integrate and rapidly transmit permissive and inhibitory metabolic cues to gonadotropin-releasing hormone neurons. Endocrinology. 2004a;145:1194–1202. doi: 10.1210/en.2003-1374. [DOI] [PubMed] [Google Scholar]
- Sullivan SD, Moenter SM. Prenatal androgens alter GABAergic drive to gonadotropin-releasing hormone neurons: implications for a common fertility disorder. Proc Natl Acad Sci USA. 2004b;101:7129–7134. doi: 10.1073/pnas.0308058101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan SD, Moenter SM. GABAergic integration of progesterone and androgen feedback to gonadotropin-releasing hormone neurons. Biol Reprod. 2005;72:33–41. doi: 10.1095/biolreprod.104.033126. [DOI] [PubMed] [Google Scholar]
- Sullivan SD, DeFazio RA, Moenter SM. Metabolic regulation of fertility through presynaptic and postsynaptic signaling to gonadotropin-releasing hormone neurons. J Neurosci. 2003;23:8578–8585. doi: 10.1523/JNEUROSCI.23-24-08578.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suter KJ. Control of firing by small (S)-alpha-amino-3-hydroxy-5-methyl-isoxazolepropionic acid-like inputs in hypothalamic gonadotropin releasing-hormone (GnRH) neurons. Neuroscience. 2004;128:443–450. doi: 10.1016/j.neuroscience.2004.06.044. [DOI] [PubMed] [Google Scholar]
- Suter KJ, Song WJ, Sampson TL, Wuarin JP, Saunders JT, Dudek FE, Moenter SM. Genetic targeting of green fluorescent protein to gonadotropin-releasing hormone neurons: characterization of whole-cell electrophysiological properties and morphology. Endocrinology. 2000a;141:412–419. doi: 10.1210/endo.141.1.7279. [DOI] [PubMed] [Google Scholar]
- Suter KJ, Wuarin JP, Smith BN, Dudek FE, Moenter SM. Whole-cell recordings from preoptic/hypothalamic slices reveal burst firing in gonadotropin-releasing hormone neurons identified with green fluorescent protein in transgenic mice. Endocrinology. 2000b;141:3731–3736. doi: 10.1210/endo.141.10.7690. [DOI] [PubMed] [Google Scholar]
- Swensen AM, Bean BP. Ionic mechanisms of burst firing in dissociated Purkinje neurons. J Neurosci. 2003;23:9650–9663. doi: 10.1523/JNEUROSCI.23-29-09650.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taddese A, Bean BP. Subthreshold sodium current from rapidly inactivating sodium channels drives spontaneous firing of tuberomammillary neurons. Neuron. 2002;33:587–600. doi: 10.1016/s0896-6273(02)00574-3. [DOI] [PubMed] [Google Scholar]
- Terasawa E, Keen KL, Grendell RL, Golos TG. Possible role of 5′-adenosine triphosphate in synchronization of Ca2+ oscillations in primate luteinizing hormone-releasing hormone neurons. Mol Endocrinol. 2005;19:2736–2747. doi: 10.1210/me.2005-0034. [DOI] [PubMed] [Google Scholar]
- Tse FW, Tse A, Hille B. Cyclic Ca2+ changes in intracellular stores of gonadotropes during gonadotropin-releasing hormone-stimulated Ca2+ oscillations. Proc Natl Acad Sci USA. 1994;91:9750–9754. doi: 10.1073/pnas.91.21.9750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witkin JW. Morphology of luteinizing hormone-releasing hormone neurons as a function of age and hormonal condition in the male rat. Neuroendocrinology. 1989;49:344–348. doi: 10.1159/000125138. [DOI] [PubMed] [Google Scholar]
- Xu C, Xu XZ, Nunemaker CS, Moenter SM. Dose-dependent switch in response of gonadotropin-releasing hormone (GnRH) neurons to GnRH mediated through the type I GnRH receptor. Endocrinology. 2004;145:728–735. doi: 10.1210/en.2003-0562. [DOI] [PubMed] [Google Scholar]
- Young SR, Chuang S-C, Wong RKS. Modulation of afterpotentials and firing pattern in guinea pig CA3 neurones by group I metabotropic glutamate receptors. J Physiol (Lond) 2004;554:371–385. doi: 10.1113/jphysiol.2003.051847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue C, Yaari Y. KCNQ/M channels control spike afterdepolarization and burst generation in hippocampal neurons. J Neurosci. 2004;24:4614–4624. doi: 10.1523/JNEUROSCI.0765-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue C, Remy S, Su H, Beck H, Yaari Y. Proximal peristent Na+ channels drive spike afterdepolarizations and associated bursting in adult CA1 pyramidal cells. J Neuorsci. 2005;25:9704–9720. doi: 10.1523/JNEUROSCI.1621-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]