

Abstract
G-protein-coupled receptors (GPCRs) mediate slow synaptic transmission and many other effects of small molecule and peptide neurotransmitters. In the standard model of GPCR signaling, receptors and G-proteins diffuse laterally within the plane of the plasma membrane and encounter each other by random collision. This model predicts that signaling will be most efficient if both GPCRs and G-proteins are free to diffuse, thus maximizing collision frequency. However, neuronal GPCRs are often recruited to and enriched at specific synaptic locations, suggesting receptor mobility is restricted in these cells. Here, we test the hypothesis that restricting GPCR mobility impairs signaling in neurons by limiting the frequency of collisions between receptors and G-proteins. μ-Opioid receptors (MORs) were immobilized on the surface of cerebellar granule neurons by avidin-mediated cross-linking, and inwardly rectifying potassium (GIRK) channels were used as rapid indicators of G-protein activation. Mobile and immobile MORs activated GIRK channels with the same onset kinetics and agonist sensitivity in these neurons. In a heterologous expression system, GFP (green fluorescent protein)-tagged GαoA subunits remained mobile after cross-linking, but their mobility was reduced in the presence of immobile MORs, suggesting that these receptors and subunits were transiently precoupled. In addition, channel activation could be reconstituted with immobile GPCRs, G-protein heterotrimers, and GIRK channels. These results show that collision frequency is not rate-limiting for G-protein activation in CNS neurons, and are consistent with the idea that signaling components are compartmentalized or preassembled.
Keywords: GPCR, GIRK channels, metabotropic, diffusion, μ-opioid receptor, G-protein
Introduction
Slow synaptic transmission in the CNS is mediated by metabotropic, G-protein-coupled receptors (GPCRs), which respond to a variety of small molecule and peptide neurotransmitters. GPCRs must directly contact heterotrimeric G-proteins to transduce signals, and this contact is thought to occur by random collision of GPCRs and G-proteins. The best understood G-protein signaling system is that which underlies mammalian phototransduction, in which rapid and sensitive signaling depends on the frequent interaction of GPCRs (rhodopsin) and G-proteins (transducin) in rod outer segment disks (Arshavsky et al., 2002). It is thought that G-protein activation in this and other systems is diffusion-limited, and thus depends on the lateral mobility of GPCRs (Rimon et al., 1978; Calvert et al., 2001). In contrast, the GPCRs that mediate and modulate synaptic transmission in CNS neurons are often enriched at specific (e.g., perisynaptic) locations, which implies that their lateral mobility is limited by interactions with other proteins (Serge et al., 2002; Choquet and Triller, 2003). It has been suggested that such restriction could impair signaling at synapses by limiting collision frequency (Hille, 1992), but this possibility has not been experimentally tested. However, it has also been suggested that synaptic GPCRs function as part of preassembled signaling complexes containing G-proteins and effectors (Rebois and Hebert, 2003), in which case random collision would not occur. We have directly manipulated the lateral mobility of GPCRs to determine whether efficient signal transduction in CNS neurons depends on the frequency of receptor–G-protein collisions. Immobilization of GPCRs in neurons had no effect on the speed or sensitivity of responses mediated by inwardly rectifying potassium (GIRK) channels, suggesting that signaling in this system is not limited by receptor diffusion. In addition, immobile GPCRs decreased the lateral mobility of G-protein heterotrimers, suggesting that these proteins may be transiently preassembled (precoupled). Finally, immobile GIRK channels could be activated by immobile receptors and G-protein heterotrimers. These results do not support an essential role for receptor–G-protein collision in signaling.
Materials and Methods
cDNA constructs, cell culture, and transfection.
pH-MOR and C-MOR (ECFP-MOR) were created by adding a cleavable signal peptide sequence from human growth hormone to the N terminus of either superecliptic pHluorin (pH) (Yuste et al., 2000) or enhanced cyan fluorescent protein (ECFP), and adding the resulting sequence to the N terminus of the rat μ-opioid receptor (MOR). pH-A1R was constructed in an analogous manner using the rat A1 adenosine receptor. GαoA-EGFP was created by fusing enhanced green fluorescent protein (EGFP) (derived from EGFP-N1; Clontech, Palo Alto, CA) in frame between amino acids 91 and 92 of human GαoA with an additional glycine–glycine linker at each junction (Bunemann et al., 2003). ECFP-TM-Gα subunits were created by adding a cleavable signal peptide sequence to the N terminus of enhanced cyan fluorescent protein, fusing this sequence to the amino-terminal 103 aa [including the first transmembrane (TM) domain] of the rat MOR, and fusing this sequence to the N terminus of Gα subunits. pH-TM-GIRK4* was created by adding a cleavable signal peptide sequence, superecliptic pHluorin, and the first TM domain of the MOR to GIRK4(S143T) (GIRK4*) (Chan et al., 1996). All constructs were made using an adaptation of QuikChange (Stratagene, La Jolla, CA) mutagenesis (Geiser et al., 2001), were expressed from pcDNA3.1 (Invitrogen), and were verified by automated sequencing.
HEK 293 cells (American Type Culture Collection, Manassas, VA) were propagated in plastic flasks and on polylysine-coated glass coverslips according to the supplier's protocol. Cerebellar granule neurons (CGNs) were prepared from postnatal day 5–8 Sprague Dawley rats and maintained as described previously (Chen et al., 2004) and in accordance with institutional guidelines. Cells were transfected using polyethyleneimine as described previously (Chen et al., 2004) after 1–7 d in culture, and were used for experiments 12–48 h later.
Avidin cross-linking.
Cells were rinsed three times in buffer containing 150 mm NaCl, 2.5 mm KCl, 10 mm HEPES, 12 mm glucose, 0.5 mm CaCl2, and 0.5 mm MgCl2 (BE buffer; pH 8.0), and incubated at room temperature for 15 min in BE containing 0.5 mg ml−1 NHS-sulfo-LC-LC-biotin (Pierce, Rockford, IL). Cells were washed an additional three times and incubated for 15 min in 0.1 mg ml−1 avidin (Pierce). Control cells were biotinylated but were not exposed to avidin. Avidin-cross-linked cells remained viable and endocytosed avidin after several hours; thus, all experiments were performed within 1 h of avidin exposure.
Imaging.
Coverslips bearing control or avidin-cross-linked cells were transferred to the stage of either a Zeiss (Oberkochen, Germany) LSM510 or Leica (Nussloch, Germany) SP2 scanning confocal microscope and visualized using a 63×, 1.4 numerical aperture objective. All imaging was performed at room temperature. Cells were scanned at 488 nm and 5% transmission, and after a control recording period a circular region of interest (ROI) centered on the plasma membrane edge was irreversibly photobleached using 100% transmission. Recovery of fluorescence into the ROI was monitored using 5% transmission. Average pixel intensity in the plasma membrane region of the ROI was corrected for background photobleaching and plotted versus time. Diffusion coefficients (D) were derived by fitting fluorescence recovery curves to the following empirical equation: F(t) = Fi + Fm(1 − [w2(w2 + 4πDt)−1]0.5), where Fi is the fluorescence intensity immediately after photobleaching, Fm is the intensity recovered, w is the width of the bleached plasma membrane, and D is the effective one-dimensional diffusion coefficient (Ellenberg et al., 1997). Fluorescence intensity just before the bleach was normalized to 1; thus, the mobile fraction (M) was calculated as follows: Fm(1 − Fi)−1.
Electrophysiology and statistics.
Whole-cell voltage-clamp recordings were made using standard procedures (Chen et al., 2004) from transfected (fluorescent) CGNs on the heated (37°C) stage of an Olympus (Tokyo, Japan) IX70 inverted fluorescence microscope. Neurons were bathed in a solution containing 122.5 mm NaCl, 30 mm KCl, 10 mm HEPES, 10 mm glucose, 1.5 mm CaCl2, and 2.5 mm MgCl2, pH 7.2 and ∼320 mOsm kg−1 H2O. Membrane potential was held at −60 mV. [d-Ala(2),N-Me-Phe(4),Gly(5)-ol]enkephalin (DAMGO) or adenosine was applied by perfusion from a multiport attachment, or by pressure application from a patch pipette positioned within 1 μm of the cell under study. Lag time was defined as the time between the start of the pressure application and the point when the activated current deviated by >2 SDs from the mean baseline current. The activation time constant τ was derived by fitting the rising phase of DAMGO-evoked currents to the equation: I(t) = A[1 − exp(−t/τ)]3. Results are reported as means ± SEM. Statistical significance was evaluated with Student's t test or one-way ANOVA with post hoc comparisons.
Results
Immobilization of GPCRs in living CNS neurons
To study the movement of GPCRs on the surface of cells, we fused the extracellular N terminus of the MOR to a pH-sensitive variant of the green fluorescent protein [superecliptic pHluorin (pH)] (Yuste et al., 2000). After transfection, the resulting receptors (pH-MORs) were efficiently expressed in HEK 293 cells or CGNs and trafficked appropriately to the plasma membrane, as indicated by confocal microscopy (Fig. 1A). The plasma membrane localization and transmembrane orientation of these receptors were confirmed by rapid application of acidic buffer, which immediately decreased membrane-associated fluorescence. Expressed pH-MORs were fully functional, as indicated by robust activation of GIRK channels by the peptide agonist DAMGO in HEK cells and CGNs (see below), both of which lack native MORs.
Figure 1.
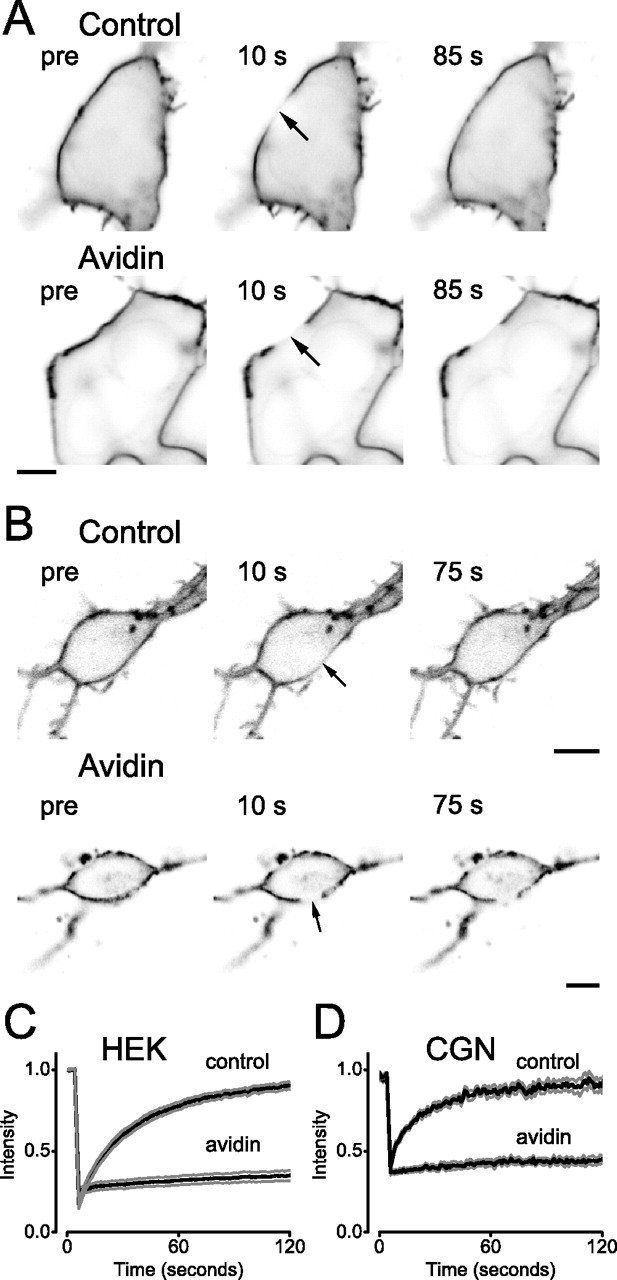
Immobilization of pH-MOR by avidin-mediated cross-linking. A, B, Confocal images of HEK 293 cells (A) or CGNs (B) expressing pH-MOR after cell surface biotinylation (Control) or cell surface biotinylation and avidin cross-linking (Avidin). Cells are shown before and at the indicated times, after photobleaching of a 5-μm-diameter ROI (arrow) centered on the plasma membrane. Scale bars, 5 μm. C, D, Fluorescence intensity in the bleached ROI is plotted versus time during FRAP experiments on control and avidin cross-linked HEK cells (C) and CGNs (D). The black traces represent the mean fluorescence intensity from 10 cells, and the gray traces represent the mean ± SEM.
We first measured the lateral mobility of pH-MORs in HEK cells using fluorescence recovery after photobleaching (FRAP). Fluorescence intensity was monitored using confocal microscopy in a 5 μm length of plasma membrane before and after photobleaching (Fig. 1A,C). Recovery of fluorescence after photobleaching reflects the lateral movement of unbleached pH-MORs into the bleached region rather than insertion of intracellular pH-MORs. This assertion was verified by photobleaching different lengths of plasma membrane, and showing that the time to half fluorescence recovery (T1/2) was proportional to the bleached area. For example, a 1 μm bleached region recovered with a T1/2 of 5.9 ± 0.4 s (n = 4), whereas in the same cells a 3 μm bleached region recovered with a T1/2 of 21 ± 2.1 s (p < 0.01). Fluorescence recovery curves were fitted with an empirical function to derive an effective one-dimensional diffusion coefficient (D), which in these cells was 0.15 ± 0.02 μm2 s−1 (n = 10). The fraction (M) of pH-MORs that was mobile was 0.88 ± 0.03 (calculated 2 min after photobleaching). These values are comparable with those reported for other GPCRs and for transmembrane proteins in general (e.g., 0.1–0.4 μm2 s−1) (Poo and Cone, 1974; Kenworthy et al., 2004).
We next sought a method to experimentally restrict the lateral mobility of pH-MORs so that we could test the role of mobility in signaling. We found that biotinylating cell surface proteins with a membrane-impermeant amine-reactive reagent and then applying soluble avidin greatly reduced the mobility of pH-MORs and other cell surface proteins. Because avidin is a tetramer with four biotin binding sites and biotin binding is extremely stable (Kd ∼ 10−15 m), this procedure likely cross-links membrane proteins in a nonspecific and practically irreversible manner. Cells remained viable indefinitely after avidin-mediated cross-linking, and surface-bound avidin was endocytosed over the course of hours (data not shown). Fluorescence recovery after photobleaching of pH-MORs was virtually abolished by avidin-mediated cross-linking (Fig. 1A,C). The mobile fraction M of pH-MORs was reduced to 0.14 ± 0.02 (n = 10; p < 10−8), and a meaningful diffusion coefficient could not be derived by curve fitting. We suspected that the small amount of fluorescence recovery observed after avidin cross-linking was an artifact of correction for background photobleaching. This appeared to be the case, because a similar amount of recovery was apparent after covalent cross-linking (fixation) using 4% paraformaldehye, which should completely immobilize proteins (M = 0.17 ± 0.02; n = 5; p = 0.40 compared with avidin). Avidin-mediated cross-linking also immobilized pH-MORs expressed in CGNs; D was reduced from 0.44 ± 0.09 μm2 s−1 (n = 10) to a value below the resolution of the method, and M was reduced from 0.87 ± 0.06 to 0.13 ± 0.02 (n = 10; p < 10−8) (Fig. 1B,D).
Rapid activation of G-proteins and GIRK channels by immobile GPCRs
Having found a method to immobilize GPCRs, we then asked whether immobile receptors could activate heterotrimeric G-proteins and downstream effectors as rapidly as mobile receptors. GIRK channels open with virtually no delay after activation of G-proteins (Bunemann et al., 2003); thus, they serve as rapid indicators of G-protein activity. Cultured CGNs express endogenous GIRK channels that can be activated with kinetics similar to those observed during synaptic activation (Otis et al., 1993). For this reason, we compared activation of GIRK channels by mobile and immobile pH-MORs in transfected CGNs after rapid pressure application of DAMGO (Fig. 1A). In an attempt to approximate the physiological situation, we performed these experiments at 37°C and transfected either 0.5 or 0.1 μg of pH-MOR cDNA to express greater or lesser amounts of pH-MOR at the cell surface. These concentrations were chosen after control experiments showed that GIRK channel onset kinetics were dependent on the amount of transfected cDNA, and that at 0.1 μg per dish the density of pH-MORs limited the rate of GIRK activation (Fig. 2B). Synaptic or agonist-mediated activation of GIRK channels begins after a delay (lag time) of tens of milliseconds, and proceeds with a sigmoidal time course to peak activation within ∼100 ms (Otis et al., 1993). We observed similar onset kinetics in CGNs expressing pH-MORs (Fig. 2A). Neither onset kinetics nor response amplitudes were significantly changed by avidin-mediated cross-linking at either level of pH-MOR expression (Fig. 2C). Experiments measuring changes in holding current after rapid application of low K+ solution indicated that these lag times included 8.2 ± 0.5 ms (n = 9) of delay introduced by dead time in the application system; thus, these measurements were not limited by solution exchange time. Responses fitted to a third-power exponential function activated with time constants (τ) that were comparable with those measured from synaptic GIRK currents (Otis et al., 1993). This result suggests that G-proteins and GIRK channels can be activated with kinetics sufficient for synaptic transmission even when the lateral mobility of the upstream GPCRs is severely restricted.
Figure 2.
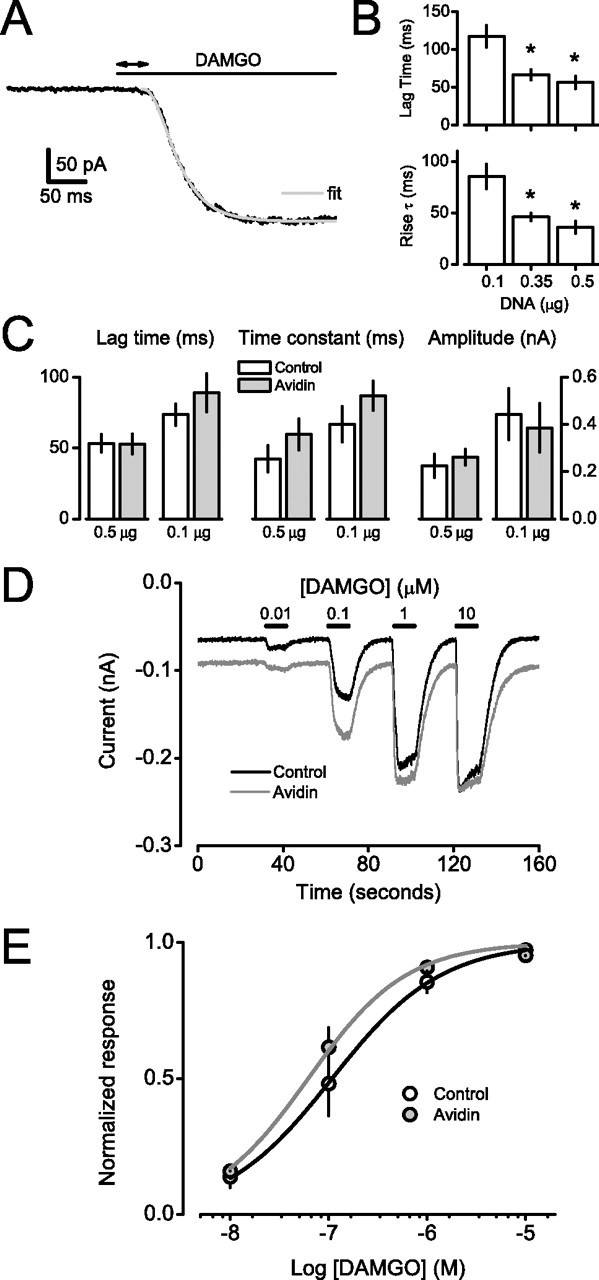
Mobile and immobile pH-MORs activate GIRK channels with the same onset kinetics and agonist sensitivity. A, Whole-cell current recorded from a cerebellar granule neuron is shown during rapid pressure application of a saturating concentration of the MOR agonist DAMGO (10 μm). DAMGO is applied where indicated by the horizontal bar. The double-headed arrow represents the lag time for current activation. The current was fitted with a third-order exponential function, and the resulting fit is shown superimposed as a gray line. B, Summary of GIRK activation lag times and activation time constants recorded from cerebellar granule neurons after rapid application of DAMGO. Neurons were transfected with the indicated amount of pH-MOR cDNA (per dish). Activation lag times and time constants were significantly faster (*p < 0.05; n = 12 each) in cells transfected with 0.35 and 0.5 μg of cDNA compared with those transfected with 0.1 μg, suggesting pH-MOR abundance was limiting in cells transfected with less cDNA. C, Summary lag time, time constant, and amplitude data from control and avidin-cross-linked neurons transfected with either 0.5 μg of pH-MOR cDNA per dish (n = 10 each) or 0.1 μg of cDNA per dish (n = 7 each). Results are means ± SEM; p > 0.05 for all comparisons within transfection groups. D, Mean whole-cell current recorded from control (n = 7) and avidin-cross-linked (n = 7) cerebellar granule neurons during perfusion with increasing concentrations of DAMGO, which was applied where indicated by the horizontal bars. E, Concentration–response curves for the same cells shown in A. Results are means ± SEM; smooth curves are logistic fits to the data. EC50 values were not significantly different (p = 0.07).
We then examined the steady-state sensitivity of pH-MOR activation of GIRK channels. Because steady-state GIRK activity depends on the rates of G-protein activation and deactivation, a change in either would be expected to affect agonist sensitivity. The G-protein (and hence GIRK channel) deactivation rate reflects the rate of GTP hydrolysis, which depends on the interaction of Gα-GTP with regulators of G-protein signaling (Doupnik et al., 1997; Saitoh et al., 1997). This interaction is entirely intracellular, and is thus unlikely to be affected by avidin cross-linking. Therefore, steady-state agonist sensitivity can be considered a more direct indicator of the G-protein activation rate than the rate of channel activation after rapid agonist application, because the latter could be limited by the rates of ligand binding and receptor activation. Concentration–response curves were generated by applying a range of agonist concentrations to control (biotinylated) and avidin cross-linked neurons (Fig. 2D). Agonist sensitivity was not significantly altered by cross-linking. EC50 values were 215 ± 68 nm in control neurons (n = 7) and 74 ± 15 nm in avidin cross-linked neurons (n = 7; p = 0.07) (Fig. 2E). These EC50 values are similar to those reported for neurons that express endogenous MORs and GIRK channels (Ingram et al., 1997), suggesting that the number of pH-MORs expressed in CGNs after transient transfection was within the physiologically relevant range. At intermediate concentrations (e.g., 0.1 μm), the abundance of ligand-actived pH-MORs limits the response; thus, unchanged agonist sensitivity implies that we would not have missed a change in signaling efficiency because of receptor overexpression. Together, these results suggest that the rate of G-protein activation in this system does not depend on the lateral mobility of GPCRs, and by extension on the frequency of collisions between GPCRs and G-proteins.
Effect of avidin cross-linking on G-protein and GIRK channel mobility
Because GIRK channel activation is thought to involve the movement of G-proteins (and possibly GIRK channels) as well as the movement of GPCRs, we were interested in determining the mobility of G-proteins and GIRK channels after avidin-mediated cross-linking. Therefore, we performed FRAP experiments on fluorescently labeled G-proteins and GIRK channels in HEK 293 cells.
Because Gα subunits and heterotrimers are attached to the plasma membrane inner leaflet by lipid modification and have no transmembrane domains, we expected that the mobility of G-proteins would be unaffected by avidin-mediated cross-linking. To test this expectation, EGFP was fused within the α-helical domain of GαoA in a position shown previously to allow for heterotrimer formation and functional activation (Bunemann et al., 2003). This construct (GαoA-EGFP) was transfected together with unlabeled Gβ1 and Gγ2 subunits, and FRAP experiments were performed to measure the mobility of the resulting heterotrimers (Fig. 3A). In HEK cells expressing GαoA-EGFP alone, D was found to be 0.47 ± 0.05 μm2 s−1 (n = 10) for biotinylated controls and 0.35 ± 0.06 μm2 s−1 (n = 12) after avidin-mediated cross-linking (p = 0.13) (Fig. 3B). Again, these values were similar to those observed for other membrane-associated, inner leaflet proteins at room temperature (e.g., 0.5 μm2 s−1) (Kenworthy et al., 2004).
Figure 3.
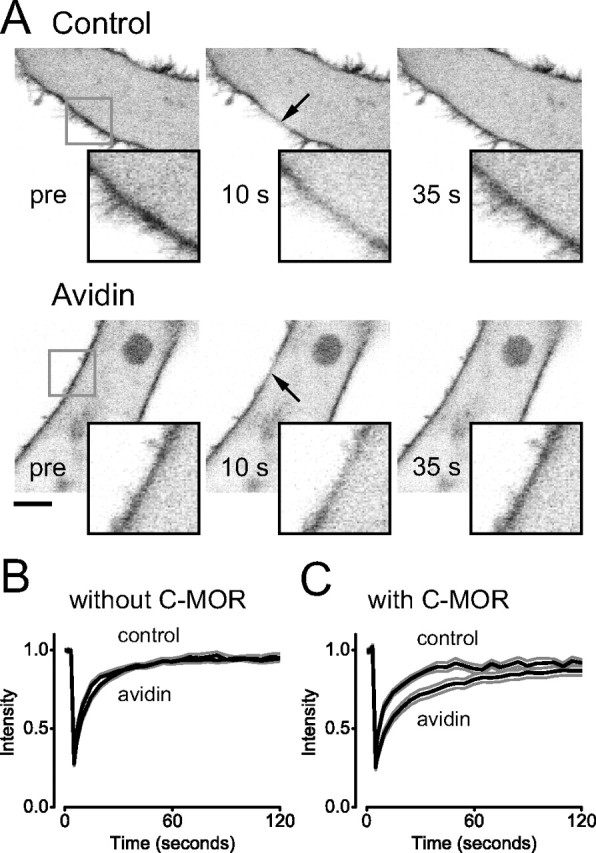
Immobile C-MORs restrict the mobility of G-proteins. A, Confocal images of HEK 293 cells expressing GαoA-EGFP together with unlabeled Gβ1 and Gγ2 subunits. Images are taken from cells after biotinylation (Control) or biotinylation and avidin cross-linking (Avidin), and show plasma membrane localization of GαoA-EGFP before and after photobleaching (arrow). Scale bar, 5 μm. B, Fluorescence intensity in the bleached ROI is plotted versus time during FRAP experiments on control (n = 10) and avidin-cross-linked (n = 12) HEK cells expressing GαoA-EGFP only. C, Fluorescence intensity in the bleached ROI is plotted versus time during FRAP experiments on control (n = 8) and avidin-cross-linked (n = 13) HEK cells expressing GαoA-EGFP together with ECFP-MOR (C-MOR). In B and C, the black traces represent the mean intensity, and the gray traces represent the mean ± SEM.
Several studies have suggested that GPCRs and G-proteins interact (precouple) before receptor activation, and can thus function as preassembled signaling complexes (Rebois and Hebert, 2003; Gales et al., 2005; Nobles et al., 2005). If inactive pH-MORs were precoupled to Gα subunits, we expected that immobilization of the former would decrease the lateral mobility of the latter. To test this idea, we constructed ECFP-MOR (C-MOR), and coexpressed this receptor together with GαoA-EGFP, Gβ1, and Gγ2. Control experiments documented that, like pH-MOR, C-MOR was fully functional and was completely immobilized by avidin-mediated cross-linking (data not shown). Immobilization of C-MOR significantly decreased the mobility of GαoA-EGFP from 0.44 ± 0.04 μm2 s−1 (n = 8) to 0.17 ± 0.02 μm2 s−1 (n = 13; p < 10−4) (Fig. 3C). A similar decrease was observed after overnight treatment with pertussis toxin (PTX) (data not shown). This result is consistent with the formation of transient precoupled complexes between C-MOR and GαoA-EGFP-containing heterotrimers. The degree to which immobile C-MORs decreased GαoA mobility would be expected to depend on the affinity of interaction between the two molecules and their relative abundance. However, recovery after photobleaching was complete either in the absence or presence of immobile C-MORs (i.e., none of the GαoA-EGFP subunits was completely immobilized, as might be expected if these two molecules formed highly stable complexes).
To test the effect of avidin cross-linking on the lateral mobility of GIRK channels, we expressed enhanced yellow fluorescent protein (EYFP) fused to the N terminus of GIRK1 (Kir3.1) subunits together with unlabeled GIRK4 (Kir3.4) subunits. As shown in Figure 4, the resulting channels were immobilized by avidin-mediated cross-linking. Because the extracellular domains of these channels and native GIRK channels are virtually identical, it is likely that avidin-mediated cross-linking also immobilized native GIRK channels in cerebellar granule neurons. This result also shows that immobilization by avidin-mediated cross-linking does not require an extracellular fluorescent protein.
Figure 4.

Avidin cross-linking immobilizes EYFP-GIRK1. A, Confocal images of HEK 293 cells expressing EYFP-GIRK1 and unlabeled GIRK4 after cell surface biotinylation (Control) or cell surface biotinylation and avidin cross-linking (Avidin). Cells are shown before and after photobleaching of a 5-μm-diameter ROI (arrow) centered on the plasma membrane. Scale bar, 5 μm. B, Fluorescence intensity in the bleached ROI is plotted versus time during FRAP experiments on control (n = 9) and avidin-cross-linked (n = 9) HEK cells. The black traces represent the mean fluorescence intensity, and the gray traces represent the mean ± SEM.
Activation of immobile GIRK channels by immobile GPCRs and G-protein heterotrimers
Because our results suggested that GPCR and GIRK channel mobility were not essential for channel activation, we sought to extend this line of investigation by limiting the mobility of G-proteins. To this end, we extended Gα subunits with a TM domain and extracellular ECFP, thus providing a means to immobilize heterotrimeric G-proteins. These ECFP-TM-Gα subunits also incorporated a mutation rendering them insensitive to PTX, which allowed us to restrict signaling to these subunits by inactivating native Gα subunits with PTX. To express fluorescent homomeric GIRK channels, we extended the N terminus of GIRK4* subunits with a TM domain and extracellular pHluorin. These pH-TM-GIRK4* subunits formed functional homomeric GIRK channels (Fig. 5A), and both they and ECFP-TM-Gα subunits were immobilized by avidin-mediated cross-linking (data not shown). Attempts to activate GIRK channels in cerebellar granule neurons using ECFP-TM-Gα subunits were unsuccessful; thus, experiments were performed in HEK 293 cells. We found that the combination of the pHluorin-tagged adenosine A1 receptor (pH-A1R), ECFP-TM-Gαi1, and pH-GIRK4* most reliably reconstituted agonist-induced channel activation after overnight PTX treatment (500 ng/dish). Activation of pH-GIRK4* was not detectably different in biotinylated control cells and avidin-cross-linked cells (n = 9 each) (Fig. 5B). This result suggests that signal transduction proceeds even when the lateral mobility of the proteins involved is severely limited. Unfortunately, the slow kinetics of GIRK activation in these cells precludes any conclusion regarding the speed of signaling mediated by immobile GPCRs, G-proteins, and channels.
Figure 5.
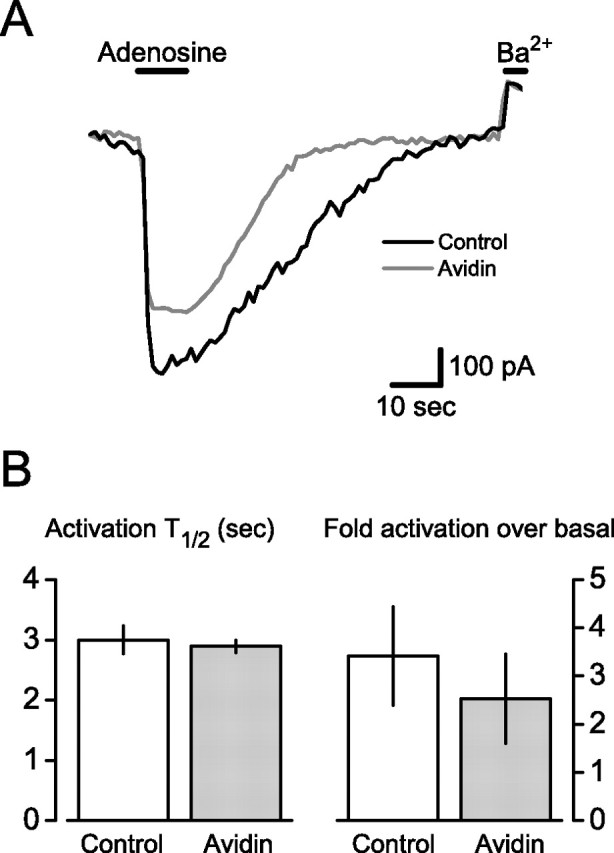
Activation of immobile GIRK channels by immobile GPCRs and G-protein heterotrimers. A, Normalized current traces recorded from HEK 293 cells expressing pH-A1R, ECFP-TM-Gαi1, Gβ1, Gγ2, and pH-GIRK4*; cells were treated overnight with PTX (500 ng/dish) to inactivate native G-proteins. Application of adenosine (50 μm; horizontal bar) reversibly activated pH-GIRK4* in both control and avidin-cross-linked cells. Application of barium (Ba2+; 200 μm) blocked basal current pH-GIRK4*, and currents are normalized as fold-activation over basal. B, Summary data from experiments identical with that shown in A. Activation half-time (T1/2) and fold activation over basal were not significantly different in control (n = 9) and avidin-cross-linked (n = 9) cells (p = 0.68 and 0.74). The time to half current recovery after agonist washout was also not significantly different between the two groups (biotin, 36 ± 5 s, vs avidin, 27 ± 7 s; p = 0.26). Error bars indicate SEM.
Discussion
Previous work has indicated that membrane protein collision frequency sets the speed of phototransduction in rod outer segment disks (Calvert et al., 2001) and turkey erythrocytes (Rimon et al., 1978). However, neither of these studies measured the diffusivity of receptors, G-proteins, or effectors, and more recent studies have raised doubts about the role of receptor mobility in phototransduction (Fotiadis et al., 2003; Liang et al., 2004). GPCRs in CNS neurons are known to be concentrated at specific subcellular sites, often in perisynaptic regions (Baude et al., 1993; Shigemoto et al., 1996; Kulik et al., 2003). It is thought that this distribution is maintained by protein interactions that produce clusters of GPCRs and/or link GPCRs to cytoskeletal scaffolds, either of which would be expected to restrict lateral mobility (Choquet and Triller, 2003). Although this arrangement would position receptors for efficient detection of synaptically released neurotransmitter, the decrease in diffusivity imposed by localization would necessarily decrease the frequency of collisions with G-proteins and other signaling molecules, and thus might impair signaling. Our results comparing responses mediated by mobile and immobile GPCRs indicate that receptor mobility is not necessary for rapid and sensitive signaling in CNS neurons. Because the frequency of collisions between two molecules diffusing in two dimensions is proportional to the sum of their diffusion coefficients (Pugh and Lamb, 1993), immobilizing pH-MORs would decrease the frequency of collisions between pH-MORs and G-proteins by ∼40% in CGNs. Thus, if GPCRs couple to G-proteins in these neurons by random collision, a decrease in collision frequency of this magnitude is not sufficient to limit the rate of G-protein and effector activation. The situation might be very different if G-protein mobility is also restricted at synaptic sites, in which case receptor mobility might be much more important. Our results showing GIRK activation mediated by immobile heterotrimers suggests that macroscopic G-protein diffusion is not absolutely required for signaling, but unequivocal assessment of this possibility must await measurements of G-protein diffusion at identified sites of GPCR activity in native cells.
Previous studies have addressed the role of receptor–G-protein collision in GIRK activation by fusing GPCRs and G-proteins and by varying the abundance of these proteins to either eliminate or manipulate the frequency of receptor–G-protein collision. One of these studies concluded that receptor–G-protein collision was rate-limiting (Vorobiov et al., 2000), whereas the other reached the opposite conclusion (Benians et al., 2003). In both of these studies, the GIRK activation rate was limited by the heterologous expression system, and it has since been shown that fusing receptor and G-protein does not eliminate intermolecular collision coupling (Molinari et al., 2003). Collision coupling in general is supported by a recent report showing that the receptor–G-protein interaction rate approaches the receptor activation rate as G-protein abundance is increased (Hein et al., 2005), although this study did not identify collision as the rate-limiting step. Similarly, biochemical studies have suggested that receptor–G-protein collision does not limit the rate of G-protein activation (Waldhoer et al., 1999). The results of our experiments add significantly to previous work by directly manipulating receptor mobility (and thus collision frequency) in a native system. We conclude that receptor–G-protein collision frequency is not rate-limiting for activation of GIRK channels in CNS neurons. By extension, CNS neurons would be free to restrict the movement of GPCRs at synaptic sites without compromising signaling efficacy.
It has been suggested that GPCRs and G-proteins (Gales et al., 2005; Nobles et al., 2005) together with some effectors (such as GIRK channels) (see below) (Huang et al., 1995; Lavine et al., 2002; Rebois and Hebert, 2003; Clancy et al., 2005) can preassemble into stable signaling complexes, thus obviating the need for collision coupling altogether. Our results do not provide evidence for highly stable complexes in CNS neurons. They do, however, suggest that receptors and G-proteins interact before receptor activation. We find that immobile GPCRs restrict the mobility of G-proteins, as expected if the two transiently bind each other. This possibility raises several questions, such as the role of transient precoupling in signaling specificity and the possible persistence of complexes throughout signaling. Several recent studies have addressed dynamic interactions between receptors and G-proteins in intact cells (Gales et al., 2005, 2006; Hein et al., 2005; Nobles et al., 2005), but as yet there is no clear consensus regarding the functional significance of precoupled receptor–G-protein complexes.
Finally, although we were unable to assess the mobility of native GIRK channels in CGNs, our results with heterologously expressed GIRK1-YFP in HEK cells suggest that the native extracellular domains of these channels render them susceptible to avidin-mediated cross-linking. If it is assumed that native GIRK channels in CGNs were also immobilized by avidin-mediated cross-linking, then our results further suggest that channel mobility is unimportant for rapid signaling. This implies that collision between free Gβγ subunits and GIRK channels also is not rate-limiting for GIRK activation. One possible reason for this outcome is that inactive G-protein heterotrimers are bound to GIRK channels in a macromolecular signaling complex, as suggested by recent biochemical and biophysical studies (Clancy et al., 2005; Riven et al., 2006). Additional experiments will be required to determine the precise arrangement of signaling molecules in native cells, and the rearrangements that occur during physiological signaling.
In summary, our results demonstrate that lateral mobility of GPCRs is not necessary for rapid signaling (i.e., for signaling as rapid as synaptic transmission). This result suggests that the frequency of collisions between inactive GPCRs and inactive G-proteins is not rate-limiting for G-protein activation. Our results further suggest that GPCRs and heterotrimeric G-proteins bind each other transiently (but not stably) in CNS neurons. Finally, signal transduction can proceed even when the macroscopic diffusion of GPCRs, G-protein heterotrimers, and effectors is restricted. Thus, the rates of random collisional encounters between signaling molecules do not dictate signaling efficiency.
Footnotes
This work was supported by Ruth L. Kirschstein National Research Service Award NS052070 (R.M.L.), American Heart Association Predoctoral Fellowship 0515176B (R.M.L.), National Institutes of Health Grant NS36455 (N.A.L.), and National Science Foundation Grant MCB060024 (N.A.L.). We thank G. Miesenbock for cDNA encoding superecliptic pHluorin, R. J. Miller for cDNA encoding the rat MOR, K. J. Harrison-Lavoie for cDNA encoding the human growth hormone signal sequence, E. Reuveny for cDNA encoding YFP-GIRK1, D. Logothetis for cDNA encoding GIRK4*, and J. Dempster for WinWCP electrophysiology software.
References
- Arshavsky VY, Lamb TD, Pugh EN., Jr G proteins and phototransduction. Annu Rev Physiol. 2002;64:153–187. doi: 10.1146/annurev.physiol.64.082701.102229. [DOI] [PubMed] [Google Scholar]
- Baude A, Nusser Z, Roberts JD, Mulvihill E, McIlhinney RA, Somogyi P. The metabotropic glutamate receptor (mGluR1 alpha) is concentrated at perisynaptic membrane of neuronal subpopulations as detected by immunogold reaction. Neuron. 1993;11:771–787. doi: 10.1016/0896-6273(93)90086-7. [DOI] [PubMed] [Google Scholar]
- Benians A, Leaney JL, Milligan G, Tinker A. The dynamics of formation and action of the ternary complex revealed in living cells using a G-protein-gated K+ channel as a biosensor. J Biol Chem. 2003;278:10851–10858. doi: 10.1074/jbc.M212299200. [DOI] [PubMed] [Google Scholar]
- Bunemann M, Frank M, Lohse MJ. Gi protein activation in intact cells involves subunit rearrangement rather than dissociation. Proc Natl Acad Sci USA. 2003;100:16077–16082. doi: 10.1073/pnas.2536719100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvert PD, Govardovskii VI, Krasnoperova N, Anderson RE, Lem J, Makino CL. Membrane protein diffusion sets the speed of rod phototransduction. Nature. 2001;411:90–94. doi: 10.1038/35075083. [DOI] [PubMed] [Google Scholar]
- Chan KW, Sui JL, Vivaudou M, Logothetis DE. Control of channel activity through a unique amino acid residue of a G protein-gated inwardly rectifying K+ channel subunit. Proc Natl Acad Sci USA. 1996;93:14193–14198. doi: 10.1073/pnas.93.24.14193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Clark MA, Lambert NA. Endogenous RGS proteins regulate presynaptic and postsynaptic function: functional expression of RGS-insensitive Galpha subunits in central nervous system neurons. Methods Enzymol. 2004;389:190–204. doi: 10.1016/S0076-6879(04)89012-8. [DOI] [PubMed] [Google Scholar]
- Choquet D, Triller A. The role of receptor diffusion in the organization of the postsynaptic membrane. Nat Rev Neurosci. 2003;4:251–265. doi: 10.1038/nrn1077. [DOI] [PubMed] [Google Scholar]
- Clancy SM, Fowler CE, Finley M, Suen KF, Arrabit C, Berton F, Kosaza T, Casey PJ, Slesinger PA. Pertussis-toxin-sensitive Galpha subunits selectively bind to C-terminal domain of neuronal GIRK channels: evidence for a heterotrimeric G-protein-channel complex. Mol Cell Neurosci. 2005;28:375–389. doi: 10.1016/j.mcn.2004.10.009. [DOI] [PubMed] [Google Scholar]
- Doupnik CA, Davidson N, Lester HA, Kofuji P. RGS proteins reconstitute the rapid gating kinetics of gbetagamma-activated inwardly rectifying K+ channels. Proc Natl Acad Sci USA. 1997;94:10461–10466. doi: 10.1073/pnas.94.19.10461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellenberg J, Siggia ED, Moreira JE, Smith CL, Presley JF, Worman HJ, Lippincott-Schwartz J. Nuclear membrane dynamics and reassembly in living cells: targeting of an inner nuclear membrane protein in interphase and mitosis. J Cell Biol. 1997;138:1193–1206. doi: 10.1083/jcb.138.6.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fotiadis D, Liang Y, Filipek S, Saperstein DA, Engel A, Palczewski K. Atomic-force microscopy: Rhodopsin dimers in native disc membranes. Nature. 2003;421:127–128. doi: 10.1038/421127a. [DOI] [PubMed] [Google Scholar]
- Gales C, Rebois RV, Hogue M, Trieu P, Breit A, Hebert TE, Bouvier M. Real-time monitoring of receptor and G-protein interactions in living cells. Nat Methods. 2005;2:177–184. doi: 10.1038/nmeth743. [DOI] [PubMed] [Google Scholar]
- Gales C, Van Durm JJ, Schaak S, Pontier S, Percherancier Y, Audet M, Paris H, Bouvier M. Probing the activation-promoted structural rearrangements in preassembled receptor-G protein complexes. Nat Struct Mol Biol. 2006;13:778–786. doi: 10.1038/nsmb1134. [DOI] [PubMed] [Google Scholar]
- Geiser M, Cebe R, Drewello D, Schmitz R. Integration of PCR fragments at any specific site within cloning vectors without the use of restriction enzymes and DNA ligase. Biotechniques. 2001;31:88–90. doi: 10.2144/01311st05. 92. [DOI] [PubMed] [Google Scholar]
- Hein P, Frank M, Hoffmann C, Lohse MJ, Bunemann M. Dynamics of receptor/G protein coupling in living cells. EMBO J. 2005;24:4106–4114. doi: 10.1038/sj.emboj.7600870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B. G protein-coupled mechanisms and nervous signaling. Neuron. 1992;9:187–195. doi: 10.1016/0896-6273(92)90158-a. [DOI] [PubMed] [Google Scholar]
- Huang CL, Slesinger PA, Casey PJ, Jan YN, Jan LY. Evidence that direct binding of G beta gamma to the GIRK1 G protein-gated inwardly rectifying K+ channel is important for channel activation. Neuron. 1995;15:1133–1143. doi: 10.1016/0896-6273(95)90101-9. [DOI] [PubMed] [Google Scholar]
- Ingram S, Wilding TJ, McCleskey EW, Williams JT. Efficacy and kinetics of opioid action on acutely dissociated neurons. Mol Pharmacol. 1997;52:136–143. doi: 10.1124/mol.52.1.136. [DOI] [PubMed] [Google Scholar]
- Kenworthy AK, Nichols BJ, Remmert CL, Hendrix GM, Kumar M, Zimmerberg J, Lippincott-Schwartz J. Dynamics of putative raft-associated proteins at the cell surface. J Cell Biol. 2004;165:735–746. doi: 10.1083/jcb.200312170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulik A, Vida I, Lujan R, Haas CA, Lopez-Bendito G, Shigemoto R, Frotscher M. Subcellular localization of metabotropic GABAB receptor subunits GABAB1a/b and GABAB2 in the rat hippocampus. J Neurosci. 2003;23:11026–11035. doi: 10.1523/JNEUROSCI.23-35-11026.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavine N, Ethier N, Oak JN, Pei L, Liu F, Trieu P, Rebois RV, Bouvier M, Hebert TE, Van Tol HH. G protein-coupled receptors form stable complexes with inwardly rectifying potassium channels and adenylyl cyclase. J Biol Chem. 2002;277:46010–46019. doi: 10.1074/jbc.M205035200. [DOI] [PubMed] [Google Scholar]
- Liang Y, Fotiadis D, Maeda T, Maeda A, Modzelewska A, Filipek S, Saperstein DA, Engel A, Palczewski K. Rhodopsin signaling and organization in heterozygote rhodopsin knockout mice. J Biol Chem. 2004;279:48189–48196. doi: 10.1074/jbc.M408362200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molinari P, Ambrosio C, Riitano D, Sbraccia M, Gro MC, Costa T. Promiscuous coupling at receptor-Galpha fusion proteins. The receptor of one covalent complex interacts with the alpha-subunit of another. J Biol Chem. 2003;278:15778–15788. doi: 10.1074/jbc.M300731200. [DOI] [PubMed] [Google Scholar]
- Nobles M, Benians A, Tinker A. Heterotrimeric G proteins precouple with G protein-coupled receptors in living cells. Proc Natl Acad Sci USA. 2005;102:18706–18711. doi: 10.1073/pnas.0504778102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otis TS, De Koninck Y, Mody I. Characterization of synaptically elicited GABAB responses using patch-clamp recordings in rat hippocampal slices. J Physiol (Lond) 1993;463:391–407. doi: 10.1113/jphysiol.1993.sp019600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poo M, Cone RA. Lateral diffusion of rhodopsin in the photoreceptor membrane. Nature. 1974;247:438–441. doi: 10.1038/247438a0. [DOI] [PubMed] [Google Scholar]
- Pugh EN, Jr, Lamb TD. Amplification and kinetics of the activation steps in phototransduction. Biochim Biophys Acta. 1993;1141:111–149. doi: 10.1016/0005-2728(93)90038-h. [DOI] [PubMed] [Google Scholar]
- Rebois RV, Hebert TE. Protein complexes involved in heptahelical receptor-mediated signal transduction. Receptors Channels. 2003;9:169–194. [PubMed] [Google Scholar]
- Rimon G, Hanski E, Braun S, Levitzki A. Mode of coupling between hormone receptors and adenylate cyclase elucidated by modulation of membrane fluidity. Nature. 1978;276:394–396. doi: 10.1038/276394a0. [DOI] [PubMed] [Google Scholar]
- Riven I, Iwanir S, Reuveny E. GIRK channel activation involves a local rearrangement of a preformed G protein channel complex. Neuron. 2006;51:561–573. doi: 10.1016/j.neuron.2006.08.017. [DOI] [PubMed] [Google Scholar]
- Saitoh O, Kubo Y, Miyatani Y, Asano T, Nakata H. RGS8 accelerates G-protein-mediated modulation of K+ currents. Nature. 1997;390:525–529. doi: 10.1038/37385. [DOI] [PubMed] [Google Scholar]
- Serge A, Fourgeaud L, Hemar A, Choquet D. Receptor activation and homer differentially control the lateral mobility of metabotropic glutamate receptor 5 in the neuronal membrane. J Neurosci. 2002;22:3910–3920. doi: 10.1523/JNEUROSCI.22-10-03910.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigemoto R, Kulik A, Roberts JD, Ohishi H, Nusser Z, Kaneko T, Somogyi P. Target-cell-specific concentration of a metabotropic glutamate receptor in the presynaptic active zone. Nature. 1996;381:523–525. doi: 10.1038/381523a0. [DOI] [PubMed] [Google Scholar]
- Vorobiov D, Bera AK, Keren-Raifman T, Barzilai R, Dascal N. Coupling of the muscarinic m2 receptor to G protein-activated K+ channels via Galpha(z) and a receptor-Galpha(z) fusion protein. Fusion between the receptor and Galpha(z) eliminates catalytic (collision) coupling. J Biol Chem. 2000;275:4166–4170. doi: 10.1074/jbc.275.6.4166. [DOI] [PubMed] [Google Scholar]
- Waldhoer M, Wise A, Milligan G, Freissmuth M, Nanoff C. Kinetics of ternary complex formation with fusion proteins composed of the A(1)-adenosine receptor and G protein alpha-subunits. J Biol Chem. 1999;274:30571–30579. doi: 10.1074/jbc.274.43.30571. [DOI] [PubMed] [Google Scholar]
- Yuste R, Miller RB, Holthoff K, Zhang S, Miesenbock G. Synapto-pHluorins: chimeras between pH-sensitive mutants of green fluorescent protein and synaptic vesicle membrane proteins as reporters of neurotransmitter release. Methods Enzymol. 2000;327:522–546. doi: 10.1016/s0076-6879(00)27300-x. [DOI] [PubMed] [Google Scholar]