

Abstract
Insufficient sleep impairs cognitive functions in humans and animals. However, whether long-term synaptic plasticity, a cellular substrate of learning and memory, is compromised by sleep loss per se remains unclear because of confounding factors related to sleep deprivation (SD) procedures in rodents. Using an ex vivo approach in C57BL/6J mice, we show that sleep loss rapidly and reversibly alters bidirectional synaptic plasticity in the CA1 area of the hippocampus. A brief (∼4 h) total SD, respecting the temporal parameters of sleep regulation and maintaining unaltered low corticosterone levels, shifted the modification threshold for long-term depression/long-term potentiation (LTP) along the stimulation frequency axis (1–100 Hz) toward the right. Reducing exposure to sensory stimuli by whisker trimming did not affect the SD-induced changes in synaptic plasticity. Recovery sleep reversed the effects induced by SD. When SD was combined with moderate stress, LTP induction was not only impaired but also occluded. Both electrophysiological analysis and immunoblotting of purified synaptosomes revealed that an alteration in the molecular composition of synaptically activated NMDA receptors toward a greater NR2A/NR2B ratio accompanied the effects of SD. This change was reversed after recovery sleep. By using an unparalleled, particularly mild form of SD, this study describes a novel approach toward dissociating the consequences of insufficient sleep on synaptic plasticity from nonspecific effects accompanying SD in rodents. We establish a framework for cellular models of cognitive impairment related to sleep loss, a major problem in modern society.
Keywords: sleep deprivation, stress free, corticosterone, ex vivo, LTP/LTD, NMDA receptor, NR2A/NR2B ratio
Introduction
It is well established that insufficient sleep strongly affects neurocognitive performance in humans (Durmer and Dinges, 2005). In rodents, sleep deprivation (SD) is known to impair cognitive behavior, particularly in hippocampus-dependent tasks such as spatial learning (Graves et al., 2001, 2003). However, the neurobiological mechanisms underlying these deficits remain unclear. Long-term potentiation (LTP) in the hippocampus is a prominent cellular model for learning and memory (Lynch, 2004). Previous in vitro studies focused on the effects of total or partial SD on LTP in the CA1 area of rat hippocampus (Campbell et al., 2002; Davis et al., 2003; McDermott et al., 2003). The impairment of LTP after 72 h of partial SD was recently proposed to be caused by a reduction in NMDA receptor (NMDAR) function, without alteration of receptor subunit composition (McDermott et al., 2006).
However, whether LTP and NMDAR function were specifically compromised by sleep loss during SD remains unclear because of prominent confounding factors (Miyamoto and Hensch, 2003). In particular, the SD procedures used were accompanied by elevated stress levels, but also by altered locomotor activity and constrained exposure to unfamiliar environments. Manipulation of the animals was long-lasting (12–72 h), forcing them either to run in a wheel (Campbell et al., 2002) or to face an aversive situation, such as falling into water when entering rapid-eye movement sleep (Davis et al., 2003; McDermott et al., 2003, 2006). Elevated levels of the stress hormone corticosterone, which is known to impair LTP (Kim and Diamond, 2002), were reported (Campbell et al., 2002; McDermott et al., 2003).
In addition to LTP, bidirectional modifications of synaptic efficacy, allowing synapses to be not only strengthened but also weakened, is considered an important mechanism underlying learning and memory (Abraham and Bear, 1996; Abraham and Tate, 1997). Learning and experience increase synaptic strength and the threshold for further potentiation, while facilitating long-term depression (LTD) (Rioult-Pedotti et al., 2000; Quinlan et al., 2004). Conversely, experimentally induced modifications in bidirectional synaptic plasticity severely affect learning and memory (Bach et al., 1995; Migaud et al., 1998; Zeng et al., 2001). However, the effects of SD on bidirectional plasticity have not been addressed, although LTD induced by 3 Hz stimulation in the CA1 area was unaltered after 72 h of SD (McDermott et al., 2003).
Here, we follow a novel approach toward dissociating the role of sleep loss in bidirectional synaptic plasticity in mice from nonspecific effects of SD. We adopted an SD procedure designed to completely avoid stress and to last for a short duration that was determined by the spontaneous behavior of the animals (∼4 h), yet significantly affects electroencephalographic markers of sleep regulation (Huber et al., 2000a). We found that sleep loss, independently of stress and somatosensory stimuli, shifted the LTD/LTP crossover point along the frequency axis and altered the subunit composition of synaptically activated NMDARs. After 3 h of recovery following SD, the effects of SD on synaptic plasticity and NMDARs were reversed, substantiating a specific role for sleep loss.
Materials and Methods
Animals.
Eight-week-old C57BL/6J mice were kept in standard Macrolon cages (26 × 20 cm) and maintained on a 12 h light/dark cycle (light from 7:00 A.M. to 7:00 P.M.) with food and water ad libitum. All experiments were performed in agreement with the guidelines of the Veterinary Institute of the Canton Basel-Stadt.
Mild SD procedure.
On the day before SD, the cage of the animal was placed in the middle of a larger box (53 × 30 cm) and kept closed. SD started between 7:30 and 9:30 A.M. and was performed in individual animals under continuous visual observation. Exceptionally, two animals were sleep deprived simultaneously; blood was collected in both mice for corticosterone measurements, and one brain was used for electrophysiology. Wakefulness was maintained by allowing ad libitum access to a larger box and by gently knocking at the cage or providing the mouse with new nesting material when it started adopting a sleeping position. The mouse was not touched during the entire procedure. SD was halted when the mouse no longer reacted to the stimuli and fell asleep (i.e., after 4.3 ± 0.1 h; n = 69 mice in total).
Control mice (n = 85 in total) were left totally undisturbed and were killed at the same time of day as the sleep-deprived animals. The control mice were asleep when they were taken to be killed. The procedure lasted <2 min, during which the animals were awake as a result of the manipulation. As reported previously (Kapfhamer et al., 2002), sleeping time approximated by the time spent resting (distance moved = 0 mm per second) was monitored in a subset of the animals for 4 h before they were killed using the video tracking system Ethovision 3.0 (Noldus Information Technology, Wageningen, The Netherlands).
Stressful SD procedure.
The mice (n = 17 in total) were kept in the same environmental conditions as described above for the mild form of SD. Stressful SD was performed for 4.5 h at the same time of day as the mild SD and was achieved by direct handling of the animal at 30 min intervals.
Measurement of plasma corticosterone concentrations.
Blood samples were obtained from trunk blood after brief isoflurane anesthesia. Samples were obtained within 2 min of touching the animal's cage. Plasma corticosterone was measured using a kit (catalog #07120102; MP Biomedicals, Orangeburg, NY), according to the manufacturer's instructions.
Slice preparation.
Mice were decapitated after a brief isoflurane anesthesia. Parasagittal slices containing the hippocampal formation were prepared on a vibratome in an ice-cold oxygenated solution containing (in mm) 126 NaCl, 2.5 KCl, 1.25 NaH2PO4, 26 NaHCO3, 18 dextrose, 1.7 l(+)-ascorbic acid, 1.2 MgCl2, 2 CaCl2, and 2 kynurenic acid. A cut was applied between CA3 and CA1 to prevent epileptic discharges.
Extracellular recordings and stimulation.
The slices were placed in an interface recording chamber at 35 ± 0.1°C and allowed to recover for 1 h. The same solution as for slicing, but containing 0.1 mm picrotoxin instead of kynurenic acid, was used to isolate glutamatergic responses. A bipolar tungsten stimulation electrode (115 μm spacing; Frederick Haer, Bowdoinham, ME) was positioned in the Schaffer-collateral pathway, and a glass recording electrode (2–3 MΩ) filled with extracellular solution was positioned in the CA1 stratum radiatum. Field EPSPs (fEPSPs) were evoked every 20 s with 100 μs current pulses (Stimulus Isolator A360; World Precision Instruments, Berlin, Germany), and stimulation intensity was adjusted to elicit 50–60% of the maximal fEPSP (range, −100 to −600 μV/ms). The signals were amplified 10,000 times and filtered between 0.3 and 3 kHz (Differential Amplifier DP301; Warner Instruments, Hamden, CT) before being digitally acquired at 10 kHz (Clampex 9.2; Molecular Devices, Sunnyvale, CA). Bath application of kynurenic acid (1 mm) reversibly blocked the second wave of the response, whereas the first wave remained intact, indicating that a glutamatergic field response preceded by a fiber volley was evoked. Synaptic plasticity was tested after a 10 min stable baseline by giving high-frequency stimulation (HFS; two 1 s pulses at 100 Hz separated by 30 s) or low-frequency stimulation (LFS; 900 pulses at 1, 5, or 10 Hz) trains. fEPSPs were recorded for 30 min after conditioning, and the level of LTP/LTD was quantified in the last 10 min of recording, which showed stable postsynaptic response levels. Saturated levels of LTP were induced by repeating HFS two to five times at 10–20 min intervals and were quantified in the last 10 min of recording after the last stimulation. Data were collected between 2 and 6 h after slicing, and no correlation was found between the time of recording and the magnitude of long-term synaptic plasticity (Pearson's correlation coefficient; 1 Hz LFS: r = −0.12, p = 0.67; 5 Hz LFS: r = −0.03, p = 0.93; 10 Hz LFS: r = 0.20, p = 0.49; 100 Hz HFS: r = −0.13, p = 0.51).
NMDAR-mediated fEPSPs were isolated pharmacologically by altering the cation concentration to 0.1 mm MgCl2 and 3 mm CaCl2 and by adding 20 μm 6,7-dinitriquinoxaline-2,3-dione (DNQX) and 1 μm glycine (Quinlan et al., 1999a). Four fEPSPs were evoked at 30 s intervals every 10 min (Quinlan et al., 1999a). Stimulation intensity was adjusted to elicit subthreshold stable responses (∼30% of the maximal response).
Synaptic strength of the postsynaptic response was assessed by the 10–90% slope of the fEPSP measured in ClampFit and normalized to the corresponding mean baseline value.
Whole-cell voltage-clamp recordings.
The slices were allowed to recover in a home-made submersion chamber at 35°C in a kynurenate-free slicing solution. After 30 min, they were maintained at room temperature for an additional 45 min before they were transferred to a recording chamber and perfused with kynurenate-free slicing solution at 3.5 ml/min. Patch pipettes (3.5–4.5 MΩ) were pulled from borosilicate glass (TW150F-4; World Precision Instruments) and filled with the following (in mm): 127.5 cesium gluconate, 10 HEPES, 5 cesium BAPTA, 6 MgCl2, 2 Mg-ATP, and 10 phosphocreatine, adjusted to pH 7.25–7.28 with gluconic acid and to 290–295 mOsm. K-GTP (0.2 mm) and QX-314 (1 mm) were freshly added daily, and the solution was filtered and maintained on ice.
Whole-cell recordings were obtained under visual control using near-infrared differential interference contrast microscopy (BX51WI; Olympus, Volketswil, Switzerland) at 33.5–35°C using an in-line solution heating system (Warner Instruments). After obtaining whole-cell access, synaptic currents were evoked at −60 mV every 5 s by brief current pulses (100 μs, 100–400 μA) delivered via a bipolar tungsten stimulation electrode placed in the CA1 stratum radiatum, and the glutamatergic EPSC was isolated by bath-applying 0.1 mm picrotoxin and 1 μm glycine. The NMDA component was isolated by bath-applying DNQX (20 μm) and by holding the cell at +40 mV. The reversal potential of NMDAR-mediated responses did not differ between recordings from control and SD experiments (p > 0.05). The NMDA/non-NMDA ratio was defined as the ratio of the peak current obtained at +40 mV and of the DNQX-sensitive current at −60 mV. For measuring the action of ifenprodil (3 μm), NMDAR-mediated EPSCs were elicited once every 5 min at +30 to +40 mV. The holding potential was ramped to these values within 30 s, starting 45 s before EPSC acquisition. Holding current levels at +30 to +40 mV ranged between 200 and 450 pA and changed by <60 pA during the entire recordings. In between acquisition of EPSCs, the holding potential was returned to −50 mV. Ifenprodil was applied after obtaining a stable baseline of at least 15 min, and steady-state effects of ifenprodil were measured as the average of current amplitudes at 10 and 15 min after bath application had started. Series resistance was monitored continuously for stability (<20% change). A measured liquid junction potential of 10 mV was taken into account when setting the holding potentials. Data were collected through an Axopatch 200B amplifier (Molecular Devices), low-pass filtered at 2 kHz, and acquired at 5 kHz using pClamp 9.2. software. Analysis was performed using Clampfit 9.2.
Immunoblot analysis.
Mice were decapitated after a brief isoflurane anesthesia. The hippocampi were rapidly dissected in the same ice-cold oxygenated slicing solution as the one used for the electrophysiology. Purified synaptosomes were prepared as described by Nakamura et al. (1993), from both hippocampi of n = 8 control and n = 8 sleep-deprived mice in four independent preparations for each group. The hippocampi were homogenized in 10 vol of ice-cold homogenization buffer (0.32 m sucrose and 10 mm Tris, pH 7.4) containing standard protease inhibitor mixture (Sigma, St. Louis, MO), using a glass Teflon tissue homogenizer (Glas-Col, Terre Haute, IN). The homogenate was centrifuged at 1000 × g for 5 min to remove nuclei and cellular debris, and the supernatant was gently stratified on discontinuous Percoll density gradient (2, 6, 10, and 20% v/v in Tris-buffered sucrose) and centrifuged at 33,500 × g for 5 min. The synaptosomal fraction from the 10–20% Percoll interface was removed and washed to eliminate Percoll, and synaptosomes were isolated from the supernatant by centrifugation at 12,000 × g for 15 min. Synaptosomes were lysed in 1 ml of ice-cold water and sonicated, and the suspension was centrifuged at 16,000 × g for 10 min. The resulting pellet was solubilized in a lysis buffer (1 mm EGTA, 1 mm EDTA, 1 mm PMSF, 1 mm Na3VO4, 50 mm HEPES, 150 mm NaCl, 10% glycerol, 1% Triton X-100, and 0.5% Nonidet P-40, pH 7.4) containing a standard protease inhibitor mixture and placed on a head-over-head shaker for 2 h.
Samples of 30 μl corresponding to equal amounts of synaptosome protein, determined using the BCA assay (Pierce, Rockford, IL), were resolved on 8% polyacrylamide gels and transferred to nitrocellulose membranes. After transfer, nonspecific binding sites were blocked for 2 h at room temperature with Tris-buffered saline Tween (t-TBS; 20 mm Tris, 137 mm NaCl, and 0.05% Tween 20) containing 3% non-fat dried milk. Nitrocellulose membranes were then incubated with primary antibodies overnight at 4°C. The primary antibodies and dilutions used were as follows: mouse anti-β-actin monoclonal antibody, 1:2500 (clone AC-15; Sigma); rabbit anti-NR2A monoclonal antibody, 1:2500 (clone A12W; Upstate Biotechnology, Lake Placid, NY); rabbit anti-NR2B polyclonal antibody, 1:250 (Upstate Biotechnology). Membranes were washed three times with t-TBS and incubated for 2 h at room temperature, with the appropriated secondary antibody coupled to horseradish peroxidase (1:2500; Biosources, Camarillo, CA). Visualization of immunoreactive bands was produced by enhanced chemiluminescence (Amersham Biosciences, Piscataway, NJ) captured on autoradiography film (Super RX; Fujifilm, Valhalla, NY). Autoradiographs were scanned with ScanJet 7400C (Hewlett Packard, Palo Alto, CA), and digital images were quantified by densitometric analysis using Odyssey version 1.2 software.
Drugs and chemicals.
Picrotoxin, d,l-3-amino-5-phosphonopentanoic acid (d,l-APV), DNQX, and ifenprodil were purchased from Tocris Cookson (Ellisville, MO); kynurenic acid, gluconic acid, and glycine were from Sigma-Aldrich (St. Louis, MO).
Statistics.
Unless specified otherwise, data are represented as mean ± SEM, and statistical significance was assessed using two-tailed paired and unpaired t test for within and between group comparisons, respectively. p < 0.05 was considered statistically significant.
Results
An SD procedure that is stress free and elicits a rebound in resting time
To circumvent stress, a mild form of SD was based on the spontaneous exploratory behavior of C57BL/6J mice, while constraining and directly manipulating the animals was avoided (see Materials and Methods). SD was performed in the first half of the light period, when the mice showed their main resting period and lowest level of motor activity in a 24 h cycle (Fig. 1A). SD was halted when the mouse no longer reacted to the stimuli and fell asleep (i.e., after 4.3 ± 0.1 h; n = 61 mice in total). Control mice were left undisturbed and were resting for 73.9 ± 2.0% (measured in a subset of controls; n = 12) of the time during which SD was performed. Note that these values, as well as the distribution of resting behavior monitored during an undisturbed 24 h cycle (Fig. 1A), are close to the amount of total sleep time measured by electroencephalography and electromyography in the C57BL/6J mouse strain (Franken et al., 1999; Huber et al., 2000a). The absence of stress after SD was confirmed by a plasma corticosterone radioimmunoassay, which yielded virtually the same levels of corticosterone in SD and control groups (Fig. 1B). These low values are typical for the time of day when the mice were killed (Le Minh et al., 2001) and are in marked contrast with the high values of corticosterone observed in animals submitted to a stressful SD procedure (see Materials and Methods) for an equivalent amount of time (Fig. 1B).
Figure 1.

Sleep–wake behavior and corticosterone levels in mice during SD and undisturbed conditions. A, Hourly time course of home-cage motor activity in meters and resting behavior (percentage of recording time) during an undisturbed 24 h baseline (n = 9). The solid line indicates the mean level of activity per hour of SD; the top and bottom dashed lines indicate the corresponding SEM (n = 12). The black bar at the top indicates the 12 h dark period of the light/dark cycle. B, Plasma corticosterone measurements in control (CTRL; n = 9) condition, after SD (n = 8) and after stressful SD (SSD; n = 8). *p < 0.01, compared with CTRL and SD (Tukey's HSD post hoc test after significance in a one-way ANOVA with factor “group”). C, Resting time (percentage of recording time) during the recovery period after SD (SD/R; n = 7) and control conditions (CTRL/R; n = 8). In sleep-deprived mice, the resting time was enhanced in the first hour compared with controls (hour 1, *p < 0.02; hour 2, p = 0.09; hour 3, p = 0.62).
In mice, as in other mammalian species, sleep regulation shows a well established relationship to previous waking duration (Franken et al., 1999, 2001; Huber et al., 2000a), and 4 h of wakefulness induce a near-maximal sleep rebound in C57BL/6J mice (Huber et al., 2000a). At the behavioral level, this rebound could be monitored as an ∼10% increase in resting time during the initial phase of the recovery period (Fig. 1C). These data show that the SD procedure applied in the present study elicited compensatory processes within the time frame expected from the electroencephalographic markers of sleep regulation, consistent with other studies that have validated a noninvasive behavioral monitoring of sleep–wake behavior in rodents (Kapfhamer et al., 2002).
A brief and stress-free SD alters bidirectional synaptic plasticity
We investigated whether stress-free SD altered the full frequency dependence of bidirectional synaptic plasticity between 1 and 100 Hz in the CA1 region of the hippocampus using extracellular field potential recordings (see Materials and Methods). There was no difference between SD and control slices during baseline recordings in the 10–90% slope of fEPSPs (control: −467.2 ± 38.4 μV/ms, n = 63; SD: −511.8 ± 34.9 μV/ms, n = 64; p = 0.39). In control slices, recordings of fEPSPs revealed a reliable LTD (∼35%) after 1 Hz LFS, whereas no long-term changes were found after 5 Hz LFS (Fig. 2A,B). Field potentials tended to be potentiated after the 10 Hz conditioning, and a marked LTP was present after HFS (Fig. 2C,D). After SD, a reliable LTD was obtained after 5 Hz LFS (∼20%) rather than after 1 Hz LFS, and no long-term change was found after stimulation at 10 Hz or HFS (Fig. 2A,D). SD and control groups were most distinguishable after 5 Hz LFS and HFS (Fig. 2E). However, repeated HFS led to similar levels of saturated LTP in both groups (Fig. 2F), and the magnitude of LTD after 1 Hz LFS in control slices and after 5 Hz LFS in the SD condition did not significantly differ from each other (p = 0.20), indicating that SD did not occlude LTP or affect the dynamic range of synaptic plasticity. Notably, the probability of occurrence of LTD/LTP across the frequency axis was changed after SD, partially accounting for the averaged effects on synaptic plasticity (Table 1). Together, these results show that SD induced a rightward shift of the LTD/LTP crossover point of synaptic plasticity in hippocampal slices, favoring the induction of LTD by stimulations in the theta frequency range (5–10 Hz).
Figure 2.
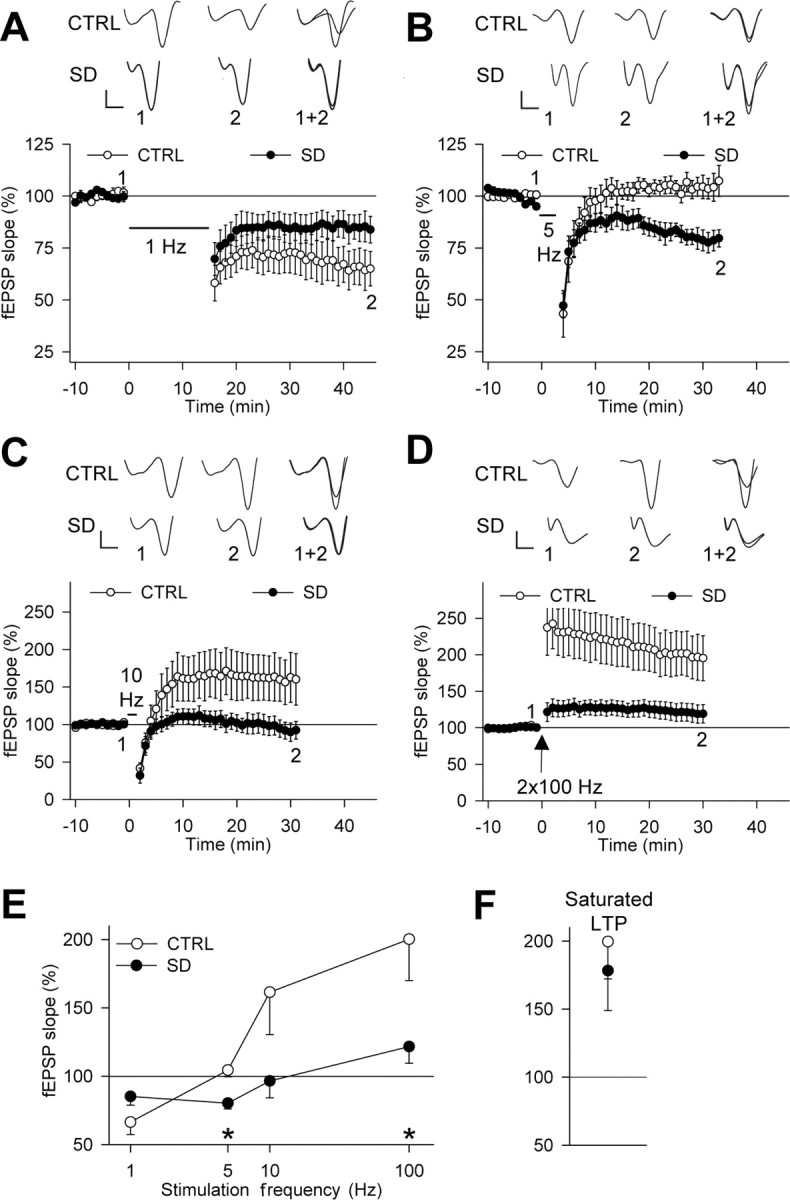
SD shifts the frequency dependence of bidirectional synaptic plasticity. A–D, Effects of 1, 5, and 10 Hz LFS and HFS on fEPSP slope (percentage of baseline) in SD experiments (A–C, n = 7; D, n = 13) and controls (A–C, n = 7; D, n = 13). Sample traces are shown above each figure for the times indicated by the numbers. Calibration: 0.2 mV, 2 ms. E, In control conditions, persistent changes in synaptic strength were significant for 1 Hz LFS (p < 0.01) and HFS (p < 0.01) but not for 5 Hz (p = 0.39) and 10 Hz LFS (p = 0.09). After SD, LTD was reliably induced by 5 Hz LFS (p < 0.01) rather than by 1 Hz LFS (p = 0.06). A 10 Hz LFS and HFS had no significant effect (p = 0.60 and p = 0.10, respectively). The effect of 5 Hz LFS and HFS differed between the groups (*p < 0.05). F, Saturated LTP was induced in all experiments, except one SD (control: n = 6, p < 0.02; SD: n = 6, p < 0.05). No difference was found between the groups (p = 0.61). CTRL, Control.
Table 1.
Probability of LTP/LTD induction after stimulation trains between 1 and 100 Hz in SD and control (CTRL) experiments
| 1 Hz | 5 Hz | 10 Hz | 2 × 100 Hz | |
|---|---|---|---|---|
| LTP | ||||
| CTRL | 0/7 | 1/7 | 4/7 | 9/13 |
| SD | 0/7 | 0/7 | 3/7 | 5/13* |
| LTD | ||||
| CTRL | 5/7 | 2/7 | 0/7 | 0/13 |
| SD | 3/7 | 5/7* | 4/7* | 0/13 |
After SD, LTD was obtained less frequently after 1 Hz but more frequently after 5 or 10 Hz stimulation, whereas LTP was less often induced after 2 × 100 Hz stimulation (*p < 0.05 compared with CTRL; two-tailed χ2 test). The criterion for LTP/LTD induction was >15% change from baseline.
SD effects on synaptic plasticity are independent of somatosensory stimuli
Exploratory behavior occurs during SD and is accompanied by prolonged sensory activity and theta activity in the hippocampus (Buzsaki, 2002), which could contribute to the effects of SD on synaptic plasticity. Olfaction constitutes the primary sensory system of rats and mice. Training in olfactory discrimination tasks reduces LTP and increases LTD in vitro (Lebel et al., 2001; Quinlan et al., 2004). However, these changes were absent in pseudo-trained animals that explored the same environment and were exposed to the same olfactory stimuli without having to learn a rule. In addition, changes in plasticity attributable to olfactory learning take weeks to develop. Thus, the absence of a learning rule in our SD procedure and the shorter time scale on which the effects of SD are observed make it unlikely that olfactory inputs would contribute to the SD-related changes in synaptic plasticity. This is not necessarily the case for somatosensory stimuli. Indeed, whisking activity and the amplitude of hippocampal activity in the theta frequency band are correlated in the rat (Berg and Kleinfeld, 2003), which might directly affect bidirectional synaptic plasticity (Abraham and Tate, 1997).
To clarify the role played by somatosensory activity during SD, we sleep deprived mice ∼20 h after bilateral whisker trimming and tested long-term synaptic plasticity after 5 Hz LFS and HFS. Monitoring of spontaneous motor activity in the home cage in the hours after the whisker trim showed a rise of activity at dark onset and indicated no disturbance in the daily distribution of motor activity (Fig. 3A). During nighttime, the amount of activity per hour did not differ significantly between mice with (distance traveled, 51.8 ± 9.0 m; n = 13) or without (distance traveled, 38.3 ± 3.4 m; n = 18; p = 0.18) whiskers. Remarkably, in control mice, the whisker trim did not alter either resting behavior (whiskers: 73.9 ± 2.0% of resting time, n = 12; no whiskers, 72.0 ± 2.9% of resting time, n = 8; p = 0.58) or the extent of synaptic plasticity (compare Figs. 2E, 3D) (5 Hz LFS, p = 0.36; HFS, p = 0.38). During SD, the hourly distance traveled by the mice was longer in the absence of whiskers (whiskers: 36.4 ± 5.8 m, n = 12; no whiskers: 66.8 ± 9.9 m, n = 16; p < 0.03). This increase in distance is consistent with our observation that the whisker-trimmed mice spent less time exploring single spots of their environment. Despite this behavioral change, SD allowed the induction of a reliable LTD after 5 Hz LFS while impairing LTP after HFS (Fig. 3). Consistent with Table 1, the effects of SD were reflected in the probability of occurrence of LTD (control, 1/7; SD, 4/7) and LTP (control, 7/7; SD, 3/7). The comparable pattern of synaptic plasticity after SD with and without whiskers (Figs. 2E, 3D) excludes a primary influence of somatosensory stimuli in mediating the effects of SD.
Figure 3.
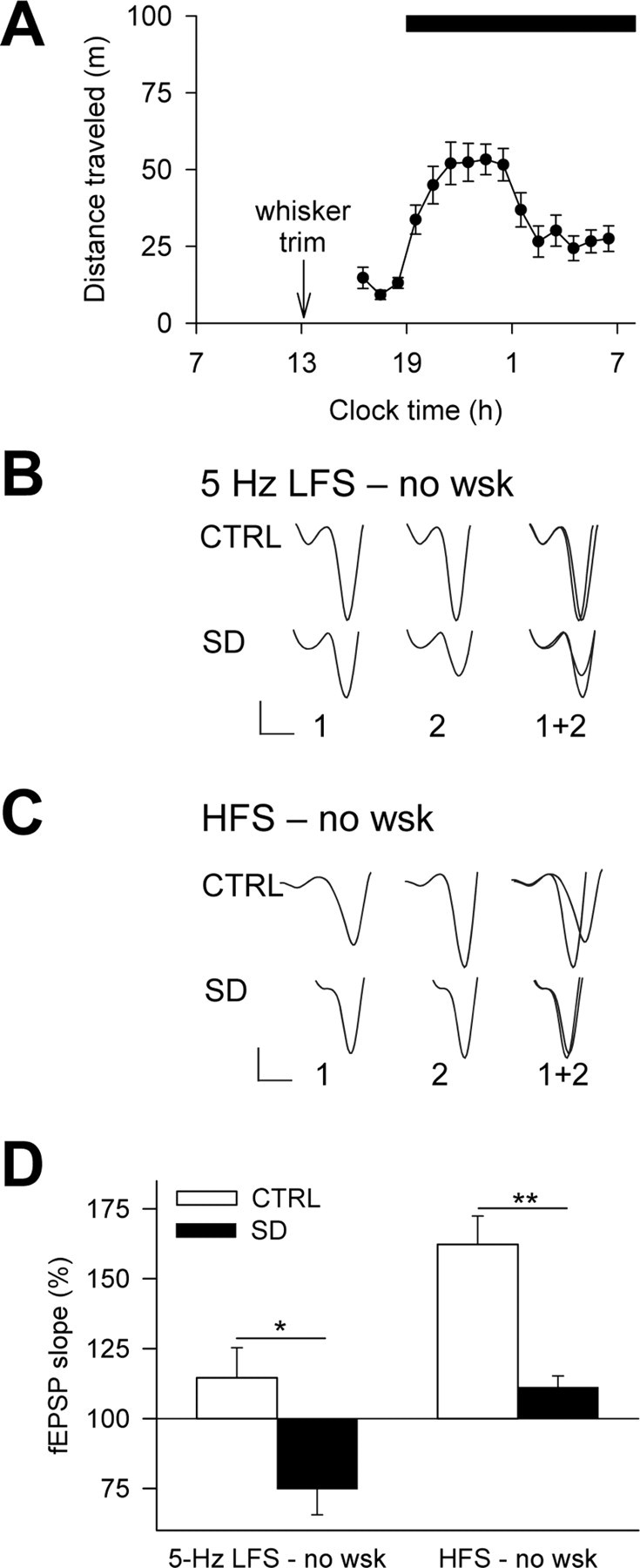
The SD effects on bidirectional synaptic plasticity are independent of somatosensory inputs. A, Hourly time course of home-cage motor activity in the hours after the whisker trim (n = 18). The black bar at the top indicates the 12 h dark period of the light/dark cycle. B, C, Representative fEPSPs at the end of the baseline (1) and during the last 10 min of recording (2) after 5 Hz LFS (B) or HFS (C) in whisker-trimmed mice. Calibration: 0.2 mV, 2 ms. D, Average long-term synaptic plasticity after SD in whisker-trimmed mice. After 5 Hz LFS, LTD was obtained in SD (n = 6; p < 0.04) but not in control experiments (n = 5; p = 0.25). After HFS, LTP was strong in control conditions (n = 7; p < 0.001) and weak after SD (n = 6; p < 0.05). The groups differed after either conditioning protocol (5 Hz LFS, *p < 0.02; HFS, **p < 0.01). CTRL, Control; no wsk, whisker-trimmed mice.
A stressful SD procedure occludes LTP
Stress is well known to reduce LTP and can facilitate LTD (Huang et al., 2005), and long-lasting, stressful SD procedures also block LTP (Campbell et al., 2002; Davis et al., 2003; McDermott et al., 2003). Therefore, we determined whether a stressful form of SD, performed at the same time of day and for an equivalent duration (see Materials and Methods), would elicit effects similar to those observed after the stress-free SD. As expected, stressful SD blocked LTP (106.7 ± 7.6% of baseline; n = 7; p = 0.41). However, LTP was also suppressed when several HFSs were given (117.9 ± 15.0% of baseline; n = 7; p = 0.27). Thus, in contrast to the mild SD, the stressful SD not only impairs but also occludes LTP.
A short recovery period reverses the effects of SD on bidirectional synaptic plasticity
If the changes in synaptic plasticity induced by SD were indeed related to the absence of sleep, then a period of recovery sleep after SD should reverse these effects. To test this hypothesis, we quantified synaptic plasticity induced by 5 Hz LFS and HFS in sleep-deprived mice that had recovered for 3 h. A period of 3 h was chosen because it is sufficient to dissipate the compensatory sleep process occurring after 4 h of wakefulness in C57BL/6J mice (Huber et al., 2000a) (Fig. 1C). Interestingly, this behavioral recovery was followed by a recovery of the synaptic plasticity profile, becoming indistinguishable in control and SD groups (Fig. 4). Also, the probability of occurrence of synaptic plasticity went back to control values (LTD: control/recovery, 0/5; SD/recovery, 0/5; LTP: control/recovery, 6/6; SD/recovery, 6/6). This result strongly corroborates the view that the SD-induced shift of long-term synaptic plasticity along the stimulation frequency axis is related to the lack of sleep in the hours preceding slice preparation.
Figure 4.
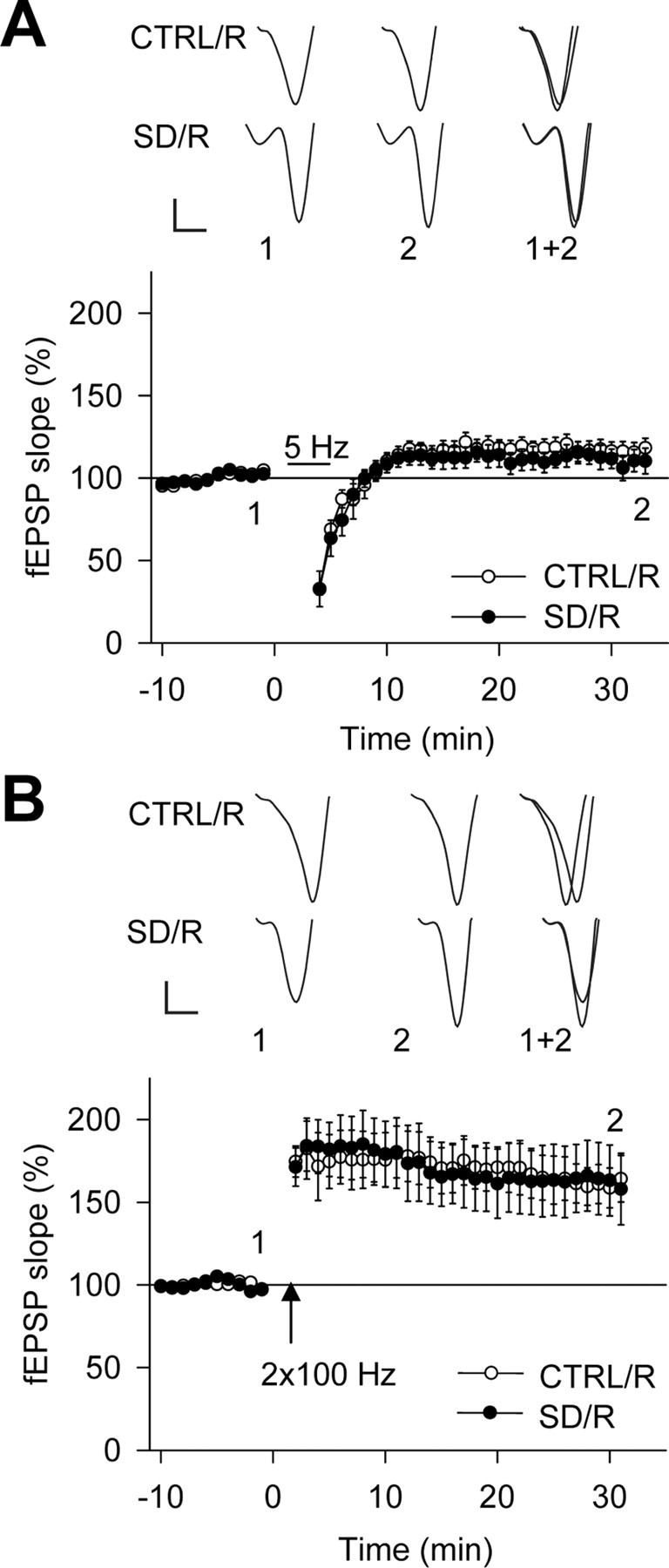
Three hours of recovery after SD restore normal bidirectional synaptic plasticity. Effects of 5 Hz LFS (A) and HFS (B) on fEPSP slope (percentage of baseline) after baseline in slices obtained after the recovery period following SD (SD/R; n = 7) and corresponding control conditions (CTRL/R; n = 8). SD/R (A, n = 5; B, n = 6) and CTRL/R (A, n = 5; B, n = 6) experiments are shown. A 5 Hz LFS induced no persistent change in synaptic strength in either group (CTRL/R: 116.3 ± 6.0% of baseline, p = 0.06; SD/R: 111.8 ± 6.3% of baseline, p = 0.14), whereas LTP was reliably induced by HFS in both groups (CTRL/R: 163.5 ± 13.1% of baseline, p < 0.01; SD/R: 163.1 ± 21.2% of baseline, p < 0.03). No difference was found between the groups (5 Hz LFS, p = 0.61; HFS, p = 0.99). Sample traces above each figure are fEPSPs at the times indicated by the numbers. Calibration: 0.2 mV, 2 ms.
SD reversibly alters the molecular composition of synaptic NMDARs
Both presynaptic and postsynaptic, as well as network, mechanisms may account for the effects of SD on bidirectional synaptic plasticity (Abraham and Tate, 1997; Bredt and Nicoll, 2003; Malenka and Bear, 2004). To test for changes in presynaptic glutamate release, we measured paired-pulse plasticity of fEPSPs at a 50 ms interpulse interval. The resulting paired-pulse facilitation did not differ between control [slope (pulse2 − pulse1)/pulse1: 115.3 ± 32.3%; n = 8; p < 0.01] and SD (92.2 ± 21.0%; n = 8; p < 0.01; p = 0.56 compared with controls) experiments. We next confirmed the requirement for NMDARs in synaptic plasticity by showing that postsynaptic responses in control slices remained unaffected by 1 Hz LFS (100.1 ± 5.5% of baseline; n = 4; p = 0.99) or HFS (96.2 ± 7.8% of baseline; n = 4; p = 0.66) in the presence of the NMDAR antagonist d,l-APV (0.1 mm). To test whether SD alters the contribution of NMDARs to glutamatergic transmission, we first assessed the ratio of NMDAR- over non-NMDAR-mediated EPSCs. This ratio was clearly reduced after SD (control: 54.6 ± 10.2%, n = 13; SD, 31.5 ± 3.6%, n = 20; p < 0.02), whereas non-NMDAR-mediated EPSCs did not differ between conditions (control: −314.5 ± 37.2 pA, n = 13; SD: −269.8 ± 33.5 pA, n = 20; p = 0.39). In contrast, when SD was accompanied by stress, the ratio of NMDAR- over non-NMDAR-mediated EPSCs was indistinguishable from controls (55.5 ± 6.9%; n = 9; p = 0.50). These results are indicative of a decreased NMDAR function caused specifically by sleep loss. In the mature hippocampus, NMDAR-mediated synaptic responses depend on both NR2A- and NR2B-type NMDARs (Scimemi et al., 2004). A switch in the NMDAR subunit composition, namely in the NR2A/NR2B ratio, can underlie shifts of the LTD/LTP crossover point (Quinlan et al., 1999a; Philpot et al., 2001; Perez-Otano and Ehlers, 2005). This switch can be assessed pharmacologically by determining the sensitivity of NMDAR-mediated fEPSPs to the NR2B-type NMDAR antagonist ifenprodil (Quinlan et al., 1999a; Philpot et al., 2001) (see Materials and Methods). There was no difference in the slope of NMDAR-mediated fEPSPs between control and SD slices (slope: control, −119.7 ± 18.2 μV/ms, n = 9; SD, −155.8 ± 33.6 μV/ms, n = 8; p = 0.34). In control slices, we found that ifenprodil (3–10 μm) reduced NMDAR-mediated fEPSPs and whole-cell EPSCs (Fig. 5) by an extent comparable to previous reports on the CA1 area (Liu et al., 2004; Scimemi et al., 2004). Moreover, ifenprodil accelerated the monoexponential decay time course of NMDAR-mediated EPSCs from τ = 48.3 ± 3.8 ms to 41.7 ± 2.9 ms (n = 8; p < 0.05). However, after SD, ifenprodil effects were smaller in both field (Fig. 5A,B) and whole-cell patch-clamp (Fig. 5C,D) recordings. The decay did not differ from the control condition during baseline (τ = 55.0 ± 4.9 ms; n = 5; p = 0.30) and was not altered by ifenprodil (τ = 52.1 ± 7.0 ms; n = 5; p = 0.58), reflecting a larger NR2A/NR2B ratio of synaptically located NMDARs. Remarkably, paralleling the changes in synaptic plasticity, the sensitivity to ifenprodil returned to control levels when SD was followed by a 3 h recovery sleep period (Fig. 5B). The fEPSPs and EPSCs remaining after treatment with ifenprodil were abolished by the NMDAR antagonist d,l-APV (data not shown), confirming the mediation of the synaptic responses by NMDARs. These data strongly suggest that SD provokes a rapid and reversible change in the molecular composition of synaptic NMDARs, paralleling the time course of the shift in synaptic plasticity.
Figure 5.

The sensitivity of NMDAR-mediated fEPSPs and whole-cell EPSCs to the NR2B-type receptor antagonist ifenprodil is reduced after SD and restored after recovery sleep. A, NMDAR-mediated fEPSP slope (percentage of baseline) during baseline, the first 40 min of ifenprodil (10 μm) application, and 20 min of steady state in SD (n = 5) and control (n = 5) experiments. Steady-state effects were reached after 64.0 ± 6.8 min in control experiments and after 62.0 ± 9.7 min in SD experiments (p = 0.87 compared with control) (Philpot et al., 2001). Traces depict individual fEPSPs at the times indicated by the numbers. Calibration: 80 μV, 2 ms. B, Ifenprodil reduced the fEPSP slope both in control (p < 0.01) and in SD (p < 0.01) experiments, but less in SD than in control conditions (*p < 0.03). In contrast, the effect did not differ between the groups after a 3 h recovery period (SD/R, n = 3; CTRL/R, n = 4; p = 0.76). C, Representative NMDAR-mediated whole-cell EPSCs during baseline and during steady-state effects of ifenprodil (3 μm). Calibration: 40 pA, 40 ms. D, Ifenprodil inhibited NMDAR-mediated EPSCs both in control (n = 9; p < 0.001) and in SD (n = 5; p < 0.02) experiments, but less in SD than in control conditions (*p < 0.03). CTRL, Control.
SD enhances the level of NR2A subunit protein at synaptic sites
To more precisely determine the composition of synaptic NMDARs at the molecular level, immunoblotting for NR2A and NR2B subunits was performed on purified synaptosomes, a subcellular fraction enriched in synaptic proteins. This biochemical analysis revealed that NR2A protein levels were higher after SD, whereas no change was detected in NR2B protein level, which led to an elevated NR2A/NR2B ratio (Fig. 6).
Figure 6.

SD alters the expression of NR2A but not NR2B protein levels at synaptic sites. A, Representative immunoblots for NR2A and NR2B subunit proteins in synaptosomes prepared from hippocampi of control and sleep-deprived mice and run on the same gel. β-Actin was used to control that equal amounts of protein (30 μg) were loaded on the gel. B, SD enhanced the level of NR2A protein (p < 0.05 compared with matched control experiments), whereas no significant change was detected for NR2B (p = 0.13), which led to a higher NR2A/NR2B ratio (p < 0.05). The optical density of each sample was first expressed as a percentage of the corresponding β-actin and then normalized to the control value run on the same gel (100%). Scatter plots depict the individual data of the SD group. CTRL, Control.
Discussion
We report that sleep loss provoked a rapid and reversible modulation of bidirectional synaptic plasticity. In contrast to previous ex vivo studies, we made an important step forward in identifying the importance of sleep loss among the confounding factors accompanying SD. The novelty of our approach consists in using a particularly mild SD protocol that (1) respects important parameters of sleep regulation, in particular its time course and its reversibility, and (2) is free of an elevation in stress hormone levels. In the spirit of the mildness of this approach, we used a noninvasive, yet electroencephalographically validated, method to monitor sleep–wake behavior (Kapfhamer et al., 2002).
By assessing the consequences of SD on bidirectional synaptic plasticity, we show that the frequency dependence of the LTP/LTD modification threshold was shifted by SD, whereas the dynamic range of plasticity was unaltered and full induction of LTP remained possible. This result excludes the important concern that synaptic strength was saturated because of the SD procedure. In contrast, we found that a stressful SD procedure not only blocks but also occludes LTP, indicating that the inhibition of LTP by non-stressful and by stressful SD (Campbell et al., 2002; Davis et al., 2003; McDermott et al., 2003) reflects distinct electrophysiological “phenotypes.” Furthermore, by recording from slices maintained in near-physiological concentrations of divalent cations, we determined the consequences of brief sleep loss and recovery sleep on NMDARs located at the synapse, while leaving presynaptic glutamate release unchanged. Previously, the actions of long-lasting and stressful SD on glutamate receptors were studied by activating synapses in low Mg2+ concentrations or by applying glutamate to outside-out patches of apical hippocampal dendrites (Chen et al., 2006; McDermott et al., 2006). Both of these approaches allow for the simultaneous activation of synaptic and extrasynaptic receptors, which show different trafficking behavior and may play opposing roles in synaptic plasticity (Neyton and Paoletti, 2006). Together, by combining an unparalleled mild form of SD with standard techniques to assess glutamatergic synaptic transmission, we have identified some of the cellular mechanisms underlying the effects of sleep loss on hippocampal synaptic plasticity and on the subunit composition of synaptic NMDARs.
The rapid trafficking of ionotropic AMPA-type receptors (AMPARs) plays a key role in the strengthening and weakening of glutamatergic synapses (Malinow and Malenka, 2002; Bredt and Nicoll, 2003). The prerequisite for this trafficking to be initiated is the activation of NMDARs, which are a primary source for the postsynaptic calcium that initiates synaptic plasticity. Both the calcium concentrations reached and the duration of these signals (Yang et al., 1999; Lisman, 2001) are important for the direction of synaptic plasticity. Distinct properties of NR2A- versus NR2B-containing receptors confer differential roles for NMDAR subtypes in shaping calcium signals and synaptic plasticity. These include distinct kinetics of the receptor currents, a differential subcellular localization of NR2A- versus NR2B-containing receptors with respect to the sites of glutamate release, and subtype-specific interactions with postsynaptic scaffolding and signaling molecules (Perez-Otano and Ehlers, 2005; Köhr, 2006). From studies in mature slice preparations, the consensus emerges that both NR2A- and NR2B-containing receptors are involved in the induction of LTP, whereas NR2B-containing receptors may also mediate LTD (for review, see Neyton and Paoletti, 2006). In the mature hippocampus, both NR2A- and NR2B-containing receptors are expressed, with a substantial NR2B-containing receptor population located extrasynaptically (Liu et al., 2004; Scimemi et al., 2004). The increase in the NR2A/NR2B ratio, detected electrophysiologically after SD, was also supported by immunoblotting confined to synaptic sites and most likely reflects a larger synaptic NR2A protein content. Thus, the enhanced presence of NR2A could either give rise to receptors containing the binary composition NR1/NR2A or may further the formation of triheteromeric receptors containing NR1/NR2A/NR2B. Both receptor configurations would contribute to a decreased ifenprodil sensitivity of the synaptic NMDAR response. Our observation that the decay kinetics of the NMDA EPSC was not noticeably accelerated after SD argues against a separate receptor population containing only NR2A and rather points toward the integration of NR2A into a more complex molecular assembly. How the SD-induced changes in synaptic plasticity relate to these differences in subunit-specific characteristics remains to be determined. In particular, it will be important to evaluate how an increase in NR2A-containing NMDARs, implicated predominantly in LTP, may cause a shift in the frequency dependence of LTD. Notably, our findings are consistent with the profile of NMDAR expression and function observed in developmental studies in visual cortex (Quinlan et al., 1999b; Philpot et al., 2003), which suggest that an elevated expression of NR2A-containing receptors correlates with a decrease of NMDAR function and a rightward shift in the frequency dependence of bidirectional synaptic plasticity (Philpot et al., 2003).
A few hours of SD and recovery sleep were sufficient to induce and reverse both functional and molecular alterations in glutamatergic transmission. The time scale of the changes observed in the present study is strikingly shorter than that observed for the shift in the LTD/LTP crossover point and in the NR2A/NR2B ratio during experience-dependent plasticity in the developing visual cortex (Philpot et al., 2001; Perez-Otano and Ehlers, 2005), during learning (Quinlan et al., 2004) or during environmental enrichment (Tang et al., 2001). Therefore, rapid forms of NMDAR trafficking (Perez-Otano and Ehlers, 2005) and associated processes controlling glutamatergic transmission may codetermine the propensity for synaptic plasticity in adulthood, depending on previous arousal. In this context, it is conceivable that substances that play a role in the regulation of sleep and wakefulness could contribute to the changes elicited by SD. Thus, orexin, which promotes arousal (Saper et al., 2005) and modulates hippocampal synaptic plasticity (Selbach et al., 2004), can control rapid NMDAR trafficking via a protein kinase C-dependent mechanism (Borgland et al., 2006). Furthermore, adenosine, which plays an important role in sleep regulation (Obal and Krueger, 2003; Rétey et al., 2005), may modulate glutamatergic receptor function (Sebastiao and Ribeiro, 1996; Dunwiddie and Masino, 2001). In this manner, the modulation of bidirectional synaptic plasticity could be determined by the on-going release of compounds characteristic for behavioral state. Furthermore, the expression of NR2A- and NR2B-NMDARs within a cortical area is pathway specific (Kumar and Huguenard, 2003), raising the possibility that the differential regulation of NMDARs may correlate with brain area-specific susceptibilities to SD (Harrison and Horne, 2000; Huber et al., 2000b).
Previous behavioral studies, based on brief (5–6 h) SD minimizing stress (Graves et al., 2003; Guan et al., 2004), reported a selective impairment of spatial memory, which depends on the hippocampus. There is now strong evidence that hippocampal and other types of memory are accompanied by shifts in the frequency dependence of bidirectional synaptic plasticity. Thus, learning is impaired after saturation of hippocampal LTP (Moser et al., 1998) and induces LTP in the CA1 region of the hippocampus, which occludes further LTP elicited by HFS (Whitlock et al., 2006). In the neocortex, motor skill training is followed by a reduced LTP and a facilitated LTD, consistent with a strengthening of synapses during learning (Rioult-Pedotti et al., 2000). In the piriform cortex, a reduction in HFS-induced LTP has also been associated with olfactory rule learning and was associated with a change in the composition of synaptic NMDARs (Quinlan et al., 2004). Conversely, genetically modified mice that show an impaired bidirectional regulation of synaptic plasticity also present severe behavioral deficits in learning and memory (Bach et al., 1995; Migaud et al., 1998; Zeng et al., 2001). Therefore, the shift in synaptic plasticity induced by sleep loss likely constitutes a crucial cellular process involved in its adverse effects on hippocampus-dependent memory. Moreover, the reversibility of SD effects after recovery sleep suggests that sleep plays a general role in maintaining glutamatergic synapses within a dynamic range and preventing them from entering states of saturated LTP or LTD, which would be deleterious for memory function (Migaud et al., 1998).
In nonhuman primates, cognitive deficits induced by SD can be counteracted by potentiating AMPAR function through allosteric modulation (Porrino et al., 2005). This pharmacological treatment may effectively counteract decreased NMDAR function because AMPAR trafficking is essential for LTD and LTP (Bredt and Nicoll, 2003) and can be differentially regulated by NMDAR subtypes (Kim et al., 2005). Therefore, identifying the factors promoting an altered synaptic NMDAR subunit composition after SD may ultimately yield insight into how synaptic plasticity, and potentially learning and memory, could be furthered in adulthood.
In conclusion, by combining behavioral and in vitro electrophysiological analysis of a particularly mild, stress-free SD in mice, we gained novel access into the relationship between sleep and excitatory synaptic transmission. Beyond the scope of this work, the approach taken here lends itself to various complementary investigations, in particular concerning the effects of SD on pharmacological aspects of sleep control in the modern society. This may further help to define how neuronal networks implicated in cognitive functions are impaired by insufficient sleep.
Footnotes
This work was supported by the Swiss National Science Foundation (3100A0-103655) and the Fonds zur Förderung von Lehre und Forschung. We thank Profs. B. Gähwiler and U. Gerber for helpful comments in the course of this study and Prof. K. G. Hofbauer and Drs. H. P. Landolt and N. Wanaverbecq for critical comments on this manuscript. We thank Dr. H. BenTekaya and F. Oliveri for help with the immunoblot analysis.
References
- Abraham WC, Bear MF. Metaplasticity: the plasticity of synaptic plasticity. Trends Neurosci. 1996;19:126–130. doi: 10.1016/s0166-2236(96)80018-x. [DOI] [PubMed] [Google Scholar]
- Abraham WC, Tate WP. Metaplasticity: a new vista across the field of synaptic plasticity. Prog Neurobiol. 1997;52:303–323. doi: 10.1016/s0301-0082(97)00018-x. [DOI] [PubMed] [Google Scholar]
- Bach ME, Hawkins RD, Osman M, Kandel ER, Mayford M. Impairment of spatial but not contextual memory in CaMKII mutant mice with a selective loss of hippocampal LTP in the range of the theta frequency. Cell. 1995;81:905–915. doi: 10.1016/0092-8674(95)90010-1. [DOI] [PubMed] [Google Scholar]
- Berg RW, Kleinfeld D. Vibrissa movement elicited by rhythmic electrical microstimulation to motor cortex in the aroused rat mimics exploratory whisking. J Neurophysiol. 2003;90:2950–2963. doi: 10.1152/jn.00511.2003. [DOI] [PubMed] [Google Scholar]
- Borgland SL, Taha SA, Sarti F, Fields HL, Bonci A. Orexin A in the VTA is critical for the induction of synaptic plasticity and behavioral sensitization to cocaine. Neuron. 2006;49:589–601. doi: 10.1016/j.neuron.2006.01.016. [DOI] [PubMed] [Google Scholar]
- Bredt DS, Nicoll RA. AMPA receptor trafficking at excitatory synapses. Neuron. 2003;40:361–379. doi: 10.1016/s0896-6273(03)00640-8. [DOI] [PubMed] [Google Scholar]
- Buzsaki G. Theta oscillations in the hippocampus. Neuron. 2002;33:325–340. doi: 10.1016/s0896-6273(02)00586-x. [DOI] [PubMed] [Google Scholar]
- Campbell IG, Guinan MJ, Horowitz JM. Sleep deprivation impairs long-term potentiation in rat hippocampal slices. J Neurophysiol. 2002;88:1073–1076. doi: 10.1152/jn.2002.88.2.1073. [DOI] [PubMed] [Google Scholar]
- Chen C, Hardy M, Zhang J, LaHoste GJ, Bazan NG. Altered NMDA receptor trafficking contributes to sleep deprivation-induced hippocampal synaptic and cognitive impairments. Biochem Biophys Res Commun. 2006;340:435–440. doi: 10.1016/j.bbrc.2005.12.021. [DOI] [PubMed] [Google Scholar]
- Davis CJ, Harding JW, Wright JW. REM sleep deprivation-induced deficits in the latency-to-peak induction and maintenance of long-term potentiation within the CA1 region of the hippocampus. Brain Res. 2003;973:293–297. doi: 10.1016/s0006-8993(03)02508-3. [DOI] [PubMed] [Google Scholar]
- Dunwiddie TV, Masino SA. The role and regulation of adenosine in the central nervous system. Annu Rev Neurosci. 2001;24:31–55. doi: 10.1146/annurev.neuro.24.1.31. [DOI] [PubMed] [Google Scholar]
- Durmer JS, Dinges DF. Neurocognitive consequences of sleep deprivation. Semin Neurol. 2005;25:117–129. doi: 10.1055/s-2005-867080. [DOI] [PubMed] [Google Scholar]
- Franken P, Malafosse A, Tafti M. Genetic determinants of sleep regulation in inbred mice. Sleep. 1999;22:155–169. [PubMed] [Google Scholar]
- Franken P, Chollet D, Tafti M. The homeostatic regulation of sleep need is under genetic control. J Neurosci. 2001;21:2610–2621. doi: 10.1523/JNEUROSCI.21-08-02610.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graves LA, Pack A, Abel T. Sleep and memory: a molecular perspective. Trends Neurosci. 2001;24:237–243. doi: 10.1016/s0166-2236(00)01744-6. [DOI] [PubMed] [Google Scholar]
- Graves LA, Heller EA, Pack AI, Abel T. Sleep deprivation selectively impairs memory consolidation for contextual fear conditioning. Learn Mem. 2003;10:168–176. doi: 10.1101/lm.48803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Z, Peng X, Fang J. Sleep deprivation impairs spatial memory and decreases extracellular signal-regulated kinase phosphorylation in the hippocampus. Brain Res. 2004;1018:38–47. doi: 10.1016/j.brainres.2004.05.032. [DOI] [PubMed] [Google Scholar]
- Harrison Y, Horne JA. The impact of sleep deprivation on decision making: a review. J Exp Psychol Appl. 2000;6:236–249. doi: 10.1037//1076-898x.6.3.236. [DOI] [PubMed] [Google Scholar]
- Huang CC, Yang CH, Hsu KS. Do stress and long-term potentiation share the same molecular mechanisms? Mol Neurobiol. 2005;32:223–235. doi: 10.1385/MN:32:3:223. [DOI] [PubMed] [Google Scholar]
- Huber R, Deboer T, Tobler I. Effects of sleep deprivation on sleep and sleep EEG in three mouse strains: empirical data and simulations. Brain Res. 2000a;857:8–19. doi: 10.1016/s0006-8993(99)02248-9. [DOI] [PubMed] [Google Scholar]
- Huber R, Deboer T, Tobler I. Topography of EEG dynamics after sleep deprivation in mice. J Neurophysiol. 2000b;84:1888–1893. doi: 10.1152/jn.2000.84.4.1888. [DOI] [PubMed] [Google Scholar]
- Kapfhamer D, Valladares O, Sun Y, Nolan PM, Rux JJ, Arnold SE, Veasey SC, Bucan M. Mutations in Rab3a alter circadian period and homeostatic response to sleep loss in the mouse. Nat Genet. 2002;32:290–295. doi: 10.1038/ng991. [DOI] [PubMed] [Google Scholar]
- Kim JJ, Diamond DM. The stressed hippocampus, synaptic plasticity and lost memories. Nat Rev Neurosci. 2002;3:453–462. doi: 10.1038/nrn849. [DOI] [PubMed] [Google Scholar]
- Kim MJ, Dunah AW, Wang YT, Sheng M. Differential roles of NR2A- and NR2B-containing NMDA receptors in Ras-ERK signaling and AMPA receptor trafficking. Neuron. 2005;46:745–760. doi: 10.1016/j.neuron.2005.04.031. [DOI] [PubMed] [Google Scholar]
- Köhr G. NMDA receptor function: subunit composition versus spatial distribution. Cell Tissue Res. 2006;326:439–446. doi: 10.1007/s00441-006-0273-6. [DOI] [PubMed] [Google Scholar]
- Kumar SS, Huguenard JR. Pathway-specific differences in subunit composition of synaptic NMDA receptors on pyramidal neurons in neocortex. J Neurosci. 2003;23:10074–10083. doi: 10.1523/JNEUROSCI.23-31-10074.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebel D, Grossman Y, Barkai E. Olfactory learning modifies predisposition for long-term potentiation and long-term depression induction in the rat piriform (olfactory) cortex. Cereb Cortex. 2001;11:485–489. doi: 10.1093/cercor/11.6.485. [DOI] [PubMed] [Google Scholar]
- Le Minh N, Damiola F, Tronche F, Schutz G, Schibler U. Glucocorticoid hormones inhibit food-induced phase-shifting of peripheral circadian oscillators. EMBO J. 2001;20:7128–7136. doi: 10.1093/emboj/20.24.7128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman JE. Three Ca2+levels affect plasticity differently: the LTP zone, the LTD zone and no man's land. J Physiol (Lond) 2001;532:285. doi: 10.1111/j.1469-7793.2001.0285f.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Wong TP, Pozza MF, Lingenhoehl K, Wang Y, Sheng M, Auberson YP, Wang YT. Role of NMDA receptor subtypes in governing the direction of hippocampal synaptic plasticity. Science. 2004;304:1021–1024. doi: 10.1126/science.1096615. [DOI] [PubMed] [Google Scholar]
- Lynch MA. Long-term potentiation and memory. Physiol Rev. 2004;84:87–136. doi: 10.1152/physrev.00014.2003. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–126. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- McDermott CM, LaHoste GJ, Chen C, Musto A, Bazan NG, Magee JC. Sleep deprivation causes behavioral, synaptic, and membrane excitability alterations in hippocampal neurons. J Neurosci. 2003;23:9687–9695. doi: 10.1523/JNEUROSCI.23-29-09687.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDermott CM, Hardy MN, Bazan NG, Magee JC. Sleep deprivation-induced alterations in excitatory synaptic transmission in the CA1 region of the rat hippocampus. J Physiol (Lond) 2006;570:553–565. doi: 10.1113/jphysiol.2005.093781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migaud M, Charlesworth P, Dempster M, Webster LC, Watabe AM, Makhinson M, He Y, Ramsay MF, Morris RG, Morrison JH, O'Dell TJ, Grant SG. Enhanced long-term potentiation and impaired learning in mice with mutant postsynaptic density-95 protein. Nature. 1998;396:433–439. doi: 10.1038/24790. [DOI] [PubMed] [Google Scholar]
- Miyamoto H, Hensch TK. Reciprocal interaction of sleep and synaptic plasticity. Mol Interv. 2003;3:404–417. doi: 10.1124/mi.3.7.404. [DOI] [PubMed] [Google Scholar]
- Moser EI, Krobert KA, Moser M-B, Morris RGM. Impaired spatial learning after saturation of long-term potentiation. Science. 1998;281:2038–2042. doi: 10.1126/science.281.5385.2038. [DOI] [PubMed] [Google Scholar]
- Nakamura Y, Iga K, Shibata T, Shudo M, Kataoka K. Glial plasmalemmal vesicles: a subcellular fraction from rat hippocampal homogenate distinct from synaptosomes. Glia. 1993;9:48–56. doi: 10.1002/glia.440090107. [DOI] [PubMed] [Google Scholar]
- Neyton J, Paoletti P. Relating NMDA receptor function to receptor subunit composition: limitations of the pharmacological approach. J Neurosci. 2006;26:1331–1333. doi: 10.1523/JNEUROSCI.5242-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obal F, Jr, Krueger JM. Biochemical regulation of non-rapid-eye-movement sleep. Front Biosci. 2003;8:d520–d550. doi: 10.2741/1033. [DOI] [PubMed] [Google Scholar]
- Perez-Otano I, Ehlers MD. Homeostatic plasticity and NMDA receptor trafficking. Trends Neurosci. 2005;28:229–238. doi: 10.1016/j.tins.2005.03.004. [DOI] [PubMed] [Google Scholar]
- Philpot BD, Sekhar AK, Shouval HZ, Bear MF. Visual experience and deprivation bidirectionally modify the composition and function of NMDA receptors in visual cortex. Neuron. 2001;29:157–169. doi: 10.1016/s0896-6273(01)00187-8. [DOI] [PubMed] [Google Scholar]
- Philpot BD, Espinosa JS, Bear MF. Evidence for altered NMDA receptor function as a basis for metaplasticity in visual cortex. J Neurosci. 2003;23:5583–5588. doi: 10.1523/JNEUROSCI.23-13-05583.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porrino LJ, Daunais JB, Rogers GA, Hampson RE, Deadwyler SA. Facilitation of task performance and removal of the effects of sleep deprivation by an ampakine (CX717) in nonhuman primates. PLoS Biol. 2005;3:1640–1652. doi: 10.1371/journal.pbio.0030299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan EM, Philpot BD, Huganir RL, Bear MF. Rapid, experience-dependent expression of synaptic NMDA receptors in visual cortex in vivo. Nat Neurosci. 1999a;2:352–357. doi: 10.1038/7263. [DOI] [PubMed] [Google Scholar]
- Quinlan EM, Olstein DH, Bear MF. Bidirectional, experience-dependent regulation of N-methyl-d-aspartate receptor subunit composition in the rat visual cortex during postnatal development. Proc Natl Acad Sci USA. 1999b;96:12876–12880. doi: 10.1073/pnas.96.22.12876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan EM, Lebel D, Brosh I, Barkai E. A molecular mechanism for stabilization of learning-induced synaptic modifications. Neuron. 2004;41:185–192. doi: 10.1016/s0896-6273(03)00874-2. [DOI] [PubMed] [Google Scholar]
- Rétey JV, Adam M, Honegger E, Khatami R, Luhmann UF, Jung HH, Berger W, Landolt HP. A functional genetic variation of adenosine deaminase affects the duration and intensity of deep sleep in humans. Proc Natl Acad Sci USA. 2005;102:15676–15681. doi: 10.1073/pnas.0505414102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rioult-Pedotti MS, Friedman D, Donoghue JP. Learning-induced LTP in neocortex. Science. 2000;290:533–536. doi: 10.1126/science.290.5491.533. [DOI] [PubMed] [Google Scholar]
- Saper CB, Cano G, Scammell TE. Homeostatic, circadian, and emotional regulation of sleep. J Comp Neurol. 2005;493:92–98. doi: 10.1002/cne.20770. [DOI] [PubMed] [Google Scholar]
- Scimemi A, Fine A, Kullmann DM, Rusakov DA. NR2B-containing receptors mediate cross talk among hippocampal synapses. J Neurosci. 2004;24:4767–4777. doi: 10.1523/JNEUROSCI.0364-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebastiao AM, Ribeiro JA. Adenosine A2 receptor-mediated excitatory actions on the nervous system. Prog Neurobiol. 1996;48:167–189. doi: 10.1016/0301-0082(95)00035-6. [DOI] [PubMed] [Google Scholar]
- Selbach O, Doreulee N, Bohla C, Eriksson KS, Sergeeva OA, Poelchen W, Brown RE, Haas HL. Orexins/hypocretins cause sharp wave- and theta-related synaptic plasticity in the hippocampus via glutamatergic, GABAergic, noradrenergic, and cholinergic signaling. Neuroscience. 2004;127:519–528. doi: 10.1016/j.neuroscience.2004.05.012. [DOI] [PubMed] [Google Scholar]
- Tang YP, Wang H, Feng R, Kyin M, Tsien JZ. Differential effects of enrichment on learning and memory function in NR2B transgenic mice. Neuropharmacology. 2001;41:779–790. doi: 10.1016/s0028-3908(01)00122-8. [DOI] [PubMed] [Google Scholar]
- Whitlock JR, Heynen AJ, Shuler MG, Bear MF. Learning induces long-term potentiation in the hippocampus. Science. 2006;313:1093–1097. doi: 10.1126/science.1128134. [DOI] [PubMed] [Google Scholar]
- Yang SN, Tang YG, Zucker RS. Selective induction of LTP and LTD by postsynaptic [Ca2+]ielevation. J Neurophysiol. 1999;81:781–787. doi: 10.1152/jn.1999.81.2.781. [DOI] [PubMed] [Google Scholar]
- Zeng H, Chattarji S, Barbarosie M, Rondi-Reig L, Philpot BD, Miyakawa T, Bear MF, Tonegawa S. Forebrain-specific calcineurin knockout selectively impairs bidirectional synaptic plasticity and working/episodic-like memory. Cell. 2001;107:617–629. doi: 10.1016/s0092-8674(01)00585-2. [DOI] [PubMed] [Google Scholar]