

Abstract
The Reelin signaling pathway controls neuronal positioning in human and mouse brain during development as well as modulation of long-term potentiation (LTP) and behavior in the adult. Reelin signals by binding to two transmembrane receptors, apolipoprotein E receptor 2 (Apoer2) and very-low-density lipoprotein receptor. After Reelin binds to the receptors, Disabled-1 (Dab1), an intracellular adaptor protein that binds to the cytoplasmic tails of the receptors, becomes phosphorylated on tyrosine residues, initiating a signaling cascade that includes activation of Src-family kinases and Akt. Here, we have created a line of mutant mice (Apoer2 EIG) in which the Apoer2 NFDNPVY motif has been altered to EIGNPVY to disrupt the Apoer2–Dab1 interaction to further study Reelin signaling in development and adult brain. Using primary neuronal cultures stimulated with recombinant Reelin, we find that normal Reelin signaling requires the wild-type NFDNPVY sequence and likely the interaction of Apoer2 with Dab1. Furthermore, examination of hippocampal, cortical, and cerebellar layering reveals that the NFDNPVY sequence of Apoer2 is indispensable for normal neuronal positioning during development of the brain. Adult Apoer2 EIG mice display severe abnormalities in LTP and behavior that are distinct from those observed for mice lacking Apoer2. In Apoer2 EIG slices, LTP degraded to baseline within 30 min, and this was prevented in the presence of Reelin. Together, these findings emphasize the complexity of Reelin signaling in the adult brain, which likely requires multiple adaptor protein interactions with the intracellular domain of Apoer2.
Keywords: signaling, neuronal migration, memory, long-term potentiation, development, phosphorylation
Introduction
The formation of the mammalian brain follows an orchestrated migration of cells that depends on several signaling pathways. In humans, disruption of the gene encoding Reelin leads to a severe developmental disorder highlighted by impaired neuronal migration, leading to lissencephaly or “smooth brain” and thickening of the cerebral cortex (Hong et al., 2000). Deletion of a 200 kb chromosomal region containing the Reelin receptor very-low-density lipoprotein receptor (VLDLR) leads to an autosomal recessive syndrome of nonprogressive cerebellar ataxia with mental retardation associated with inferior cerebellar hypoplasia and mild cerebral gyral simplification (Boycott et al., 2005). In mice, defects of individual Reelin receptors induced by conventional gene targeting leads to less severe phenotypes with regional specificity, with either mild cerebellar disturbances (Vldlr) or cortical and hippocampal mislayering with apolipoprotein E receptor 2 (Apoer2) (Trommsdorff et al., 1999). However, disruption of Reelin, Disabled-1 (Dab1), or both Apoer2 and Vldlr in mice leads to an indistinguishable Reeler phenotype, as a result of severe cell positioning defects in the cerebellum and cortical regions of the brain (D’Arcangelo et al., 1995; Howell et al., 1997; Trommsdorff et al., 1999). The similarity of the anatomical alterations in these mouse models firmly establishes that these proteins belong to a pathway crucial for neuronal migration and brain development.
In the past few years, the details of the Reelin signaling pathway have become clearer. During development, the secreted protein Reelin binds to two cell-surface receptors, Apoer2 and Vldlr (D’Arcangelo et al., 1999; Hiesberger et al., 1999), causing receptor clustering (Strasser et al., 2004) and further transmission of a signal to the cell. Dab1 is an intracellular adaptor protein that in its unphosphorylated state binds to the cytoplasmic domains of Apoer2 and Vldlr (Trommsdorff et al., 1998; Howell et al., 1999; Stolt et al., 2003). Reelin binding to the receptors induces Dab1 tyrosine phosphorylation by Src-family kinases such as Src and Fyn (Arnaud et al., 2003; Bock and Herz, 2003). Phosphorylated Dab1 subsequently interacts with other proteins known to be important for the regulation of neuronal migration including lissencephaly protein 1 (Assadi et al., 2003) and phosphatidylinositol 3-kinase (PI3K) (Bock et al., 2003). Activation of PI3K leads to further downstream signaling including activation of Akt and alterations in glycogen synthase kinase 3β (GSK3β) and tau (Beffert et al., 2002).
Through its protein interaction or phosphotyrosine-binding (PTB) domain, Dab1 binds to a highly conserved NPXY motif in LDL receptor and amyloid precursor protein family members (Trommsdorff et al., 1998; Howell et al., 1999). To obtain more detailed information on the role of Reelin signaling in development and the adult brain, we disrupted the interaction of Apoer2 and Dab1 by mutating the specific residues in Apoer2 required for Dab1 binding in the mouse, in which we have examined neuronal positioning during development as well as electrophysiology and behavior.
Materials and Methods
All protocols involving the use of animals for the following experiments were in compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and the University of Texas Southwestern and Baylor College of Medicine Institutional Animal Care and Use Committees.
Targeted insertion of mutated Apoer2 intracellular domain.
We performed site-directed mutagenesis (Stratagene, La Jolla, CA) on a 434 bp cDNA encoding the Apoer2 intracellular domain (ICD) using the following primers: EIG forward (EIGfwd; 5′-GGAAGAACACCAAGAGCATGGAAATCGGCAACCCAGTGTACAGG-3′) and EIG reverse (EIGrev; 5′-CCTGTACACTGGGTTGCCGATTTCCATGCTCTTGGTGTTCTTC-C3′). The mutant cDNA fragment was inserted into a genomic fragment containing Apoer2 exons 9–16. Additional cloning, embryonic stem cell screening, and selection were performed as described previously (Beffert et al., 2005). Genotyping of wild-type (WT) and Apoer2 EIG mice were performed with the following primers: WTfwd (5′-ATCTGGAGGAACTGGAAGCGGAAGAACACC-3′), WTrev (5′-AAAAGGTATGGGGATTGTGGATGGAGAG-3′), Dab*fwd (5′-TTAGGAAAGGACAGTGGGAGTGGCACC-3′), and Dab*rev (5′-AACACCAAGAGCATGGAAATCGGC-3′).
GST pull downs.
Vldlr and Apoer2 intracellular domains were introduced in-frame with GST using pGEX-KG vector. GST–Apoer2 EIG was created from the wild-type GST–Apoer2 using the mutagenesis primers described above. Fusion proteins were expressed in BL21 cells (Stratagene) and purified over protein Sepharose beads (Sigma, St. Louis, MO).
Reelin signaling.
Stimulation of primary cortical neurons with Reelin was performed as described previously (Beffert et al., 2002). Cell lysates were separated on SDS-PAGE gels, transferred to nitrocellulose, and blotted with antibodies against proteins affected by Reelin signaling, including Dab1, phospho-(Tyr418)-Src family kinase (Biosource, Camarillo, CA), cyclin-dependent kinase 5 (CDK5; Santa Cruz Biotechnology, Santa Cruz, CA), phospho (Ser473)-Akt (Cell Signaling Technology, Beverly, MA), Akt (Cell Signaling Technology), and phospho (Ser9)-GSK3β (Cell Signaling Technology).
Creation of chimeric LDLR–Apoer2 receptors and binding assays.
We mutated a full-length human LDLR expression plasmid (pLDLR4) at the transmembrane/cytoplasmic domain junction to contain a NarI restriction site to insert each Apoer2 ICD. Chinese hamster ovary cells deficient in LDLR (ldlA-7) were stably transfected with each chimeric receptor and then screened using an extracellular domain LDLR antibody. Human LDL iodination and binding assays were performed essentially as described previously (Davis et al., 1987).
Modeling of EIGNPVY–Dab1 complex.
The Protein Data Bank (PDB) coordinates from the Dab1 –Apoer2 peptide complex (PDB identifier, 1NTV) were used as a starting point to model the EIGNPVY sequence in place of the native Apoer2 sequence NFDNPVY. The N, F, and D side chains were mutated respectively to E, I, and G using the program O (Jones et al., 1991) and were positioned as close to the orientation of the native amino acid side chain as was possible without steric clashes. The peptide model was then refined by conjugate gradient minimization using CNS (Brunger et al., 1998). The molecular surface representation and final figure were made using PyMol (DeLano, 2002).
Hematoxylin and eosin staining.
Sublethally anesthetized mice were perfused through the left ventricle with PBS followed by 4% paraformaldehyde in PBS. Dissected brains were postfixed by immersion in 4% paraformaldehyde for 16 h and embedded in paraffin. Five micrometer sagittal sections were stained with hematoxylin and eosin (Richard-Allan Scientific, Kalamazoo, MI) for histological analysis (at least three samples per genotype).
In situ hybridization.
A 513 bp probe encompassing the 3′ untranslated region for transducin-like enhancer of split 4 (Tle4) (base pairs 3930–4442 of GenBank accession number NM_011600) was cloned from mouse brain cDNA in pBluescript (Stratagene). Probe labeling and in situ hybridization were performed essentially as described previously (Shelton et al., 2000).
Immunohistochemistry.
Indirect immunofluorescence was performed on 10-μm-thick sagittal brain sections from postnatal day 21 (P21) mice. Frozen sections were first dried and then blocked with 10% goat or horse serum in PBS. Antibodies against forkhead box P2 (FOXP2) (N-16; Santa Cruz Biotechnology), calbindin-D-28K (Sigma), microtubule-associated protein 2 (MAP2; HM-2; Sigma), affinity-purified Apoer2 (2561), and Reelin (G10) were diluted at 1:100 (except Apoer2 at 1:25) in buffer A (0.3% IgG-free BSA; Jackson ImmunoResearch, West Grove, PA) and 0.3% Triton X-100 in PBS) and incubated overnight at 4°C. After three washes in PBS with 0.1% Triton X-100, sections were incubated with secondary anti-mouse Alexa Fluor 488 and/or anti-rabbit/goat Alexa Fluor 594 (Invitrogen, Eugene, OR) antibodies. Sections were mounted with mounting medium with 4′,6′-diamidino-2-phenylindole (DAPI; Vector Laboratories, Burlingame, CA) to stain nuclei. Fluorescence images were captured using a Zeiss (Oberkochen, Germany) Axioplan 2 microscope with Apotome. Images were processed using Adobe Photoshop (Adobe Systems, San Jose, CA).
Labeling of corticospinal neurons.
The retrograde tracer (True Blue; Sigma) was injected into the corticospinal tracts at the cervical spinal cord levels of the corticospinal tract (Bonatz et al., 2000). The tracer injections (True Blue; 2% in 0.2% DMSO) are placed laterally of each corticospinal tract using a fine glass micropipette connected to a microinjection system. The coordinates of the injections are based on analyses of frontal sections through the cervical spinal cord. After a transport time of at least 3 d, the tracer can be identified in the soma of corticospinal neurons by fluorescence microscopy (373 nm).
Hippocampal slice preparation, CA1 electrophysiology, and behavior.
Hippocampal slices (400 μm) were prepared from 8- to 12-week-old mice as described previously (Weeber et al., 2002). Slices were perfused (1 ml/min) with artificial CSF [in mm: 125 NaCl, 2.5 KCl, 1.24 NaH2PO4, 25 NaHCO3, 10 d-glucose, 2 CaCl2, and 1 MgCl2 (resistance, 1–4 MΩ)] in an interface chamber maintained at 30°C. Responses are presented as an average of six individual traces. Baseline stimulus intensities were determined from the intensity that produced a field EPSP at ∼50% of the maximal response. Potentiation was measured as the increase of the mean population EPSP (pEPSP) after tetanic stimulation normalized to the mean pEPSP for the duration of the baseline recording. Experimental results were obtained from those slices that exhibited stable baseline synaptic transmission for a minimum of 20–40 min before the delivery of long-term potentiation (LTP)-inducing stimulus. Fear-conditioning and Morris water maze experiments were performed essentially as described previously (Weeber et al., 2002; Beffert et al., 2005).
Results
Generation and characterization of Apoer2–Dab1 binding site mutant mice
Dab1 binds to the cytoplasmic NPXY motif of LDL receptor family members including Apoer2 (Trommsdorff et al., 1998, 1999; Howell et al., 1999). To examine the functional importance of adaptor protein binding to Apoer2, knock-in Apoer2 mice were created, which lack the ability to interact with the Dab1 protein (hereafter described as Apoer2 EIG). This was accomplished by mutating some of the amino acids necessary for Dab1 binding to the cytoplasmic domain of Apoer2 including N893E, F894I, and D895G (996 residues total; NP_444303) (Fig. 1A). These residues were chosen because the Dab1 PTB domain showed almost undetectable binding to a peptide containing this EIGNPVY compared with the wild-type NFDNPVY sequence (Howell et al., 1999). The detailed methods used to create the Apoer2 EIG knock-in mice including targeting vector design were identical to those described by Beffert et al. (2005), except for mutagenesis of the Dab1 binding site. Mutant mice were routinely genotyped by PCR yielding a 428 bp product (Fig. 1B, lanes 1 and 4) compared with a 603 bp product for wild-type mice (lane 3).
Figure 1.

Generation and basic characterization of Apoer2 EIG mice. A, Schematic representation of mouse Apoer2 demonstrating the cytoplasmic location of the wild-type NFDNPVY sequence and the mutated sequence EIGNPVY introduced into Apoer2 EIG. The position of the alternatively spliced exon 19 is indicated. The diagram is not drawn to scale. B, PCR genotyping of Apoer2 EIG homozygous (lanes 1 and 4), heterozygous (lane 2), and wild-type (lane 3) mice. C, The Dab1 protein interacts with wild-type Apoer2 but not the Apoer2 EIG mutant receptor. GST fusion proteins containing the cytoplasmic domains of wild-type Vldlr (lane 3) and Apoer2 (lane 5), but not the mutated Apoer2 EIG (lane 4) or GST control (lane 2), bound Dab1 from transfected human embryonic kidney 293 cells. D, Reelin signaling in Apoer2 EIG embryos is disrupted. Cultured primary neurons (E16; 5 d in culture) from Vldlr−/− (lanes 1 and 2), Apoer2−/−;Vldlr−/− (lanes 3 and 4), and Apoer2 EIG;Vldlr−/− (lanes 5 and 6) mice were treated with control (lanes 1, 3, and 5) or Reelin-conditioned (lanes 2, 4, and 6) medium. Lysates were collected, run on SDS-PAGE, transferred to nitrocellulose, and immunoblotted using antibodies against the Apoer2 extracellular domain (α-ED) or C terminus (α-CT), phosphotyrosine to detect Dab1 phosphorylation (4G10), total Dab1, p-SFK, CDK5, serine phosphorylated Akt, total Akt, and serine phosphorylated GSK3β.
Apoer2 EIG mutant mice appeared outwardly phenotypically normal; however, as has been described previously in Apoer2−/− animals, male mutants demonstrated reduced fertility (data not shown), consistent with an essential role for Apoer2 in sperm development (Trommsdorff et al., 1999; Andersen et al., 2003). When combined with Vldlr deficiency, Apoer2 EIG mutants developed a Reeler phenotype analogous to homozygous Reelin- or Dab1-deficient mice, with a progressive reeling gait, tremors, imbalance, and ataxia by 2 weeks of age. Apoer2 EIG, Vldlr−/− mice were also smaller than their wild-type littermates (∼50% of wild-type at P14) and usually died between 2 and 3 weeks after birth, as a result of their severely impaired motor functions.
To verify the disruption of the Apoer2–Dab1 interaction in vitro, GST pull-down experiments were performed using GST fusion proteins of LDL receptor family cytoplasmic domains (Fig. 1C). The Dab1 protein interacted with wild-type Apoer2 (lane 5) and Vldlr (lane 3) but not with the mutated Apoer2 EIG (lane 4) or with GST protein only (lane 2). Lane 1 contained 5% of the lysate used for pull downs in lanes 2–5. GST fusion proteins with single point mutations at position 893 including N893A, N893E, and N893Q all bound Dab1 with similar affinity to wild-type GST–Apoer2 (data not shown).
To examine Reelin signaling in Apoer2 EIG mice during brain development (Fig. 1D), we used a well established primary cortical neuronal culture system (Beffert et al., 2002). Neurons were prepared from embryonic day 16 (E16) embryos from Vldlr−/− mice that express wild-type Apoer2 (lanes 1 and 2), lacking Apoer2 (lanes 3 and 4), and Apoer2 EIG (lanes 5 and 6). All mice were on a Vldlr-deficient background, because both Apoer2 and Vldlr are necessary for normal Reelin signaling. Neurons (5 d in culture) were treated with either mock (lanes 1, 3, and 5) or recombinant Reelin-conditioned medium (lanes 2, 4, and 6). Expression of Apoer2 was verified with two Apoer2 antibodies, one against the extracellular domain (α-ED) and one against the C terminus (α-CT), demonstrating normal Apoer2 expression in Apoer2 EIG mutants (lanes 5 and 6) compared with wild-type (lanes 1 and 2), whereas neurons from Apoer2−/− embryos expressed no Apoer2 (lanes 3 and 4). Application of Reelin to neurons expressing wild-type Apoer2 increased tyrosine phosphorylation of Dab1 (p-DAB1 4G10) without affecting total Dab1 levels (lanes 1 and 2). Neurons deficient in Apoer2 did not respond to Reelin with a change in Dab1 phosphorylation (lanes 3 and 4) but did show increased levels of total Dab1 relative to wild-type Apoer2. Similarly, Dab1 phosphorylation was not increased in Apoer2 EIG neurons in response to Reelin (lanes 5 and 6); however, total Dab1 levels were increased compared with neurons expressing Apoer2. Reelin also increased the tyrosine phosphorylation of Src-family kinases (p-SFK), as well as serine phosphorylation of both Akt and GSK3β (lanes 1 and 2). Total levels of the serine–threonine kinases CDK5 and Akt remained unchanged in response to Reelin. In contrast, Reelin caused no alterations in the phosphorylation status of SFK, Akt, or GSK3β in neurons deficient for Apoer2 (lanes 3 and 4) or in Apoer2 EIG mutant neurons (lanes 5 and 6), indicating that Reelin signaling in primary cortical neurons was disrupted similarly in both of these mouse models.
Effect of ICD mutation on Apoer2 endocytosis
Apoer2 is structurally closely related to the LDL receptor, a highly active endocytic receptor that also contains an NFDNPVY motif in its cytoplasmic domain (Goldstein et al., 1977). To investigate the extent to which the different forms of the Apoer2 ICD affect the ability of the receptor to undergo endocytosis, we generated stable CHO cell lines transfected with chimeric constructs in which the cytoplasmic domain of the LDL receptor had been replaced by various Apoer2 ICDs, including Apoer2 EIG. We have previously reported and characterized two other Apoer2 ICD splice variants, Apoer2 ex19, containing the full-length wild-type ICD, and Apoer2 Δex19, which lacks a 59 amino acid exon in the ICD (Beffert et al., 2005). Table 1 shows the endocytosis capability (internalization index) of the recombinant Apoer2 tails compared with the LDL receptor. All forms of Apoer2 ICDs including the full-length form were considerably less active than the LDL receptor in this assay, internalizing at a rate similar to the LDL receptor defective derived from subject J.D. (Brown and Goldstein, 1976), confirming that endocytosis and cargo transport are likely not major biological functions of Apoer2 (Li et al., 2001).
Table 1.
Internalization index for 125I-LDL in hamster cells stably transfected with chimeric human LDL receptor extracellular and Apoer2 transmembrane and mutant mouse intracellular domains
| Mutant ICD | Cell line | Surface bound | Intracellular | Degraded | Internalization index | Percentage of control |
|---|---|---|---|---|---|---|
| LDLR (Tyr807) wild type | TR 715-19 | 150 | 369 | 3995 | 29.7 | 100 |
| LDLR −/− | ldlA-7 | 7 | 29 | 0 | 4.1 | 14 |
| Tyr807–Cys807 “JD mutant” | TR 735-7 | 119 | 131 | 546 | 6.0 | 20 |
| Apoer2 (ex19) | TR 3508-1-4 | 36 | 78 | 275 | 9.8 | 33 |
| Apoer2 (Δex19) | TR 3509-1-1 | 32 | 75 | 246 | 10.0 | 34 |
| Apoer2 EIG | TR 3507-3-1 | 260 | 281 | 1064 | 5.4 | 18 |
On day 5 of cell growth, each monolayer received 2 ml of medium containing lipoprotein-deficient serum and 10 μg of protein per milliliter of 125I-LDL (140 cpm/ng protein) in the absence and presence of 500 μg/ml unlabeled LDL. After 5 h of incubation at 37°C, high-affinity values for surface-bound, intracellular, and degraded 125I-LDL were determined. The internalization index is defined as (intracellular + degraded)/surface bound.
Structural basis for disruption of the Apoer2–Dab1 interaction
To better understand why the mutation of three amino acids in the cytoplasmic domain of Apoer2 led to a loss of Reelin signaling, we modeled the structure of the mutant Apoer2 motif in silico using the previously determined structure of the Dab1 PTB domain bound to a peptide including residues 889–902 of the native Apoer2 sequence TKSMNFDNPVYRKT (Stolt et al., 2003). Based on previous convention, Y899 of Apoer2 was designated as the “0” position in the structural model of the cytoplasmic domain. In the native structure, the first four residues of the peptide (Thr −10 to Met −7) were disordered and thus not visible and were also unnecessary for binding to the Dab1 PTB domain (Stolt et al., 2003). As shown previously by Stolt et al. (2003), Asn −6 to Asp −4 of the Apoer2 peptide formed hydrogen bonds with Lys 118, Asp 58, and Arg 56 of the Dab1 PTB domain (Fig. 2A). Furthermore, Phe −5 of Apoer2 packed tightly into a hydrophobic groove bordered by Ile 151, Leu 154, Arg 155, and Phe 158 of Dab1. In the model of the mutant Apoer2 peptide, Asn −6 was replaced with Glu −6, Phe −5 with Ile −5, and Asp −4 with Gly −4 (Fig. 2B). These mutations disrupted the interactions discussed above, with only Glu −6 forming a hydrogen bond with Lys 118. Most importantly, the Ile for Phe substitution at position −5 of the Apoer2 peptide (F894I) removed the aromatic ring that made several hydrophobic contacts with the Dab1 groove.
Figure 2.
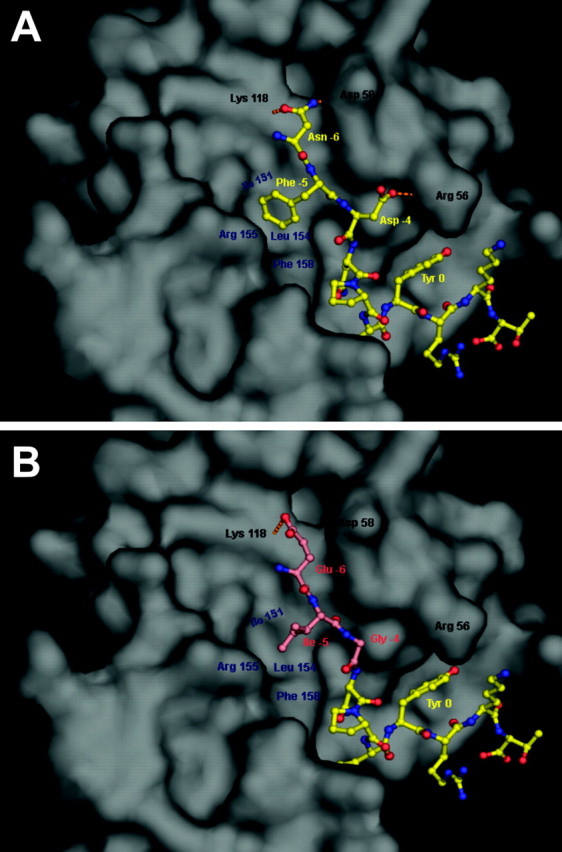
Structural differences in binding of Dab1 by native Apoer2 and the Apoer2 EIG mutant. A, Interaction between the Dab1 PTB domain and the native ApoER2 NFDNPVYRKT peptide sequence (from PDB coordinates 1NTV). Based on previous convention, Y is designated as the “0” position. The Dab1 PTB domain is shown as a molecular surface representation (gray), whereas the Apoer2 peptide is shown in ball-and-stick form (yellow; Corey–Pauling–Koltun). Amino acids in the peptide sequence are labeled in yellow. Residues of Dab1 that form hydrogen bonds with the N or D residues of the peptide are labeled in black, and the hydrogen bonds are represented by orange dashed lines. Residues of Dab1 that form hydrophobic interactions with the F residue of the peptide are labeled in blue. B, Modeled interaction of the Dab1 PTB domain and the Apoer2 EIG mutant EIGNPVYRKT sequence. The Dab1 PTB domain, its labeled residues, and the unmodified residues of the peptide are shown as in A. The mutated peptide residues and the corresponding labels are colored salmon.
Histopathology reveals neuronal positioning defects in Apoer2 EIG mice
To examine the effect of the EIGNPVY mutation on neuronal migration in vivo, we first examined the pathology of Apoer2 EIG mutant mice at P21 in the presence and absence of Vldlr compared with wild-type and receptor-deficient controls using hematoxylin and eosin staining (Fig. 3). Examination of cortical layering (Fig. 3A–F) revealed that the normally cell-free layer 1 or marginal zone was infiltrated in Apoer2 EIG, Vldlr−/− mice (Fig. 3F), similar to mice deficient in both Apoer2 and Vldlr (Fig. 3E). In contrast, wild-type, single Apoer2−/−, Vldlr−/−, and Apoer2 EIG mutant mice displayed a relatively cell-free layer 1 (Fig. 3A–D). Layering in the hippocampus (Fig. 3G–L) was perturbed in both single Apoer2−/− (Fig. 3H) and Apoer2 EIG (Fig. 3I) mutants but not in Vldlr−/− mice (Fig. 3J). Both Apoer2−/− and Apoer2 EIG displayed a splitting of the neuronal layering of region CA1 (white arrowheads), with a pronounced dispersion of neurons in CA3 and dentate gyrus (DG) (Fig. 3H,I). Mice deficient in both Vldlr and either Apoer2 (Fig. 3K) or containing Apoer2 EIG mutants (Fig. 3L) revealed a striking disorganization of the entire hippocampal region, with a more prominent splitting and disruption of CA1, CA3, and DG regions. Development of the cerebellum revealed the most striking phenotype. When combined with Vldlr deficiency, Apoer2 EIG mutant mice (Fig. 3R) presented with an indistinguishable Reeler-like phenotype to that of mice lacking both Apoer2 and Vldlr (Fig. 3Q). The cerebellum was markedly smaller, with a single band of cell nuclei arranged around the outer edge and no apparent foliation.
Figure 3.

Brain histopathology of neuronal positioning defects in Apoer2 mouse mutants in the presence and absence of Vldlr. A–R, Hematoxylin and eosin staining of sagittal brain sections illustrating neuronal positioning in P21 mouse cortex layers 1–3 (A–F), hippocampus (G–L), and cerebellum (M–R) in wild-type (A, G, M), Apoer2−/− (B, H, N), Apoer2 EIG (C, I, O), Vldlr−/− (D, J, P), Apoer2−/−; Vldlr−/− (E, K, Q), and Apoer2 EIG; Vldlr−/− (F, L, R) mice. Scale bars: (in A) cortex and cerebellum, 500 μm; (in G) hippocampus, 250 μm.
To examine neuronal migration in Apoer2 EIG mutant mice in more detail, we used a series of layer-specific markers at various ages of cortical, hippocampal, and cerebellar development. In both Apoer2−/− and Apoer2 EIG sections (Fig. 4B,C), TLE4 (a cortical layer 6 and subplate neuron marker) (Zhou et al., 1999) expression was broadened compared with wild type (Fig. 4A), infiltrating layers closer to the cortical surface. The distribution pattern of another marker, Foxp2, further reflected the abnormal cortical lamination of the Apoer2 EIG brain. Expression of the Foxp2 transcription factor is normally restricted to cortical layer 6 in the adult mouse brain (Ferland et al., 2003). Foxp2 immunohistochemistry in P21 brain sections (Fig. 4D–I) revealed distinct staining of layer 6 neurons in wild-type (Fig. 4D) and Vldlr−/− (Fig. 4F) brains. In Apoer2−/− (Fig. 4E) and Apoer2 EIG (Fig. 4H) brains, Foxp2 staining infiltrated higher layers, mostly settling into the layer 4–5 area. Mice deficient for both Apoer2 and Vldlr (Fig. 4G) and Apoer2 EIG; Vldlr−/− (Fig. 4I) mice displayed a complete disruption of cortical layering, with staining occurring in all cortical layers from 1 through to subplate. Layer 5 corticospinal neurons can be retrogradely labeled by injection of the tracer Fast Blue (Bonatz et al., 2000) (Fig. 4J). In Apoer2−/− (Fig. 4K) and Apoer2 EIG (Fig. 4L) mutants, the majority of layer 5 corticospinal neurons shifted to layer 4, with a smaller population residing in layer 6.
Figure 4.

Molecular marker analyses of cortical layering in Apoer2 mouse mutants. In situ hybridization for TLE4 (A–C) in P1 mouse cortex of wild-type (A), Apoer2−/− (B), and Apoer2 EIG (C) mutant mice demonstrating misplaced layer 6 and subplate neurons is shown. Indirect immunofluorescence detection of Foxp2-labeled cells in wild-type (D), Apoer2−/− (E), Vldlr−/− (F), Apoer2−/−; Vldlr−/− (G), Apoer2 EIG (H), and Apoer2 EIG; Vldlr−/− (I) mutant cortex is shown at P21. The relative position of individual Foxp2-expressing neurons (red) within the neocortical layers is represented by the scatter plot to the right of each section (D–I). Nuclei (blue) are labeled with DAPI. J–L, Direct immunofluorescence detection of corticospinal neurons labeled with Fast Blue highlighting cortical layer 5 neuron displacement in P28 brain. The numbers 1–6 represent cortical layers. SP, Subplate. Scale bars, 500 μm.
The large Purkinje neurons of the cerebellum expressed high levels of the calcium binding protein calbindin (green) and were organized in a well defined pattern along the cerebellar lobules, bordered by a population of granule cells (blue) (Fig. 5A). Consistent with previous reports (Trommsdorff et al., 1999), mice deficient for Vldlr displayed a clear disorganization of Purkinje cell layering in anterior lobules of the cerebellum (Fig. 5B). Mice deficient for Apoer2 (Fig. 5C) and Apoer2 EIG mutant mice (Fig. 5D) demonstrated normal Purkinje cell placement along these same anterior lobules; however, a minor population of ectopic calbindin-positive cells was also found in the cerebellar peduncle (arrow). The cerebellum of both Apoer2−/−; Vldlr−/− (Fig. 5E) and Apoer2 EIG; Vldlr−/− (Fig. 5F) mice was severely reduced in size with Purkinje cells located ectopically below an outer layer of granule cells. Furthermore, the cerebellum of these mice was unfoliated as a consequence of the failure to expand the granule cell population.
Figure 5.
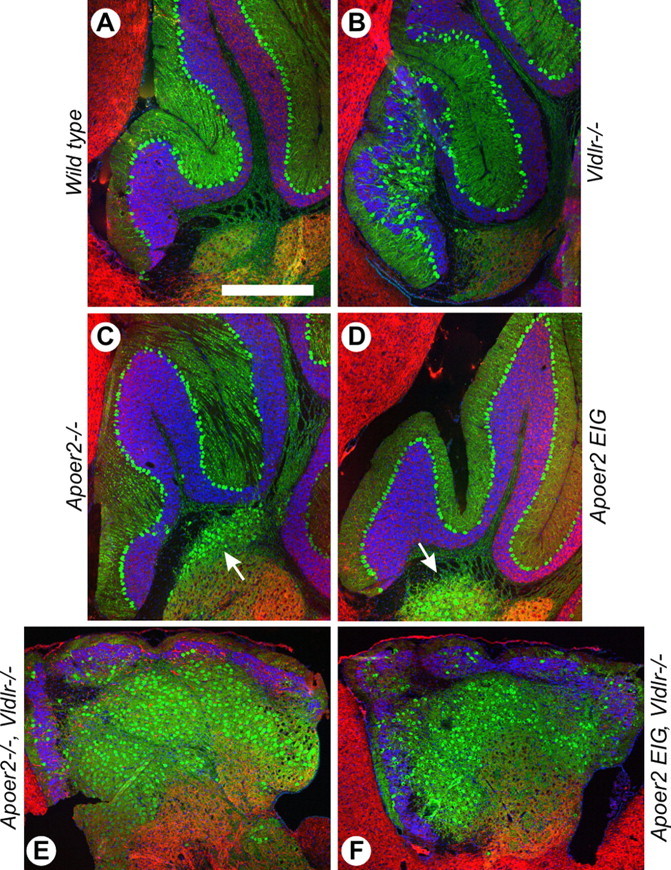
Cerebellar neuronal positioning defects in Apoer2 mouse mutants. A–F, Immunohistochemistry for the calcium binding protein calbindin (green) and MAP2 (red) in P21 mouse cerebellum in wild-type (A), Vldlr−/− (B), Apoer2−/− (C), Apoer2 EIG (D), Apoer2−/−; Vldlr−/− (E), and Apoer2 EIG; Vldlr−/− (F) mice. Nuclei are stained blue with DAPI. The arrows in C and D indicate ectopic calbindin staining in the cerebellar medulla. Rostral is to the left, and dorsal to the top. Scale bar: (in A) A–F, 500 μm.
In the hippocampus, calbindin stained predominantly the inner layer of granule cells along the dentate gyrus (Fig. 6A). The weaker staining of the granule cell dendrites in these images is not visible relative to the strong staining of the granule cell bodies. In Vldlr−/− brain, a small population of calbindin-positive cells was found in the polymorph layer, whereas others were spread into the molecular layer. In Apoer2−/− dentate gyrus, the majority of ectopic calbindin cells were located in the polymorph layer between the densely packed granule cell layers (Fig. 6C). In Apoer2 EIG mutants (Fig. 6D), a similar pattern of calbindin staining emerged, with many ectopic calbindin cells in the polymorph layer, whereas the granule cell layers appeared more dispersed than in Apoer2−/−. In both Apoer2−/−; Vldlr−/− (Fig. 6E) and Apoer2 EIG; Vldlr−/− (Fig. 6F) brains, the granule cells no longer formed a tightly packed layer, and calbindin-positive cells were scattered throughout the granule cell population. Combined, examination of neuronal positioning in the cortex, hippocampus, and cerebellum revealed that Apoer2 EIG mutant mice displayed very similar developmental abnormalities as mice deficient in the receptor Apoer2.
Figure 6.

Abnormal neuronal positioning in the dentate gyrus. A–F, Immunohistochemistry for the calcium binding protein calbindin (green) and MAP2 (red) in P21 mouse dentate gyrus in wild-type (A), Vldlr−/− (B), Apoer2−/− (C), Apoer2−/−; Vldlr−/− (D), Apoer2 EIG (E), and Apoer2 EIG; Vldlr−/− (F) mice. Nuclei are stained blue with DAPI. Scale bar, 250 μm.
Reelin is expressed in adult brain
Reelin signaling through Apoer2 and Vldlr is essential for proper neuronal positioning in the developing brain; however, recent evidence also suggests that Reelin has a fundamental role in synaptic plasticity and memory in the adult brain (Weeber et al., 2002; Beffert et al., 2005). Consistent with previous reports in wild-type brain, Reelin immunohistochemistry in P21 mouse cortex (Fig. 7) revealed a distinct cytoplasmic staining pattern in a subset of cells across all cortical layers. This pattern was not significantly altered in any of the mutant mice including Apoer2−/− (Fig. 7B), Apoer2 EIG (Fig. 7C), or Vldlr−/− (Fig. 7D), as well as those mice exhibiting a Reeler-like cortex, in which neuronal positioning was greatly disturbed, including Apoer2−/−; Vldlr−/− (Fig. 7E) and Apoer2 EIG; Vldlr−/− (Fig. 7F).
Figure 7.
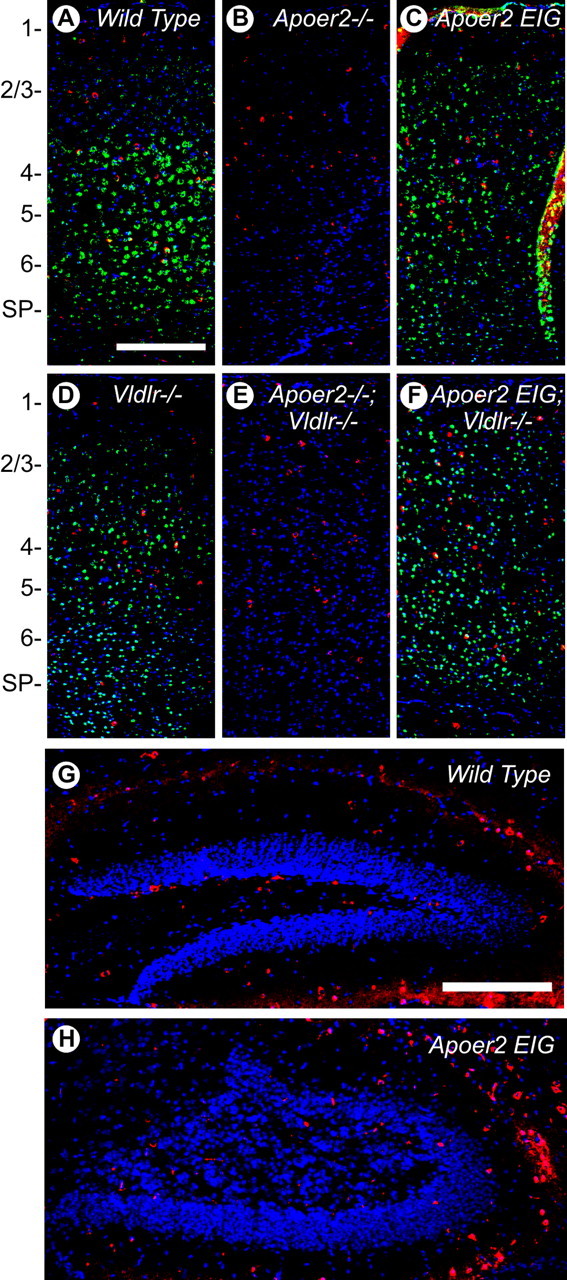
Reelin is expressed in adult cortex and hippocampus. A–F, Indirect immunofluorescence for Reelin (red) and Apoer2 (green) in wild-type (A), Apoer2−/− (B), Vldlr−/− (C), Apoer2 EIG (D), Apoer2−/−; Vldlr−/− (E), and Apoer2 EIG; Vldlr−/− (F) cortex from P21 mice. G, H, Representative sections from wild type (G) and Apoer2 EIG (H) showing indirect Reelin immunofluorescence (red) in the dentate region of the hippocampus. Nuclei are stained blue with DAPI. The numbers 1–6 represent cortical layers. SP, Subplate. Scale bars: A, 500 μm; G, 250 μm.
Coimmunostaining for Apoer2 revealed expression in all cortical layers except layer 1 (Fig. 7A), with the most intense signal occurring in layers 4–6. In Vldlr−/− (Fig. 7C) and Apoer2 EIG (Fig. 7D) cortex, Apoer2 staining infiltrated more into layers 2/3. In Apoer2 EIG; Vldlr−/− cortex (Fig. 7F), Apoer2 expression spread from layer 2 through to the subplate region. Expression of Apoer2 was absent in Apoer2−/− (Fig. 7B) and Apoer2−/−; Vldlr−/− (Fig. 7E) mice, as expected.
In the dentate gyrus region of the hippocampus of wild-type mice (Fig. 7G), Reelin expression was found predominantly in the polymorph layer, although some staining was found throughout the entire hippocampus. Some Reelin staining was also observed in small blood vessels and along the hippocampal fissure, consistent with expression of Reelin in the vascular and hematopoietic system (Ikeda and Terashima, 1997; Smalheiser et al., 2000). In Apoer2 EIG hippocampus (Fig. 7H), a similar amount of Reelin expression was observed, although the placement of cells was likely altered because of the significant alterations in neuronal positioning. Combined, these results demonstrate a significant amount of Reelin expression in the adult cortex and hippocampus that may play an important role in higher functions such as plasticity and memory.
The Apoer2 NFD motif is necessary for proper baseline and reelin-enhanced LTP
We have shown previously that Apoer2 is involved in hippocampal LTP in area CA1 but is not required for basal synaptic transmission (Weeber et al., 2002). To further explore the role of Apoer2 signaling in the hippocampus, we probed the well defined Schaffer collateral synapses for altered synaptic function and synaptic plasticity in hippocampal slices from wild-type and Apoer2 EIG mutants.
Overall, synaptic transmission was different as determined from the pEPSPs of field recordings from hippocampal area CA1 synapses and evaluated by determining the amplitude of the evoked fiber volley versus the slope of pEPSP at increasing stimulus intensities (Fig. 8A) [K, 5.298K; top of the curve (TOP), 0.9684; p < 0.001; nonlinear regression (Y = TOP × [1 − exp(−K ×X)])]. This suggests that the overall connectivity across Schaffer collateral synapses was reduced in the Apoer2 EIG mutant mice. Short-term plasticity was evaluated using paired-pulse facilitation and revealed a reduction in Apoer2 EIG compared with wild-type control mice at an interpulse interval of 50 ms (p = 0.0256; t test) (Fig. 8B). The LTP deficit in Apoer2-deficient mice was most prominently revealed using a theta burst stimulation protocol (Weeber et al., 2002). The degree of potentiation produced with this protocol was also reduced in Apoer2 EIG brains (Fig. 8C), which showed a severe LTP deficit that degraded to baseline responses within 30 min (genotype, p < 0.0001; two-way ANOVA).
Figure 8.

Electrophysiological defects in Apoer2 EIG mutants distinct from Apoer2−/−. A, Synaptic transmission is represented as the slope of the field EPSP versus the fiber volley amplitude at increasing stimulus intensities for wild type (•; n = 9) and Apoer2 EIG (□; n = 14). B, Short-term synaptic plasticity is evaluated by the amount of paired-pulse facilitation with interpulse intervals of 20, 50, 100, 200, and 300 ms in wild type (•; n = 9) and Apoer2 EIG (□; n = 9). C, LTP induced with theta-burst stimulation consisting of five trains of four pulses at 100 Hz with an interburst interval of 20 s. Apoer2 EIG mutants display reduced LTP (□; n = 6) compared with wild-type mice (•; n = 6). [For comparison, the dotted line represents LTP induced in Apoer2 −/− (Weeber et al., 2002).] D, Hippocampal slices perfused with Reelin (▪), in combination with theta-burst stimulation, caused a slight but significant increase of LTP compared with control medium (□) in Apoer2 EIG (▪, n = 9; □, n = 8). Insets, Representative pEPSP traces from Reelin perfusion experiments (mean ± SEM of 6 successive EPSPs) immediately before HFS (a) and at 60 min after tetanus (b) obtained from Reelin (top traces) or control (bottom traces) perfusion experiments.
We have reported previously that Reelin increased LTP induction in wild-type hippocampus slices. In addition, this Reelin-induced increase in potentiation was dependent on the presence of Apoer2 (Weeber et al., 2002), specifically exon 19 of Apoer2 (Beffert et al., 2005). Reelin was unable to enhance LTP in the Apoer2 EIG mutants, in which overall LTP induction was greatly reduced (Fig. 8D) (genotype, p < 0.0001; two-way ANOVA). Interestingly, however, Reelin prevented LTP degradation. Together, these data suggest that an interaction of Apoer2 with at least one adaptor protein (e.g., Dab1, but possibly others that also may bind to the Apoer2 NPXY motif) is necessary for baseline LTP induction. In contrast, the Reelin-dependent enhancement of LTP appeared to be primarily dependent on the alternatively spliced exon 19, as shown previously (Beffert et al., 2005).
Altered behavior in Apoer2 EIG mutant mice
Apoer2 is necessary for normal associative learning using a fear conditioning paradigm (Weeber et al., 2002). To better define the role of the Apoer2–Dab interaction, we assessed associative learning in wild-type and Apoer2 EIG mutants. We first performed two-trial fear condition by pairing an aversive stimulus (mild footshock) with an acoustic component [conditioned stimulus (CS); white noise] in a novel context. Fear response was assessed by the frequency at which normal motor behavior was disrupted by “freezing.” The degree of freezing to the tone and shock during training was not significantly different between animal groups (data not shown). Figure 9 shows the extent of short-term (1 h; black bars) and long-term (24 h; white bars) associative learning to the (environmental) context and the (acoustic) cue. The extent of learning was evaluated by the amount of freezing behavior of the animal. Although both contextual and cued fear-conditioned learning was dependent on proper function of the amygdala, only the contextual component of fear-conditioned learning was hippocampus dependent. Apoer2 EIG (n = 12) showed a significant reduction in freezing after the reintroduction to the context compared with wild-type mice (n = 19; 1 and 24 h; p < 0.0001; one-way ANOVA). We also performed open-field, rotorod, and hot plate tests to ensure that the observed deficits were not caused by a pre-existing physical or behavioral deficit and found no significant differences in any of the animal groups compared with wild type or to each other (data not shown). These observations indicate that Apoer2 EIG mutants show overall normal activity, perception, motor coordination, and nociception but that lack of a functional Dab1 docking motif leads to a severe contextual associative learning deficit in the absence of auditory component deficits. This is consistent with a defect in hippocampal function and indicates that the signaling mechanisms that are disrupted in these mutants are necessary for the modulation of synaptic plasticity that underlies long-term hippocampus-dependent associative learning.
Figure 9.

Normal associative and spatial learning requires Dab1 interaction with Apoer2. Fear conditioning/associative learning. Two-trial fear conditioning to assess associative learning 1 and 24 h after two CS–unconditioned stimulus pairings. A, No significant difference in the conditioned response was observed during the cue test between wild-type (n = 19) and Apoer2 EIG (n = 12) mice at either time point. B, Assessment of freezing to the context revealed a conditioned response that was greater in wild type than in Apoer2 EIG (n = 12) at both times tested. Morris water maze task/spatial learning. C–F, Results of escape latency (C), distance to platform (D), wall hugging (thigmotaxis) (E), and swim speed (F) for wild type and Apoer2 EIG during the acquisition phase in the hidden platform task. G, H, Percentage of time spent in the target or opposite quadrant (G) or the number of platform crossings (H) during a probe trial performed on day 12 (mean ± SEM; number of n for training consistent with probe trails; *p < 0.05 compared with wild type).
Our observations that hippocampus-dependent learning is impaired in the Apoer2−/− (Weeber et al., 2002) and Apoer2 Δex19 mutants (Beffert et al., 2005) prompted us to determine whether spatial learning might also be altered. Animal groups from wild-type and Apoer2 EIG mutants were trained in the Morris water task to locate a submerged platform in a circular pool filled with opaque water using distal visual cues outside of the pool. Training continued for 11 consecutive days, with each day consisting of four trials, with an interblock interval of ∼10 min. A profound learning deficit was apparent in the Apoer2 EIG mutant (n = 17), even when compared with the Apoer2−/− mutant (n = 15). For all days after the first training day, the Apoer2 EIG mice required considerably more time (Fig. 9C) and a longer swimming distance (Fig. 9D) to find the platform than wild-type or Apoer2−/− animals (p < 0.0001; one-way ANOVA). Analysis of the search strategy used by Apoer2 EIG mice showed an effect of this genotype on thigmotaxic behavior (p < 0.0001; one-way ANOVA), but the high variability in wall-hugging exhibited by the Apoer2 EIG mice resulted in a statistically significant difference only during training days 6–9 (p < 0.05; parametric analysis) (Fig. 9E). The overall swim speed of Apoer2 EIG mice steadily decreased over days 1–7, becoming significantly slower compared with wild type and Apoer2−/− after trial day 5 (Fig. 9F) (p < 0.0001; one-way ANOVA). Motor coordination and motor learning assessed with open-field and rotorod analysis were normal in all of the animal groups (data not shown), suggesting that the reduced swim speed after prolonged training of the Apoer2 EIG mutants was not caused by a physical limitation. This is supported by the observation that the Apoer2 EIG mutant mice were able to obtain swim speeds equal to that of wild-type mice on training days 1–3 (Fig. 9F).
To determine whether Apoer2 EIG mutants were capable of learning to use spatial cues to locate the escape platform, we subjected them to a probe test on day 12. All of the animal groups used a search strategy that focused on the quadrant where the escape platform had been located (Fig. 9G). However, Apoer2 EIG mutant mice showed reduced time in the training quadrant compared with wild-type mice (p < 0.01; Dunnett’s multiple comparison test). With respect to the number of platform crossings per quadrant, both animal groups demonstrated an overall trend in the probe test on day 12 (Fig. 9H) that was similar to the quadrant test time; however, a decrease in the total number of platform crossings was observed for Apoer2 EIG mutants compared with wild type (p < 0.01; Dunnett’s multiple comparison test). The deficits in latency to platform, swim speed, and thigmotaxic behavior in Apoer2 EIG mutants during training suggest an inclusive lack of a learned escape strategy, consistent with the overall poor performance during the probe tests.
Discussion
In this study, we demonstrated that the disruption of the Dab1 binding domain of Apoer2 is critical for the propagation of the Reelin signal and correct positioning of cells in the developing brains of mice. The disruption of this binding domain likely altered the ability of the receptor to make high-affinity contact with the PTB domain of Dab1. Furthermore, the Apoer2 EIG mutation disrupted LTP induction in adult brain and resulted in abnormal memory and learning in animals.
Proper neuronal positioning in the developing mouse brain is dependent on the ability of Reelin to bind and signal through two lipoprotein receptors, Apoer2 and Vldlr. The Reelin signal is further transmitted into migrating neurons by the adaptor protein Dab1, which binds directly to a highly conserved NPXY motif in the cytoplasmic domains of lipoprotein receptors. We demonstrated that mutation of key residues near the receptor NPXY motif of Apoer2 is sufficient to disrupt Dab1 binding to Apoer2 in vitro (Fig. 1C). This is consistent with previous work showing that the NFDNPXY motif of Apoer2 binds to the PTB domain of Dab1 in a hydrophobic groove (Stolt et al., 2003; Yun et al., 2003). Modeling in silico (Fig. 2) revealed that the mutated Apoer2 peptide no longer has the capacity to fill this hydrophobic groove and also lacks the ability to form several hydrogen bonds, which together were critical for binding to Dab1. These results are consistent with mutation of one particular amino acid in the Dab1 hydrophobic groove (F158V), which leads to decreased binding to Apoer2 (Herrick and Cooper, 2004). Our in vitro (Fig. 1) and pathological examination of Apoer2 EIG mice (Figs. 3–7) further imply that the functional interaction between Apoer2 and Dab1 has been disrupted.
Reelin signaling during development leads to phosphorylation of the adaptor protein Dab1. Disruption of the Apoer2–Dab1 interaction resulted in an inhibition of Dab1 phosphorylation, as demonstrated by Reelin activation of primary neuronal cultures (Fig. 1D). Furthermore, downstream signaling including activation of Src-family kinases and Akt and inactivation of GSK3β was prevented, indicating that the Apoer2 NFDNPXY motif where Dab1 interacts with the receptor is necessary for proper transmission of the Reelin signal in neurons. Although the NFDNPXY domain has also been shown to be important for normal endocytosis in LDL receptor family members, our results (Table 1) indicate that the primary role of Apoer2 is likely not the endocytosis of ligands but the propagation of signals from ligands such as Reelin and possibly others.
The effect of the Apoer2 EIG mutation on Reelin signaling was revealed by examining neuronal positioning in the brains of mice (Figs. 3–6). The phenotype of Apoer2 EIG mice mimicked that of mice deficient for Apoer2, with the exception that Apoer2 EIG mice expressed Apoer2 (Figs. 1, 7). This raised the possibility of a dominant-negative effect, in that the receptor was now able to bind ligands such as Reelin while being incapable of transmitting the signal through Dab1. However, with respect to neuronal positioning during development, no gross differences were observed between Apoer2−/− and Apoer2 EIG, either on a wild-type or a Vldlr-deficient background. The Apoer2 EIG mutant mouse by likely completely disrupting the Apoer2–Dab1 interaction provides another model to study the role of Reelin in development and the adult brain. A related study recently showed that mutating F158V in the hydrophobic groove of Dab1 led to partial disruption the Apoer2–Dab1 interaction and led to a more subtle developmental phenotype that was only apparent when heterozygous Dab1 F158V mutant mice were crossed with a deficient Dab1 allele (Herrick and Cooper, 2004). Combined, these studies provide a gradient of Apoer2–Dab1 interactivity, in which partial disruption leads to a weak loss-of-function allele and the Apoer2 EIG mutation leads to complete disruption and a phenotype very similar to Apoer2 deficiency.
In the adult brain, Reelin, its receptors Apoer2 and Vldlr, and Dab1 continue to be expressed (Alcantara et al., 1998; Drakew et al., 1998; Pesold et al., 1998; Deguchi et al., 2003; Frotscher et al., 2003). Consistent with these previous reports, we observed Reelin expression spread throughout the brains of adult mice (Fig. 7). This expression pattern did not appear significantly different in any of the mutant mice examined, although the positioning of neurons was clearly affected. We have shown previously that Reelin and its receptors Apoer2 and Vldlr play a fundamental role in modulating LTP and memory functions in adult brain (Weeber et al., 2002). Mice deficient for either Vldlr or Apoer2 displayed moderate to severe defects in LTP, whereas Reelin significantly enhanced LTP over baseline. Recently, we extended this work using two Apoer2 knock-in mouse models by demonstrating that Reelin-enhanced LTP was dependent on the splicing of the ICD of Apoer2 (Beffert et al., 2005). Specifically, when exon 19 of Apoer2 was deleted, Reelin-enhanced LTP was abolished, whereas neuronal positioning in these mice was normal. These results suggested that other adaptor proteins that bind the Apoer2 ICD in exon 19, such as mitogen-activated protein kinase 8-interacting protein 1/2 (JIP1/2) and/or PSD95 (postsynaptic density 95) (Gotthardt et al., 2000; Stockinger et al., 2000), are necessary for transmitting the Reelin signal required for enhanced LTP and modulating adult memory and behavior.
The NPXY motif in exon 18 is positioned 27 residues upstream of exon 19 in the ICD of Apoer2. The spacing of these motifs theoretically allows for the simultaneous interaction of Dab1 as well as other signaling proteins such as JIP1/2 and PSD95 to the ICD. As shown here, the NPXY motif is important for Dab1 binding and for Reelin signaling in development, whereas our previous work showed that exon 19 binds JIP1/2 and PSD95 and is crucial in adult brain although dispensable in development (Beffert et al., 2005). However, our results on Apoer2 EIG mutant mice also indicate that the NFDNPVY motif is paramount for normal synaptic transmission in adult mice (Fig. 8A). One interpretation of these findings may be simply to assume that incorrect neuronal positioning leads to defective LTP; however, we have shown previously that mice deficient in the CDK5 activator p35 display normal baseline LTP, although p35−/− mice display similar disruptions in hippocampal neuronal positioning compared with mice deficient for Apoer2 (Beffert et al., 2004). Therefore, improper neuronal positioning alone cannot account for the observed defects in LTP, implying rather that a disruption in signaling through Dab1 underlies the LTP phenotype observed in Apoer2 EIG mice.
Baseline LTP is severely reduced in Apoer2 EIG mice, even more dramatically than that observed in Apoer2−/− mice (Fig. 8C). This effect is likely caused by a dominant-negative interference of a signaling-defective Apoer2 inasmuch as LTP in Apoer2 EIG slices degraded more rapidly over time to return to prestimulation levels. Interestingly, in the presence of Reelin (Fig. 8D), LTP degradation was prevented, an effect that was not observed in Apoer2−/− slices (Weeber et al., 2002). This Reelin-induced increase in LTP in the Apoer2 EIG mice suggests that some LTP-dependent signaling can occur independently of the Apoer2–Dab1 binding site and likely involves the interaction of other adaptor proteins such as JIP1/2 and PSD95 to the exon 19 (Beffert et al., 2005) and potentially other as yet uncharacterized binding proteins. Together, our results suggest that a cooperative signaling complex assembles on the ICD of Apoer2 to facilitate Reelin-induced LTP that includes interaction of Dab1 to the NPXY domain and of other adaptor proteins to exon 19.
Reelin binding to Apoer2 and Vldlr activates nonreceptor tyrosine kinases such as Src and Fyn (Arnaud et al., 2003; Bock and Herz, 2003), which can in turn modulate the phosphorylation state of the NMDA receptor (Suzuki and Okumura-Noji, 1995; Miyakawa et al., 1997), thereby increasing the number of synaptic NMDA receptors (Prybylowski et al., 2005). Reelin further influences the maturation of NMDA receptors (Sinagra et al., 2005), leading to changes in the phosphorylation state of NR2 subunits and potentiating synaptically evoked NMDA currents (Beffert et al., 2005; Chen et al., 2005). Although the ectopic neurons in the Apoer2 EIG mutants complicate the interpretation of the field recordings, our previous results on single-cell recordings demonstrate that Reelin specifically alters NMDA receptor currents (Beffert et al., 2005; Chen et al., 2005). Combined, these results suggest that Reelin can modulate NMDA receptor function; however, the precise cellular mechanism and the role of Apoer2 remain to be elucidated.
In summary, the findings presented here establish the importance of the Apoer2 NFDNPVY motif in Reelin signaling during neuronal development. Furthermore, Reelin signaling in adult brain requires the binding of multiple adaptor proteins to distinct sites in the Apoer2 ICD.
Footnotes
This work was supported by grants from the National Institutes of Health (HL20948, HL63762, and NS43408 to J.H. and MH57014 and NS13546 to J.D.S.), the Alzheimer’s Association, the Perot Family Foundation, the Humboldt Foundation, and the American Foundation on Aging Research (E.J.W.). U.B. received fellowships from the Human Frontier Science Program and from the Canadian Institutes of Health. We thank Wen-Ling Niu, Huichuan Reyna, Elida Priscilla Rodriguez, Elizabeth Lummus, Debra Morgan, Tiina Kotti, and the University of Texas Southwestern pathology core for excellent technical support.
References
- Alcantara S, Ruiz M, D’Arcangelo G, Ezan F, de Lecea L, Curran T, Sotelo C, Soriano E (1998). Regional and cellular patterns of reelin mRNA expression in the forebrain of the developing and adult mouse. J Neurosci 18:7779–7799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen OM, Yeung CH, Vorum H, Wellner M, Andreassen TK, Erdmann B, Mueller EC, Herz J, Otto A, Cooper TG, Willnow TE (2003). Essential role of the apolipoprotein E receptor-2 in sperm development. J Biol Chem 278:23989–23995. [DOI] [PubMed] [Google Scholar]
- Arnaud L, Ballif BA, Forster E, Cooper JA (2003). Fyn tyrosine kinase is a critical regulator of disabled-1 during brain development. Curr Biol 13:9–17. [DOI] [PubMed] [Google Scholar]
- Assadi AH, Zhang G, Beffert U, McNeil RS, Renfro AL, Niu S, Quattrocchi CC, Antalffy BA, Sheldon M, Armstrong DD, Wynshaw-Boris A, Herz J, D’Arcangelo G, Clark GD (2003). Interaction of reelin signaling and Lis1 in brain development. Nat Genet 35:270–276. [DOI] [PubMed] [Google Scholar]
- Beffert U, Morfini G, Bock HH, Reyna H, Brady ST, Herz J (2002). Reelin-mediated signaling locally regulates protein kinase B/Akt and glycogen synthase kinase 3beta. J Biol Chem 277:49958–49964. [DOI] [PubMed] [Google Scholar]
- Beffert U, Weeber EJ, Morfini G, Ko J, Brady ST, Tsai LH, Sweatt JD, Herz J (2004). Reelin and cyclin-dependent kinase 5-dependent signals cooperate in regulating neuronal migration and synaptic transmission. J Neurosci 24:1897–1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beffert U, Weeber EJ, Durudas A, Qiu S, Masiulis I, Sweatt JD, Li WP, Adelmann G, Frotscher M, Hammer RE, Herz J (2005). Modulation of synaptic plasticity and memory by Reelin involves differential splicing of the lipoprotein receptor Apoer2. Neuron 47:567–579. [DOI] [PubMed] [Google Scholar]
- Bock HH, Herz J (2003). Reelin activates SRC family tyrosine kinases in neurons. Curr Biol 13:18–26. [DOI] [PubMed] [Google Scholar]
- Bock HH, Jossin Y, Liu P, Forster E, May P, Goffinet AM, Herz J (2003). Phosphatidylinositol 3-kinase interacts with the adaptor protein Dab1 in response to Reelin signaling and is required for normal cortical lamination. J Biol Chem 278:38772–38779. [DOI] [PubMed] [Google Scholar]
- Bonatz H, Rohrig S, Mestres P, Meyer M, Giehl KM (2000). An axotomy model for the induction of death of rat and mouse corticospinal neurons in vivo. J Neurosci Methods 100:105–115. [DOI] [PubMed] [Google Scholar]
- Boycott KM, Flavelle S, Bureau A, Glass HC, Fujiwara TM, Wirrell E, Davey K, Chudley AE, Scott JN, McLeod DR, Parboosingh JS (2005). Homozygous deletion of the very low density lipoprotein receptor gene causes autosomal recessive cerebellar hypoplasia with cerebral gyral simplification. Am J Hum Genet 77:477–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MS, Goldstein JL (1976). Analysis of a mutant strain of human fibroblasts with a defect in the internalization of receptor-bound low density lipoprotein. Cell 9:663–674. [DOI] [PubMed] [Google Scholar]
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL (1998). Crystallography and NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr 54:905–921. [DOI] [PubMed] [Google Scholar]
- Chen Y, Beffert U, Ertunc M, Tang TS, Kavalali ET, Bezprozvanny I, Herz J (2005). Reelin modulates NMDA receptor activity in cortical neurons. J Neurosci 25:8209–8216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Arcangelo G, Miao GG, Chen SC, Soares HD, Morgan JI, Curran T (1995). A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature 374:719–723. [DOI] [PubMed] [Google Scholar]
- D’Arcangelo G, Homayouni R, Keshvara L, Rice DS, Sheldon M, Curran T (1999). Reelin is a ligand for lipoprotein receptors. Neuron 24:471–479. [DOI] [PubMed] [Google Scholar]
- Davis CG, van Driel IR, Russell DW, Brown MS, Goldstein JL (1987). The low density lipoprotein receptor. Identification of amino acids in cytoplasmic domain required for rapid endocytosis. J Biol Chem 262:4075–4082. [PubMed] [Google Scholar]
- Deguchi K, Inoue K, Avila WE, Lopez-Terrada D, Antalffy BA, Quattrocchi CC, Sheldon M, Mikoshiba K, D’Arcangelo G, Armstrong DL (2003). Reelin and disabled-1 expression in developing and mature human cortical neurons. J Neuropathol Exp Neurol 62:676–684. [DOI] [PubMed] [Google Scholar]
- DeLano WL (2002). In: The PyMOL molecular graphics system http://delsci.com/rel/0_98/ San Carlos, CA: DeLano Scientific.
- Drakew A, Frotscher M, Deller T, Ogawa M, Heimrich B (1998). Developmental distribution of a reeler gene-related antigen in the rat hippocampal formation visualized by CR-50 immunocytochemistry. Neuroscience 82:1079–1086. [DOI] [PubMed] [Google Scholar]
- Ferland RJ, Cherry TJ, Preware PO, Morrisey EE, Walsh CA (2003). Characterization of Foxp2 and Foxp1 mRNA and protein in the developing and mature brain. J Comp Neurol 460:266–279. [DOI] [PubMed] [Google Scholar]
- Frotscher M, Haas CA, Forster E (2003). Reelin controls granule cell migration in the dentate gyrus by acting on the radial glial scaffold. Cereb Cortex 13:634–640. [DOI] [PubMed] [Google Scholar]
- Goldstein JL, Brown MS, Stone NJ (1977). Genetics of the LDL receptor: evidence that the mutations affecting binding and internalization are allelic. Cell 12:629–641. [DOI] [PubMed] [Google Scholar]
- Gotthardt M, Trommsdorff M, Nevitt MF, Shelton J, Richardson JA, Stockinger W, Nimpf J, Herz J (2000). Interactions of the low density lipoprotein receptor gene family with cytosolic adaptor and scaffold proteins suggest diverse biological functions in cellular communication and signal transduction. J Biol Chem 275:25616–25624. [DOI] [PubMed] [Google Scholar]
- Herrick TM, Cooper JA (2004). High affinity binding of Dab1 to Reelin receptors promotes normal positioning of upper layer cortical plate neurons. Brain Res Mol Brain Res 126:121–128. [DOI] [PubMed] [Google Scholar]
- Hiesberger T, Trommsdorff M, Howell BW, Goffinet A, Mumby MC, Cooper JA, Herz J (1999). Direct binding of Reelin to VLDL receptor and ApoE receptor 2 induces tyrosine phosphorylation of disabled-1 and modulates tau phosphorylation. Neuron 24:481–489. [DOI] [PubMed] [Google Scholar]
- Hong SE, Shugart YY, Huang DT, Shahwan SA, Grant PE, Hourihane JO, Martin ND, Walsh CA (2000). Autosomal recessive lissencephaly with cerebellar hypoplasia is associated with human RELN mutations. Nat Genet 26:93–96. [DOI] [PubMed] [Google Scholar]
- Howell BW, Hawkes R, Soriano P, Cooper JA (1997). Neuronal position in the developing brain is regulated by mouse disabled-1. Nature 389:733–737. [DOI] [PubMed] [Google Scholar]
- Howell BW, Lanier LM, Frank R, Gertler FB, Cooper JA (1999). The disabled 1 phosphotyrosine-binding domain binds to the internalization signals of transmembrane glycoproteins and to phospholipids. Mol Cell Biol 19:5179–5188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda Y, Terashima T (1997). Expression of reelin, the gene responsible for the reeler mutation, in embryonic development and adulthood in the mouse. Dev Dyn 210:157–172. [DOI] [PubMed] [Google Scholar]
- Jones TA, Zou J-Y, Cowan SW, Kjeldgaard M (1991). Improved methods for binding protein models in electron density maps and the location of errors in these models. Acta Crystallogr A 47:110–119. [DOI] [PubMed] [Google Scholar]
- Li Y, Lu W, Marzolo MP, Bu G (2001). Differential functions of members of the low density lipoprotein receptor family suggested by their distinct endocytosis rates. J Biol Chem 276:18000–18006. [DOI] [PubMed] [Google Scholar]
- Miyakawa T, Yagi T, Kitazawa H, Yasuda M, Kawai N, Tsuboi K, Niki H (1997). Fyn-kinase as a determinant of ethanol sensitivity: relation to NMDA-receptor function. Science 278:698–701. [DOI] [PubMed] [Google Scholar]
- Pesold C, Pisu MG, Impagnatiello F, Uzunov DP, Caruncho HJ (1998). Simultaneous detection of glutamic acid decarboxylase and reelin mRNA in adult rat neurons using in situ hybridization and immunofluorescence. Brain Res Brain Res Protoc 3:155–160. [DOI] [PubMed] [Google Scholar]
- Prybylowski K, Chang K, Sans N, Kan L, Vicini S, Wenthold RJ (2005). The synaptic localization of NR2B-containing NMDA receptors is controlled by interactions with PDZ proteins and AP-2. Neuron 47:845–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelton JM, Lee MH, Richardson JA, Patel SB (2000). Microsomal triglyceride transfer protein expression during mouse development. J Lipid Res 41:532–537. [PubMed] [Google Scholar]
- Sinagra M, Verrier D, Frankova D, Korwek KM, Blahos J, Weeber EJ, Manzoni OJ, Chavis P (2005). Reelin, very-low-density lipoprotein receptor, and apolipoprotein E receptor 2 control somatic NMDA receptor composition during hippocampal maturation in vitro. J Neurosci 25:6127–6136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smalheiser NR, Costa E, Guidotti A, Impagnatiello F, Auta J, Lacor P, Kriho V, Pappas GD (2000). Expression of reelin in adult mammalian blood, liver, pituitary pars intermedia, and adrenal chromaffin cells. Proc Natl Acad Sci USA 97:1281–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockinger W, Brandes C, Fasching D, Hermann M, Gotthardt M, Herz J, Schneider WJ, Nimpf J (2000). The reelin receptor ApoER2 recruits JNK-interacting proteins-1 and -2. J Biol Chem 275:25625–25632. [DOI] [PubMed] [Google Scholar]
- Stolt PC, Jeon H, Song HK, Herz J, Eck MJ, Blacklow SC (2003). Origins of peptide selectivity and phosphoinositide binding revealed by structures of disabled-1 PTB domain complexes. Structure 11:569–579. [DOI] [PubMed] [Google Scholar]
- Strasser V, Fasching D, Hauser C, Mayer H, Bock HH, Hiesberger T, Herz J, Weeber EJ, Sweatt JD, Pramatarova A, Howell B, Schneider WJ, Nimpf J (2004). Receptor clustering is involved in Reelin signaling. Mol Cell Biol 24:1378–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, Okumura-Noji K (1995). NMDA receptor subunits epsilon 1 (NR2A) and epsilon 2 (NR2B) are substrates for Fyn in the postsynaptic density fraction isolated from the rat brain. Biochem Biophys Res Commun 216:582–588. [DOI] [PubMed] [Google Scholar]
- Trommsdorff M, Borg JP, Margolis B, Herz J (1998). Interaction of cytosolic adaptor proteins with neuronal apolipoprotein E receptors and the amyloid precursor protein. J Biol Chem 273:33556–33560. [DOI] [PubMed] [Google Scholar]
- Trommsdorff M, Gotthardt M, Hiesberger T, Shelton J, Stockinger W, Nimpf J, Hammer RE, Richardson JA, Herz J (1999). Reeler/Disabled-like disruption of neuronal migration in knockout mice lacking the VLDL receptor and ApoE receptor 2. Cell 97:689–701. [DOI] [PubMed] [Google Scholar]
- Weeber EJ, Beffert U, Jones C, Christian JM, Forster E, Sweatt JD, Herz J (2002). Reelin and ApoE receptors cooperate to enhance hippocampal synaptic plasticity and learning. J Biol Chem 277:39944–39952. [DOI] [PubMed] [Google Scholar]
- Yun M, Keshvara L, Park CG, Zhang YM, Dickerson JB, Zheng J, Rock CO, Curran T, Park HW (2003). Crystal structures of the Dab homology domains of mouse disabled 1 and 2. J Biol Chem 278:36572–36581. [DOI] [PubMed] [Google Scholar]
- Zhou C, Qiu Y, Pereira FA, Crair MC, Tsai SY, Tsai MJ (1999). The nuclear orphan receptor COUP-TFI is required for differentiation of subplate neurons and guidance of thalamocortical axons. Neuron 24:847–859. [DOI] [PubMed] [Google Scholar]