

Abstract
Connexins (Cxs) are a family of protein subunits constituting gap junctions, which facilitate exchanges of molecules important for cellular signaling and metabolic activities intercellularly or between different regions of the cytoplasm in the same cells. Mutations in Cxs are the major cause of nonsyndromic childhood deafness, which are mostly found in Cx26 and Cx30 expressed in cochlear supporting cells and fibrocytes. So far, little is known about the functional contribution of Cxs in other types of cochlear cells. Here, we show that Cx29 was highly expressed in the cochlea. The developmental expression time course of Cx29 was similar to that of a myelin marker [myelin associate glycoprotein (MAG)]. Immunolabeling identified Cx29 exclusively in the Schwann cells myelinating the soma and fiber of spiral ganglion (SG) neurons. The absence of the Cx29 gene in mice (Cx29−/− mice), with a penetrance of ∼50%, caused a delay in the maturation of hearing thresholds, an early loss of high-frequency sensitivities, a prolongation in latency and distortion in the wave I of the auditory brainstem responses, and elevated sensitivity to noise damages. The morphology of sensory hair cells and otoacoustic emissions that depend on the integrity of hair cells were normal in Cx29−/− mice. In contrast, decreases in MAG expression and severe demyelination at the soma of SG neurons were found in Cx29−/− mice. Our findings demonstrated the requirement of Cx29 for normal cochlear functions and suggest that Cx29 is a new candidate gene for studying the auditory neuropathy.
Keywords: connexin29, gap junction, myelin, cochlear Schwann cells, spiral ganglion neuron, cochlea
Introduction
Mutations in connexin (Cx) genes are the most frequently encountered genetic defects for childhood nonsyndromic deafness, which are responsible for about one-half of all cases (Bergoffen et al., 1993; Maw et al., 1995; Morell et al., 1998; Xia et al., 1998; Grifa et al., 1999; Liu et al., 2002). Cxs oligomerize to form connexons or “hemichannels” in plasma membranes. In small areas of cell membrane called gap junction (GJ) plaques, two connexons align to form the intercellular channels, which provide conduits directly linking the cytoplasm of different cells or different regions of the same cell. About 20 Cx genes have been identified so far (Willecke et al., 2002). Four subtypes of Cxs (Cx26, -30, -29, and -43) are most prominently expressed in the cochlea (Ahmad et al., 2003). Cx26 and Cx30 are coexpressed in the supporting cells and fibrocytes (Lautermann et al., 1998; Ahmad et al., 2003; Forge et al., 2003; Sun et al., 2005), whereas Cx43 is expressed in the bone of the otic capsule (Cohen-Salmon et al., 2004).
Normal myelination in the central and peripheral nervous systems has been shown to be critically dependent on Cx47 and Cx32 expressions (Menichella et al., 2003; Odermatt et al., 2003). Cx29 is a relatively new member in the Cx protein family (Sohl et al., 2001; Altevogt et al., 2002), whose mRNA expression level was only lower than Cx26 in the cochlea (Ahmad et al., 2003). Previous reports by others (Sohl et al., 2001; Altevogt et al., 2002; Li et al., 2002) show that it is primarily expressed in myelinating glial cells in both central and peripheral nervous systems. Little is known about Cx29 cellular expression and functional roles in the cochlea. Spiral ganglion (SG) neurons are bipolar cells faithfully relaying sound signals from hair cells to the cochlear nuclei. Proper myelination is crucial for their normal functions, because a high degree of temporal precision for the conduction of action potentials is essential for the auditory perception. However, the roles of GJ-mediated intercellular communication among Schwann cells and other types of glial cells in the inner ear have not been investigated. Therefore, we have characterized Cx29 expression in the cochlea and tested its contribution to normal hearing functions using the Cx29−/− mice. We found that Cx29 was strongly and exclusively expressed in the Schwann cells myelinating the auditory nerve inside the cochlea. Absence of the Cx29 gene, with a penetrance of ∼50%, caused delay in the development of hearing sensitivities, early high-frequency hearing loss in matured cochlea, and distorted the waveform of the auditory brainstem responses (ABRs). Both inner and outer hair cell morphology and function appeared to be normal; therefore, they were unlikely to be the site responsible for hearing loss. The most likely etiology is the severe demyelination found at the soma of SG neurons of affected Cx29−/− mice. Our findings demonstrated that Cx29 is required for the normal function of the cochlea and suggest that Cx29 is a new candidate gene for studying the molecular mechanisms underlying auditory neuropathy.
Materials and Methods
Generation of Cx29 knock-out (Cx29−/−) mice.
Production of a targeted deletion of Cx29 was described previously by Altevogt and Paul (2004). Briefly, homologous arms were PCR amplified from BALB/c genomic DNA, subcloned into pCRII (Invitrogen, Carlsbad, CA) for sequencing, and then transferred to the pDTA targeting vector (gift from Frank Gertler, Massachusetts Institute of Technology, Cambridge, MA) (Altevogt and Paul, 2004). The 5′ homology arm consisted of 1536 bp immediately upstream of the Cx29 start codon. The 3′ homology arm started 800 bp downstream of the Cx29 stop codon and was 3893 bp in length. Thus, the complete coding region of Cx29 was eliminated. The coding region was replaced in frame with a nuclear-localized β-galactosidase (gift from Susan Dymecki, Harvard Medical School, Boston, MA). The targeting construct was linearized with XhoI and electroporated into J1 ES cells. G418-resistant clones were screened by PCR using a 5′ primer external to the targeting vector and a 3′ primer within the neo cassette. The drug selection cassette was not removed. A targeting efficiency of 2% was observed. Recombination at the 3′ end was verified using Southern blotting. Littermate controlled mice were generated by interbreeding of Cx29+/− mice.
Detection of Cx29 expression in the cochlea and other tissues by Western blots (WBs).
Mice were anesthetized by intraperitoneal injections of chloral hydrate (400 mg/kg; 4% solution dissolved in PBS, pH 7.4) and killed by intracardiac perfusion of PBS. While submerged in ice-cold PBS, cochlear modiolus including the spiral ganglia, facial nerve, sciatic nerve, spinal cord, brain, and liver were dissected out with the help of a stereomicroscope (Zeiss Stemi; Carl Zeiss, Shrewsbury, PA). All chemicals were purchased from Sigma (St. Louis, MO) unless otherwise indicated. Antibodies against Cx29 and myelin associate glycoprotein (MAG) were purchased from Zymed (South San Francisco, CA). Tissue samples were washed twice in cold PBS, stored in Eppendorf tubes (1.5 ml), and frozen on dry ice. Total protein was extracted using radioimmunoprecipitation assay buffer (Upstate Biotechnology Cell Signaling System, Waltham, MA) by following the manufacturer’s instructions and stored at −80°C until use. Total protein concentration was measured by using the BCA protein assay kit according to the manufacturer’s instructions (Pierce, Rockford, IL). Final protein concentration was adjusted by adding appropriate amount of Laemmli buffer. In addition to directly measuring the protein concentration for the Western blot (WB) loading, we tested the consistence of protein loading in each lane by stripping the blots after each WB with Cx or MAG in a solution containing the following: 100 mm 2-mercaptoethanol, 2% SDS, and 62.5 mm Tris, pH 6.7, at 50°C, for 30 min. The film was reprobed with a monoclonal antibody against a housekeeping protein (actin) (dilution factor, 1:1000; Chemicon, Temecula, CA).
Samples were loaded (6 μg of protein in each lane) onto SDS-polyacrylamide gels (12%), electrophoresed (90 V; at room temperature for 80 min), and transferred to nitrocellulose membranes by electroblotting (30 V; overnight at 4°C) in a transfer buffer (Bio-Rad, Richmond, CA). Nitrocellulose membranes were blocked in nonfat dry milk (5% w/v) and Tween 20 (0.1% v/v) in PBS, pH 7.4, for 1 h at room temperature. Membranes were then incubated with primary antibodies [Cx29 (0.125 μg/ml) or MAG (2 μg/ml)] in blocking buffer overnight (at 4°C). After washing three times with PBS-Tween solution, membranes were then incubated with secondary antibodies [goat anti-rabbit IgG(H+L)-HRP conjugates or goat anti-mouse IgG(H+L)-HRP conjugates; 1:5000 dilution; Bio-Rad] for 1 h at room temperature. Protein bands were visualized by reacting with a chemiluminescent substrate (SuperSignal West Femto Maximum Sensitivity Substrate; Pierce), and exposing the membrane on XAR-5 films (Eastman Kodak, Rochester, NY). Exposure times were adjusted to ensure that the densitometric intensity of measured bands was in the linear range in relation to protein concentrations. Relative density of each band on Western blots was measured and analyzed by NIH Image software (version 1.61). Background in films was subtracted from the optical density measurements. All Western blots were repeated at least four times.
Immunofluorescent labeling and colabeling of Cxs in the cochlea.
Mice at various developmental stages specified in Results [ranging from embryonic day 18 (E18) to adult] were used. Animals older than postnatal day 4 (P4) were fixed by cardiac perfusion of 4% paraformaldehyde dissolved in PBS, pH 7.4. Dissected cochleas were fixed again in paraformaldehyde (4%; at 4°C) for 1 h, and decalcified in 10% EDTA solution for 72 h. Decalcified cochleas were immersed in 20% sucrose (in PBS, pH 7.4) overnight followed by embedding in OCT compound (Sakura Finetek, Torrance, CA) overnight. The tissues were frozen by first dipping in liquid nitrogen for 5 s and then maintaining on the surface between liquid nitrogen and air for 1 min. Sections were cut (7 μm) using a cryostat (2800 Frigocut E; Cambridge Instruments, Nussloch, Germany). For immunostaining, cochlear samples were permeabilized by incubating in Triton (0.1% v/v in PBS) for 30 min. Nonspecific bindings were reduced by first incubating with goat serum (5% in PBS) for 1 h at room temperature. Antibodies used were as follows (concentrations used are given in parentheses): (1) Cx29 (0.625 μg/ml), (2) Cx32 (5 μg/ml), (3) MAG (5 μg/ml), (4) neurofilament (NF) (5 μg/ml), (5) glial fibrillary acidic protein (GFAP) (7.5 μg/ml), and (6) myosinVI (5 μg/ml; Proteus Bioscience, Ramona, CA). Tissue sections were labeled with primary antibodies overnight at 4°C. Control immunolabeling was performed by either omitting the primary antibodies, or labeling with cochlear sections obtained from Cx29−/− mice. After washing in PBS six times, sections were incubated with secondary antibodies at room temperature for 1 h. Cy2- or Cy3-conjugated secondary antibodies (Jackson ImmunoResearch, West Grove, PA) were used to visualize the labeling results. Labeled samples were mounted in a SlowFade anti-fading mounting kit (Electron Microscopy Sciences, Hatfield, PA) and observed with either an optical microscope (Axiovert 135TV; Carl Zeiss) or a confocal microscope (Zeiss LSM 510). DAPI (4′,6-diamidino-2-phenylindole dihydrochloride) contained in the mounting solution provided counterstain for the cochlea sections. The immunolabeling procedures were repeated using at least four cochlear sections at each developmental stage. The percentage of yellow pixels in coimmunolabeled samples, which represented the area colabeled by both antibodies, was analyzed using an image analysis software customer programmed using the Labview (version 6i; National Instruments, Austin, TX). The results are defined as number of yellow pixels divided by total number of pixels that showed either red or green labeling.
Protocol for electron-microscopic observations of myelination in the cochlea.
Animal-handling procedures were the same as those described above. Tissues were fixed by cardiac perfusion of glutaraldehyde (2.5% in phosphate buffer). The cochleas were dissected out and treated with the same fixative overnight. The round window and the apex of the cochlea were opened to allow access of fixative. Samples were decalcified with 10% EDTA at 4°C for 72 h. After decalcification, specimens were rinsed with 0.1 m phosphate buffer, pH 7.4, and fixed again in osmium tetroxide (1% dissolved in 0.2 m sodium cacadolate buffer) for 1.5 h at room temperature. The membranous labyrinth was dissected out and separated into smaller pieces including the basal membrane, the modiolus containing the soma of SG neurons, and modiolus containing the glial juncture. The specimens were dehydrated in graded alcohol and embedded in epoxy resin for 48 h. Ultrathin sections (60–70 nm) were cut with an ultramicrotome (Leica, Nussloch, Germany). Sections were counterstained with uranyl acetate and lead citrate, and then examined under a transmission electron microscope (H-800; Hitachi, Tokyo, Japan).
Protocol for noise exposure, ABR, and otoacoustic emission measurements.
ABRs measure evoked responses elicited by sounds. Distortion products of the otoacoustic emission are results of the nonlinearity of basilar membrane motion, which reflects motility function of the outer hair cells. Both techniques are widely used in both research and clinical practices to assess the hearing functions (Whitehead et al., 1996). Details of the ABR and noise exposure protocols can be found in our previous publication (Peng et al., 2004b). Briefly, mice were anesthetized with ketamine (80 mg/kg) and xylazine (10 mg/kg). Surgical level of anesthesia was maintained by a supplementary half-dose of ketamine and xylazine every 60 min or as needed if toe pinch elicited reflexive body movements. Sounds, consisting of either click or tone bursts of various frequencies (10 ms in duration and a rise–fall time of 0.5 ms), were generated by Tucker-Davis System II hardware and software (Tucker-Davis Technologies, Alachua, FL). For testing noise sensitivities, wild-type and Cx29−/− mice were exposed to white-band noise [110 dB sound pressure level (SPL)] for 1 h to create temporary threshold shifts (TTSs) (Peng et al., 2004b). The long-term effect of noise exposures on hearing sensitivity was monitored by measuring ABR thresholds for 4 weeks after noise exposures. The ABR threshold was defined visually from a series of repeatable ABR responses obtained at various sound intensities. The genotype of mice was kept confidential to the ABR tester. The probe used for recording distortion production otoacoustic emissions (DPOAEs) was ER-10C manufactured by Etymotic Research (Elk Grove Village, IL). Two primary tones were given at the same intensity, and f2 = 1.2f1. Data acquisition, time, and frequency domain averaging were accomplished by Tucker Davis System III hardware and software.
Results
Western blotting and immunolabeling results revealed that Cx29 was highly and exclusively expressed in Schwann cells in the cochlea
In our previous studies, we detected Cx29 mRNA expression in the cochlea by cDNA dot-blot hybridizations (Ahmad et al., 2003). These results prompted us to pursue this project. We first assessed the level of Cx29 protein expression in the cochlea at various developmental stages by Western blotting (Fig. 1A). An equal amount of total protein (6 μg) was loaded in each lane, and the actual protein loading was normalized to the Western blot intensity of actin (see labels in Fig. 1). Consistent with findings of others (Altevogt et al., 2002), we usually detected monomeric Cx29 together with its higher order multimers in the denaturing SDS-PAGE gels (Fig. 1A,B). The specificity of the antibody we used was confirmed by the disappearance of all bands corresponding to Cx29 in the Western blots when cochlear tissues from Cx29−/− mice were used, whereas they were clearly detected from wild-type mice (Fig. 1B). At E18, Cx29 was not detectable in the cochlea. After birth, Cx29 expression was gradually increased (Fig. 1A). The relative expression densities of Cx29 in the auditory nerve (AN), facial nerve (FN), sciatic nerve (SN), spinal cord (SC), brain (BR), and liver (LR) were compared by the density of their corresponding bands in Western blots (Fig. 1C) (6 μg of total protein was loaded in each lane). Results normalize to the amount of actin showed that the auditory nerve (modiolus tissues) had the highest expression level of Cx29 among the six tissues we compared (Fig. 1D).
Figure 1.

Characterization of Cx29 expression in the cochlea by Western blots. A, Western blots of Cx29 at five different developmental stages. The amount of protein loading in each lane was checked by Western blot with actin. B, Western blots of Cx29 obtained from cochlear tissues of the wild-type and Cx29−/− mice. C, Western blots comparing Cx29 expression levels in six different tissues. AN, Auditory nerve; FN, facial nerve; SN, sciatic nerve; SC, spinal cord; BR, brain (gray matter); LR, liver. D, Histogram quantifying the Cx29 protein expression level in six different tissues. E, Western blots of MAG at five different developmental stages obtained from the wild-type (top panel) and Cx29−/− (bottom panel) mice. Corresponding Western blot results with actin are given under each data panel. F, Histogram showing relative protein expression levels of Cx29 or MAG at different development stages normalized to their respective adult levels. Error bars indicate SE.
Published work by others demonstrated that Cx29 is largely expressed by myelinating glial cells (Sohl et al., 2001; Altevogt et al., 2002). We therefore compared the developmental expression of Cx29 with MAG, which is a glial cell marker (Fig. 1E,F). Results showed that Cx29 (Fig. 1F, white bars) and MAG (Fig. 1F, black bars) protein expression levels increased monotonically after birth for wild-type mice (Fig. 1E, top panel). The expression of MAG in Cx29−/− mice (normalized to that of actin) was initially increased until P7, and then significantly reduced at P13 and adult stages (Fig. 1E, bottom panel; F, gray bars). The myelin sheath along the auditory nerve develop postnatally, and gradually mature after mice were born (Romand and Romand, 1985). Therefore, these results are consistent with the notion that Cx29 contributed to the development of normal myelination process of the auditory nerve.
We next investigated the cellular distribution of Cx29 in the adult cochlea by immunolabeling. Observed at a low magnification (Fig. 2A), we found that all tracts traveled by the auditory and vestibular nerves inside the cochlea were strongly labeled by the antibody against Cx29. We consistently observed a dramatic reduction in the Cx29 immunoreactivity beyond the glial junction (marked by dashed lines in Fig. 2A,B) of the VIIIth nerve in the brainstem, indicating that Cx29 was highly expressed only within the cochlea. Data presented later (Figs. 3, 4) localize the Cx29 to Schwann cells myelinating the auditory nerve inside the cochlea. In contrast, Cx29 expression in glial cells myelinating the central portion of the auditory nerve in the brainstem was below the detection level. Faint labeling was also observed in the stria vascularis (Fig. 2A, double arrows). The Cx29 coding exon was replaced by β-galactosidase (β-gal) in Cx29−/− mice; therefore the expression of β-gal should reflect the endogenous Cx29 expression. Although giving strong signals in the Schwann cells (Fig. 4C), an antibody against β-gal never labeled any cells in the stria vascularis in the Cx29−/− mice. In addition, the weak labeling in the stria vascularis usually remained, whereas the strong Cx29 immunoreactivity disappeared in the cochlea of Cx29−/− mice. Therefore, we considered the weak Cx29 immunolabeling in the stria vascularis to be nonspecific.
Figure 2.

Immunolabeling results showing patterns of Cx29 and Cx32 expression in the cochlea and the auditory nerve bundle in the brainstem. Adult mouse (3 months) cochleas were used. A, A low-magnification view of the entire cochlear section labeled with an antibody against Cx29. The dashed lines mark the border at the glial juncture. Landmark structures of the cochlea were labeled. SG, Spiral ganglion; SV, scala vestibule; SM, scala media; ST, scala tympani; SVas, stria vascularis. B, Immunolabeling results showing the expression pattern of Cx32 in the cochlea and the auditory nerve bundle in the brainstem. The dashed lines mark the border at the glial juncture.
Figure 3.
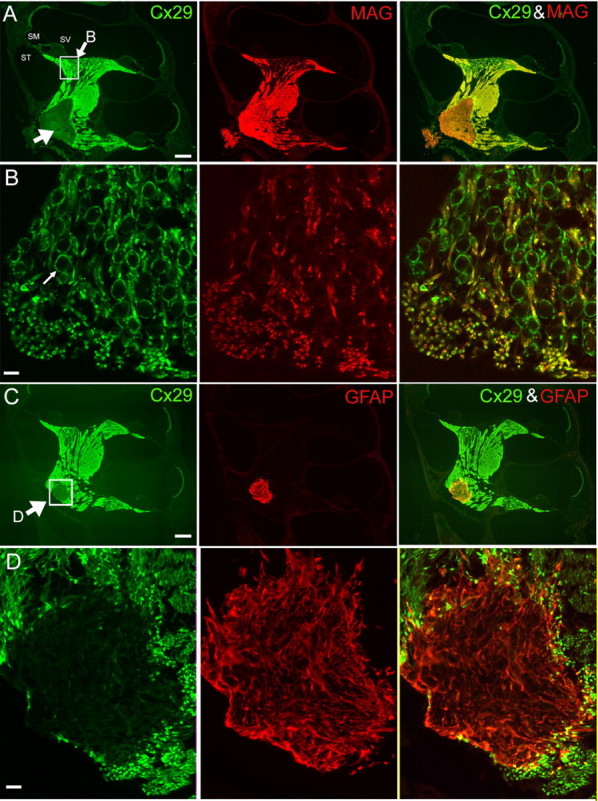
Coimmunolabeling results of Cx29 with either MAG (A, B) or GFAP (C, D). Adult mouse cochleas (2 months) were used. The images in B and D are enlarged views of the boxed areas in the left panels of A and C, respectively. A, Cochlear sections were colabeled with antibodies against Cx29 (green; left panel) and MAG (red; middle panel). A superimposed image is given in the right panel. An arrow in the left panel points to the area of weak Cx29 staining outside the glial junction in the brainstem. ST, Scala tympani; SM, scala media; SV, scala vestibule. C, Cochlear sections were colabeled with antibodies against Cx29 (green; left panel) and GFAP (red; middle panel). A superimposed image is given in the right panel. An arrow in the left panel points to the boxed area where immunoreactivities of Cx29 and GFAP around the glial juncture were compared. Scale bars: A, C, ∼400 μm; B, D, 20 μm.
Figure 4.

Cx29 (green) coimmunolabeled with either MAG (red; A2, A3) or NF (red; B2, B3). Adult mouse cochleas (2 months) were used. Overlapped images are given in A3 and B3. C shows immunolabeling results obtained with an antibody against β-galactosidase (β-gal; red) superimposed on an image obtained with differential interference contrast optics to show the SG neurons (arrows). Scale bars: A1–A3, B1–B3, 1 μm; C, 20 μm.
It is known that mutations in Cx32 underlie the X-linked Charcot–Marie–Tooth disease (CMTX). In some CMTX patient families, hearing loss manifests as one of the symptoms of the CMTX neuropathy (Bergoffen et al., 1993). We therefore investigated whether Cx32 was also expressed in the cochlea. Results showed that the Cx32 was expressed along the central portion of the auditory nerve tract (Fig. 2B; dashed line indicates the glial juncture) outside the cochlea. The expression of Cx32 in axonal myelin in the cochlea was below the detection level. Observed at higher magnifications, we found that Cx32 was only sparsely expressed in the myelin wrapping around the soma of SG neurons (data not shown). Combined with our previous finding that the cochlear Cx32 mRNA expression was below the detection level by cDNA dot-blot hybridizations (Ahmad et al., 2003), these data indicate that Cx32 is not a major myelinating Cx inside the cochlea.
The cellular localization of Cx29 was further explored by double immunostaining of Cx29 together with a general marker for glial cells (MAG) and a specific marker for astrocytes (GFAP) (Altevogt et al., 2002). As with the results presented in Figure 1A, the antibody against Cx29 labeled tracts where the auditory nerve travels inside the cochlea until the border marked by the glial juncture (indicated by big arrow in Fig. 3A, left panel). Views obtained at larger magnification showing the SG neuron soma area (outlined by a white box in Fig. 3A, left panel) demonstrated that MAG and Cx29 immunoreactivities overlapped only in the fibers of SG neurons (Fig. 3B, area labeled yellow in the right panel). Strong Cx29 immunoreactivity formed many relatively large circles where Cx29 and MAG labeling did not overlap (Fig. 3B, left and right panels, green circles). MAG immunoreactivity was absent in the myelin sheath wrapping the cell body of SG neurons (one example is pointed out by a small arrow in Fig. 3B, left panel), suggesting a particular importance of Cx29 for the myelin wrapping the soma of SG neurons. This was further supported by demyelination patterns we observed in Cx29−/− mice (see Fig. 8). As expected, the general glial cell marker MAG labeled the entire tract of the VIIIth nerve (Fig. 3A, middle panel) including the region beyond the glial junction and inside the brainstem where oligodendrocytes and astrocytes are located (indicated by large arrows in Fig. 3A,C, left panels). In a sharp contrast, dramatically weaker Cx29 immunoreactivities were observed in this area where it was exclusively labeled by an antibody against GFAP (Fig. 3C, middle panel). An enlarged view for an area outlined by the white box in Figure 3C, left panel, shows that immunoreactivities of Cx29 and GFAP were largely nonoverlapping (Fig. 3D), suggesting that the Cx29 expression was restricted in Schwann cells of the cochlea.
Figure 8.

Myelin morphology of wild-type and Cx29−/− mice observed with a transmission electron microscope. A, Myelin around the soma of a SG neuron obtained from a wild-type animal. The right panel presents an enlarged view of myelin outlined by a white box in the left panel. B, Myelin around the soma of a SG neuron obtained from a Cx29−/− mouse (6 months of age). The right panel presents an enlarged view of myelin outlined by a white box in the left panel. The arrows give examples of detachments between myelin and cell membrane. C, D, Examples of abnormal myelination around the soma of SG neurons observed from noise-exposed mice.
To further examine whether Cx29 was expressed exclusively by the Schwann cells in the cochlea, we colabeled Cx29 in the cochlear sections with antibodies against MAG and NF. Confocal images obtained at high magnification demonstrated that both Cx29 and MAG gave circular immunostaining patterns (Fig. 4A1,A2, hollow rings) in cross sections of the auditory nerve, consistent with immunoreactivities at the myelin sheath. Pixels colabeled by the two antibodies overlapped for 38.6 ± 4.2% (n = 10). In sharp contrast, the antibody against neurofilament gave an immunostaining pattern of solid dots (labeled red in Fig. 4B2), which are encircled by Cx29-positive pixels (labeled green in Fig. 4B1,B3). Unlike the colabeling pattern demonstrated by Cx29 and MAG, the Cx29- and NF-colabeled pixels showed only 6.2 ± 2.1% of overlap (n = 10). As an additional test to better differentiate whether Cx29 is expressed by Schwann cells or SG neurons, we labeled cochlear sections using an antibody against β-gal in Cx29−/− mice in which Cx29 gene was replaced by a reporter gene coding for nuclear-bound LacZ. Only Schwann cells along the tract of the auditory nerve were labeled (Fig. 4C, small red cells). In contrast, none of the SG neurons (examples are indicated by arrows in Fig. 4C) was β-gal positive, confirming that circling Cx29 staining patterns in the location of SG neurons was contributed by myelin of Schwann cells rather than cell membrane of SG neurons (Figs. 3B, left and right panels; 4A1,B1).
Mice lacking Cx29 gene showed delayed maturation of hearing thresholds, high-frequency hearing losses, elevated sensitivity to noise damages, and demyelination at the soma of SG neurons
Requirement of Cx29 for normal cochlear functions was examined using the Cx29−/− mice. Otoacoustic emissions, which reflect functions of the outer hair cells, were not significantly different between the Cx29−/− and the wild-type animals (Fig. 5A, n = 10). ABRs, which measure the hearing threshold, in contrast, revealed abnormalities in littermate-controlled Cx29−/− mice. The hearing sensitivity of mice is fully developed to the adult level by 3 weeks after birth (Walsh and Romand, 1992). Three weeks postnatally, 11 of the 20 Cx29−/− mice (scattered symbols outside the shaded area) tested showed hearing threshold elevation across all frequencies tested (Fig. 5B). Six weeks after birth, the hearing thresholds of most mice (13 of 16) developed into the shaded area representing the controls (symbols inside the shaded area in Fig. 5C). These data suggested that Cx29−/− mice were delayed in developing their hearing sensitivities and the phenotype showed ∼55% of penetrance at 3 weeks postnatally. Tested again at 6 months after birth, one-half of Cx29−/− mice (7 of 14) (Fig. 5D, scattered symbols outside the shaded area) showed hearing threshold elevations at high frequencies. Notice that low-frequency hearing thresholds at 4–8 kHz of almost all animals fell within the shaded area. This pattern of hearing loss was dramatically different from that displayed by 3-week-old mice (Fig. 5B). In addition to hearing threshold elevations, the waveform of click-evoked ABRs was greatly distorted. Figure 5E compares click-evoked ABRs (80 dB SPL) obtained from the wild-type (solid curve) and Cx29−/− (dashed curve) mice. Remaining microphonics (the beginning is indicated by an arrow in Fig. 5E) in data traces showed that there were no differences in the onset of sound stimuli in the cochlea. However, the wave I latency of the ABRs in Cx29−/− mice was significantly delayed at all sound intensities we tested (Fig. 5F, n = 10). Additionally, the wave I and II in the Cx29−/− mice often merged to give a single wave I. The single wave obtained from Cx29−/− mice was significantly broader than their wild-type counterpart. The width of wave I measured at 50% of the peak amplitude was 0.42 ± 0.05 and 0.57 ± 0.1 ms for wild-type and Cx29−/− mice, respectively (n = 10). Broader ABR waveform is consistent with the notion that dyssynchrony in the firing of the auditory nerve underlies the distorted waveform.
Figure 5.

Assessments of the cochlear function for the Cx29−/− mice. A, Growth of the distortion product measured at 2f1 − f2, as a function of the intensity of primary tones (f1). Two-month-old adult mice were used. B–D, Hearing thresholds across a frequency range of 4–32 kHz measured by tone-burst-evoked ABRs for mice 3 weeks (B), 6 weeks (C), and 6 months (D) after birth. The scattered symbols are raw data obtained from Cx29−/− mice, and the individual animals are represented by different symbols. The plots give averaged ABR threshold obtained from littermate-controlled wild-type mice (filled squares connected by solid lines). The shaded areas represent a data range twice of the value of the SD above and below the averaged data. E, F, Comparison of click-evoked ABR waveforms obtained from wild-type (sold line) and Cx29−/− (dashed line) mice. Mice were between 3 and 4 weeks of age. The Roman numbers indicate the peaks in the ABR waveform. F gives the latency of the wave I as a function of sound intensity for wild-type (solid line) and Cx29−/− (dashed line) mice. Error bars indicate SE.
White-band noises given at a dosage of 110 dB SPL for 1 h cause only TTS (Peng et al., 2004a). We used such a test of noise susceptibility as a method to test how cochleas respond to stress. Six-week-old mice were used because differences in the hearing thresholds were statistically insignificant for most animals at this developmental stage (Fig. 5C), thus ensuring a similar degree of noise exposure. Results (n = 10 for each groups) revealed that Cx29−/− mice were more vulnerable to noise damages (Fig. 6). Consistent with a high-frequency hearing loss pattern demonstrated in Figure 5D, threshold shifts at 8 and 12 kHz of both wild-type and Cx29−/− mice all returned to the baseline (Fig. 6A,B) 4 weeks after noise exposures, although the initial threshold shifts measured at 12 kHz was statistically larger (Fig. 6C, asterisks, unpaired Student’s t tests). The hearing losses for Cx29−/− mice at 18 and 24 kHz, however, were more severe than the wild-type controls throughout the 4 week period. In contrast to control animals whose hearing losses were temporary, significant threshold shifts at 18 and 24 kHz remained 4 weeks after noise exposure (Fig. 6C,D).
Figure 6.

Hearing threshold shifts after noise exposures measured at 4 (A), 8 (B), 18 (C), and 24 (D) kHz. Data from wild-type and Cx29KO mice are represented by open and filled squares, respectively. The threshold changes were followed for 4 weeks by ABR measurements after noise exposures. Error bars indicate SE.
To investigate the cellular sources that caused the hearing loss, we examined the morphology of hair cells of Cx29−/− mice. Basal cochlear hair cells (corresponding to high-frequency range) of Cx29−/− mice that demonstrated high-frequency hearing loss (Fig. 5D, n = 4) appeared to be normal at the optical microscope level (Fig. 7A). Whole-mount views of basal hair cells labeled by myosin VI obtained from noise-exposed Cx29−/− mice also showed no hair cell loss (examples are indicated by arrows in Fig. 7B). Consistent with the DPOAE measurements (Fig. 5A), these data suggested that hair cells were not the source of hearing losses. Western blot and immunolabeling results presented above indicate that Cx29 was expressed by the Schwann cells myelinating the VIIIth nerve traveling inside the cochlea. We therefore examined whether myelination of SG neurons was affected in the Cx29−/− mice. Observed at the electron microscope level, the myelin sheath wrapping the auditory nerve fibers of Cx29−/− mice was little affected (Fig. 7C,D). The average ratios of myelin thickness to the diameter of the fiber were 0.22 ± 0.001 (n = 56) and 0.22 ± 0.0008 (n = 78) for wild-type and Cx29−/− mice, respectively. Gross demyelination and vacuolation at the auditory nerve fiber were never observed. In contrast, demyelination was observed in the myelin around the soma of SG neurons of Cx29−/− mice that demonstrated high-frequency hearing losses. Compared with the perikaryal myelin in wild-type controls, which showed tightly layered and smoothly contoured myelin sheath around the soma of SG neurons (Fig. 8A, area outlined by a white box is enlarged), the myelin sheath around the soma of basal SG neurons obtained from Cx29−/− mice (6 months old) were much less regular and in some areas showed disintegrated myelin layers (one example is outlined by a white box in Fig. 8B). In addition, vacuolation just below the cell membrane (Fig. 8B, arrows) was observed. In noise-exposed mice (n = 3) that showed permanent high-frequency hearing losses, abnormal myelination (vacuolation) either inside the myelin sheath (examples given by arrows in Fig. 8C) or between the myelin and cell membrane (examples given by arrows in Fig. 8D) were observed from basal SG neurons. These data were consistent with the notion that defects in SG neurons underlie the hearing losses observed in the Cx29−/− mice.
Figure 7.
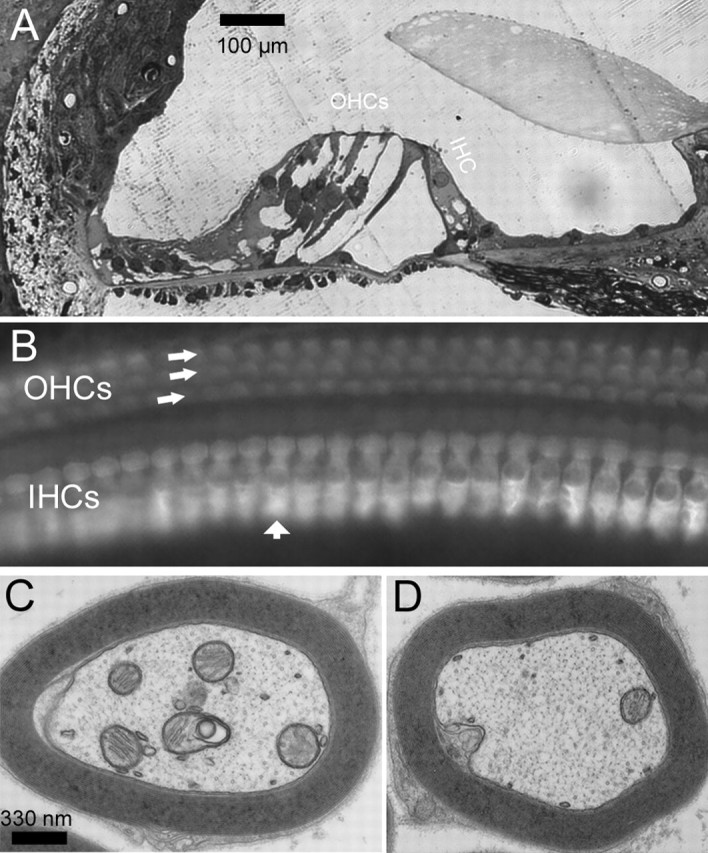
Morphological examinations of hair cells and myelin around the fibers of Cx29−/− mice. A, A cochlear section showing the organ of Corti obtained from the basal region of the cochlea. B, Whole-mount view of basal inner and outer hair cells visualized by immunolabeling with an antibody against myosinVI. IHC, Inner hair cell; OHC, outer hair cell. C, D, Cross sections of myelin around auditory nerve fibers obtained from Cx29−/− (C) and wild-type (D) mice, observed with an electron microscope.
Discussion
Many subtypes of Cxs are expressed by myelinating glial cells in the central and peripheral nervous systems. Oligodendrocytes express Cx32 (Dermietzel et al., 1989; Scherer et al., 1995), Cx47 (Menichella et al., 2003; Odermatt et al., 2003), and Cx29 (Altevogt et al., 2002; Altevogt and Paul, 2004). Astrocytes express Cx30, Cx43, and possibly Cx26 as well (Nagy and Rash, 2000; Filippov et al., 2003). Prominent mRNA expression of Cx26, Cx30, Cx43, and Cx29 are detected in the cochlea, and the mRNA expression levels of the four Cxs in the cochlea are at least an order of magnitude higher than other subtypes of Cxs (Ahmad et al., 2003). Cx26, Cx30, and Cx43 are not detected in the tract along the auditory nerve (Lautermann et al., 1998; Cohen-Salmon et al., 2004; Sun et al., 2005), leaving Cx29 as a strong candidate for Cxs expressed in myelinating glial cells in the cochlea. This is further supported by studies of others showing that Cx29 is exclusively expressed by myelinating glial cells in both peripheral and central nervous systems (Sohl et al., 2001; Altevogt et al., 2002). Following these clues, we demonstrated for the fist time that Cx29 was highly and exclusively expressed in the Schwann cells myelinating both the soma and processes of the SG neurons inside the cochlea until the glial juncture. We further revealed the functional importance of Cx29 in hearing by characterizing Cx29−/− mice. ABR, DPOAE measurements, and examinations of cochlear histology at both the optical and electron-microscope levels indicated that the severe demyelination found at the soma of SG neurons of Cx29−/− mice most likely underlies hearing impairments. In addition, noise exposures at a dosage that caused only TTS in wild-type mice resulted in a permanent high-frequency hearing loss in Cx29−/− mice (Fig. 6), indicating they are more vulnerable to noise-induced hearing losses. Therefore, Cx29 is required for normal cochlear functions.
Cx29−/− mice is a new model for studying molecular mechanisms of demyelinating type of the auditory neuropathy
Auditory neuropathy was first described ∼10 years ago for hearing-impaired patients who show normal otoacoustic emissions and abnormal ABRs (Starr et al., 1996). Patients with auditory neuropathy can usually hear sounds, and they show sound-evoked brain-imaging activities and cortical potentials (Rance et al., 2002). Usually, patients show a variable degree of pure-tone hearing losses, and the performance in speech understanding is worse than that predicted from the pure-tone audiogram (Doyle et al., 1998). Common psychoacoustic findings of patients with auditory neuropathy are profound abnormalities in their auditory temporal processing capability in detecting gap in continuous sound, amplitude modulation detection, as well as speech discrimination (Zeng et al., 2005). Most patients with auditory neuropathy are not helped by hearing aids, which simply amplify the sound intensity. The cellular and molecular basis of the pathophysiology of auditory neuropathy remains unknown. The preservation of otoacoustic emissions suggests that outer hair cells function normally. Therefore, the sites of cellular disorders are either in the inner hair cells, SG neurons, central auditory centers, or a combination of these sites (Starr et al., 2000). Genetic, as well as toxic and metabolic factors (e.g., anoxia, hyperbilirubinemia), are suspected contributing factors to auditory neuropathy.
Genes currently linked to the auditory neuropathy include Cx32 (Scherer et al., 1998), Cx31 (Lopez-Bigas et al., 2001), and otoferlin (Yasunaga et al., 2000). The gene for Cx29 (GJE1) is located on human chromosome 7 (7q22) and close to both the prestin and pendrin genes. Our findings that the Cx29−/− mice demonstrated (1) normal hair cell morphology (Fig. 7A,B) and DPOAE (Fig. 5A), (2) variable degree of hearing losses measured by tone-burst ABRs (Fig. 5B–D), (3) abnormal ABR waveforms and longer latency (Fig. 5E,F), and (4) demyelination of SG neuron soma (Fig. 8), are consistent with the symptoms displayed in patients suffering the auditory neuropathy. Therefore, Cx29−/− mice seemed to represent at least one type of auditory neuropathy with SG neuron demyelination as underlying pathology. In contrast to the immunoreactivity of MAG, which labeled only the myelin in the processes (Fig. 3A), Cx29 immunoreactivity was uniformly strong in the myelin wrapping both processes and soma of SG neurons. The MAG-immunolabeling pattern reported here was consistent with a previous report by Martini (1994). These results suggest that heterogeneous types of Schwann cells ensheathed the soma and processes of the SG neurons, which is consistent with a selective demyelination we observed only at the soma of SG neurons.
Possible molecular mechanisms of the Cx29 deficiency caused hearing impairments
The absence of one Cx gene is often compensated by another Cx, as in the case for Cx47 (Menichella et al., 2003; Odermatt et al., 2003) and Cx32 (Sutor et al., 2000), whereas concurrent losses of two Cxs cause severe demyelination phenotype and early mortality (Menichella et al., 2003; Odermatt et al., 2003). However, we found specific demyelination in the perikaryal myelin of SG neurons by a single loss of Cx29, suggesting unique contributions of Cx29 to the myelination of the soma of SG neurons. In contrast to Cx32 knock-out mice, which showed mild demyelination with late onset in peripheral nerve without significant functional consequences (Anzini et al., 1997; Scherer et al., 1998) and a mild decrease in thickness of central axonal myelin sheaths (Sutor et al., 2000), lack of Cx29 resulted in perikaryal myelin degeneration and hearing loss. The difference in the hearing phenotype may be explained by our observations that Cx32 is primarily expressed outside the cochlea (Fig. 2B). The different cellular distribution patterns of Cx29 and Cx32 make coassembly of Cx29 and Cx32 unlikely. Thus, it is doubtful that Cx29 and Cx32 are able to compensate functionally for each other. The mRNA of another myelinating Cx, Cx47, was minimally expressed in the cochlea (Ahmad et al., 2003). Unless it is dramatically upregulated in the absence of either Cx29, it is unlikely that Cx47 would be able to provide redundant functions rescuing either Cx29 in the cochlea.
Myelinating Schwann cells are usually not connected intercellularly by GJs. Rather, they contain reflexive gap junctions between the layers of the myelin sheath (Bergoffen et al., 1993). These reflexive gap junctions allow ions and small molecules to diffuse directly across the myelin sheath (Balice-Gordon et al., 1998). Cx29, together with other Cxs, may form reflexive GJs. Such radial channels for movement of intracellular molecules directly across the layers of myelin sheath provides a much shorter pathway than a circumferential route. Disruption of this radial pathway, therefore resulting in a much reduced efficiency for transporting molecules, might be a reason that demyelination was found in the soma of SG neurons of Cx29−/− mice (Fig. 8). The lack of rescuing effect by other remaining Cxs further suggested that the function of Cx29 in perikaryal myelin of SG neurons was nonredundant.
Based on the observation that Cx29 rarely colocalized in the same gap junction plaque with any of the other myelinating glial Cxs, it is possible that one major Cx29 function is conducted by its hemichannels located in the innermost layer of myelin facing SG neuron membranes. Hemichannel activities have been reported for a number of Cxs, including Cx43 (Li et al., 1996) and Cx32 (Castro et al., 1999). However, Cx29 hemichannel activity has not yet been demonstrated. The abnormal detachment between the cell membrane and myelin at the soma of SG neuron (Fig. 8B–D) observed in Cx29−/− mice would be consistent with this notion. The uniform subcellular distribution in of Cx29 in Schwann cells (Figs. 2–4) would be consistent with a hemichannel function allowing glial uptake of potassium from the extracellular space between axon and the myelin. We speculate that Cx29 hemichannels could comprise a pathway for the removal of K+ accumulated in periaxonal space during high-frequency firings of the auditory nerve.
Why would demyelination at the soma of SG neurons cause a delay in development of hearing sensitivity and hearing loss? In explaining demyelinating neuropathies, one prevailing theory is that axonal cytoskeleton is altered and axonal transport is disrupted when myelin is dissociated (Suter and Scherer, 2003). Therefore, the ability of neurons to transport cargo efficiently along their axons is compromised. Perikaryal demyelination in SG neurons, in contrast, may directly affect many cellular activities of the entire SG neuron. Therefore, the crucial questions are how do myelinating glial cells communicate with soma and axons (and vice versa) and how is this process altered by demyelination? Possibilities include increased cellular energetic costs of maintaining normal spontaneous and sound-evoked firings by SG neurons, decreased neurotrophic support from Schwann cells to sustain normal function of SG neurons, and abnormal signaling activities emanating from the affected myelin sheath. Other possible contributing factors to demyelination and/or axonal loss include inflammatory changes initiated by demyelination process. Clearly, additional investigations are needed to study the interactions between Schwann cells and SG neurons in the cochlea.
Footnotes
This work was supported by grants from the National Institute on Deafness and Other Communication Disorders (R01-DC04709 and R01-DC006483) (X.L.), the Woodruff Foundation (X.L.), and the National Institutes of Health [GM37751 (D.L.P.); P30-HD18655 (MRRC at Children’s Hospital, Boston, MA)]. We thank Paul Chen for expert assistance in maintaining and genotyping mouse breeding colonies.
References
- Ahmad S, Chen S, Sun J, Lin X (2003). Connexins 26 and 30 are co-assembled to form gap junctions in the cochlea of mice. Biochem Biophys Res Commun 307:362–368. [DOI] [PubMed] [Google Scholar]
- Altevogt BM, Paul DL (2004). Four classes of intercellular channels between glial cells in the CNS. J Neurosci 24:4313–4323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altevogt BM, Kleopa KA, Postma FR, Scherer SS, Paul DL (2002). Connexin29 is uniquely distributed within myelinating glial cells of the central and peripheral nervous systems. J Neurosci 22:6458–6470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anzini P, Neuberg DH, Schachner M, Nelles E, Willecke K, Zielasek J, Toyka KV, Suter U, Martini R (1997). Structural abnormalities and deficient maintenance of peripheral nerve myelin in mice lacking the gap junction protein connexin 32. J Neurosci 17:4545–4551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balice-Gordon RJ, Bone LJ, Scherer SS (1998). Functional gap junctions in the Schwann cell myelin sheath. J Cell Biol 142:1095–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergoffen J, Scherer SS, Wang S, Scott MO, Bone LJ, Paul DL, Chen K, Lensch MW, Chance PF, Fischbeck KH (1993). Connexin mutations in X-linked Charcot-Marie-Tooth disease. Science 262:2039–2042. [DOI] [PubMed] [Google Scholar]
- Castro C, Gomez-Hernandez JM, Silander K, Barrio LC (1999). Altered formation of hemichannels and gap junction channels caused by C-terminal connexin-32 mutations. J Neurosci 19:3752–3760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen-Salmon M, Maxeiner S, Kruger O, Theis M, Willecke K, Petit C (2004). Expression of the connexin43- and connexin45-encoding genes in the developing and mature mouse inner ear. Cell Tissue Res 316:15–22. [DOI] [PubMed] [Google Scholar]
- Dermietzel R, Traub O, Hwang TK, Beyer E, Bennett MV, Spray DC, Willecke K (1989). Differential expression of three gap junction proteins in developing and mature brain tissues. Proc Natl Acad Sci USA 86:10148–10152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle KJ, Sininger Y, Starr A (1998). Auditory neuropathy in childhood. Laryngoscope 108:1374–1377. [DOI] [PubMed] [Google Scholar]
- Filippov MA, Hormuzdi SG, Fuchs EC, Monyer H (2003). A reporter allele for investigating connexin 26 gene expression in the mouse brain. Eur J Neurosci 18:3183–3192. [DOI] [PubMed] [Google Scholar]
- Forge A, Marziano NK, Casalotti SO, Becker DL, Jagger D (2003). The inner ear contains heteromeric channels composed of cx26 and cx30 and deafness-related mutations in cx26 have a dominant negative effect on cx30. Cell Commun Adhes 10:341–346. [DOI] [PubMed] [Google Scholar]
- Grifa A, Wagner CA, D’Ambrosio L, Melchionda S, Bernardi F, Lopez-Bigas N, Rabionet R, Arbones M, Monica MD, Estivill X, Zelante L, Lang F, Gasparini P (1999). Mutations in GJB6 cause nonsyndromic autosomal dominant deafness at DFNA3 locus. Nat Genet 23:16–18. [DOI] [PubMed] [Google Scholar]
- Lautermann J, ten Cate WJ, Altenhoff P, Grummer R, Traub O, Frank H, Jahnke K, Winterhager E (1998). Expression of the gap-junction connexins 26 and 30 in the rat cochlea. Cell Tissue Res 294:415–420. [DOI] [PubMed] [Google Scholar]
- Li H, Liu TF, Lazrak A, Peracchia C, Goldberg GS, Lampe PD, Johnson RG (1996). Properties and regulation of gap junctional hemichannels in the plasma membranes of cultured cells. J Cell Biol 134:1019–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Lynn BD, Olson C, Meier C, Davidson KG, Yasumura T, Rash JE, Nagy JI (2002). Connexin29 expression, immunocytochemistry and freeze-fracture replica immunogold labelling (FRIL) in sciatic nerve. Eur J Neurosci 16:795–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XZ, Xia XJ, Ke XM, Ouyang XM, Du LL, Liu YH, Angeli S, Telischi FF, Nance WE, Balkany T, Xu LR (2002). The prevalence of connexin 26 (GJB2) mutations in the Chinese population. Hum Genet 111:394–397. [DOI] [PubMed] [Google Scholar]
- Lopez-Bigas N, Olive M, Rabionet R, Ben-David O, Martinez-Matos JA, Bravo O, Banchs I, Volpini V, Gasparini P, Avraham KB, Ferrer I, Arbones ML, Estivill X (2001). Connexin 31 (GJB3) is expressed in the peripheral and auditory nerves and causes neuropathy and hearing impairment. Hum Mol Genet 10:947–952. [DOI] [PubMed] [Google Scholar]
- Martini R (1994). Myelin-associated glycoprotein is not detectable in perikaryal myelin of spiral ganglion neurons of adult mice. Glia 10:311–314. [DOI] [PubMed] [Google Scholar]
- Maw MA, Allen-Powell DR, Goodey RJ, Stewart IA, Nancarrow DJ, Hayward NK, Gardner RJ (1995). The contribution of the DFNB1 locus to neurosensory deafness in a Caucasian population. Am J Hum Genet 57:629–635. [PMC free article] [PubMed] [Google Scholar]
- Menichella DM, Goodenough DA, Sirkowski E, Scherer SS, Paul DL (2003). Connexins are critical for normal myelination in the CNS. J Neurosci 23:5963–5973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morell R, Kim H, Hood L, Goforth L, Friderici K, Fisher R, Van CG, Berlin C, Oddoux C, Ostrer H, Keats B, Friedman T (1998). Mutations in the connexin 26 gene (GJB2) among Ashkenazi Jews with nonsyndromic recessive deafness. N Engl J Med 339:1500–1505. [DOI] [PubMed] [Google Scholar]
- Nagy JI, Rash JE (2000). Connexins and gap junctions of astrocytes and oligodendrocytes in the CNS. Brain Res Brain Res Rev 32:29–44. [DOI] [PubMed] [Google Scholar]
- Odermatt B, Wellershaus K, Wallraff A, Seifert G, Degen J, Euwens C, Fuss B, Bussow H, Schilling K, Steinhauser C, Willecke K (2003). Connexin 47 (Cx47)-deficient mice with enhanced green fluorescent protein reporter gene reveal predominant oligodendrocytic expression of Cx47 and display vacuolized myelin in the CNS. J Neurosci 23:4549–4559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng BG, Ahmad S, Chen S, Chen P, Price MP, Lin X (2004a). Acid-sensing ion channel 2 contributes a major component to acid-evoked excitatory responses in spiral ganglion neurons and plays a role in noise susceptibility of mice. J Neurosci 24:10167–10175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng BG, Li QX, Ren TY, Ahmad S, Chen SP, Chen P, Lin X (2004b). Group I metabotropic glutamate receptors in spiral ganglion neurons contribute to excitatory neurotransmissions in the cochlea. Neuroscience 123:221–230. [DOI] [PubMed] [Google Scholar]
- Rance G, Cone-Wesson B, Wunderlich J, Dowell R (2002). Speech perception and cortical event related potentials in children with auditory neuropathy. Ear Hear 23:239–253. [DOI] [PubMed] [Google Scholar]
- Romand R, Romand MR (1985). Qualitative and quantitative observations of spiral ganglion development in the rat. Hear Res 18:111–120. [DOI] [PubMed] [Google Scholar]
- Scherer SS, Deschenes SM, Xu YT, Grinspan JB, Fischbeck KH, Paul DL (1995). Connexin32 is a myelin-related protein in the PNS and CNS. J Neurosci 15:8281–8294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherer SS, Xu YT, Nelles E, Fischbeck K, Willecke K, Bone LJ (1998). Connexin32-null mice develop demyelinating peripheral neuropathy. Glia 24:8–20. [DOI] [PubMed] [Google Scholar]
- Sohl G, Eiberger J, Jung YT, Kozak CA, Willecke K (2001). The mouse gap junction gene connexin29 is highly expressed in sciatic nerve and regulated during brain development. Biol Chem 382:973–978. [DOI] [PubMed] [Google Scholar]
- Starr A, Picton TW, Sininger Y, Hood LJ, Berlin CI (1996). Auditory neuropathy. Brain 119:741–753. [DOI] [PubMed] [Google Scholar]
- Starr A, Sininger YS, Pratt H (2000). The varieties of auditory neuropathy. J Basic Clin Physiol Pharmacol 11:215–230. [DOI] [PubMed] [Google Scholar]
- Sun J, Ahmad S, Chen S, Tang W, Zhang Y, Chen P, Lin X (2005). Cochlear gap junctions coassembled from Cx26 and 30 show faster intercellular Ca2+ signaling than homomeric counterparts. Am J Physiol 288:C613–C623. [DOI] [PubMed] [Google Scholar]
- Suter U, Scherer SS (2003). Disease mechanisms in inherited neuropathies. Nat Rev Neurosci 4:714–726. [DOI] [PubMed] [Google Scholar]
- Sutor B, Schmolke C, Teubner B, Schirmer C, Willecke K (2000). Myelination defects and neuronal hyperexcitability in the neocortex of connexin 32-deficient mice. Cereb Cortex 10:684–697. [DOI] [PubMed] [Google Scholar]
- Walsh EJ, Romand R (1992). Functional development of the cochlea and cochlear nerve. In: Development of auditory and vestibular systems (Romand R, ed.) , pp. 161–219. Amsterdam: Elsevier.
- Whitehead WL, Lonsbury-Martin BL, Martin GK, McCoy MJ (1996). Otoacoustic emissions: animal models and clinical observations. In: Clinical aspects of hearing (Van De Water TR, Popper AN, Fay RR, eds) , pp. 199–254. New York: Springer.
- Willecke K, Eiberger J, Degen J, Eckardt D, Romualdi A, Guldenagel M, Deutsch U, Sohl G (2002). Structural and functional diversity of connexin genes in the mouse and human genome. Biol Chem 383:725–737. [DOI] [PubMed] [Google Scholar]
- Xia JH, Liu CY, Tang BS, Pan Q, Huang L, Dai HP, Zhang BR, Xie W, Hu DX, Zheng D, Shi XL, Wang DA, Xia K, Yu KP, Liao XD, Feng Y, Yang YF, Xiao JY, Xie DH, Huang JZ (1998). Mutations in the gene encoding gap junction protein beta-3 associated with autosomal dominant hearing impairment. Nat Genet 20:370–373. [DOI] [PubMed] [Google Scholar]
- Yasunaga S, Grati M, Chardenoux S, Smith TN, Friedman TB, Lalwani AK, Wilcox ER, Petit C (2000). OTOF encodes multiple long and short isoforms: genetic evidence that the long ones underlie recessive deafness DFNB9. Am J Hum Genet 67:591–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng FG, Kong YY, Michalewski HJ, Starr A (2005). Perceptual consequences of disrupted auditory nerve activity. J Neurophysiol 93:3050–3063. [DOI] [PubMed] [Google Scholar]