

Abstract
The mechanisms through which circulating ghrelin relays hunger signals to the CNS are not yet fully understood. In this study, we have examined the potential role of the subfornical organ (SFO), a circumventricular structure that lacks the normal blood–brain barrier, as a CNS site in which ghrelin acts to influence the hypothalamic centers controlling food intake. We report that ghrelin increased intracellular calcium concentrations in 28% (12 of 43) of dissociated SFO neurons and that the SFO expresses mRNA for the growth hormone secretagogue receptor. Whole-cell patch recordings from SFO neurons demonstrated that in 29% (9 of 31) of neurons tested ghrelin induced a mean depolarization of 7.4 ± 0.69 mV, accompanied by an increase in action potential frequency. Voltage-clamp recordings revealed that ghrelin activates a putative nonselective cationic conductance.
Previous reports that the satiety signal amylin exerts similar excitatory effects on SFO neurons led us to examine whether these two peptides influence different subpopulations of SFO neurons. Concentration-dependent depolarizing effects of amylin were observed in 59% (28 of 47) of SFO neurons (mean depolarization, 8.32 ± 0.60 mV). In contrast to ghrelin, voltage-clamp recordings suggest that amylin influences a voltage-dependent current activated at depolarized potentials. We tested single SFO neurons with both peptides and identified cells responsive only to ghrelin (n = 9) and only to amylin (n = 7) but no cells that responded to both peptides. These data support a role for the SFO as a center at which ghrelin and amylin may influence separate subpopulations of neurons to influence the hypothalamic regulation of feeding.
Keywords: ghrelin, amylin, subfornical organ, circumventricular organ, electrophysiology, feeding
Introduction
Ghrelin, a 28 amino acid peptide (Kojima et al., 1999) and endogenous ligand for the growth hormone secretagogue receptor (GHSR), has significant roles in food intake, independent from its actions on growth hormone release (Tschop et al., 2000; Wren et al., 2000). Studies showing the effects of peripheral and central ghrelin administration on weight gain (Tschop et al., 2000) and food intake (Wren et al., 2000), respectively, suggest important integrated roles for this peptide in controlling energy balance.
Ghrelin, originally isolated from stomach (Kojima et al., 1999), is expressed in various tissues (Date et al., 2000; Mori et al., 2000; Korbonits et al., 2001; Volante et al., 2002; Caminos et al., 2003), including brain (Lu et al., 2002), in which ghrelin-expressing neurons have been described (Cowley et al., 2003); however, the predominant expression of ghrelin in the stomach suggests important roles for circulating ghrelin as a signal to hypothalamic feeding centers.
In contrast to ghrelin, the 37 amino acid peptide amylin is an anorexigenic signal cosecreted with insulin from pancreatic β cells. Amylin increases after a meal (Butler et al., 1990) and inhibits feeding (Morley and Flood, 1991; Lutz et al., 1994, 1995; Morley et al., 1994; Rushing et al., 2000). Circulating amylin levels are correlated with long-term changes in body fat (Pieber et al., 1994).
Although these two peptides influence food intake (Rushing et al., 2000, 2001; Seoane et al., 2000; Tschop et al., 2000; Kamegai et al., 2001; Nakazato et al., 2001; Wren et al., 2001), how they access the CNS remains to be clearly established. Partial roles for vagal afferents in mediating ghrelin signaling have been suggested (Date et al., 2002; le Roux et al., 2005), whereas amylin effects were unchanged after vagal manipulations (Morley et al., 1994). The existence of specific transporters for ghrelin (Banks et al., 2002) and amylin (Banks and Kastin, 1998) suggest another mechanism through which these peptides may cross the blood–brain barrier (Banks and Kastin, 1992), although their physiological relevance remains unclear.
Studies showing binding sites for amylin in two circumventricular organs (CVOs), the area postrema and the subfornical organ (SFO) (Sexton et al., 1994), combined with demonstrated roles for these structures in mediating amylin actions on feeding (Lutz et al., 2001) and food-associated drinking (Perez-Velazquez and Zhang, 1994), suggest that CVOs are a potential target for these circulating peptides. The CVOs lack the normal blood–brain barrier and have been implicated extensively in autonomic regulation, including the control of feeding (McKinley et al., 1998; Cottrell and Ferguson, 2004). Neurons in the CVOs are bathed continuously by all constituents of the circulation, and amylin activates area postrema (Riediger et al., 2001) and SFO (Riediger et al., 1999) neurons. SFO efferents project to hypothalamic centers regulating feeding (Cottrell and Ferguson, 2004), including arcuate (Gruber et al., 1987), paraventricular, and supraoptic nuclei, and lateral hypothalamus (Miselis, 1981, 1982; Silverman et al., 1981; Lind et al., 1982).
In this study we investigated the potential role of SFO neurons as a target where ghrelin and amylin act to influence brain centers involved in the control of food intake.
Materials and Methods
Subfornical organ neuron preparation.
All experiments were approved by the Queen's University Animal Care Committee. SFO neurons were dissociated as described previously (Ferguson et al., 1997). Male Sprague Dawley rats (125–175 g) were decapitated, and the brains were removed immediately and immersed in ice-cold HBSS (Invitrogen, Grand Island, NY; Ca2+ and Mg2+ free; 0.03 m sucrose). A tissue block containing the hippocampal commissure was dissected from the brain, and the SFO was separated from the surrounding tissue by means of microdissection techniques. The isolated SFO was placed in HBSS containing 1 mg/ml trypsin and incubated at 37°C in 5% CO2–95%O2 for 20 min. Cells were then suspended in ice-cold HBSS containing Ca2+ and 4 μg/ml BSA, triturated with a 20 gauge needle with a 1 ml syringe, and centrifuged at 900 × g for 5 min. The supernatant was removed, and cells were resuspended with Neurobasal A Medium (Invitrogen) and centrifuged again for 5 min. Cells were suspended a second time and plated on plastic culture dishes (Corning, Corning, NY) with Neurobasal A supplemented with 100 U/ml penicillin–streptomycin and 0.5 mm l-glutamine. Cells were incubated for a minimum of 24 h, and experiments were performed 1–2 d after dissociation.
Calcium-imaging techniques.
Measurements of intracellular calcium ([Ca2+]i) were obtained by microfluorescent imaging of SFO neurons loaded with fura-2 (Invitrogen, Eugene, OR). Neurons were loaded by incubation (30 min; 37°C) with the acetomethoxy ester form of fura-2 (3 μm in HBSS), followed by an HBSS wash and a second incubation to permit esterase conversion to free fura-2. Imaging was performed with an InCyt dual-wavelength imaging system (Intracellular Imaging, Cincinnati, OH) with a PixelFly CCD camera 12-bit dynamic range; 1360 × 1024 pixel resolution; Cooke Corporation, Auburn Hills, MI). [Ca2+]i was calculated from images collected at 0.5 Hz with ratiometric comparison of emissions (505 nm) from excitation at 340 and 380 nm wavelengths and by comparing these ratios with those obtained from known Ca2+ calibration standards.
The standard experimental protocol involved a 5 min baseline recording of [Ca2+]i followed by bath exchange by gravity perfusion of HBSS supplemented with ghrelin. The ghrelin treatment period lasted 8 min and was followed by bath exchange with HBSS and a recovery period of ∼20 min. The experiments were concluded by perfusion with HBSS containing 40 mm KCl to confirm cell viability. To determine whether ghrelin-induced increases in [Ca2+]i were secondary to depolarization-induced opening of voltage-gated Ca2+ channels, a second identical application of ghrelin in HBSS containing tetrodotoxin (TTX) (5 μm; Alomone Labs. Jerusalem, Israel) was performed, and the effects on calcium mobilization were assessed.
RNA extraction and reverse transcriptase-PCR.
The SFO was acutely dissected from rat brains as above. Total RNA was extracted with TRIzol reagent (Invitrogen) per the manufacturer's directions. The RNA from a single SFO was treated with reverse transcriptase (RT)-PCR-grade DNase I (Sigma, St. Louis, MO). Oligo-dT-primed cDNA was synthesized with all of the RNA from an SFO in a 20 μl cDNA synthesis reaction according to directions (Retroscript; Ambion, Austin, TX).
RT-PCR was performed with 1 μl of cDNA in a standard reaction containing 1× PCR buffer, 1.5 mm MgCl2, 0.2 mm each dNTP, 0.2 μm each primer, and 2 U of Platinum Taq (Invitrogen). The reactions were cycled in an Eppendorf Mastercycler as follows: initial denaturation at 94°C for 3 min; 35 cycles at 94°C for 60 s, 55°C for 60 s, and 72°C for 60 s; and a final extension at 72°C for 5 min. Two sets of primers were used to detect expression of growth hormone secretagogue receptor cDNA (Barreiro et al., 2003). One set specifically detected cDNA encoding the active form of the receptor (GHSR1a: sense 5′-TTC TTT CTA CCG GTC TTC TGC CTC-3′ and antisense 5′-GGA CAC CAG GTT GCA GTA CTG GCT-3′), and a second set detected both the active and inactive forms (GHSR1b: sense 5′-GTC GAG CAT CAA CAC ATG AC-3′ and antisense 5′-GGT TGC ACA TGA GTG CTC TG-3′). A primer set specific to synaptotagmin-1 was used as a positive control (sense 5′-AGG GGC TTT CCT ATC TAA GGG-3′ and antisense 5′-GTT GGC AGT GTT GCA AGA GA-3′). PCR products were electrophoresed on a 2% agarose gel and sequenced to confirm identity (Robarts, London, Ontario, Canada).
Electrophysiological techniques.
Whole-cell patch-clamp recordings were performed as reported previously (Washburn et al., 1999; Desson and Ferguson, 2003; Cottrell et al., 2004). Electrodes were made from 1.2 mm filament glass (A-M Systems, Carlsborg, WA) on a Sutter Instruments (Novato, CA) puller, polished to a resistance of 6–10 MΩ, and filled with the following (in mm): 130 potassium gluconate, 10 HEPES, 10 EGTA, 1 MgCl2, 4 Na2ATP, and 0.1 GTP (290–300 mOsm, pH 7.2, with KOH). Cells were bathed in artificial CSF (ACSF) containing the following (in mm): 140 NaCl, 5 KCl, 1 MgCl2, 10 HEPES, 10 glucose, and 2 CaCl2 (290–300 mOsm, pH 7.4, with NaOH).
Whole-cell recordings were obtained from SFO cells identified as neurons by the presence of voltage-gated Na+ currents when under voltage-clamp conditions and at least 80 mV action potentials induced from a holding potential of −55 mV during current-clamp recordings. Signals were amplified with an Axopatch 200B amplifier (Molecular Devices, Foster City, CA) filtered with an eight-pole Bessel filter at 1 kHz and digitized with a Cambridge Electronics Design (Cambridge, UK) micro1401 interface at 8 kHz. Data were collected with Signal (voltage clamp) or Spike 2 (current clamp) software packages (Cambridge Electronics Design).
Peptides and drugs.
Unless stated otherwise, all chemicals were obtained from Sigma. Ghrelin (Phoenix Pharmaceuticals, Belmont, CA) was stored in a stock solution at −80°C and diluted to the appropriate concentrations on the day of the experiment. The GHSR antagonist, [d-Lys-3]-growth hormone-releasing peptide (GHRP)-6 (Bachem Bioscience, King of Prussia, PA), was stored in a stock solution at −80°C and diluted to 100 μm for use in patch-clamp and calcium-imaging experiments (Cheng et al., 1989; Smith et al., 1993). Amylin (Phoenix Pharmaceuticals) was rehydrated in 200 μl of 0.1 m acetic acid and subsequently stored in a stock solution at −80°C that was diluted to the appropriate concentrations on the day of the experiment.
Statistical analysis.
In the calcium-imaging experiments, baseline [Ca2+]i was averaged over the 100 s immediately before ghrelin or control HBSS application. To assess a treatment effect, [Ca2+]i was averaged over the last 100 s of the treatment period. Cells were then classified as responders if the mean [Ca2+]i increased by >20% of baseline levels during the treatment period. Control and treatment values were compared with a Student's t test.
In current-clamp recordings, cells were classified as responders if their membrane potential changed by >5 mV within 200 s from the start of peptide perfusion and if the changes were sustained for a minimum of 1 min and followed by return toward baseline membrane potential. The change in membrane potential was measured relative to the 100 s control period before application of the peptide. Changes in membrane potential and action potential frequency were compared between control and treatment groups with a Student's t test.
In voltage-clamp recordings, cells were classified as responders if they exhibited an increase in inward current >5 pA at a membrane potential of −80 mV and the presence of at least a partial recovery toward baseline after removal of ghrelin from the bath. Peak current change was compared between control and treatment groups with a Student's t test. All values are reported as means ± SEM with p < 0.05.
Results
Ghrelin influences SFO neurons through actions at the GHSR
Previous evidence suggests that ghrelin influences [Ca2+]i in other neuronal populations (Kohno et al., 2003). The effects of ghrelin on [Ca2+]i in SFO neurons were assessed with microfluorometry in fura-2-loaded cells. Ghrelin (10 nm) induced reversible transient increases in [Ca2+]i in 28% (12 of 43) of neurons imaged (Fig. 1A,B). In the responding neurons, the mean increase from baseline was 78.6 ± 19.6%, which was significantly greater than that of control cells treated with HBSS wash (4.2 ± 3.4%; n = 43; Student's t test; p < 0.05), and it was abolished in the presence of 100 μm [d-Lys-3]-GHRP-6, a GHSR antagonist (−6.3 ± 4.5%; n = 17) (Fig. 1C). These initial screening studies therefore identified a subset of SFO neurons capable of responding to ghrelin with an increase in [Ca2+]i effects that were abolished by the GHSR antagonist, suggesting an action at this receptor.
Figure 1.

Ghrelin increases [Ca2+]i in SFO neurons. A, B, [Ca2+]i measured in individual SFO neurons by microfluorometry of the Ca2+-sensitive dye fura-2 demonstrates that ghrelin treatment causes transient increases in [Ca2+]i that are reversible after removal of ghrelin from the bath solution. The amplitude and duration of responses varied among cells. In some SFO neurons, the effect was repeatable, although of smaller magnitude, thus suggesting that some desensitization may be occurring (B). C, Mean change in [Ca2+]i measured relative to baseline in control cells treated with HBSS (n = 43), neurons responding to 10 nm ghrelin (n = 12; *p < 0.05), cells treated with 10 nm ghrelin in the presence of [d-Lys-3]-GHRP-6 (lined bar; n = 17; p > 0.05), and cells treated with 10 nm ghrelin in the presence of TTX (n = 6; p > 0.05). D, Ethidium bromide-stained agarose gel of the RT-PCR products generated with the GHSR and synaptotagmin-1 (synap-1) primers. Lane 1, DNA marker; lane 2, GHSR1a receptor (261 bp; specifically detects only the active GHSR1a receptor); lane 3, GHSR receptor (321 bp; detects both GHSR1a and GHSR1b receptors); lane 4, synaptotagmin-1 (199 bases); lane 5, negative control.
We next attempted to confirm expression of the GHSR in the SFO. Previous work has shown that there are two forms of the ghrelin receptor: the active form, named GHSR1a, and the inactive truncated form, named GHSR1b (McKee et al., 1997). We therefore examined whether the active GHSR1a was expressed in the SFO by performing RT-PCR on acutely dissected SFOs with primers specific to the receptor. Figure 1D shows a representative ethidium bromide-stained agarose gel of the RT-PCR products generated with the GHSR and synaptotagmin-1 (as a positive control) primers. The results showing that RT-PCR detected cDNA encoding the GHSR1a indicate that the active form of the receptor is expressed in the SFO.
Electrophysiology
Whole-cell recordings were obtained from 174 dissociated rat SFO neurons in either current-clamp or voltage-clamp mode. All of these cells elicited action potentials of >80 mV and had a mean resting membrane potential of −49.0 ± 2.1 mV and a mean input resistance of 1.5 ± 0.09 GΩ, values similar to those reported previously for dissociated SFO neurons (Anderson et al., 2001; Williams et al., 2003).
Ghrelin depolarizes SFO neurons
Continuous current-clamp recordings were performed to assess any changes in membrane potential and firing frequency of SFO neurons in response to ghrelin. Ghrelin was administered to the bath by gravity perfusion for 200 s, followed by perfusion with ACSF. Of the neurons tested at varying concentrations of ghrelin, 37% (21 of 57) were considered responsive, as demonstrated by a clear reversible depolarization from baseline membrane potential.
Plasma ghrelin levels have been shown to vary significantly in rats fed ad libitum, from as low as 0.380 ± 4.22 nm (Tschop et al., 2000) to 556.2 ± 43.8 nm (Date et al., 2000). In accordance with this wide range of values, ghrelin was initially applied at a concentration of 1 nm. As illustrated in Figure 2, A and B, 1 nm ghrelin induced a depolarization of 7.4 ± 0.69 mV in 9 of 31 neurons, effects that were reversible after removal of ghrelin from the bath. The magnitude of the depolarization was significantly different from changes seen in control cells treated with ACSF (−0.23 ± 0.26 mV; p < 0.001; Student's t test) (Fig. 2C).
Figure 2.

Ghrelin has an excitatory effect on SFO neurons. A, B, Single-cell recordings showing depolarization and an increase in action potential frequency induced by application of 1 nm ghrelin (black bar) and return to baseline membrane potential and frequency after removal of ghrelin from the bath. C, Mean membrane potential change in ACSF (white bar) compared with 1 nm ghrelin (black bar); *p < 0.001. D, Peak action potential frequency measured over 10 s in the presence of ACSF before ghrelin treatment (white bar) compared with 1 nm ghrelin (black bar) in the same cells; *p < 0.05.
The excitatory effect of ghrelin was accompanied by an increase in action potential firing frequency in spontaneously active cells as well as in relatively quiescent cells. Firing frequency was assessed by comparing changes in peak frequency over a 10 s period during the 100 s control period before ghrelin application and during 100 s of the peak excitatory response. Ghrelin significantly increased firing from 0.52 ± 0.16 to 3.2 ± 0.97 Hz in the nine responding neurons (p < 0.05; Student's paired t test) (Fig. 2D). In unaffected neurons, peak firing frequency was 0.70 ± 0.21 Hz during control perfusion with ACSF and was unchanged at 0.77 ± 0.23 Hz in the presence of 1 nm ghrelin (p > 0.05; Student's paired t test). Similarly, no significant change was seen in the firing frequency of control cells when treated with ACSF (control = 0.28 ± 0.09 Hz vs ACSF = 0.33 ± 0.14 Hz; Student's paired t test; p > 0.05). These effects on action potential frequency also raised the possibility that the effects of ghrelin described above on [Ca2+]i may simply be a consequence of increased action potential-induced voltage-gated calcium channel opening, as opposed to a true effect on intracellular calcium-based signal transduction. This possibility was tested in additional [Ca2+]i measurements from SFO cells with and without the sodium channel blocker TTX in the medium. These experiments demonstrated that such treatment completely blocked all calcium responses of SFO neurons to bath application of ghrelin (control 81 ± 28% vs TTX 19 ± 6%; n = 6) as illustrated in Figure 1C, confirming that such effects were likely secondary to the effects of ghrelin causing increases in action potential frequency.
Cells tested with 1 nm ghrelin in the presence of 100 μm [d-Lys-3]-GHRP-6 showed no significant change in membrane potential compared with control cells (−1.69 ± 2.02 mV; p > 0.05; n = 9), indicating that the GHSR antagonist was capable of blocking the effects of ghrelin on SFO neurons.
Ghrelin effects are concentration dependent
We also examined the effects of concentrations of ghrelin ranging from 10 nm to 100 pm to determine whether ghrelin activated SFO neurons in a concentration-dependent manner. At a concentration of 10 nm, ghrelin induced a mean depolarization of 7.3 ± 1.3 mV in five of nine neurons tested (Fig. 3A). This excitatory response was similar in magnitude to that seen at 1 nm. Most of the responding neurons tested with the higher concentration of ghrelin demonstrated sustained responses, with no recovery seen after 45 min of recording. Responses to low concentrations (100 pm) of ghrelin were also examined in 17 SFO neurons, none of which showed >5 mV responses to the peptide. To identify the ghrelin-responsive subpopulation of these SFO neurons, they were then tested with 1 nm ghrelin to determine whether the cell was responsive, as illustrated in Figure 3B. Of the 17 neurons tested at 100 pm, 7 were classified as responders (membrane potential change >5 mV in response to 1 nm ghrelin); however, the mean membrane potential change of responding neurons after application of 100 pm ghrelin was 1.2 ± 1.8 mV, which was not significantly different from the membrane potential change in unaffected cells (−0.89 ± 1.1 mV; p > 0.05) or in control cells treated with ACSF (−0.23 ± 0.26 mV; p > 0.05; data not shown). A summary of the mean membrane potential changes induced by different concentrations of ghrelin is shown in Figure 3C, with these data suggesting an all-or-none membrane potential response to ghrelin with maximal effects observed at 1 nm.
Figure 3.

Effects of ghrelin on SFO neurons are concentration dependent. A, Single-cell recording demonstrating depolarization and increase in action potential frequency after application of 10 nm ghrelin (black bar). The response was not reversible after removal of ghrelin from the bath. B, Trace showing no change in membrane potential after application of 100 pm ghrelin (black bar); however, application of 1 nm ghrelin (black bar) in the same cell induced a reversible depolarization. C, Bar graph illustrates significant difference between mean depolarization at 100 pm and 1 nm (*p < 0.05) and 100 pm (*p < 0.01) and 10 nm. There was no significant difference between the magnitudes of depolarization at 1 and 10 nm (p > 0.05).
Ghrelin activates nonselective cation channels
There is considerable literature indicating that many peptides exert their depolarizing effects by activation of a nonselective cationic conductance (NSCC) (Oliet and Bourque, 1993; Yang and Ferguson, 2002; Desson and Ferguson, 2003). Conductance changes in response to ghrelin administration were assessed with slow (10 mV/s) depolarizing voltage ramps (−100 to +20 mV) initiated after a hyperpolarizing prepulse to −100 mV for 500 ms. The currents recorded from a single SFO neuron in response to such voltage ramps before ghrelin application and in the presence of 1 nm ghrelin are illustrated in Figure 4A (each trace is a mean of five ramps). The ghrelin-induced current is shown in the inset, calculated by subtracting control current from current in the presence of 1 nm ghrelin. Activation of a voltage-independent current, characteristic of an NSCC, was seen in 60% (9 of 15) of the cells that were tested. Partial recovery back to control current was seen in 55% of affected neurons. The data presented in Figure 4B show the mean ghrelin-induced current in responding neurons. The slope of the line is 0.88 ± 0.048 pA, and the mean reversal potential is −49.4 ± 2.64 mV.
Figure 4.
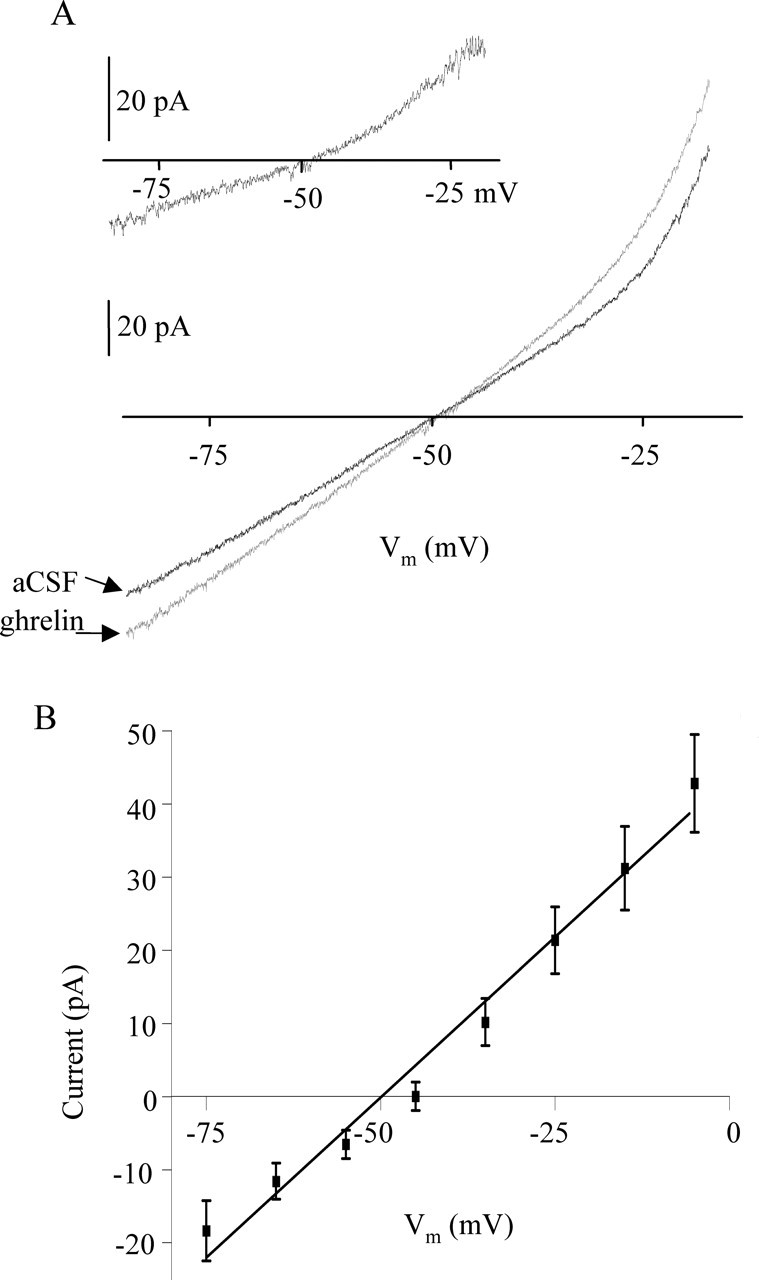
Ghrelin activates a nonselective cationic conductance. A, Graph represents the current recorded in a single cell during voltage ramps from −100 to −30 mV (each trace is a mean of 5 ramps) in the presence of ACSF (black) and 10 nm ghrelin (red). The inset represents the difference current calculated by subtracting the ghrelin-induced current from the control current (ACSF) in the same cell. B, Graph represents the mean (±SEM) difference current measured in nine responding cells.
Amylin depolarizes SFO neurons
Continuous current-clamp recordings were performed to assess any changes in membrane potential and firing frequency of SFO neurons in response to gravity perfusion of amylin for 200 s, followed by perfusion with ACSF. Of the neurons tested at varying concentrations of amylin, 59% (47 of 79) were considered responsive, as demonstrated by a clear depolarization from baseline membrane potential (Fig. 5A,B).
Figure 5.

Amylin has an excitatory effect on SFO neurons. A, B, Current-clamp recordings showing a depolarization and an increase in action potential frequency induced by the application of 1 nm amylin (gray bar). Note the return to baseline membrane potential and spike frequency after removal of amylin from the bath. C, Mean membrane potential change in the presence of 1 nm amylin (black bar) compared with controls treated with ACSF (white bar); *p < 0.0001. D, Mean peak action potential frequency in the presence of ACSF (white bar) compared with peak frequency in the same cells in the presence of 1 nm amylin (black bar); *p < 0.001.
Amylin was initially applied at a concentration of 1 nm, inducing a mean depolarization of 8.32 ± 0.60 mV (n = 11 of 16), significantly different from control cells treated with ACSF (−0.23 ± 0.26 mV; n = 10; p < 0.001; Student's t test) (Fig. 5C). These excitatory effects were repeatable and reversible after removal of amylin from the bath.
Accompanying the depolarizing effect of amylin was an increase in action potential frequency in responding neurons. Peak action potential frequency increased significantly from 0.68 ± 0.30 Hz in ACSF to 2.54 ± 0.41 Hz in the presence of 1 nm amylin (Student's paired t test; p < 0.001) (Fig. 5D).
Amylin effects are concentration dependent
Amylin was applied at concentrations ranging from 100 nm to 1 pm to determine whether the depolarizing effects of amylin were concentration related. When neurons were tested with 100 nm amylin, 61% (11 of 18) of the neurons responded with a mean depolarization of 9.77 ± 1.14 mV. Of the neurons tested with 10 nm amylin, 54% (7 of 13) responded with a mean depolarization of 11.79 ± 1.01 mV (Fig. 6A); 100 pm amylin induced a mean change of 8.49 ± 0.78 mV in 53% (10 of 19) of the neurons tested (Fig. 6B). A response to 10 pm amylin was observed in 67% (6 of 9) of the neurons tested, with a mean change of 9.10 ± 2.50 mV. In cells tested with 1 pm amylin, if the membrane potential change was <5 mV, the cell was tested with a higher concentration of amylin to further clarify whether the cell had the capability to respond to amylin (Fig. 6C). Fifty percent (2 of 4) of neurons tested were considered amylin responsive, and the mean change observed with 1 pm amylin was −0.92 ± 2.64 mV, indicating that at this low concentration SFO neurons are much less sensitive to amylin.
Figure 6.
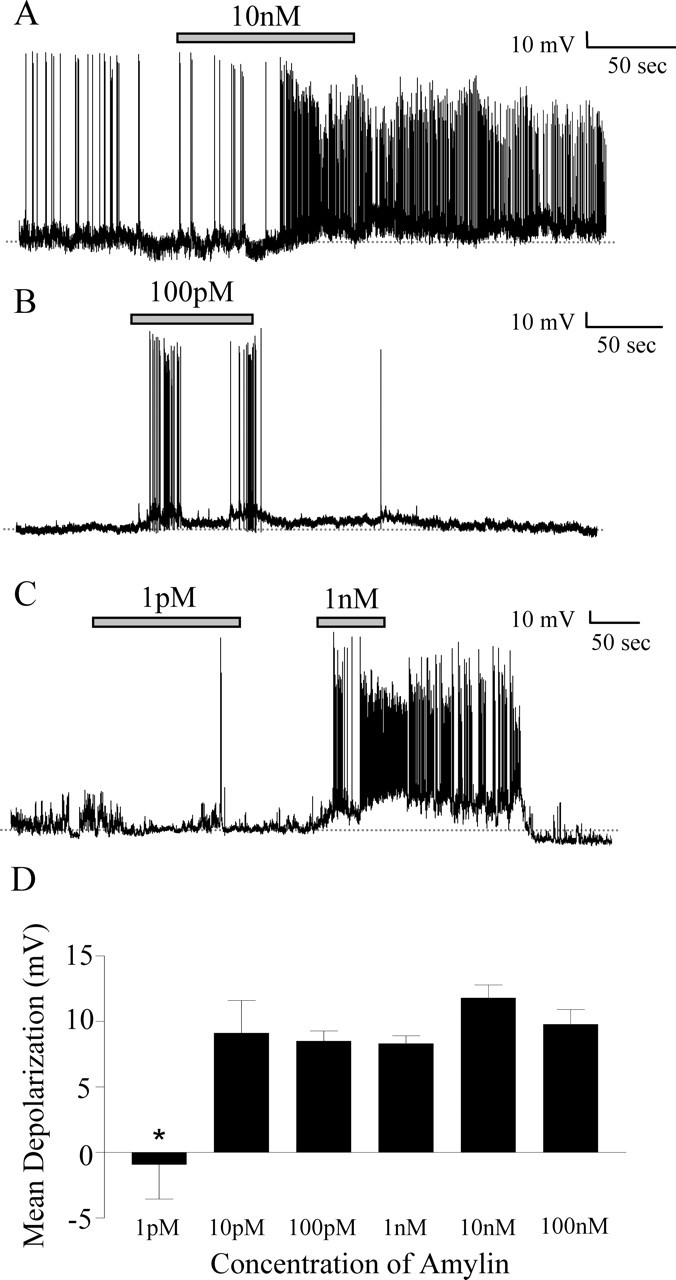
Effects of amylin are observed at different concentrations. A, Current-clamp recording showing a sustained depolarization in response to 10 nm amylin (gray bar). B, Current-clamp recording showing a response to 100 pm amylin (gray bar) that is smaller in magnitude than that seen at 10 nm (A). C, Current-clamp recording showing no change in membrane potential in response to 1 pm amylin (gray bar) and a subsequent reversible depolarization in response to 1 nm amylin (gray bar). D, The mean membrane potential changes in response to concentrations of amylin ranging from 100 nm to 1 pm. There was a significant difference between the mean depolarization at 1 pm and all other doses (*p < 0.05); however, there were no significant differences between doses ranging from 10 pm to 100 nm.
Interestingly, the magnitude of the amylin-induced depolarization in response to all concentrations ≥10 pm was similar, and there were no significant differences between the mean depolarization values at any of these concentrations (one-way ANOVA; p > 0.05) (Fig. 6D). The mean change at 1 pm amylin was significantly different from all other concentrations tested (one-way ANOVA; p < 0.05). The populations of responding neurons at each dose were not significantly different (χ2 test; p > 0.05), suggesting, as for ghrelin, an all-or-none membrane potential response to amylin with maximal effects observed at 10 pm.
Amylin does not influence NSCC in SFO neurons
Voltage-clamp experiments similar to those described above for ghrelin were performed to determine whether amylin effects on SFO neurons were also the result of activation of NSCC. Conductance changes in response to amylin were assessed with similar slow (10 mV/s) depolarizing voltage ramps (−100 to +20 mV) initiated after a hyperpolarizing prepulse to −100 mV for 500 ms. The currents recorded from a single SFO neuron in response to such voltage ramps in ACSF and in the presence of 10 nm amylin are illustrated in Figure 7A (each trace is a mean of five ramps). The amylin-induced current, calculated by subtracting control current from current in the presence of amylin, is shown in the inset. In contrast to the effects of ghrelin, the voltage-independent currents activated by this peptide were unaffected. A small increase in the net inward current (with a peak at approximately −5 mV), as well as a reduction in the net outward current from 0 to 20 mV, was observed in 4 of 11 neurons tested, with partial recovery in two affected neurons. The data presented in Figure 7B represent the mean amylin-induced current recorded from responsive neurons.
Figure 7.
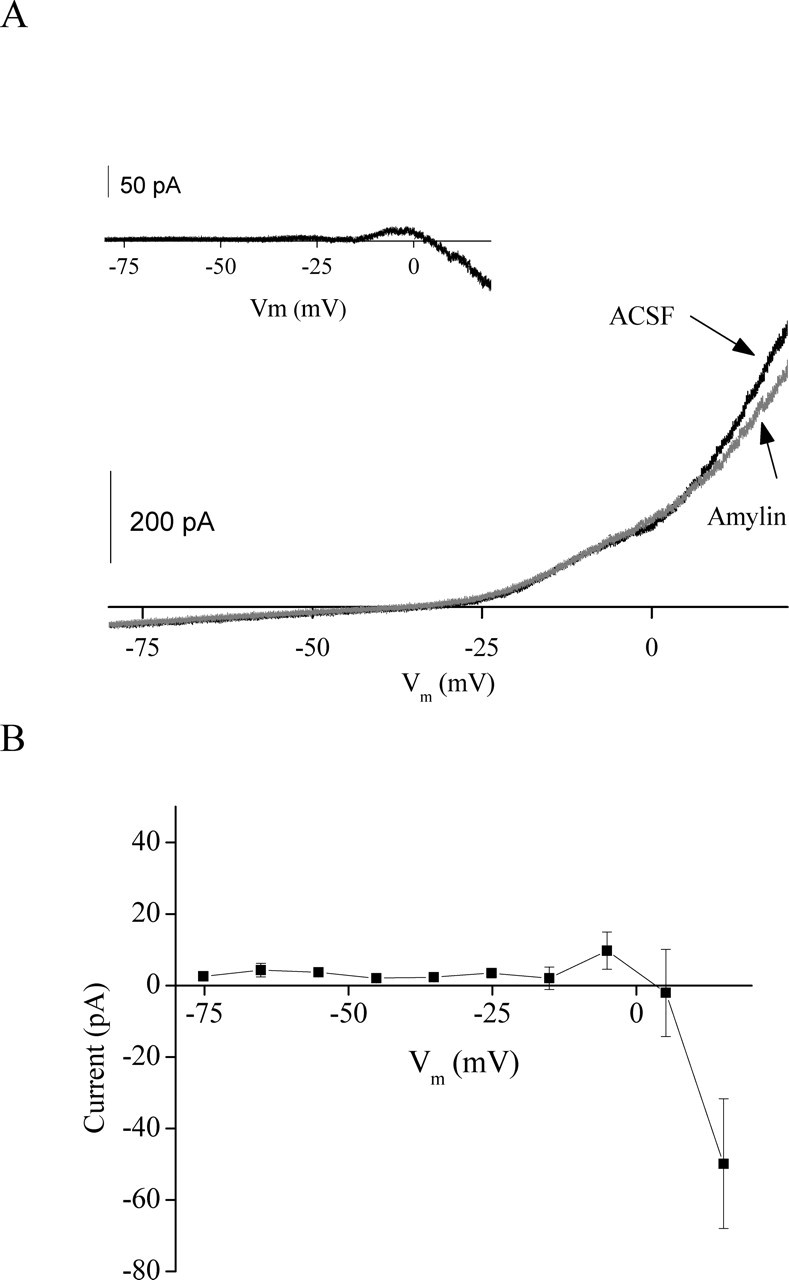
Amylin causes a net decrease in outward conductance at depolarized potentials. A, Graph represents current recorded in a single cell during voltage ramps from −80 to +20 mV (each trace is a mean of 5 ramps) in the presence of ACSF (black) and 10 nm amylin. The inset represents the difference current calculated by subtracting the amylin-induced current from the control current (ACSF) in the same cell. B, Graph represents the mean (±SEM) difference current measured in four responding cells.
Amylin and ghrelin activate separate subpopulations of SFO neurons
The experiments described above established that SFO neurons respond to both amylin and ghrelin with excitatory effects of similar magnitude, a somewhat surprising observation in view of the opposing roles that these substances have been proposed to play as feeding–satiety signals. We therefore hypothesized that separate subpopulations of SFO neurons may be ghrelin versus amylin responsive and tested this hypothesis by treating single SFO neurons with both 1 nm ghrelin and 1 nm amylin (doses that elicited maximal reversible responses in the above experiments) applied in either order. Recordings from nine ghrelin-responsive SFO neurons showed that none of these cells responded to amylin, whereas similar recordings from seven amylin-responsive neurons showed no effects of ghrelin (Fig. 8).
Figure 8.

Ghrelin- and amylin-responsive neurons represent two different populations of SFO neurons. A, Current-clamp recording shows a neuron that displays no change in membrane potential in the presence of 1 nm ghrelin (black bar); however, application of 1 nm amylin (gray bar) induces a depolarization. B, Current-clamp recording shows a neuron that depolarizes in response to 1 nm ghrelin (black bar) but does not demonstrate any change in response to 1 nm amylin (gray bar).
Discussion
The data presented in this study demonstrate that ghrelin and amylin influence the excitability of SFO neurons, supporting a role for SFO in sensing peripheral feeding signals. Our initial calcium-imaging studies identified actions of ghrelin on SFO neurons to increase [Ca2+]I, and our molecular identification of the mRNA for the GHSR in SFO supports the potential physiological relevance by demonstrating a potential for ghrelin receptor expression in SFO neurons in vivo as well as in culture. Our electrophysiological studies demonstrated depolarizing effects of ghrelin on a subpopulation of SFO neurons that appear to be a consequence of the opening of a nonselective cation channel. Amylin elicited similar excitatory effects on SFO neurons in concordance with previous extracellular recording studies (Riediger et al., 1999). The observation that these two peptides, which exert opposite effects on food intake, had similar effects on the excitability of SFO neurons suggested effects on separate subpopulations of SFO neurons. Our recordings testing the effects of both peptides on single neurons confirmed this hypothesis by showing that in all cases single cells were responsive to either ghrelin or amylin but never responded to both peptides. These results suggest the existence of two distinct populations of SFO neurons responsive to peptides with opposing effects on food intake. Interestingly, we found that the sensitivity of SFO neurons to different concentrations of ghrelin correlated well with plasma ghrelin levels in rats fed ad libitum (Date et al., 2000; Tschop et al., 2000), levels that increase in fasted rats and decrease after food intake in rats (Tschop et al., 2000; Bagnasco et al., 2002) and humans (Cummings et al., 2001; Tschop et al., 2001).
In our experiments, we used the calcium-imaging technique as an initial screening tool to assess the responsiveness of SFO neurons to ghrelin. The increase in [Ca2+]i seen in this study would be expected to be associated with neuronal activation, and, in support of our results, Kohno et al. (2003) demonstrated a similar increase in [Ca2+]i that is coupled to the activation of protein kinase A in neurons of the arcuate nucleus. Our observation that these effects are abolished in TTX supports the conclusion that such increases in [Ca2+]i result from action potential-induced opening of voltage-gated calcium channels and should not be interpreted as indicative of a prerequisite role for calcium in the signal transduction mechanisms underlying these effects.
Our hypothesis that the SFO is able to monitor changes in circulating ghrelin–amylin and in turn relay these signals to other hypothalamic centers is also supported by the concentration-dependent nature of the depolarizing effects of these peptides on single SFO neurons. We believe that the “all-or-none” nature of our concentration–response curves is most likely suggestive of the active ranges in which these peptides exert their physiological effects (100 pm–1 nm for ghrelin and 1–10 pm for amylin), although additional in vivo experiments would be necessary to confirm such a conclusion. In addition, the reversibility and repeatability of these responses in the same SFO neurons suggests that it is possible for information regarding changes in circulating ghrelin to be rapidly transmitted to other CNS sites. Concentration-dependent activation permits the SFO to sense the magnitude of changes in circulatory ghrelin levels that are coordinated with hunger and to regulate the strength of its input to the hypothalamus accordingly. The ability of the antagonist [d-Lys-3]-GHRP-6 to block the depolarizing response suggests that these excitatory effects are receptor mediated, whereas our RT-PCR data provide the first definitive data localizing the GHSR1a receptor in the SFO.
We examined the effects of ghrelin on specific ion channels with voltage-clamp techniques to further elucidate the mechanism through which ghrelin exerts its depolarizing effects. Currents recorded in response to slow voltage ramps demonstrate that ghrelin activates a voltage-independent current with a reversal potential of −49 mV, which is characteristic of an NSCC (Oliet and Bourque, 1993; Desson and Ferguson, 2003; Yang et al., 2004). On the basis of the properties of the conductance seen in our study, we can estimate that in a cell resting at −55 mV, ghrelin would induce a depolarization of 5–10 mV, depending on the input resistance of the cell. These theoretical values are similar to the magnitude of depolarization seen in this study. The activation of an NSCC could also theoretically act as a potential site of entry of calcium into the cell, which may contribute at least partly to the demonstrated increases in intracellular calcium. Amylin, in contrast to ghrelin, had no effect on such voltage-independent currents in response to slow voltage ramps; however, amylin did influence what appears to be a voltage-activated inward current at depolarized potentials (Fig. 7), which may represent actions on the persistent Na+ current that we have described previously in SFO neurons (Washburn et al., 2000). Importantly, these observations demonstrate separate mechanisms of action for these two peptides.
The opposing nature of ghrelin and amylin on feeding suggested that these peptides would exert excitatory effects on different SFO neurons. Electrophysiological recordings supported this hypothesis, indicating that the SFO consists of two distinct populations of neurons responsive to either peptide. It could be speculated that these populations may project to different anatomical regions within the hypothalamus, thus contributing to the regulation of feeding in an opposing manner. We are not currently able to identify the types of neurons that respond to these peptides, but similar research with angiotensin II (ANG), vasopressin, and interleukin-1β also demonstrated subpopulations of responsive SFO neurons (Ferguson and Bains, 1997; Washburn et al., 1999; Desson and Ferguson, 2003).
The physiological relevance of this responsiveness of SFO neurons to ghrelin–amylin has yet to be clearly elucidated. We speculate that ghrelin and amylin influence separate SFO neurons that project to hypothalamic areas such as the paraventricular nucleus and lateral hypothalamus or the arcuate nucleus, all of which are involved in regulating feeding. Further studies to determine the projection sites of affected SFO neurons will be necessary to test these hypotheses.
Date et al. (2002) demonstrated a prominent role for the vagus nerve in the transmission of gut signals to the brain in regulating food intake. Inhibition of this pathway showed a significant reduction, but not a complete abolishment, of ghrelin-induced food intake. An additional study has demonstrated that although vagal inputs are important for fasting-induced increases in circulating ghrelin, they do not play a role in short-term reductions in ghrelin levels after food intake (Williams et al., 2003). It is not surprising that such a sensitive and intricate network of physiological processes may be linked to more than one signaling pathway. It would seem logical that the SFO may be essential in sensing circulating ghrelin levels and in modifying its activity to reflect these short-term changes.
Until now, the SFO has been studied most extensively with regard to its role in the regulation of water intake and body fluid balance. Kozaka et al. (2003) demonstrated a reduction in water intake after intracranial injection of ghrelin in the eel model; however, this is the only study to date to examine the role of ghrelin in water intake. Further studies are required to demonstrate the potential role of ghrelin in controlling fluid balance through the SFO.
In contrast, amylin has been hypothesized to play a role in water intake by stimulating ANG-sensitive neurons that are well known for their dipsogenic influence on body fluid homeostasis (Simpson and Routtenberg, 1975; Simpson et al., 1978). Subcutaneous injections of amylin triggered a dipsogenic response that is likely to be mediated through these ANG-sensitive SFO neurons, thus leading to the conclusion that the excitatory effects of amylin mediate drinking (Riediger et al., 1999). Because of the strong association of these two ingestive behaviors (Fitzsimons, 1979), it is not surprising that peptides known to be secreted in correlation to feeding behaviors may also influence prandial drinking. The sites at which amylin has been shown to reduce food intake include the area postrema (Lutz et al., 2001) and nucleus accumbens (Baldo and Kelley, 2001).
In conclusion, we have demonstrated that ghrelin depolarizes and increases [Ca2+]i in SFO neurons, an effect that may be mediated through the activation of an NSCC. Amylin has similar depolarizing effects on SFO neurons; however, ghrelin and amylin affect different subpopulations of SFO neurons. These observations suggest an important role for the SFO in sensing circulating levels of peptides involved in regulating energy balance. In turn, the SFO may relay these gastrointestinal signals to many of the CNS centers that modulate feeding and drinking responses.
Footnotes
This work was supported by a grant to A.V.F. from the Canadian Institutes for Health Research. We thank Pauline Smith for expert technical assistance.
References
- Anderson JW, Smith PM, Ferguson AV (2001). Subfornical organ neurons projecting to paraventricular nucleus: whole-cell properties. Brain Res 921:78–85. [DOI] [PubMed] [Google Scholar]
- Bagnasco M, Kalra PS, Kalra SP (2002). Ghrelin and leptin pulse discharge in fed and fasted rats. Endocrinology 143:726–729. [DOI] [PubMed] [Google Scholar]
- Baldo BA, Kelley AE (2001). Amylin infusion into rat nucleus accumbens potently depresses motor activity and ingestive behaviour. Am J Physiol Regul Integr Comp Physiol 281:R1232–R1242. [DOI] [PubMed] [Google Scholar]
- Banks WA, Kastin AJ (1992). Bidirectional passage of peptides across the blood-brain barrier. Prog Brain Res 91:139–148. [DOI] [PubMed] [Google Scholar]
- Banks WA, Kastin AJ (1998). Differential permeability of the blood-brain barrier to two pancreatic peptides: insulin and amylin. Peptides 19:883–889. [DOI] [PubMed] [Google Scholar]
- Banks WA, Tschop M, Robinson SM, Heiman ML (2002). Extent and direction of ghrelin transport across the blood-brain barrier is determined by its unique primary structure. J Pharmacol Exp Ther 302:822–827. [DOI] [PubMed] [Google Scholar]
- Barreiro ML, Suominen JS, Gaytan F, Pinilla L, Chopin LK, Casanueva FF, Dieguez C, Aguilar E, Toppari J, Tena-Sempere M (2003). Developmental, stage-specific, and hormonally regulated expression of growth hormone secretagogue receptor messenger RNA in rat testis. Biol Reprod 68:1631–1640. [DOI] [PubMed] [Google Scholar]
- Butler PC, Chou J, Carter WB, Wang YN, Bu BH, Chang D, Chang JK, Rizza RA (1990). Effects of meal ingestion on plasma amylin concentration in NIDDM and nondiabetic humans. Diabetes 39:752–756. [DOI] [PubMed] [Google Scholar]
- Caminos JE, Tena-Sempere M, Gaytan F, Sanchez-Criado JE, Barreiro ML, Nogueiras R, Casanueva FF, Aguilar E, Dieguez C (2003). Expression of ghrelin in the cyclic and pregnant rat ovary. Endocrinology 144:1594–1602. [DOI] [PubMed] [Google Scholar]
- Cheng K, Chan WW, Barreto A Jr, Convey EM, Smith RG (1989). The synergistic effects of His-d-Trp-Ala-Trp-d-Phe-Lys-NH2 on growth hormone (GH)-releasing factor-stimulated GH release and intracellular adenosine 3′,5′-monophosphate accumulation in rat primary pituitary cell culture. Endocrinology 124:2791–2798. [DOI] [PubMed] [Google Scholar]
- Cottrell GT, Ferguson AV (2004). Sensory circumventricular organs: central roles in integrated autonomic regulation. Regul Pept 117:11–23. [DOI] [PubMed] [Google Scholar]
- Cottrell GT, Zhou QY, Ferguson AV (2004). Prokineticin 2 modulates the excitability of subfornical organ neurons. J Neurosci 24:2375–2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowley MA, Smith RG, Diano S, Tschop M, Pronchuk N, Grove KL, Strasburger CJ, Bidlingmaier M, Esterman M, Heiman ML, Garcia-Segura LM, Nillni EA, Mendez P, Low MJ, Sotonyi P, Friedman JM, Liu H, Pinto S, Colmers WF, Cone RD, et al (2003). The distribution and mechanism of action of ghrelin in the CNS demonstrates a novel hypothalamic circuit regulating energy homeostasis. Neuron 37:649–661. [DOI] [PubMed] [Google Scholar]
- Cummings DE, Purnell JQ, Frayo RS, Schmidova K, Wisse BE, Weigle DS (2001). A preprandial rise in plasma ghrelin levels suggests a role in meal initiation in humans. Diabetes 50:1714–1719. [DOI] [PubMed] [Google Scholar]
- Date Y, Kojima M, Hosoda H, Sawaguchi A, Mondal MS, Suganuma T, Matsukura S, Kangawa K, Nakazato M (2000). Ghrelin, a novel growth hormone-releasing acylated peptide, is synthesized in a distinct endocrine cell type in the gastrointestinal tracts of rats and humans. Endocrinology 141:4255–4261. [DOI] [PubMed] [Google Scholar]
- Date Y, Murakami N, Toshinai K, Matsukura S, Niijima A, Matsuo H, Kangawa K, Nakazato M (2002). The role of the gastric afferent vagal nerve in ghrelin-induced feeding and growth hormone secretion in rats. Gastroenterology 123:1120–1128. [DOI] [PubMed] [Google Scholar]
- Desson SE, Ferguson AV (2003). Interleukin 1beta modulates rat subfornical organ neurons as a result of activation of a non-selective cationic conductance. J Physiol (Lond) 550:113–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson AV, Bains JS (1997). Actions of angiotensin in the subfornical organ and area postrema: implications for long term control of autonomic output. Clin Exp Pharmacol Physiol 24:96–101. [DOI] [PubMed] [Google Scholar]
- Ferguson AV, Bicknell RJ, Carew MA, Mason WT (1997). Dissociated adult rat subfornical organ neurons maintain membrane properties and angiotensin responsiveness for up to 6 days. Neuroendocrinology 66:409–415. [DOI] [PubMed] [Google Scholar]
- Fitzsimons JT (1979). In: The physiology of thirst and sodium appetite Cambridge, UK: Cambridge UP. [PubMed]
- Gruber K, McRae-Degueurce A, Wilkin LD, Mitchell LD, Johnson AK (1987). Forebrain and brainstem afferents to the arcuate nucleus in the rat: potential pathways for the modulation of hypophyseal secretions. Neurosci Lett 75:1–5. [DOI] [PubMed] [Google Scholar]
- Kamegai J, Tamura H, Shimizu T, Ishii S, Sugihara H, Wakabayashi I (2001). Chronic central infusion of ghrelin increases hypothalamic neuropeptide Y and Agouti-related protein mRNA levels and body weight in rats. Diabetes 50:2438–2443. [DOI] [PubMed] [Google Scholar]
- Kohno D, Gao HZ, Muroya S, Kikuyama S, Yada T (2003). Ghrelin directly interacts with neuropeptide-Y-containing neurons in the rat arcuate nucleus: Ca2+ signaling via protein kinase A and N-type channel-dependent mechanisms and cross-talk with leptin and orexin. Diabetes 52:948–956. [DOI] [PubMed] [Google Scholar]
- Kojima M, Hosoda H, Date Y, Nakazato M, Matsuo H, Kangawa K (1999). Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature 402:656–660. [DOI] [PubMed] [Google Scholar]
- Korbonits M, Bustin SA, Kojima M, Jordan S, Adams EF, Lowe DG, Kangawa K, Grossman AB (2001). The expression of the growth hormone secretagogue receptor ligand ghrelin in normal and abnormal human pituitary and other neuroendocrine tumors. J Clin Endocrinol Metab 86:881–887. [DOI] [PubMed] [Google Scholar]
- Kozaka T, Fujii Y, Ando M (2003). Central effects of various ligands on drinking behavior in eels acclimated to seawater. J Exp Biol 206:687–692. [DOI] [PubMed] [Google Scholar]
- le Roux CW, Neary NM, Halsey TJ, Small CJ, Martinez-Isla AM, Ghatei MA, Theodorou NA, Bloom SR (2005). Ghrelin does not stimulate food intake in patients with surgical procedures involving vagotomy. J Clin Endocrinol Metab 90:4521–4524. [DOI] [PubMed] [Google Scholar]
- Lind RW, Van Hoesen GW, Johnson AK (1982). An HRP study of the connections of the subfornical organ of the rat. J Comp Neurol 210:265–277. [DOI] [PubMed] [Google Scholar]
- Lu S, Guan JL, Wang QP, Uehara K, Yamada S, Goto N, Date Y, Nakazato M, Kojima M, Kangawa K, Shioda S (2002). Immunocytochemical observation of ghrelin-containing neurons in the rat arcuate nucleus. Neurosci Lett 321:157–160. [DOI] [PubMed] [Google Scholar]
- Lutz TA, Del Prete E, Scharrer E (1994). Reduction of food intake in rats by intraperitoneal injection of low doses of amylin. Physiol Behav 55:891–895. [DOI] [PubMed] [Google Scholar]
- Lutz TA, Geary N, Szabady MM, Del Prete E, Scharrer E (1995). Amylin decreases meal size in rats. Physiol Behav 58:1197–1202. [DOI] [PubMed] [Google Scholar]
- Lutz TA, Mollet A, Rushing PA, Riediger T, Scharrer E (2001). The anorectic effect of a chronic peripheral infusion of amylin is abolished in area postrema/nucleus of the solitary tract (AP/NTS) lesioned rats. Int J Obes Relat Metab Disord 25:1005–1011. [DOI] [PubMed] [Google Scholar]
- McKee KK, Palyha OC, Feighner SD, Hreniuk DL, Tan CP, Phillips MS, Smith RG, Van der Ploeg LH, Howard AD (1997). Molecular analysis of rat pituitary and hypothalamic growth hormone secretagogue receptors. Mol Endocrinol 11:415–423. [DOI] [PubMed] [Google Scholar]
- McKinley MJ, Allen AM, Burns P, Colvill LM, Oldfield BJ (1998). Interaction of circulating hormones with the brain: the roles of the subfornical organ and the organum vasculosum of the lamina terminalis. Clin Exp Pharmacol Physiol [Suppl] 25:S61–S67. [DOI] [PubMed] [Google Scholar]
- Miselis RR (1981). The efferent projections of the subfornical organ of the rat: a circumventricular organ within a neural network subserving water balance. Brain Res 230:1–23. [DOI] [PubMed] [Google Scholar]
- Miselis RR (1982). The subfornical organ's neural connections and their role in water balance. Peptides 3:501–502. [DOI] [PubMed] [Google Scholar]
- Mori K, Yoshimoto A, Takaya K, Hosoda K, Ariyasu H, Yahata K, Mukoyama M, Sugawara A, Hosoda H, Kojima M, Kangawa K, Nakao K (2000). Kidney produces a novel acylated peptide, ghrelin. FEBS Lett 486:213–216. [DOI] [PubMed] [Google Scholar]
- Morley JE, Flood JF (1991). Amylin decreases food intake in mice. Peptides 12:865–869. [DOI] [PubMed] [Google Scholar]
- Morley JE, Flood JF, Horowitz M, Morley PM, Walter MJ (1994). Modulation of food intake by peripherally administered amylin. Am J Physiol 267:R178–R184. [DOI] [PubMed] [Google Scholar]
- Nakazato M, Murakami N, Date Y, Kojima M, Matsuo H, Kangawa K, Matsukura S (2001). A role for ghrelin in the central regulation of feeding. Nature 409:194–198. [DOI] [PubMed] [Google Scholar]
- Oliet SHR, Bourque CW (1993). Steady-state osmotic modulation of cationic conductance in neurons of rat supraoptic nucleus. Am J Physiol 265:R1475–R1479. [DOI] [PubMed] [Google Scholar]
- Perez-Velazquez JL, Zhang L (1994). In vitro hypoxia induces expression of the NR2C subunit of the NMDA receptor in rat cortex and hippocampus. J Neurochem 63:1171–1173. [DOI] [PubMed] [Google Scholar]
- Pieber TR, Roitelman J, Lee Y, Luskey KL, Stein DT (1994). Direct plasma radioimmunoassay for rat amylin-(1–37): concentrations with acquired and genetic obesity. Am J Physiol 267:E156–E164. [DOI] [PubMed] [Google Scholar]
- Riediger T, Rauch M, Schmid HA (1999). Actions of amylin on subfornical organ neurons and on drinking behavior in rats. Am J Physiol 276:R514–R521. [DOI] [PubMed] [Google Scholar]
- Riediger T, Schmid HA, Lutz T, Simon E (2001). Amylin potently activates AP neurons possibly via formation of the excitatory second messenger cGMP. Am J Physiol Regul Integr Comp Physiol 281:R1833–R1843. [DOI] [PubMed] [Google Scholar]
- Rushing PA, Hagan MM, Seeley RJ, Lutz TA, Woods SC (2000). Amylin: a novel action in the brain to reduce body weight. Endocrinology 141:850–853. [DOI] [PubMed] [Google Scholar]
- Rushing PA, Hagan MM, Seeley RJ, Lutz TA, D’Alessio DA, Air EL, Woods SC (2001). Inhibition of central amylin signaling increases food intake and body adiposity in rats. Endocrinology 142:5035. [DOI] [PubMed] [Google Scholar]
- Seoane LM, Tovar S, Baldelli R, Arvat E, Ghigo E, Casanueva FF, Dieguez C (2000). Ghrelin elicits a marked stimulatory effect on GH secretion in freely-moving rats. Eur J Endocrinol 143:R7–R9. [DOI] [PubMed] [Google Scholar]
- Sexton PM, Paxinos G, Kenney MA, Wookey PJ, Beaumont K (1994). In vitro autoradiographic localization of amylin binding sites in rat brain. Neuroscience 62:553–567. [DOI] [PubMed] [Google Scholar]
- Silverman AJ, Hoffman DL, Zimmerman EA (1981). The descending afferent connections of the paraventricular nucleus of the hypothalamus (PVN). Brain Res Bull 6:47–61. [DOI] [PubMed] [Google Scholar]
- Simpson JB, Routtenberg JB (1975). Subfornical organ lesions reduce intravenous angiotensin-induced drinking. Brain Res 88:154–161. [DOI] [PubMed] [Google Scholar]
- Simpson JB, Epstein AN, Camardo JS (1978). Localization of receptors for dipsogenic action of angiotensin II in subfornical organ of rat. J Comp Physiol Psychol 92:581–608. [DOI] [PubMed] [Google Scholar]
- Smith RG, Cheng K, Schoen WR, Pong SS, Hickey G, Jacks T, Butler B, Chan WW, Chaung LY, Judith F (1993). A nonpeptidyl growth hormone secretagogue. Science 260:1640–1643. [DOI] [PubMed] [Google Scholar]
- Tschop M, Smiley DL, Heiman ML (2000). Ghrelin induces adiposity in rodents. Nature 407:908–913. [DOI] [PubMed] [Google Scholar]
- Tschop M, Wawarta R, Riepl RL, Friedrich S, Bidlingmaier M, Landgraf R, Folwaczny C (2001). Post-prandial decrease of circulating human ghrelin levels. J Endocrinol Invest 24:RC19–RC21. [DOI] [PubMed] [Google Scholar]
- Volante M, Allia E, Gugliotta P, Funaro A, Broglio F, Deghenghi R, Muccioli G, Ghigo E, Papotti M (2002). Expression of ghrelin and of the GH secretagogue receptor by pancreatic islet cells and related endocrine tumors. J Clin Endocrinol Metab 87:1300–1308. [DOI] [PubMed] [Google Scholar]
- Washburn DLS, Beedle AM, Ferguson AV (1999). Inhibition of subfornical organ neuronal potassium channels by vasopressin. Neuroscience 93:349–359. [DOI] [PubMed] [Google Scholar]
- Washburn DLS, Anderson JW, Ferguson AV (2000). A subthreshold persistent sodium current mediates bursting in rat subfornical organ neurones. J Physiol (Lond) 529:359–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams DL, Grill HJ, Cummings DE, Kaplan JM (2003). Vagotomy dissociates short- and long-term controls of circulating ghrelin. Endocrinology 144:5184–5187. [DOI] [PubMed] [Google Scholar]
- Wren AM, Small CJ, Ward HL, Murphy KG, Dakin CL, Taheri S, Kennedy AR, Roberts GH, Morgan DG, Ghatei MA, Bloom SR (2000). The novel hypothalamic peptide ghrelin stimulates food intake and growth hormone secretion. Endocrinology 141:4325–4328. [DOI] [PubMed] [Google Scholar]
- Wren AM, Small CJ, Abbott CR, Dhillo WS, Seal LJ, Cohen MA, Batterham RL, Taheri S, Stanley SA, Ghatei MA, Bloom SR (2001). Ghrelin causes hyperphagia and obesity in rats. Diabetes 50:2540–2547. [DOI] [PubMed] [Google Scholar]
- Yang B, Ferguson AV (2002). Orexin-A depolarizes dissociated rat area postrema neurons through activation of a nonselective cationic conductance. J Neurosci 22:6303–6308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang B, Goulet M, Boismenu R, Ferguson AV (2004). Secretin depolarizes nucleus tractus solitarius neurons through activation of a nonselective cationic conductance. Am J Physiol Regul Integr Comp Physiol 286:R927–R934. [DOI] [PubMed] [Google Scholar]