

Abstract
Consumption of alcoholic beverages produces alterations in motor coordination and equilibrium that are responsible for millions of accidental deaths. Studies indicate that ethanol produces these alterations by affecting the cerebellum, a brain region involved in the control of motor systems. Purkinje neurons of the cerebellar cortex have been shown to be particularly important targets of ethanol. However, its mechanism of action at these neurons is poorly understood. We hypothesized that ethanol could modulate Purkinje neuron function by altering the excitatory input provided by the climbing fiber from the inferior olive, which evokes a powerful all-or-none response denoted as the complex spike. To test this hypothesis, we performed whole-cell patch-clamp electrophysiological and Ca2+ imaging experiments in acute slices from rat cerebella. We found that ethanol potently inhibits the late phase of the complex spike and that this effect is the result of inhibition of type-1 metabotropic glutamate receptor-dependent responses at the postsynaptic level. Moreover, ethanol inhibited climbing fiber long-term depression, a form of synaptic plasticity that also depends on activation of these metabotropic receptors. Our findings identify the climbing fiber→Purkinje neuron synapse as an important target of ethanol in the cerebellar cortex and indicate that ethanol significantly affects cerebellar circuits even at concentrations as low as 10 mm (legal blood alcohol level in the United States is below 0.08 g/dl = 17 mm).
Keywords: cerebellum, electrophysiology, slice, glutamate, motor, ethanol
Introduction
Acute and chronic ethanol (EtOH) exposure produces profound impairments in motor functioning. Acutely, EtOH causes motor control alterations, ataxia, and vertigo, increasing the risk of accidental injuries, which are responsible for millions of deaths around the world (Dinh-Zarr et al., 2004). Individuals with lower sensitivity to the acute motor impairing effects of EtOH have an increased risk of developing alcohol dependence and abuse (Schuckit, 1994), and infants with subtle delays in motor coordination development may have an increased risk for subsequently developing alcoholism (Manzardo et al., 2005). Chronically, EtOH produces alterations in the mature nervous system leading to progressive impairments in stance and gait (Diamond and Messing, 1994), and developmental exposure induces long-lasting motor deficits that cause significant disability and require intensive rehabilitation (Klintsova et al., 1998, 2002; Autti-Ramo et al., 2002; Coffin et al., 2005). Thus, understanding the mechanism by which EtOH disrupts motor functioning is paramount.
Studies suggest that the motor impairing effects of EtOH are, in part, a consequence of cerebellar function alterations. The Purkinje neuron (PN) is an important target of EtOH (Siggins and French, 1979; Rogers et al., 1980; Sorensen et al., 1980; Basile et al., 1983), and evidence indicates that its effects are in part mediated by an increase in GABAergic transmission (Lin et al., 1994; Lee et al., 1995; Freund and Palmer, 1997; Yang et al., 2000). Recent studies indicate that EtOH increases GABAergic tone at molecular layer interneuron→PN (Criswell and Breese, 2005; Mameli et al., 2005a) and Golgi→granule cell synapses (Carta et al., 2004; Hanchar et al., 2005; Valenzuela et al., 2005). Thus, alcohol affects mossy fiber–parallel fiber excitatory input to PNs via an increase in inhibitory neurotransmission at different relay points along this pathway.
A mature PN also receives excitatory input from the inferior olive via a single climbing fiber (CF). This pathway provides powerful input via ∼1500 synapses that release glutamate with high release probability at the proximal portion of the dendritic tree (Schmolesky et al., 2002). CF activation elicits a unique response known as the complex spike (CS) (see Fig. 1). This response is triggered by activation of AMPA receptors (AMPARs), which evoke an all-or-none EPSP that activates dendritic voltage-gated Ca2+ channels (VGCCs). Depolarization then spreads to the soma and axon, activating voltage-gated Na+ channels and generating the first spike of the CS. Evidence suggests that the depolarization plateau and spikelets that follow the first spike are mediated by Ca2+ and resurgent Na+ currents (Schmolesky et al., 2002). However, the precise identity of the slow CS components remains to be determined.

EtOH modulates the late phase of the CS. A1, B1, Sample traces illustrating the effect of 50 mm EtOH on the late phase of the CS. The corresponding cumulative CS integrals are shown in the insets. A2, B2, The same traces are shown at a more compressed time scale to further illustrate the effect of EtOH. Note that the timing but not the amplitude of the afterhyperpolarization is altered. Ctrl, Control. C, Time course of the effect of 50 mm EtOH (n = 16–18 neurons). D, Summary of the effect of increasing concentrations of EtOH. n = 6 (10 mm), 6 (25 mm), 18 (50 mm), 7 (75 mm). *p < 0.05; ***p < 0.001 by one-sample t test versus a theoretical mean of 100. Error bars represent SEM. See Results for one-way ANOVA.
CF activation of PNs plays a central role in motor adaptation and learning by communicating an error signal that depresses concomitantly active parallel fiber inputs, allowing appropriate motor activity to occur (Hansel et al., 2001). CF input is thought to encode the unconditioned stimulus in pavlovian eyeblink conditioning, and EtOH acutely modulates this paradigm (Hernandez et al., 1986). We hypothesized that CF input to PNs could be affected by acute EtOH exposure. To test this, we used patch-clamp electrophysiological and Ca2+ imaging techniques in rat cerebellar slices.
Materials and Methods
Unless indicated, all chemicals were from Sigma (St. Louis, MO). Experiments were performed in parasagittal vermis cerebellar slices, which were prepared from 20- to 30-d-old male Sprague Dawley rats (Harlan, Indianapolis, IN) as described previously (Carta et al., 2004). Artificial CSF (ACSF) contained the following (in mm): 126 NaCl, 3 KCl, 1.25 NaH2PO4, 1 MgSO4, 26 NaHCO3, 2 CaCl2, and 10 glucose equilibrated with 95% O2 plus 5% CO2. After a recovery time of ≥80 min, slices were transferred to a chamber perfused with ACSF containing 20 μm bicuculline methiodide at a rate of 2–3 ml/min. When indicated, EtOH (AAPER Chemical, Shelbyville, KY), 2,3-dihydroxy-6-nitro-7-sulfamoylbenzo[f]quinoxaline (NBQX), (+)-2-methyl-4-carboxyphenylglycine (LY367385), and/or dl-threo-β-benzyloxyaspartate (TBOA) (all from Tocris Cookson, Ellisville, MO) were added to the ACSF.
Whole-cell patch-clamp electrophysiological recordings were performed under infrared-differential interference contrast microscopy at 31–32°C with Axopatch 200B or Multiclamp 700B amplifiers (Molecular Devices, Sunnyvale, CA). Data were acquired and analyzed with pClamp-9 (Molecular Devices). The effect of ethanol on membrane proteins is sensitive to changes in intracellular signaling pathways, which are affected during whole-cell patch-clamp recordings. This uncertainty must be kept in mind when interpreting our results. Stimulation was performed with a concentric bipolar electrode (Frederick Haer Company, Bowdoinham, ME) placed in the granule cell layer near the patched PN. Constant-current steps (50–100 μA per 100 μs) were delivered every 20 s. The location of the stimulating electrode was gently adjusted until all-or-none CF responses were observed. Patch pipettes had resistances of 3–5 MΩ. Access resistances were between 20 and 35 MΩ; if access resistance changed >20%, the recording was discarded.
CSs were recorded in the whole-cell current-clamp configuration using an internal solution containing the following (in mm): 135 K-gluconate, 10 MgCl2, 0.1 CaCl2, 1 EGTA, 10 HEPES, pH 7.3, and 2 Na2-ATP. Whole-cell voltage-clamp recordings of type-1 metabotropic glutamate receptor (mGluR1)-dependent EPSCs were recorded using the same internal solution at −65 mV. Whole-cell voltage-clamp recordings of AMPAR-mediated EPSCs were performed at a holding potential between −40 and −10 mV using an internal solution containing the following (in mm): 60 CsCl, 30 Cs d-gluconate, 20 tetraethylammonium-Cl, 20 EGTA, 4 MgCl2, 4 ATP, and 30 HEPES, pH 7.3, adjusted with CsOH.
For the long-term depression (LTD) studies, the internal solution contained the following (in mm): 128 CsOH, 111 gluconic acid, 4 NaOH, 10 CsCl, 2 MgCl2, 10 HEPES, 4 Na2ATP, 30 sucrose, and 4 Lidocaine N-ethyl bromide quaternary salt, pH 7.25. LTD was induced by stimulating the CF at 5 Hz for 30 s, as described previously (Weber et al., 2003).
Paired-pulse experiments were performed at an interpulse interval of 50 ms; the paired-pulse ratio (PPR) was calculated as the ratio between the second and first EPSCs. For some PPR determinations, the concentrations of CaCl2 and MgSO4 in the ACSF were changed to 0.5 and 2.5 mm, respectively, to lower glutamate release probability.
Calcium imaging experiments were performed with patch pipettes back-filled with an internal solution containing the following (in mm): 9 KCl, 10 KOH, 120 K-gluconate, 3.48 MgCl2, 10 HEPES, pH 7.25, 4 NaCl, 4 Na2ATP, 17.5 sucrose, and fura-2 AM (100–150 μm; Invitrogen, Carlsbad, CA). Twenty to 30 min were allowed for the dye to diffuse distally and reach a steady-state level. Ca2+ measurements were made by ratio imaging of fura-2 AM using 350/380 nm excitation from a monochrometer (TILL Photonics, Gräfelfig, Germany). Emission was collected using a cooled CCD camera system using DCLP 410 dichroic and LP510/80 emission filters (Chroma Technology, Rockingham, VT). Images were acquired every minute. Each acquisition consisted of ∼20 frames (individual exposure time was 20 ms) at an acquisition frequency of 10 Hz. Data acquisition and analysis were performed with TILLvisION imaging software (TILL Photonics). Fluorescence changes were normalized to resting levels and expressed as ΔF/F, where ΔF = F350/F380, and F is the fluorescence obtained during baseline before CF activation (average of 8–12 frames).
Each slice was exposed once to a single EtOH concentration, and the duration of EtOH exposure was limited to 5 min in all cases to avoid the development of rapid tolerance (Miyakawa et al., 1997). In all cases, effects of EtOH were calculated with respect to the average of control and washout responses. Data are presented as mean ± SEM.
Results
Stimulation of CFs elicited CSs with duration of 140 ± 10 ms and amplitude of the fast Na+ spike of 72 ± 0.8 mV (n = 30). The average number of secondary spikelets was 2.6 ± 0.2 per CS (n = 30). Acute exposure to 50 mm EtOH decreased the area of the CS plateau without affecting the amplitude of the fast Na+ spike (Fig. 1A1,B1). Traces at a more compressed time further illustrate the EtOH-induced decrease in CS area (Fig. 1A2,B2) and also show that the afterhyperpolarization that typically follows the CS (amplitude, −3.2 ± 0.2 mV; n = 22) was not significantly affected by 50 mm EtOH (−8.02 ± 6.8% of control; n = 9). The onset of the effect of EtOH was evident as early as 1 min after the start of application, reaching a maximum 5 min later (Fig. 1C). CS area returned to baseline levels ∼10 min after termination of the application. The effect of EtOH was dose dependent, producing a significant effect even at 10 mm (Fig. 1D). One-way ANOVA followed by Dunnett’s post hoc test revealed statistically significant differences between 10 and 75 mm EtOH (p < 0.05). EtOH (10–75 mm) did not significantly affect the resting membrane potential or the amplitude of the fast Na+ spike (data not shown).
To assess whether EtOH produced changes in the Ca2+ transients associated with the CS, simultaneous Ca2+ imaging and electrophysiological recordings were performed in a subset of neurons used to collect the data shown in Figure 1C. We measured Ca2+ transients in soma, proximal dendrites, and distal dendrites (Fig. 2A) and found that application of 50 mm EtOH did not significantly affect the amplitude or kinetics of these transients (Fig. 2B,C).

EtOH does not affect Purkinje cell Ca2+ transients elicited by CF stimulation. A, Image of a PN filled with 100–150 μm fura-2 AM via the patch electrode illustrating the areas used for analysis. B1–B4, Averaged Ca2+ transients elicited in the soma, a proximal dendrite, and two distal dendrites in the absence and presence of EtOH (n = 6 neurons). The averages of transients obtained 1–4 min [control (ctrl)] and 6–9 min (EtOH) after initiation of recording are shown. Electrophysiological recordings were simultaneously obtained from these six neurons, and these revealed an effect of EtOH on CS area in all cases (data shown as part of Fig. 1C). C, Lack of an effect of EtOH on Ca2+ transient amplitude in a distal dendrite. Error bars represent SEM.
It was shown recently that CF glutamate release activates mGluR1, which mediates an EPSC (Dzubay and Otis, 2002). We hypothesized that this EPSC could contribute to the late phase of the CS. Consequently, we examined the effect of the mGluR1 antagonist LY367385 (50 μm) on the CS. Like EtOH, this agent reduced the CS area and duration without affecting the fast Na+ spike (Fig. 3A–C). The effect of this antagonist on the CS area was greater than that of 50 mm EtOH (compare Figs. 1C,D, 3B,C). Importantly, LY367385 occluded the effect of EtOH (Fig. 3B,C).
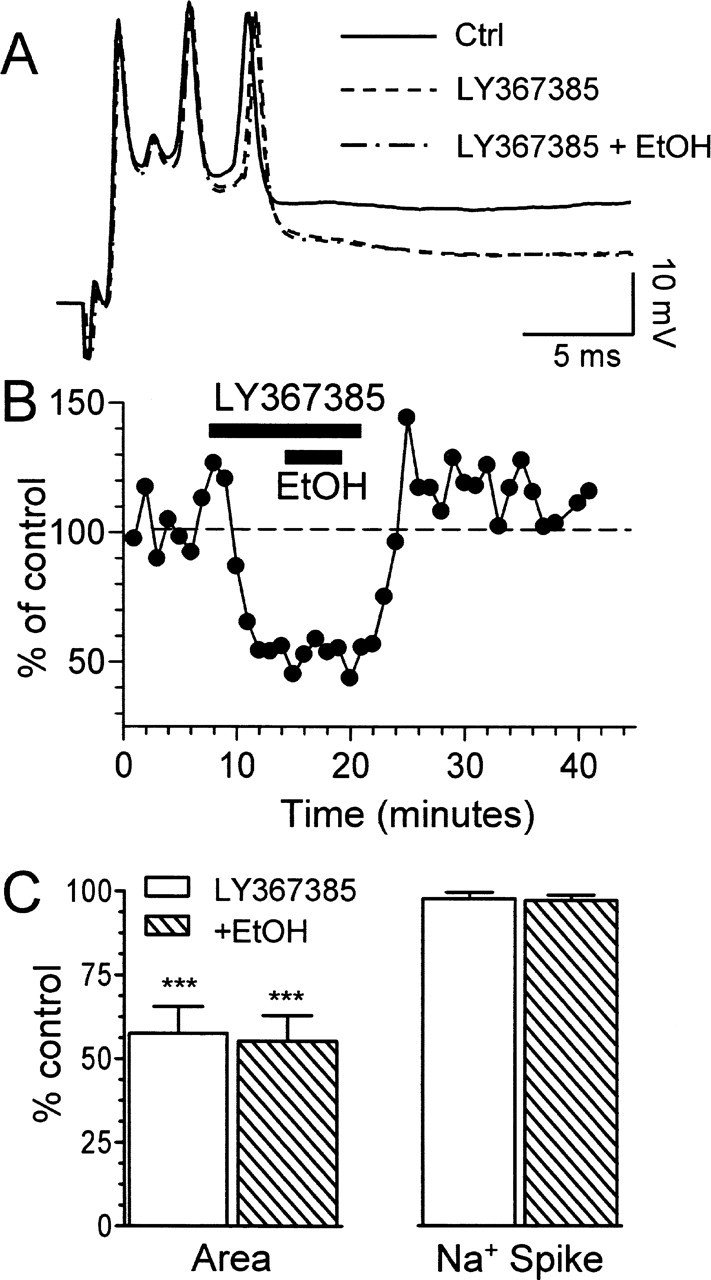
Blockade of mGluR1 mimics and occludes the effect of EtOH on CS area. A, Effect of the mGluR1 antagonist LY367385 (50 μm) in the absence and presence of 50 mm EtOH. Ctrl, Control. B, Time course graph corresponding to the same recording shown in A. C, Effects of LY367385 on CS area and amplitude of the fast Na+ spike in the absence and presence of 50 mm EtOH (n = 4–6; ***p < 0.001 by one-sample t test vs a theoretical mean of 100). Error bars represent SEM.
We next measured the effect of EtOH on EPSCs mediated by mGluR1. In agreement with a previous report (Dzubay and Otis, 2002), these EPSCs could be evoked by stimulating the CF with a train of four pulses at a frequency of 20 Hz in the presence of 10 μm NBQX and 20 μm bicuculline (Fig. 4). These EPSCs were abolished by LY367385, confirming that they depend on mGluR1 activation (Fig. 4A,B,D). Application of 50 mm EtOH decreased the amplitude of these events by ∼70% (Fig. 4A,B,D). As reported previously (Dzubay and Otis, 2002), the glutamate transporter inhibitor TBOA (100 μm) increased the amplitude of the mGluR1-dependent EPSCs (Fig. 4C). EtOH decreased the amplitude of the mGluR1-dependent EPSCs to a similar extent in the absence and presence of TBOA (Fig. 4D). In contrast to a recent report (Zhu et al., 2005), we did not observe enhancement of the EPSC under conditions of mGluR1 antagonism in any of the cells tested (Fig. 4D).

EtOH inhibits mGluR1-dependent EPSCs evoked by CF stimulation in PNs. A, Stimulation of CFs with a train of four pulses at a frequency of 20 Hz in the presence of 10 μm NBQX and 20 μm bicuculline elicited EPSCs that were inhibited by EtOH (50 mm). These EPSCs were abolished by the mGluR1 antagonist LY367385 (50 μm). B, Time course of the effect of EtOH and LY367385 for the same cell shown in A. C, Sample traces of recordings obtained as described in A but in the presence of the glutamate transporter inhibitor TBOA (100 μm). D, Effect of 50 mm EtOH on the amplitude of mGluR1-dependent EPSCs in the absence (n = 5) and presence (n = 4) of TBOA. The effect of 50 μm LY367385 is also shown. Because the effect of LY367385 was the same in the absence and presence of TBOA, the data were combined (n = 9). **p < 0.01 versus EtOH and EtOH plus TBOA by one-way ANOVA followed by Tukey’s post hoc test. A one-sample t test revealed that all sets of data were significantly different from 100% (p < 0.006; data not shown). ctrl, Control. Error bars represent SEM.
We next investigated whether EtOH modulates mGluR1 function via a presynaptic mechanism by measuring its effect on paired-pulse plasticity. Pharmacologically isolated AMPAR-mediated fast EPSCs evoked by CF stimulation displayed paired-pulse depression, consistent with the high release probability of these fibers (Fig. 5). The EPSCs were abolished by 10 μm NBQX (n = 4; data not shown). Application of 50 mm EtOH did not significantly affect either the amplitude of the EPSCs or the PPR (Fig. 5A,B). An effect of 50 mm EtOH on the PPR or EPSC amplitude could not be observed, even under conditions of low probability of glutamate release (i.e., low extracellular Ca2+ levels) (Fig. 5C–F).

EtOH does not affect the PPR of AMPAR-mediated EPSCs evoked by CF stimulation. A, CF stimulation with pairs of stimuli at an interpulse interval of 50 ms elicited EPSCs that exhibited paired-pulse depression. These events were inhibited by NBQX (10 μm; data not shown). EtOH did not affect either the PPR or the amplitude of the first EPSC. B, Lack of an effect of EtOH on these parameters (n = 8). Events were normalized with respect to the initial PPR or amplitude (i.e., first 3 min of recording). C, Lowering the extracellular Ca2+ concentration to 0.5 mm decreased paired-pulse depression. EtOH did not affect either the PPR or the amplitude of the first EPSC, even under these conditions of low glutamate release probability. D, Effect of lowering extracellular Ca2+ levels on the PPR (n = 7). E, Lack of an effect of EtOH on the PPR under low Ca2+ conditions (n = 7). The error bar for the control group is not visible. F, Effect of lowering extracellular Ca2+ levels on the amplitude of the first EPSC (n = 7) and lack of an effect of EtOH on EPSC amplitude under low Ca2+ conditions (n = 7). Ctrl, Control; Reg, regular; W/O, washout. Error bars represent SEM.
Finally, we assessed the effect of EtOH on CF LTD, a form of synaptic plasticity that requires activation of mGluR1 (Hansel and Linden, 2000). Shown in Figure 6 are recordings of CF-evoked AMPAR-mediated fast EPSCs obtained before and after CF tetanization for 30 s at 5 Hz. Tetanization induced a long-lasting decrease in EPSC amplitude, and this effect could not be observed in the presence of 50 mm EtOH (Fig. 6A,B).

EtOH inhibits CF LTD. A, AMPAR-mediated EPSCs evoked by CF stimulation during baseline (solid line) and 20 min after tetanization (Post 20′; dotted line; 5 Hz for 30 s). Note that tetanization induced LTD of the amplitude of these events under control conditions (left). Traces from a different PN illustrating that LTD could not be induced in presence of 50 mm EtOH are shown on the right. B, CF EPSC amplitude changes (percentage) in control and EtOH-exposed neurons (n = 6). The duration of the EtOH exposure is represented by the bar. The time of tetanization is represented by the arrow. Error bars represent SEM.
Discussion
We report here that EtOH potently modulates CF→PN synapses even at a concentration of 10 mm, placing them among a select group that is significantly affected by levels near the legal intoxication limit in the United States (17 mm). Examples of synapses belonging to this group include those in the cerebellar granule cell layer (Carta et al., 2004; Hanchar et al., 2005), CA1 and CA3 hippocampal regions (Carta et al., 2003; Galindo et al., 2005), and amygdala (Roberto et al., 2003).
EtOH does not modulate the initial phase of the CS
The fast voltage-gated Na+ channel-mediated spike was unaffected by EtOH, in agreement with the literature indicating that these channels are insensitive to subanesthetic EtOH concentrations (Shiraishi and Harris, 2004). This finding also indicates that EtOH does not affect AMPARs, which are responsible for initiating the CS (Schmolesky et al., 2002). This was confirmed by the finding that EtOH does not decrease the amplitude of CF AMPAR-mediated EPSCs. These results are in agreement with those from other studies indicating that AMPARs are relatively insensitive to EtOH in the hippocampus (Lovinger et al., 1989, 1990; Weiner et al., 1999; Carta et al., 2003; Hendricson et al., 2003) and cerebellum (Carta et al., 2004). However, hippocampal AMPARs can be very sensitive to EtOH under some circumstances, such as early in development (Lu and Yeh, 1999; Moykkynen et al., 2003; Mameli et al., 2005b), and this could also be the case at developing CF→PN synapses.
EtOH does not affect the Ca2+ transients associated with the CS
EtOH significantly reduced the late phase of the CS, and based on the literature (Llinas and Sugimori, 1980a,b; Schmolesky et al., 2002), we hypothesized that it might act by inhibiting VGCCs. This hypothesis was not supported by our finding that EtOH did not alter Ca2+ transients associated with the CS. Although the identity of the VGCCs involved in CS generation is currently unknown, dendritic P/Q-type and T-type channels have been implicated in this process (Schmolesky et al., 2002). Thus, our findings suggest that these channels are insensitive to EtOH in PNs. In agreement with this suggestion, P-type channels were shown to be insensitive to EtOH concentrations as high as 200 mm in acutely dissociated rat cerebellar PNs (Hall et al., 1994; Walter and Messing, 1999). However, the effect of EtOH on T-type channel function in PNs has not been tested, and we cannot eliminate the possibility that EtOH modulates these channels in light of the recent demonstration that it potently regulates them in thalamic neurons (Mu et al., 2003). Voltage-clamp electrophysiological studies, which have faster temporal resolution than our Ca2+ imaging studies, may reveal effects of ethanol on VGCCs in PNs.
mGluR1-dependent EPSCs contribute to CS generation and are targets of EtOH
Our finding that LY367385 decreases the late phase of the CS suggests that mGluR1 is involved in the generation of this response. This is consistent with the slow timing of the mGluR1-mediated EPSC (Dzubay and Otis, 2002). It was generally believed that voltage-gated Na+ and Ca2+ channel-mediated currents were the principal components of the slow phase of this response (Schmolesky et al., 2002). Our findings indicate that the late portion of this slow phase is mediated in part by mGluR1, which has been shown to be physically coupled to the type-1 transient receptor potential cation (TRPC1) channel that ultimately mediates the EPSC in PNs (Kim et al., 2003). Future experiments will be required to clarify the precise mechanism by which TRPC1 channels mediate the slow components of the CS.
Three pieces of evidence suggest that EtOH inhibits the slow phase of the CS in an mGluR1-dependent manner. First, the effect of EtOH is mimicked by the mGluR1 antagonist LY367385. Second, this antagonist occludes the effect of EtOH. Third, EtOH inhibits the mGluR1-dependent EPSC. The EtOH-induced inhibition of the mGluR1-dependent EPSC is not significantly different in the presence or absence of TBOA, indicating that it is not the result of an indirect action of EtOH at glutamate transporters. The lack of an effect of EtOH on the PPR suggests that its mechanism of action does not involve a decrease in glutamate release probability. Therefore, we hypothesize that EtOH acts at the level of the mGluR1 receptor itself or the intracellular signaling pathways that it activates. Netzeband and Gruol (1995) found that, in cerebellar PNs cultured for 21–37 d, EtOH (33 mm) had little effect on changes in firing rates evoked by the nonselective mGluR agonist (1S,3R)-1-aminocyclopentane-1,3-dicarboxylic acid (ACPD). At 66 mm, it increased the duration of the ACPD-induced response. Subsequently, Gruol et al. (1997) reported that acute EtOH exposure (33–66 mm) had little or no effect on Ca2+ signals to ACPD and the mGluR1/5 agonist (R,S)-3,5-dihydroxyphenylglycine (DHPG). Interestingly, lower concentrations of EtOH (10 mm) significantly enhanced the Ca2+ signals to DHPG. Based on the results of these studies and those of our study, it cannot be discerned whether EtOH modulates PN function via direct modulation of mGluRs or a downstream indirect mechanism. However, a downstream effect of EtOH is more likely, given that mGluR1-driven Ca2+-dependent Cl− currents were found to be insensitive to 10–100 mm EtOH in Xenopus oocytes (Minami et al., 1998). Recent reports indicate that members of the TRP family, such as TRPV1 and TRPM8, are modulated by EtOH and that this may be in part responsible for EtOH-induced sensory and inflammatory responses in the skin, digestive, and respiratory tracts (Trevisani et al., 2002, 2004; Lyall et al., 2005a,b). Thus, EtOH might affect the mGluR1-dependent EPSC by altering TRPC1 channel function or coupling of this channel with the mGluR1 receptor–Gq–homer complex (Yuan et al., 2003).
Given that PN TRPC1 channels are Ca2+ permeable (Dzubay and Otis, 2002; Canepari et al., 2004), it might have been expected for EtOH to reduce the CS-associated Ca2+ transient. However, evidence indicates that VGCCs, as well as Ca2+ internal stores, are the predominant sources for these transients (for review, see Schmolesky et al., 2002). The large contribution of these sources likely masks that of TRPC channels, which is expected to be relatively small.
Acute EtOH exposure inhibits CF LTD
Hansel and Linden (2000) demonstrated that antagonism of group I mGluRs blocks CF LTD. Therefore, the effect of EtOH on CF LTD is likely to be a consequence of its inhibitory effect on mGluR1-dependent signaling. Our findings are in general agreement with those of Overstreet et al. (1997), who reported that 10 mm EtOH inhibits LTD induced by the nonselective mGluR agonist ACPD in CA1 pyramidal hippocampal neurons from immature rats.
The EtOH-induced inhibition of CF LTD could have an impact at different levels. First, this form of LTD controls the direction of plasticity at parallel fiber→PN synapses. CF-dependent Ca2+ elevations induce parallel fiber LTD, which has a higher Ca2+ threshold than parallel fiber long-term potentiation (LTP) (Coesmans et al., 2004). CF LTD is associated with decreased dendritic Ca2+ transients in PNs, and this increases the probability of parallel fiber LTP induction (Weber et al., 2003; Coesmans et al., 2004). By inhibiting CF LTD, EtOH could increase the probability of parallel fiber LTD induction. Second, CF LTD might be a neuroprotective mechanism that prevents excessive Ca2+ elevations in PNs (Hansel and Linden, 2000). The inhibitory effects of EtOH on this mechanism could trigger Purkinje cell loss in alcoholics (Sullivan et al., 2003). Third, the associative interaction between parallel and climbing fibers enhances endocannabinoid-mediated retrograde inhibition of parallel fiber→PN synapses that lasts for several seconds (Brenowitz and Regehr, 2005). Previous CF LTD induction is likely to modulate this short-term form of synaptic plasticity, and EtOH might affect this regulatory process, leading to alterations in ongoing motor activity and fine motor control.
Functional implications
CF input to Purkinje cells controls simple spike activity driven by mossy fiber–parallel fiber input or an intrinsic spike generator (Cerminara and Rawson, 2004). CF input produces a pause in simple spike activity, which could be impacted both by the EtOH-induced decrease in CS area that is shown here and by the increase in CF bursts that was demonstrated previously by Rogers et al. (1980, 1986). Moreover, not all individual spikelets in the CS are propagated down the PN axon (Khaliq and Raman, 2005; Monsivais et al., 2005). On average, only two spikes, corresponding to the fast Na+ spike and a late spikelet occurring at an interspike interval >4 ms, are propagated to deep cerebellar nuclei (Monsivais et al., 2005). This axonal doublet might relay important information to and/or control the firing properties of deep cerebellar nuclear neurons. Given that EtOH alters the number of secondary spikelets in some cases (Fig. 1B1), it is possible that CF axonal propagation is altered by this agent.
Although the physiological relevance of changes in axonal firing or synaptic strength triggered by the CS is presently unknown, it has been shown that CF input to PNs participates both in movement generation and refinement (Kitazawa et al., 1998). The data presented here indicate that the powerful CF input is diminished by EtOH and this could contribute to the motor impairing actions of this widely abused substance.
Footnotes
*M.C. and M.M. contributed equally to this work.
M. Carta’s present address: Department of Experimental Biology, Section of Neuroscience, University of Cagliari, 09123 Cagliari, Italy.
This work was supported by National Institutes of Health Grant AA014973. We thank L. D. Partridge for critically reading this manuscript and assistance with calcium imaging studies. We are also grateful to W. Muller and J. A. Connor for invaluable comments.
References
- Autti-Ramo I, Autti T, Korkman M, Kettunen S, Salonen O, Valanne L (2002). MRI findings in children with school problems who had been exposed prenatally to alcohol. Dev Med Child Neurol 44:98–106. [DOI] [PubMed] [Google Scholar]
- Basile A, Hoffer B, Dunwiddie T (1983). Differential sensitivity of cerebellar Purkinje neurons to ethanol in selectively outbred lines of mice: maintenance in vitro independent of synaptic transmission. Brain Res 264:69–78. [DOI] [PubMed] [Google Scholar]
- Brenowitz SD, Regehr WG (2005). Associative short-term synaptic plasticity mediated by endocannabinoids. Neuron 45:419–431. [DOI] [PubMed] [Google Scholar]
- Canepari M, Auger C, Ogden D (2004). Ca2+ ion permeability and single-channel properties of the metabotropic slow EPSC of rat Purkinje neurons. J Neurosci 24:3563–3573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carta M, Ariwodola OJ, Weiner JL, Valenzuela CF (2003). Alcohol potently inhibits the kainate receptor-dependent excitatory drive of hippocampal interneurons. Proc Natl Acad Sci USA 100:6813–6818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carta M, Mameli M, Valenzuela CF (2004). Alcohol enhances GABAergic transmission to cerebellar granule cells via an increase in Golgi cell excitability. J Neurosci 24:3746–3751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerminara NL, Rawson JA (2004). Evidence that climbing fibers control an intrinsic spike generator in cerebellar Purkinje cells. J Neurosci 24:4510–4517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coesmans M, Weber JT, De Zeeuw CI, Hansel C (2004). Bidirectional parallel fiber plasticity in the cerebellum under climbing fiber control. Neuron 44:691–700. [DOI] [PubMed] [Google Scholar]
- Coffin JM, Baroody S, Schneider K, O’Neill J (2005). Impaired cerebellar learning in children with prenatal alcohol exposure: a comparative study of eyeblink conditioning in children with ADHD and dyslexia. Cortex 41:389–398. [DOI] [PubMed] [Google Scholar]
- Criswell HE, Breese GR (2005). A conceptualization of integrated actions of ethanol contributing to its GABAmimetic profile: a commentary. Neuropsychopharmacology 30:1407–1425. [DOI] [PubMed] [Google Scholar]
- Diamond I, Messing RO (1994). Neurologic effects of alcoholism. West J Med 161:279–287. [PMC free article] [PubMed] [Google Scholar]
- Dinh-Zarr T, Goss C, Heitman E, Roberts I, DiGuiseppi C (2004). Interventions for preventing injuries in problem drinkers. Cochrane Database Syst Rev CD001857. [DOI] [PMC free article] [PubMed]
- Dzubay JA, Otis TS (2002). Climbing fiber activation of metabotropic glutamate receptors on cerebellar Purkinje neurons. Neuron 36:1159–1167. [DOI] [PubMed] [Google Scholar]
- Freund RK, Palmer MR (1997). Beta adrenergic sensitization of gamma-aminobutyric acid receptors to ethanol involves a cyclic AMP/protein kinase A second-messenger mechanism. J Pharmacol Exp Ther 280:1192–1200. [PubMed] [Google Scholar]
- Galindo R, Zamudio P, Valenzuela C (2005). Alcohol is a potent stimulant of immature neuronal networks: implications for fetal alcohol spectrum disorder. J Neurochem 94:1500–1511. [DOI] [PubMed] [Google Scholar]
- Gruol DL, Parsons KL, DiJulio N (1997). Acute ethanol alters calcium signals elicited by glutamate receptor agonists and K+ depolarization in cultured cerebellar Purkinje neurons. Brain Res 773:82–89. [DOI] [PubMed] [Google Scholar]
- Hall AC, Lieb WR, Franks NP (1994). Insensitivity of P-type calcium channels to inhalational and intravenous general anesthetics. Anesthesiology 81:117–123. [DOI] [PubMed] [Google Scholar]
- Hanchar HJ, Dodson PD, Olsen RW, Otis TS, Wallner M (2005). Alcohol-induced motor impairment caused by increased extrasynaptic GABAA receptor activity. Nat Neurosci 8:339–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansel C, Linden DJ (2000). Long-term depression of the cerebellar climbing fiber–Purkinje neuron synapse. Neuron 26:473–482. [DOI] [PubMed] [Google Scholar]
- Hansel C, Linden DJ, D’Angelo E (2001). Beyond parallel fiber LTD: the diversity of synaptic and non-synaptic plasticity in the cerebellum. Nat Neurosci 4:467–475. [DOI] [PubMed] [Google Scholar]
- Hendricson AW, Thomas MP, Lippmann MJ, Morrisett RA (2003). Suppression of L-type voltage-gated calcium channel-dependent synaptic plasticity by ethanol: analysis of miniature synaptic currents and dendritic calcium transients. J Pharmacol Exp Ther 307:550–558. [DOI] [PubMed] [Google Scholar]
- Hernandez LL, Valentine JD, Powell DA (1986). Ethanol enhancement of Pavlovian conditioning. Behav Neurosci 100:494–503. [DOI] [PubMed] [Google Scholar]
- Khaliq ZM, Raman IM (2005). Axonal propagation of simple and complex spikes in cerebellar Purkinje neurons. J Neurosci 25:454–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SJ, Kim YS, Yuan JP, Petralia RS, Worley PF, Linden DJ (2003). Activation of the TRPC1 cation channel by metabotropic glutamate receptor mGluR1. Nature 426:285–291. [DOI] [PubMed] [Google Scholar]
- Kitazawa S, Kimura T, Yin PB (1998). Cerebellar complex spikes encode both destinations and errors in arm movements. Nature 392:494–497. [DOI] [PubMed] [Google Scholar]
- Klintsova AY, Cowell RM, Swain RA, Napper RM, Goodlett CR, Greenough WT (1998). Therapeutic effects of complex motor training on motor performance deficits induced by neonatal binge-like alcohol exposure in rats. I. Behavioral results. Brain Res 800:48–61. [DOI] [PubMed] [Google Scholar]
- Klintsova AY, Scamra C, Hoffman M, Napper RM, Goodlett CR, Greenough WT (2002). Therapeutic effects of complex motor training on motor performance deficits induced by neonatal binge-like alcohol exposure in rats: II. A quantitative stereological study of synaptic plasticity in female rat cerebellum. Brain Res 937:83–93. [DOI] [PubMed] [Google Scholar]
- Lee RS, Smith SS, Chapin JK, Shimizu N, Waterhouse BD, Maddus BN, Woodward DJ (1995). Effects of systemic and local ethanol on responses of rat cerebellar Purkinje neurons to iontophoretically applied norepinephrine and gamma-aminobutyric acid. Brain Res 687:12–21. [DOI] [PubMed] [Google Scholar]
- Lin AM, Freund RK, Hoffer BJ, Palmer MR (1994). Ethanol-induced depressions of cerebellar Purkinje neurons are potentiated by beta-adrenergic mechanisms in rat brain. J Pharmacol Exp Ther 271:1175–1180. [PubMed] [Google Scholar]
- Llinas R, Sugimori M (1980a). Electrophysiological properties of in vitro Purkinje cell somata in mammalian cerebellar slices. J Physiol (Lond) 305:171–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llinas R, Sugimori M (1980b). Electrophysiological properties of in vitro Purkinje cell dendrites in mammalian cerebellar slices. J Physiol (Lond) 305:197–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovinger DM, White G, Weight FF (1989). Ethanol inhibits NMDA-activated ion current in hippocampal neurons. Science 243:1721–1724. [DOI] [PubMed] [Google Scholar]
- Lovinger DM, White G, Weight FF (1990). NMDA receptor-mediated synaptic excitation selectively inhibited by ethanol in hippocampal slice from adult rat. J Neurosci 10:1372–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu SM, Yeh HH (1999). Ethanol modulates AMPA-induced current responses of primary somatosensory cortical neurons. Neurochem Int 35:175–183. [DOI] [PubMed] [Google Scholar]
- Lyall V, Heck GL, Phan TH, Mummalaneni S, Malik SA, Vinnikova AK, DeSimone JA (2005a). Ethanol modulates the VR-1 variant amiloride-insensitive salt taste receptor. I. Effect on TRC volume and Na+ flux. J Gen Physiol 125:569–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyall V, Heck GL, Phan TH, Mummalaneni S, Malik SA, Vinnikova AK, Desimone JA (2005b). Ethanol modulates the VR-1 variant amiloride-insensitive salt taste receptor. II. Effect on chorda tympani salt responses. J Gen Physiol 125:587–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mameli M, Zamudio P, Carta M, Valenzuela C (2005a). Effect of ethanol on GABAergic transmission in cerebellar Purkinje neurons. Soc Neurosci Abstr 31:955–2. [Google Scholar]
- Mameli M, Zamudio PA, Carta M, Valenzuela CF (2005b). Developmentally regulated actions of alcohol on hippocampal glutamatergic transmission. J Neurosci 25:8027–8036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzardo AM, Penick EC, Knop J, Nickel EJ, Hall S, Jensen P, Gabrielli WF Jr. (2005). Developmental differences in childhood motor coordination predict adult alcohol dependence: proposed role for the cerebellum in alcoholism. Alcohol Clin Exp Res 29:353–357. [DOI] [PubMed] [Google Scholar]
- Minami K, Gereau RW, Minami M, Heinemann SF, Harris RA (1998). Effects of ethanol and anesthetics on type 1 and 5 metabotropic glutamate receptors expressed in Xenopus laevis oocytes. Mol Pharmacol 53:148–156. [DOI] [PubMed] [Google Scholar]
- Miyakawa T, Yagi T, Kitazawa H, Yasuda M, Kawai N, Tsuboi K, Niki H (1997). Fyn-kinase as a determinant of ethanol sensitivity: relation to NMDA-receptor function. Science 278:698–701. [DOI] [PubMed] [Google Scholar]
- Monsivais P, Clark BA, Roth A, Hausser M (2005). Determinants of action potential propagation in cerebellar Purkinje cell axons. J Neurosci 25:464–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moykkynen T, Korpi ER, Lovinger DM (2003). Ethanol inhibits alpha-amino-3-hydyroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor function in central nervous system neurons by stabilizing desensitization. J Pharmacol Exp Ther 306:546–555. [DOI] [PubMed] [Google Scholar]
- Mu J, Carden WB, Kurukulasuriya NC, Alexander GM, Godwin DW (2003). Ethanol influences on native T-type calcium current in thalamic sleep circuitry. J Pharmacol Exp Ther 307:197–204. [DOI] [PubMed] [Google Scholar]
- Netzeband JG, Gruol DL (1995). Modulatory effects of acute ethanol on metabotropic glutamate responses in cultured Purkinje neurons. Brain Res 688:105–113. [DOI] [PubMed] [Google Scholar]
- Overstreet LS, Pasternak JF, Colley PA, Slater NT, Trommer BL (1997). Metabotropic glutamate receptor mediated long-term depression in developing hippocampus. Neuropharmacology 36:831–844. [DOI] [PubMed] [Google Scholar]
- Roberto M, Madamba SG, Moore SD, Tallent MK, Siggins GR (2003). Ethanol increases GABAergic transmission at both pre- and postsynaptic sites in rat central amygdala neurons. Proc Natl Acad Sci USA 100:2053–2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers J, Siggins GR, Schulman JA, Bloom FE (1980). Physiological correlates of ethanol intoxication tolerance, and dependence in rat cerebellar Purkinje cells. Brain Res 196:183–198. [DOI] [PubMed] [Google Scholar]
- Rogers J, Madamba SG, Staunton DA, Siggins GR (1986). Ethanol increases single unit activity in the inferior olivary nucleus. Brain Res 385:253–262. [DOI] [PubMed] [Google Scholar]
- Schmolesky MT, Weber JT, De Zeeuw CI, Hansel C (2002). The making of a complex spike: ionic composition and plasticity. Ann NY Acad Sci 978:359–390. [DOI] [PubMed] [Google Scholar]
- Schuckit MA (1994). Low level of response to alcohol as a predictor of future alcoholism. Am J Psychiatry 151:184–189. [DOI] [PubMed] [Google Scholar]
- Shiraishi M, Harris RA (2004). Effects of alcohols and anesthetics on recombinant voltage-gated Na+ channels. J Pharmacol Exp Ther 309:987–994. [DOI] [PubMed] [Google Scholar]
- Siggins GR, French E (1979). Central neurons are depressed by iontophoretic and micropressure application of ethanol and tetrahydropapaveroline. Drug Alcohol Depend 4:239–243. [DOI] [PubMed] [Google Scholar]
- Sorensen S, Palmer M, Dunwiddie T, Hoffer B (1980). Electrophysiological correlates of ethanol-induced sedation in differentially sensitive lines of mice. Science 210:1143–1145. [DOI] [PubMed] [Google Scholar]
- Sullivan EV, Harding AJ, Pentney R, Dlugos C, Martin PR, Parks MH, Desmond JE, Chen SH, Pryor MR, De Rosa E, Pfefferbaum A (2003). Disruption of frontocerebellar circuitry and function in alcoholism. Alcohol Clin Exp Res 27:301–309. [DOI] [PubMed] [Google Scholar]
- Trevisani M, Smart D, Gunthorpe MJ, Tognetto M, Barbieri M, Campi B, Amadesi S, Gray J, Jerman JC, Brough SJ, Owen D, Smith GD, Randall AD, Harrison S, Bianchi A, Davis JB, Geppetti P (2002). Ethanol elicits and potentiates nociceptor responses via the vanilloid receptor-1. Nat Neurosci 5:546–551. [DOI] [PubMed] [Google Scholar]
- Trevisani M, Gazzieri D, Benvenuti F, Campi B, Dinh QT, Groneberg DA, Rigoni M, Emonds-Alt X, Creminon C, Fischer A, Geppetti P, Harrison S (2004). Ethanol causes inflammation in the airways by a neurogenic and TRPV1-dependent mechanism. J Pharmacol Exp Ther 309:1167–1173. [DOI] [PubMed] [Google Scholar]
- Valenzuela CF, Carta M, Mameli M (2005). Letter to the editor. Alcohol Clin Exp Res 29:1356–1357. [DOI] [PubMed] [Google Scholar]
- Walter HJ, Messing RO (1999). Regulation of neuronal voltage-gated calcium channels by ethanol. Neurochem Int 35:95–101. [DOI] [PubMed] [Google Scholar]
- Weber JT, De Zeeuw CI, Linden DJ, Hansel C (2003). Long-term depression of climbing fiber-evoked calcium transients in Purkinje cell dendrites. Proc Natl Acad Sci USA 100:2878–2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner JL, Dunwiddie TV, Valenzuela CF (1999). Ethanol inhibition of synaptically evoked kainate responses in rat hippocampal CA3 pyramidal neurons. Mol Pharmacol 56:85–90. [DOI] [PubMed] [Google Scholar]
- Yang X, Criswell HE, Breese GR (2000). Ethanol modulation of gamma-aminobutyric acid (GABA)-mediated inhibition of cerebellar Purkinje neurons: relationship to GABAb receptor input. Alcohol Clin Exp Res 24:682–690. [PubMed] [Google Scholar]
- Yuan JP, Kiselyov K, Shin DM, Chen J, Shcheynikov N, Kang SH, Dehoff MH, Schwarz MK, Seeburg PH, Muallem S, Worley PF (2003). Homer binds TRPC family channels and is required for gating of TRPC1 by IP3 receptors. Cell 114:777–789. [DOI] [PubMed] [Google Scholar]
- Zhu L, Strata P, Andjus PR (2005). Pharmacology of the metabotropic glutamate receptor mediated current at the climbing fiber to Purkinje cell synapse. Prog Brain Res 148:299–306. [DOI] [PubMed] [Google Scholar]